

Abstract
Neutrophil extracellular traps (NETs) evolved to protect the host against microbial infections, and are formed by a web-like structure of DNA that is decorated with anti-microbial effectors. Due to their potent inflammatory functions, NETs also cause tissue damage, and can favor and/or aggravate inflammatory diseases. This multi-pronged activity of NETs requires that the induction, release, and degradation of NETs are tightly regulated. Here we describe the key pathways that are intrinsic to neutrophils and regulate NETosis, and we review the most recent findings on how neutrophil extrinsic factors participate in the formation of NETs. In particular, we emphasize how bystander cells contribute to modifying the capacity of neutrophils to undergo NETosis. Finally, we discuss how these neutrophil extrinsic processes can be harnessed to protect the host against the excessive inflammation elicited by uncontrolled NET release.
Introduction.
Neutrophils represent the most abundant immune cell in the blood and, in case of infection or sterile injury, are among the first immune cells recruited to the inflamed tissue [1]. Neutrophils are activated through receptors that recognize proinflammatory signals such as pathogen-associated molecular patterns (PAMPs), which originate from an invading pathogen, or damaged-associated molecular patterns (DAMPs) that derive from the host. Activation of neutrophils triggers a signaling cascade that induces a variety of effector functions. The importance of neutrophils in fighting infections is highlighted by the recurrent bacterial and fungal infections that characterize patients with congenital or acquired conditions that alter neutrophil numbers or functionality [2,3]. Besides releasing cytokines and chemokines to maximize the host’s immune response, neutrophils also exert three major antimicrobial effector functions: release of granules that are packed with antimicrobial peptides, internalization and neutralization of pathogens by phagocytosis, and release of neutrophil extracellular traps (NETs). To maintain tissue homeostasis, neutrophil responses must be commensurate to the threat, in order to effectively eliminate an invading pathogen, without damaging the host. Thus, understanding how neutrophil activities are modulated is a prerequisite to intervene against infection-driven and/or inflammatory diseases sustained by unbalanced neutrophil responses.
Neutrophil extracellular traps.
The production and release of NETs, referred to as NETosis, was first described as a suicidal tool employed by neutrophils to kill bacteria extracellularly [4]. NETs are long filamentous extracellular web-like structures of decondensed DNA characterized by the presence of citrullinated histone H3, and proteins usually stored in azurophilic granules, such as neutrophil elastase (NE), cathepsin G and myeloperoxidase (MPO). The majority of DNA in NETs originates from the nucleus of cells that produce them, although NETs can also contain mitochondrial DNA [5]. The release of nuclear DNA follows the rupture of the nuclear and plasma membranes, and results in death of the neutrophil within 3–8 hours. An alternative mechanism of rapid chromatin release happens within minutes of neutrophil activation, does not lead to membrane rupture and lytic death, and is referred to as “vital NETosis” (as opposed to “suicidal NETosis) [6,7].
NETosis was first described as an antimicrobial mechanism for entrapping and killing bacteria, but NETs are also induced by a large variety of stimuli [8] that include fungi [9,10] and viruses [11–15], as well as parasites [16] (Table 1). NETosis can occur during non-infectious sterile inflammation, and NETs are also involved in the development of immune-mediated disorders (BOX 1).
Table 1.
Stimuli that induce NETosis and cell-intrinsic regulators utilized.
| Stimulus | PAD4 requirement | ROS requirement | Calcium requirement | NE requirement | Refs. |
|---|---|---|---|---|---|
| PMA | Yes | Yes, by NADPH oxidase | Yes | n.d. | [34] |
| Ionomycin | Yes | Yes, by mitochondria | Yes | n.d. | [38] |
| Immune complexes | n.d. | Yes, by mitochondria | n.d. | n.d. | [34] |
| S. aureus | Yes | Yes, by NADPH oxidase | n.d. | n.d. | [72]; [34] |
| C. albicans | No | Yes | No | n.d. | [10]; [34] |
| P. aeruginosa | Yes | n.d. | Yes | Yes | [56]; [34] |
| T. gondii | n.d. | n.d. | n.d. | n.d. | [16] |
| L. amazonensis | n.d | Yes | Yes | n.d | [60] |
| Cytosolic Gram-negative bacteria/ligand | No | No | n.d. | No | [40] |
| Group B Streptococcus | No | No, by NADPH oxidase | n.d. | n.d. | [34] |
| Methicillin-resistant S. aureus | Yes | n.d. | n.d. | Yes | [34,57] |
| Glycans | n.d. | No | n.d. | n.d. | [39] |
| LPS | Yes | n.d. | Yes | n.d. | [47] |
| S. flexneri | Yes | n.d. | n.d. | n.d. | [47] |
| SARS-CoV-2 | n.d. | Yes, by NADPH oxidase | n.d. | n.d. | [12–14]; [122] |
| Influenza virus | n.d. | n.d. | n.d. | n.d. | [15] |
| HIV | n.d. | n.d. | n.d. | n.d. | [11] |
| Sendai virus | Yes | n.d. | n.d. | Yes | [123] |
(n.d. = not determined)
BOX 1. NETosis beyond infections: NETs as drivers of inflammatory diseases.
In a mouse model of transfusion-related acute lung injury, activated platelets exacerbate the release of NETs in the lungs, cause endothelial damage, and facilitate platelet accumulation and thrombus formation in the microcirculation, which in turn further increase lung injury [23]. Uncontrolled NETs formation also occludes the vasculature, and promotes deep vein thrombosis: the filamentous DNA structure of the NETs promotes platelet adhesion, activation and aggregation, and facilitates thrombus formation and growth. Histones present on NETs also induce the release of platelet mediators that then induce NETosis, and generate a detrimental feedback loop [24].
NETs are involved in the development of atherosclerosis: they sustain the inflammatory milieu that is characteristic of atherosclerosis [25] and, once the plaques are established, histones released within NETs become cytotoxic to smooth muscle cells, damage arterial tissue and destabilize the plaque [26]. NETs are also implicated in autoimmune-related diseases, where neutrophil-derived proteins serve as self-antigens that trigger the production of autoantibodies. NETs are also found in kidney biopsy tissue from patients with antineutrophil cytoplasmic autoantibody (ANCA)-associated vasculitis that shows antibodies against MPO and proteinase 3 [27]. In patients with rheumatoid arthritis (RA), antibodies against citrullinated histone are abundant in the synovial fluid [28], and similarly, patients with systemic lupus erythematosus (SLE) also show high levels of antiribonucleoprotein antibodies and autoantibody against citrullinated epitopes [29,30]. Likewise, lesions of plaque psoriasis present an elevated concentration of NETs that are rich in antimicrobial peptide LL37 complexed with DNA that induces a strong immune response; this response is considered to be the major cause of monocyte activation in psoriasis [31]. In patients with sterile and non-autoimmune diseases such as gout, monosodium urate (MSU) crystals induce the release of high levels of NETs in fluid from acutely inflamed joints [32]. Lastly, given the key functions played by neutrophils during tumorigenesis [33], a role for NETs is emerging in the context of tumors [19].
Given that a large variety of pathogen-derived and host-derived ligands stimulate NETosis [17], the cellular mechanism that drive NETosis has recently received much attention, nevertheless, the molecular mechanisms that determine whether or not NETs will be released by neutrophils in response to an infection remain a matter of debate. An intriguing model hypothesizes that the size of the infecting microorganism is a major factor in determining the release of NETs, due to a competition between NETs and phagocytosis to access NE: if the pathogen is too large to be phagocytosed, neutrophils preferentially release NETs, whereas smaller microorganisms are engulfed into phagosomes that fuse with azurophilic granules, sequester NE away from the nucleus, and thereby prevent chromatin decondensation [18]. However, many smaller extracellular - or even intracellular - pathogens also induce NETosis, pointing to the virulence of these pathogens as the discriminating factor that determines the induction of NETs [19]. A more recent programmed “proteome disarming” model proposes that the neutrophil proteome is modified via a cell-intrinsic program that causes a progressive loss of granule content and NET-forming capacity. This process is controlled by the CXCR2 receptor and by regulators of the circadian clock, which determine a diurnal loss of the ability of neutrophils to release NETs in tissues [20]. In keeping with this hypothesis, aged neutrophils that express a low level of Cxcl2 are more prone to the release of NETs [21]. Overall, it is possible that different subsets of neutrophils, defined by their activation state, protein content, and/or the circadian rhythm, are differently inclined to release NETs, thus dictating the choice between NET release or another response.
When NETosis is dysregulated, NETs may contribute to the pathogenesis of immune-related diseases: as such, NETosis is regulated at multiple levels. Two major types of programs that regulate the release of NETs have been described: i) those that are intrinsic to neutrophils, such as the level of activation of specific key enzymes or molecules; and ii) those that are extrinsic to the neutrophils, and involve interactions with other cells or mediators that potentiate or diminish the levels of NETs. Numerous novel intrinsic and extrinsic programs have been proposed as being involved in the control of NETosis, and point to a complex network of NET modulators. Here, we summarize the most important and most recently introduced ones.
Regulators of NETosis that are intrinsic to neutrophils.
The specific pathways that cause NET release are a function of activating stimuli, but several key processes initiated in neutrophils that regulate NETosis have been identified (Figure 1). One of the best-described pathways that lead to NETosis involves production of reactive oxygen species (ROS) via NADPH oxidase. ROS stimulate MPO, enable translocation of the NE from azurophilic granules into the cytosol, and thence into the nucleus; here, NE starts to degrade histones, unpacks the chromatin, and releases DNA. ROS also activate protein-arginine deiminase 4 (PAD4), which converts the arginine on histones to citrulline, further supporting chromatin decondensation and DNA expansion, which then leads to nuclear membrane disruption. Chromatin is released into the cytosol, where it is decorated with a variety of granular and cytosolic proteins [19]. The exact mechanism whereby NETs are terminally released into the extracellular space has not been delineated, but an active and programmed disassembling of the plasma membrane (as opposed to a passive membrane rupture due to DNA expansion) has been suggested [22].
Figure 1. Neutrophil intrinsic regulators of NETosis.
