

ABSTRACT
Combined pre−/postcapillary pulmonary hypertension (Cpc‐PH), a complication of left heart failure, is associated with higher mortality rates than isolated postcapillary pulmonary hypertension alone. Currently, knowledge gaps persist on the mechanisms responsible for the progression of isolated postcapillary pulmonary hypertension (Ipc‐PH) to Cpc‐PH. Here, we review the biomechanical and mechanobiological impact of left heart failure on pulmonary circulation, including mechanotransduction of these pathological forces, which lead to altered biological signaling and detrimental remodeling, driving the progression to Cpc‐PH. We focus on pathologically increased cyclic stretch and decreased wall shear stress; mechanotransduction by endothelial cells, smooth muscle cells, and pulmonary arterial fibroblasts; and signaling‐stimulated remodeling of the pulmonary veins, capillaries, and arteries that propel the transition from Ipc‐PH to Cpc‐PH. Identifying biomechanical and mechanobiological mechanisms of Cpc‐PH progression may highlight potential pharmacologic avenues to prevent right heart failure and subsequent mortality.
Keywords: biomechanics, mechanotransduction, pulmonary hypertension due to left heart failure, pulmonary vascular remodeling
Subject Categories: Pulmonary Hypertension, Heart Failure, Remodeling, Valvular Heart Disease
Nonstandard Abbreviations and Acronyms
- BAECs
bovine arterial endothelial cells
- Cpc‐PH
combined pre−/postcapillary pulmonary hypertension
- dPAP
diastolic pulmonary artery pressure
- EC
endothelial cell
- HUVEC
human umbilical vein endothelial cell
- Ipc‐PH
isolated postcapillary pulmonary hypertension
- LAP
left atrial pressure
- LHF
left heart failure
- mPAP
mean pulmonary artery pressure
- PAC
pulmonary artery compliance
- PAEC
pulmonary artery endothelial cell
- PAH
pulmonary arterial hypertension
- PASMC
pulmonary artery smooth muscle cell
- PH‐LHF
pulmonary hypertension‐left heart failure
- PP
pulse pressure
- PVR
pulmonary vascular resistance
- sPAP
systolic pulmonary artery pressure
- TPG
transpulmonary pressure gradient
- WSS
wall shear stress
Left heart failure (LHF) impacts nearly 5.9 million adults and contributes to 1 out of every 9 deaths in the United States. 1 Pulmonary hypertension (PH) occurs in 36% to 83% of those with LHF (PH‐LHF) 2 and dramatically increases morbidity and mortality. 3 , 4 PH‐LHF begins as a passive process termed isolated postcapillary PH (Ipc‐PH), diagnosed by elevated mean pulmonary artery pressure (mPAP) with normal pulmonary vascular resistance (PVR). Mortality significantly increases once Ipc‐PH transitions to combined pre−/postcapillary PH (Cpc‐PH), with increased PVR, which also typically marks a change from reversible to irreversible disease. 5 Although genetic, environmental, and metabolic factors likely impact disease progression in individual patients, biomechanical forces and mechanobiological signaling may be common drivers of this key pathophysiological transition.
Vascular biomechanical forces such as cyclic stretch, which acts on all cells in the vascular wall, and shear stress, which acts on the cells that line the lumen of the vascular wall, are transduced into biological signals in a process termed mechanotransduction. Mechanotransduction pathways contribute to the pathophysiology of cardiovascular diseases including atherosclerosis, arteriovenous malformations, and type 2 diabetes, among others. 6 , 7 , 8 , 9 Specifically, mechanotransduction induces biological signals that drive vascular remodeling including hypertrophy, hyperplasia, apoptosis, and extracellular matrix (ECM) synthesis and degradation. 10 This remodeling in turn alters the mechanical function of the vessels. 11 For example, increased collagen synthesis (and less degradation) in the vessel wall will decrease vessel compliance and pulse wave dampening. 12 Within a less compliant vessel, cells stretch less with each pressure pulse, which alters the biomechanical forces on those cells. Although this self‐perpetuating process—biomechanical forces transduced into biological signals that cause vascular remodeling, which in turn change biomechanical forces—can be homeostatic and adaptive, it can also be maladaptive, especially for tissues upstream and downstream. In PH caused by LHF, the ultimate outcome is right heart failure (Figure 1).
Figure 1. Mechanotransduction is the key step in the pathophysiologic progression of pulmonary vascular disease in the setting of left heart failure.
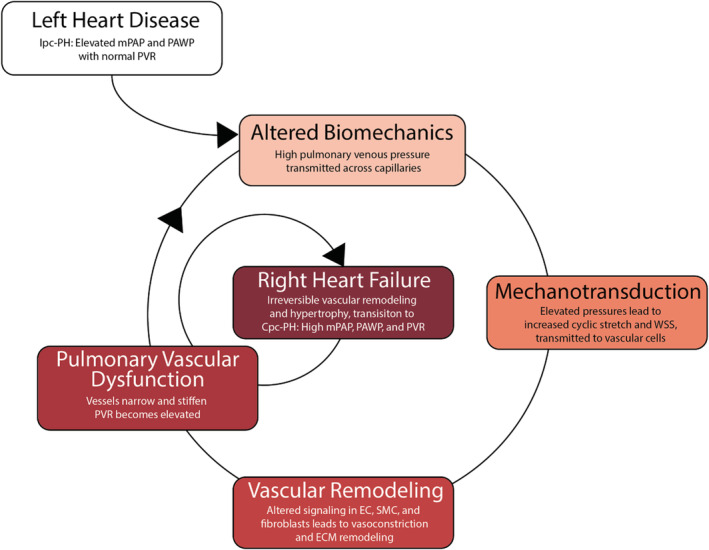
The transition from isolated postcapillary pulmonary hypertension to combined pre−/postcapillary pulmonary hypertension is characterized by pulmonary vascular remodeling and results in right heart failure. Cpc‐PH, combined pre−/postcapillary pulmonary hypertension; EC, endothelial cell; Ipc‐PH, isolated postcapillary pulmonary hypertension; mPAP, mean pulmonary artery pressure; PAWP, pulmonary artery wedge pressure; PVR, pulmonary vascular resistance; SMC, smooth muscle cell; and WSS, wall shear stress.
This review examines the known and suspected roles of LHF‐induced hemodynamic changes in altering 2 key vascular biomechanical forces: cyclic stretch and wall shear stress (WSS), which activate mechanotransduction pathways; drive pulmonary venous, capillary, and arterial remodeling; and characterize the transition from Ipc‐PH to Cpc‐PH and subsequent right heart failure.
Clinical Definitions of Ipc‐PH and Cpc‐PH
As left ventricular function declines, left ventricular filling pressures rise, inducing a concomitant elevation in left atrial pressure (LAP). The passive transmission of elevated LAP into the pulmonary veins is characteristic of Ipc‐PH. Increased pulmonary venous pressures are transmitted across the capillaries and arteries in a 1‐to‐1 fashion such that the rise in mPAP is proportional to the rise in LAP. The 2022 European Society of Cardiology/European Respiratory Society Guidelines for the Diagnosis and Treatment of Pulmonary Hypertension defined Ipc‐PH as mPAP >20 mm Hg, pulmonary artery wedge pressure >15 mm Hg with PVR <2 Woods units. 13 As the disease progresses, pulmonary vasoconstriction contributes to a >1‐to‐1 rise in mPAP. 14 In this reactive phase, the increase in mPAP can be reversed with adequate left ventricular afterload reduction. 15
Twelve percent to 38% of patients with PH‐LHF progress through this reversible reactive phase of Ipc‐PH to irreversible Cpc‐PH. 15 The clinical definition of Cpc‐PH is mPAP >20 mm Hg, pulmonary artery wedge pressure >15 mm Hg, and PVR >2 Woods units. 13 The elevation in PVR in Cpc‐PH is associated with a greater risk of right heart failure and mortality in comparison with patients with Ipc‐PH alone. 15 Postmortem studies have demonstrated that increased PVR is associated with pulmonary vascular remodeling, including pulmonary venous and arterial medial hypertrophy, diffuse lung intimal fibrosis, and distal arterial luminal occlusion. 16
In addition to increasing PVR, Cpc‐PH is associated with decreased pulmonary arterial compliance (PAC), which is calculated as the pulse pressure (systolic pulmonary artery pressure minus diastolic pulmonary artery pressure) divided by stroke volume and represents the ability of the pulmonary arterial compartment to absorb and dampen hemodynamic pulsatility. 17 Large clinical studies have found that PAC is more predictive of mortality than mPAP or PVR in PH‐LHF. 18 , 19 , 20 Importantly, decreased PAC has consequences distinct from increased PVR on the mechanical forces imposed on the upstream right ventricle (RV) and downstream pulmonary capillaries via altered pulse wave reflection and transmission, respectively. 17
Biomechanics: The Missing Link
The initial insult to the pulmonary vasculature in Ipc‐PH is increased pulmonary venous, capillary, and arterial pressure. The impact of increased pressure on the 2 key vascular biomechanical forces, cyclic stretch and wall shear stress, in the pulmonary vasculature depends, in part, on the mechanical properties of the pulmonary vasculature. Because each compartment in the pulmonary vasculature, namely arteries, capillaries, and veins, has a different structure, 21 the cells in each compartment will be exposed to different biomechanical stimuli.
Healthy pulmonary arteries are rich in elastin and have concentric layers of smooth muscle cells (SMCs), yielding a highly compliant structure. 22 They are populated with pulmonary arterial fibroblasts in the adventitia, which are responsible for ECM remodeling. As the distance from the main pulmonary artery (PA) increases, the relative amount of ECM protein decreases, and the percentage of SMC increases, reaching a maximum at the arterioles. 23 The capillary system is a vast but fragile network consisting only of endothelial cells (ECs) and ECM. Although thin‐walled and lacking in SMCs and elastic fibers, the capillaries derive tensile strength from type IV collagen. 24 The pulmonary venules consist of elastic fibers and connective tissue, with minimal SMCs. As pulmonary venules become veins approaching the left atrium, the number of SMCs and amount of elastin increases. 25 Overall, pulmonary veins are less compliant than pulmonary arteries. 26
Cyclic stretch occurs in a compliant vessel in which pressure is pulsatile; it is defined as the change in diameter from systole to diastole divided by the diameter at diastole (Figure 2). Thus, increased pulse pressure (PP) (defined as systolic pressure minus diastolic pressure) can drive increases in cyclic stretch. In the pulmonary vasculature, PP is highest in large proximal arteries and drops precipitously at the pulmonary arterioles. 27 Although difficult to measure, PP (ie, pulsatility) in the capillaries in a healthy state is thought to be minimal; pulsatility in the pulmonary veins is also low. Therefore, cyclic stretch in the capillaries and veins in the healthy state is likely negligible.
Figure 2. Schematic representation of wall shear stress (WSS) and cyclic stretch in a vessel.
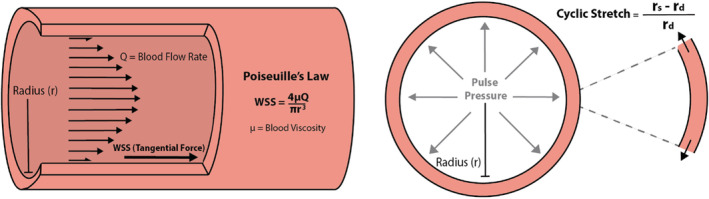
WSS is directly proportional to blood flow rate (Q) and blood viscosity (μ) and is inversely proportional to the radius of the vessel lumen (r). Cyclic stretch is the difference between lumen radius at systole (rs) and the lumen radius at diastole (rd) normalized by the radius at diastole.
