

Abstract
Traumatic brain injury (TBI) leads to lasting brain dysfunction with chronic neuroinflammation typified by nucleotide-binding domain leucine-rich repeat and pyrin domain-containing receptor 3 (NLRP3) inflammasome activation in microglia. This study probed whether a single intranasal (IN) administration of human mesenchymal stem cell-derived extracellular vesicles (hMSC-EVs) naturally enriched with activated microglia-modulating miRNAs can avert chronic adverse outcomes of TBI. Small RNA sequencing confirmed the enrichment of miRNAs capable of modulating activated microglia in hMSC-EV cargo. IN administration of hMSC-EVs into adult mice ninety minutes after the induction of a unilateral controlled cortical impact injury resulted in their incorporation into neurons and microglia in both injured and contralateral hemispheres. A single higher dose hMSC-EV treatment also inhibited NLRP3 inflammasome activation after TBI, evidenced by reduced NLRP3, apoptosis-associated speck-like protein containing a CARD, activated caspase-1, interleukin-1 beta, and IL-18 levels in the injured brain. Such inhibition in the acute phase of TBI endured in the chronic phase, which could also be gleaned from diminished NLRP3 inflammasome activation in microglia of TBI mice receiving hMSC-EVs. Proteomic analysis and validation revealed that higher dose hMSC-EV treatment thwarted the chronic activation of the p38 mitogen-activated protein kinase (MAPK) signaling pathway by IL-18, which decreased the release of proinflammatory cytokines. Inhibition of the chronic activation of NLRP3-p38/MAPK signaling after TBI also prevented long-term cognitive and mood impairments. Notably, the animals receiving higher doses of hMSC-EVs after TBI displayed better cognitive and mood function in all behavioral tests than animals receiving the vehicle after TBI. A lower dose of hMSC-EV treatment also partially improved cognitive and mood function. Thus, an optimal IN dose of hMSC-EVs naturally enriched with activated microglia-modulating miRNAs can inhibit the chronic activation of NLRP3-p38/MAPK signaling after TBI and prevent lasting brain dysfunction.
Keywords: human mesenchymal stem cell-derived extracellular vesicles, interleukin-18, traumatic brain injury, mitogen-activated protein kinase signaling, NLRP3 inflammasomes, proinflammatory microglia
Graphical Abstract
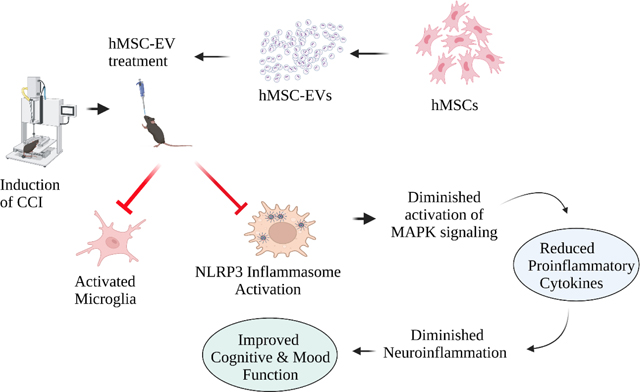
1. Introduction
A moderate traumatic brain injury (TBI) leads to significant morbidity in survivors due to enduring cognitive and neuropsychiatric problems (Stocchetti and Zanier, 2016). Studies have also suggested that TBI is a significant risk factor for developing dementia (Fann et al., 2018; Gorgoraptis et al., 2019). The long-term outcomes of TBI’s depend not only on the extent of initial brain tissue loss but also on the intensity of downstream secondary neuropathological sequelae (Needham et al., 2019; Semple et al., 2019). A chronic neuroinflammatory state typified by nucleotide-binding domain leucine-rich repeat and pyrin domain-containing receptor 3 (NLRP3) inflammasome activation in microglia is one of the striking neuropathological changes observed after TBI (O’Brien et al., 2020). Furthermore, NLRP3 inflammasome activation leads to hyperactivation of p38 mitogen-activated protein kinase (p38/MAPK) signaling, leading to a continuous release of multiple proinflammatory cytokines in the injured brain (O’Brien et al., 2020; Bachstetter et al., 2013; Jiang et al., 2021). There is also evidence that microglia promote enduring neuropathology, neuronal dysfunction, and functional deficits after TBI (Witcher et al., 2021).
Activation of NLRP3 inflammasomes in microglia after TBI is triggered by the increased concentration of danger-associated molecular patterns resulting in activation of nuclear factor-kappa B (NF-kB) (Cassel and Sutterwala, 2010). NLRP3 activation involves the assembly of the pyrin domain (PYD) of NLRP3 with the PYD of apoptosis-associated speck-like protein containing a CARD (ASC), resulting in ASC linking with caspase-1 precursor protein. Such association leads to the activation of caspase-1, which cleaves pro-forms of interleukin 1β (IL-1β) and IL-18 to their active and secreted forms resulting in potentiating inflammatory responses (Heneka et al., 2018; Ising et al., 2019). The continuous release of proinflammatory cytokines IL-1β and IL-18 by activated NLRP3 inflammasomes in microglia can chronically activate the p38/MAPK signaling (Rex, et al., 2020). Indeed, the role of p38/MAPK in creating a chronic proinflammatory milieu after TBI has been observed (Bachstetter et al., 2013; Morganti et al., 2019). Such lingering inflammatory milieu can adversely affect neuronal function and sustain cognitive and mood dysfunction (Ryan and Nolan, 2016). Therefore, therapeutic interventions robustly inhibiting NLRP3 inflammasome activation in microglia are needed to prevent the hyperactivated p38/MAPK signaling from releasing multiple proinflammatory cytokines and causing long-term cognitive and mood impairments after TBI (Henry et al., 2020).
Several studies have reported that intravenous administration of human mesenchymal stem cells (hMSCs) after TBI can curtail acute neuroinflammation (Cozene et al., 2021; Caplan et al., 2021). However, given the recent evidence that MSCs mediate antiinflammatory effects mainly through paracrine actions of their secretome packaged into nanosized extracellular vesicles (EVs) released by them (Baraniak and McDevitt, 2010; Kim et al., 2016), hMSC-EV therapy has received considerable attention for treating neuroinflammation in brain injury and neurodegenerative diseases (Kim et al., 2016; Zhang et al., 2017, 2020b; Long et al., 2017; Patel et al., 2018; Williams et al., 2020; Cone et al., 2021; Moss et al., 2021). However, it is unknown whether hMSC-EV intervention after TBI can thwart chronic neuroinflammation associated with the activation of NLRP3 inflammasomes and p38/MAPK signaling and prevent long-term cognitive and mood impairments.
This study employed EVs spontaneously shed by hMSCs grown in well-standardized culture conditions and isolated through anion-exchange chromatography (AEC). Furthermore, through small RNA sequencing, we validated that such hMSC-EVs were naturally enriched with multiple miRNAs capable of modulating the proinflammatory microglia and inhibiting NLRP3 inflammasome activation. Our results provide novel evidence that a single intranasal (IN) administration of an optimal dose of hMSC-EVs naturally enriched with microglia-modulating miRNAs 90 minutes after a controlled cortical impact injury (CCI) can inhibit NLRP3 inflammasome activation and the release of IL-1β and IL-18. Remarkably, NLRP3 inflammasome inhibition in the acute phase of TBI endured in the chronic phase and prevented a chronic inflammatory state typified by hyperactivated NLRP3-p38/MAPK signaling, releasing an increased concentration of multiple proinflammatory cytokines. Inhibiting NLRP3-p38/MAPK signaling also prevented TBI-induced long-term cognitive and mood impairments.
2. Materials and Methods
2.1. Animals and study design
Young adult male C57BL/6J mice acquired from Jackson Laboratories (Bar Harbor, ME, USA) were employed. All procedures conducted in the study were approved by the Animal Care and Use Committee of Texas A&M University. A schematic displaying the short-term and long-term studies performed after CCI induction is illustrated in Fig. 1. In short-term studies, we investigated the incorporation of intranasally (IN) administered hMSC-EVs by neural cells 6 hours post-EV administration and the efficacy of IN-administered EVs for inhibiting NLRP3 inflammasome activation 48 hours post-EV administration (Fig. 1). In both studies, hMSC-EVs were administered 90 minutes post-TBI with hyaluronidase application at 60 minutes post-TBI. We employed 10 × 109 EVs/animal in the hMSC-EV tracking experiment to determine whether IN-administered hMSC-EVs get incorporated into neurons and glia in the TBI brain (n=4). The dose of EVs and the choice to evaluate EV incorporation at 6 hours post-EV administration were based on our previous findings that examined the biodistribution of intranasally administered EVs in other mouse models (Long et al., 2017; Upadhya et al., 2020). We employed 25.6 × 109 EVs/animal to determine whether a single IN dose of hMSC-EVs would inhibit NLRP3 inflammasome activation in the acute phase of TBI (n=5–6/group). The choice to evaluate the effects of hMSC-EVs 48 hours post-EV administration in this experiment was based on previous findings from both mouse and rat models of CCI that the major components of NLRP3 inflammasome complex (NLRP3, ASC, and caspase-1) increase between 24 hours and 72 hours post-TBI (Liu et al., 2013; Ismael et al., 2018; Xu et al., 2018). The animals for EV tracking studies were perfused with 4% paraformaldehyde, and brain tissue sections were processed for immunofluorescence staining of neuronal and glial cell antigens. The animals for NLRP3 inflammasome studies were euthanized to harvest fresh brain tissues for biochemical assays.
Figure 1: A schematic displaying the various in vivo experiments and analyses performed in the study.

The sequence comprises the induction of controlled cortical impact injury (CCI), intranasal (IN) administration of hyaluronidase at 60 minutes post-TBI, IN administration of hMSC-EVs at 90 minutes post-TBI, and the timeline of studies in the acute and chronic phases of TBI. The experiments in the acute phase of TBI include analyses of hMSC-EV incorporation into neurons and glia at 6 hours post-EV administration and NLRP3 inflammasome activation in the injured brain at 48 hours after vehicle or hMSC-EV administration. The experiments in the chronic phase of TBI comprise cognitive and mood function tests at 63–83 days post-TBI and brain tissue harvesting at 84 days after vehicle or hMSC-EV administration.
For studies in the chronic phase of TBI, we tested the effects of the larger dose used in the acute study (25.6 × 109 EVs) and two smaller doses of hMSC-EVs (6.4 or 12.8 × 109 EVs). The animals were randomly assigned to naïve control (n=15), sham surgery (n=14), or TBI groups (n=59). Animals in TBI groups received an IN dose of vehicle (referred to as TBI or TBI+VEH group, n=15/group) or hMSC-EVs (6.4, 12.8, or 25.6 × 109, n=14–15/group) ninety minutes after the induction of CCI (Fig. 1). The range of doses chosen for this dose-response study was based on our previous study showing that IN administration of 15 × 109 EVs can suppress acute neuroinflammation and maintain better cognitive function after status epilepticus-induced brain injury (Long et al., 2017). Sixty-three days after vehicle or hMSC-EV treatment, animals in all groups were subjected to a series of neurobehavioral tests (Fig. 1). Because the chronic study aimed to assess the effects of a single dose of hMSC-EVs in the acute phase of TBI (i.e., at 90 minutes postTBI) on long-term cognitive and mood impairments, an extended delay between hMSC-EV administration and the commencement of behavioral tests was employed in this study. The animals in the naïve control and sham surgery groups did not receive treatment but were subjected to similar behavioral testing at time points matching the TBI groups. After behavioral tests, subgroups of animals were perfused with 4% paraformaldehyde for immunohistochemical studies (n=7–8/group) or euthanized to harvest fresh brain tissues for biochemical and molecular biological studies (n=7–8/group) (Fig. 1). The investigators who performed the behavioral tests, immunohistochemical and immunofluorescence quantifications, and biochemical and molecular biological analysis were blinded to group identification of animals or samples.