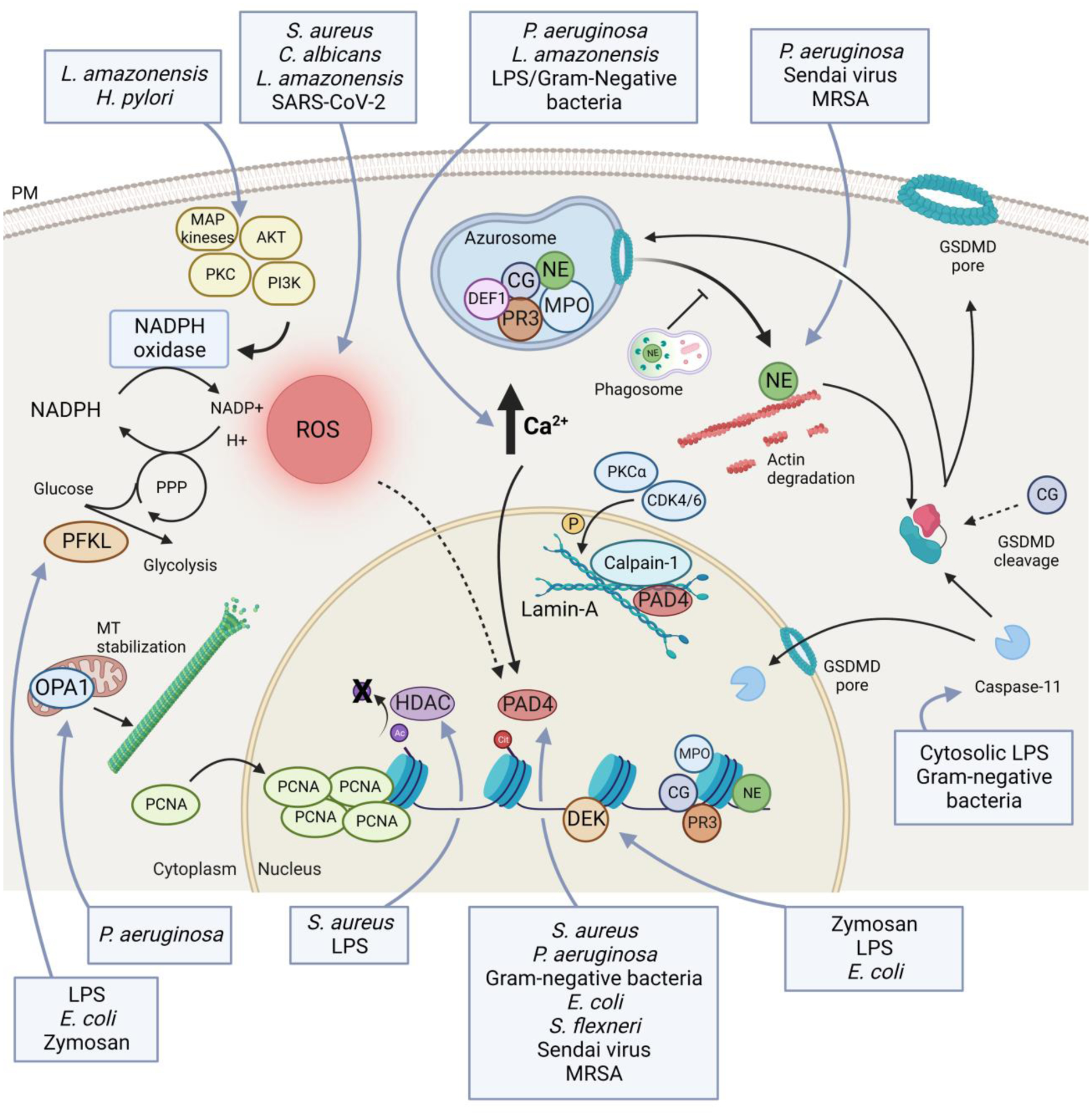
Schematic representation of the main cell-intrinsic regulators of NETosis. ROS production has an important role in regulating the release of NETs. ROS are mainly generated by the NADPH oxidase, that is regulated by several kinases, such as AKT, PI3K, PKC and MAPKs. Amount of ROS is also determined by levels of NADPH that are indirectly controlled by phosphofructokinase-1 liver type (PFKL) [70]. An increase in intracellular calcium levels is required for NETosis. Increased calcium levels, possibly in association with production of ROS, activate protein-arginine deiminase 4 (PAD4) that mediates histone citrullination and facilitates chromatin decondensation. PAD4, in association with calpain-1, PKCα and CDK4/6, mediates the disruption of the structural rigidity of the nucleus governed by the lamin-A network [51,73,74]. Histone citrullination, required to chromatin decondensation, occurs after deacetylation by class I/IIb histone deacetylase (HDAC) [72]. Other enzymes central to the formation of NETs are contained in the azurosome, and are mainly represented by myeloperoxidase (MPO), neutrophil elastase (NE), cathepsin G (CG), proteinase 3 (PR3) and defensin-1 (DEF1). Upon neutrophil activation, these enzymes translocate into the nucleus where they promote chromatin decondensation. Before moving to nuclei, NE is released in the cytoplasm and binds and degrades F-actin to arrest actin dynamics and to inhibit neutrophil migration [22]. The release of NE into the cytosol is mediated by ROS and MPO, and it is facilitated by gasdermin-D (GSDMD), but it does not require granule disruption [22,66]. GSDMD is also necessary to allow caspase-11 translocation into the nucleus, where it mediates histone clipping [40]. Moreover, GSDMD facilitates DNA release into the cytosol first, and then extracellularly by creating membranes pores. GSDMD is activated upon being cleaved by NE, caspase-11, and, potentially, by CG. Mitochondrial optic atrophy 1 (OPA1) is key to maintain ATP production and, consequently, for microtubule stability that is essential for NET release [71]. Lastly, further chromatin disassembly is accelerated by massive nuclear translocation of proliferating cell nuclear antigen (PCNA) [75] and by chromatin-binding protein DEK [59]. Blue boxes indicate pathogens and/or PAMPs that drive specific steps in the induction of NETs.
ROS and NADPH oxidase are key players during NETosis as underscored by the report that neutrophils from patients who suffer from chronic granulomatous disease (CGD) and in whom NADPH oxidase is non-functional, are unable to release NETs in response to S. aureus or PMA [34]. Similarly, NADPH synthesis is essential for maintaining the ROS burst: in neutrophils, NADPH is generated from glucose 6-phosphate in the oxidative branch of the pentose phosphate pathway (oxPPP). In fact, patients with defective glucose 6-phosphate dehydrogenase (the first enzyme of the oxPPP) exhibit low levels of NADPH and ROS, and a much higher susceptibility to bacterial infection [35]. Lungs have a high availability of oxygen that can be transformed into ROS, making this tissue particularly susceptible to the detrimental effects of uncontrolled NETosis; this is a factor that contributes to the pathogenesis of numerous chronic inflammatory lung diseases such as cystic fibrosis, chronic obstructive pulmonary disease (COPD) and allergic asthma [36]. Another example of the importance of NETosis in the lungs is represented by the infection with severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2). During lung infection with SARS-CoV-2, induction of NETosis is a key feature of severe COVID-19 [12–14], although whether ROS are directly involved in this process remains to be resolved [37]. Depending on the stimulus, NADPH may also be dispensable, as seen in the responses to immune complexes and ionomycin that instead rely on mitochondrial ROS [38]. Of note, NETs are induced independently of ROS in some conditions, including during non-lytic NETosis as well as when NETs are induced by glycans in the saliva [39], by cytoplasmic lipopolysaccharide (LPS), or by cytosolic Gram-negative bacteria [40]. How oxygen availability modulates NET release is still a matter of debate, complicated by the fact that in in vitro settings the levels of oxygen used to define normoxia and hypoxia are often very different from those at the tissue level [41]. Neutrophil activation and ROS production can lead to low oxygen availability at tissue level, however hypoxia has been associated both with increased as well as reduced NETosis [42–45]. A more comprehensive analysis of the impact of oxygen levels on neutrophil response will be critical to better predict the responses of neutrophils in different tissues (e.g., lungs, intestine) and/or environments (e.g., inflamed tissue, tumor microenvironment) characterized by different levels of oxygen availability.
Calcium is another important chemical mediator of NETosis. An increase in cytosolic calcium concentration is a direct consequence of neutrophil activation via classical receptors such as G protein-coupled receptors (GPCRs), Fcγ receptors, Toll-like receptors (TLRs) and integrins, which trigger calcium release from the endoplasmic reticulum (ER). Chelation of intracellular calcium inhibits release of NETs induced by chemical inducers as well as LPS, but not by Candida albicans [34,40]; nevertheless, chelation of extracellular calcium also impairs production of NETosis [34], suggesting that a sustained high level of calcium over time is critical for completion of NET release. Although it has been shown that the spike in intracellular calcium concentration activates the key enzyme PAD4 (see below) to drive NET induction [46], we still lack a comprehensive understanding of how calcium controls NETosis.
As mentioned above, PAD4 is a key enzyme for the induction of NETs [46–48]. PAD4 citrullinates the core histones H3, H4 and H2A [49], and converts amine groups to ketones, which results in the loss of a positive charge and facilitates chromatin decondensation [50]; PAD4 also achieves DNA decondensation by decreasing the affinity between DNA and histones. In ionomycin-induced NETosis, the increased calcium concentration not only activates PAD4, but also triggers the activity of calpain-1, which, together with PAD4, is necessary to fully break down the nuclear envelope and the chromatin-bound proteins such as lamin-A and high mobility group protein B1 (HMGB1) [51]. These findings suggest that PAD4-dependent citrullination of histones is required not only for disassembling DNA, but also for mediating the activity of other proteases on histones and nuclear proteins, which together assist nuclear rupture and NET release. Pad4-deficient neutrophils are unable to citrullinate histone H3 and cannot release NETs when stimulated with PMA, LPS, Shigella flexneri, calcium ionophores or methicillin-resistant S. aureus infection, although PAD4 is dispensable in response to C. albicans or Group B Streptococcus infection [34]. The level of activation of PAD4 is also important in the context of sterile inflammation. In fact, genetic absence of PAD4 ameliorates deep vein thrombosis [52], while PAD4 expression is upregulated in patients with diabetes [53]. But how PAD4 is activated is still a matter of debate. The peak concentration of intracellular calcium in activated neutrophils is low relative to the levels required to activate PAD4 in vitro [34]; thus other pathways (e.g., ROS), have been implicated in PAD4 activation in vivo. NETosis also occurs in the absence of histone citrullination, and NE and MPO are directly involved in chromatin decondensation (see below) [54]. Thus, although PAD4 is a key enzyme in the induction of NET release, more details about how this enzyme is activated and about its interactions with other enzymes still need to be revealed.
