

Abstract
The use of deep machine learning (ML) in protein structure prediction has made it possible to easily access a large number of annotated conformations that can potentially compensate for missing experimental structures in structure-based drug discovery (SBDD). However, it is still unclear whether the accuracy of these predicted conformations is sufficient for screening chemical compounds that will effectively interact with a protein target for pharmacological purposes. In this opinion, we examine the potential benefits and limitations of using state-annotated conformations for ultra-large library screening (ULLS) in light of the growing size of ultra-large libraries (ULL). We believe that targeting different conformational states of common drug targets like G-protein-coupled receptors (GPCRs), which can regulate human physiology by switching between different conformations, can offer multiple advantages.
Keywords: drug discovery, structure-based drug discovery, AlphaFold, ultra-large library, GPCR, biased ligands
Ultra-large libraries for drug discovery: limits and opportunities
It has been estimated that there are up to 1060 potential molecules with drug-like properties [1]. The extraction of promising drug scaffolds from this gigantic chemical space has been a long-sought goal in drug discovery. Compared to classical libraries that have been historically limited to 1 million compounds, DNA-encoded libraries (DELs) significantly expanded the number of experimentally assessable molecules at a reasonable cost and resulted in some successful hit discovery campaigns [2–6]. Complementary to DELs, the advent of make-on-demand ultra-large libraries (ULLs) made available hundreds of millions to several billions of REadily AccessibLe (REAL) molecules that can be visualized in silico and are chemically diverse, affordable and rapidly synthesized [12–14]. The availability of ULLs such as ZINC15, ZINC20 and Enamine triggered the development of structure-based drug discovery (SBDD) technologies aiming to identify high-affinity molecules that can be purchased at one of the corresponding website [7–11]. Ultra-large library screening (ULLS) methods need to conjugate speed of sampling with an accuracy high enough to enrich the selection of compounds that will be confirmed active against a protein target (Box1).
Box 1. Ultra-large library screening technologies.
ULLS technologies aim to retain scoring accuracy while speeding up sampling. To do so, three methods with different features have been developed (Figure I). DOCK3.7 exploits a simple physics-based scoring function composed of the sum of Van der Waals, electrostatic, and desolvation energy terms [68]. Because DOCK3.7 is a fully rigid docking method, a 3D conformer library representing different ligand conformations and orientations needs to be precalculated. Recently, a practical guide to HTS has been published [69], providing a convenient receipt that users can follow to perform ULLS. While DOCK3.7 has been used to explore chemical spaces in the order of several million compounds, standard ULLs reached a scale of billions and expanded so rapidly that they will likely reach trillion molecules in the next future. In this scenario, VirtualFlow exploits a sophisticated parallelization mechanism compatible with widely-used cluster systems and can potentially be used with multiple docking programs [70]. VirtualFlow has been tested on the exploration of more than one billion molecules. Alternatively, V-SYNTHES was developed to keep the size of the docked library equal to the number of building blocks composing the REAL space of the combinatorial library [21]. The key step is the creation of a library in which building blocks have only one of the two reactive groups free to react, whereas the other one remains capped. Then, the resulting fragments are docked to score and select an ensemble of promising fragments that are redocked upon the substitution of the capped reactive group with all the remaining building blocks in the library. This modular fragment-based approach has been tested in the exploration of an 11 billion ULL.
Due to their ability to respond to a variety of extracellular signaling molecules, G-protein-coupled receptors (GPCRs) are an ideal drug target class [15,16]. Different ligands can trigger distinct conformations linked to specific signaling cascades and physiological effects. While the number of REAL molecules will soon reach astonishing numbers, the number of experimental structures with detailed functional annotation will struggle to keep pace (Figure 1) [17]. Therefore, ULLS for hit discovery is in urgent need of alternative workflows that can serve as a surrogate for missing experimental structural models.
Figure 1.