2.2. Induction of CCI
Animals in the TBI group received a unilateral CCI using a device (eCCI Model 6.3; Custom Design and Fabrication at Virginia Commonwealth University Medical Center, Richmond, VA, USA) (Watanabe et al., 2013; Kim et al., 2016). Briefly, the head of each deeply anesthetized mouse was mounted in a horizontal position on a stereotactic frame. A 4 mm diameter craniotomy was performed on the right side of the cranial vault following a midline incision. The center of the craniotomy was at the midpoint between bregma and lambda and 2 mm lateral to the midline overlying the temporoparietal cortex. Animals received a single impact from the device, which delivered a deformation of 0.8 mm depth with a velocity of 5 m/sec and a dwell time of 300 milliseconds using a 3 mm diameter impactor tip. After the injury, a disk made from dental cement was adhered to the skull using tissue adhesive. The scalp was closed with surgical skin clips. The animal was transferred to a heated recovery cage and monitored.
2.3. hMSC cultures, isolation of EVs, and characterization of the number, size, markers, antiinflammatory activity, and morphology of hMSC-EVs
We employed protocols detailed in our previous reports for culturing passage 4 (P4) human bone-marrow-derived MSCs (TAMU-IRM generated MSCs, donor #:6015) and isolating hMSC-EVs from the spent media using AEC (Fig. 2 [A]). Additional methodological details are available in the supplemental file. The concentration and the size of EVs were measured by nanoparticle tracking analysis using NanoSight. The EV marker ALIX and the deep cellular marker GM130 in hMSC-EVs preparations were examined through Western blotting (Upadhya et al., 2020). The primary antibodies were purchased from Santa Cruz (anti-ALIX, 1:1000) and BD Biosciences (anti-GM130, 1:1000). The anti-inflammatory activity of hMSC-EVs was determined using lipopolysaccharide (LPS)-stimulated mouse macrophage assay (Kim et al., 2016; Long et al., 2017). The ultrastructure of EVs was visualized using transmission electron microscopy (TEM) (Upadhya et al., 2020). Additional methodological details are available in the supplemental file.
Figure 2: Human mesenchymal stem cells (hMSC) cultures and isolation and characterization of hMSC-derived extracellular vesicles (EVs) using various methods.

Cartoon A depicts the timelines employed for culturing hMSCs and collecting spent media, hMSC-EV isolation using anion-exchange chromatography (AEC), and characterization of hMSC-EVs using various methods. Graph B depicts the concentration and size of hMSC-EVs obtained through NanoSight analysis. The blots in C demonstrate the presence of EV-specific protein ALIX and the absence of the cytoplasmic protein GM130 in hMSC-EVs. The image in D displays the morphology and size of hMSC-EVs revealed through transmission electron microscopy. Scale bar, 50 nm. The bar chart in E demonstrates the robust antiinflammatory activity of hMSC-EVs on macrophages stimulated with lipopolysaccharide (LPS) compared to the dexamethasone (DEX) treatment. **, p<0.01; ***, p<0.001; ****, p<0.0001. The pie chart in F shows different fractions of small RNAs in hMSC-EVs identified through small RNA sequencing, and bar chart G illustrates the eight validated miRNAs.
2.4. Labeling of hMSC-EVs with PKH26 and IN administration of hMSC-EVs
For labeling hMSC-EVs with PKH26 (Sigma), we followed the manufacturer’s instructions. Briefly, ~40 × 109 EVs in 100 μl of PBS were mixed with 1 ml of diluent C containing 4 μl of PKH26 dye. Following 20 minutes of incubation at 37 degrees C in a shaker, the free dye was removed from the bound dye through ultrafiltration using 10 kDa MWCO filter columns (Vivaspin, Sigma). In parallel, 100 μl of PBS mixed with 1 ml of diluent C containing 4 μl of PKH26 dye was processed with identical incubation and filtration steps as the PKH26-labeled EVs. This solution was employed in neuronal cell culture studies to determine any free dye presence. No red particles (free PKH26 particles) were seen in human neuronal cultures incubated with PKH26 dye solution undergoing the identical incubation and filtration steps as the PKH26-labeled EVs. See supplemental figure 1. IN administration of hMSC-EVs was performed bilaterally, as described in our recent reports (Upadhya et al., 2020; Kodali et al., 2020).
2.5. Small RNA-sequencing of hMSC EVs and miRNA pathway analysis
Texas A&M Institute for Genome Sciences and Society Genomics Core (College Station, TX, USA) performed small RNA (smRNA) sequencing hMSC-EVs from 2 biological replicates. Details on the isolation of smRNA, and sequencing methodology, including the creation, size selection, quantification, trimming, and filtering of smRNA libraries, are detailed in the supplemental file. The top 20 miRs exhibiting ≥42 reads in two biological replicates were subjected to enrichment pathway analysis using DIANA-279 miRPath v.3 (Vlachos et al., 2015). Since many predicted targets are false positives, we included only experimentally validated human target genes using the Tarbase v7.0 database. The miR-ID was based on Mirbase version 21. The alignments were visually assessed for miRs with ambiguous IDs, and the correct designations were determined. The functional annotation was performed according to the Gene Ontology databases. We used the “Pathways/Categories Union” algorithm to assess the combined action of selected miRs. Fisheŕs exact test was chosen for enrichment analysis, with a micro threshold of 0.8, false discovery rate (FDR) correction, and p-value threshold at ≤ 0.01. The heat map of miRs versus pathways was based on significance values, where darker colors represent lower p values.
2.6. Validation of selected miRNA using quantitative real-time PCR
Among the top 20 enriched miRs from RNA-Seq data, eight miRs that are known to modulate activated microglia and/or NLRP3 inflammasomes were chosen and validated through quantitative real-time PCR. The total RNA was isolated from two biological replicates of hMSC-EVs (10 × 109 each) using the SeraMir Exosome RNA amplification kit (System Biosciences, Palo Alto, CA, USA). miRCURY LNA RT Kit (Qiagen, Germantown, MD, USA) was employed for converting 5 ng/μl of total RNA into cDNA. miRCURY LNA miRNA SYBR Green PCR kit (Qiagen) and miRCURY LNA miRNA PCR assay primer mix (Qiagen) were used to measure the comparative expression of 8 different miRs.
2.7. Measurement of PKH26-labeled hMSC-EVs within neurons and microglia
Single and dual immunofluorescence methods and Z-section analysis in a confocal microscope were employed to visualize and quantify neurons and microglia containing PKH26+ hMSC-EVs (red particles). The sections from animals euthanized 6 hours after an IN administration of PKH-26 labeled hMSC-EVs were employed. The brain sections were processed for neuron-specific nuclear antigen (NeuN) staining alone, NeuN and CD63 or CD81 (markers of EVs), ionized calcium-binding adaptor protein 1 (IBA-1) alone, or IBA-1 and CD63 or CD81. Every 15th 30 μm thick section through the entire forebrain was used from each animal (n=4) for NeuN or IBA-1 immunofluorescence studies. Representative sections from each animal were utilized for NeuN and CD63 or CD81 or IBA-1 and CD63 or CD81 dual immunofluorescence studies. The methods and antibodies used are detailed in the supplemental file.
2.8. Analyses of cognitive and mood function using behavioral tests
The mice from all groups were first probed with an open field test (OFT) to ascertain the presence of any motor impairments in TBI groups compared to other groups. Then, the animals were probed with two behavioral tests to measure cognitive function. An object location test (OLT) assesses the proficiency of animals to discern subtle changes in their immediate environment, a cognitive function dependent on the hippocampus (Warburton and Brown, 2015). A pattern separation test (PST) measured the ability of animals to transform highly similar sensory inputs into distinct, dissimilar representations, a function dependent on the dentate gyrus and the extent of neurogenesis (Jain et al., 2012; van Goethem et al., 2018). Mood function was assessed through a sucrose preference test (SPT) examining anhedonia or depression-like behavior and a novelty-suppressed feeding test (NSFT) probing anxiety-like behavior. Anhedonia in SPT is evident from a decreased preference for sweet fluids such as sucrose or saccharin solution (Liu et al., 2018). Anxiety-like behavior in an NSFT is apparent from an increased latency to taking the first bite of food in a novel environment following fasting for 24 hours (Samuels and Hen, 2011). The detailed protocols employed for OLT, PST, SPT, and NSFT are available in our previous reports (Long et al., 2017; Upadhya et al., 2019; Shetty et al., 2020) and the supplemental file.
2.9. Quantification of microglia and astrocytes
The detailed methods employed for brain tissue processing are available in our previous reports (Rao et al., 2006; 2008; Hattiangady et al., 2011). The supplemental file details the immunohistochemical staining of brain tissue sections using specific primary antibodies. Three 30 μm thick sections separated by ~450 μm were employed from each animal (n=5–6/group) to quantify the numbers of glial fibrillary acidic protein (GFAP)-positive astrocytes and IBA-1-positive microglia in the corpus callosum (CC), the somatosensory cortex (SSC), and different subfields of the hippocampus. Both GFAP+ and IBA-1+ cell counts were expressed as the number per 0.1 mm3. All cell counts utilized the optical fractionator method using the StereoInvestigator system (Microbrightfield, Williston, Vermont, USA). The detailed stereological methods employed are described in our earlier reports (Hattiangady et al., 2007; 2011; Kodali et al., 2015; 2016).
2.10. Quantification of markers of activated NLRP3 inflammasomes and pro-and anti-inflammatory cytokines
The lysates from the hippocampus and/or the frontoparietal cortex and individual enzyme-linked immunoassay (ELISA) kits were employed for quantifying the various markers of NLRP3 inflammasomes and proinflammatory cytokines (n=6/group), as detailed in our previous study (Madhu et al., 2021). The lysates from regions ipsilateral to the CCI were employed in TBI groups. The methodology used for preparing lysates is described in the supplemental file. The kits for NF-kB and nuclear subunit of NF-kB (NF-kB p65; Aviva Systems Biology, San Diego, CA, USA), NLRP3 (Abcam, Cambridge, MA, USA), ASC (MyBioSource, San Diego, CA, USA), cleaved caspase-1 (BioVision Inc, Milpitas, CA, USA), IL-18, IL-1β, IL-4, and IL-10 (R&D Systems, Minneapolis, MN, USA) were utilized.
2.11. Measurement of proinflammatory microglia and NLRP3 inflammasomes in microglia
Dual or triple immunofluorescence staining and Z-section analysis in a Nikon confocal microscope or Leica THUNDER 3D Imager were employed for the following measures. 1) Percentages of proinflammatory microglia expressing IBA-1 and CD68 (Madhu et al., 2021). 2) Number of NLRP3 inflammasomes per 216 μm2 area (i.e., structures co-expressing NLRP3 and ASC) and percentages of microglia with NLRP3 inflammasomes (i.e., cells positive for IBA-1, NLRP3, and ASC) in SSC and the CA1 stratum radiatum (Madhu et al., 2021). The methods and antibodies employed for these studies are detailed in the supplemental file. Three to six representative sections from each animal (n=5–6/group) were used for these measurements.
2.12. Proteome profiling of hippocampal tissues
Snap frozen hippocampal tissues from naïve control mice and mice receiving vehicle or hMSC-EVs after TBI were employed (n=3/group). In TBI groups, the tissues ipsilateral to the CCI were utilized. The preparation and loading of samples and the various methods, software, and analyses employed to select peptides in proteome profiling of hippocampal tissues are detailed in the supplemental file. The Peptide Spectrum Matches (PSMs) output from PD2.1 was used to group peptides onto gene levels using the ‘gpGrouper’ algorithm (Saltzman et al., 2018). An in-housed program, gpGrouper, utilizes a universal peptide grouping logic to accurately allocate and provide MS1-based quantification across multiple gene products. Gene-protein products (GPs) quantification was performed using the label-free, intensity-based absolute quantification (iBAQ) approach and then normalized to iFOT (a fraction of the total protein iBAQ amount per experiment). iFOT was defined as an individual protein’s iBAQ divided by the total iBAQ of all identified proteins within one experiment. Furthermore, selected GPs were subjected to String analysis (Szklarczyk D, et al. 2019) to dissociate the functional correlation of significantly altered proteins.