In addition to PAD4, other enzymes such as MPO, NE, proteinase 3 (PR3) and cathepsin G (CTSG) also participate in the formation of NETs. These enzymes are contained in the azurophilic granules along with other antimicrobial proteins such as defensin-1; together, these constitute the so-called “azurosome”. Upon induction of NETosis, components of the azurosome move from the granules into the cytosol and then into the nucleus, where they contribute to the disassembly of chromatin. How this movement occurs is not completely clear. NE translocation to the nucleus does not require granule disruption or fusion, but occurs in a ROS-dependent, MPO-dependent manner [22]. Initially, NE is released into the cytosol, where it binds F-actin and mediates F-Actin degradation, in order to arrest actin dynamics. This process may be essential for blocking neutrophil chemotaxis and also for immobilizing neutrophils that are present close to the site of the infection, where NETs need to be released. Once in the nucleus, NE partners with MPO to process histones and allow chromatin decondensation, and results in nuclear swelling [22]. At the same time, once the proteinases PR3 and CTSG are in the nucleus, they contribute to histone processing and, when released in the extracellular space together with DNA, they function as inflammatory mediators and antimicrobial molecules. The importance of the MPO-NE axis in NET release is supported by numerous studies on neutrophils harvested from patients who are deficient in MPO activity [55], and also based on observations in NE-deficient mice, or in neutrophils that are treated with NE inhibitors [54]. Patients with Papillon-Lefevre syndrome are characterized by defects in cathepsin C (CTSC), an enzyme that processes NE and CTSG to their mature forms. These patients present with defects in NET formation, and CTSC-deficient mice do not undergo NETosis in models of aortic aneurism and Sendai virus infection [19]. As with PAD4, the requirement of NE for NETosis differs depending on the stimulus, the cells, and/or the experimental model used. For instance, genetic absence of NE impairs the release of NETs in vivo upon exposure to P. aeruginosa [56] or methicillin-resistant S. aureus [57], and in response to non-infectious stimulus such as cholesterol crystals [58], but NE deficiency does not impact NETosis in response to PMA or ionomycin [34,56].
More recently, the chromatin-binding protein DEK in the nucleus has also been implicated in NETosis. Dek-deficient neutrophils are impaired in NET release, and ameliorate the outcome in a mouse model of joint inflammation. Absence of the enzymatic activity of DEK suggests that its binding to the chromatin sustains the decondensation process in a manner similar to that of MPO [59].
A number of kinases are implicated in the early steps that drive ROS production, and are involved in NETosis. The Raf-MEK-ERK MAP kinase pathway, protein kinase C (PKC), phospho-inositide 3-kinase (PI3K), AKT and IL-1 receptor-associated kinase (IRAK) are linked to NETosis in response to stimuli such as PMA, microorganisms, and parasites [60–62]. Studies of the role of these kinases in inducing NETosis have also revealed a role for autophagy machinery in the release of NETs: PI3K is required for NETosis and also for the autophagy process, and genetic absence of ATG7 (a key autophagy-associated protein) reduces the release of extracellular DNA from promyelocytes [63]. Thus, autophagy and ROS production are simultaneously required for NET induction, and inhibition of autophagy or NADPH oxidase prevents intracellular chromatin decondensation, which is vital for NET formation [64].
Besides the neutrophil intrinsic regulators of NETosis described thus far, recently other pathways have been implicated in the control of NETosis as reviewed below (Figure 1).
Inflammasome-dependent induction of NETosis.
Activation of the non-canonical and canonical inflammasome has recently been described as an additional mechanism that regulates NET induction. The inflammasome is a supramolecular organizing center (SMOC) [65], which often culminates in the cleavage and activation of gasdermin D (GSDMD). Genetic absence or inhibition of GSDMD prevents NET formation [40,66]. GSDMD is a pore-forming protein that is essential for the release of cleaved interleukin (IL)-1β, as well as for a form of cell death called pyroptosis, which occurs downstream of inflammasome activation. In neutrophils, GSDMD is cleaved and activated by two pathways. When triggered by classical stimuli such as PMA or extracellular pathogens, GSDMD is cleaved by NE; and in response to LPS or cytosolic Gram-negative bacteria, GSDMD is activated by caspase-11. The cleavage sites for NE are in positions 268 and 255, upstream of the caspase cleavage site, and they generate an active N-terminus domain [66,67]. GSDMD is required for the release of NE from the granules but it also forms pores on the plasma membrane that enable membrane disruption and the release of NETs [66]. Simultaneously, with caspase-11, GSDMD, is also involved in nuclear permeabilization and chromatin relaxation. In particular, GSDMD creates pores in the nuclear membrane that enable caspase-11 to access to the chromatin and act in a similar manner to NE, thereby mediating histone clipping and facilitating DNA expansion [40]. During pyroptosis, GSDMD in neutrophils can also be cleaved by CTSG, but whether this GSDMD-CTSG axis plays a role in modulation of NET extrusion still remains a mystery. Thus, GSDMD represents a player that is shared between pyroptosis and NETosis, two forms of cell death that are mechanistically different but that protect against intracellular and extracellular threats, respectively. Beside the activation that occurs in response to microbial encounter, during sterile inflammation PAD4 post-transcriptionally regulates the levels of NLR family pyrin domain containing 3 (NLRP3) and apoptosis-associated speck-like protein containing a CARD (ASC), as well as the assembly of the inflammasome, further linking the processes of NETosis and inflammasome activation [68]. Lastly, early during formation of the atherosclerosis plaque, cholesterol accumulation in myeloid cells has been shown to facilitate neutrophil recruitment and NETosis via inflammasome activation [69].
Metabolic regulation of NETosis
NETosis is additionally modulated via metabolic pathways, for example those that regulate the levels of NADPH that are critical for inducing NETs [35]. Recently, phosphofructokinase-1 liver type (PFKL) was identified as a modulator of glycolysis in neutrophils: it negatively regulates the glycolytic flux through the PPP, which determines NADPH levels, and tunes NADPH oxidase-dependent ROS production and NETosis [70]. When neutrophils are stimulated, PKC activation inhibits PFKL activity, to fully engage the release of NETs. Whether PKC regulates the functions of PFKL, and if so, whether it does so directly or indirectly, is not known [70].
Analogously, mitochondrial activity is required to produce NAD+, which fuels ATP production via glycolysis. Dysfunction of optic atrophy 1 (OPA1), a protein of the mitochondrial inner membrane, has recently been linked to reduced activity of the mitochondrial electron transport complex 1 in neutrophils [71]: this defect compromises NAD+ level and thus affects ATP production, leading to a severe defect in the microtubule network and the formation of NETs. The inability to release NETs means that mice lacking Opa1 fail to clear P. aeruginosa in the lungs [71].
Chromatin decondensation and nuclear dynamics controls NETosis
Disassembling of the chromatin is a fundamental step for NETosis. Normally, DNA is packed around histones, and post-translational modifications on the histones dictate access to the DNA. For example, acetylation of histones plays a key role in allowing gene transcription. A new role for histone acetylation in the control of NETosis was recently revealed [72]. In particular, chemical inhibition of class I and class IIb histone deacetylases (HDACs) has been shown to dampen NET formation and protect mice during sepsis, as well as during virus-induced lung damage, or during S. aureus bacterial lung infections [72]. The presence of the acetyl groups on histone H3 prevents its citrullination, and thereby impedes DNA expansion and NET release, without affecting other neutrophil functions such as phagocytosis or ROS production [72]. These findings spotlight the activity of class I/IIb HDACs downstream of ROS production but before histone H3 citrullination, and document that HDACs serve as another key checkpoint (besides PAD4) in the process of NET release.
The release of DNA outside the nucleus requires disruption of the nuclear membrane. A lamin network contributes to the structure of the nuclear envelope and plays a major role in protecting the nucleus from mechanical forces. During NETosis, the release of DNA into the cytosol requires a major remodeling of the lamin network, forming holes and then completely rupturing the membrane. To start this process, lamin is phosphorylated by PKCα and cyclin-dependent kinase (CDK) 4/6, leading to the disruption of the structural rigidity of the nucleus [73]. Lamin remodeling appears to be the last active step in DNA release: after this, the mechanical forces due to chromatin expansion lead to the definitive rupture of the nuclear envelope and the release of cytosolic DNA [73,74].
DNA expansion during NETosis is also facilitated by the activity of the DNA repair protein proliferating cell nuclear antigen (PCNA). ROS production leads to extensive DNA damage, and thus induces a massive translocation of PCNA from the cytosol to the nucleus, where the DNA-repair-complex assembly on DNA further supports chromatin decondensation. As a result, the DNA expands, and in turn accelerates rupture of the nuclear envelope [75].
Neutrophil extrinsic regulators of NETosis.
The level of NETs released in the circulation is critical for the host and high levels of circulating NETs are linked to severity in various infectious and non-infectious diseases [14,76,77]. Besides the above-mentioned pathways that regulate NETosis in neutrophils, other cells - or inflammatory mediators released by neighboring cells - can also enhance or reduce the release of NET by neutrophils (Figure 2). Although we mostly focus on the activity of bystander cells in regulating NETosis, it is important to note that also non-cellular factors have been involved in the regulation of the induction of NETs. In particular, the clotting factor fibrin [78] as well as substrate elasticity [79] have been shown to impact NETosis.
Figure 2. Neutrophil extrinsic regulators of NETosis.
