

Abstract
As crucial mediators and regulators of our immune system, cytokines are involved in a broad range of biological processes and are implicated in various disease pathologies. The field of cytokine therapeutics has gained much momentum from the maturation of conventional protein engineering methodologies such as structure-based designs and/or directed evolution, which is further aided by the advent of in silico protein designs and characterization. Just within the past five years, there has been an explosion of proof-of-concept, preclinical, and clinical studies that utilize an armory of protein engineering methods to develop cytokine-based drugs. Here we highlight the key engineering strategies undertaken by recent studies that aim to improve the pharmacodynamic and pharmacokinetic profile of interferons and other cytokines as therapeutics.
Keywords: Interferons, cytokines, interleukins, protein engineering, therapeutics, pleiotropy
Cytokines are promising therapeutic candidates, but engineering endows improvements for success in the clinic
Cytokines represent a family of secreted proteins that influence a broad range of physiological processes that regulate innate and adaptive immunity [1]. Over 130 known cytokines are categorized into six superfamilies that include interferons (IFNs), interleukins (ILs), chemokines, colony-stimulating factors (CSFs), tumor-necrosis factors (TNFs), and transforming growth factors (TGFs) [2–4]. With their broad implications in various disease pathologies, cytokines were used as part of the first generation of immunotherapeutics for cancers and autoimmune disorders. For example, in 1986, recombinant IFNα received U.S. Food and Drug Administration (FDA) approval for treating hematological malignancies such as hairy cell leukemia and chronic myeloid leukemia, AIDS-related Kaposi sarcoma, melanoma, and chronic viral hepatitis B and C infections [5]. IFNγ was then approved for chronic granulomatous disease in 1990, high dose IL-2 therapy for the treatment of metastatic renal cell carcinoma in 1992, and IFNβ for multiple sclerosis in 1996 [6–8]. IFNs alone have continued to be explored as therapies, with more than 20 clinical trials initiated since 2017 (Supplemental Table 1). However, due to limited efficacy and toxicity concerns, the use of therapeutic cytokines has since been superseded or relegated to second-line therapy by advanced immunotherapeutics such as checkpoint inhibitors or monoclonal antibodies, and small-molecule drugs.
Most cytokine monotherapies are restricted by three critical issues. First is the biological pleiotropism inherent to many therapeutic cytokines [9]. The context-dependent and highly dynamic nature of responses induced by these cytokines contribute to their suboptimal specificity and efficacy [10, 11]. To this end, decoupling the dual immunosuppressive and immunostimulatory or anti-proliferative and antiviral properties of a specific cytokine is necessary to engineer tailored variants with biased signaling and selective activities. Second, the poor drug-like properties of these proteins impose another layer of limitations [12, 13]. The short circulating half-life of most cytokines and narrow therapeutic windows reduce the efficacy of cytokine monotherapies. Although high doses and frequent administrations allow therapeutic concentrations of cytokines at targeted sites, this approach incurs the third issue – acute toxicity and morbidity [14, 15]. IFN use is associated with flu-like symptoms and neuropsychiatric issues, as well as more severe conditions such as thrombocytopenia, leukopenia, and neutropenia, that require either a dose reduction or cessation [16–18]. Collectively, these challenges have represented a major roadblock in the mainstream clinical applications of cytokines.
Recent technological advances in structural biology and protein engineering, bolstered by the advent of artificial intelligence and machine learning, have revolutionized our understanding of protein sequence-structure-functional relationships. Consequently, the field of protein therapeutics, in general, has seen a tremendous uptick in both research efforts and preclinical/clinical explorations in recent years. With their inherent issues, cytokine-based therapeutics have benefited most from these recent developments. Here, we review a set of promising cytokines – primarily IFNs and ILs due to their widespread study and use – with a focus on specific engineering strategies undertaken to enhance their therapeutic applicability. This review categorizes representative studies mainly over the past five years into three groups based on the primary issue the studies aim to resolve. They include: (1) deconvoluting the pleiotropism, (2) targeting specific cells or tissues, and (3) enhancing drug-like properties of cytokines.
Deconvoluting the Pleiotropism of Cytokines
An important attribute of cytokines is their broad physiological properties, spanning antiviral, anti-proliferative, and immunomodulatory activities [19] (Figure 1, Figure 2). The high degree of pleiotropism, however, has greatly limited their therapeutic efficacy. Key approaches to target the pleiotropy of IFNs and other cytokines are outlined here, which utilize a combination of structure-guided engineering, de novo design, and/or directed evolution.
Figure 1. IFN signaling pathway and downstream cellular effects.
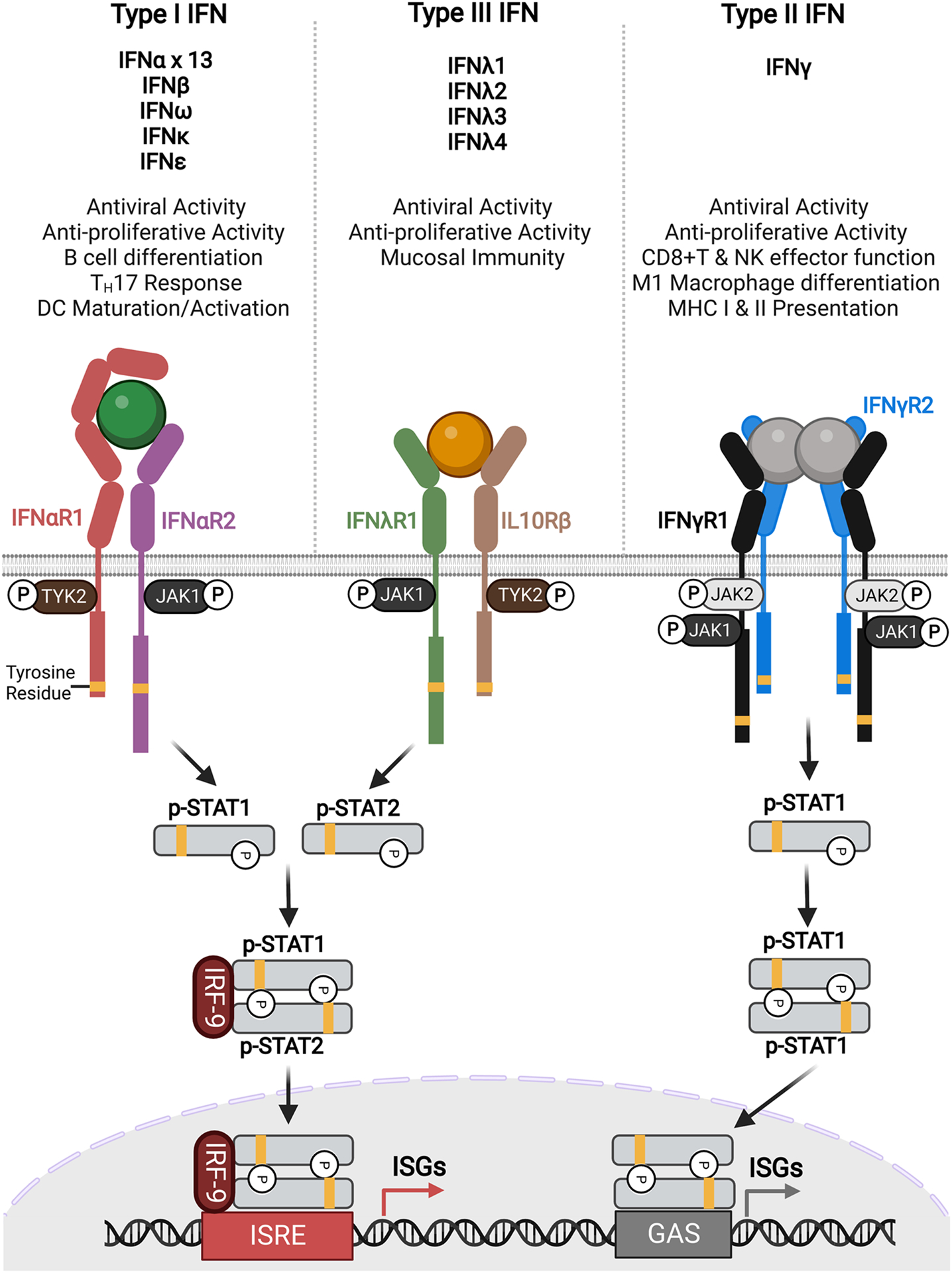
Type I, II, and III IFNs signal via intracellular JAK/STAT pathway. For type I and III IFNs, ligand binding dimerizes the receptors, resulting in the phosphorylation of STAT1 and STAT2 proteins (signal transducer and activator of transcription) that dimerize and form a complex with IFN regulatory factor 9 (IRF-9). After translocating to the nucleus, the complex then binds to the IFN stimulated response element (ISRE) to activate the transcription of IFN stimulated genes (ISGs). Type II IFN acts as a homodimer and recruits two sets of receptors, phosphorylating STAT1 proteins which then dimerize and translocate to the nucleus where the complex binds gamma IFN activation site (GAS) elements to initiate transcription of a distinct set of ISGs.
Figure 2. Signaling pathways of Common gamma chain cytokines and their downstream functions.