2.13. Validation of proteomics data on the activation of p38 MAPK signaling pathway
The lysates from the frontoparietal cortex and the hippocampus were utilized for quantifying the various markers involved in IL-18 and IL-1β mediated activation of the p38 MAPK signaling pathway and related cytokines using individual ELISA kits (n=6/group). The lysates from regions ipsilateral to the CCI were employed in TBI groups. The kits for myeloid differentiation primary response 88 (MyD88, Aviva Systems Biology), a small GTPase rat sacrcoma virus (Ras, MyBioSource), p38 MAPK (Cell Signaling, Danvers, MA, USA), activator protein-1 (AP-1, Novus Biologicals, Centennial, CO; USA), IL-6 (R&D systems), and IL-8 (Biomatik, Wilmington, DE, USA) were employed. The ELISA methods used are followed as per manufacture instructions.
2.14. Statistical analyses
Comparisons between two datasets employed a two-tailed, unpaired Student’s t-test or Mann-Whitney U-test when standard deviations differed significantly between the groups. Comparisons involving three or more datasets used one-way analysis of variance (ANOVA) with Tukey’s multiple comparison post hoc tests. When individual groups failed the normality test (Shapiro-Wilk test), we performed the Kruskal Wallis test followed by Dunn’s post hoc tests. In all comparisons, p<0.05 was considered a statistically significant value.
3. Results
3.1. AEC-purified hMSC-EVs expressed the EV-specific marker ALIX, lacked the cytoplasmic protein GM130, and displayed small EV morphology in TEM
The timelines and protocols employed for culturing and expanding bone marrow-derived hMSCs, isolation of EVs from spent media using AEC, and characterization of hMSC-EVs are presented in Fig. 2 [A]. NanoSight analysis suggested that the size of hMSC-EVs ranged from 60–200 nm in diameter with a mean diameter of 102 nm (Fig. 2 [B]). Our previously published flow cytometry investigations have already demonstrated the presence of tetraspanins CD63 and CD81 on the surface of these EVs (Kim et al., 2016). In this study, western blot analysis also demonstrated the expression of the EV-specific marker ALIX but not the cytoplasmic protein GM130 in our EV preparations (Fig. 2 [C]). Furthermore, TEM demonstrated both clusters of EVs and individual EVs with characteristic small EV morphology, i.e., vesicles ranging in diameter from 60–200 nm and enclosed by double membranes (Fig. 2 [D]). Thus, EV preparations employed in the study comprised bonafide EVs.
3.2. AEC-purified hMSC-EVs are antiinflammatory and enriched with miRNAs capable of modulating proinflammatory microglia
Evaluation of hMSC-EVs in LPS-stimulated mouse macrophage cultures demonstrated their antiinflammatory activity. LPS exposure alone greatly enhanced the release of IL-6 from macrophages (Fig. 2 [E]). However, elevated IL-6 released by activated macrophages could be inhibited by dexamethasone or low doses (1–2 × 109) of two different biological replicates of hMSC-EVs (Fig. 2 [E]). Analysis of biological replicates of hMSC-EVs with small RNA (smRNA) sequencing revealed that both preparations contained similar fractions of miRNAs and other RNAs (Fig. 2 [F] and supplemental Table 1). Furthermore, the most enriched miRNAs, i.e., the top 20 miRNAs having 42 or more reads and having antiinflammatory activity, were found in both replicates. The miRNAs showing ≥5 reads are listed in supplemental Table 2, and the top 20 miRs with ≥42 reads with their biological functions are illustrated in supplemental Fig. 2. Many enriched miRNAs found in hMSC-EVs have robust antiinflammatory and neuroprotective activity, including NLRP3 inflammasome inhibition, modulating the proinflammatory microglia, neuroprotection, or BBB repair (Table 1). Among those, we validated the occurrence of 8 miRNAs in two biological replicates of hMSC-EVs through qPCR (Fig. 2 [G]). These include a miRNA proficient for inhibiting NLRP3 inflammasomes (miR-30e-3p; Li et al., 2018) and many miRNAs capable of modulating proinflammatory microglia, including miR-654–3p (Tu et al., 2019), miR-26b-5p (Kang et al., 2018), let-7c-5p (Lv et al., 2018) and let-7g-5p (Rom et al., 2015; Bernstein and Rom, 2020). The other validated miRNAs have neuroprotective properties (miR-335–5p and miR-494–3p; Liu et al., 2015; Zhao et al., 2019) or can repair BBB (miR-98–5p; Bernstein et al., 2020).
Table 1.
Top 20 miRNAs with ≥42 reads in two biological replicates of hMSC-EVs
| miRNA | Known Biological Functions |
|---|---|
|
| |
| 1) hsa-miR-100-3p | Alleviates neurodegeneration and activation of microglia (Li et al., 2019) |
| 2) hsa-miR-26b-5p | Regulates microglial inflammatory response by targeting IL-6 after brain injury (Kang et al., 2018). |
| 3) hsa-let-7c-5p | Inhibits microglial activation and alleviates neuroinflammation (Li et al., 2018). |
| 4) hsa-miR-335-5p | Reduces neurodegeneration after brain injury (Liu et al., 2015). |
| 5) hsa-miR-423-3p | Exerts antiapoptotic effects by inhibiting caspase-6 (Li et al., 2014) |
| 6) has-let-7i-5p | Protects brain microvascular endothelial cells against oxygen glucose deprivation by decreasing toll-like receptor 4 expression (Xiang et al., 2017). |
| 7) hsa-miR-409-3p | Promotes microglial migration and activation (Hu et al. 2021) |
| 8) hsa-let-7g-5p | Reduces the production of proinflammatory cytokines and repairs leaky blood-brain barrier after brain injury (Rom et al. 2015; Bernstein and Rom, 2020). |
| 9) hsa-miR-494-3p | Reduces neurodegeneration by inhibiting histone deacetylase 3 after brain injury (Zhao et al., 2019) |
| 10) hsa-miR-654-3p | Regulates macrophage migration inhibitor factor (MIF), inhibits the phosphorylation of ERK and AKT and reduces downstream inflammatory cytokine production (Tu et al., 2019). |
| 11) hsa-miR-10b-5p | Regulates brain-derived neurotrophic factor expression (Varendi et al., 2014). |
| 12) hsa-miR-320a | Regulates reactive astrocyte-mediated neuronal damage by regulating ATP release (Fatima et al., 2017). |
| 13) hsa-miR-152-3p | Protects neurons from oxygen-glucose deprivation by upregulating NRF-2 (Zhang et al., 2019). |
| 14) hsa-miR-98-5p | Reduces endothelial dysfunction, protects blood-brain barrier, and improves neurological outcomes after brain injury (Bernstein et al., 2020). |
| 15) hsa-miR-10a-5p | Regulates brain-derived neurotrophic factor expression (Zhang et al., 2020a) |
| 16) hsa-miR-30a-3p | Loss of miR-30a-3p after controlled cortical injury in rats induces neuropathic pain via elevated E-cadherin transcriptional activator enhancing BDNF (Tan et al., 2020). |
| 17) hsa-miR-30e-5p | Targets the 3'UTR region of Nlrp3 to negatively regulate Nlrp3 mRNA and protein expression (Li et al., 2018). |
| 18) hsa-miR-30d-5p | Inhibits autophagy-mediated microglial polarization into M1 phenotype (Jiang et al.,2018). |
| 19) hsa-miR-30e-3p | Targets the 3'UTR region of Nlrp3 to negatively regulate Nlrp3 mRNA and protein expression (Li et al., 2018). |
| 20) hsa-miR-30a-5p | Targets MAPK/ERK pathway and ameliorates inflammatory injury (Fu et al., 2018). |
3.3. IN administered hMSC-EVs incorporated into neurons and microglia bilaterally in the brain
In mice subjected to CCI, bilateral IN administration of PKH26-labeled hMSC-EVs resulted in their incorporation into neurons and microglia bilaterally in virtually all brain regions by six hours, as observed earlier for naïve mice and mice with status epilepticus-induced brain injury (Long et al., 2017; Upadhya et al., 2020). Incorporation of EVs by NeuN+ neurons and IBA-1+ microglia in the SSC and CA1 and CA3 subfields of the hippocampus, both ipsilateral and contralateral to the CCI are illustrated (Fig. 3 [A–L]). The percentages of neurons and microglia incorporating EVs on the ipsilateral and contralateral sides of the CCI were mostly similar with minor differences observed for neurons (Fig. 3 [M–N]). However, measurement of the area fraction of PKH26+ structures in hippocampal CA1 and CA3 cell layers revealed a higher density of EVs ipsilateral to the CCI (Fig. 3 [O]). Analysis using CD63 and CD81 confirmed that PKH26+ red particles found within neurons and microglia are indeed IN administered EVs (Fig. 4 [A–D]).
Figure 3: Intranasally administered hMSC-EVs incorporated into neurons and microglia in the brain on both ipsilateral and contralateral sides of the controlled cortical impact injury (CCI).

Figures A-L show the incorporation of PKH26-labeled hMSC-EVs (red particles, some indicated by arrows) into NeuN+ neurons (A-F) and IBA-1+ microglia (G-L) in the somatosensory cortex (A-B, G-H), the hippocampal CA1 subfield (C-D, I-J), and the CA3 subfield (E-F, K-L) on both ipsilateral and contralateral sides of the CCI at 6 hours post-administration. The bar charts in M-N compare the percentage of neurons (M) and microglia (N) incorporating hMSC-EVs in the SSC, CA1, and CA3 regions between the ipsilateral and contralateral sides of the CCI. The bar chart in O compares the area fraction (AF) of PKH26+ hMSC-EVs in the hippocampal CA1 and CA3 pyramidal cell layers between the ipsilateral and contralateral sides of the CCI. Scale bar, A-L = 25μm. *, p<0.05. NS, not significant.
Figure 4: Panels A-D: Intranasally administered hMSC-EVs incorporating into neurons and microglia in the TBI brain expressed CD63 and CD81.

The image panels in A-B demonstrate that virtually PKH26+ structures (red) found within NeuN+ neurons and IBA-1+ microglia (blue) are positive for the EV-specific marker CD63 (green). The image panels C-D show that virtually all PKH26+ structures (red) in NeuN+ neurons and IBA-1+ microglia (blue) are positive for the EV-specific marker CD81 (green). Arrows in panels A-D point to some PKH26+ structures that are positive for CD63 and CD81. Scale bar, 10 μm. Bar charts E-K: Intranasal administration of 25.6 × 109 hMSC-EVs after TBI blocked the activation of NLRP3 inflammasomes and reduced the release of IL-1β and IL-18 in the acute phase of TBI. The bar charts in E-I compare the concentration of NF-kB (E) NF-kB p65 (F) and the various markers of NLRP3 inflammasome activation, including NLRP3 (G), ASC (H), and cleaved caspase-1 (I) between naïve, TBI and TBI+EV groups. The bar charts J-K compare the concentration of IL-1β (J) and IL-18 (K) across the three groups. *, p<0.05; **, p<0.01; ***, p<0.001; ****, p<0.0001; NS, not significant.