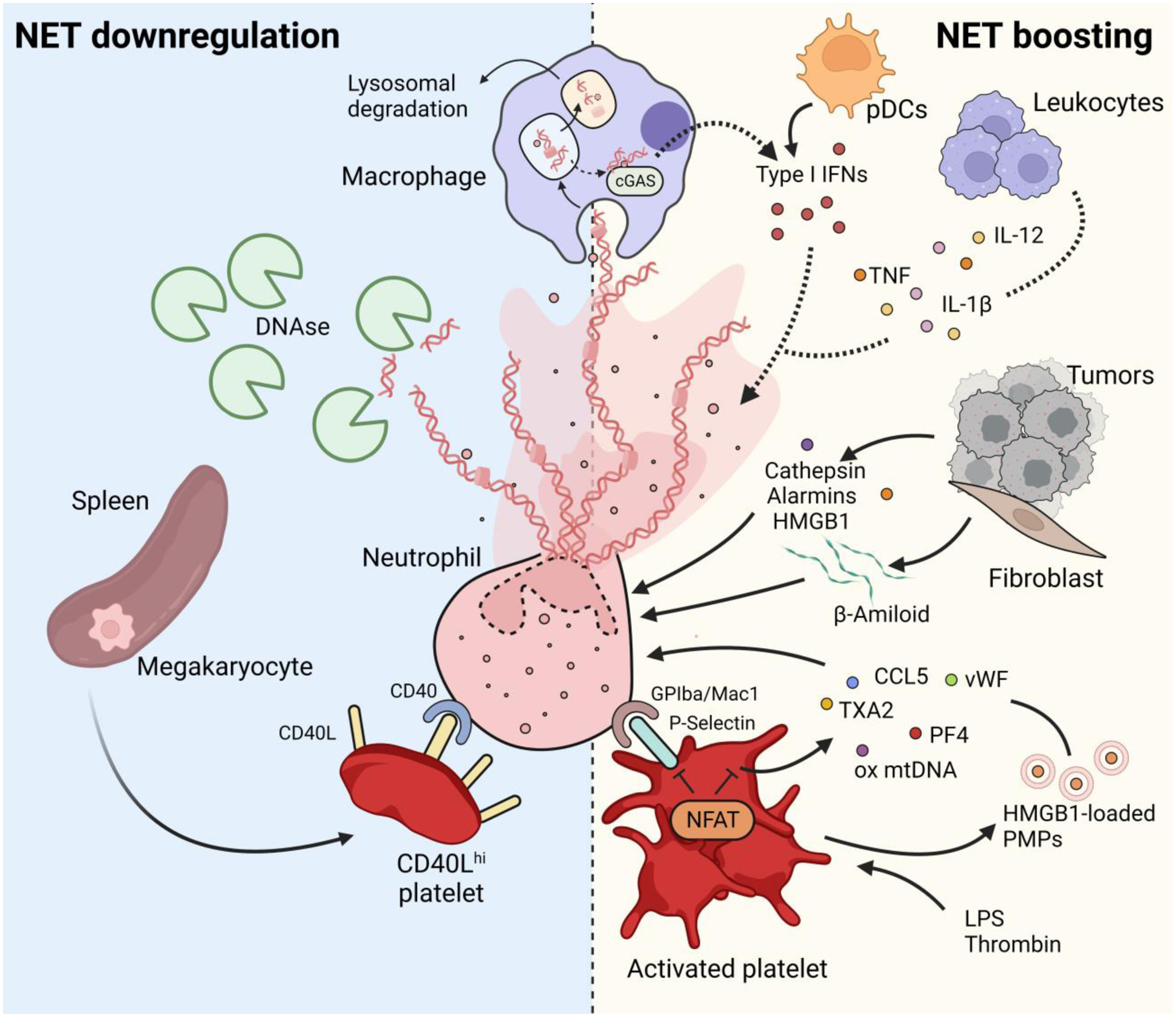
NETs are removed from the circulation by DNases [80] and/or by macrophage phagocytosis and subsequent lysosomal degradation [81]. NET release can be negatively modulated by a population of CD40Lhi platelets, produced by megakaryocytes in the spleen during severe sepsis [120]. Macrophages can also boost NETosis. The cGAS pathway senses the presence of NETs and allows the production of type I IFNs. Macrophage-derived, as well as plasmacytoid dendritic cells (pDCs)-derived [30] type I IFNs, in turn, favor NET release. Proinflammatory cytokines, such as TNF, IL-1β and IL-12, that are released by leukocytes during inflammation, also boost NETosis. Tumors also contribute to NET formation either directly by releasing cathepsin C and HMGB1, or indirectly by favoring the release of β-amyloid by tumor-associated fibroblasts [102]. Platelets, activated by either pathogens or via their activating receptors, potently induce NETs. Platelets stimulate NETosis either via direct contact with neutrophils that is mediated by the P-selectin-GPIba/Mac1 axis, or via release of soluble mediators such as thromboxane A2 (TXA2), platelet factor 4 (PF4), von Willebrand Factor (vWF), or CCL5. Platelets can also release oxidized mitochondrial DNA [116] and microparticles (PMPs) containing HMGB1 [96,97], that stimulate NETosis. The transcription factor NFAT is fundamental to modulate platelet activation by tuning P-selectin upregulation and granule release, and to prevent an exaggerated inflammatory response [113].
NET removal
Although how NETs are removed from the circulation is still an unsolved question, host DNases have been implicated. Mice that are defective in DNase1 as well as DNase1L3 are susceptible to the formation of clots and vascular occlusion during septicemia [80]. Since physiological concentrations of DNase in vitro do not effectively clear the DNA filaments, other factors must be also involved in vivo, and macrophages appear to play a key role in the process of NET removal. Macrophages phagocytose NETs, using an active process that is facilitated by DNase and NET opsonization [81]. NETs that are engulfed by macrophages are then degraded in the lysosomal compartment. While such degradation per se does not induce pro-inflammatory cytokine production by macrophages, in the presence of LPS, this process boosts the release of IL-1β, IL-6, and TNF by macrophages [81]. When NETs translocate from the phagosome to the cytosol, the DNA can be recognized by the DNA sensor cyclic GMP-AMP synthase (cGAS), and induces production of type I interferons both in vitro and in vivo. Stimulation of the cGAS/STING pathway relies on DNA-associated NE, and inhibition of NE activity diminishes interferon production by half [82]. Interferons, in turn, positively regulate NETosis (see below), highlighting once more the complex balance between the induction, sensing, and degradation of NETs.
A decrease in the ability of macrophages to clear NETs is linked to sustained inflammation in ARDS patients [83], and a recent study also suggests that polarization of macrophages determines their ability to eliminate NETs [84]. In particular, macrophage polarization toward a proinflammatory phenotype enhances the removal of NETs by these cells, through micropinocytosis. The inhibition of this mechanism increases the level of circulating NETs and is associated with a poor outcome in a murine model of vena cava thrombosis [84]. An inverse correlation between the local density of macrophages and the surrounding NETs has also been reported in the context of abdominal aortic aneurysm, confirming an important role of macrophages in controlling the number of NETs in vivo [84].
NET boosting
In addition to pathogen ligands, several host molecules, DAMPs, and pro-inflammatory cytokines induce and/or boost the release of NETs. For example, type I interferons exacerbate the formation of NETs in multiple chronic infections [30,85–87]. In contrast to type I interferons, type III interferons, in keeping with their capacity to decrease ROS production by neutrophils [88], have an inhibitory effect on NETosis in neonatal mice in a model of viral bronchiolitis [89]. It is important to note that the ability of neutrophils to sense and respond to interferons changes during their maturation. Mature neutrophils express higher level of the receptor of type I interferons, thus potentially increasing their ability to release NETs, compared to immature neutrophils [90]. Moreover, pro-inflammatory cytokines such as TNF, IL-1 β and IL-12 also induce NETosis in a NADPH oxidase-dependent manner [91,92]. The DAMP protein HMGB1 induces NETosis both in vitro and in vivo via the TLR4/MyD88 pathway [93], is produced in multiple settings, and can be actively or passively released by host cells and tumors [94,95]. Platelets release this molecule or present it, thereby inducing autophagy in neutrophils and, in turn, promoting NETosis and inhibiting apoptosis [96]. This mechanism has also been recently observed in patients with systemic sclerosis who show an enrichment in HMGB1-loaded platelet microparticles and NETs in the blood [97]. These results further underscore that balancing the production of NETs is a tightly regulated process.
Due to the extensive roles played by neutrophils during tumorigenesis [33], NETosis is also implicated in the context of tumor progression and the formation of metastases [98–100]. Tumors release several molecules that stimulate the release of NETs. HMGB1, alarmins and cathepsin C are released by tumor cells, and they induce NET release in human and murine neutrophils [95,101]. β-amiloid released by tumor-associated fibroblasts induces NADPH oxidase-dependent NET formation that, in turn, further favors tumor progression, whereas inhibition of NET formation prevents tumor growth [102]. Additionally, tumors release factors that stimulate NETosis and promote the recruitment and polarization of neutrophils [99,103,104]. Induction of NETs by tumors facilitates tumor progression and metastasis, and leads to a negative feedback loop that worsens the pathology. For this reason, it has been suggested that NETosis components can serve as prognostic markers in patients with cancer [100,105].
The interaction between neutrophils and platelets favors NETosis.
Platelets are major extrinsic neutrophil regulators and, in recent years, the interaction between platelets and neutrophils has been studied extensively [106–108]. Platelet-neutrophil complexes (PNCs) also contribute to the etiology of several thrombotic and infectious disease [109], in which the number of PCNs in the bloodstream serves as a hallmark of the severity of the pathology [110]. These data demonstrate that a deeper understanding of the interaction between neutrophils and platelets is a critical prerequisite for developing innovative treatments against microbial and non-microbial diseases.
Platelets that are activated by pathogens or sterile threats can efficiently induce NET formation, which in turn further contributes to inflammation. Formation of platelet-induced NETs requires physical interaction between P-selectin (which is upregulated on platelet surface upon activation) and P-selectin glycoprotein ligand 1 (PSGL-1) (which is expressed on neutrophils) [111]. The interaction between P-selectin and GPIbα/Mac-1 on platelets also serves to regulate NETosis [112]. Beside P-selectin, platelets simultaneously also release soluble factors (such as chemokines, PF4, CCL5, von Willebrand Factor (vWF), and thromboxane A2), to recruit and sustain the formation of NETs [24,112].