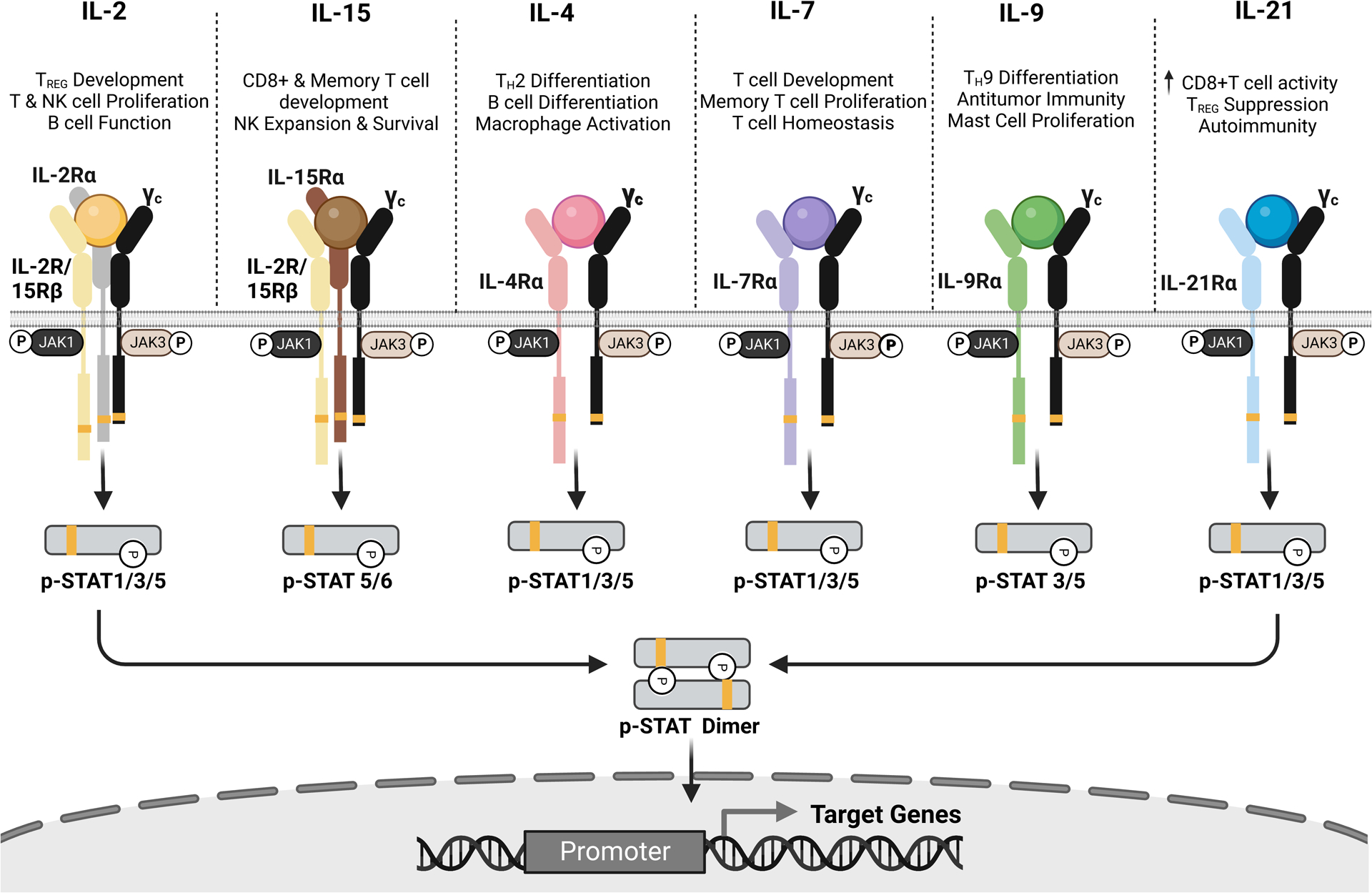
Members of the common gamma chain (cytokine family include IL-2, IL-15, IL-4, IL-7, IL-9, and IL-21) hold important roles in driving the immune cell landscape of many disease pathologies. Upon ligand binding, the common gamma chain is recruited to the ligand-bound cognate receptors to direct lymphocytes’ responses. Current engineering themes to potentiate the therapeutic applications of these cytokines include de novo designs (IL-2/IL-15), half-life extensions such as albumin fusion (IL-2, IL-4, IL-15), Fc fusion (IL-2, IL7, IL-12, IL-15), and PEGylation (IL-2, IL-12, IL-15), tumor antigen or lymphocyte targeting using antibody fusions (IL-2, IL-12, IL-21).
Structure-based design of partial agonists
With structural knowledge of a protein complex, engineers can selectively bias a cytokine’s interactions with its receptors to influence cell signaling (Figure 3A). In a notable example involving IFNγ, Mendoza et. al. reported the structure of the complete hexameric IFNγ-IFNγR1-IFNγR2 complex, which enabled the rational design of partial agonists [20]. Despite existing as natural homodimers, a series of mutant heterodimeric IFNγ variants were engineered as single-chained constructs, each with a precisely controlled number of recruited receptors within a signaling complex. Subsequently, these variants exhibited tunable differences in downstream phosphorylated STAT1 (pSTAT1) signaling and gene expression profiles (Figure 1, right). In cancer cells treated with engineered IFNγ, programmed death-ligand 1 (PD-L1) expression was greatly reduced whereas MHC class I remained highly expressed; this uncoupling of MHC I and PD-L1 expression indicates not only that a major activity of IFNγ and contributing factor to pleiotropy can be decoupled, but also these biased agonists could enhance tumor antigen presentation without the immunosuppression stimulated by WT IFNγ.
Figure 3. Structure-derived approaches to designing and engineering cytokine therapies.
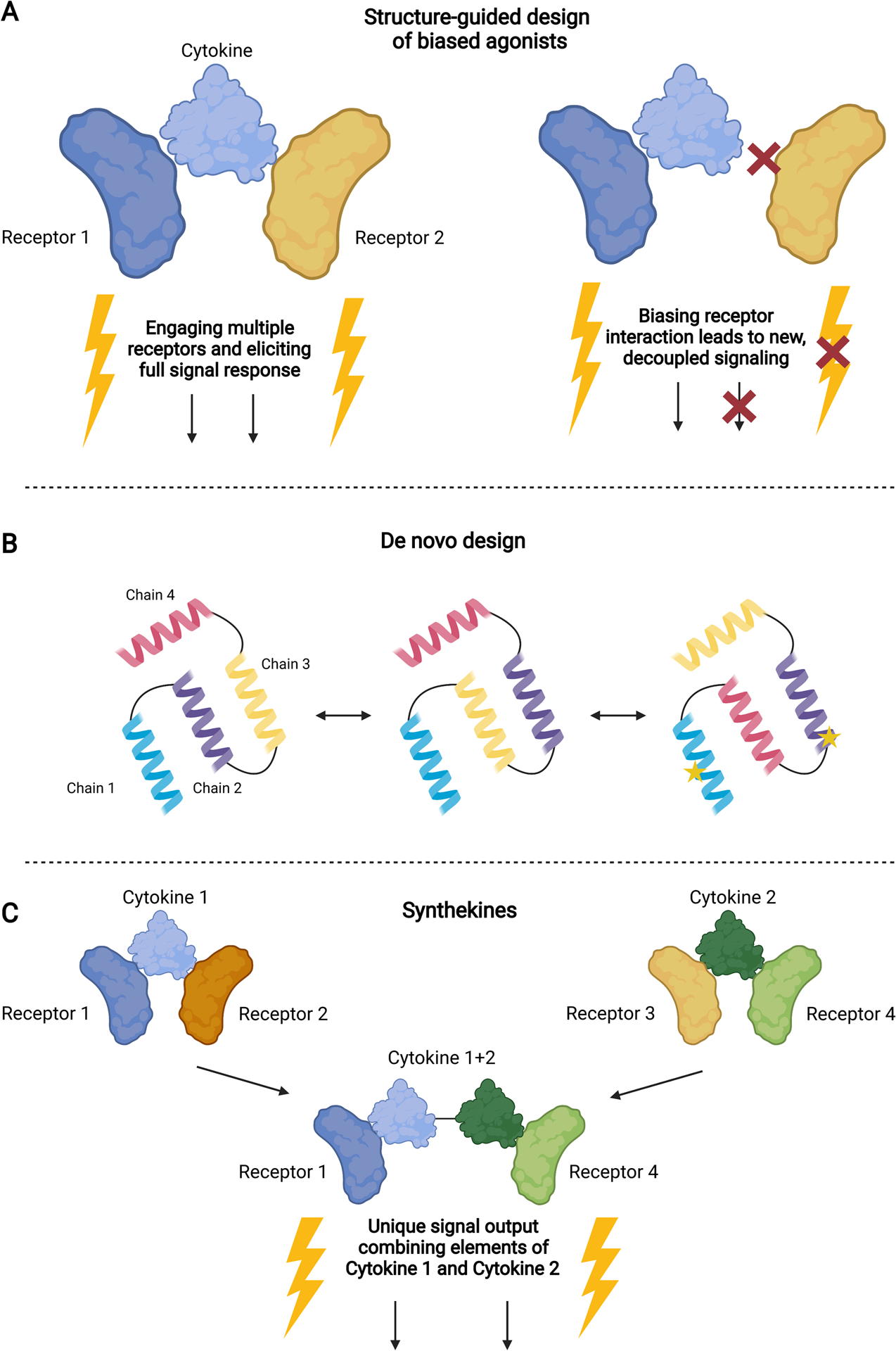
A) Using structural information about how a cytokine engages with its receptors, biased agonists can be designed by rationally mutating cytokine-receptor interfaces to tune gene expression and cell signaling. B) De novo design of novel cytokines can be accomplished using computational methods, such as structure prediction and modeling, to either design entirely new proteins or rearrange existing protein structures to selectively engage with targets. Site-directed mutagenesis can also be employed in combination with this technique to engineer specific residues. C) By covalently linking two different cytokines, synthekines can selectively engage pathways that are not engaged by natural proteins, leading to new and unique combinations of receptor signaling.