As noted above, with Ipc‐PH, increased LAP is transmitted from the veins across the capillaries to arteries in a 1‐to‐1 fashion, such that the rise in mPAP is proportional to the rise in LAP. For no change in cardiac output or PVR, the increase in mPAP is equal to the increase in LAP. How this affects cyclic stretch in the 3 compartments is not precisely known. A common feature of all vessels is nonlinear compliance; arteries, capillaries, and veins are more compliant at low pressures than at high pressures. 28 Uniquely, in the pulmonary circulation, there is a linear relationship between mPAP and PP, such that as mPAP increases, PP increases proportionally. 29 Thus, increased mean pressure decreases compliance and increases PP, which increases cyclic stretch on vascular cells. 30 In 2000, West hypothesized that high mPAP injures pulmonary capillaries and leads to stress failure 31 ; alternatively, excessive cyclic stretch could be the mechanism. Whether this putatively increased pulsatility is then transmitted downstream of capillaries to pulmonary veins is unknown. In sum, the cyclic stretch imposed on cells in the pulmonary vasculature depends on PP (which itself depends on mean pressure), and vessel wall structure, both of which depend on compartment (artery, capillary, vein) and distance from the heart.
WSS in the pulmonary vasculature also depends on the compartment and distance from the heart. On the basis of Poiseuille's law, WSS is proportional to blood flow velocity and blood viscosity, and inversely proportional to the lumen radius 32 (Figure 2). The branching pattern of the pulmonary arteries and veins is thought to keep time‐averaged WSS relatively constant with distance from the heart, because flow rate decreases in proportion to radius cubed. 33 WSS in the capillaries is difficult to define and measure. With Ipc‐PH, increased mean pressure increases diameter, which should decrease WSS in all compartments (again, dependent on vascular compliance). Moreover, when cardiac output decreases because of LHF, blood flow velocities, and thus WSS, will decrease in all compartments. Decreased pulmonary venous systolic velocity has been found in subjects with LHF, 34 which supports decreased WSS in the pulmonary veins.
Cyclic stretch, which acts directly on ECs, SMCs, and fibroblasts in the vessel wall, and WSS, which acts directly on ECs and can have consequences for SMCs and fibroblasts, are potent mechanical stimuli for vascular remodeling. Below, we review the pathways through which these biomechanical stimuli act on each cell type and provide evidence that biomechanics and mechanobiology incite key pulmonary vascular events in the transition from Ipc‐PH to Cpc‐PH.
Mechanotransduction of Cyclic Stretch and WSS
Increased Cyclic Stretch
Increases in pulmonary pressure caused by LHF and Ipc‐PH can increase cyclic stretch, which stimulates EC signaling pathways that cause vasodilation, inflammation, and pathologic vessel remodeling through ECM turnover and SMC proliferation (Figure 3). Because both pressures and wall mechanical properties vary throughout the pulmonary vasculature, with pressures decreasing from the arteries to the capillaries and veins, and mechanical properties reflecting their differing functions, the impact of Ipc‐PH on cyclic stretch is distinct in each compartment and not entirely known. In the systemic circulation, a low magnitude cyclic stretch in the range of 5% to 10% is considered to be physiological, and a high magnitude stretch >20% is considered pathological. 8 The physiological and pathological stretch levels in the pulmonary vasculature are not well defined. In particular, the mechanical forces in PH‐LHF have not been characterized, and further investigations are needed to fully understand the abnormal biomechanical forces generated by this disease. Recent computational modeling simulations from Bartolo et al suggest that physiological cyclic stretch is 15% to 20% in the pulmonary arteries and <5% in the capillaries and veins; in the setting of Ipc‐PH (an increase in LAP from 2 to 20 mm Hg), cyclic stretch is predicted to increase to up to 60% in the arteries, 10% in the capillaries, and 40% in the veins. 35 To understand the impact of these mechanical stimuli on biological changes in each cell type in the pulmonary vasculature, below we review key findings from in vitro studies examining the impact of cyclic stretch on ECs, SMCs, and fibroblasts, with the caveat that most of these studies use nonpulmonary cell sources. Table 1 provides the subset of these study results conducted using pulmonary vascular ECs, SMCs, and fibroblasts.
Figure 3. Mechanotransduction of increased cyclic stretch because of left heart failure in endothelial cells, fibroblasts, and smooth muscle cells triggers a cascade of pathologic vascular remodeling.
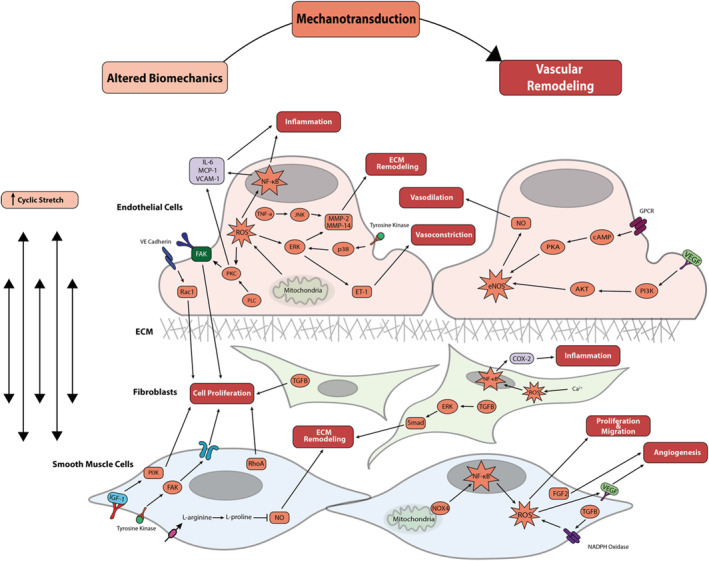
Boxes within the cell signaling cascade are colored to match their corresponding box at the top of the figure (altered biomechanics, mechanotransduction, or vascular remodeling). AKT indicates protein kinase B; cAMP indicates cyclic adenosine monophosphate; Cox‐2 indicates cyclooxygenase‐2; ECM, extracellular matrix; eNOS, endothelial nitric oxide synthase; ERK, extracellular signal‐regulated kinase; FAK, focal adhesion kinase; FGF‐2, fibroblast growth factor‐2; GPCR, G protein‐coupled receptor; IGF‐1, insulin‐like growth factor‐1; IL‐6, interleukin‐6; JNK, c‐Jun N‐terminal kinase; MCP‐1, myocyte chemoattractant protein‐1; MMP, matrix metalloproteinase; NF‐κB, nuclear factor‐κB; NOX‐4, NADPH oxidase‐4; P13K, p13 kinase; PKA, protein kinase A; PKC, protein kinase C; PLC, phospholipase C; Rac1, Ras‐related C3 botulinum toxin substrate 1; RhoA, ras homolog family member A; ROS, reactive oxygen species; TGFβ, transforming growth factor beta; TNF‐α, tumor necrosis factor‐α; VCAM‐1, vascular cell adhesion protein 1; VE cadherin, vascular endothelial cadherin; and VEGF, vascular endothelial growth factor.
Table 1.
Cyclic Stretch in Pulmonary Vascular ECs, SMCs, and Fibroblasts
| Cell type | Stimulus vs static | Mechanotransduction | Biological response | References |
|---|---|---|---|---|
| Bovine PAEC and PASMC | 20% cyclic stretch at 1 Hz for 0–24 h | VE‐cadherin– and Rac1‐dependent EC proliferation; RhoA kinase‐dependent SMC proliferation | Vessel remodeling | Liu 72 |
| Bovine PAEC | 25% cyclic stretch at 0.25 Hz for 24 h | Mitochondrial complex III‐stimulated increase in ROS leading to increased FAK activation | Angiogenesis | Ali 66 |
| Human PMVEC | 20% cyclic stretch at 0.83 Hz for 24 h | Increase of IL‐8 synthesis and release via p38 activation | Inflammation | Iwaki 63 |
| Human PMVEC | 18% cyclic stretch at 0.5 Hz for 4 d | Increased MMP‐2, 14, increased activity of tissue inhibitor of metalloproteinase‐2 | ECM remodeling | Haseneen 53 |
| Rabbit PASMC | 15% cyclic stretch at 1 Hz for 24 h | Increased tyrosine kinase phosphorylation of FAK leading to increased PDGF and PDGF‐R expression | SMC proliferation | Tanabe 74 |
| Ovine PASMC | 5%–25% cyclic stretch at 1 Hz 48 h | Increased VEGF and FGF‐2 | SMC proliferation/angiogenesis | Quinn 75 |
| Ovine PASMC | 20% cyclic stretch at 1 Hz for 8 h | Increased TGF‐β1 led to NADPH oxidase– and ROS‐dependent increase in VEGF | Angiogenesis | Mata‐Greenwood 73 |
| Ovine PASMC | 15% cyclic stretch at 1 Hz for 24 h | Increased ROS via NOX‐4 | SMC proliferation and migration | Wedgwood 77 |
| Rat PAAF | 10% equibiaxial static stretch for 24 h | Increased myofibroblast differentiation and increased Col1a1, Col3a1, Eln | Vessel remodeling | Wang 85 |
| Human lung fibroblasts | 20% cyclic stretch at 1 Hz for 30 min | Increased intracellular Ca2+, increased production of ROS leading to NF‐κB activation and increased COX‐2 | Inflammation | Amma 91 |
COX‐2 indicates cyclooxygenase‐2; EC, endothelial cell; ECM, extracellular matrix; FAK, focal adhesion kinase; FGF‐2, fibroblast growth factor‐2; IL, interleukin; MMP, matrix metalloproteinase; NF‐κB, nuclear factor‐κB; NOX‐4, NADPH oxidase‐4; PAAF, pulmonary artery adventitial fibroblast; PAEC, pulmonary artery endothelial cell; PASMC, pulmonary arterial smooth muscle cell; PDGF, platelet derived growth factor; PDGF‐R, platelet derived growth factor receptor; PMVEC, pulmonary microvascular endothelial cells; Rac1, Ras‐related C3 botulinum toxin substrate 1; RhoA, ras homolog family member A; ROS, reactive oxygen species; SMC, smooth muscle cell; TGF‐β1, transforming growth factor‐β1; VE cadherin, vascular endothelial cadherin; and VEGF, vascular endothelial growth factor.
Impact of Cyclic Stretch on ECs in Veins, Capillaries, and Arteries
High cyclic stretch on human umbilical vein endothelial cells (HUVECs) in vitro increases endothelial nitric oxide synthase (eNOS) phosphorylation through the PKA (protein kinase A) and P13K (p13 kinase)/Akt (protein kinase B) pathways. 36 , 37 Consequent vasodilation may contribute to the decreased pulmonary blood flow velocities found in LHF. 34 HUVECs subjected to pathological cyclic stretch also increase release of interleukin‐6 via NF‐κB (nuclear factor‐κB)‐dependent pathway. 38 Increased activation of the NF‐κB pathway triggered by EC cyclic stretch leads to reactive oxygen species (ROS) stress and cytokine release, resulting in inflammation. 39 , 40 Cyclic stretch–triggered ROS production has been further shown to lead to ET‐1 (endothelin‐1) production, a potent vasoconstrictor, in both HUVECs and bovine arterial endothelial cells (BAECs). 41 Cyclic stretch also triggers production of inflammatory cytokines including MCP‐1 (myocyte chemoattractant protein‐1) and interleukin‐8. 42 , 43 In addition to vasodilation and inflammation, increased cyclic stretch stimulates ECM remodeling through matrix metalloproteinase (MMP) production and activation. 44 , 45 , 46 Stretch leads to EC stiffening via cytoskeleton remodeling, characterized by increased actin fiber bundle thickness and fiber reorientation. 47 , 48 This remodeling could contribute to increased intimal thickness, which is a key histologic feature of PH‐LHF remodeling and has been demonstrated throughout the pulmonary vascular bed in human patients. 16 , 49 , 50 These findings highlight the potentially important role of EC‐stimulated remodeling in Cpc‐PH progression.