Interactions between platelets and neutrophils can exacerbate bacterial and viral infections. In influenza infected patients, platelets induce NETosis via TLR7-dependent complement C3 release [15]. Platelet-induced NETs are detrimental in bacterial sepsis [108,113], a life-threatening syndrome that is caused by a dysregulated immune response [114]. LPS-activated platelets induce the release of NETs through TLR4, and thereby enhance bacterial trapping in a mouse model of endotoxemia [115]. Notably, activation of a family of transcription factors collectively known as NFATs was recently shown to dampen platelet activation, and to prevent the initiation of a detrimental feedback loop that potentiates inflammation and aggravates septic shock and sepsis [113]. Also, GSDMD cleavage is elevated in patients with severe sepsis; S100A8/A9 released by neutrophils in a mouse model of sepsis induces a TLR4-dependent pathway in platelets that then leads to GSDMD-dependent pyroptosis: pyroptotic platelets release oxidized mitochondrial DNA that strongly promote NETosis, generating a detrimental feedback loop [116]. These data underscore the importance of identifying neutrophil extrinsic pathways that potentiate NETosis and possibly also aggravate pathogen-driven diseases. In fact, exaggerated platelet activation determines excessive recruitment of leukocytes and induction of inflammation, and boosts NETs release. During the initial phase of an infection, NETs are vital for ensnaring bacteria and preventing their spread in the bloodstream [117]. But NETs also facilitate platelet aggregation, thrombin activation and fibrin clot formation, which then cause liver damage and organ failure [108]. Severe sepsis is associated with thrombocytopenia and altered coagulation, which in turn leads to disseminated intravascular coagulation (DIC), ultimately impairs tissue oxygenation and results in patient death [118]. Additionally, plasma from patients with severe sepsis triggers the formation of NETs in the presence of platelets in vitro [115]. In this context, NET components further stimulate platelet activation, fueling a negative loop that often leads to a poor outcome [119]. However, during acute inflammatory states such as sepsis, the spleen produces a population of platelets that express high levels of CD40 ligand and have a strong immunomodulatory function [120], suggesting that specific pools of platelets could each drive a different neutrophil response. Further studies are needed to identify possible molecular targets that can be used to favor anti-inflammatory interactions between platelets and neutrophils, and at the same time can disadvantage pro-inflammatory ones.
Concluding Remarks.
Understanding how NETs are induced and released has been challenging due to the variety of stimuli (bacterial, viral, fungal, or chemical), types of neutrophils (human, murine, adult or neonate), and assays employed to study this question. Although several mechanisms have been posited, multiple questions still remain to be resolved. The relatively recent concept that neutrophils are a heterogeneous population [90,121] further complicates this scenario, and indicates that not all NETs are created equally, and that differences in their composition have different effects on inflammation. A deeper knowledge of neutrophil intrinsic and extrinsic mechanisms that regulate NET formation, and the spatial and temporal distribution of NETs, will be valuable in informing the design of better therapeutic approaches (Box 2, Outstanding questions). Since NETs have evolved to protect the host against microbial infections, a fundamental property of any novel therapy must be that the antibacterial role of NETs is preserved, while their detrimental effects are prevented.
Box 2, Outstanding Questions.
How do neutrophils take the decision to encounter NETosis or to engage a different functional program?
Is pathogen size the solely factor that determines the decision to encounter NETosis or to release granules?
What are the exact factors associated with the proteome disarming model that influence the capacity of neutrophils to undertake or not NETosis?
Is the balance of different types of cell death, e.g.: NETosis vs. pyroptosis, regulated in a stimulus-dependent or -independent manner? And how is the involvement of effectors common to multiple death pathways, such as caspases and gasdermins, regulated?
How do multiple cell types (e.g.: platelets, tumor cells, etc.) that interact with neutrophils modify their capacity to release NETs?
How does the heterogeneous nature of neutrophils influence their capacity to undergo NETosis in response to different microbial ligands?
What are the factors and pathways that drive suicidal versus vital NETosis?
What is the relative contribution to NET induction of histone modifications driven by PAD4, HDACs, and possibly other histone modifiers?
How can the spatial and temporal distribution of NETs be harnessed in order to design new effective ways of therapeutic intervention against microbially-driven or inflammatory diseases sustained by excessive or altered NETosis?
Highlights.
Release of NETs by neutrophils is regulated by neutrophil-intrinsic and -extrinsic factors and pathways.
Modifiers of histones play fundamental roles in driving NETosis. For example, histone deacetylation by class I and class IIb HDACs is required to allow histone H3 citrullination by the enzyme PAD4.
Regulation of GSDMD activation has been recently identified as a key process that controls the permeability of the nuclear, plasma, and granule membranes, thus allowing NET release.
Metabolic pathways that regulate glycolysis and energy supply are tightly linked to the capacity of neutrophils to produce NETs.
Macrophages work as either positive or negative regulators of NETosis.
Platelets are major drivers of NETosis and PNCs controls an inflammatory feedback loop that needs to be tightly regulated to avoid excessive NET release and to prevent thrombosis.
Multiple exogenous and endogenous factors control the capacity of platelets to interact with neutrophils and to form PNCs. Among these factors, NFAT expressed by platelets has been shown to dampen platelet activation and induction of NETosis, thus protecting against excessive inflammation and DIC during sepsis.
ACKNOWLEDGEMENTS
IZ is supported by NIH grants R01AI121066, R01DK115217, R01AI165505 and contract no. 75N93019C00044, Lloyd J. Old STAR Program CRI3888, and holds an Investigators in the Pathogenesis of Infectious Disease Award from the Burroughs Wellcome Fund. Figures were created with BioRender.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References.
- 1.Stark K, Eckart A, Haidari S, Tirniceriu A, Lorenz M, von Bruhl ML, Gartner F, Khandoga AG, Legate KR, Pless R, et al. : Capillary and arteriolar pericytes attract innate leukocytes exiting through venules and ‘instruct’ them with pattern-recognition and motility programs. Nat Immunol 2013, 14:41–51. [DOI] [PubMed] [Google Scholar]
- 2.Newburger PE: Autoimmune and other acquired neutropenias. Hematology Am Soc Hematol Educ Program 2016, 2016:38–42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Dinauer MC: Primary immune deficiencies with defects in neutrophil function. Hematology Am Soc Hematol Educ Program 2016, 2016:43–50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Brinkmann V, Reichard U, Goosmann C, Fauler B, Uhlemann Y, Weiss DS, Weinrauch Y, Zychlinsky A: Neutrophil extracellular traps kill bacteria. Science 2004, 303:1532–1535. [DOI] [PubMed] [Google Scholar]
- 5.Lood C, Blanco LP, Purmalek MM, Carmona-Rivera C, De Ravin SS, Smith CK, Malech HL, Ledbetter JA, Elkon KB, Kaplan MJ: Neutrophil extracellular traps enriched in oxidized mitochondrial DNA are interferogenic and contribute to lupus-like disease. Nat Med 2016, 22:146–153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Pilsczek FH, Salina D, Poon KK, Fahey C, Yipp BG, Sibley CD, Robbins SM, Green FH, Surette MG, Sugai M, et al. : A novel mechanism of rapid nuclear neutrophil extracellular trap formation in response to Staphylococcus aureus. J Immunol 2010, 185:7413–7425. [DOI] [PubMed] [Google Scholar]
- 7.Yipp BG, Petri B, Salina D, Jenne CN, Scott BN, Zbytnuik LD, Pittman K, Asaduzzaman M, Wu K, Meijndert HC, et al. : Infection-induced NETosis is a dynamic process involving neutrophil multitasking in vivo. Nat Med 2012, 18:1386–1393. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Branzk N, Papayannopoulos V: Molecular mechanisms regulating NETosis in infection and disease. Semin Immunopathol 2013, 35:513–530. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Smolarz M, Zawrotniak M, Satala D, Rapala-Kozik M: Extracellular Nucleic Acids Present in the Candida albicans Biofilm Trigger the Release of Neutrophil Extracellular Traps. Front Cell Infect Microbiol 2021, 11:681030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Guiducci E, Lemberg C, Kung N, Schraner E, Theocharides APA, LeibundGut-Landmann S: Candida albicans-Induced NETosis Is Independent of Peptidylarginine Deiminase 4. Front Immunol 2018, 9:1573. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Saitoh T, Komano J, Saitoh Y, Misawa T, Takahama M, Kozaki T, Uehata T, Iwasaki H, Omori H, Yamaoka S, et al. : Neutrophil extracellular traps mediate a host defense response to human immunodeficiency virus-1. Cell Host Microbe 2012, 12:109–116. [DOI] [PubMed] [Google Scholar]