Similar structure-based approaches have been applied successfully to other cytokines. Like IFNγ, IL-10 is a dimeric cytokine with both immunosuppressive and immunostimulatory functions [21]. To decouple the pleiotropy and boost the immunomodulatory functions, Gorby et. al. engineered high-affinity versions of both monomeric and dimeric IL-10 using yeast display-based affinity maturation [22]. By varying the stability and/or the number of receptors within a complex, the authors tuned the activation of STAT1 and STAT3 in human monocytes and CD8+ T cells. In a subsequent study, Saxton et. al. engineered their high-affinity version of IL-10 and utilized it to solve the structure of the hexameric IL-10-IL-10Rα-IL-10Rβ complex [23]. As with IFNγ, rational mutations were introduced to WT IL-10 to weaken the interaction with IL-10Rβ, generating partial agonists which exhibited pronounced monocyte-biased signaling activity. Similarly, Mo et. al. used a high-affinity IL-2 to generate partial agonist H9T, with reduced binding at the H9T- IL-2Rγ interface [24]. H9T expands activated mouse CD8+ T cells to a similar extent to IL-2 and enhances CD8+ T cell anti-tumor activity in mouse models of melanoma and acute lymphoblastic leukemia by modulating/reprogramming CD8+ T cell transcription and metabolism [24]. Together, these examples show how structure-guided engineering can maintain the desired effects of a cytokine therapeutic while reducing or eliminating the undesired effects.
Design of novel proteins
Structural or functional knowledge of cytokines can also be used to design completely novel protein mimetics which make use of known or predicted interactions to design proteins that outperform their natural counterparts or have specifically tailored properties (Figure 3B).
In one striking example leveraging computational design, Silva et. al. utilized Rosetta, a protein structure prediction and design tool, to create a potent and selective mimic of IL-2 and IL-15 [25, 26] (Figure 2, left). The potential of IL-2 for cancer treatment due to its central role in the immune system has led to extensive efforts to improve its therapeutic capabilities by increasing its efficacy or chemically modifying the protein, with prior approaches that have largely failed to limit its clinical toxicity. Previous studies identified, however, that toxicity is noticeably reduced in animals deficient in IL-2Rα [27–30]. With this information, Rosetta was used to design an IL-2 mimetic that ablates any interaction with IL-2Rα but maintains contact with the other two receptor components, IL-2Rβ (also called IL-15Rβ) and IL-2Rγ (the common gamma chain). Using the structure of IL-2 as a template, the 4 helices (H1–4) of the protein were used as a structural framework to find unique ligands and sequences with desired binding behaviors while also improving the packing of the original protein; the best-predicted designs were characterized via yeast display [31–33].
A second generation of design and optimization to improve thermal stability and binding yielded the final product termed Neoleukin-2/15 (Neo-2/15), which does not interact with IL-2Rα while having higher affinities for IL-2Rβγ and higher activity than both native and previously engineered versions of IL-2 [34]. Neo-2/15 is also supremely thermostable, resistant to mutation, and does not require additional protein modifications or fusions to bias activity among immune cell populations as desired. Treatment with Neo-2/15 in CT26 (colon cancer) and B16F10 (melanoma) mouse tumor models led to dose-dependent tumor regressions and overall improved efficacy [34]. Further studies have found that Neo-2/15 also improves the expansion of natural killer (NK) cells, demonstrating its ability to synergize with other immunotherapies [35]. This example of de novo protein design touts the great potential for cytokine therapeutics in general but has also been used to design several other novel binding proteins with selective properties [36]. Neo-2/15 represents the first example of a de novo cytokine, however many other computational methods, including PROSS and FireProt, have also found success in cytokine engineering and design [37, 38]. As computational design methods continue to improve and gain traction in the experimental community, the diversity of novel cytokines which can be designed and their potential efficacy in the clinic will greatly increase.
While the computational design of de novo proteins represents an exciting new frontier in the field of cytokine therapeutics, experimental engineering methods still serve as excellent tools to develop novel protein mimetics which elicit cytokine-like responses in cells. Of particular interest are synthetic cytokines known as synthekines, which selectively dimerize novel receptor pairs, initiating new signaling pathways (Figure 3C). Moraga et. al. first engineered IL-2, IL-4, and type I IFNs that engage only with their high-affinity cognate receptor [39]. They are then linked covalently via a glycine-serine peptide chain to selectively couple their signaling abilities and engage with these new receptor pairs. While the signaling ability of these synthekines is reduced compared to their natural counterparts, this method leads to unique cellular and signaling signatures not possible using natural cytokines, creating the opportunity for custom cellular responses [39]. A similar approach was taken by Ng et. al. to create a recombinant protein composed of protein domains from IL-15, IL-15Rα, and a TGFβ receptor domain to overcome immunosuppressive TGFβ while stimulating NK and CD8+ T cells [40].
Other studies have focused on harnessing the signaling ability of cytokines by creating surrogate agonists [41, 42]. Kolarova et. al. used yeast display to evolve a general protein scaffold for affinity towards the type III IFN receptors to create a type III IFN mimic with similar signaling abilities [41]. On the other hand, Yen et. al. used a combination of small antibody domains (VHH and/or scFv) to create libraries of receptor dimerizing ligands which exhibit functional activities similar to IL-2/15, type I IFN, and IL-10 [42]. Synthekines and mimetics demonstrate the power of novel protein design using experimental methods and its ability to potentiate cytokine-like therapeutics.
Engineering cytokines to improve their therapeutic capabilities
A strong example of how protein engineering has effectively improved cytokines as therapeutics is the directed evolution of type III IFNs (Figure 4A). Type III IFNs, also known as IFNλs, offer exciting therapeutic potential because they signal through the same pathway as type I IFNs but their receptor expression is limited to immune cell subsets and barrier tissues, avoiding the negative side effects associated with the ubiquitous expression of type I IFN receptors (Figure 1, left and middle) [43]. However, their signaling potency is lower than type I IFNs, leading to limited clinical efficacy. Mendoza et. al. utilized yeast surface display to engineer a high-affinity version of the type III IFN IFNλ3, called H11, which improved overall complex affinity by ~150-fold and increased antiviral and antiproliferative abilities relative to the wild type [20]. These engineering efforts also elucidated previously unappreciated elements of the type III IFN signaling paradigm which, despite engineering of the type III IFNs, remains less potent than type I IFN signaling. This suggests some unknown aspects of signaling contribute to differences in signaling and potency, necessitating continued studies. However, this family of IFNs still represents an exciting group of naturally-targeted therapeutics that can serve as an alternative to type I IFN, the side effects of which lead to problems in clinics.
Figure 4. Engineering cytokines to improve their activity and targeting ability in the body.
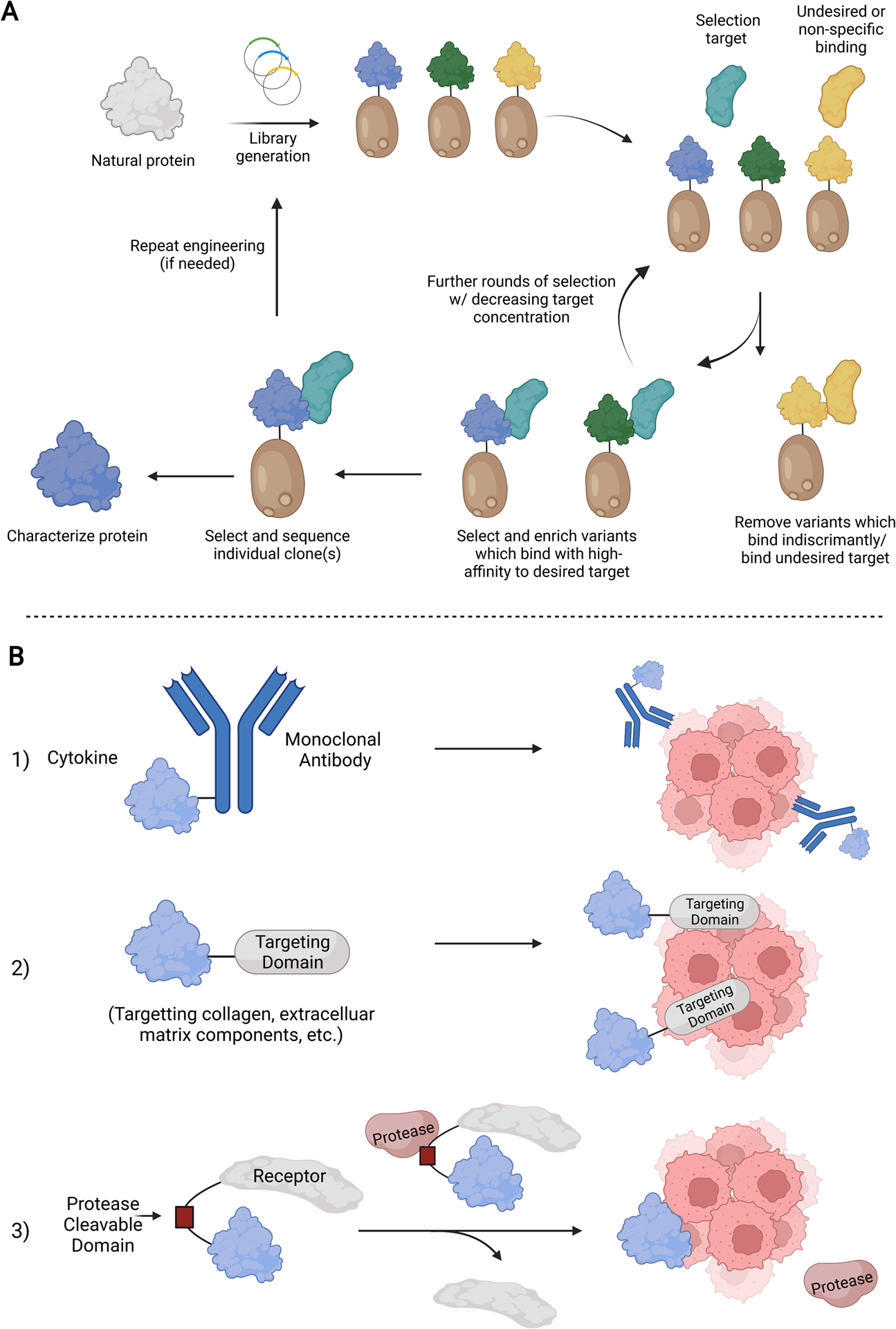
A) Schematic diagram showing directed evolution using yeast display, a common method for cytokine engineering. Following library generation, variants are selected for their desired characteristics (increased affinity, activity, etc.) and subjected to increasing selection pressure. Individual clones are then isolated and characterized. B) Targeting motifs that increase cytokine delivery to the tissues of interest and reduce off-target effects. 1) Conjugating to a monoclonal antibody 2) Conjugating to a targeting domain for a specific tissue or environment 3) Linkage to a receptor by a protease-cleavable domain.