In the capillary bed, increased EC cyclic stretch has been hypothesized to cause alveolar‐capillary stress failure. 24 As first proposed by West, alveolar‐capillary stress failure is the physical disruption of the alveolar‐capillary membrane in response to elevated capillary pressure and volume. 3 Mechanical breakdown of the capillary membrane is theorized to increase permeability, stimulate remodeling, and release factors that further alter the function of the membrane. 51 Microvascular remodeling and dysfunction could be a critical link in the transition of Ipc‐PH to Cpc‐PH. Consistent with this hypothesis, pulmonary microvascular remodeling, including thickened alveolar septa and collapsed airspaces, were demonstrated in a mouse model of LHF. 52 Human lung microvascular ECs subjected to cyclic stretch demonstrated an increase in MMP‐2, leading to degradation of the basement membrane, which can result in leakage of intraluminal fluid. 53 This mechanism is consistent with increased systemic levels of MMP‐2 and MMP‐9 in subjects with heart failure with preserved ejection fraction. 54 Additionally, components of the EC cytoskeleton undergo rearrangement in response to pathologic stretch, which weakens junctional protein complexes with neighboring cells and reduces the integrity of the endothelium. 55 A rat model of LHF provided further support for the alveolar‐capillary stress failure theory, because altered capillary EC membrane permeability and cytoskeletal rearrangement were revealed as additional signs of capillary EC dysfunction. 56 These studies demonstrate that cyclic stretch stimulates ECM turnover, cytoskeleton rearrangement, and endothelial dysfunction that is similar to the irreversible pathological remodeling demonstrated in animal models of LHF and subsequent Cpc‐PH. 52 , 57 , 58 Thus, mechanotransduction of increased cyclic stretch occurring in the capillaries because of Ipc‐PH likely drives pathologic remodeling that characterizes the transition to Cpc‐PH. The overall effects of increased cyclic stretch leading to altered capillary EC function, ECM remodeling, and inflammation, which contribute to further pulmonary vascular remodeling, may be key to inciting the development of Cpc‐PH from Ipc‐PH. 59
In the arterial compartment, increased cyclic stretch similarly triggers vasodilation, inflammation, and ECM remodeling, although through some different pathways than in the venous and capillary compartments. Unlike in HUVECs, cyclic stretch applied to pulmonary artery endothelial cells (PAECs) increased proliferation and eNOS phosphorylation while reducing NO via the P13K pathway. 37 , 60 In arterial ECs, cyclic stretch activates VEGFR2 (vascular endothelial growth factor receptor 2), which leads to Src‐dependent vascular endothelial (VE)‐cadherin tyrosine phosphorylation, resulting in proliferation and migration. 61 , 62 Dynamically stretching human PAECs (20% versus 5%) for 24 hours led to time‐dependent increases in interleukin‐8 production. 63 Cyclically stretching PAECs also activates the interleukin‐6 release pathways observed in the venous cells in addition to JNK (c‐Jun N‐terminal kinase), ERK (extracellular signal‐regulated kinase), and p38 pathways unique to the arterial cells. 64 Cyclically stretching PAECs further induces the upregulation of TSP‐1 (thrombospondin‐1), which inhibits NO‐stimulated pulmonary arterial smooth muscle cell (PASMC) growth and proliferation. 65 Similar to in venous and capillary ECs, in BAECs, 10% cyclic stretch induced a 9‐fold increase in MMP‐2 compared with static culture. 46 This study identified the stimulation of p38‐ and ERK‐dependent pathways as the mechanisms responsible for the MMP increase. 46 Additional studies have demonstrated that when vascular ECs experience pathological cyclic stretch, the cytoskeleton transmits the force to the subcellular mitochondria. Dynamically stretched PAECs increase mitochondrial release of ROS and activation of protein kinase C and FAK (focal adhesion kinase), a key regulator of angiogenesis. 66 , 67 Given these findings, it is likely that the increased cyclic stretch associated with LHF contributes to intracellular mitochondrial‐driven metabolic dysfunction.
Previous reviews compiled systemic and umbilical endothelial cell response to cyclic stretch, 8 , 68 but EC characteristics depend on location. 69 Nonpulmonary cell studies present targetable pathways, which should be investigated in pulmonary EC‐specific experiments. The differing stretch‐induced vasodilation, inflammation, and remodeling pathways observed in venous, capillary, and arterial EC further motivates studies to understand the biomechanical complexity of Ipc‐PH progression to Cpc‐PH. Key knowledge gaps include mechanotransduction pathways in pulmonary specific cell lines as well as the mechanical forces imposed on these cell types in both the physiological and pathological conditions in each compartment of the pulmonary circulation.
Impact of Cyclic Stretch on SMCs in Veins and Arteries
Cyclic stretch is a key regulator of SMC function impacting gene expression and cell signaling pathways to regulate proliferation, apoptosis, and remodeling. 70 Like ECs, pulmonary SMCs respond to pathologic cyclic stretch by increasing proliferation, ECM remodeling, and inflammation (Figure 4). In the systemic venous compartment, cyclic stretch activates the IGF‐1 (insulin‐like growth factor 1) pathway to induce SMC proliferation. 71 In the pulmonary arterial compartment, 20% biaxial stretch induces a ras homolog family member A (RhoA)‐dependent increase in SMC proliferation. 72 Additionally, pulmonary arterial SMCs demonstrate stretch‐induced dysfunction through increased VEGF (vascular endothelial growth factor) expression via ROS‐dependent TGF‐β1 (transforming growth factor‐β1) signaling promoting angiogenesis and inflammation. 73 Beyond VEGF, cyclic stretch stimulates overexpression of other growth factors and their receptors in PASMCs, including PDGF (platelet‐derived growth factor) and their receptors (PDGF‐R) via phosphorylation of FAK. Overexpression of PDGF‐R is also present in rat models of pulmonary artery hypertension (PAH), indicating its likely role in pathological pulmonary vascular remodeling. 74 , 75 Dynamically stretched rat aortic vascular smooth muscle cells show significant time‐dependent increases in L‐arginine activity and transport velocity, as well as L‐arginine‐dependent products such as L‐proline, putrescine, and L‐ornithine. 76 Increased levels of L‐proline products result in decreased NO and increased collagen deposition by SMCs, 76 a combination of effects that could drive remodeling associated with the transition from Ipc‐PH to Cpc‐PH. Similar to ECs, when PASMCs experience pathological stretch, activity of mitochondrial complex III increases, leading to elevated cytosolic ROS and NOX‐4 (NADPH oxidase‐4) activity, both of which contribute to vascular remodeling. 77 Pathological remodeling of PASMCs caused by increased cyclic stretch is characterized by proliferation, collagen deposition, and inflammation, which likely contribute to the decreased pulmonary arterial compliance associated with the progression to Cpc‐PH. Thus, arterial stiffening because of cyclic stretch–stimulated pathological remodeling by SMCs and ECs may compound the dysfunction and remodeling in the pulmonary capillaries and veins, driving the progression from Ipc‐PH to Cpc‐PH.
Figure 4. Mechanotransduction of low wall shear stress because of left heart failure in endothelial cells and smooth muscle cells results in pathologic vascular remodeling throughout the vessel wall.
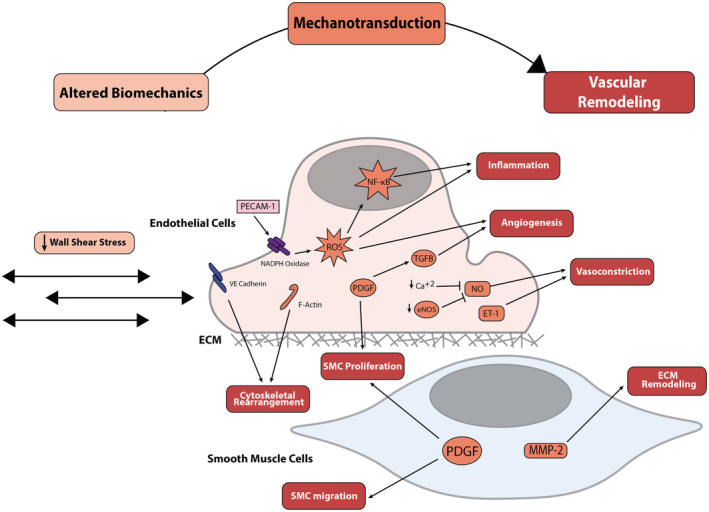
Boxes within the cell signaling cascade are colored to match their corresponding box at the top of the figure (altered biomechanics, mechanotransduction, or vascular remodeling). ECM indicates extracellular matrix; eNOS, endothelial nitric oxide synthase; ET‐1, endothelin‐1; F‐Actin, anti‐filamentous actin; MMP, matrix metalloproteinase; NF‐κB, nuclear factor‐κB; PDGF, platelet‐derived growth factor; PECAM‐1, platelet endothelial cell adhesion molecule‐1; ROS, reactive oxygen species; SMC, smooth muscle cell; TGFβ, transforming growth factor beta; and VE cadherin, vascular endothelial cadherin.
Impact of Cyclic Stretch on Fibroblasts in Arteries
The activation of fibroblasts and their differentiation into proinflammatory myofibroblasts is a major contributor to the arterial stiffening, alveolar membrane thickening, and interstitial fibrosis, which characterize the irreversible remodeling seen in Cpc‐PH. 51 Several studies have shown pulmonary artery adventitial fibroblasts (PAAFs) act as direct biomechanical transducers in response to stretch and injury. Increased cyclic stretch stimulates PAAF differentiation into myofibroblasts and increases expression of collagen and elastin messenger RNAs in vitro. 78 Cyclic stretch directly activates latent TGF‐β1, which sustains the myofibroblast phenotype 79 via activation of Smad proteins 80 and the mitogen‐activated protein kinase/extracellular signal‐regulated kinase (MAPK/ERK) signaling pathway. 81 Indirectly, fibroblasts can act as mediators of pathological stress from SMCs and ECs induced by altered biomechanics. For example, cyclic stretch increases the expression of FGF‐2 (fibroblast growth factor‐2) 75 and NOX‐4 77 in pulmonary vascular SMCs. Notably, NOX‐4 has been shown to regulate TGF‐β1, 82 which may act as a feedback mechanism for myofibroblast differentiation. Additionally, uniaxial cyclic stretch has been shown upregulate COX‐2 (cyclooxygenase‐2) in fibroblasts via an increase in intracellular Ca2+, 83 introducing yet another mechanism of indirect biomechanical transduction via NF‐κB activation. Thus, the differentiation of fibroblasts into myofibroblasts and their proliferation stimulated by cyclic stretch that lead to upregulation of ROS and other proinflammatory proteins and cytokines are important drivers of pathological fibrosis and remodeling observed in Cpc‐PH.