- 12.Veras FP, Pontelli MC, Silva CM, Toller-Kawahisa JE, de Lima M, Nascimento DC, Schneider AH, Caetite D, Tavares LA, Paiva IM, et al. : SARS-CoV-2-triggered neutrophil extracellular traps mediate COVID-19 pathology. J Exp Med 2020, 217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Aymonnier K, Ng J, Fredenburgh LE, Zambrano-Vera K, Munzer P, Gutch S, Fukui S, Desjardins M, Subramaniam M, Baron RM, et al. : Inflammasome activation in neutrophils of patients with severe COVID-19. Blood Adv 2022, 6:2001–2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Zuo Y, Yalavarthi S, Shi H, Gockman K, Zuo M, Madison JA, Blair C, Weber A, Barnes BJ, Egeblad M, et al. : Neutrophil extracellular traps in COVID-19. JCI Insight 2020, 5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Koupenova M, Corkrey HA, Vitseva O, Manni G, Pang CJ, Clancy L, Yao C, Rade J, Levy D, Wang JP, et al. : The role of platelets in mediating a response to human influenza infection. Nat Commun 2019, 10:1780. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Abi Abdallah DS, Lin C, Ball CJ, King MR, Duhamel GE, Denkers EY: Toxoplasma gondii triggers release of human and mouse neutrophil extracellular traps. Infect Immun 2012, 80:768–777. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Kenny EF, Herzig A, Kruger R, Muth A, Mondal S, Thompson PR, Brinkmann V, Bernuth HV, Zychlinsky A: Diverse stimuli engage different neutrophil extracellular trap pathways. Elife 2017, 6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Branzk N, Lubojemska A, Hardison SE, Wang Q, Gutierrez MG, Brown GD, Papayannopoulos V: Neutrophils sense microbe size and selectively release neutrophil extracellular traps in response to large pathogens. Nat Immunol 2014, 15:1017–1025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Papayannopoulos V: Neutrophil extracellular traps in immunity and disease. Nat Rev Immunol 2018, 18:134–147. [DOI] [PubMed] [Google Scholar]
- 20.Adrover JM, Aroca-Crevillen A, Crainiciuc G, Ostos F, Rojas-Vega Y, Rubio-Ponce A, Cilloniz C, Bonzon-Kulichenko E, Calvo E, Rico D, et al. : Programmed ‘disarming’ of the neutrophil proteome reduces the magnitude of inflammation. Nat Immunol 2020, 21:135–144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Zhang D, Chen G, Manwani D, Mortha A, Xu C, Faith JJ, Burk RD, Kunisaki Y, Jang JE, Scheiermann C, et al. : Neutrophil ageing is regulated by the microbiome. Nature 2015, 525:528–532. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Metzler KD, Goosmann C, Lubojemska A, Zychlinsky A, Papayannopoulos V: A myeloperoxidase-containing complex regulates neutrophil elastase release and actin dynamics during NETosis. Cell Rep 2014, 8:883–896. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Caudrillier A, Kessenbrock K, Gilliss BM, Nguyen JX, Marques MB, Monestier M, Toy P, Werb Z, Looney MR: Platelets induce neutrophil extracellular traps in transfusion-related acute lung injury. J Clin Invest 2012, 122:2661–2671. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Brill A, Fuchs TA, Savchenko AS, Thomas GM, Martinod K, De Meyer SF, Bhandari AA, Wagner DD: Neutrophil extracellular traps promote deep vein thrombosis in mice. J Thromb Haemost 2012, 10:136–144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Liu Y, Carmona-Rivera C, Moore E, Seto NL, Knight JS, Pryor M, Yang ZH, Hemmers S, Remaley AT, Mowen KA, et al. : Myeloid-Specific Deletion of Peptidylarginine Deiminase 4 Mitigates Atherosclerosis. Front Immunol 2018, 9:1680. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Silvestre-Roig C, Braster Q, Wichapong K, Lee EY, Teulon JM, Berrebeh N, Winter J, Adrover JM, Santos GS, Froese A, et al. : Externalized histone H4 orchestrates chronic inflammation by inducing lytic cell death. Nature 2019, 569:236–240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Sangaletti S, Tripodo C, Chiodoni C, Guarnotta C, Cappetti B, Casalini P, Piconese S, Parenza M, Guiducci C, Vitali C, et al. : Neutrophil extracellular traps mediate transfer of cytoplasmic neutrophil antigens to myeloid dendritic cells toward ANCA induction and associated autoimmunity. Blood 2012, 120:3007–3018. [DOI] [PubMed] [Google Scholar]
- 28.Khandpur R, Carmona-Rivera C, Vivekanandan-Giri A, Gizinski A, Yalavarthi S, Knight JS, Friday S, Li S, Patel RM, Subramanian V, et al. : NETs are a source of citrullinated autoantigens and stimulate inflammatory responses in rheumatoid arthritis. Sci Transl Med 2013, 5:178ra140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Odqvist L, Jevnikar Z, Riise R, Oberg L, Rhedin M, Leonard D, Yrlid L, Jackson S, Mattsson J, Nanda S, et al. : Genetic variations in A20 DUB domain provide a genetic link to citrullination and neutrophil extracellular traps in systemic lupus erythematosus. Ann Rheum Dis 2019, 78:1363–1370. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Garcia-Romo GS, Caielli S, Vega B, Connolly J, Allantaz F, Xu Z, Punaro M, Baisch J, Guiducci C, Coffman RL, et al. : Netting neutrophils are major inducers of type I IFN production in pediatric systemic lupus erythematosus. Sci Transl Med 2011, 3:73ra20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Di Domizio J, Gilliet M: Psoriasis Caught in the NET. J Invest Dermatol 2019, 139:1426–1429. [DOI] [PubMed] [Google Scholar]
- 32.Chatfield SM, Grebe K, Whitehead LW, Rogers KL, Nebl T, Murphy JM, Wicks IP: Monosodium Urate Crystals Generate Nuclease-Resistant Neutrophil Extracellular Traps via a Distinct Molecular Pathway. J Immunol 2018, 200:1802–1816. [DOI] [PubMed] [Google Scholar]
- 33.Jaillon S, Ponzetta A, Di Mitri D, Santoni A, Bonecchi R, Mantovani A: Neutrophil diversity and plasticity in tumour progression and therapy. Nat Rev Cancer 2020, 20:485–503. [DOI] [PubMed] [Google Scholar]
- 34.Thiam HR, Wong SL, Wagner DD, Waterman CM: Cellular Mechanisms of NETosis. Annu Rev Cell Dev Biol 2020, 36:191–218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Siler U, Romao S, Tejera E, Pastukhov O, Kuzmenko E, Valencia RG, Meda Spaccamela V, Belohradsky BH, Speer O, Schmugge M, et al. : Severe glucose-6-phosphate dehydrogenase deficiency leads to susceptibility to infection and absent NETosis. J Allergy Clin Immunol 2017, 139:212–219 e213. [DOI] [PubMed] [Google Scholar]
- 36.Liu S, Su X, Pan P, Zhang L, Hu Y, Tan H, Wu D, Liu B, Li H, Li H, et al. : Neutrophil extracellular traps are indirectly triggered by lipopolysaccharide and contribute to acute lung injury. Sci Rep 2016, 6:37252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Silva CM, Wanderley CWS, Veras FP, Gonçalves AV, Lima MHF, Toller Kawahisa JE, Gomes GF, Nascimento DC, Silva Monteiro VV, Paiva IM, et al. : Gasdermin-D activation by SARS-CoV-2 trigger NET and mediate COVID-19 immunopathology. medRxiv 2022:2022.2001.2024.22269768. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Douda DN, Khan MA, Grasemann H, Palaniyar N: SK3 channel and mitochondrial ROS mediate NADPH oxidase-independent NETosis induced by calcium influx. Proc Natl Acad Sci U S A 2015, 112:2817–2822. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Mohanty T, Sjogren J, Kahn F, Abu-Humaidan AH, Fisker N, Assing K, Morgelin M, Bengtsson AA, Borregaard N, Sorensen OE: A novel mechanism for NETosis provides antimicrobial defense at the oral mucosa. Blood 2015, 126:2128–2137. [DOI] [PubMed] [Google Scholar]
- 40.Chen KW, Monteleone M, Boucher D, Sollberger G, Ramnath D, Condon ND, von Pein JB, Broz P, Sweet MJ, Schroder K: Noncanonical inflammasome signaling elicits gasdermin D-dependent neutrophil extracellular traps. Sci Immunol 2018, 3. [DOI] [PubMed] [Google Scholar]
- 41.Place TL, Domann FE, Case AJ: Limitations of oxygen delivery to cells in culture: An underappreciated problem in basic and translational research. Free Radic Biol Med 2017, 113:311–322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Lodge KM, Vassallo A, Liu B, Long M, Tong Z, Newby PR, Agha-Jaffar D, Paschalaki K, Green CE, Belchamber KBR, et al. : Hypoxia Increases the Potential for Neutrophil-mediated Endothelial Damage in Chronic Obstructive Pulmonary Disease. Am J Respir Crit Care Med 2022, 205:903–916. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Dolling M, Eckstein M, Singh J, Schauer C, Schoen J, Shan X, Bozec A, Knopf J, Schett G, Munoz LE, et al. : Hypoxia Promotes Neutrophil Survival After Acute Myocardial Infarction. Front Immunol 2022, 13:726153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Khawaja AA, Chong DLW, Sahota J, Mikolasch TA, Pericleous C, Ripoll VM, Booth HL, Khan S, Rodriguez-Justo M, Giles IP, et al. : Identification of a Novel HIF-1alpha-alphaMbeta2 Integrin-NET Axis in Fibrotic Interstitial Lung Disease. Front Immunol 2020, 11:2190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Talla U, Bozonet SM, Parker HA, Hampton MB, Vissers MCM: Prolonged exposure to hypoxia induces an autophagy-like cell survival program in human neutrophils. J Leukoc Biol 2019, 106:1367–1379. [DOI] [PubMed] [Google Scholar]