Another clever approach using engineering to improve therapeutic capability has been showcased with IL-18, which is a member of the IL-1 cytokine family and mediates inflammation downstream of the NLRP1 and NLRP3 inflammasomes [44]. Recombinant IL-18 has been explored as a cancer therapeutic in clinical trials; however, its development has been complicated by a lack of efficacy attributed to the presence of IL-18 binding protein (IL-18BP) which is upregulated in the tumor microenvironment (TME). To overcome this limitation, Zhou et. al. engineered a decoy-resistant IL-18 (DR-18) which retains full signaling capacity for the IL-18 receptor but is not inhibited by binding to IL-18BP [45]. Using directed evolution with yeast surface display, more than 250 million variants, randomized at 13 receptor contact positions, were screened and selected for affinity towards IL-18Rα and counter-selected against IL-18BP. The final product DR-18 is active against MC38 mouse colon tumors, acting on T and NK cells to drive anti-tumor responses while similarly remodeling the TME to immune checkpoint inhibitors [44].
Targeting Cytokines to Specific Cells or Tissues
Strategies against off-target signaling involve the covalent linkage of a cytokine to its receptor via a protease-cleavable peptide and to a targeting moiety that displays high affinities toward a specific cell or microenvironment, encouraging site-directed accumulation and activation (Figure 4B). Recent studies showed that IL-2/anti-IL2-monoclonal antibody (mAb) immunocomplexes, as well as IL-2 conjugated to a tumor-targeting antibody against human epidermal growth factor receptor (EGFR) or carcinoembryonic antigen (CEA), enhances tumor binding and specific activation of cytotoxic T lymphocytes [34, 46–48]. In vivo tumor regression studies in melanoma, colon, and pancreatic tumor models also indicated superior antitumoral efficacies of IL-2/antibody conjugates over their recombinant counterparts. Similarly, antibody-protein conjugates of IL-21/anti-PD-1 and IFNγ/anti-HER2/neu have much-improved pharmacodynamics [49].
Targeting strategies that take advantage of the TME, mainly the unique protein makeup, the low pH, or hypoxia, have also seen success with cytokine therapeutics. Mansurov et. al. leveraged the high amounts of exposed collagen present in disordered tumor vasculature by fusing a collagen-binding domain (CBD) to IL-12 (CBD-IL-12) to target tumor sites [50]. Compared to unmodified IL-12, CBD-IL-12 was preferentially retained in tumors, displayed substantially reduced systemic levels, and provided superior anticancer efficiency. In another study, Cao et. al. linked type I IFNs to their natural receptors to further effectively target tumors, reduce systemic toxicity, and increase half-life [51]. These “masked” IFNs, dubbed ProIFNs, are connected to their receptors via a matrix metalloproteinase (MMP)-cleavable domain such that the IFNs are only active at the tumor sites where MMPs are enriched, limiting the toxicity associated with type I IFN use in the clinic. They reported a reduction in IFN activity by several orders of magnitude in tissues not expressing MMPs and found that treatment with these ProIFNs led to an enhanced immune response and improved outcomes in mice. Masked variants of cytokines have also been explored for IL-12, IL-2 and IFNβ [52] [53, 54].
While much of the focus of cytokine therapies are on cancer treatment, it is important to note that specific targetable domains and targeting moieties have also been identified to potentiate cytokine therapeutics beyond cancers. Alterations in the extracellular matrix (ECM) are implicated in wound environments and inflamed joints and tissues [55, 56], with the heparin-binding domain of placental growth factor-2 (PlGF-2123–144) binding promiscuously to ECM proteins [57]. By conjugating growth factors such as platelet-derived growth factor-BB (PDGF-BB) and vascular endothelial growth factor-A (VEGF-A) to PIGF-2123–144, White et. al. showed that improved wound healing was achieved through longer residence time at the site of administration and favorable changes in the immune-cells’ milieu of the wound in the type 1 diabetic (T1D) NOD mouse model [58]. Similarly, local administration of therapeutic α-TNF antibodies conjugated to PIGF-2123–144 suppressed arthritis development almost completely in a collagen antibody-induced arthritis (CAIA) mouse model compared to unmodified α-TNF controls [59].
Improving the Pharmacokinetics and Safety of Cytokines
Multiple engineering approaches have been developed to circumvent the short serum half-life of native cytokines (Figure 5). However, polyethylene glycol (PEG) conjugation, PEGylation, remains the only clinically approved strategy [60, 61]. PEGylation increases the hydrodynamic radii of proteins to reduce renal clearance, improves in vivo stability, and reduces immunogenicity [62, 63]. Currently, PEGylated IFNα has been approved for the treatment of cancer with PEGylated IFNγ, IFNλ, IL-2, IL-12, and IL-15 in various stages of clinical trials [64–71]. However, PEGylation poses some inherent challenges including reduced site selectivity of PEGylated cytokines, high production costs associated with multiple-step reactions, complex purification processes, heterogeneity due to a lack of precisely defined polymer length, and safety concerns regarding the potential renal toxicity of PEG metabolites and/or allergic reactions to PEG via anti-PEG antibodies [72–77]. Next-generation polymer conjugation methods seek to overcome these challenges by direct conjugation to specific sites as well as supplanting PEGs with well-defined synthetic polymers or polypeptides such as XTEN or PAS [78–82] (Figure 5A). However, further work is required to match the half-life extension afforded by these new approaches to that of conventional PEGylation.
Figure 5. Engineering strategies are being explored preclinically to enhance the drug-like properties of cytokines.
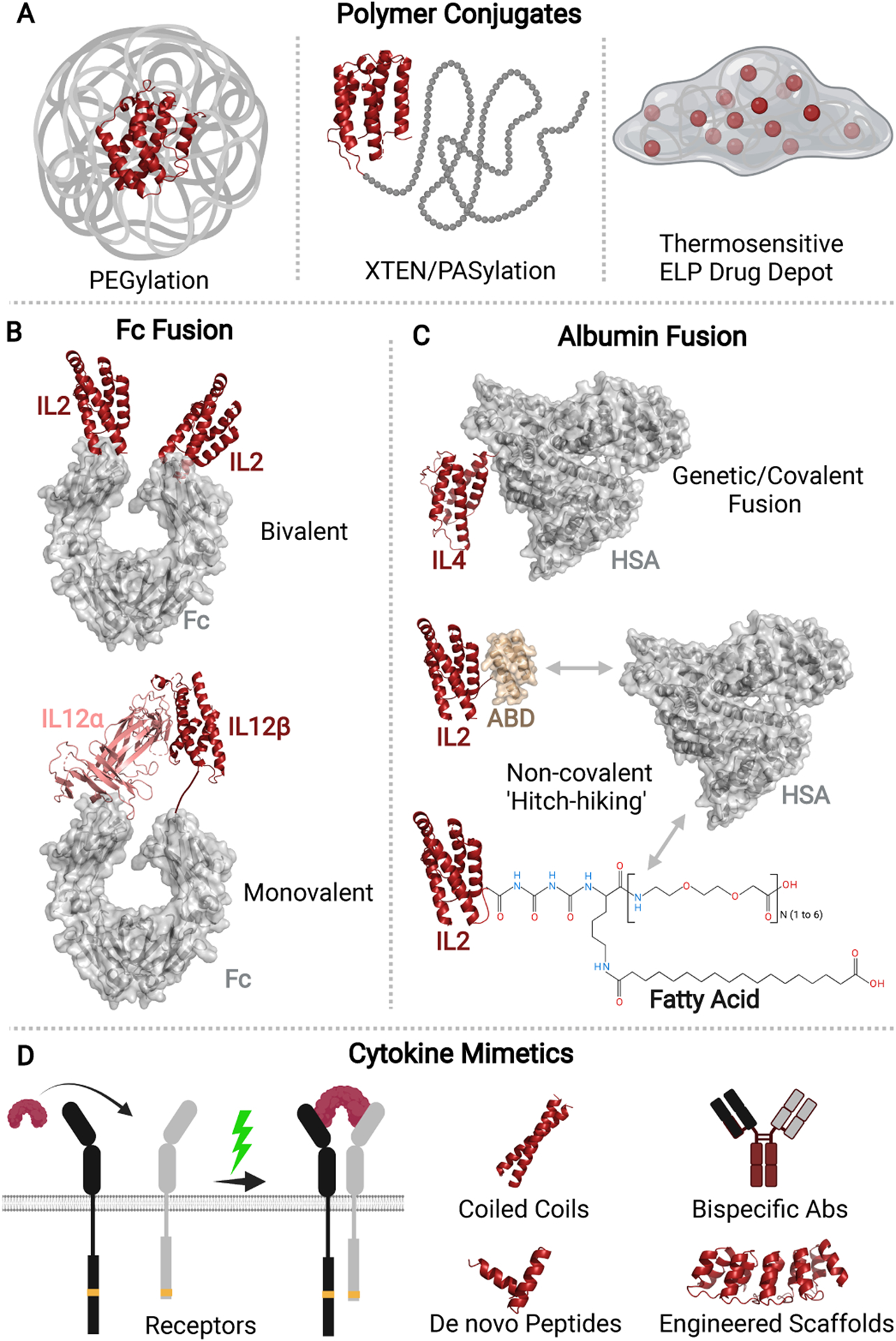
(A) Both site-specific and non-specific polymer conjugation methods have been explored to extend the serum half-lives of cytokines or to create a drug depot for controllable sustained release. (B) The bivalent (Top) and monovalent (Bottom) formats of Fc-fused cytokines have been engineered. (C) Cytokines are conjugated to human serum albumin (HSA) directly via covalent or genetic fusions (Top). Alternatively, they are fused to the albumin-binding domain (ABD) (Middle) or fatty acid moieties (Bottom) that can non-covalently bind to endogenous HSA upon administration. (D) Cytokine mimetics such as peptides, bispecific antibodies, zipper-like coiled-coil constructs, and other engineered protein scaffolds have been studied as potential therapeutics.