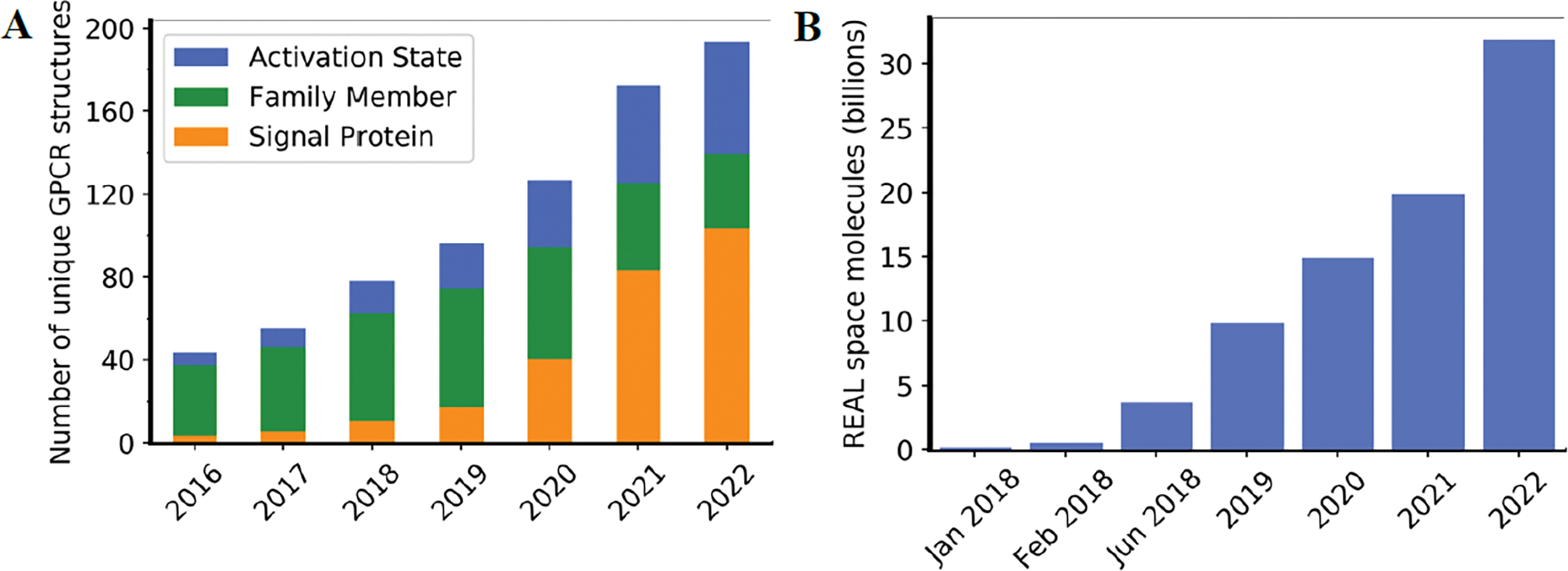
Number of virtual small molecules and unique GPCR structures available over time. A) Number of unique GPCR structures available in the GPCRdb (most recent structure deposited: July 27, 2022) with different annotation details. The sum of unique protein activation states is shown in blue. The sum of unique GPCR members invariant to any annotation is shown in green. The sum of proteins solved with either a G protein or Arrestin bound is shown in orange. B) Number of Enamine REadily AccessibLe (REAL) molecules.
Here, we first summarize recent successful efforts in hit discovery of GPCR modulators. Then, we discuss features, benefits and limitations of modern computational methods developed for sampling multiple conformational states, with a focus on the breakthrough technology AlphaFold2 (AF2). We envision that soon machine learning will make the prediction of distinct GPCR conformations fast, easy, broadly accessible, and with an accuracy rivaling experimental structures. This will have a profound impact on GPCR hit discovery.
Ultra-large library screens targeting G-protein-coupled receptors
In the last few years, ULLS campaigns targeting GPCRs have been applied to identify compounds with high potency and target selectivity [18,19]. Furthermore, the identification of binders with chemical scaffolds different from known ligands captured interest for their potential therapeutic utility as selective modulators [20,21]. A new chemical scaffold can make peculiar interactions, thereby improving potency and selectivity against other receptor subtypes. However, the identification of subtype-specific structural features is not always apparent. In a ULLS against the MT1 co-crystal structure [19], Stein R.M. and colleagues were able to extract only a few active ligands selective for MT1 over MT2 from the first docking screen of hundreds of million compounds. To improve potency while retaining selectivity, they searched for analogs of the active chemotypes in the full library by using the Tanimoto coefficient. Alternatively, a structure-activity relationship (SAR) search can be used to identify compounds with similar chemical properties. SAR was chosen by Sadybekov and colleagues in their search for potent compounds that are selective for CB2 over CB1 [21]. Out of a billion compounds, three hits were found with CB2 submicromolar affinity and were used to extract low-nM molecules with higher CB2 selectivity from the whole library.
Besides being useful to discriminate among receptor subtypes, novel chemotypes can translate into functional selectivity through biased recruitment of a subset of signaling proteins (Figure 2). Fink and colleagues targeted the α2BA GPCR crystal structure to extract molecules able to selectively engage a subset of G proteins [22]. Functional selectivity resulted in pain relief without sedation, thereby providing the intended therapeutic effects while reducing unwanted symptoms. With a similar scope, Kaplan and colleagues created a library of 75 million tetrahydropyridines (THPs) to target the 5-HT2A receptor [23]. Interestingly, no crystal structure was available at the time of the work, forcing them to build homology models using the 5-HT2B X-ray structure bound to LSD as a template. The models were then ranked for their ability to enrich known 5-HT2A ligands versus inactive molecules. ULLS against the selected computational model identified four molecules with agonistic or antagonistic activity. The subsequent SAR search in the full 4.3-billion-compound ULL was instrumental to identify new agonists recruiting a G protein instead of β-arrestin associated with psychedelic effects. Functional selectivity paved the way for the development of new therapeutics against depression, anxiety and post-traumatic stress disorder.