Decreased WSS
WSS is the drag force (per unit area) exerted by blood on ECs throughout the vasculature. 60 Endothelial shear stress is a key regulator of vascular tone, structure, gene expression, and remodeling. 84 , 85 , 86 Regional variations in shear stress are associated with systemic vascular pathologies such as atherosclerosis and aneurysm development. 85 , 86 , 87 Physiological WSS in the pulmonary arteries of healthy adults has been estimated to range from 15 to 20 dyn/cm2, whereas patients with severe pulmonary hypertension have lower WSS in the range of 5 to 8 dyn/cm2. 88 Altered WSS has been shown to occur in a rat model of LHF; echocardiography showed increased PA luminal size and blood flow analysis found reduced WSS. 58 Bartolo et al predicted a 50% or more decrease in WSS in PH‐LHF from 10 to 25 dyn/cm2 (in the healthy state) to 2 to 5 dyn/cm2 (in PH‐LHF) using a computational fluid dynamics model. 35 Moreover, in humans with PH, computational fluid dynamics and phase‐contrast cardiac magnetic resonance imaging have demonstrated lower WSS in the proximal arteries than controls. 89 Pathological alterations in WSS activate EC mechanotransduction pathways, leading to a chronic proinflammatory state characterized by disorganized alignment, vasoconstriction, increased vascular permeability, and maladaptive ECM remodeling. 85 , 87
In vitro studies have investigated the mechanisms by which altered shear stress triggers EC‐driven remodeling. The molecular pathways involved in the resulting pathological inflammation, vasoconstriction, and vessel remodeling are illustrated in Figure 4. As with cyclic stretch, the majority of studies evaluating the impact of WSS have used HUVECs or arterial ECs as the prototype EC; however, some studies have specifically evaluated the impact of this mechanical stimulus in PAECs, and these are detailed in Table 2. To date, findings highlight WSS as a potent mechanical stimulus that is transduced into a wide array of biological signals influencing intracellular energetics, cytoskeletal structure, and vascular tone. 90 , 91 , 92 Future work specifically evaluating these mechanotransduction pathways in EC from throughout the pulmonary vascular bed is essential to understanding disease progression and identifying therapeutic targets.
Table 2.
Shear Stress in Pulmonary Vascular EC
| Cell type | Stimulus vs control | Mechanotransduction | Biological response | References |
|---|---|---|---|---|
| Physiological shear stress | ||||
| Human PAEC | 3–8 dyn/cm2 vs static for 10 min | Caveolin‐mediated mitochondrial ATP generation | EC homeostasis | Yamamoto 91 |
| Bovine and human PAEC | 10 dyn/cm2 vs static for 24 h | Rac/PAK‐dependent myosin light chain phosphorylation and actin polymerization | EC cytoskeleton rearrangement | Birukov 92 |
| Ovine PAEC | 20 dyn/cm2 vs static for 8 h | Akt‐dependent eNOS phosphorylation and NO production | Vasodilation | Wedgwood 90 |
| Low shear stress | ||||
| Bovine PAEC | 5 vs 20–60 dyn/cm2 for 20 h | Reduced eNOS phosphorylation NO, PGF‐1α, and VEGF, increased ET‐1, F‐Actin, and VE‐cadherin rearrangement | Vasoconstriction and cytoskeleton rearrangement | Li 88 |
| Mouse PMVEC | Loss of flow vs 5 dyn/cm2 for 1 h | NADPH oxidase–dependent ROS production | Inflammation and angiogenesis | Milovanova 104 |
| Mouse PMVEC | Loss of flow vs 5 dyn/cm2 for 1 h | PECAM‐1–dependent ROS production and proliferation | Inflammation and angiogenesis | Noel 103 |
| High shear stress | ||||
| Ovine PAEC | 30–100 dyn/cm2 vs 5–20 dyn/cm2 for 4 h | Catalase inhibition increasing ROS, Akt‐mediated eNOS phosphorylation increasing NO production | Inflammation and vasodilation | Kumar 97 |
Akt indicates protein kinase B; EC, endothelial cell; ECM, extracellular matrix; eNOS, endothelial nitric oxide synthase; ET‐1, endothelin‐1; F‐Actin, anti‐filamentous actin; PAEC, pulmonary artery endothelial cell; PAK, p21 activated kinase; PECAM‐1, platelet endothelial cell adhesion molecule‐1; PGF‐1α, prostoglandin F1‐α; PMVEC, pulmonary microvascular endothelial cell; Rac, ras‐related C3 botulinum toxin substrate; ROS, reactive oxygen species; VE‐cadherin, vascular endothelial cadherin; and VEGF, vascular endothelial growth factor.
Impact of WSS on ECs in Veins and Arteries
Vascular ECs sense WSS and transduce it into biochemical signals resulting in synthesis and release of the potent vasodilator NO. 93 , 94 Under physiological WSS, the production of NO is regulated through both calcium‐independent eNOS phosphorylation and calcium‐dependent pathways in ECs. 90 , 95 , 96 , 97 However, this process is dysregulated under pathologically low WSS (Figure 4). Compared with physiological WSS, pathologically low WSS reduces the release of vasodilators such as NO and PGF1α (prostaglandin F1‐α) while increasing the release of vasoconstrictors such as ET‐1 by up to 40% in cultured PAECs 88 and HUVECs. 98 Moreover, total eNOS expression under pathologically low WSS is reduced by 65% compared with physiological WSS, and downstream Akt phosphorylation is reduced by 81%. 88 Consistent with this mechanism, patients with World Health Organization Group 2 PH, including PH‐LHF, have reduced PA eNOS expression, which correlates with the degree of vascular remodeling. 99 Infusion of NOS inhibitors such as N(omega)‐monomethyl‐L‐argenine acetate (L‐NMMA) into the pulmonary arteries of subjects with LHF caused a dose‐dependent reduction in pulmonary blood flow velocity with no change in PA pressure, 100 demonstrating that NO‐dependent pulmonary vasoconstriction was a key contributor to increased PVR in this cohort. These studies suggest that low WSS drives increased ET‐1 and reduced NO, both of which have been demonstrated in subjects with PH‐LHF. 100 , 101 , 102
Beyond vasoconstriction, chronically low WSS can drive pathological remodeling via altered EC, and subsequently altered SMC, structure, and function. Under physiological WSS conditions, EC alignment is parallel to flow. However, low WSS is associated with disorganized EC cytoskeletal alignment in bovine PAECs. 88 Low WSS, as modeled by in vivo and in vitro cessation of flow in mouse pulmonary microvasculature, triggers ROS production via PECAM‐1 (platelet endothelial cell adhesion molecule‐1) and NADPH oxidase, leading to inflammation and angiogenesis. 103 , 104 In coculture of rat aortic ECs and SMCs, low WSS upregulates PDGF release in ECs, which increases SMC proliferation and migration, 105 2 hallmarks of pathological pulmonary arteriole remodeling. 106 Additionally, ECs exposed to low WSS stimulate SMC migration via MMP‐2 activation and increased PDGF. 107 , 108 Because MMP‐2 activation degrades the extracellular matrix through type IV collagen proteolysis, increased expression promotes integrin detachment and SMC migration. 109 ECM remodeling driven by MMP activation is a key feature of pathologic changes in PH. 106 , 110 Both low WSS and elevated MMPs have been found in PH‐LHF. 54 , 58 The in vitro studies cited above provide a potential mechanism that links the observed pathological mechanical stimulus of low WSS resulting from LHF to the observed molecular changes that result in the pathological vascular remodeling observed in the transition from Ipc‐PH to Cpc‐PH.
Both chronically impaired NO production and ECM remodeling driven by chronically low WSS in PH‐LHF result in increased arterial stiffness. Arterial stiffening ultimately causes key hemodynamic changes that are the hallmark of Cpc‐PH, such as decreased PAC. 17 Reduced compliance in the arterial compartment results in highly pulsatile flow downstream in pulmonary arterioles, which further stimulates EC dysregulation and SMC hypertrophy, ultimately driving the cycle of disease progression (Figure 1). In a vascular mimetic coculture model with PAECs and SMCs, high pulsatility flow increased SMC size and elevated expression of contractile proteins, such as SMA (smooth muscle actin) and smooth muscle myosin heavy chain (SM‐MHC). 111 Moreover, high pulsatility flow reduced eNOS expression and increased ET‐1, angiotensin‐converting enzyme, and TGF‐β1, all of which have been associated with SMC hypertrophy and vasoconstriction 111 that drive increased PVR, the pathological development marking the transition from Ipc‐PH to Cpc‐PH.
Role of Biomechanics in Transition to Cpc‐PH and RVF
The presence of pulmonary arterial remodeling is the defining characteristic of Cpc‐PH. Irreversible pulmonary arterial muscularization, diffuse vascular fibrosis, and distal arterial luminal narrowing characterize Cpc‐PH. 16 Multiple animal studies have demonstrated that these structural features are associated with the functional change, namely elevation in PVR, in Cpc‐PH. 52 , 112 As chronic elevated pulmonary pressure and decreased flow change capillary and venous mechanical properties downstream, these same altered biomechanical factors can induce further pulmonary arterial remodeling. Because the structural and functional changes that occur in the pulmonary vasculature with Cpc‐PH are similar to those that occur with PAH, computational simulations of pulmonary vascular blood flow dynamics in PAH subjects have been used to shed light on the impact of altered biomechanics in Cpc‐PH subjects. Using a combined magnetic resonance imaging–computational fluid dynamics approach, Tang and colleagues demonstrated a profound reduction in WSS by a factor of 6 in the proximal and distal pulmonary arteries of subjects with PAH compared with control subjects. 113 Similarly, a significant inverse relationship was found among WSS, mPAP, and PVR, and a significant positive relationship was found between WSS and capacitance in the main PA in subjects with PH. 89 Extensive muscularization of proximal and distal PAs associated with increased PVR and TPG has been confirmed in postmortem human studies. 112 Thus, once Ipc‐PH occurs, pressures increase in the pulmonary arteries, leading to dilation and decreased shear stress. Subsequent remodeling increases PVR and lowers compliance, which in turn alters hemodynamics downstream and promotes a vicious cycle of disease progression (Figure 1).
The irreversible pulmonary arterial remodeling that marks the transition to Cpc‐PH is associated with and defined by PVR. In the setting of LHF, increased PVR dramatically increases risk of RV failure and mortality. 114 In a clinical study measuring RV function and mortality in Ipc‐PH and Cpc‐PH subjects, the prevalence of RV enlargement, RV dysfunction, and all cause‐mortality increased with higher PVR. 115 Moreover, reduced PAC contributes to RV decline. 17 , 116 Because the clinical outcomes of subjects with Cpc‐PH depend on RV function, understanding the mechanisms by which mechanotransduction drives the transition from Ipc‐PH to Cpc‐PH is critical in determining therapeutic targets.
Therapeutic Implications
Currently, there are no Food and Drug Administration–approved pharmacotherapies in the clinician's armamentarium for Cpc‐PH. Therapeutic strategies beyond treatment of LHF are limited and largely have consisted of trials of pharmacologics developed for PAH, the vast majority of which target the altered biomechanics of PH. Current clinical management of Cpc‐PH involves the use of vasodilators, diuretics, angiotensin‐converting enzyme inhibitors, angiotensin receptor blockers, and phosphodiesterase type 5 (PDE5) inhibitors. PAH drugs, including endothelin‐1 antagonists, have shown variable effectiveness because of their potent systemic effects, which are generally not tolerated in the setting of LHF. 117 , 118 The purpose of current treatments are to relieve dyspnea, improve exercise capacity, and define eligibility for heart transplantation. 119 Targeting the mechanotransduction pathways in PH‐LHF is a novel and potentially powerful therapeutic strategy that could disrupt a critical link in the transition from Ipc‐PH to Cpc‐PH. Thus, biomechanical and mechanobiological mechanisms of disease progression and their transduction pathways should be future targets for Cpc‐PH therapies.
Bridging the Knowledge Gaps: Directions for Future Work
Here we have reviewed the known and hypothesized altered biomechanical forces in pulmonary veins, capillaries, and arteries that occur because of Ipc‐PH and the mechanobiological mechanisms that may drive transition to Cpc‐PH and subsequent RHF. There is an urgent clinical need for improved understanding of this disease pathophysiology and progression as well as for novel therapeutic interventions to improve patient outcomes. As highlighted in this review, critical knowledge gaps remain both in our understanding of pulmonary biomechanics in PH‐LHF as well as in mechanotransduction of these signals in the context of the 3 pulmonary vascular compartments.