An alternative method is the covalent fusion of cytokines to the fragment crystallizable (Fc) domain of an immunoglobulin G (IgG) [83–85] (Figure 5B). In addition to effective evasion of renal filtration owing to their increased size (>60kDa), cytokine-Fc domain conjugates are further protected from intracellular catabolism via a pH-dependent binding of Fc-fragment to neonatal Fc receptor (FcRn) present in endosomes and subsequent recycling back into the bloodstream [86, 87]. In several recent studies, engineered Fc-fusions of IL-2, IL-7, IL-15, and IL-22 were reported to exhibit significantly improved safety, pharmacokinetics, and biodistribution profiles compared to their recombinant forms when evaluated in murine models as well as human subjects [88–91]. While cytokine-Fc-fusions are typically limited by the bivalent nature of the IgG-Fc domain, which can potentially increase potency and toxicity via avidity, the study by Jung et. al. in 2018 addressed this issue [92]. Here, the two polypeptide chains of murine IL-12 (mIL-12) heterodimer were covalently fused to two different Fc variants to construct a monovalent mIL-12-Fc fusion unit (mono-mIL-12-Fc). Compared to recombinant mIL-12 and bivalent mIL-12-Fc fusion, the monovalent fusion construct displayed 5–10 times longer circulation half-lives. When given as intraperitoneal injections to the colon-rectal tumor (CT26) bearing mice on a twice-weekly basis, 73% of treated mice achieved tumor-free survival 35 days post tumor inoculation. However, Fc fusion risks immunogenicity caused by Fc-effector functions such as antibody-dependent cellular cytotoxicity and complement-dependent cytotoxicity, reduced activity, and heterogeneous glycosylation [93, 94]. Another recent strategy has focused on the addition of the C-terminal peptide of human chorionic gonadotropin and an N-glycosylation sequence. This addition increased the half-lives of IFNλ and IFNα by 2–3 times and maintained the antiviral and anti-proliferative activities of the IFNs, however, in the case of IFNα, the activities were reduced compared to those induced by the commercial antibody [95, 96].
Recently, there has been an explosion of research into albumin fusion as a strategy to improve the half-lives of cytokines (Figure 5C). Human serum albumin (HSA), the most abundant plasma protein in the blood, has an extraordinarily long circulatory half-life (~19 days in humans) [97, 98]. Compared to recombinant IFNα (4–8h) and PEGylated IFNα (77h), HSA fused IFNα exhibited a notably longer serum half-life (144h) [99–101]. Fc fusion and albumin fusion are similar in terms of FcRn recycling and reduced renal clearance, however, albumin does not engage Fc effector functions in immune cells. In addition, given the lymphatic dwelling nature of HSA, albumin-fusion cytokines may display altered biodistribution profiles through preferential accumulation in the spleen and lymph nodes [102, 103]. This may directly impact dendritic cell (DC) maturation, priming, and T cell activation, which has important implications for cancer and autoimmune therapeutics.
Compared to their pristine forms, engineered albumin fusions of IL-4 and IL-10 have been shown to improve therapeutic outcomes in murine models of multiple sclerosis and rheumatoid arthritis respectively through a combination of improved serum half-lives, increased trafficking to, and prolonged residence in secondary lymphoid organs [104, 105]. However, the steric hindrance created by the added bulk of HSA (~67kDa) to cytokines significantly reduces their in vivo bioactivities [106, 107]. For instance, IFNα-HSA fusion retained only 1.4% of the in vitro specific activity of recombinant IFN and displayed weak antiviral activity against encephalomyocarditis virus (EMCV) in WISH cells [106]. The increased sterics also restrict the tumor penetration of albumin-fused proteins, resulting in low anticancer efficacy. To this end, albumin-binding domains, or ABDs, small three-helical structures of ~5kDa, have been used to create cytokine-fusions [108]. Their circulatory half-life is extended here by ABD ‘hitch-hiking’ on endogenous serum albumin. Using this approach, ABD fusions of IL-2 and IL-15 have been reported to display markedly improved half-lives in vivo and potent anti-tumoral activities in murine cancer models [109, 110]. Interestingly, Guo et. al. engineered a cyclic IFNα2-ABD (c-IFN-ABD) by head-to-tail macrocyclization approach using a one-step sortase-mediated protein ligation. Compared to linear ABD fusion and recombinant IFNα2, c-IFN-ABD showed improved bioactivity and in vivo anti-tumoral activity, which was attributed to improved half-life, thermal stability, tumor penetration, and distribution [111]. While the study provides an interesting proof of concept, it remains to be seen whether the approach works broadly for other six-helical class II and four-helical class I cytokines.
Other engineering approaches to improve the pharmacokinetic properties and safety profiles of cytokines include but are not limited to polymer conjugates with thermoresponsive polypeptides and modification with fatty acids. Previous studies on IFNα fusion proteins with temperature-responsive elastin-like polypeptide (ELP) or poly-di (ethylene glycol) methyl ether methacrylate (PDEGMA) showed that half-life extension with thermoresponsive controlled release of IFNs can be achieved by in situ creation of a site-specific drug depot upon subcutaneous injection [112, 113]. The depot, formed as a result of the phase transition temperature (Tt) of IFN-polymer conjugates being below the body temperature, gradually releases the soluble drug over a prolonged period in response to the concentration-dependent Tt (Figure 5C). The sustained and controlled drug release has been shown to provide protection against remitting/relapsing in murine models of encephalomyelitis [112]. Alternatively, in a 2021 study by Qian et. al., recombinant IL-2 was modified with fatty acids that formed high-affinity non-covalent interactions with HSA in the circulation [114]. Compared to direct conjugation with HSA, fusion with fatty acids using sortase-mediated ligation offered reduced costs due to a short production cycle, deeper tissue penetration, and higher activity-to-mass ratio due to the smaller size of the bioconjugates.
Thirdly, peptide mimetics of IFNα and IFNβ have also been developed to resolve issues of high immunogenicity and systemic toxicity of parent IFNs [115, 116] (Figure 5D). IFNα–C peptide, which is a peptide from the C-terminus of IFNα1 conjugated to palmitoyl lysine for cell penetration, was shown to retain all the activities of pristine IFNα but lacked the inherent toxicity when treated for experimental autoimmune uveitis (EAU) in rodents [115]. Similarly, a Rosetta-designed peptide mimetic of IFNβ showed protective effects in a murine model of multiple sclerosis by reducing immune dysfunction and neuroinflammation [116]. Finally, as outlined in the study by Lee et. al., a glycoengineered IFNβ variant showed increased strength and duration of biological responses by altering the binding kinetics of the ligand to its receptors [117]. As a result, the engineered mutein achieved long-lasting signal activation and gene expression, which are potentially beneficial for multiple sclerosis patients.
Concluding Remarks
Although many proofs-of-concept, preclinical, and clinical studies discussed in this review are conducted on IFNs, ILs, and to a lesser extent, growth factors, it should be noted that the engineering framework, tools, and insights derived from these studies are nonetheless greatly applicable to most other types of cytokines and even small, soluble protein therapeutics. For instance, the de novo design of neoleukins based on IL-2 and IL-15 not only effectively integrates machine learning into protein engineering, but also establishes a blueprint for deconvoluting the functional pleiotropism of cytokines and other proteins through biased receptor recruitment and signaling. The study holds crucial clinical implications for future designs of cytokines that selectively engage receptors, as the receptors for many therapeutic cytokines such as type I IFNs are ubiquitously expressed, and even with site-directed delivery approaches, systemic toxicity remains a hurdle. Further application of these systems can also assist in the more high-throughput design of cytokines with tunable antiviral, anti-proliferative, and/or immunostimulatory capabilities, as many current approaches rely on time-consuming experimental techniques.
While improved design and engineering techniques will enhance candidate cytokines, clinical efficacy remains a bottleneck for therapeutic cytokines. As such, methods that continue to improve site-specific delivery and reduce the toxicity profile of many of these treatments are incredibly important. While we have covered several promising techniques in this review, methods that have not yet been applied to cytokines but have found success with other therapeutics like antibodies and small molecules, such as pH-dependent release, non-cleavable, reversible inhibitors, or disease-agnostic targeting domains could provide new and improved methods for cytokine delivery [118–120]. Encapsulated delivery of IFNs has also been explored but is plagued with issues that dampen enthusiasm for use; future work which resolves these issues may re-invigorate interest in IFN delivery methods [121].
Even as the field of cytokine therapeutics is experiencing a resurgence of financial investment, research work, and clinical interests, it remains to be seen if the emerging strategies can be leveraged to overcome the inherent poor drug-like qualities of cytokines; however, based on the sheer volume and sophistication of preclinical studies on novel cytokines within the past five years, advancement of more cytokine therapeutics into clinical trials appears imminent. These clinical results will further inform and improve our engineering efforts to unleash the full therapeutic potential of many cytokines against cancers, infections, and autoimmune disorders in the future. Moving forward, it is paramount that the insights gleaned from these existing studies be incorporated into the design of next-generation cytokines to impart them with enhanced functionality, improved tissue selectivity, and better drug-like properties.
Supplementary Material
Outstanding Questions.
The clinical application of protein therapeutics is limited by the large doses required to reach therapeutically relevant concentrations. How can existing methodologies for protein expression, purification, and functional modifications be optimized to overcome the prohibitive costs of production?