Figure 2.
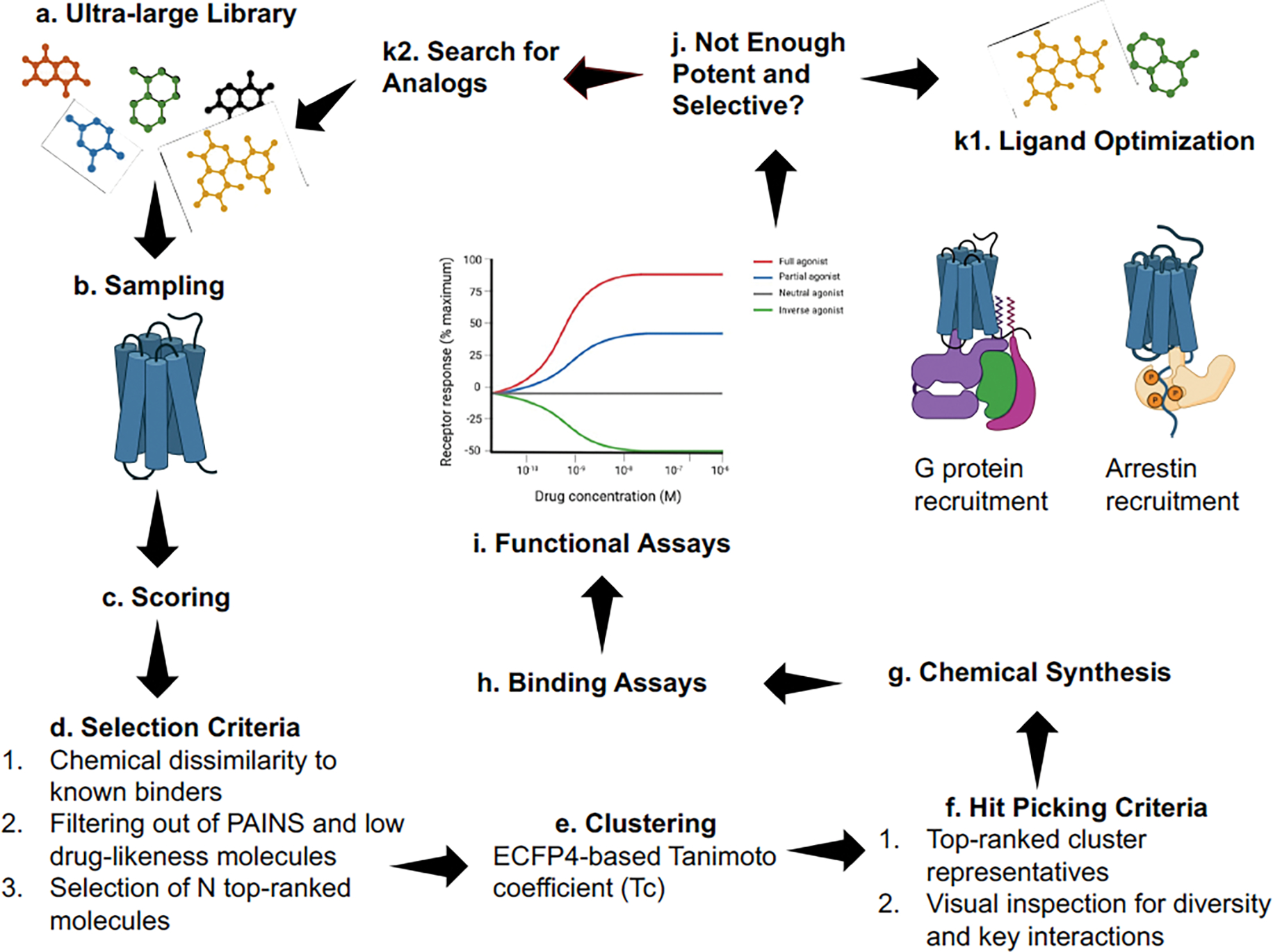
Schematic representation of an ultra-large library screening targeting a GPCR structure for detecting compounds carrying functional selectivity. a) An ultra-large library with million to billion molecules is prepared. b) Ligands are docked in a pocket of the target structure to sample optimal interactions with protein residues. c) Ligand-Protein poses are ranked in accord to a scoring function. d) Thousand compounds are selected in accord to multiple criteria such as top-ranked scores, and Tanimoto distance from ligands already known to bind the receptor or deposited in CHEMBL [71]. Pan-assay interference compounds (PAINS) and low drug-likeness molecules are discharged. e) The selected subset of molecules is clustered with a structure similarity metric, usually the ECFP4-based Tanimoto coefficient. f) Top-ranked clusters and representative compounds are visually inspected to pick diverse chemical scaffolds making new or key interactions in the pocket. g) A small subset (usually < 100) of compounds is synthesized. h) Compounds successfully synthesized are assessed for binding affinity to the target receptor. i) Ligands below low-micromolar affinity are assessed for their ability to modulate receptor activity. In looking for functional selectivity, diverse agonists or partial agonists can potentially recruit specific signaling proteins. j) Out of binding and functional assays, molecules are ranked for their ability to tightly bind the receptor and to activate the intended signaling cascade. If these criteria are only partially satisfied, k1) compounds can be optimized with medicinal chemistry approaches or k2) searching for analogs (SAR or Tanimoto similarity) of the most promising molecules in the whole library.