Robust clinical and animal studies that quantify the mechanical forces acting on ECs and SMCs in both Ipc‐PH and Cpc‐PH are integral to understanding the WSS and cyclic stretch distribution in Ipc‐PH and transition to Cpc‐PH. Invasive pressure measurements coupled with noninvasive flow and anatomic imaging with sufficient resolution will enable computation or estimation of these mechanical stimuli. In turn, knowledge of these mechanical stimuli will facilitate high quality, impactful in vitro mechanistic studies that clarify the mechanotransduction pathways in this disease process.
Critically, these mechanistic studies should be performed in pulmonary cell types. Research to date has been concentrated in systemic vascular‐derived cell lines with mechanical stimuli that model disturbed and oscillatory flow conditions that drive atherosclerosis but are not relevant to either Ipc‐PH or Cpc‐PH. Beyond in vitro studies modeling physiological and pathological WSS or cyclic stretch in pulmonary vascular cells, both stresses need to be applied simultaneously. A limited number of studies have evaluated the impact of combined WSS and cyclic stretch. One such study, performed with systemic artery‐derived cells, demonstrated potentiation of some mechanotransduction responses and inhibition of others, highlighting the need to study both mechanical forces together. 120 Importantly, future studies should also consider the effects of the biological environment, including sex, sex hormone status, age, and systemic diseases such as metabolic syndrome and diabetes on mechanotransduction.
In vitro studies that interrogate the intersections between known risk factors and pulmonary vascular cell mechanotransduction will accelerate research breakthroughs. We posit that patient‐specific factors such as sex, genetics, and comorbidities impact cellular mechanotransduction and may be key to outcomes and responses to treatment. Women are known to be at higher risk of developing PAH than men, 121 although in other contexts estrogen is considered vasculo‐protective. 122 , 123 Although sex differences in mechanotransduction have not been identified in vascular and ECs and SMCs, estrogen has been shown to affect mechanotransduction in bone cell networks. 124 In terms of genetics, CAV1, a gene responsible for encoding caveolin‐1 protein, regulates mechanotransduction of vascular shear stress 125 in pulmonary vascular ECs and has been shown to be dysfunctional in PH. 126 CAV1 mutations are also associated with lipid disorders such as type 2 diabetes, 127 which itself has been shown to be an independent risk factor for PH. 128 Previous studies have shown that diabetes may cause defects in the mechanotransduction of arterial SMCs via alterations in ECM composition, which lead to increased stiffness and decreased arteriolar compliance. 129 Other comorbidities of Cpc‐PH, such as obesity and age, have also been shown to alter mechanotransduction in ECs and SMCs. 130 , 131 Thus, sex and its consequences for sex steroid hormones, certain genetic mutations, obesity, and age may modulate mechanotransduction and thereby drive the transition from Ipc‐PH to Cpc‐PH. These relationships warrant further investigation as both mechanisms of disease progression and potential therapeutic targets.
Conclusions
PH‐LHF alters pulmonary vascular biomechanical forces resulting in increased cyclic stretch and decreased WSS, which may drive transition from Ipc‐PH to Cpc‐PH. These mechanical stimuli and their biological consequences need further investigation to identify targetable mechanisms to prevent progression of this disease. Understanding the mechanisms of mechanotransduction in pulmonary circulation will deepen our understanding of Ipc‐PH and Cpc‐PH and could open doors to new pharmacologic therapies.
Sources of Funding
This work was supported by the National Institutes of Health (R01 HL154624, R01 HL147590, and T32HL110853).
Disclosures
None.
Acknowledgments
Author contributions: Dr Allen: concept development, literature review, and manuscript writing. H. Frye: concept development, literature review, manuscript writing, and table preparation. R. Ramanathan: concept development, literature review, manuscript writing, and figure development. Dr Caggiano: manuscript review and editing, and figure development. Dr Tabima: concept development and manuscript review and editing. Dr Chesler: concept development, literature review, manuscript writing, manuscript review and editing, and figure development. Dr Philip: literature review, manuscript writing, manuscript review and editing, and figure development.
For Sources of Funding and Disclosures, see page 12.
Contributor Information
Naomi C. Chesler, Email: nchesler@uci.edu.
Jennifer L. Philip, Email: philip@surgery.wisc.edu.
References
- 1. Mozaffarian D, Benjamin EJ, Go AS, Arnett DK, Blaha MJ, Cushman M, Das SR, de Ferranti S, Després JP, Fullerton HJ, et al. Heart disease and stroke statistics‐2016 update: a report from the American Heart Association. Circulation. 2016;133:e38–e360. doi: 10.1161/CIR.0000000000000350 [DOI] [PubMed] [Google Scholar]
- 2. Lam CSP, Roger VL, Rodeheffer RJ, Borlaug BA, Enders FT, Redfield MM. Pulmonary hypertension in heart failure with preserved ejection fraction. J Am Coll Cardiol. 2009;53:1119–1126. doi: 10.1016/j.jacc.2008.11.051 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Guazzi M. Alveolar‐capillary membrane dysfunction in heart failure: evidence of a pathophysiologic role. Chest. 2003;124:1090–1102. doi: 10.1378/chest.124.3.1090 [DOI] [PubMed] [Google Scholar]
- 4. Miller WL, Grill DE, Borlaug BA. Clinical features, hemodynamics, and outcomes of pulmonary hypertension due to chronic heart failure with reduced ejection fraction: pulmonary hypertension and heart failure. JACC Heart Fail. 2013;1:290–299. doi: 10.1016/j.jchf.2013.05.001 [DOI] [PubMed] [Google Scholar]
- 5. Vanderpool RR, Saul M, Nouraie M, Gladwin MT, Simon MA. Association between hemodynamic markers of pulmonary hypertension and outcomes in heart failure with preserved ejection fraction. JAMA Cardiol. 2018;3:298–306. doi: 10.1001/jamacardio.2018.0128 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Ding Z, Friedman MH. Dynamics of human coronary arterial motion and its potential role in coronary atherogenesis. J Biomech Eng. 2000;122:488–492. doi: 10.1115/1.1289989 [DOI] [PubMed] [Google Scholar]
- 7. Caro CG. Discovery of the role of wall shear in atherosclerosis. Arterioscler Thromb Vasc Biol. 2009;29:158–161. doi: 10.1161/ATVBAHA.108.166736 [DOI] [PubMed] [Google Scholar]
- 8. Jufri NF, Mohamedali A, Avolio A, Baker MS. Mechanical stretch: physiological and pathological implications for human vascular endothelial cells. Vascular Cell. 2015;7:7. doi: 10.1186/s13221-015-0033-z [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Baker PN, Stranko CP, Davidge ST, Davies PS, Roberts JM. Mechanical stress eliminates the effects of plasma from patients with preeclampsia on endothelial cells. Am J Obstet Gynecol. 1996;174:730–736. doi: 10.1016/s0002-9378(96)70457-x [DOI] [PubMed] [Google Scholar]
- 10. Chien S. Mechanotransduction and endothelial cell homeostasis: the wisdom of the cell. Am J Physiol Heart Circ. 2007;292:H1209–H1224. doi: 10.1152/ajpheart.01047.2006 [DOI] [PubMed] [Google Scholar]
- 11. Fernandes DC, Araujo TLS, Laurindo FRM, Tanaka LY. Chapter 7 ‐ Hemodynamic forces in the endothelium: From mechanotransduction to implications on development of atherosclerosis. In: Da Luz PL, Libby P, Chagas ACP, Laurindo FRM, eds. Endothelium and Cardiovascular Diseases. Cambridge, MA: Academic Press; 2018:85–95. [Google Scholar]
- 12. Xie H, Wu L, Deng Z, Huo Y, Cheng Y. Emerging roles of YAP/TAZ in lung physiology and diseases. Life Sci. 2018;214:176–183. doi: 10.1016/j.lfs.2018.10.062 [DOI] [PubMed] [Google Scholar]
- 13. Humbert M, Kovacs G, Hoeper MM, Badagliacca R, Berger RMF, Brida M, Carlsen J, Coats AJS, Escribano‐Subias P, Ferrari P, et al. ESC/ERS guidelines for the diagnosis and treatment of pulmonary hypertension. Eur Heart J. 2022;2022:3618–3731. doi: 10.1093/eurheartj/ehac237 [DOI] [PubMed] [Google Scholar]
- 14. Ghio S, Schirinzi S, Pica S. Pulmonary arterial compliance: how and why should we measure it? Glob Cardiol Sci Pract. 2015;2015:58. doi: 10.5339/gcsp.2015.58 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Rosenkranz S, Gibbs JS, Wachter R, De Marco T, Vonk‐Noordegraaf A, Vachiery JL. Left ventricular heart failure and pulmonary hypertension. Eur Heart J. 2016;37:942–954. doi: 10.1093/eurheartj/ehv512 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Fayyaz AU, Edwards WD, Maleszewski JJ, Konik EA, DuBrock HM, Borlaug BA, Frantz RP, Jenkins SM, Redfield MM. Global pulmonary vascular remodeling in pulmonary hypertension associated with heart failure and preserved or reduced ejection fraction. Circulation. 2018;137:1796–1810. doi: 10.1161/CIRCULATIONAHA.117.031608 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Thenappan T, Prins KW, Pritzker MR, Scandurra J, Volmers K, Weir EK. The critical role of pulmonary arterial compliance in pulmonary hypertension. Ann Am Thorac Soc. 2016;13:276–284. doi: 10.1513/AnnalsATS.201509-599FR [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Dupont M, Mullens W, Skouri HN, Abrahams Z, Wu Y, Taylor DO, Starling RC, Tang WH. Prognostic role of pulmonary arterial capacitance in advanced heart failure. Circ Heart Fail. 2012;5:778–785. doi: 10.1161/CIRCHEARTFAILURE.112.968511 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Al‐Naamani N, Preston IR, Paulus JK, Hill NS, Roberts KE. Pulmonary arterial capacitance is an important predictor of mortality in heart failure with a preserved ejection fraction. JACC Heart Fail. 2015;3:467–474. doi: 10.1016/j.jchf.2015.01.013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Dragu R, Rispler S, Habib M, Sholy H, Hammerman H, Galie N, Aronson D. Pulmonary arterial capacitance in patients with heart failure and reactive pulmonary hypertension. Eur J Heart Fail. 2015;17:74–80. doi: 10.1002/ejhf.192 [DOI] [PubMed] [Google Scholar]