Native structures of full-length cytokine signaling complexes remain evasive to the field, rendering the finer aspects of intracellular signaling pathways and their functions inaccessible. How can cytokine engineering innovate to address this gap in our knowledge? What alternative methods can be implemented to probe the subtle differences in the intracellular structures of cytokine signaling units and how they contribute to cytokine functions?
One potential issue with the de novo proteins and cytokines is that they may be recognized by the host immune system as foreign antigens, triggering an immune response. What strategies can be implemented at the sequence level and/or post-purification level to mitigate this issue?
Such issues concerning the immunogenicity of protein therapeutics usually arise during the clinical trials, leading to premature termination of the studies and incurring high costs. How can current cell-based assays and/or animal models be improved to more reliably predict the immune responses of these therapeutics in human subjects?
Highlights.
Cytokines influence a broad range of physiological processes including inflammatory responses to injurious stimuli, regulation of cellular proliferation, differentiation and cell death, and homeostasis post molecular perturbation via repair and remodeling processes.
Several known cytokines including interferons, interleukins, and growth factors are implicated in human diseases, presenting as potential therapeutic targets.
Developing successful cytokine drugs entails deconvolution of inherent pleiotropy, reduction of off-target toxicity, and/or improved drug-like properties.
Recent studies highlight the importance of protein engineering in cytokine therapeutics, encompassing many varied approaches from structure-guided and in silico protein designs to site-directed drug delivery and half-life extension as conjugates.
Despite the early promise from proof-of-concept and preclinical research, it remains to be seen how these novel cytokine-based drugs will perform in clinical trials against various diseases.
Acknowledgments
The funding to support J.L.M. is provided by the National Institutes of Health (NIH) grant 1R35GM147179-01 and the Pritzker School of Molecular Engineering at the University of Chicago.
Glossary
- Biased agonists
A protein that selectively activates only some of the activities or functions capable of being induced by the wild-type ligands.
- Chemokines
Chemotactic cytokines act as signaling proteins to induce cellular migration. Cells follow these signals from low concentration to high concentration such that appropriate cells can be recruited to sites of infection, injury, or tumor growth.
- Colony-stimulating factors (CSFs)
Secreted cellular proteins that induce growth and differentiation in hematopoietic progenitor cells.
- Directed evolution
A method of protein engineering designed to alter protein-protein interactions and thus their function. This is achieved by generating a library of mutants by the introduction of mutations (such as through random mutagenesis or the use of degenerate codons) into the protein sequence and then selecting variants of interest based on fitness for further study.
- Interferons (IFNs)
A family of innate immune proteins that have a diverse array of functions, including antiviral and anti-proliferative activities. They are used clinically in the treatment of viral infections, such as hepatitis B and hepatitis C, multiple sclerosis, and in the treatment of many types of cancers.
- Interleukins (ILs)
Cell-secreted proteins which induce various cellular responses and play a diverse role in the immune system, including stimulating cell activity, maturation, and cell adhesion, among many other functions. They can have both pro- and anti-inflammatory activities depending on the interleukin.
- Partial agonists
Proteins that only partially stimulate a response relative to the wild-type ligands.
- PEGylation
The addition of polyethylene glycol (PEG) chains to extend the circulating half-life of proteins.
- Protein therapeutics
Proteins that are used in the clinical treatment of disease.
- Surrogate agonists
Proteins that stimulate the activity of receptors that are not the natural ligand of that receptor.
- Synthekines
Engineered, synthetic cytokines that activate cytokine signaling through a pair of receptors that no known natural cytokine dimerize.
- Transforming growth factors (TGFs)
Family of secreted proteins that act in a diverse set of roles from embryonic development to apoptosis. Their role in cell growth and differentiation makes them an attractive candidate for use in the treatment of cancer.
- Tumor-necrosis factors (TNFs)
A family of transmembrane proteins that can be cleaved to become soluble proteins and play a role in inflammation and apoptosis.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Declaration of Competing Interest
J.L.M. is a co-inventor on patent applications US11198717B2, US20210309707A1, and US62/878,574, 2019, and has shares in Synthekine.
References
- 1.Kany S, Vollrath JT, and Relja B, Cytokines in Inflammatory Disease. International journal of molecular sciences, 2019. 20(23): p. 6008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Oppenheim JJ, Cytokines: past, present, and future. Int J Hematol, 2001. 74(1): p. 3–8. [DOI] [PubMed] [Google Scholar]
- 3.Turner MD, et al. , Cytokines and chemokines: At the crossroads of cell signalling and inflammatory disease. Biochimica et Biophysica Acta (BBA) - Molecular Cell Research, 2014. 1843(11): p. 2563–2582. [DOI] [PubMed] [Google Scholar]
- 4.Ait-Oufella H, et al. , Recent Advances on the Role of Cytokines in Atherosclerosis. Arteriosclerosis, Thrombosis, and Vascular Biology, 2011. 31(5): p. 969–979. [DOI] [PubMed] [Google Scholar]
- 5.Castro LS, et al. , Interferon-Based Biopharmaceuticals: Overview on the Production, Purification, and Formulation. Vaccines, 2021. 9(4): p. 328. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Ahlin A, et al. , Gamma interferon treatment of patients with chronic granulomatous disease is associated with augmented production of nitric oxide by polymorphonuclear neutrophils. Clinical and diagnostic laboratory immunology, 1999. 6(3): p. 420–424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Klapper JA, et al. , High-dose interleukin-2 for the treatment of metastatic renal cell carcinoma : a retrospective analysis of response and survival in patients treated in the surgery branch at the National Cancer Institute between 1986 and 2006. Cancer, 2008. 113(2): p. 293–301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Filipi M and Jack S, Interferons in the Treatment of Multiple Sclerosis: A Clinical Efficacy, Safety, and Tolerability Update. International journal of MS care, 2020. 22(4): p. 165–172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Zhang J-M and An J, Cytokines, inflammation, and pain. International anesthesiology clinics, 2007. 45(2): p. 27–37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Wang X, et al. , Structural biology of shared cytokine receptors. Annual review of immunology, 2009. 27: p. 29–60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Foster JR, The functions of cytokines and their uses in toxicology. International journal of experimental pathology, 2001. 82(3): p. 171–192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Khodabakhsh F, et al. , Challenges and advancements in the pharmacokinetic enhancement of therapeutic proteins. Preparative Biochemistry & Biotechnology, 2021. 51(6): p. 519–529. [DOI] [PubMed] [Google Scholar]
- 13.Leader B, Baca QJ, and Golan DE, Protein therapeutics: a summary and pharmacological classification. Nature Reviews Drug Discovery, 2008. 7(1): p. 21–39. [DOI] [PubMed] [Google Scholar]
- 14.Lerner DM, Stoudemire A, and Rosenstein DL, Neuropsychiatric toxicity associated with cytokine therapies. Psychosomatics, 1999. 40(5): p. 428–35. [DOI] [PubMed] [Google Scholar]
- 15.Baldo BA, Side effects of cytokines approved for therapy. Drug safety, 2014. 37(11): p. 921–943. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Sleijfer S, et al. , Side effects of interferon-alpha therapy. Pharm World Sci, 2005. 27(6): p. 423–31. [DOI] [PubMed] [Google Scholar]
- 17.Kirkwood JM, et al. , Mechanisms and Management of Toxicities Associated With High-Dose Interferon Alfa-2b Therapy. Journal of Clinical Oncology, 2002. 20(17): p. 3703–3718. [DOI] [PubMed] [Google Scholar]
- 18.Ravaud A, et al. , Toxicity and feasibility of adjuvant high-dose interferon alpha-2b in patients with melanoma in clinical oncologic practice. British journal of cancer, 1999. 80(11): p. 1767–1769. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Vilček J and Feldmann M, Historical review: Cytokines as therapeutics and targets of therapeutics. Trends in Pharmacological Sciences, 2004. 25(4): p. 201–209. [DOI] [PubMed] [Google Scholar]
- 20.Mendoza JL, et al. , The IFN-λ-IFN-λR1-IL-10Rβ Complex Reveals Structural Features Underlying Type III IFN Functional Plasticity. Immunity, 2017. 46(3): p. 379–392. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Saraiva M and O’Garra A, The regulation of IL-10 production by immune cells. Nature Reviews Immunology, 2010. 10(3): p. 170–181. [DOI] [PubMed] [Google Scholar]