- 46.Wang Y, Li M, Stadler S, Correll S, Li P, Wang D, Hayama R, Leonelli L, Han H, Grigoryev SA, et al. : Histone hypercitrullination mediates chromatin decondensation and neutrophil extracellular trap formation. J Cell Biol 2009, 184:205–213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Li P, Li M, Lindberg MR, Kennett MJ, Xiong N, Wang Y: PAD4 is essential for antibacterial innate immunity mediated by neutrophil extracellular traps. J Exp Med 2010, 207:1853–1862. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Lewis HD, Liddle J, Coote JE, Atkinson SJ, Barker MD, Bax BD, Bicker KL, Bingham RP, Campbell M, Chen YH, et al. : Inhibition of PAD4 activity is sufficient to disrupt mouse and human NET formation. Nat Chem Biol 2015, 11:189–191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Nakashima K, Arai S, Suzuki A, Nariai Y, Urano T, Nakayama M, Ohara O, Yamamura K, Yamamoto K, Miyazaki T: PAD4 regulates proliferation of multipotent haematopoietic cells by controlling c-myc expression. Nat Commun 2013, 4:1836. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Rohrbach AS, Slade DJ, Thompson PR, Mowen KA: Activation of PAD4 in NET formation. Front Immunol 2012, 3:360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Gosswein S, Lindemann A, Mahajan A, Maueroder C, Martini E, Patankar J, Schett G, Becker C, Wirtz S, Naumann-Bartsch N, et al. : Citrullination Licenses Calpain to Decondense Nuclei in Neutrophil Extracellular Trap Formation. Front Immunol 2019, 10:2481. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Martinod K, Demers M, Fuchs TA, Wong SL, Brill A, Gallant M, Hu J, Wang Y, Wagner DD: Neutrophil histone modification by peptidylarginine deiminase 4 is critical for deep vein thrombosis in mice. Proc Natl Acad Sci U S A 2013, 110:8674–8679. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Wong SL, Wagner DD: Peptidylarginine deiminase 4: a nuclear button triggering neutrophil extracellular traps in inflammatory diseases and aging. FASEB J 2018:fj201800691R. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Papayannopoulos V, Metzler KD, Hakkim A, Zychlinsky A: Neutrophil elastase and myeloperoxidase regulate the formation of neutrophil extracellular traps. J Cell Biol 2010, 191:677–691. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Metzler KD, Fuchs TA, Nauseef WM, Reumaux D, Roesler J, Schulze I, Wahn V, Papayannopoulos V, Zychlinsky A: Myeloperoxidase is required for neutrophil extracellular trap formation: implications for innate immunity. Blood 2011, 117:953–959. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Thanabalasuriar A, Scott BNV, Peiseler M, Willson ME, Zeng Z, Warrener P, Keller AE, Surewaard BGJ, Dozier EA, Korhonen JT, et al. : Neutrophil Extracellular Traps Confine Pseudomonas aeruginosa Ocular Biofilms and Restrict Brain Invasion. Cell Host Microbe 2019, 25:526–536 e524. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Kolaczkowska E, Jenne CN, Surewaard BG, Thanabalasuriar A, Lee WY, Sanz MJ, Mowen K, Opdenakker G, Kubes P: Molecular mechanisms of NET formation and degradation revealed by intravital imaging in the liver vasculature. Nat Commun 2015, 6:6673. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Warnatsch A, Ioannou M, Wang Q, Papayannopoulos V: Inflammation. Neutrophil extracellular traps license macrophages for cytokine production in atherosclerosis. Science 2015, 349:316–320. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Mor-Vaknin N, Saha A, Legendre M, Carmona-Rivera C, Amin MA, Rabquer BJ, Gonzales-Hernandez MJ, Jorns J, Mohan S, Yalavarthi S, et al. : DEK-targeting DNA aptamers as therapeutics for inflammatory arthritis. Nat Commun 2017, 8:14252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.DeSouza-Vieira T, Guimaraes-Costa A, Rochael NC, Lira MN, Nascimento MT, Lima-Gomez PS, Mariante RM, Persechini PM, Saraiva EM: Neutrophil extracellular traps release induced by Leishmania: role of PI3Kgamma, ERK, PI3Ksigma, PKC, and [Ca2+]. J Leukoc Biol 2016, 100:801–810. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Hakkim A, Fuchs TA, Martinez NE, Hess S, Prinz H, Zychlinsky A, Waldmann H: Activation of the Raf-MEK-ERK pathway is required for neutrophil extracellular trap formation. Nat Chem Biol 2011, 7:75–77. [DOI] [PubMed] [Google Scholar]
- 62.Douda DN, Yip L, Khan MA, Grasemann H, Palaniyar N: Akt is essential to induce NADPH-dependent NETosis and to switch the neutrophil death to apoptosis. Blood 2014, 123:597–600. [DOI] [PubMed] [Google Scholar]
- 63.Ma R, Li T, Cao M, Si Y, Wu X, Zhao L, Yao Z, Zhang Y, Fang S, Deng R, et al. : Extracellular DNA traps released by acute promyelocytic leukemia cells through autophagy. Cell Death Dis 2016, 7:e2283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Remijsen Q, Vanden Berghe T, Wirawan E, Asselbergh B, Parthoens E, De Rycke R, Noppen S, Delforge M, Willems J, Vandenabeele P: Neutrophil extracellular trap cell death requires both autophagy and superoxide generation. Cell Res 2011, 21:290–304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Kagan JC, Magupalli VG, Wu H: SMOCs: supramolecular organizing centres that control innate immunity. Nat Rev Immunol 2014, 14:821–826. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Sollberger G, Choidas A, Burn GL, Habenberger P, Di Lucrezia R, Kordes S, Menninger S, Eickhoff J, Nussbaumer P, Klebl B, et al. : Gasdermin D plays a vital role in the generation of neutrophil extracellular traps. Sci Immunol 2018, 3. [DOI] [PubMed] [Google Scholar]
- 67.Kambara H, Liu F, Zhang X, Liu P, Bajrami B, Teng Y, Zhao L, Zhou S, Yu H, Zhou W, et al. : Gasdermin D Exerts Anti-inflammatory Effects by Promoting Neutrophil Death. Cell Rep 2018, 22:2924–2936. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Munzer P, Negro R, Fukui S, di Meglio L, Aymonnier K, Chu L, Cherpokova D, Gutch S, Sorvillo N, Shi L, et al. : NLRP3 Inflammasome Assembly in Neutrophils Is Supported by PAD4 and Promotes NETosis Under Sterile Conditions. Front Immunol 2021, 12:683803. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Westerterp M, Fotakis P, Ouimet M, Bochem AE, Zhang H, Molusky MM, Wang W, Abramowicz S, la Bastide-van Gemert S, Wang N, et al. : Cholesterol Efflux Pathways Suppress Inflammasome Activation, NETosis, and Atherogenesis. Circulation 2018, 138:898–912. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Amara N, Cooper MP, Voronkova MA, Webb BA, Lynch EM, Kollman JM, Ma T, Yu K, Lai Z, Sangaraju D, et al. : Selective activation of PFKL suppresses the phagocytic oxidative burst. Cell 2021, 184:4480–4494 e4415. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Amini P, Stojkov D, Felser A, Jackson CB, Courage C, Schaller A, Gelman L, Soriano ME, Nuoffer J-M, Scorrano L, et al. : Neutrophil extracellular trap formation requires OPA1-dependent glycolytic ATP production. Nature Communications 2018, 9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Poli V, Pui-Yan Ma V, Di Gioia M, Broggi A, Benamar M, Chen Q, Mazitschek R, Haggarty SJ, Chatila TA, Karp JM, et al. : Zinc-dependent histone deacetylases drive neutrophil extracellular trap formation and potentiate local and systemic inflammation. iScience 2021, 24:103256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Amulic B, Knackstedt SL, Abu Abed U, Deigendesch N, Harbort CJ, Caffrey BE, Brinkmann V, Heppner FL, Hinds PW, Zychlinsky A: Cell-Cycle Proteins Control Production of Neutrophil Extracellular Traps. Dev Cell 2017, 43:449–462 e445. [DOI] [PubMed] [Google Scholar]
- 74.Neubert E, Meyer D, Rocca F, Gunay G, Kwaczala-Tessmann A, Grandke J, Senger-Sander S, Geisler C, Egner A, Schon MP, et al. : Chromatin swelling drives neutrophil extracellular trap release. Nat Commun 2018, 9:3767. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Azzouz D, Khan MA, Palaniyar N: ROS induces NETosis by oxidizing DNA and initiating DNA repair. Cell Death Discov 2021, 7:113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Masucci MT, Minopoli M, Del Vecchio S, Carriero MV: The Emerging Role of Neutrophil Extracellular Traps (NETs) in Tumor Progression and Metastasis. Front Immunol 2020, 11:1749. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Zucoloto AZ, Jenne CN: Platelet-Neutrophil Interplay: Insights Into Neutrophil Extracellular Trap (NET)-Driven Coagulation in Infection. Front Cardiovasc Med 2019, 6:85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Silva LM, Doyle AD, Greenwell-Wild T, Dutzan N, Tran CL, Abusleme L, Juang LJ, Leung J, Chun EM, Lum AG, et al. : Fibrin is a critical regulator of neutrophil effector function at the oral mucosal barrier. Science 2021, 374:eabl5450. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Erpenbeck L, Gruhn AL, Kudryasheva G, Gunay G, Meyer D, Busse J, Neubert E, Schon MP, Rehfeldt F, Kruss S: Effect of Adhesion and Substrate Elasticity on Neutrophil Extracellular Trap Formation. Front Immunol 2019, 10:2320. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Jimenez-Alcazar M, Rangaswamy C, Panda R, Bitterling J, Simsek YJ, Long AT, Bilyy R, Krenn V, Renne C, Renne T, et al. : Host DNases prevent vascular occlusion by neutrophil extracellular traps. Science 2017, 358:1202–1206. [DOI] [PubMed] [Google Scholar]
- 81.Farrera C, Fadeel B: Macrophage clearance of neutrophil extracellular traps is a silent process. J Immunol 2013, 191:2647–2656. [DOI] [PubMed] [Google Scholar]
- 82.Apel F, Andreeva L, Knackstedt LS, Streeck R, Frese CK, Goosmann C, Hopfner KP, Zychlinsky A: The cytosolic DNA sensor cGAS recognizes neutrophil extracellular traps. Sci Signal 2021, 14. [DOI] [PubMed] [Google Scholar]
- 83.Gregoire M, Uhel F, Lesouhaitier M, Gacouin A, Guirriec M, Mourcin F, Dumontet E, Chalin A, Samson M, Berthelot LL, et al. : Impaired efferocytosis and neutrophil extracellular trap clearance by macrophages in ARDS. Eur Respir J 2018, 52. [DOI] [PubMed] [Google Scholar]
- 84.Haider P, Kral-Pointner JB, Mayer J, Richter M, Kaun C, Brostjan C, Eilenberg W, Fischer MB, Speidl WS, Hengstenberg C, et al. : Neutrophil Extracellular Trap Degradation by Differently Polarized Macrophage Subsets. Arterioscler Thromb Vasc Biol 2020, 40:2265–2278. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Moreira-Teixeira L, Stimpson PJ, Stavropoulos E, Hadebe S, Chakravarty P, Ioannou M, Aramburu IV, Herbert E, Priestnall SL, Suarez-Bonnet A, et al. : Type I IFN exacerbates disease in tuberculosis-susceptible mice by inducing neutrophil-mediated lung inflammation and NETosis. Nat Commun 2020, 11:5566. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Gul E, Sayar EH, Gungor B, Eroglu FK, Surucu N, Keles S, Guner SN, Findik S, Alpdundar E, Ayanoglu IC, et al. : Type I IFN-related NETosis in ataxia telangiectasia and Artemis deficiency. J Allergy Clin Immunol 2018, 142:246–257. [DOI] [PubMed] [Google Scholar]