- 21. Townsley MI. Structure and composition of pulmonary arteries, capillaries, and veins. Compr Physiol. 2012;2:675–709. doi: 10.1002/cphy.c100081 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. He S, Zhu T, Fang Z. The role and regulation of pulmonary artery smooth muscle cells in pulmonary hypertension. Int J Hypertens. 2020;2020:1478291–1478210. doi: 10.1155/2020/1478291 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Burgstaller G, Oehrle B, Gerckens M, White ES, Schiller HB, Eickelberg O. The instructive extracellular matrix of the lung: basic composition and alterations in chronic lung disease. Eur Respir J. 2017;50:50. doi: 10.1183/13993003.01805-2016 [DOI] [PubMed] [Google Scholar]
- 24. West JBTK, Odile MC, Prediletto R. Stress failure in pulmonary capillaries. J Appl Physiol. 1985;1991(70):1731–1742. doi: 10.1152/jappl.1991.70.4.1731 [DOI] [PubMed] [Google Scholar]
- 25. Horsfield KG, Gordon WI. Morphometry of pulmonary veins in man. Lung. 1981;159:211–218. doi: 10.1007/BF02713917 [DOI] [PubMed] [Google Scholar]
- 26. Maloney JER, Rooholamini SA, Wexler L. Pressure‐diameter relations of small blood vessels in isolated dog lung. Microvasc Res. 1970;2:1–12. doi: 10.1016/0026-2862(70)90048-8 [DOI] [PubMed] [Google Scholar]
- 27. Attinger EO. Hydrodynamics of blood flow. Adv Hydrosci. 1966;3:111–152. [Google Scholar]
- 28. Stergiopulos N, Meister JJ, Westerhof N. Evaluation of methods for estimation of total arterial compliance. Am J Physiol Heart Circ. 1995;268:H1540–H1548. doi: 10.1152/ajpheart.1995.268.4.H1540 [DOI] [PubMed] [Google Scholar]
- 29. Saouti N, Westerhof N, Postmus PE, Vonk‐Noordegraaf A. The arterial load in pulmonary hypertension. Eur Respir Rev. 2010;19:197–203. doi: 10.1183/09059180.00002210 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. London GM, Pannier B, Safar ME. Arterial stiffness gradient, systemic reflection coefficient, and pulsatile pressure wave transmission in essential hypertension. Hypertension. 2019;74:1366–1372. doi: 10.1161/HYPERTENSIONAHA.119.13387 [DOI] [PubMed] [Google Scholar]
- 31. West JB. Cellular responses to mechanical stress invited review: pulmonary capillary stress failure. J Appl Physiol. 2000;89:2483–2489. doi: 10.1152/jappl.2000.89.6.2483 [DOI] [PubMed] [Google Scholar]
- 32. Reneman RS, Arts T, Hoeks APG. Wall shear stress – an important determinant of endothelial cell function and structure – in the arterial system in vivo. J Vasc Res. 2006;43:251–269. doi: 10.1159/000091648 [DOI] [PubMed] [Google Scholar]
- 33. Tang BT, Fonte TA, Chan FP, Tsao PS, Feinstein JA, Taylor CA. Three‐dimensional hemodynamics in the human pulmonary arteries under resting and exercise conditions. Ann Biomed Eng. 2011;39:347–358. doi: 10.1007/s10439-010-0124-1 [DOI] [PubMed] [Google Scholar]
- 34. Masuyama T, Lee JM, Nagano R, Nariyama K, Yamamoto K, Naito J, Mano T, Kondo H, Hori M, Kamada T. Doppler echocardiographic pulmonary venous flow‐velocity pattern for assessment of the hemodynamic profile in acute congestive heart failure. Am Heart J. 1995;129:107–113. doi: 10.1016/0002-8703(95)90050-0 [DOI] [PubMed] [Google Scholar]
- 35. Bartolo MA, Qureshi MU, Colebank MJ, Chesler NC, Olufsen MS. Numerical predictions of shear stress and cyclic stretch in pulmonary hypertension due to left heart failure. Biomech Model Mechanobiol. 2022;21:363–381. doi: 10.1007/s10237-021-01538-1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Hu Z, Xiong Y, Han X, Geng C, Jiang B, Huo Y, Luo J. Acute mechanical stretch promotes eNOS activation in venous endothelial cells mainly via PKA and Akt pathways. PLoS One. 2013;8:e71359. doi: 10.1371/journal.pone.0071359 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Takeda H, Komori K, Nishikimi N, Nimura Y, Sokabe M, Naruse K. Bi‐phasic activation of eNOS in response to uni‐axial cyclic stretch is mediated by differential mechanisms in BAECs. Life Sci. 2006;79:233–239. doi: 10.1016/j.lfs.2005.12.051 [DOI] [PubMed] [Google Scholar]
- 38. Kobayashi S, Nagino M, Komatsu S, Naruse K, Nimura Y, Nakanishi M, Sokabe M. Stretch‐induced IL‐6 secretion from endothelial cells requires NF‐κB activation. Biochem Biophys Res Commun. 2003;308:306–312. doi: 10.1016/s0006-291x(03)01362-7 [DOI] [PubMed] [Google Scholar]
- 39. Xiao L, Liu Y, Wang N. New paradigms in inflammatory signaling in vascular endothelial cells. Am J Physiol Heart Circ. 2014;306:H317–H325. doi: 10.1152/ajpheart.00182.2013 [DOI] [PubMed] [Google Scholar]
- 40. Du W, Mills I, Sumpio BE. Cyclic strain causes heterogeneous induction of transcription factors, AP‐1, CRE binding protein and NF‐kB, in endothelial cells: species and vascular bed diversity. J Biomech. 1995;28:1485–1491. doi: 10.1016/0021-9290(95)00096-8 [DOI] [PubMed] [Google Scholar]
- 41. Cheng T‐H, Shih N‐L, Chen S‐Y, Loh S‐H, Cheng P‐Y, Tsai C‐S, Liu S‐H, Wang DL, Chen J‐J. Reactive oxygen species mediate cyclic strain‐induced endothelin‐1 gene expression via Ras/Raf/extracellular signal‐regulated kinase pathway in endothelial cells. J Mol Cell Cardiol. 2001;33:1805–1814. doi: 10.1006/jmcc.2001.1444 [DOI] [PubMed] [Google Scholar]
- 42. Wung B, Cheng J, Chao Y, Lin J, Shyy Y‐J, Wang DL. Cyclical strain increases monocyte chemotactic protein‐1 secretion in human endothelial cells. Am J Physiol Heart Circ. 1996;270:H1462–H1468. [DOI] [PubMed] [Google Scholar]
- 43. Okada M, Matsumori A, Ono K, Furukawa Y, Shioi T, Iwasaki A, Matsushima K, Sasayama S. Cyclic stretch upregulates production of interleukin‐8 and monocyte chemotactic and activating factor/monocyte chemoattractant protein‐1 in human endothelial cells. Arterioscler Thromb Vasc Biol. 1998;18:894–901. doi: 10.1161/01.ATV.18.6.894 [DOI] [PubMed] [Google Scholar]
- 44. Wang B, Chang H, Lin S, Kuan P, Shyu K. Induction of matrix metalloproteinases‐14 and ‐2 by cyclical mechanical stretch is mediated by tumor necrosis factor‐alpha in cultured human umbilical vein endothelial cells. Cardiovasc Res. 2003;59:460–469. doi: 10.1016/s0008-6363(03)00428-0 [DOI] [PubMed] [Google Scholar]
- 45. Meng X, Mavromatis K, Galis ZS. Mechanical stretching of human saphenous vein grafts induces expression and activation of matrix‐degrading enzymes associated with vascular tissue injury and repair. Exp Mol Pathol. 1999;66:227–237. doi: 10.1006/exmp.1999.2260 [DOI] [PubMed] [Google Scholar]
- 46. von Offenberg SN, Cummins PM, Birney YA, Cullen JP, Redmond EM, Cahill PA. Cyclic strain‐mediated regulation of endothelial matrix metalloproteinase‐2 expression and activity. Cardiovasc Res. 2004;63:625–634. doi: 10.1016/j.cardiores.2004.05.008 [DOI] [PubMed] [Google Scholar]
- 47. Hatami J, Tafazzoli‐Shadpour M, Haghighipour N, Shokrgozar MA, Janmaleki M. Influence of cyclic stretch on mechanical properties of endothelial cells. Experimental Mechanics. 2013;53:1291–1298. doi: 10.1007/s11340-013-9744-3 [DOI] [Google Scholar]
- 48. Omidvar R, Tafazzoli‐Shadpour M, Mahmoodi‐Nobar F, Azadi S, Khani MM. Quantifying effects of cyclic stretch on cell‐collagen substrate adhesiveness of vascular endothelial cells. Proc Inst Mech Eng H. 2018;232:531–541. doi: 10.1177/0954411918767477 [DOI] [PubMed] [Google Scholar]
- 49. Naeije R, Gerges M, Vachiery JL, Caravita S, Gerges C, Lang IM. Hemodynamic phenotyping of pulmonary hypertension in left heart failure. Circ Heart Fail. 2017;10:10. doi: 10.1161/CIRCHEARTFAILURE.117.004082 [DOI] [PubMed] [Google Scholar]
- 50. Hunt JM, Bethea B, Liu X, Gandjeva A, Mammen PP, Stacher E, Gandjeva MR, Parish E, Perez M, Smith L. Pulmonary veins in the normal lung and pulmonary hypertension due to left heart disease. A J Physiol Lung Cell Molr Physiol. 2013;305:L725–L736. doi: 10.1152/ajplung.00186.2013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Dayeh NR, Ledoux J, Dupuis J. Lung capillary stress failure and arteriolar remodelling in pulmonary hypertension associated with left heart disease (group 2 PH). Prog Cardiovasc Dis. 2016;59:11–21. doi: 10.1016/j.pcad.2016.05.002 [DOI] [PubMed] [Google Scholar]
- 52. Chen Y, Guo H, Xu D, Xu X, Wang H, Hu X, Lu Z, Kwak D, Xu Y, Gunther R. Left ventricular failure produces profound lung remodeling and pulmonary hypertension in mice: heart failure causes severe lung disease. Hypertension. 2012;59:1170–1178. doi: 10.1161/HYPERTENSIONAHA.111.186072 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Haseneen NAV, Vaday GG, Zucker S, Foda HD. Mechanical stretch induces MMP‐2 release and activation in lung endothelium: role of EMMPRIN. Am J Physiol Lung Cell Mol Physiol. 2003;284:541–547. doi: 10.1152/ajplung.00290.2002 [DOI] [PubMed] [Google Scholar]
- 54. Farrero M, Blanco I, Batlle M, Santiago E, Cardona M, Vidal B, Castel MA, Sitges M, Barbera JA, Perez‐Villa F. Pulmonary hypertension is related to peripheral endothelial dysfunction in heart failure with preserved ejection fraction. Circ Heart Fail. 2014;7:791–798. doi: 10.1161/CIRCHEARTFAILURE.113.000942 [DOI] [PubMed] [Google Scholar]
- 55. Tsukimoto KO, Mathieu‐Costello O, Prediletto R, Elliott AR, West JB. Ultrastructural appearances of pulmonary capillaries at high transmural pressures. J Appl Physiol (1985). 1991;71:573–582. doi: 10.1152/jappl.1991.71.2.573 [DOI] [PubMed] [Google Scholar]
- 56. Kerem A, Yin J, Kaestle SM, Hoffmann J, Schoene AM, Singh B, Kuppe H, Borst MM, Kuebler WM. Lung endothelial dysfunction in congestive heart failure: role of impaired Ca2+ signaling and cytoskeletal reorganization. Circ Res. 2010;106:1103–1116. doi: 10.1161/circresaha.109.210542 [DOI] [PubMed] [Google Scholar]
- 57. Philip JL, Murphy TM, Schreier DA, Stevens S, Tabima DM, Albrecht M, Frump AL, Hacker TA, Lahm T, Chesler NC. Pulmonary vascular mechanical consequences of ischemic heart failure and implications for right ventricular function. Am J Physiol Heart Circ. 2019;316:H1167–H1177. doi: 10.1152/ajpheart.00319.2018 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Driss AB, Devaux C, Henrion D, Duriez M, Thuillez C, Levy BI, Michel J‐B. Hemodynamic stresses induce endothelial dysfunction and remodeling of pulmonary artery in experimental compensated heart failure. Circulation. 2000;101:2764–2770. doi: 10.1161/01.CIR.101.23.2764 [DOI] [PubMed] [Google Scholar]