- 22.Gorby C, et al. , Engineered IL-10 variants elicit potent immunomodulatory effects at low ligand doses. Sci Signal, 2020. 13(649). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Saxton Robert A, et al. , Structure-based decoupling of the pro- and anti-inflammatory functions of interleukin-10. Science, 2021. 371(6535): p. eabc8433. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Mo F, et al. , An engineered IL-2 partial agonist promotes CD8+ T cell stemness. Nature, 2021. 597(7877): p. 544–548. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Silva D-A, et al. , De novo design of potent and selective mimics of IL-2 and IL-15. Nature, 2019. 565(7738): p. 186–191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Baek M, et al. , Accurate prediction of protein structures and interactions using a three-track neural network. Science, 2021. 373(6557): p. 871–876. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Boyman O and Sprent J, The role of interleukin-2 during homeostasis and activation of the immune system. Nature Reviews Immunology, 2012. 12(3): p. 180–190. [DOI] [PubMed] [Google Scholar]
- 28.Blattman JN, et al. , Therapeutic use of IL-2 to enhance antiviral T-cell responses in vivo. Nat Med, 2003. 9(5): p. 540–7. [DOI] [PubMed] [Google Scholar]
- 29.Siegel JP and Puri RK, Interleukin-2 toxicity. J Clin Oncol, 1991. 9(4): p. 694–704. [DOI] [PubMed] [Google Scholar]
- 30.Levin AM, et al. , Exploiting a natural conformational switch to engineer an interleukin-2 ‘superkine’. Nature, 2012. 484(7395): p. 529–533. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Fleishman SJ, et al. , RosettaScripts: a scripting language interface to the Rosetta macromolecular modeling suite. PLoS One, 2011. 6(6): p. e20161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Leaver-Fay A, et al. , ROSETTA3: an object-oriented software suite for the simulation and design of macromolecules. Methods Enzymol, 2011. 487: p. 545–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Chaudhury S, Lyskov S, and Gray JJ, PyRosetta: a script-based interface for implementing molecular modeling algorithms using Rosetta. Bioinformatics, 2010. 26(5): p. 689–91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Sahin D, et al. , An IL-2-grafted antibody immunotherapy with potent efficacy against metastatic cancer. Nature Communications, 2020. 11(1): p. 6440. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Guo M, et al. , Proliferation of Highly Cytotoxic Human Natural Killer Cells by OX40L Armed NK-92 With Secretory Neoleukin-2/15 for Cancer Immunotherapy. Front Oncol, 2021. 11: p. 632540. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Cao L, et al. , Design of protein-binding proteins from the target structure alone. Nature, 2022. 605(7910): p. 551–560. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Weinstein JJ, et al. , PROSS 2: a new server for the design of stable and highly expressed protein variants. Bioinformatics, 2020. 37(1): p. 123–125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Bednar D, et al. , FireProt: Energy- and Evolution-Based Computational Design of Thermostable Multiple-Point Mutants. PLOS Computational Biology, 2015. 11(11): p. e1004556. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Moraga I, et al. , Synthekines are surrogate cytokine and growth factor agonists that compel signaling through non-natural receptor dimers. eLife, 2017. 6: p. e22882. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Ng S, et al. , Stimulation of Natural Killer Cell-Mediated Tumor Immunity by an IL15/TGFβ-Neutralizing Fusion Protein. Cancer Res, 2016. 76(19): p. 5683–5695. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Kolářová L, et al. , De novo developed protein binders mimicking Interferon lambda signaling. The FEBS Journal, 2022. 289(9): p. 2672–2684. [DOI] [PubMed] [Google Scholar]
- 42.Yen M, et al. , Facile discovery of surrogate cytokine agonists. Cell, 2022. 185(8): p. 1414–1430.e19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Lasfar A, et al. , Characterization of the mouse IFN-lambda ligand-receptor system: IFN-lambdas exhibit antitumor activity against B16 melanoma. Cancer Res, 2006. 66(8): p. 4468–77. [DOI] [PubMed] [Google Scholar]
- 44.Mantovani A, et al. , Interleukin-1 and Related Cytokines in the Regulation of Inflammation and Immunity. Immunity, 2019. 50(4): p. 778–795. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Zhou T, et al. , IL-18BP is a secreted immune checkpoint and barrier to IL-18 immunotherapy. Nature, 2020. 583(7817): p. 609–614. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Sun Z, et al. , A next-generation tumor-targeting IL-2 preferentially promotes tumor-infiltrating CD8+ T-cell response and effective tumor control. Nature Communications, 2019. 10(1): p. 3874. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Klein C, et al. , Cergutuzumab amunaleukin (CEA-IL2v), a CEA-targeted IL-2 variant-based immunocytokine for combination cancer immunotherapy: Overcoming limitations of aldesleukin and conventional IL-2-based immunocytokines. OncoImmunology, 2017. 6(3): p. e1277306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Tomala J, et al. , Chimera of IL-2 Linked to Light Chain of anti-IL-2 mAb Mimics IL-2/anti-IL-2 mAb Complexes Both Structurally and Functionally. ACS Chemical Biology, 2013. 8(5): p. 871–876. [DOI] [PubMed] [Google Scholar]
- 49.Li Y, et al. , Targeting IL-21 to tumor-reactive T cells enhances memory T cell responses and anti-PD-1 antibody therapy. Nature Communications, 2021. 12(1): p. 951. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Mansurov A, et al. , Collagen-binding IL-12 enhances tumour inflammation and drives the complete remission of established immunologically cold mouse tumours. Nature Biomedical Engineering, 2020. 4(5): p. 531–543. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Cao X, et al. , Next generation of tumor-activating type I IFN enhances anti-tumor immune responses to overcome therapy resistance. Nature Communications, 2021. 12(1): p. 5866. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Mansurov A, et al. , Masking the immunotoxicity of interleukin-12 by fusing it with a domain of its receptor via a tumour-protease-cleavable linker. Nat Biomed Eng, 2022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Nirschl CJ, et al. , Discovery of a Conditionally Activated IL-2 that Promotes Antitumor Immunity and Induces Tumor Regression. Cancer Immunol Res, 2022. 10(5): p. 581–596. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Adams G, et al. , Targeting cytokines to inflammation sites. Nat Biotechnol, 2003. 21(11): p. 1314–20. [DOI] [PubMed] [Google Scholar]
- 55.Olczyk P, Mencner Ł, and Komosinska-Vassev K, The Role of the Extracellular Matrix Components in Cutaneous Wound Healing. BioMed Research International, 2014. 2014: p. 747584. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Maldonado M and Nam J, The role of changes in extracellular matrix of cartilage in the presence of inflammation on the pathology of osteoarthritis. BioMed research international, 2013. 2013: p. 284873–284873. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Martino MM, et al. , Growth factors engineered for super-affinity to the extracellular matrix enhance tissue healing. Science, 2014. 343(6173): p. 885–8. [DOI] [PubMed] [Google Scholar]
- 58.White MJV, et al. , VEGF-A, PDGF-BB and HB-EGF engineered for promiscuous super affinity to the extracellular matrix improve wound healing in a model of type 1 diabetes. npj Regenerative Medicine, 2021. 6(1): p. 76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Katsumata K, et al. , Conferring extracellular matrix affinity enhances local therapeutic efficacy of anti-TNF-α antibody in a murine model of rheumatoid arthritis. Arthritis Res Ther, 2019. 21(1): p. 298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Quesada JR, et al. , Treatment of hairy cell leukemia with recombinant alpha-interferon. Blood, 1986. 68(2): p. 493–7. [PubMed] [Google Scholar]
- 61.Bohn J-P, Gastl G, and Steurer M, Long-term treatment of hairy cell leukemia with interferon-α: still a viable therapeutic option. Memo, 2016. 9: p. 63–65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Harris JM and Chess RB, Effect of pegylation on pharmaceuticals. Nature reviews Drug discovery, 2003. 2(3): p. 214–221. [DOI] [PubMed] [Google Scholar]
- 63.Veronese FM, Peptide and protein PEGylation: a review of problems and solutions. Biomaterials, 2001. 22(5): p. 405–417. [DOI] [PubMed] [Google Scholar]
- 64.Fam CM, et al. , PEGylation improves the pharmacokinetic properties and ability of interferon gamma to inhibit growth of a human tumor xenograft in athymic mice. Journal of interferon & cytokine research : the official journal of the International Society for Interferon and Cytokine Research, 2014. 34(10): p. 759–768. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Eiger B, Phase 3 Study to Evaluate the Efficacy and Safety of Peginterferon Lambda for 48 Weeks in Patients With Chronic HDV. 2024.
- 66.Johns Hopkins U and Eiger B, Peginterferon Lambda-1a for the Prevention and Treatment of SARS-CoV-2 (COVID-19) Infection. 2021.
- 67.National Institute of, D., et al. , Treatment of Chronic Delta Hepatitis With Lonafarnib, Ritonavir and Lambda Interferon. 2020.
- 68.Raymond C, Eiger B, and H. Massachusetts General, Pegylated Interferon Lambda Treatment for COVID-19. 2021.