3.4. hMSC-EV administration inhibited NLRP3 inflammasome activation in the acute phase of TBI
We ascertained whether a single IN administration of 25.6 × 109 hMSC-EVs ninety minutes after TBI can inhibit NLRP3 inflammasome activation in the acute inflammatory phase of TBI. We compared the various markers of NLRP3 inflammasome activation in the SSC and hippocampus (SSC+HPS) tissues across naïve, TBI+VEH, and TBI+EV groups at 48 hours post-EV administration. The concentrations of NF-kB, NF-kB p65, the different components of the NLRP3 inflammasome complex (NLRP3, ASC, and cleaved caspase-1), and the end products of NLRP3 inflammasome activation (IL-1β and IL-18) were measured for this comparison. One-way ANOVA analyses revealed significant differences between the three groups for concentrations of both total NF-kB and NF-kB p65, with the TBI group showing upregulation compared to the naive control group and the TBI-EVs group exhibiting downregulation compared to the TBI group (Fig. 4 [E–F]). Tukey’s posthoc tests revealed statistically significant differences between naïve and TBI groups and TBI and TBI+EVs groups for the total NF-kB but not for the NF-kB p65 (Fig. 4 [E–F]). Nonetheless, NLRP3, ASC, and cleaved caspase-1 levels showed significant differences between groups (Fig. 4 [G–I]). All three proteins were significantly upregulated in the TBI group compared to the naïve control group and were significantly reduced in the TBI+EVs group compared to the TBI group (Fig. 4 [G–I]), implying a robust inhibition of NLRP3 inflammasomes. Such inhibition resulted in reduced IL-1β and IL-18 levels in the TBI+EVs group compared to the TBI group (Fig. 4 [J–K]). Also, the concentrations of NLRP3, cleaved caspase-1, IL-1β, and IL-18 in the TBI+EV group were normalized to naïve control levels (Fig. 4 [G, I–K]). Additional statistical details such as means, confidence intervals, degrees of freedom, and ANOVA p values for different comparisons are detailed in Table 3 of the supplemental file. Thus, a single higher dose of hMSCEV treatment after TBI can inhibit NLRP3 inflammasome activation in the acute phase of TBI.
3.5. Studies on the effects of hMSC-EV administration 90 minutes after TBI on long-term cognitive and mood impairments associated with NLRP3-p38/MAPK signaling
We next determined whether NLRP3 inflammasome inhibition observed in the acute phase of TBI after a higher dose of hMSC-EV treatment is long-lasting. We focused on testing whether such inflammasome inhibition in the acute phase would be efficacious in preventing the TBI-induced long-term cognitive and mood impairments allied with chronic neuroinflammation in the injured brain typified by hyperactivated NLRP3-p38/MAPK signaling, releasing an increased concentration of multiple proinflammatory cytokines. In these studies, to determine the dose-dependent effects of hMSC-EVs, adult mice received IN treatment of different doses of hMSC-EVs (6.4, 12.8, or 25.6 × 109 hMSC-EVs) ninety minutes after the induction of TBI. The animals were investigated with a battery of behavioral tests 63 days post-TBI. The brain tissues were then evaluated for the extent of neuroinflammation, particularly the activation of NLRP3 inflammasomes and p38/MAPK signaling.
3.6. hMSC-EV administration after TBI prevented long-term cognitive and mood impairments
Animals in all groups were first evaluated for motor impairments using an OFT 63 days after the induction of TBI and IN administration of VEH or hMSC-EVs. One-ANOVA analysis with Tukey’s posthoc tests for distances traveled across groups revealed that neither the sham TBI group nor any TBI groups significantly differed from the naïve control group (Fig. 5 [A]). This finding implied no significant motor impairments were present in TBI groups during cognitive function testing. Next, we investigated hippocampus-dependent cognitive functions in animals with an OLT and a PST. The OLT measures the proficiency of animals to discern minor changes in their immediate environment (Fig. 5 [B]), and competency in this task relies heavily on hippocampal activity, notably the dorsal CA1 region (Assini et al., 2009). Animals proficient in object location memory display an increased propensity to inspect the object repositioned to a new location (i.e., the object in a novel place, OINP) compared to the object in the familiar place (OIFP) in the OLT. While naïve animals and animals with sham TBI surgery showed such competence, the animals receiving VEH or a lower dose of EVs (6.4 × 109) after TBI were impaired (Fig. 5 [C]). Notably, animals receiving higher doses of hMSC-EVs (12.8 or 25.6 × 109) after TBI displayed competency in discriminating the OINP from the OIFP (Fig. 5 [C]). Furthermore, a comparison of the OINP discrimination index (DI) across groups using one-way ANOVA with post hoc tests revealed that the DI in TBI mice receiving all doses of EVs was similar to naïve mice. However, the DI values in the TBI+VEH was lower than the DI in naïve mice (Fig. 5 [D]). The total object exploration times (TOETs), the total distances traveled, and the velocities of movement in T2 and TOETs in T3 are illustrated in the supplemental Fig. 3 [A–D]. Animals were next investigated for pattern separation ability using a PST, as described in our previous report (Long et al., 2017; Fig. 5 [E]). A competence for discriminating similar but not identical experiences by storing similar representations in a non-overlapping manner is required for pattern separation (Leutgeb et al., 2007). Such function depends on the integrity of the DG-CA3 circuitry in the hippocampus (Gandy et al., 2017). Animals proficient in pattern separation function explore the novel object on pattern 2 (NO on P2) for longer durations than the familiar object on pattern 2 (FO on P2) in the PST. Such competence was intact in naïve animals and animals with sham TBI surgery but was impaired in animals receiving VEH or lower EV dose (6.4 × 109 EVs) after TBI (Fig. 5 [F]). Remarkably, the animals receiving higher EV doses (12.8 or 25.6 × 109 EVs) exhibited proficiency in discriminating NO on P2 from FO on P2 (Fig. 5 [F]). A comparison of the NO on P2 discrimination across groups using one-way ANOVA with post hoc tests revealed that the DI in TBI mice receiving higher doses of EVs was similar to naïve mice (Fig. 5 [G]). However, the DI values in the TBI+VEH and TBI+6.4 × 109 EV groups were significantly lower than the DI in naïve mice (Fig. 5 [G]). The TOETs, total distances traveled, and velocities of movement in T2 and T3, and TOETs in T4 are illustrated in the supplemental Fig. 3 [E–K].
Figure 5: Intranasal hMSC-EV treatment after TBI prevented long-term cognitive and mood impairments.

Bar chart A compares total distances traveled in an open field test between naïve control, TBI, and different TBI+EV groups. Cartoon B depicts different trials (T1-T3) in an object location test. The bar charts in C compare percentages of object exploration times spent with the object in the familiar place (OIFP) vis-à-vis the object in the novel place (OINP) in naïve (N), sham surgery (Sham), TBI, and TBI+6.4, 12.8, or 25.6 × 109 EV groups. Bar chart D compares the OINP discrimination index across different groups. Cartoon E depicts different trials (T1-T4) in a pattern separation test. The bar charts in F compare percentages of object exploration times spent with the familiar object on pattern 2 (FO on P2) vis-à-vis the novel object on P2 (NO on P2) in naïve (N), sham surgery (Sham), TBI, and TBI+6.4, 12.8, or 25.6 × 109 EV groups. Bar chart G compares the NO on P2 discrimination index across different groups.
The bar charts H and I compare the sucrose preference rate (SPR, H) and total fluid consumption (I) across different groups. The bar chart J compares latencies to the first bite of food in a novelty-suppressed feeding test between different groups. *, p<0.05; **, p<0.01; ***, p<0.001; and ****, p<0.0001; NS, not significant.
The animals were also investigated for mood function using an SPT and an NSFT. The SPT was employed to evaluate the extent of anhedonia (Fig. 5 [H]), which is a condition typified by a diminished ability to perceive pleasure in activities that are pleasurable in normal healthy conditions and serves as a measure of depressive-like behavior. Animals exhibiting anhedonia do not prefer sucrose-containing water over the standard water; hence, the sucrose preference rate (SPR) is measured in this test. Comparison across groups using one-way ANOVA with post hoc tests revealed that the SPRs in TBI mice receiving different doses of EVs were similar to naïve mice. However, the SPR in the TBI+VEH group was significantly lower than the SPR in naïve mice (Fig. 5 [H]). The overall fluid consumption did not vary between different groups (Fig. 5 [I]), implying that the results of the SPT were not due to variable consumption of the liquid by animals in different groups. The comparative consumption of sucrose-containing water and standard water in different groups is illustrated in the supplemental Fig. 3. The NSFT was used to evaluate the extent of anxiety-like behavior. Animals exhibiting anxiety-like behavior will have longer latencies to eating familiar food in a novel environment (Merali et al., 2003). Comparison across groups using one-way ANOVA with post hoc tests revealed that latencies to the first bite in TBI mice receiving higher doses of EVs (12.8 or 25.6 × 109 EVs) were similar to naïve mice. However, the latencies to the first bite in the TBI+VEH and TBI+6.4 × 109 EV groups were significantly higher than those observed for naïve mice (Fig. 5 [J]). Additional statistical details such as means, confidence intervals, degrees of freedom, and ANOVA p values for different comparisons are detailed in Table 4 of the supplemental file. Thus, IN administration of moderate to higher hMSC-EV doses after TBI preventfig ed long-term cognitive and mood impairments.
3.7. hMSC-EV administration after TBI reduced densities of astrocytes and proinflammatory microglia in the chronic phase
We first measured densities of glial-fibrillary acidic protein-positive (GFAP+) astrocytes and ionized calcium binding adaptor molecule 1-positive (IBA-1+) microglia in the somatosensory cortex (SSC), corpus callosum (CC), and the hippocampal CA1 subfield. Compared to the naïve control group, the density of GFAP+ astrocytes and IBA-1+ microglia in the TBI+VEH group was increased in all regions. However, in TBI groups receiving 12.8 or 25.6 × 109 EVs, the densities of astrocytes and microglia were similar to naïve control levels in most areas. See supplemental Fig. 4 and supplemental table 9 for additional information. Next, we visualized proinflammatory microglia through dual immunofluorescence for IBA-1 and CD68 in the SSC (Fig. 6 [A–D]) and the hippocampal CA1 subfield (Fig. 6 [E–H]). In TBI groups, measurements were performed on regions ipsilateral to the injury. ANOVA with post hoc tests revealed that in both SSC and the CA1 subfield, compared to the naïve control group, the TBI+VEH and TBI+6.4 × 109 EV groups exhibited increased percentages of the proinflammatory phenotype (Fig. 6 [I–J]). Remarkably, the percentages of the proinflammatory phenotype in TBI+12.8 or 25.6 × 109 EV groups matched the percentage observed in the naïve control group. Also, compared to the TBI+VEH group, percentages of the proinflammatory phenotype were significantly reduced in the TBI+12.8 or 25.6 × 109 EV groups (Fig. 6 [I–J]). Additional statistical details such as means, confidence intervals, degrees of freedom, and ANOVA p values for different comparisons are detailed in Table 4 of the supplemental file.
Figure 6: Intranasal hMSC-EV treatment after TBI led to a lower ratio of proinflammatory microglia in the chronic phase.

The image panels in A-H illustrate examples of IBA-1+ microglia (green) expressing CD68 (red) in the somatosensory cortex (SSC; A-D) and the hippocampal CA1 subfield (E-H). Panels A and E, naïve group; B and F, TBI group; C and G, TBI+6.4 × 109 EVs group; D and H, TBI+25.6 × 109 EVs group. The bar charts in I and J compare percentages of IBA-1+ microglia with CD68 in the SSC (I) and the hippocampal CA1 subfield (J) between different groups. Scale bar, A-H = 25 μm; ****, p<0.0001; NS, not significant.
3.8. hMSC-EV administration after TBI prevented the chronic activation of NLRP3 inflammasomes and enhanced antiinflammatory cytokines
We first visualized the activation of NLRP3 inflammasomes in microglia within the SSC (Fig. 7 [A–D]) and the CA1 subfield (Fig. 7 [E–H]) through triple immunofluorescence for NLRP3, ASC, and IBA-1. In TBI groups, measurements were performed on regions ipsilateral to the injury. In SSC, compared to the naïve group, the microglia with inflammasomes increased in the TBI+VEH, TBI+6.4 or 12.8 × 109 EV groups, but not in the TBI+25.6 × 109 EV group (Fig. 7 [I]). In the CA1 subfield, the percentage of microglia with inflammasomes was increased in the TBI+VEH group but not in TBI groups receiving higher doses of EVs (Fig. 7 [J]). Thus, the highest dose of hMSC-EVs reduced the percentage of microglia containing NLRP3 inflammasomes in the SSC. In contrast, both moderate and highest (12.8 or 25.6 × 109 EVs) doses of EVs tested in the study effectively reduced the percentage of microglia with NLRP3 inflammasomes in the CA1 subfield.
Figure 7: Intranasal hMSC-EV treatment after TBI blocked the chronic inflammasome activation in microglia and increased the concentration of antiinflammatory cytokines.