- 59. Guazzi M, Naeije R. Pulmonary hypertension in heart failure: pathophysiology, pathobiology, and emerging clinical perspectives. J Am Coll Cardiol. 2017;69:1718–1734. doi: 10.1016/j.jacc.2017.01.051 [DOI] [PubMed] [Google Scholar]
- 60. Lu D, Kassab GS. Role of shear stress and stretch in vascular mechanobiology. J R Soc Interface. 2011;8:1379–1385. doi: 10.1098/rsif.2011.0177 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Terman B, Dougher‐Vermazen M, Carrion ME, Dimitrov D, Armellino DC, Gospodarowicz D, Böhlen P. Identification of the KDR tyrosine kinase as a receptor for vascular endothelial cell growth factor. Biochem Biophys Res Commun. 1992;187:187–1586. doi: 10.1016/0006-291x(92)90483-2 [DOI] [PubMed] [Google Scholar]
- 62. Tian Y, Gawlak G, O'Donnell JJ 3rd, Birukova AA, Birukov KG. Activation of vascular endothelial growth factor (VEGF) receptor 2 mediates endothelial permeability caused by cyclic stretch. J Biol Chem. 2016;291:10032–10045. doi: 10.1074/jbc.M115.690487 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Iwaki M, Ito S, Morioka M, Iwata S, Numaguchi Y, Ishii M, Kondo M, Kume H, Naruse K, Sokabe M, et al. Mechanical stretch enhances IL‐8 production in pulmonary microvascular endothelial cells. Biochem Biophys Res Commun. 2009;389:531–536. doi: 10.1016/j.bbrc.2009.09.020 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Birukova AA, Tian X, Cokic I, Beckham Y, Gardel ML, Birukov KG. Endothelial barrier disruption and recovery is controlled by substrate stiffness. Microvasc Res. 2013;87:50–57. doi: 10.1016/j.mvr.2012.12.006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Ochoa CD, Baker H, Hasak S, Matyal R, Salam A, Hales CA, Hancock W, Quinn DA. Cyclic stretch affects pulmonary endothelial cell control of pulmonary smooth muscle cell growth. Am J Respir Cell Mol Biol. 2008;39:105–112. doi: 10.1165/rcmb.2007-0283OC [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Ali MH, Mungai PT, Schumacker PT. Stretch‐induced phosphorylation of focal adhesion kinase in endothelial cells: role of mitochondrial oxidants. Am J Physiol Lung Cell Mol Physiol. 2006;291:L38–L45. doi: 10.1152/ajplung.00287.2004 [DOI] [PubMed] [Google Scholar]
- 67. Ali MH, Pearlstein DP, Mathieu CE, Schumacker PT. Mitochondrial requirement for endothelial responses to cyclic strain: Implications for mechanotransduction. Am J Physiol Lung Cell Mol Physiol. 2004;287:486–496. doi: 10.1152/ajplung.00389.2003 [DOI] [PubMed] [Google Scholar]
- 68. Ramella M, Bertozzi G, Fusaro L, Talmon M, Manfredi M, Catoria MC, Casella F, Porta CM, Boldorini R, Fresu LG, et al. Effect of cyclic stretch on vascular endothelial cells and abdominal aortic aneurysm (AAA): role in the inflammatory response. Int J Mol Sci. 2019;20:20. doi: 10.3390/ijms20020287 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. dela Paz NG, D'Amore PA. Arterial versus venous endothelial cells. Cell Tissue Res. 2009;335:5–16. doi: 10.1007/s00441-008-0706-5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70. Haga JH, Li YS, Chien S. Molecular basis of the effects of mechanical stretch on vascular smooth muscle cells. J Biomech. 2007;40:947–960. doi: 10.1016/j.jbiomech.2006.04.011 [DOI] [PubMed] [Google Scholar]
- 71. Cheng J, Du J. Mechanical stretch simulates proliferation of venous smooth muscle cells through activation of the insulin‐like growth factor‐1 receptor. Arterioscler Thromb Vasc Biol. 2007;27:1744–1751. doi: 10.1161/ATVBAHA.107.147371 [DOI] [PubMed] [Google Scholar]
- 72. Liu WF, Nelson CM, Tan JL, Chen CS. Cadherins, RhoA, and Rac1 are differentially required for stretch‐mediated proliferation in endothelial versus smooth muscle cells. Circ Res. 2007;101:e44–e52. doi: 10.1161/CIRCRESAHA.107.158329 [DOI] [PubMed] [Google Scholar]
- 73. Mata‐Greenwood E, Grobe A, Kumar S, Noskina Y, Black SM. Cyclic stretch increases VEGF expression in pulmonary arterial smooth muscle cells via TGF‐beta1 and reactive oxygen species: a requirement for NAD(P)H oxidase. Am J Physiol Lung Cell Mol Physiol. 2005;289:L288–L289. doi: 10.1152/ajplung.00417.2004 [DOI] [PubMed] [Google Scholar]
- 74. Tanabe Y, Saito M, Ueno A, Nakamura M, Takeishi K, Nakayama K. Mechanical stretch augments PDGF receptor β expression and protein tyrosine phosphorylation in pulmonary artery tissue and smooth muscle cells. Mol Cell Biochem. 2000;215:103–113. doi: 10.1023/A:1026506801659 [DOI] [PubMed] [Google Scholar]
- 75. Quinn TP, Schlueter M, Soifer SJ, Gutierrez JA. Cyclic mechanical stretch induces VEGF and FGF‐2 expression in pulmonary vascular smooth muscle cells. Am J Physiol Lung Cell Mol Physiol. 2002;282:L897–L903. doi: 10.1152/ajplung.00044.2001 [DOI] [PubMed] [Google Scholar]
- 76. Durante W, Liao L, Reyna SV, Peyton KJ, Schafer AI. Physiological cyclic stretch directs L‐arginine transport and metabolism to collagen synthesis in vascular smooth muscle. FASEB J. 2000;14:1775–1783. doi: 10.1096/fj.99-0960com [DOI] [PubMed] [Google Scholar]
- 77. Wedgwood S, Lakshminrusimha S, Schumacker PT, Steinhorn RH. Cyclic stretch stimulates mitochondrial reactive oxygen species and Nox4 signaling in pulmonary artery smooth muscle cells. Am J Physiol Lung Cell Mol Physiol. 2015;309:L196–L203. doi: 10.1152/ajplung.00097.2014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78. Wang A, Cao S, Stowe JC, Valdez‐Jasso D. Substrate stiffness and stretch regulate profibrotic mechanosignaling in pulmonary arterial adventitial fibroblasts. Cell. 2021;10:10. doi: 10.3390/cells10051000 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79. Walker M, Godin M, Pelling AE. Mechanical stretch sustains myofibroblast phenotype and function in microtissues through latent TGF‐beta1 activation. Integr Biol (Camb). 2020;12:199–210. doi: 10.1093/intbio/zyaa015 [DOI] [PubMed] [Google Scholar]
- 80. Evans RA, Tian YC, Steadman R, Phillips AO. TGF‐beta1‐mediated fibroblast‐myofibroblast terminal differentiation‐the role of Smad proteins. Exp Cell Res. 2003;282:90–100. doi: 10.1016/s0014-4827(02)00015-0 [DOI] [PubMed] [Google Scholar]
- 81. Midgley AC, Rogers M, Hallett MB, Clayton A, Bowen T, Phillips AO, Steadman R. Transforming growth factor‐beta1 (TGF‐beta1)‐stimulated fibroblast to myofibroblast differentiation is mediated by hyaluronan (HA)‐facilitated epidermal growth factor receptor (EGFR) and CD44 co‐localization in lipid rafts. J Biol Chem. 2013;288:14824–14838. doi: 10.1074/jbc.M113.451336 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82. Cucoranu I, Clempus R, Dikalova A, Phelan PJ, Ariyan S, Dikalov S, Sorescu D. NAD(P)H oxidase 4 mediates transforming growth factor‐beta1‐induced differentiation of cardiac fibroblasts into myofibroblasts. Circ Res. 2005;97:900–907. doi: 10.1161/01.RES.0000187457.24338.3D [DOI] [PubMed] [Google Scholar]
- 83. Amma H, Naruse K, Ishiguro N, Sokabe M. Involvement of reactive oxygen species in cyclic stretch‐induced NF‐kappaB activation in human fibroblast cells. Br J Pharmacol. 2005;145:364–373. doi: 10.1038/sj.bjp.0706182 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84. Wragg JW, Durant S, McGettrick HM, Sample KM, Egginton S, Bicknell R. Shear stress regulated gene expression and angiogenesis in vascular endothelium. Microcirculation. 2014;21:290–300. doi: 10.1111/micc.12119 [DOI] [PubMed] [Google Scholar]
- 85. Chatzizisis YS, Coskun AU, Jonas M, Edelman ER, Feldman CL, Stone PH. Role of endothelial shear stress in the natural history of coronary atherosclerosis and vascular remodeling: molecular, cellular, and vascular behavior. J Am Coll Cardiol. 2007;49:2379–2393. doi: 10.1016/j.jacc.2007.02.059 [DOI] [PubMed] [Google Scholar]
- 86. Resnick N, Yahav H, Shay‐Salit A, Shushy M, Schubert S, Zilberman LC, Wofovitz E. Fluid shear stress and the vascular endothelium: for better and for worse. Prog Biophys Mol Biol. 2003;81:177–199. doi: 10.1016/S0079-6107(02)00052-4 [DOI] [PubMed] [Google Scholar]
- 87. Baeyens N, Bandyopadhyay C, Coon BG, Yun S, Schwartz MA. Endothelial fluid shear stress sensing in vascular health and disease. J Clin Invest. 2016;126:821–828. doi: 10.1172/jci83083 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88. Li M, Stenmark KR, Shandas R, Tan W. Effects of pathological flow on pulmonary artery endothelial production of vasoactive mediators and growth factors. J Vasc Res. 2009;46:561–571. doi: 10.1159/000226224 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89. Schafer M, Kheyfets VO, Schroeder JD, Dunning J, Shandas R, Buckner JK, Browning J, Hertzberg J, Hunter KS, Fenster BE. Main pulmonary arterial wall shear stress correlates with invasive hemodynamics and stiffness in pulmonary hypertension. Pulm Circ. 2016;6:37–45. doi: 10.1086/685024 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90. Wedgwood S, Mitchell CJ, Fineman JR, Black SM. Developmental differences in the shear stress‐induced expression of endothelial NO synthase: changing role of AP‐1. Am J Physiol Lung Cell Mol Physiol. 2003;284:L650–L662. doi: 10.1152/ajplung.00252.2002 [DOI] [PubMed] [Google Scholar]
- 91. Yamamoto K, Imamura H, Ando J. Shear stress augments mitochondrial ATP generation that triggers ATP release and Ca2+ signaling in vascular endothelial cells. Am J Physiol Heart Circ. 2018;315:H1477–H1485. doi: 10.1152/ajpheart.00204.2018 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92. Birukov KG, Birukova AA, Dudek SM, Verin AD, Crow MT, Zhan X, DePaola N, Garcia JG. Shear stress‐mediated cytoskeletal remodeling and cortactin translocation in pulmonary endothelial cells. Am J Respir Cell Mol Biol. 2002;26:453–464. doi: 10.1165/ajrcmb.26.4.4725 [DOI] [PubMed] [Google Scholar]
- 93. Fels B, Kusche‐Vihrog K. It takes more than two to tango: mechanosignaling of the endothelial surface. Pflugers Arch. 2020;472:419–433. doi: 10.1007/s00424-020-02369-2 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94. Davies PF. Flow‐mediated endothelial mechanotransduction. Physiol Rev. 1995;75:519–560. doi: 10.1152/physrev.1995.75.3.519 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95. Fineman JR, Black SM. Pressure vs flow‐induced pulmonary hypertension. Advances in Pulmonary Hypertension. 2019;18:19–24. doi: 10.21693/1933-088x-18.1.19 [DOI] [Google Scholar]