- 69.Yang JC, et al. , The use of polyethylene glycol-modified interleukin-2 (PEG-IL-2) in the treatment of patients with metastatic renal cell carcinoma and melanoma. A phase I study and a randomized prospective study comparing IL-2 alone versus IL-2 combined with PEG-IL-2. Cancer, 1995. 76(4): p. 687–94. [DOI] [PubMed] [Google Scholar]
- 70.Rouanne M, Zitvogel L, and Marabelle A, Pegylated Engineered IL2 plus Anti–PD-1 Monoclonal Antibody: The Nectar Comes from the Combination. Cancer Discovery, 2020. 10(8): p. 1097–1099. [DOI] [PubMed] [Google Scholar]
- 71.Robinson TO, et al. , NKTR-255 is a polymer-conjugated IL-15 with unique mechanisms of action on T and natural killer cells. The Journal of Clinical Investigation, 2021. 131(19). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Mathews JM, Parker MK, and Matthews HB, Metabolism and disposition of diethylene glycol in rat and dog. Drug metabolism and disposition, 1991. 19(6): p. 1066–1070. [PubMed] [Google Scholar]
- 73.Herold DA, et al. , Toxicity of topical polyethylene glycol. Toxicology and Applied Pharmacology, 1982. 65(2): p. 329–335. [DOI] [PubMed] [Google Scholar]
- 74.Ulbricht J, Jordan R, and Luxenhofer R, On the biodegradability of polyethylene glycol, polypeptoids and poly (2-oxazoline) s. Biomaterials, 2014. 35(17): p. 4848–4861. [DOI] [PubMed] [Google Scholar]
- 75.Turecek PL, et al. , PEGylation of biopharmaceuticals: a review of chemistry and nonclinical safety information of approved drugs. Journal of pharmaceutical sciences, 2016. 105(2): p. 460–475. [DOI] [PubMed] [Google Scholar]
- 76.Mima Y, et al. , Anti-PEG IgM is a major contributor to the accelerated blood clearance of polyethylene glycol-conjugated protein. Molecular pharmaceutics, 2015. 12(7): p. 2429–2435. [DOI] [PubMed] [Google Scholar]
- 77.Muneeruddin K, et al. , Characterization of a PEGylated protein therapeutic by ion exchange chromatography with on-line detection by native ESI MS and MS/MS. Analyst, 2017. 142(2): p. 336–344. [DOI] [PubMed] [Google Scholar]
- 78.Schellenberger V, et al. , A recombinant polypeptide extends the in vivo half-life of peptides and proteins in a tunable manner. Nature Biotechnology, 2009. 27(12): p. 1186–1190. [DOI] [PubMed] [Google Scholar]
- 79.Kierstead PH, et al. , The effect of polymer backbone chemistry on the induction of the accelerated blood clearance in polymer modified liposomes. J Control Release, 2015. 213: p. 1–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Schlapschy M, et al. , PASylation: a biological alternative to PEGylation for extending the plasma half-life of pharmaceutically active proteins. Protein Eng Des Sel, 2013. 26(8): p. 489–501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Shamloo A, Rostami P, and Mahmoudi A, PASylation Enhances the Stability, Potency, and Plasma Half-Life of Interferon α-2a: A Molecular Dynamics Simulation. Biotechnology Journal, 2020. 15(8): p. 1900385. [DOI] [PubMed] [Google Scholar]
- 82.Harari D, et al. , Enhanced in vivo efficacy of a long-life type I Interferon superagonist in a mouse model of multiple sclerosis. Journal of Neuroimmunology, 2014. 275(1): p. 219. [Google Scholar]
- 83.Andersen JT and Sandlie I, The versatile MHC class I-related FcRn protects IgG and albumin from degradation: implications for development of new diagnostics and therapeutics. Drug metabolism and pharmacokinetics, 2009. 24(4): p. 318–332. [DOI] [PubMed] [Google Scholar]
- 84.Stapleton NM, et al. , Competition for FcRn-mediated transport gives rise to short half-life of human IgG3 and offers therapeutic potential. Nature communications, 2011. 2(1): p. 1–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Sockolosky JT and Szoka FC, The neonatal Fc receptor, FcRn, as a target for drug delivery and therapy. Advanced Drug Delivery Reviews, 2015. 91: p. 109–124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Wu B and Sun YN, Pharmacokinetics of Peptide-Fc fusion proteins. J Pharm Sci, 2014. 103(1): p. 53–64. [DOI] [PubMed] [Google Scholar]
- 87.Rath T, et al. , Fc-fusion proteins and FcRn: structural insights for longer-lasting and more effective therapeutics. Critical reviews in biotechnology, 2015. 35(2): p. 235–254. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Vazquez-Lombardi R, et al. , Potent antitumour activity of interleukin-2-Fc fusion proteins requires Fc-mediated depletion of regulatory T-cells. Nature Communications, 2017. 8(1): p. 15373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Kim J-H, et al. , Hybrid Fc-fused interleukin-7 induces an inflamed tumor microenvironment and improves the efficacy of cancer immunotherapy. Clinical & Translational Immunology, 2020. 9(9): p. e1168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Liu B, et al. , Evaluation of the biological activities of the IL-15 superagonist complex, ALT-803, following intravenous versus subcutaneous administration in murine models. Cytokine, 2018. 107: p. 105–112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Tang K-Y, et al. , Safety, pharmacokinetics, and biomarkers of F-652, a recombinant human interleukin-22 dimer, in healthy subjects. Cellular & Molecular Immunology, 2019. 16(5): p. 473–482. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Jung K, et al. , Heterodimeric Fc-fused IL12 shows potent antitumor activity by generating memory CD8+ T cells. OncoImmunology, 2018. 7(7): p. e1438800. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Hussell T and Openshaw PJ, Intracellular IFN-gamma expression in natural killer cells precedes lung CD8+ T cell recruitment during respiratory syncytial virus infection. Journal of General Virology, 1998. 79(11): p. 2593–2601. [DOI] [PubMed] [Google Scholar]
- 94.Czajkowsky DM, et al. , Fc-fusion proteins: new developments and future perspectives. EMBO molecular medicine, 2012. 4(10): p. 1015–1028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Yuan W. m., et al. , The generation and biological activity of a long-lasting recombinant human interferon-λ1. Protein Engineering, Design and Selection, 2018. 31(9): p. 355–360. [DOI] [PubMed] [Google Scholar]
- 96.Zhang Q, et al. , Development and biological activity of long-acting recombinant human interferon-α2b. BMC Biotechnology, 2020. 20(1): p. 16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Peters T Jr., Serum albumin. Adv Protein Chem, 1985. 37: p. 161–245. [DOI] [PubMed] [Google Scholar]
- 98.Sleep D, Cameron J, and Evans LR, Albumin as a versatile platform for drug half-life extension. Biochimica et Biophysica Acta (BBA)-General Subjects, 2013. 1830(12): p. 5526–5534. [DOI] [PubMed] [Google Scholar]
- 99.Subramanian GM, et al. , Albinterferon alpha-2b: a genetic fusion protein for the treatment of chronic hepatitis C. Nat Biotechnol, 2007. 25(12): p. 1411–9. [DOI] [PubMed] [Google Scholar]
- 100.Zeuzem S, et al. , Albinterferon alfa‐2b dosed every two or four weeks in interferon‐naïve patients with genotype 1 chronic hepatitis C. Hepatology, 2008. 48(2): p. 407–417. [DOI] [PubMed] [Google Scholar]
- 101.Heathcote EJ, et al. , Peginterferon alfa-2a in patients with chronic hepatitis C and cirrhosis. New England Journal of Medicine, 2000. 343(23): p. 1673–1680. [DOI] [PubMed] [Google Scholar]
- 102.Liu H, et al. , Structure-based programming of lymph-node targeting in molecular vaccines. Nature, 2014. 507(7493): p. 519–522. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Rakhra K, et al. , Exploiting albumin as a mucosal vaccine chaperone for robust generation of lung-resident memory T cells. Sci Immunol, 2021. 6(57). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Ishihara A, et al. , Prolonged residence of an albumin-IL-4 fusion protein in secondary lymphoid organs ameliorates experimental autoimmune encephalomyelitis. Nat Biomed Eng, 2021. 5(5): p. 387–398. [DOI] [PubMed] [Google Scholar]
- 105.Yuba E, et al. , Suppression of Rheumatoid Arthritis by Enhanced Lymph Node Trafficking of Engineered Interleukin-10 in Murine Models. Arthritis & Rheumatology, 2021. 73(5): p. 769–778. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Huang Y-S, et al. , Preparation and characterization of a potent, long-lasting recombinant human serum albumin-interferon-α2b fusion protein expressed in Pichia pastoris. European journal of pharmaceutics and biopharmaceutics, 2007. 67(2): p. 301–308. [DOI] [PubMed] [Google Scholar]
- 107.Zhao HL, et al. , Balancing the Pharmacokinetics and Pharmacodynamics of Interferon-α2b and Human Serum Albumin Fusion Protein by Proteolytic or Reductive Cleavage Increases Its in Vivo Therapeutic Efficacy. Molecular Pharmaceutics, 2012. 9(3): p. 664–670. [DOI] [PubMed] [Google Scholar]
- 108.Nilvebrant J and Hober S, The albumin-binding domain as a scaffold for protein engineering. Computational and structural biotechnology journal, 2013. 6: p. e201303009–e201303009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Adabi E, et al. , Evaluation of an Albumin-Binding Domain Protein Fused to Recombinant Human IL-2 and Its Effects on the Bioactivity and Serum Half-Life of the Cytokine. Iranian biomedical journal, 2017. 21(2): p. 77–83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Hsu F-T, et al. , Preclinical Evaluation of Recombinant Human IL15 Protein Fused with Albumin Binding Domain on Anti-PD-L1 Immunotherapy Efficiency and Anti-Tumor Immunity in Colon Cancer and Melanoma. Cancers, 2021. 13(8): p. 1789. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Guo J, et al. , Head-to-tail macrocyclization of albumin-binding domain fused interferon alpha improves the stability, activity, tumor penetration, and pharmacology. Biomaterials, 2020. 250: p. 120073. [DOI] [PubMed] [Google Scholar]
- 112.Wang Z, et al. , One-month zero-order sustained release and tumor eradication after a single subcutaneous injection of interferon alpha fused with a body-temperature-responsive polypeptide. Biomaterials Science, 2019. 7(1): p. 104–112. [DOI] [PubMed] [Google Scholar]
- 113.Gao X.-y.L.J.H.J.-w.G.G.-l.W.W.-p., Temperature-responsive Polymer Conjugation of Interferon-α Enhances Antitumor Efficacy. CNKI, 2018. [Google Scholar]
- 114.Qian M, et al. , Long-Acting Human Interleukin 2 Bioconjugate Modified with Fatty Acids by Sortase A. Bioconjugate Chemistry, 2021. 32(3): p. 615–625. [DOI] [PubMed] [Google Scholar]
- 115.Ahmed CM, et al. , A Cell Penetrating Peptide from Type I Interferon Protects the Retina in a Mouse Model of Autoimmune Uveitis. bioRxiv, 2019: p. 2019.12.23.886986. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Poorebrahim M, et al. , Immunomodulatory effects of a rationally designed peptide mimetic of human IFNβ in EAE model of multiple sclerosis. Progress in Neuro-Psychopharmacology and Biological Psychiatry, 2018. 82: p. 49–61. [DOI] [PubMed] [Google Scholar]
- 117.Lee S, et al. , A Glycoengineered Interferon-β Mutein (R27T) Generates Prolonged Signaling by an Altered Receptor-Binding Kinetics. Frontiers in Pharmacology, 2019. 9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Engelen W, et al. , Programmable Bivalent Peptide–DNA Locks for pH-Based Control of Antibody Activity. ACS Central Science, 2020. 6(1): p. 22–31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Zhao Y, et al. , Switchable immune modulator for tumor-specific activation of anticancer immunity. Science Advances, 2021. 7(37): p. eabg7291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Trang VH, et al. , A coiled-coil masking domain for selective activation of therapeutic antibodies. Nature Biotechnology, 2019. 37(7): p. 761–765. [DOI] [PubMed] [Google Scholar]
- 121.Ramos TI, et al. , Forms and Methods for Interferon’s Encapsulation. Pharmaceutics, 2021. 13(10). [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.