The image panels in A-H illustrate examples of nucleotide-binding domain leucine-rich repeat and pyrin domain-containing receptor 3 (NLRP3) inflammasome complexes co-expressing NLRP3 (green) and apoptosis-associated speck-like protein containing a CARD (ASC, red) in IBA-1+ microglia from the somatosensory cortex (SSC; A-D) and the CA1 stratum radiatum (E-H). Panels A and E, naïve group; B and F, TBI group; C and G, TBI+6.4 × 109 EVs group; D and H, TBI+25.6 × 109 EVs group. The bar charts I-J compare the percentages of microglia with inflammasomes in the SSC (I) and CA1 subfield (J) between different groups. The bar charts K-O compare concentrations of the NF-kB (K), NF-kB p65 (L), NLRP3 (M), ASC (N), and cleaved caspase-1 (O) between different groups. The bar charts P-Q compare the levels of end products of NLRP3 inflammasomes interleukin-1 beta (IL-1β; P) and IL-18 (Q), whereas the bar charts R-S compare the concentration of antiinflammatory cytokines IL-4 (R) and IL-10 (S) between different groups. Scale bar, A-H = 25 μm; *, p<0.05; **, p<0.01; ***, p<0.001; ****, p<0.0001; NS, not significant.
We next measured the concentrations of NF-kB, NF-kB p65, NLRP3, ASC, cleaved caspase-1, IL-1β, and IL-18 in the SSC ipsilateral to the CCI. One-way ANOVA analyses revealed significant differences between groups for concentrations of both total NF-kB and NF-kB p65, with the TBI group showing upregulation compared to the naive control group and the TBI-EVs group exhibiting downregulation compared to the TBI group (Fig. 7 [K–L]). Tukey’s posthoc tests revealed statistically significant differences between naïve and TBI groups and TBI and TBI+25.6 × 109 EV groups for the total NF-kB but not for the p65 NF-kB (Fig. 7 [K–L]). One-way ANOVA analyses also demonstrated significant differences between the five groups for concentrations of NLRP3, ASC, and cleaved caspase-1 (Fig. 7 [M–O]). Tukey’s posthoc tests revealed statistically significant differences between the naïve and TBI groups and the TBI and TBI+25.6 × 109 EV groups for NLRP3 and ASC (Fig. 7 [M–N]). However, cleaved caspase-1 levels in TBI groups receiving 6.4, 12.8, or 25.6 × 109 EVs were reduced compared to the TBI group (Fig. 7 [O]). Also, in TBI groups receiving 6.4, 12.8, or 25.6 × 109 EVs, NF-kB, ASC, and cleaved caspase-1 were normalized to naive control levels (Fig. 7 [M–O]). Compared to the naïve control group, the IL-1β and IL-18 levels were elevated in the TBI group but not in the TBI+6.4, 12.8, or 25.6 × 109 EV groups (Fig. 7 [P–Q]). The IL-1β concentration was also reduced in the TBI+25.6 × 109 EV group compared to the TBI group (Fig 7 [P]). In summary, the TBI group displayed significantly higher levels of NF-kB, NLRP3, ASC, IL-1β, and IL-18 than the naïve control group. Notably, levels of these proteins and caspase-1 were similar to naïve control levels in TBI groups receiving 6.4, 12.8, or 25.6 × 109 EVs, implying that all doses of EVs tested in the study were proficient in reducing inflammasome activation.
Furthermore, compared to the naïve group, the antiinflammatory cytokine IL-4 in the SSC ipsilateral to the CCI was decreased in the TBI+VEH, TBI+6.4, or 12.8 × 109 EV groups but not in the TBI+ 25.6 × 109 EV group (Fig. 7 [R]). Also, the antiinflammatory cytokine IL-10 was decreased in the TBI+VEH group compared to the naïve group but not in TBI groups receiving different doses of EVs (Fig. 7 [S]). Thus, a higher dose (25.6 × 109) of hMSC-EV treatment in the acute phase of TBI can induce the secretion of antiinflammatory cytokines IL-4 and IL-10 for prolonged periods. Additional statistical details such as means, confidence intervals, degrees of freedom, and ANOVA p values for different comparisons presented in Fig. 7 are detailed in Table 5 of the supplemental file.
3.9. hMSC-EV administration after TBI prevented a chronic inflammatory proteome in the hippocampus
We analyzed hippocampal tissues from naive control, TBI+VEH, and TBI+25.6 × 109 EV groups for protein profiling (n=3/group). In TBI+VEH and TBI+25.6 × 109 EV groups, the hippocampi ipsilateral to TBI were utilized. The results showed good consistency in proteome identification and quantification (Supplemental Fig. 5) and identified 7,142 gene protein products (GPs). To increase reliability, we selected 5,702 GPs that were detected at least three times in a group for principal component analysis (PCA) and clustering (Supplemental Fig. 5). The PCA plot and clustering demonstrated the similarity of bio-repeats from the same group and a clear distinction between the different treatment groups, revealing an altered proteomic landscape induced by TBI and EV treatment (Supplemental Fig. 5). We selected the 35 GPs that were significantly altered after TBI and normalized to naïve control levels following EV treatment (Supplemental Fig. 6). A comparison of naïve versus TBI and TBI versus TBI+25.6 × 109 EV groups revealed that 18 GPs were upregulated with TBI and normalized to naïve control levels after hMSC-EV administration (Supplemental Fig. 6). These comprise Kras, Pak1, Shc3, Prkacb, Rasgrf2, Ntrk2, Jund, Stmn1, Map3k4, Ecsit, Cacng3, Chrm3, Hrh1, Plcb4, Taok3, Atp2b4, Gng3, and Gng13. Supplemental Table 7 details the biological functions of these proteins when upregulated in the brain. Furthermore, 17 GPs were downregulated with TBI and normalized to naïve control levels after hMSC-EV treatment (Supplemental Fig. 6). These include Prkca, Ppp3r1, Braf, Ptk2b, Insr, Rasgrf1, Rasa1, Ppm1b, Cacng8, Stim2, Grin2a, Adcy9, Camk2B, Ryr2, Orai2, Ksr1, and Gng11. Supplemental Table 8 details the adverse effects of these proteins when downregulated in the brain. Next, using the above 35 GPs, we performed string analysis (Szklarczyk et al., 2019) for known protein-protein associated network analysis to decipher the functional relation of those significantly altered proteins (Fig. 8 [A]). FDR sorting and KEGG pathway analysis revealed that 35 GPs were involved mainly in p38/MAPK signaling (FDR, 1.47e-22), Ras signaling (FDR, 5.33e-19), and calcium signaling (FDR, 9.81e-19) pathways. Supplementary figure 7 illustrates MAPK signaling cascade (panel A), its activation after TBI (panel B), and modulation after EV treatment (panel C).
Figure 8: String analysis and validation of the activation of MAPK signaling pathway after TBI and its modulation by hMSC-EVs.

Figure A shows the three most enriched signaling pathways after TBI identified through the KEGG pathway and string network analysis, using 35 GPs that showed significant alterations. The bar charts in B-E and H-K compare the concentrations of various proteins involved in activating the MAPK signaling pathway in the hippocampus (B-E) and the somatosensory cortex (SSC, H-K) between naïve, TBI+VEH, and TBI+25.6 × 109 EV treatment groups. The proteins include myeloid differentiation primary response 88 (MyD88; B, H), a small GTPase Ras (C, I), p38 mitogen-activated protein kinase (p38 MAPK; D, J), activator protein 1 (AP-1; E, K). The bar charts F-G and L-M compare the concentrations of IL-6 (F, L) and IL-8 (G, M) (i.e., some of the end products of activated MAPK signaling pathway) in the hippocampus (F-G) and SSC (L-M) of naïve, TBI+VEH, and TBI+25.6 × 109 EV treatment groups. *, p<0.05; **, p<0.01; ****, p<0.0001; NS, not significant.
3.10. Validation of hMSC-EV administration preventing TBI-induced activation of p38/MAPK signaling
While p38/MAPK signaling activation can occur from either IL-18 or IL-1β secreted by activated inflammasomes in microglia, we focused on measuring IL-18 mediated p38/MAPK activation in this study. We first measured the concentrations of different components of IL-18 mediated p38/MAPK signaling, including MyD88, Ras, phospho-p38 MAPK, and AP-1 in the hippocampus and SSC of naïve, TBI+VEH, and TBI+EVs (25.6 × 109 EVs) groups (Fig. 8 [B–E, H–K]). Next, we measured two known end products of such signaling, including IL-6 and IL-8. In the hippocampus, one-way ANOVA analyses revealed that MyD88, pMAPK, AP-1, IL-6, and IL-8 levels significantly differed between groups (Fig. 8 [B, D–G]). Tukey’s posthoc tests revealed that 1) levels of pMAPK, AP-1, and IL-6 were upregualted in the TBI group compared to the naïve control group; 2) concentrations of MyD88, pMAPK, AP-1, IL-6 and IL-8 were normalized to naïve control levels in TBI+EVs group; and 3) amounts of MyD88, AP-1, IL-6 and IL-8 were reduced in the TBI+EVs group compared to the TBI group (Fig. 8 [B, D–G]). In the SSC, one-way ANOVA analyses revealed that MyD88, Ras, pMAPK, AP-1, and IL-6 levels significant differed between groups (Fig. 8 [H–L]). Tukey’s posthoc tests revealed that 1) levels of MyD88, pMAPK, Ras, AP-1, and IL-6 were upregualted in the TBI group compared to the naïve control group; 2) concentrations of Ras, pMAPK, AP-1, and IL-6 were normalized to naïve control levels in TBI+EVs group; and 3) levels of MyD88 and IL-6 were reduced in the TBI+EVs group compared to the TBI group (Fig. 8 [H, L]). Additional statistical details such as means, confidence intervals, degrees of freedom, and ANOVA p values for different comparisons are detailed in Table 6 of the supplemental file. Figure 9 illustrates how NLRP3 inflammasome activation in microglia after TBI leads to enhanced p38/MAPK signaling via upregulation of IL-18, MyD88, and Ras, and mechanisms by which activated p38/MAPK signaling leads to sustained release of higher concentrations of proinflammatory cytokines in the chronic phase of TBI. The figure also shows how the inhibition of NLRP3 inflammasome activation by hMSC-EVs in the acute phase of TBI can retard this process and reduce the activation of p38/MAPK signaling and the release of proinflammatory cytokines in the chronic phase of TBI.
4. Discussion
This study provides novel evidence that a single IN treatment of an optimal dose of hMSC-EVs enriched with activated microglia-modulating miRNAs in the acute phase of TBI is adequate for preventing a chronic neuroinflammatory state and long-term cognitive and mood impairments. Remarkably, robust inhibition of NLRP3 inflammasome activation by hMSC-EV treatment in the acute phase of TBI restrained the enduring activation of p38/MAPK inflammatory cascade, resulting in a diminished release of multiple proinflammatory cytokines in the chronic phase of TBI. Improved brain function in hMSC-EV treated TBI animals was evident from enhanced proficiency for object location memory and pattern separation, absence of anhedonia, and reduced anxiety-like behavior.
Acute neuroinflammation after TBI-induced brain tissue loss or damage comprises invasion of neutrophils, lymphocytes, and monocyte-derived macrophages from the periphery through the damaged BBB and activation of resident microglia (Irrera et al., 2020). Such combined effort by peripheral and resident immune cells is beneficial for removing the dead cell debris and triggering an antiinflammatory response to brain damage (Schimmel et al., 2017). However, sustained activation of microglia after the resolution of initial precipitating injury can activate many inflammatory cascades. Such cascades result in a chronic neuroinflammatory state typified by an enhanced concentration of proinflammatory cytokines in the microenvironment (Semple et al., 2019). Close links between chronic neuroinflammation and cognitive and mood impairments have been recognized (Collins-Praino and Corrigan, 2017). Given these, strategies that modulate proinflammatory microglia in the acute phase of TBI have received attention in reducing the chronic repercussions of TBI, including post-TBI dementia (Gorgoraptis et al., 2009). One of the mechanisms by which activated microglia contribute to a chronic neuroinflammatory state of the brain is the activation of NLRP3 inflammasomes (O’Brien et al., 2020). NLRP3 inflammasome activation after TBI is triggered through multiple danger-associated molecular patterns, including the increased concentration of reactive oxygen species (Bauernfeind et al., 2011) and the high mobility group box 1 (Frank et al., 2016) mediating toll-like receptor and NF-kB signaling (O’Brien et al., 2020). NLRP3 activation sequentially involves the oligomerization of NLRP3 protein, the binding of NLRP3 to ASC, cleavage of the pro-caspase-1 into activated caspase-1 by ASC, and binding of the activated caspase-1 with the NLRP3-ASC complex (O’Brien et al., 2020). The activated caspase-1 then promotes the maturation of IL-1β and IL-18 by cleaving pro-IL-1β and pro-IL-18 (Jha et al., 2010). Sustained release of these cytokines results in a chronic proinflammatory environment.