Computational methods for predicting physiological GPCR conformations
Given that high-accuracy structures are a crucial component of successful ULLS campaigns, there is a strong interest in developing computational methods able to detect one or more GPCR conformational states that can complement missing experimental structures.
AlphaFold-based prediction of multiple functional states
Recent advances in machine learning (ML) methods for protein-structure prediction have had an impressive impact on the number of high-quality protein models available [24,25], that now cover the full human proteome and beyond [26,27]. In particular, AF2 demonstrated the ability to model difficult protein targets with comparable accuracy to experimental structures [28]. However, models in either the AlphaFold protein structure database or out of the AF2 algorithm represent a single conformational ground state. In the last year, a number of AF2 workflows have been developed and validated in their ability to predict alternative functional states of GPCRs that differ from the AF2 ground state (Figure 3).
Figure 3.
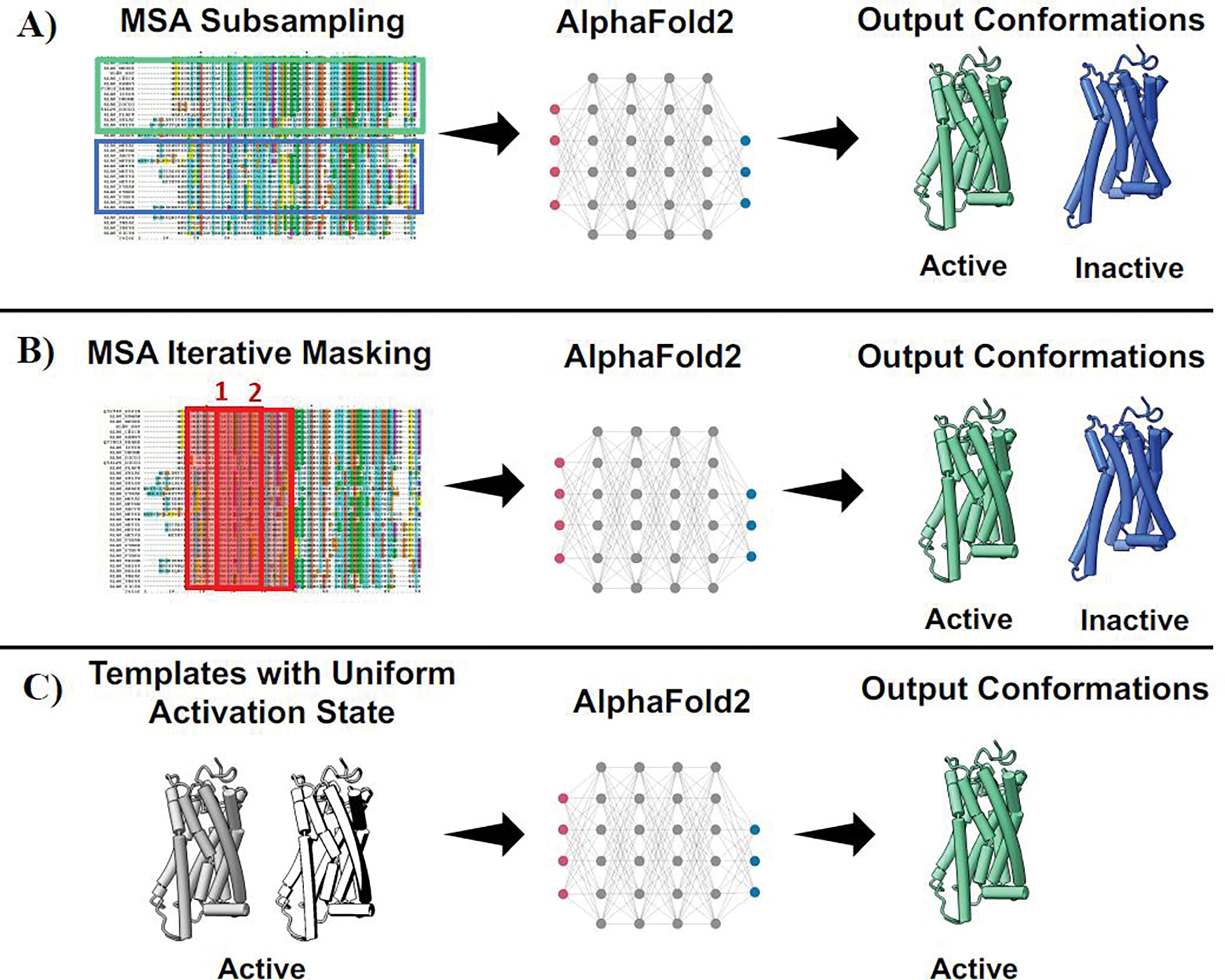
Main features of the AF2 workflows developed to sample GPCR conformational states. The default AF2 implementation takes three inputs: 1. the protein sequence, 2. the protein MSA and 3. template structures of protein homologs. All three workflows in the plot tackle the input MSA and templates to expand or drive AF2 sampling. A) Templates are removed and a small subset of MSA sequences is randomly selected to sample different conformations. B) Templates are removed and a sliding window is used to mask a portion of the MSA for each prediction. The masked window may hide the region that biases the prediction toward a unique conformational state, thereby increasing the chance of detecting alternative conformations. C) The MSA is removed and a local database of structures is used to feed AF2 with conformational homogenous templates of homologous proteins that can drive the prediction toward a specific conformational state.