- 87.Byrd AS, Carmona-Rivera C, O’Neil LJ, Carlucci PM, Cisar C, Rosenberg AZ, Kerns ML, Caffrey JA, Milner SM, Sacks JM, et al. : Neutrophil extracellular traps, B cells, and type I interferons contribute to immune dysregulation in hidradenitis suppurativa. Sci Transl Med 2019, 11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Broggi A, Tan Y, Granucci F, Zanoni I: IFN-lambda suppresses intestinal inflammation by non-translational regulation of neutrophil function. Nat Immunol 2017, 18:1084–1093. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Sebina I, Rashid RB, Sikder MAA, Rahman MM, Ahmed T, Radford-Smith DE, Kotenko SV, Hill GR, Bald T, Phipps S: IFN-lambda Diminishes the Severity of Viral Bronchiolitis in Neonatal Mice by Limiting NADPH Oxidase-Induced PAD4-Independent NETosis. J Immunol 2022, 208:2806–2816. [DOI] [PubMed] [Google Scholar]
- 90.Montaldo E, Lusito E, Bianchessi V, Caronni N, Scala S, Basso-Ricci L, Cantaffa C, Masserdotti A, Barilaro M, Barresi S, et al. : Cellular and transcriptional dynamics of human neutrophils at steady state and upon stress. Nat Immunol 2022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Zhang Y, Chandra V, Riquelme Sanchez E, Dutta P, Quesada PR, Rakoski A, Zoltan M, Arora N, Baydogan S, Horne W, et al. : Interleukin-17-induced neutrophil extracellular traps mediate resistance to checkpoint blockade in pancreatic cancer. J Exp Med 2020, 217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Keshari RS, Jyoti A, Dubey M, Kothari N, Kohli M, Bogra J, Barthwal MK, Dikshit M: Cytokines induced neutrophil extracellular traps formation: implication for the inflammatory disease condition. PLoS One 2012, 7:e48111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Zhan Y, Ling Y, Deng Q, Qiu Y, Shen J, Lai H, Chen Z, Huang C, Liang L, Li X, et al. : HMGB1-Mediated Neutrophil Extracellular Trap Formation Exacerbates Intestinal Ischemia/Reperfusion-Induced Acute Lung Injury. J Immunol 2022, 208:968–978. [DOI] [PubMed] [Google Scholar]
- 94.Chen R, Kang R, Tang D: The mechanism of HMGB1 secretion and release. Exp Mol Med 2022, 54:91–102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Boone BA, Orlichenko L, Schapiro NE, Loughran P, Gianfrate GC, Ellis JT, Singhi AD, Kang R, Tang D, Lotze MT, et al. : The receptor for advanced glycation end products (RAGE) enhances autophagy and neutrophil extracellular traps in pancreatic cancer. Cancer Gene Ther 2015, 22:326–334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Maugeri N, Campana L, Gavina M, Covino C, De Metrio M, Panciroli C, Maiuri L, Maseri A, D’Angelo A, Bianchi ME, et al. : Activated platelets present high mobility group box 1 to neutrophils, inducing autophagy and promoting the extrusion of neutrophil extracellular traps. J Thromb Haemost 2014, 12:2074–2088. [DOI] [PubMed] [Google Scholar]
- 97.Maugeri N, Capobianco A, Rovere-Querini P, Ramirez GA, Tombetti E, Valle PD, Monno A, D’Alberti V, Gasparri AM, Franchini S, et al. : Platelet microparticles sustain autophagy-associated activation of neutrophils in systemic sclerosis. Sci Transl Med 2018, 10. [DOI] [PubMed] [Google Scholar]
- 98.Ronchetti L, Boubaker NS, Barba M, Vici P, Gurtner A, Piaggio G: Neutrophil extracellular traps in cancer: not only catching microbes. J Exp Clin Cancer Res 2021, 40:231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Demers M, Krause DS, Schatzberg D, Martinod K, Voorhees JR, Fuchs TA, Scadden DT, Wagner DD: Cancers predispose neutrophils to release extracellular DNA traps that contribute to cancer-associated thrombosis. Proc Natl Acad Sci U S A 2012, 109:13076–13081. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Rayes RF, Mouhanna JG, Nicolau I, Bourdeau F, Giannias B, Rousseau S, Quail D, Walsh L, Sangwan V, Bertos N, et al. : Primary tumors induce neutrophil extracellular traps with targetable metastasis promoting effects. JCI Insight 2019, 5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Xiao Y, Cong M, Li J, He D, Wu Q, Tian P, Wang Y, Yang S, Liang C, Liang Y, et al. : Cathepsin C promotes breast cancer lung metastasis by modulating neutrophil infiltration and neutrophil extracellular trap formation. Cancer Cell 2021, 39:423–437 e427. [DOI] [PubMed] [Google Scholar]
- 102.Munir H, Jones JO, Janowitz T, Hoffmann M, Euler M, Martins CP, Welsh SJ, Shields JD: Stromal-driven and Amyloid beta-dependent induction of neutrophil extracellular traps modulates tumor growth. Nat Commun 2021, 12:683. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Teijeira A, Garasa S, Gato M, Alfaro C, Migueliz I, Cirella A, de Andrea C, Ochoa MC, Otano I, Etxeberria I, et al. : CXCR1 and CXCR2 Chemokine Receptor Agonists Produced by Tumors Induce Neutrophil Extracellular Traps that Interfere with Immune Cytotoxicity. Immunity 2020, 52:856–871 e858. [DOI] [PubMed] [Google Scholar]
- 104.Park J, Wysocki RW, Amoozgar Z, Maiorino L, Fein MR, Jorns J, Schott AF, Kinugasa-Katayama Y, Lee Y, Won NH, et al. : Cancer cells induce metastasis-supporting neutrophil extracellular DNA traps. Sci Transl Med 2016, 8:361ra138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Snoderly HT, Boone BA, Bennewitz MF: Neutrophil extracellular traps in breast cancer and beyond: current perspectives on NET stimuli, thrombosis and metastasis, and clinical utility for diagnosis and treatment. Breast Cancer Res 2019, 21:145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Sreeramkumar V, Adrover JM, Ballesteros I, Cuartero MI, Rossaint J, Bilbao I, Nacher M, Pitaval C, Radovanovic I, Fukui Y, et al. : Neutrophils scan for activated platelets to initiate inflammation. Science 2014, 346:1234–1238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Perdomo J, Leung HHL, Ahmadi Z, Yan F, Chong JJH, Passam FH, Chong BH: Neutrophil activation and NETosis are the major drivers of thrombosis in heparin-induced thrombocytopenia. Nat Commun 2019, 10:1322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.McDonald B, Davis RP, Kim SJ, Tse M, Esmon CT, Kolaczkowska E, Jenne CN: Platelets and neutrophil extracellular traps collaborate to promote intravascular coagulation during sepsis in mice. Blood 2017, 129:1357–1367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Lisman T: Platelet-neutrophil interactions as drivers of inflammatory and thrombotic disease. Cell Tissue Res 2018, 371:567–576. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Zhou J, Xu E, Shao K, Shen W, Gu Y, Li M, Shen W: Circulating platelet-neutrophil aggregates as risk factor for deep venous thrombosis. Clin Chem Lab Med 2019, 57:707–715. [DOI] [PubMed] [Google Scholar]
- 111.Etulain J, Martinod K, Wong SL, Cifuni SM, Schattner M, Wagner DD: P-selectin promotes neutrophil extracellular trap formation in mice. Blood 2015, 126:242–246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Carestia A, Kaufman T, Rivadeneyra L, Landoni VI, Pozner RG, Negrotto S, D’Atri LP, Gomez RM, Schattner M: Mediators and molecular pathways involved in the regulation of neutrophil extracellular trap formation mediated by activated platelets. J Leukoc Biol 2016, 99:153–162. [DOI] [PubMed] [Google Scholar]
- 113.Poli V, Di Gioia M, Sola-Visner M, Granucci F, Frelinger AL 3rd, Michelson AD, Zanoni I: Inhibition of transcription factor NFAT activity in activated platelets enhances their aggregation and exacerbates gram-negative bacterial septicemia. Immunity 2022, 55:224–236 e225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Singer M, Deutschman CS, Seymour CW, Shankar-Hari M, Annane D, Bauer M, Bellomo R, Bernard GR, Chiche JD, Coopersmith CM, et al. : The Third International Consensus Definitions for Sepsis and Septic Shock (Sepsis-3). JAMA 2016, 315:801–810. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Clark SR, Ma AC, Tavener SA, McDonald B, Goodarzi Z, Kelly MM, Patel KD, Chakrabarti S, McAvoy E, Sinclair GD, et al. : Platelet TLR4 activates neutrophil extracellular traps to ensnare bacteria in septic blood. Nat Med 2007, 13:463–469. [DOI] [PubMed] [Google Scholar]
- 116.Su M, Chen C, Li S, Li M, Zeng Z, Zhang Y, Xia L, Li X, Zheng D, Lin Q, et al. : Gasdermin D-dependent platelet pyroptosis exacerbates NET formation and inflammation in severe sepsis. Nat Cardiovasc Res 2022, 1:732–747. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.McDonald B, Urrutia R, Yipp BG, Jenne CN, Kubes P: Intravascular neutrophil extracellular traps capture bacteria from the bloodstream during sepsis. Cell Host Microbe 2012, 12:324–333. [DOI] [PubMed] [Google Scholar]
- 118.Angus DC, van der Poll T: Severe sepsis and septic shock. N Engl J Med 2013, 369:840–851. [DOI] [PubMed] [Google Scholar]
- 119.Elaskalani O, Abdol Razak NB, Metharom P: Neutrophil extracellular traps induce aggregation of washed human platelets independently of extracellular DNA and histones. Cell Commun Signal 2018, 16:24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Valet C, Magnen M, Qiu L, Cleary SJ, Wang KM, Ranucci S, Grockowiak E, Boudra R, Conrad C, Seo Y, et al. : Sepsis promotes splenic production of a protective platelet pool with high CD40 ligand expression. J Clin Invest 2022, 132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Ng LG, Ostuni R, Hidalgo A: Heterogeneity of neutrophils. Nat Rev Immunol 2019, 19:255–265. [DOI] [PubMed] [Google Scholar]
- 122.Arcanjo A, Logullo J, Menezes CCB, de Souza Carvalho Giangiarulo TC, Dos Reis MC, de Castro GMM, da Silva Fontes Y, Todeschini AR, Freire-de-Lima L, Decote-Ricardo D, et al. : The emerging role of neutrophil extracellular traps in severe acute respiratory syndrome coronavirus 2 (COVID-19). Sci Rep 2020, 10:19630. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Akk A, Springer LE, Pham CT: Neutrophil Extracellular Traps Enhance Early Inflammatory Response in Sendai Virus-Induced Asthma Phenotype. Front Immunol 2016, 7:325. [DOI] [PMC free article] [PubMed] [Google Scholar]