The current study quantified total NF-kB, NF-kB p65, and concentrations of various proteins involved in NLRP3 inflammasome activation, in both the acute and chronic phases of TBI. In animals receiving the vehicle after TBI, robust activation of NLRP3 inflammasomes was evident in both acute and chronic TBI phases from higher concentrations of NF-kB, NF-kB p65, NLRP3, ASC, activated caspase-1, IL-1β, and IL-18. Remarkably, a single IN treatment of hMSC-EVs, including lower doses, considerably reduced the concentration of all proteins linked to NLRP3 inflammasome activation. Such finding implied that hMSC-EV mediated inhibition of NLRP3 inflammasome activation in the acute phase has long-lasting effects. We next probed proinflammatory markers in microglia and changes in astrocytes to comprehend the enduring antiinflammatory effects of a single hMSC-EV treatment in the acute phase of TBI. The results demonstrated increased percentages of proinflammatory microglia expressing CD68 and NLRP3 inflammasomes in the SSC and the hippocampus ipsilateral to CCI in animals receiving the vehicle after TBI. In contrast, animals receiving hMSC-EVs after TBI, particularly those receiving higher doses, displayed reduced percentages of proinflammatory microglia expressing CD68 or presenting NLRP3 inflammasomes. Interestingly, a lower dose (6.4 X 109) hMSC-EV treatment was found insufficient for reducing microglia with NLRP3 inflammasomes in the somatosensory cortex and the hippocampus. However, biochemical assays suggested that such a lower dose of hMSC-EV treatment in TBI mice could reduce the various markers of inflammasomes and bring them closer to naïve control levels. Furthermore, the low-dose hMSCEV treatment improved simple cognitive tasks like the object location test but not the more complex cognitive task involved in a pattern separation test. The low-dose hMSC-EV treatment also reversed anhedonia but failed to reverse anxiety-like behavior. Overall, it appeared that relatively milder antiinflammatory effects mediated by the lower dose of hMSC-EVs resulted in only a partial reversal of neurobehavioral impairments.
Furthermore, the proteomic analysis uncovered activation of the p38/MAPK signaling pathway in the chronic phase of TBI in animals receiving the vehicle. Since the downstream signaling of IL-18 released by activated NLRP3 inflammasomes can also lead to the activation of p38/MAPK, we investigated p38/MAPK signaling in detail. Indeed, animals receiving the vehicle after TBI displayed activation of p38 MAPK kinase signaling by IL-18 and increased release of various proinflammatory cytokines in the chronic phase of TBI. Such outcomes were evident from higher concentrations of multiple proteins involved in p38/MAPK signaling, including MyD88, Ras, p38 MAPK, AP-1, and proinflammatory cytokines, IL-1β, IL-6, and IL-8. Activation of p38/MAPK signaling observed after TBI in this study is consistent with the earlier finding that deletion of p38/MAPK in microglia could blunt TBI-induced inflammatory response (Bachstetter, et al. 2013; Morganti et al., 2019). Remarkably, animals receiving hMSC-EV treatment after TBI exhibited normalized or reduced levels of MyD88, Ras, p38 MAPK, AP-1, IL-1β, IL-6, and IL-8. These results underscore that robust inhibition of NLRP3 inflammasome activation by hMSC-EVs in the acute phase of TBI prevented the IL-18 mediated sustained p38/MAPK signaling in the chronic phase of TBI. Consistent with the above changes, the SSC of hMSC-EV treated animals also displayed higher levels of antiinflammatory cytokines IL-4 and IL-10 in the chronic phase of TBI, further supporting the promotion of an antiinflammatory milieu following hMSC-EV treatment.
How does a single dose of hMSC-EV treatment transform the proinflammatory microglia into noninflammatory or antiinflammatory phenotypes? We analyzed the miRNA cargo of hMSC-EVs through small-RNA sequencing to obtain insights on this issue, as miRNAs carried by EVs have been shown to play critical roles in modulating the phenotype or the cellular processes in recipient cells (Cocucci and Meldolesi, 2015; Vogel et al., 2018). Investigation of the known functions of top 20 miRNAs within hMSC-EVs from published studies revealed that most enriched miRNAs are proficient in mediating multiple beneficial effects in the injured brain. First, hMSC-EVs were enriched with miR-30e-5p, which can suppress NLRP3 mRNA and protein expression (Li et al., 2018). Second, multiple miRNAs enriched in hMSC-EVs are proficient in modulating proinflammatory microglia into noninflammatory or antiinflammatory phenotypes. These include miR-100–3p (Li et al., 2019), miR-26b-5p (Kang et al., 2018), let-7c-5p (Lv et al., 2018), let-7g-5p (Bernstein and Rom, 2020), miR-654–3p (Tu et al., 2019), miR-30d-5p (Jiang et al., 2018), and miR-30a-5p (Fu et al., 2018). Third, hMSC-EVs were also enriched with miRNAs capable of reducing neurodegeneration after brain injury (miR-335–5p and miR-494–3p, Liu et al., 2015; Zhao et al., 2019), antiapoptotic effects (miR-423–3p, Li et al., 2014), protecting neurons and brain vascular endothelial cells against oxygen-glucose deprivation (let-7i-5p and miR-152–3p; Xiang et al.,2017; Zhang et al., 2019), regulating reactive astrocytes (miR-320a, Fatima et al., 2017), reducing endothelial dysfunction, and protecting BBB (miR-98–5p and let7g-5p, Bernstein et al., 2020). To validate the above miRNAs identified through small-RNA sequencing in hMSC-EVs, we performed qPCR and confirmed the occurrence of 8 miRNAs implicated in modulating NLRP3 inflammasomes or proinflammatory microglia, mediating neuroprotective effects, or protecting BBB. The validated miRNAs include miR-30e-3p, miR-654–3p, miR-26b-5p, let-7c-5p, let-7g-5p, miR-335–5p, miR-494–3p; miR-98–5p. Thus, hMSC-EVs employed in the study were naturally enriched with many miRNAs capable of inhibiting the activation of microglia, astrocytes, and NLRP3 inflammasomes, protecting neurons and endothelial cells and maintaining BBB integrity. Such a composition of miRs explains the potent suppressing effects of a single dose of hMSC-EVs in the acute phase of TBI on the activation of NLRP3 inflammasomes and p38/MAPK inflammatory cascade during the chronic phase of TBI in this study. Moreover, the incorporation of IN-administered hMSC-EVs by microglia in both injured and contralateral sides of CCI in the study raises the possibility that hMSC-EVs directly modulate microglia by releasing their cargo. Single-cell RNAseq studies in the future will be helpful to precisely ascertain the specific transcriptomic changes induced by hMSC-EVs in microglia.
The proteomic analysis uncovered 18 GPs that were significantly upregulated after TBI and normalized to naïve control levels following hMSC-EV treatment. The adverse effects promoted by these GPs are detailed in the supplemental Table 7, which comprises the activation of the p38/MAPK pathway, BBB dysfunction after TBI, tau phosphorylation, Aβ-induced toxicity, chronic neuroinflammation, synaptic dysfunction, abnormal functional connectivity in the brain, epilepsy, depression, and anxiety. Given the adverse effects of these GPs upon upregulation, their normalization following hMSC-EV treatment appeared beneficial for promoting functional recovery in the chronic phase of TBI. The proteomic analysis also revealed 17 GPs that were downregulated with TBI and normalized to levels seen in naïve controls after EV treatment. The beneficial effects promoted by these GPs are described in supplemental Table 8, which includes improved memory, long-term potentiation, synaptic plasticity, mood, and angiogenesis, reduced tau aggregation, and protection against neurodegenerative disorders. Considering the beneficial effects of these GPs, their downregulation observed in the chronic phase of TBI is detrimental to functional recovery. Therefore, normalization of the expression of these GPs following hMSC-EV treatment likely contributed to improved cognition and mood function in the chronic phase of TBI.
In conclusion, IN administration of a single optimal dose of hMSC-EV treatment in the acute phase of TBI blocked the progression of acute inflammation into a chronic neuroinflammatory state characterized by continuous activation of NLRP3 inflammasomes and the p38/MAPK inflammatory signaling. A robust inhibition of NLRP3 inflammasome activation in the acute phase of TBI by hMSC-EVs seemed to be the primary mechanism underlying such an effect. This concept is supported by studies in CCI models of TBI showing that inhibition of NLRP3 inflammasome activation alone by specific small molecules can ease neuroinflammation and brain dysfunction (Xu et al., 2018; Kuwar et al., 2019). Also, preserved cognitive function and reduced brain damage observed in NLRP3−/− mice following TBI support the above possibility (Irrera et al., 2020). Although small molecules capable of inhibiting NLRP3 inflammasome activation have the promise to ease TBI-mediated cognitive impairments, their safety, side effects, long-term efficacy, or mechanisms by which they inhibit inflammasome activation are unknown (O’Brien et al., 2020). From this perspective, a single dose of hMSC-EV treatment after TBI curbing the development of a chronic neuroinflammatory state by blocking the activation of NLRP3 inflammasomes in microglia observed in this study is appealing for clinical translation in the future.
Supplementary Material
Figure 9: MAPK activation and its modulation by hMSC-EVs treatment.

The cartoon presents the mechanisms by which activation of MAPK signaling occurs after TBI and how hMSC-EV treatment after TBI prevents the chronic activation of MAPK signaling and the enduring increased release of proinflammatory cytokines. The upward red arrows indicate upregulation, and the downward green arrows imply the downregulation of different proteins involved in p38 MAPK signaling. The complexes indicated with question marks suggest the involvement of additional pathways.
Highlights.
Human mesenchymal stem cell-derived extracellular vesicles (hMSC-EVs) are naturally enriched with activated microglia-modulating miRNAs
Intranasally administered hMSC-EVs quickly incorporate into neurons and microglia in the injured brain
An optimal intranasal dose of hMSC-EVs after traumatic brain injury (TBI) prevents enduring cognitive and mood impairments
An optimal intranasal dose of hMSC-EVs after TBI inhibits NLRP3 inflammasome activation in microglia
Inhibition of NLRP3 inflammasome activation by hMSC-EVs after TBI thwarts activation of p38 MAPK signaling
4. Acknowledgments
This work was primarily supported by a grant from the National Institute of Neurological Disorders and Stroke (1R01NS106907 to A.K.S.). The authors thank Dr. Darwin Prockop for insightful suggestions on culturing human mesenchymal stem cells and isolating and characterizing the therapeutic properties of human mesenchymal cell-derived extracellular vesicles. The SmallRNA sequencing was performed at the Institute for Genome Sciences and Society, Texas A&M University. The mass spectrometry analyses were performed at the Mass Spectrometry based proteomics core at the Baylor College of Medicine, supported by the N.I.H. Grant P30CA125123. The TEM images of hMSC-EVs were performed in the Image Analysis Laboratory, Texas A&M Veterinary Medicine & Biomedical Sciences (RRIS: SCR_022479).