Del Alamo and colleagues implemented the first AF2 pipeline aiming to sample alternative conformations with high accuracy [29]. In the proposed workflow, the removal of templates used in combination with a shallower multiple sequence alignment (MSA) was instrumental to extract an ensemble of dissimilar models. The rationale behind a shallow MSA relies on randomly subsampling a subset of sequences that can potentially shift the prediction toward alternative conformations, a process that can also be fine-tuned through sequence similarity clustering or by using templates in a uniform activation state [30,31]. On different GPCR class targets, AF2 was able to sample conformations corresponding to either active or inactive functional states. This workflow has been already widely used to validate experimental data and expand the portion of conformational space known [32–34]. The prediction of alternative conformations with AF2 can also be achieved through in silico mutagenesis of a subset of MSA positions [35]. By mutating a portion of the MSA to alanine, AF2 looks for alternative structures that can potentially match the remaining MSA information content. This approach successfully sampled multiple functional conformations for GPCRs at high accuracy. Without prior knowledge of the positions to mutate for predicting alternative structures, an iterative sliding window must be used. Interestingly, Heo and coworkers managed to use AF2 as a comparative modeling method [36]. To bias the prediction toward the intended conformational state, the MSA of a target protein was removed and a local state-annotated GPCR structure database was used as a unique source of templates in a uniform functional state. Highly accurate models for both active and inactive state structures were generated, although some active targets were missed. Models were further validated in reproducing correct ligand poses with protein-ligand docking, showing that in targeting either active or inactive conformations models were more accurate than those predicted with either an alternative template-based approach or the default AF2 implementation.
The described methods usually sample an ensemble of models made of either a highly homogeneous conformational state or multiple conformational states. Different approaches have been successfully applied for minimizing noise and picking representative models. Models that are misfolded or predicted with low confidence can be discharged by ranking them based on predicted confidence scores and Molprobity score [35,37,38]. The resulting ensemble usually has a smaller structural variance than physics-based ensembles, allowing to pick representative conformations through simple analyses such as Principal Component Analysis or visual inspection [29]. Inspecting known microswitches can also help estimate the accuracy of specific functional states in greater detail [39,40].
Molecular dynamics to detect intermediate states
While AF2 has shown impressive performances in the prediction of active or inactive GPCR conformations, intermediate metastable states typically escape experimental detection and are also hard to predict using ML due to their transient nature. However, intermediate states may have unique structural features and functional purposes making them potentially useful as targets for ULLS. In this regard, molecular dynamics (MD) can complement experimental and ML methods to provide unique insights into intermediate protein-specific physiological states. Progress in computational tools and resources has enabled the implementation of microseconds to milliseconds MD simulations at atomic resolution. Starting from experimental structures, MD has been exploited to investigate structural perturbations of GPCRs upon binding/unbinding of various ligands [40,41]. The generated trajectories can be analyzed to detect multiple energetically accessible conformations along the activation pathway or to elucidate allostery and cooperativity of GPCR ligands (Figure 4) [42]. For instance, Suomivuori and colleagues investigated the molecular mechanism of prototypical GPCR AT1R binding to design ligands with desired biased signaling profiles [43]. They proposed two conformations induced by extracellular agonists which favor either G protein or arrestin signaling. Their results offer a detailed mechanism for biased signaling of AT1R, providing signal-specific conformations that can be used for ULLS. In another work, Lu et al. collected 300 μs of MD trajectories to study the activation mechanism of the angiotensin II type 1 receptor (AT1) [44]. They identified and validated with site-direct mutagenesis an intracellular cryptic pocket of an apo conformational intermediate that can potentially be used as a target for allosteric drug discovery. By mutating residues in the allosteric site, the endogenous agonist was incapable of stabilizing the active conformation and promoting the binding of transducers.