- 96. Zhao Y, Vanhoutte PM, Leung SW. Vascular nitric oxide: beyond eNOS. J Pharmacol Sci. 2015;129:83–94. doi: 10.1016/j.jphs.2015.09.002 [DOI] [PubMed] [Google Scholar]
- 97. Kumar S, Sud N, Fonseca FV, Hou Y, Black SM. Shear stress stimulates nitric oxide signaling in pulmonary arterial endothelial cells via a reduction in catalase activity: role of protein kinase Cδ. Am J Physiol Lung Cell Mol Physiol. 2010;298:L105–L116. doi: 10.1152/ajplung.00290.2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98. Masatsugu K, Itoh H, Chun T‐H, Ogawa Y, Tamura N, Yamashita J, Doi K, Inoue M, Fukunaga Y, Sawada N. Physiologic shear stress suppresses endothelin‐converting enzyme‐1 expression in vascular endothelial cells. J Cardiovasc Pharmacol. 1998;31:S42–S45. doi: 10.1097/00005344-199800001-00014 [DOI] [PubMed] [Google Scholar]
- 99. Giaid A, Saleh D. Reduced expression of endothelial nitric oxide synthase in the lungs of patients with pulmonary hypertension. N Engl J Med. 1995;333:214–221. doi: 10.1056/NEJM199507273330403 [DOI] [PubMed] [Google Scholar]
- 100. Cooper CJ, Jevnikar FW, Walsh T, Dickinson J, Mouhaffel A, Selwyn AP. The influence of basal nitric oxide activity on pulmonary vascular resistance in patients with congestive heart failure. Am J Cardiol. 1998;82:609–614. doi: 10.1016/S0002-9149(98)00400-7 [DOI] [PubMed] [Google Scholar]
- 101. Moraes DL, Colucci WS, Givertz MM. Secondary pulmonary hypertension in chronic heart failure: the role of the endothelium in pathophysiology and management. Circulation. 2000;102:1718–1723. doi: 10.1161/01.CIR.102.14.1718 [DOI] [PubMed] [Google Scholar]
- 102. Tsutamoto T, Wada A, Maeda Y, Adachi T, Kinoshita M. Relation between endothelin‐1 spillover in the lungs and pulmonary vascular resistance in patients with chronic heart failure. J Am Coll Cardiol. 1994;23:1427–1433. doi: 10.1016/0735-1097(94)90387-5 [DOI] [PubMed] [Google Scholar]
- 103. Noel J, Wang H, Hong N, Tao J‐Q, Yu K, Sorokina EM, DeBolt K, Heayn M, Rizzo V, Delisser H. PECAM‐1 and caveolae form the mechanosensing complex necessary for NOX2 activation and angiogenic signaling with stopped flow in pulmonary endothelium. Am J Physiol Lung Cell Mol Physiol. 2013;305:L805–L818. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104. Milovanova T, Chatterjee S, Manevich Y, Kotelnikova I, DeBolt K, Madesh M, Moore JS, Fisher AB. Lung endothelial cell proliferation with decreased shear stress is mediated by reactive oxygen species. Am J Physiol Cell Physiol. 2006;290:C66–C76. doi: 10.1152/ajpcell.00094.2005 [DOI] [PubMed] [Google Scholar]
- 105. Qi Y‐X, Jiang J, Jiang X‐H, Wang X‐D, Ji S‐Y, Han Y, Long D‐K, Shen B‐R, Yan Z‐Q, Chien S. PDGF‐BB and TGF‐β1 on cross‐talk between endothelial and smooth muscle cells in vascular remodeling induced by low shear stress. Proc Natl Acad Sci. 2011;108:1908–1913. doi: 10.1073/pnas.1019219108 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106. Crosswhite P, Sun Z. Molecular mechanisms of pulmonary arterial remodeling. Mol Med. 2014;20:191–201. doi: 10.2119/molmed.2013.00165 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107. Palumbo R, Gaetano C, Antonini A, Pompilio G, Bracco E, Ronnstrand L, Heldin CH, Capogrossi MC. Different effects of high and low shear stress on platelet‐derived growth factor isoform release by endothelial cells: consequences for smooth muscle cell migration. Arterioscler Thromb Vasc Biol. 2002;22:405–411. doi: 10.1161/hq0302.104528 [DOI] [PubMed] [Google Scholar]
- 108. Garanich JS, Pahakis M, Tarbell JM. Shear stress inhibits smooth muscle cell migration via nitric oxide‐mediated downregulation of matrix metalloproteinase‐2 activity. Am J Physiol Heart Circ. 2005;288:H2244–H2252. doi: 10.1152/ajpheart.00428.2003 [DOI] [PubMed] [Google Scholar]
- 109. Belo VA, Guimaraes DA, Castro MM. Matrix metalloproteinase 2 as a potential mediator of vascular smooth muscle cell migration and chronic vascular remodeling in hypertension. J Vasc Res. 2015;52:221–231. doi: 10.1159/000441621 [DOI] [PubMed] [Google Scholar]
- 110. Humbert M, Morrell NW, Archer SL, Stenmark KR, MacLean MR, Lang IM, Christman BW, Weir EK, Eickelberg O, Voelkel NF. Cellular and molecular pathobiology of pulmonary arterial hypertension. J Am Coll Cardiol. 2004;43:S13–S24. doi: 10.1016/j.jacc.2004.02.029 [DOI] [PubMed] [Google Scholar]
- 111. Scott D, Tan Y, Shandas R, Stenmark KR, Tan W. High pulsatility flow stimulates smooth muscle cell hypertrophy and contractile protein expression. Am J Physiol Lung Cell Mol Physiol. 2013;304:L70–L81. doi: 10.1152/ajplung.00342.2012 [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 112. Delgado JF, Conde E, Sanchez V, Lopez‐Rios F, Gomez‐Sanchez MA, Escribano P, Sotelo T, Gomez de la Camara A, Cortina J, de la Calzada CS. Pulmonary vascular remodeling in pulmonary hypertension due to chronic heart failure. Eur J Heart Fail. 2005;7:1011–1016. doi: 10.1016/j.ejheart.2004.10.021 [DOI] [PubMed] [Google Scholar]
- 113. Tang BT, Pickard SS, Chan FP, Tsao PS, Taylor CA, Feinstein JA. Wall shear stress is decreased in the pulmonary arteries of patients with pulmonary arterial hypertension: an image‐based, computational fluid dynamics study. Pulm Circ. 2012;2:470–476. doi: 10.4103/2045-8932.105035 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114. Gorter TM, van Veldhuisen DJ, Voors AA, Hummel YM, Lam CSP, Berger RMF, van Melle JP, Hoendermis ES. Right ventricular‐vascular coupling in heart failure with preserved ejection fraction and pre‐ vs. post‐capillary pulmonary hypertension. Eur Heart J Cardiovasc Imaging. 2018;19:425–432. doi: 10.1093/ehjci/jex133 [DOI] [PubMed] [Google Scholar]
- 115. Caravita S, Faini A, Carolino D'Araujo S, Dewachter C, Chomette L, Bondue A, Naeije R, Parati G, Vachiery JL. Clinical phenotypes and outcomes of pulmonary hypertension due to left heart disease: role of the pre‐capillary component. PLoS One. 2018;13:e0199164. doi: 10.1371/journal.pone.0199164 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116. Assad TR, Hemnes AR, Larkin EK, Glazer AM, Xu M, Wells QS, Farber‐Eger EH, Sheng Q, Shyr Y, Harrell FE, et al. Clinical and biological insights into combined post‐ and pre‐capillary pulmonary hypertension. J Am Coll Cardiol. 2016;68:2525–2536. doi: 10.1016/j.jacc.2016.09.942 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117. Alfraidi H, Qanash S, Bshouty Z. Pulmonary arterial hypertension specific therapy in patients with combined post‐and precapillary pulmonary hypertension. Pulm Med. 2018;2018:1–7. doi: 10.1155/2018/7056360 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118. Al‐Omary MS, Sugito S, Boyle AJ, Sverdlov AL, Collins NJ. Pulmonary hypertension due to left heart disease: diagnosis, pathophysiology, and therapy. Hypertension. 2020;75:1397–1408. doi: 10.1161/HYPERTENSIONAHA.119.14330 [DOI] [PubMed] [Google Scholar]
- 119. Sahay S, Khirfan G, Tonelli AR. Management of combined pre‐and post‐capillary pulmonary hypertension in advanced heart failure with reduced ejection fraction. Respir Med. 2017;131:94–100. doi: 10.1016/j.rmed.2017.08.006 [DOI] [PubMed] [Google Scholar]
- 120. Qiu Y, Tarbell JM. Interaction between wall shear stress and circumferential strain affects endothelial cell biochemical production. J Vasc Res. 2000;37:147–157. doi: 10.1159/000025726 [DOI] [PubMed] [Google Scholar]
- 121. Pugh ME, Hemnes AR. Pulmonary hypertension in women. Expert Rev Cardiovasc Ther. 2010;8:1549–1558. doi: 10.1586/erc.10.137 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122. Davezac M, Buscato M, Zahreddine R, Lacolley P, Henrion D, Lenfant F, Arnal J‐F, Fontaine C. Estrogen receptor and vascular aging. Front Aging. 2021;2:2. doi: 10.3389/fragi.2021.727380 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123. Baird GL, Walsh T, Aliotta J, Allahua M, Andrew R, Bourjeily G, Brodsky AS, Denver N, Dooner M, Harrington EO, et al. Insights from the menstrual cycle in pulmonary arterial hypertension. Ann Am Thorac Soc. 2021;18:218–228. doi: 10.1513/AnnalsATS.202006-671OC [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124. Yavropoulou MP, Yovos JG. The molecular basis of bone mechanotransduction. J Musculoskelet Neuronal Interact. 2016;16:221–236. [PMC free article] [PubMed] [Google Scholar]
- 125. Yu JMT, Alp I, Bauer M, Lin M, Drab M, Kurzchalia T, Stan R, Sessa W. Direct evidence for the role of caveolin‐1 and caveolae in mechanotransduction and remodeling of blood vessels. J Clin Investig. 2006;116:1284–1291. doi: 10.1172/jci27100 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126. Mathew R. Critical role of caveolin‐1 loss/dysfunction in pulmonary hypertension. Med Sci. 2021;9:58. doi: 10.3390/medsci9040058 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127. Méndez‐Giménez L, Rodríguez A, Balaguer I, Frühbeck G. Role of aquaglyceroporins and caveolins in energy and metabolic homeostasis. Mol Cell Endocrinol. 2014;397:78–92. doi: 10.1016/j.mce.2014.06.017 [DOI] [PubMed] [Google Scholar]
- 128. Luongo F, Miotti C, Scoccia G, Papa S, Manzi G, Cedrone N, Toto F, Malerba C, Papa G, Caputo A, et al. Future perspective in diabetic patients with pre‐ and post‐capillary pulmonary hypertension. Heart Fail Rev. 2022. doi: 10.1007/s10741-021-10208-4 [DOI] [PubMed] [Google Scholar]
- 129. Yu G, Zou H, Prewitt RL, Hill MA. Impaired arteriolar Mechanotransduction in experimental diabetes mellitus. J Diabetes Complications. 1999;13:235–242. doi: 10.1016/S1056-8727(99)00050-1 [DOI] [PubMed] [Google Scholar]
- 130. Bajpai A, Li R, Chen W. The cellular mechanobiology of aging: from biology to mechanics. Ann N Y Acad Sci. 2021;1491:3–24. doi: 10.1111/nyas.14529 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131. Gliemann L, Rytter N, Lindskrog M, Slingsby MHL, Åkerström T, Sylow L, Richter EA, Hellsten Y. Endothelial mechanotransduction proteins and vascular function are altered by dietary sucrose supplementation in healthy young male subjects. J Physiol. 2017;595:5557–5571. doi: 10.1113/jp274623 [DOI] [PMC free article] [PubMed] [Google Scholar]