Footnotes
Disclosure of interest: The authors report no conflicts of interest.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
5. References
- Assini FL, Duzzioni M, Takahashi RN, 2009. Object location memory in mice: pharmacological validation and further evidence of hippocampal CA1 participation. Behav Brain Res 204, 206–211. [DOI] [PubMed] [Google Scholar]
- Bachstetter AD, Rowe RK, Kaneko M, Goulding D, Lifshitz J, Van Eldik LJ, 2013. The p38α MAPK regulates microglial responsiveness to diffuse traumatic brain injury. The Journal of neuroscience : the official journal of the Society for Neuroscience 33, 6143–6153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baraniak PR, McDevitt TC, 2010. Stem cell paracrine actions and tissue regeneration. Regen Med 5, 121–143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bauernfeind F, Bartok E, Rieger A, Franchi L, Núñez G, Hornung V, 2011. Cutting edge: reactive oxygen species inhibitors block priming, but not activation, of the NLRP3 inflammasome. J Immunol 187, 613–617. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bernstein DL, Zuluaga-Ramirez V, Gajghate S, Reichenbach NL, Polyak B, Persidsky Y, Rom S, 2020. miR-98 reduces endothelial dysfunction by protecting blood-brain barrier (BBB) and improves neurological outcomes in mouse ischemia/reperfusion stroke model. J Cereb Blood Flow Metab 40, 1953–1965. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bernstein DL, Rom S, 2020. Let-7g* and miR-98 Reduce Stroke-Induced Production of Proinflammatory Cytokines in Mouse Brain. Front Cell Dev Biol 8, 632. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Caplan HW, Prabhakara KS, Toledano Furman NE, Zorofchian S, Kumar A, Martin C, Xue H, Olson SD, Cox CS Jr., 2021. Combination therapy with Treg and mesenchymal stromal cells enhances potency and attenuation of inflammation after traumatic brain injury compared to monotherapy. Stem Cells 39, 358–370. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cassel SL, Sutterwala FS, 2010. Sterile inflammatory responses mediated by the NLRP3 inflammasome. Eur J Immunol 40, 607–611. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cocucci E, Meldolesi J, 2015. Ectosomes and exosomes: shedding the confusion between extracellular vesicles. Trends Cell Biol 25, 364–372. [DOI] [PubMed] [Google Scholar]
- Collins-Praino LE, Corrigan F, 2017. Does neuroinflammation drive the relationship between tau hyperphosphorylation and dementia development following traumatic brain injury? Brain Behav Immun 60, 369–382. [DOI] [PubMed] [Google Scholar]
- Cone AS, Yuan X, Sun L, Duke LC, Vreones MP, Carrier AN, Kenyon SM, Carver SR, Benthem SD, Stimmell AC, Moseley SC, Hike D, Grant SC, Wilber AA, Olcese JM, Meckes DG Jr., 2021. Mesenchymal stem cell-derived extracellular vesicles ameliorate Alzheimer’s disease-like phenotypes in a preclinical mouse model. Theranostics 11, 8129–8142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cozene B, Sadanandan N, Farooq J, Kingsbury C, Park YJ, Wang ZJ, Moscatello A, Saft M, Cho J, Gonzales-Portillo B, Borlongan CV, 2021. Mesenchymal Stem Cell-Induced Anti-Neuroinflammation Against Traumatic Brain Injury. Cell Transplant 30, 9636897211035715. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fann JR, Ribe AR, Pedersen HS, Fenger-Grøn M, Christensen J, Benros ME, Vestergaard M, 2018. Long-term risk of dementia among people with traumatic brain injury in Denmark: a population-based observational cohort study. Lancet Psychiatry 5, 424–431. [DOI] [PubMed] [Google Scholar]
- Fatima M, Prajapati B, Saleem K, Kumari R, Mohindar Singh Singal C, Seth P, 2017. Novel insights into role of miR-320a-VDAC1 axis in astrocyte-mediated neuronal damage in neuroAIDS. Glia 65, 250–263. [DOI] [PubMed] [Google Scholar]
- Frank MG, Weber MD, Fonken LK, Hershman SA, Watkins LR, Maier SF, 2016. The redox state of the alarmin HMGB1 is a pivotal factor in neuroinflammatory and microglial priming: A role for the NLRP3 inflammasome.Brain Behav Immun 55, 215–224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fu X, Shen Y, Wang W, Li X, 2018. MiR-30a-5p ameliorates spinal cord injury-induced inflammatory responses and oxidative stress by targeting Neurod 1 through MAPK/ERK signalling. Clinical and experimental pharmacology & physiology 45, 68–74. [DOI] [PubMed] [Google Scholar]
- Gandy K, Kim S, Sharp C, Dindo L, Maletic-Savatic M, Calarge C, 2017. Pattern Separation: A Potential Marker of Impaired Hippocampal Adult Neurogenesis in Major Depressive Disorder. Front Neurosci 11, 571. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gorgoraptis N, Li LM, Whittington A, Zimmerman KA, Maclean LM, McLeod C, Ross E, Heslegrave A, Zetterberg H, Passchier J, Matthews PM, Gunn RN, McMillan TM, Sharp DJ, 2019. In vivo detection of cerebral tau pathology in long-term survivors of traumatic brain injury. Sci Transl Med 11. [DOI] [PubMed] [Google Scholar]
- Hattiangady B, Shuai B, Cai J, Coksaygan T, Rao MS, Shetty AK, 2007. Increased dentate neurogenesis after grafting of glial restricted progenitors or neural stem cells in the aging hippocampus. Stem Cells 25, 2104–2117. [DOI] [PubMed] [Google Scholar]
- Hattiangady B, Kuruba R, Shetty AK, 2011. Acute Seizures in Old Age Leads to a Greater Loss of CA1 Pyramidal Neurons, an Increased Propensity for Developing Chronic TLE and a Severe Cognitive Dysfunction. Aging Dis 2, 1–17. [PMC free article] [PubMed] [Google Scholar]
- Heneka MT, McManus RM, Latz E, 2018. Inflammasome signalling in brain function and Neurodegenerative disease. Nat Rev Neurosci 19, 610–621. [DOI] [PubMed] [Google Scholar]
- Henry RJ, Ritzel RM, Barrett JP, Doran SJ, Jiao Y, Leach JB, Szeto GL, Wu J, Stoica BA, Faden AI, Loane DJ, 2020. Microglial Depletion with CSF1R Inhibitor During Chronic Phase of Experimental Traumatic Brain Injury Reduces Neurodegeneration and Neurological Deficits. The Journal of neuroscience : the official journal of the Society for Neuroscience 40, 2960–2974. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hu L, Si L, Dai X, Dong H, Ma Z, Sun Z, Li N, Sha H, Chen Y, Qian Y, Zhang Z, 2021. Exosomal miR-409–3p secreted from activated mast cells promotes microglial migration, activation and neuroinflammation by targeting Nr4a2 to activate the NF-κB pathway. J Neuroinflammation 18, 68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Irrera N, Russo M, Pallio G, Bitto A, Mannino F, Minutoli L, Altavilla D, Squadrito F, 2020. The Role of NLRP3 Inflammasome in the Pathogenesis of Traumatic Brain Injury. Int J Mol Sci 21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ising C, Venegas C, Zhang S, Scheiblich H, Schmidt SV, Vieira-Saecker A, Schwartz S, Albasset S, McManus RM, Tejera D, Griep A, Santarelli F, Brosseron F, Opitz S, Stunden J, Merten M,Kayed R, Golenbock DT, Blum D, Latz E, Buée L, Heneka MT, 2019. NLRP3 inflammasome activation drives tau pathology. Nature 575, 669–673. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ismael S, Nasoohi S, Ishrat T, 2018. MCC950, the selective inhibitor of nucleotide oligomerization domain-like receptor protein-3 inflammasome, protects mice against traumatic brain injury. J Neurotrauma 35, 1294–303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jain S, Yoon SY, Zhu L, Brodbeck J, Dai J, Walker D, Huang Y, 2012. Arf4 determines dentate gyrus-Mediated pattern separation by regulating dendritic spine development. PLoS One 7, e46340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jha S, Srivastava SY, Brickey WJ, Iocca H, Toews A, Morrison JP, Chen VS, Gris D, Matsushima GK, Ting JP 2010. The inflammasome sensor, NLRP3, regulates CNS inflammation and demyelination via caspase-1 and interleukin-18. The Journal of neuroscience : the official journal of the Society for Neuroscience 30, 15811–15820. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jiang M, Wang H, Jin M, Yang X, Ji H, Jiang Y, Zhang H, Wu F, Wu G, Lai X, Cai L, Hu R, Xu L, Li L, 2018. Exosomes from MiR-30d-5p-ADSCs Reverse Acute Ischemic Stroke-Induced, Autophagy-Mediated Brain Injury by Promoting M2 Microglial/Macrophage Polarization. Cell Physiol Biochem 47, 864–878. [DOI] [PubMed] [Google Scholar]
- Jiang Y, Chen Y, Huang C, Xia A, Wang G, Liu S, 2021. Hyperbaric oxygen therapy improves neurological function via the p38-MAPK/CCL2 signaling pathway following traumatic brain injury. Neuroreport 32, 1255–1262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kang YC, Zhang L, Su Y, Li Y, Ren WL, Wei WS, 2018. MicroRNA-26b Regulates the Microglial Inflammatory Response in Hypoxia/Ischemia and Affects the Development of Vascular Cognitive Impairment. Front Cell Neurosci 12, 154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim DK, Nishida H, An SY, Shetty AK, Bartosh TJ, Prockop DJ, 2016. Chromatographically isolated CD63+CD81+ extracellular vesicles from mesenchymal stromal cells rescue cognitive impairments after TBI.Proc Natl Acad Sci U S A 113, 170–175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kodali M, Parihar VK, Hattiangady B, Mishra V, Shuai B, Shetty AK, 2015. Resveratrol prevents age-related memory and mood dysfunction with increased hippocampal neurogenesis and microvasculature, and reduced glial activation. Sci Rep 5, 8075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kodali M, Megahed T, Mishra V, Shuai B, Hattiangady B, Shetty AK, 2016. Voluntary Running Exercise-Mediated Enhanced Neurogenesis Does Not Obliterate Retrograde Spatial Memory. The Journal of neuroscience : the official journal of the Society for Neuroscience 36, 8112–8122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kodali M, Castro OW, Kim DK, Thomas A, Shuai B, Attaluri S, Upadhya R, Gitai D, Madhu LN, Prockop DJ, Shetty AK, 2020. Intranasally Administered Human MSC-Derived Extracellular Vesicles Pervasively Incorporate into Neurons and Microglia in both Intact and Status Epilepticus Injured Forebrain. Int J Mol Sci, 21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuwar R, Rolfe A, Di L, Xu H, He L, Jiang Y, Zhang S, Sun D, 2019. A novel small molecular NLRP3 inflammasome inhibitor alleviates neuroinflammatory response following traumatic brain injury. J Neuroinflammation 16, 81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leutgeb JK, Leutgeb S, Moser MB, Moser EI, 2007. Pattern separation in the dentate gyrus and CA3 of the hippocampus. Science 315, 961–966. [DOI] [PubMed] [Google Scholar]
- Li D, Yang H, Ma J, Luo S, Chen S, Gu Q, 2018. MicroRNA-30e regulates neuroinflammation in MPTP model of Parkinson’s disease by targeting Nlrp3. Hum Cell 31, 106–115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li MM, Jiang T, Sun Z, Zhang Q, Tan CC, Yu JT, Tan L, 2014. Genome-wide microRNA expression profiles in hippocampus of rats with chronic temporal lobe epilepsy. Sci Rep 4, 4734. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li XH, Fu NS, Xing ZM, 2019. MiR-100 suppresses inflammatory activation of microglia and neuronal Apoptosis following spinal cord injury via TLR4/NF-κB pathway. Eur Rev Med Pharmacol Sci 23, 8713–8720. [DOI] [PubMed] [Google Scholar]
- Liu HD, Li W, Chen ZR, Hu YC, Zhang DD, Shen W, Zhou ML, Zhu L, Hang CH 2013. Expression of the NLRP3 inflammasome in cerebral cortex after traumatic brain injury in a rat model. Neurochem Res 38, 2072–2083. [DOI] [PubMed] [Google Scholar]