Figure 4.
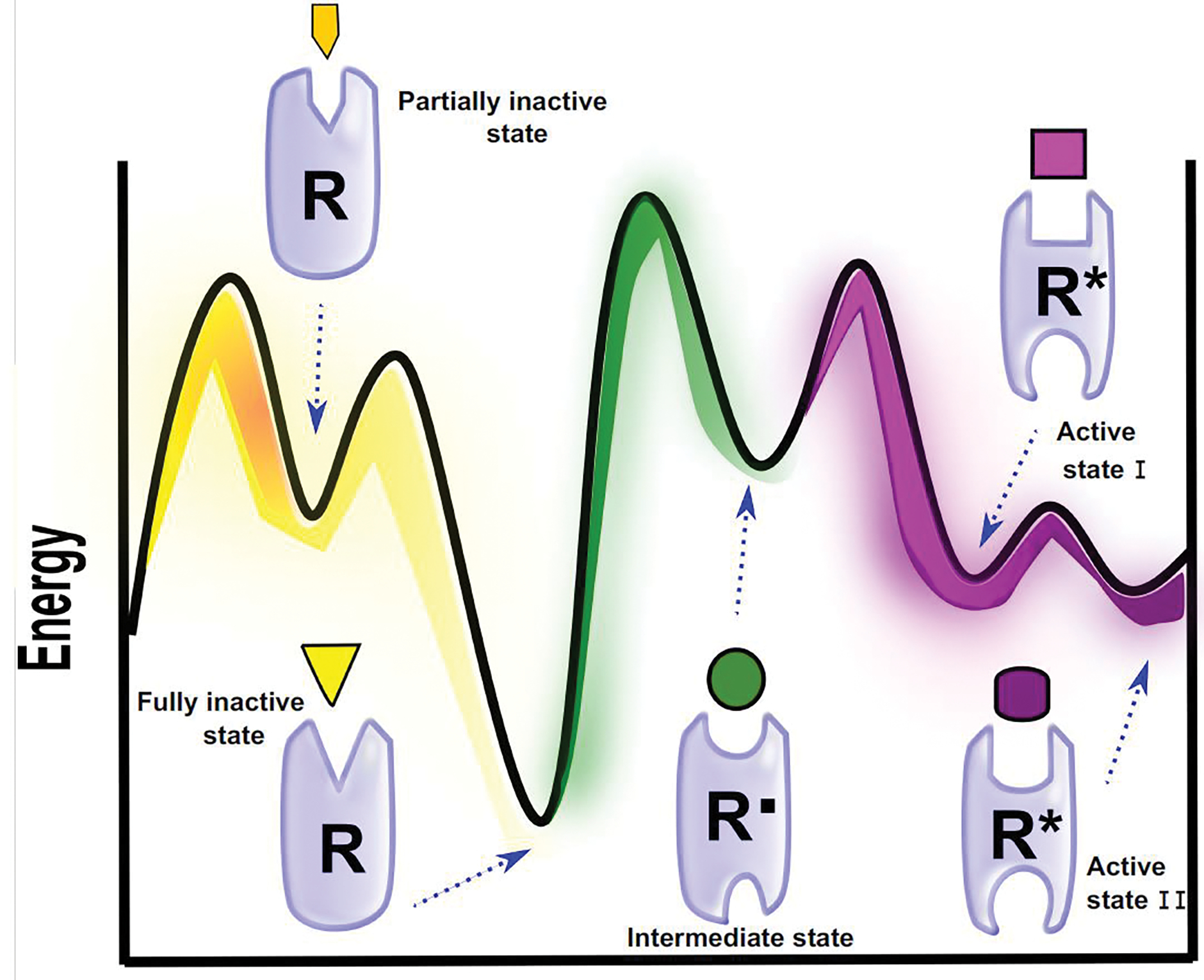
One dimensional energy landscape of class A GPCRs. Along the activation pathway different holo and apo inactive (R), intermediate (R’) and active (R*) receptor conformations exist. Each receptor state features specific ligand binding properties, indicated here with different geometries. Active receptors transmit the extracellular signal to downstream signaling proteins with ligand-dependent efficiencies, as indicated here with different size of the intracellular binding pocket.
Despite some successful stories, one of the main restrictions in applying MD is represented by simulation time scales that often are shorter than those of the investigated events, preventing their visualization. Li et al. developed a new enhanced sampling approach that was assessed in the exploration of the full activation mechanism of A1R, including endogenous ligand–receptor recognition, receptor pre-activation, and receptor–G protein recognition [45]. They observed the G protein binding to the intracellular side of A1R propagating a reduction in the volume of the orthosteric site that stabilizes the receptor-ligand binding. Starting from both active and inactive conformations they have identified a number of metastable intermediate states through a new MD technique, thereby providing a further tool that can play a role in SBDD.