- Liu FJ, Kaur P, Karolina DS, Sepramaniam S, Armugam A, Wong PT, Jeyaseelan K, 2015. MiR-335 Regulates Hif-1α to Reduce Cell Death in Both Mouse Cell Line and Rat Ischemic Models. PLoS One 10, e0128432. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu MY, Yin CY, Zhu LJ, Zhu XH, Xu C, Luo CX, Chen H, Zhu DY, Zhou QG, 2018. Sucrose preference test for measurement of stress-induced anhedonia in mice. Nat Protoc 13, 1686–1698. [DOI] [PubMed] [Google Scholar]
- Long Q, Upadhya D, Hattiangady B, Kim DK, An SY, Shuai B, Prockop DJ, Shetty AK, 2017. Intranasal MSC-derived A1-exosomes ease inflammation, and prevent abnormal neurogenesis and memory dysfunction after status epilepticus. Proc Natl Acad Sci U S A 114, E3536–e3545. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lv J, Zeng Y, Qian Y, Dong J, Zhang Z, Zhang J, 2018. MicroRNA let-7c-5p improves neurological outcomes in a murine model of traumatic brain injury by suppressing neuroinflammation and regulating microglial activation. Brain Res 1685, 91–104. [DOI] [PubMed] [Google Scholar]
- Madhu LN, Kodali M, Attaluri S, Shuai B, Melissari L, Rao X, Shetty AK, 2021. Melatonin improves Brain function in a model of chronic Gulf War Illness with modulation of oxidative stress, NLRP3 inflammasomes, and BDNF-ERK-CREB pathway in the hippocampus. Redox Biol 43, 101973. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Merali Z, Levac C, Anisman H, 2003. Validation of a simple, ethologically relevant paradigm for assessing anxiety in mice. Biol Psychiatry 54, 552–565. [DOI] [PubMed] [Google Scholar]
- Morganti JM, Goulding DS, Van Eldik LJ, 2019. Deletion of p38α MAPK in microglia blunts trauma-Induced inflammatory responses in mice. J Neuroinflammation 16, 98. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moss LD, Sode D, Patel R, Lui A, Hudson C, Patel NA, Bickford PC, 2021. Intranasal delivery of exosomes from human adipose derived stem cells at forty-eight hours post injury reduces motor and cognitive impairments following traumatic brain injury. Neurochem Int 150, 105173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Needham EJ, Helmy A, Zanier ER, Jones JL, Coles AJ, Menon DK, 2019. The immunological response to traumatic brain injury. J Neuroimmunol 332, 112–125. [DOI] [PubMed] [Google Scholar]
- O’Brien WT, Pham L, Symons GF, Monif M, Shultz SR, McDonald SJ, 2020. The NLRP3 inflammasome in traumatic brain injury: potential as a biomarker and therapeutic target. J Neuroinflammation 17, 104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Patel NA, Moss LD, Lee JY, Tajiri N, Acosta S, Hudson C, Parag S, Cooper DR, Borlongan CV, Bickford PC 2018. Long noncoding RNA MALAT1 in exosomes drives regenerative function and modulates inflammation-linked networks following traumatic brain injury. J Neuroinflammation 15, 204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rao MS, Hattiangady B, Shetty AK, 2006. The window and mechanisms of major age-related decline in the production of new neurons within the dentate gyrus of the hippocampus. Aging Cell 5, 545–558. [DOI] [PubMed] [Google Scholar]
- Rao MS, Hattiangady B, Shetty AK, 2008. Status epilepticus during old age is not associated with Enhanced hippocampal neurogenesis. Hippocampus 18, 931–944. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rex DAB, Agarwal N, Prasad TSK, Kandasamy RK, Subbannayya Y, Pinto SM, 2020. A comprehensive pathway map of IL-18-mediated signalling. J Cell Commun Signal 14, 257–266. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rom S, Dykstra H, Zuluaga-Ramirez V, Reichenbach NL, Persidsky Y, 2015. miR-98 and let-7g* protect the blood-brain barrier under neuroinflammatory conditions. J Cereb Blood Flow Metab 35, 1957–1965. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ryan SM, Nolan YM, 2016. Neuroinflammation negatively affects adult hippocampal neurogenesis and cognition:can exercise compensate? Neuroscience and biobehavioral reviews 61, 121–131. [DOI] [PubMed] [Google Scholar]
- Saltzman AB, Leng M, Bhatt B, Singh P, Chan DW, Dobrolecki L, Chandrasekaran H, Choi JM, Jain A, Jung SY, Lewis MT, Ellis MJ, Malovannaya A, 2018. gpGrouper: A Peptide Grouping Algorithm for Gene-Centric Inference and Quantitation of Bottom-Up Proteomics Data. Mol Cell Proteomics 17, 2270–2283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Samuels BA, Hen R, (2011). Novelty-Suppressed Feeding in the Mouse. In: Gould T (eds) Mood and Anxiety-Related Phenotypes in Mice. Neuromethods 63:107–121. [Google Scholar]
- Schimmel SJ, Acosta S, Lozano D, 2017. Neuroinflammation in traumatic brain injury: A chronic response to an acute injury. Brain Circ 3, 135–142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Semple BD, Zamani A, Rayner G, Shultz SR, Jones NC, 2019. Affective, neurocognitive and Psychosocial disorders associated with traumatic brain injury and post-traumatic epilepsy. Neurobiol Dis 123, 27–41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shetty AK, Attaluri S, Kodali M, Shuai B, Shetty GA, Upadhya D, Hattiangady B, Madhu LN, Upadhya R, Bates A, Rao X, 2020. Monosodium luminol reinstates redox homeostasis, improves cognition, mood and neurogenesis, and alleviates neuro- and systemic inflammation in a model of Gulf War Illness. Redox Biol 28, 101389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stocchetti N, Zanier ER, 2016. Chronic impact of traumatic brain injury on outcome and quality of life: a Narrative review. Crit Care 20, 148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Szklarczyk D, Gable AL, Lyon D, Junge A, Wyder S, Huerta-Cepas J, Simonovic M, Doncheva NT, Morris JH, Bork P, Jensen LJ, Mering CV, 2019. STRING v11: protein-protein association networks with increased coverage, supporting functional discovery in genome-wide experimental datasets. Nucleic Acids Res 47, D607–d613. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tan M, Shen L, Hou Y, 2020. Epigenetic modification of BDNF mediates neuropathic pain via miR-30a-3p/EP300 axis in CCI rats. Biosci Rep 40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tu Y, Guo R, Li J, Wang S, Leng L, Deng J, Bucala R, Lu L, 2019. MiRNA Regulation of MIF in SLE and Attenuation of Murine Lupus Nephritis With miR-654. Front Immunol 10, 2229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Upadhya D, Hattiangady B, Castro OW, Shuai B, Kodali M, Attaluri S, Bates A, Dong Y, Zhang SC, Prockop DJ, Shetty AK, 2019. Human induced pluripotent stem cell-derived MGE cell grafting after status epilepticus attenuates chronic epilepsy and comorbidities via synaptic integration. Proc Natl Acad Sci U S A 116, 287–296. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Upadhya R, Madhu LN, Attaluri S, Gitaí DLG, Pinson MR, Kodali M, Shetty G, Zanirati G, Kumar S, Shuai B, Weintraub ST, Shetty AK, 2020. Extracellular vesicles from human iPSC-derived neural stem cells: miRNA and protein signatures, and anti-inflammatory and neurogenic properties. J Extracell Vesicles 9, 1809064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van Goethem NP, van Hagen BTJ, Prickaerts J, 2018. Assessing spatial pattern separation in rodents using the object pattern separation task. Nat Protoc 13, 1763–1792. [DOI] [PubMed] [Google Scholar]
- Varendi K, Kumar A, Härma MA, Andressoo JO, 2014. miR-1, miR-10b, miR-155, and miR-191 are novel regulators of BDNF. Cell Mol Life Sci 71, 4443–4456. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vlachos IS, Zagganas K, Paraskevopoulou MD, Georgakilas G, Karagkouni D, Vergoulis T, Dalamagas T, Hatzigeorgiou AG, 2015. DIANA-miRPath v3.0: deciphering microRNA function with experimental support. Nucleic Acids Res 43, W460–466. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vogel A, Upadhya R, Shetty AK, 2018. Neural stem cell derived extracellular vesicles: Attributes and prospects for treating neurodegenerative disorders. EBioMedicine 38, 273–282. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Warburton EC, Brown MW, 2015. Neural circuitry for rat recognition memory. Behav Brain Res 285, 131–139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Watanabe J, Shetty AK, Hattiangady B, Kim DK, Foraker JE, Nishida H, Prockop DJ, 2013. Administration of TSG-6 improves memory after traumatic brain injury in mice. Neurobiol Dis 59, 86–99. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Williams AM, Bhatti UF, Brown JF, Biesterveld BE, Kathawate RG, Graham NJ, Chtraklin K, Siddiqui AZ, Dekker SE, Andjelkovic A, Higgins GA, Buller B, Alam HB, 2020. Early single-dose treatment with exosomes provides neuroprotection and improves blood-brain barrier integrity in swine model of traumatic brain injury and hemorrhagic shock. J Trauma Acute Care Surg 88, 207–218. [DOI] [PubMed] [Google Scholar]
- Witcher KG, Bray CE, Chunchai T, Zhao F, O’Neil SM, Gordillo AJ, Campbell WA, McKim DB, Liu X, Dziabis JE, Quan N, Eiferman DS, Fischer AJ, Kokiko-Cochran ON, Askwith C, Godbout JP, 2021. Traumatic Brain Injury Causes Chronic Cortical Inflammation and Neuronal Dysfunction Mediated by Microglia. J Neurosci 41, 1597–1616. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xiang W, Tian C, Peng S, Zhou L, Pan S, Deng Z, 2017. Let-7i attenuates human brain microvascular Endothelial cell damage in oxygen glucose deprivation model by decreasing toll-like receptor 4 expression. Biochem Biophys Res Commun 493, 788–793. [DOI] [PubMed] [Google Scholar]
- Xu X, Yin D, Ren H, Gao W, Li F, Sun D, Wu Y, Zhou S, Lyu L, Yang M, Xiong J, Han L, Jiang R, Zhang J, 2018. Selective NLRP3 inflammasome inhibitor reduces neuroinflammation and improves long-term neurological outcomes in a murine model of traumatic brain injury. Neurobiol Dis 117, 15–27. [DOI] [PubMed] [Google Scholar]
- Zhang A, Qian Y, Qian J, 2019. MicroRNA-152–3p protects neurons from oxygen-glucose-deprivation/reoxygenation-induced injury through upregulation of Nrf2/ARE antioxidant signaling by targeting PSD-93. Biochem Biophys Res Commun 517, 69–76. [DOI] [PubMed] [Google Scholar]
- Zhang T, Liu C, Chi L, 2020a. Suppression of miR-10a-5p in bone marrow mesenchymal stem cells enhances the therapeutic effect on spinal cord injury via BDNF. Neurosci Lett 714, 134562. [DOI] [PubMed] [Google Scholar]
- Zhang Y, Chopp M, Zhang ZG, Katakowski M, Xin H, Qu C, Ali M, Mahmood A, Xiong Y, 2017. Systemic administration of cell-free exosomes generated by human bone marrow derived mesenchymal stem cells cultured under 2D and 3D conditions improves functional recovery in rats after traumatic brain injury. Neurochem Int 111, 69–81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Y, Zhang Y, Chopp M, Zhang ZG, Mahmood A, Xiong Y, 2020b. Mesenchymal Stem Cell-Derived Exosomes Improve Functional Recovery in Rats After Traumatic Brain Injury: A Dose-Response and Therapeutic Window Study. Neurorehabil Neural Repair 34, 616–626. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao H, Li G, Zhang S, Li F, Wang R, Tao Z, Ma Q, Han Z, Yan F, Fan J, Li L, Ji X, Luo Y, 2019. Inhibition of histone deacetylase 3 by MiR-494 alleviates neuronal loss and improves neurological recovery in experimental stroke. J Cereb Blood Flow Metab 39, 2392–2405. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.