Modeling GPCRs complexity
Structural modeling and functional annotations of GPCRs can be far more complex than the simple separation between active, intermediate and inactive states. As signal transducers, GPCRs interact with multiple effectors, can be activated by extracellular proteins like proteases or transactivated by receptor tyrosine kinases (RTKRs), and can form hetero- or homodimers [46–48]. In the field of interactome modeling, recent technologies like AlphaFold-multimer have shown promising accuracy [49–51]. However, higher resolution is needed to capture either atomic interactions or the subtle structural changes that characterize the binding of different signaling effectors. Additionally, GPCRs are highly dynamic, can contain disordered regions and can be modulated by multiple external factors like membrane composition, cofactors and post-translational modifications (PTMs) [52,53]. To properly investigate these factors, a combination of ML and physics-based methods, along with experimental data from sources such as nuclear magnetic resonance (NMR), electron paramagnetic resonance (EPR), mass spectrometry (MS) and others is needed [54–57]. However, using multiple software programs to model these challenging structural features can be time-consuming and laborious. In the future, a modular platform that can easily incorporate various types of input data and integrate ML and physics-based methods with experimental data will make the process more accessible. Eventually, models of GPCRs that include molecular partners, cofactors, and PTMs at high resolution, along with detailed annotations of their functional and biological states, would be crucial for fully understanding the behavior of these receptors and advancing the field of SBDD.
Benefits and limitations of high-accuracy computational models for hit discovery
The size of ultra-large libraries of chemicals is constantly increasing, as a result, the number of detectable drug-like molecules follows the same trend. ULLS technologies able to explore significant portions of those chemical spaces already exist and are more and more used [58]. One of the main restrictions of applying ULLS on GPCRs is currently represented by the limited conformational portfolio of structures experimentally solved at high accuracy. We propose that modern ML methods like AF2 and its customizations can bridge this gap. Because such predicted structures rival experimentally determined structures in accuracy and can be generated in a high-throughput manner at low-cost, researchers will be able to predict multiple conformational states for a large number of proteins.
Discovery of conformation-selective hit compounds
As a result of being able to predict one or multiple protein conformational states at high accuracy, researchers can now merge computational technologies for the discovery of conformation-selective hit compounds. ML-based workflows will be a crucial component for ensemble state sampling coupled with ULLS to build fully computational workflows that go from a protein sequence directly to hit discovery, without the need for a predetermined experimental structure. As such, access to enriched portfolios of predicted structural targets and diversified chemical scaffolds represents exciting prospects for hit discovery.
Targeting multiple orthosteric pocket configurations can increase both the number and chemical diversity of compounds active on a specific protein target or on multiple members of the same protein family. An obvious benefit is a relatively higher chance of identifying new chemotypes able to increase potency and/or selectivity. In addition, new chemotypes making previously unobserved pocket interactions can expand the number of lead compounds with distinct pharmacology with respect to known ligands, thus leading to new therapeutics [22,23]. In this regard, there is a strong interest in the identification of agonists or partial agonists able to bias GPCR signaling cascades by recruiting a subset of effectors [59]. The availability of predicted specific biased signaling conformational states can potentially enrich a screening with such molecules.
Another important aspect of applying ULLS for hit discovery concerns the identification of chemical scaffolds more likely to become inverse agonists or antagonists [60]. While in the first case they must induce a conformational change interrupting or biasing the downstream signaling cascade, a condition difficult to predict a priori, in the latter case they can act as structural stabilizers. To do so, predicted full or partial inactive conformations can provide a pocket configuration better suited to host an antagonist.
In addition of the orthosteric pocket, GPCRs have a number of druggable allosteric sites. Those pockets are usually less conserved in sequence space but are well localized in 3D space and made of residues with similar physiochemical properties that make conserved interactions linked to a specific activation state, at least for class A and B1 [61]. These features suggest that specific pocket configurations can be better suited to be screened with the aim of identifying positive or negative allosteric modulators with fine-tuned specificity. The availability of a broad range of pocket configurations would help to increase the change of either collecting a physiological state or picking a configuration of interest to bias ULLS toward the identification of a specific type of allosteric modulators.
In general, the virtual generation of protein conformations at high accuracy can promote the identification of protein binders that in turn may aid experimental structures determination. Experimental structures will iteratively improve hits identification and will inform SBDD and design.
As a last perspective, the generation of GPCR complexes at high accuracy can also have a deep impact on hit identification of compounds targeting protein-protein interactions, one of the most challenging drug discovery tasks [62].
Limitations
Experimental information is still a crucial component to drive modeling toward a higher resolution structure or to validate the accuracy of structures, eventually being determinant for successful hit discovery campaigns (Box 2). The main limitation of predicted structures is the expected relatively lower resolution with respect to experimental structures. GPCRs present very complex multifaceted activation mechanisms characterized by diverse conformations that differ to various extents protein by protein, upon binding of various ligands and can be stabilized by external factors [63]. High-resolution structural details are in general harder to predict, as well as protein-specific intermediate states. Modeling methods able to predict large portions of protein-specific conformational space under different conditions are still missing. Given that ULLS is usually carried out with rigid side chains, even small errors in their orientation can lead to misleading results. Despite predicted GPCR models exhibited a significant accuracy in reproducing protein-ligand docking poses [36], an additional benchmark on 28 common drug targets showed that a refining step with physics-based approaches is often helpful in enriching the selection of active compounds, reducing the gap from co-crystal structures hit rates [64]. Indeed, hit discovery campaigns are usually carried out on protein co-crystal structures, co-crystallized with other ligands to detect residue rotamers more favorable to ligand binding. However, predicted conformations correspond to apo structures and thereby often miss all the induced fit effects upon ligand binding. If the induced fit effects mainly involve side chains and small backbone rearrangements, the availability of a number of known ligands to perform physics-based refinement can potentially recover them [65]. On the contrary, large conformational induced-fit changes are hard to capture and may be retrieved only with the assistance of experimentally-supported data or enhanced sampling techniques [66].
Box 2. Impact of experimental information on Modeling and ULLS.
This Box presents four scenarios ordered by decreasing amount of experimental data available. Each scenario explores how modeling can support ULLS and identifies areas that still pose challenges and require further advancement in computational modeling techniques.
A lot of information is available: there is a vast amount of data available about different experimental protein structures and their functional states, as well as ligands interactions and their corresponding pharmacological effects. Using modeling techniques, it is possible to analyze local structural flexibility and identify alternative conformations that maintain important structural elements while also offering potential targets for ligand binding. This can be done by using ML to generate a set of state-specific conformations which are then used as starting points for MD simulations to determine the likelihood of a particular state or configuration. By determining the probability and the structural features of each state or configuration, it becomes possible to bias the screen more accurately. Data derived from known ligands will help to make informed decisions about which ligands to focus on.
A significant amount of information is available: one state-annotated protein structure is available with a ligand linked to the receptor activity. As in the previous scenario, ML and MD can expand the number of configurations visible. However, more advanced modeling methods are needed to accurately capture high-resolution structural details for new or different functional states. In addition, a single known ligand offers limited data to refine and pick a meaningful model for ULLS as well as to select promising hits.
Little information is available: only one structure is available without ligands bound. In this scenario, computational modeling can still sample both an ensemble of conformations of the same activation state or alternative to that. However, missing information on alternative functional states and induce-fit effects may prevent the sampling of pocket configurations able to enrich ULLS of active compounds.
No information available: no structures are available and the receptor is orphan. Despite AF2 can still be used to capture an apo backbone conformation with high accuracy, small inaccuracies can result in a very low or absent hit rate. In the future, with methods able to capture induced-fit effects and to detect key pocket interactions that propagate the signal to the intracellular region, also this scenario can be tackled.
Concluding remarks and future perspectives
In this Opinion, we have discussed how recent breakthroughs in protein structure prediction can contribute to hit discovery (see Outstanding Questions). While MD methods are usually elaborate and time-consuming thereby preventing their application to a large number of proteins, ML and docking are usually faster and more user-friendly. However, all the ML-based methods presented here that are able to sample multiple conformational states of GPCRs can be seen as a sort of AF2 “hacks” rather than neural networks specifically trained to explore the conformational space or to get all the global energetic minima structures of a protein of interest. Both goals share the problem of assigning subsets of evolutionary, physical and geometric constraints to separate conformations representing one of multiple physiological states. The separation of evolutionary couplings representative of distinct global energetic minima or functional states should be easier to tackle than retrieving less representative evolutionary information, like transient contacts corresponding to higher-energy metastable structures. As a result, ML methods specifically developed to sample functional conformations at experimental accuracy will probably emerge sooner than methods able to explore a large portion of conformational space for proteins. In addition, none of the actual released ML algorithms can predict any ligand-protein induced-fit effect. This will be a very intensive research area in the near future.
Ultimately, advances in ML technologies will make conformational modeling fast, accurate, broadly accessible and bespoke thereby fueling ULLS campaigns. A higher number or more functionally tailored active molecules will translate in a higher chance to develop drugs with improved potency, specificity and/or fine-tuned spectrum of activity. For pharmacological targets like GPCRs that exhibit a wide variety of different conformational states with state-specific ligand binding affinities and signaling properties [67], identifying small molecules that specifically signal through a particular pathway, so-called biased ligands, will result in modulators with lower side effects. In this regard, structure-prediction methods able to capture the subtle conformational differences that lead a receptor to recruit specific molecular partners will have a huge impact.
Figure I.
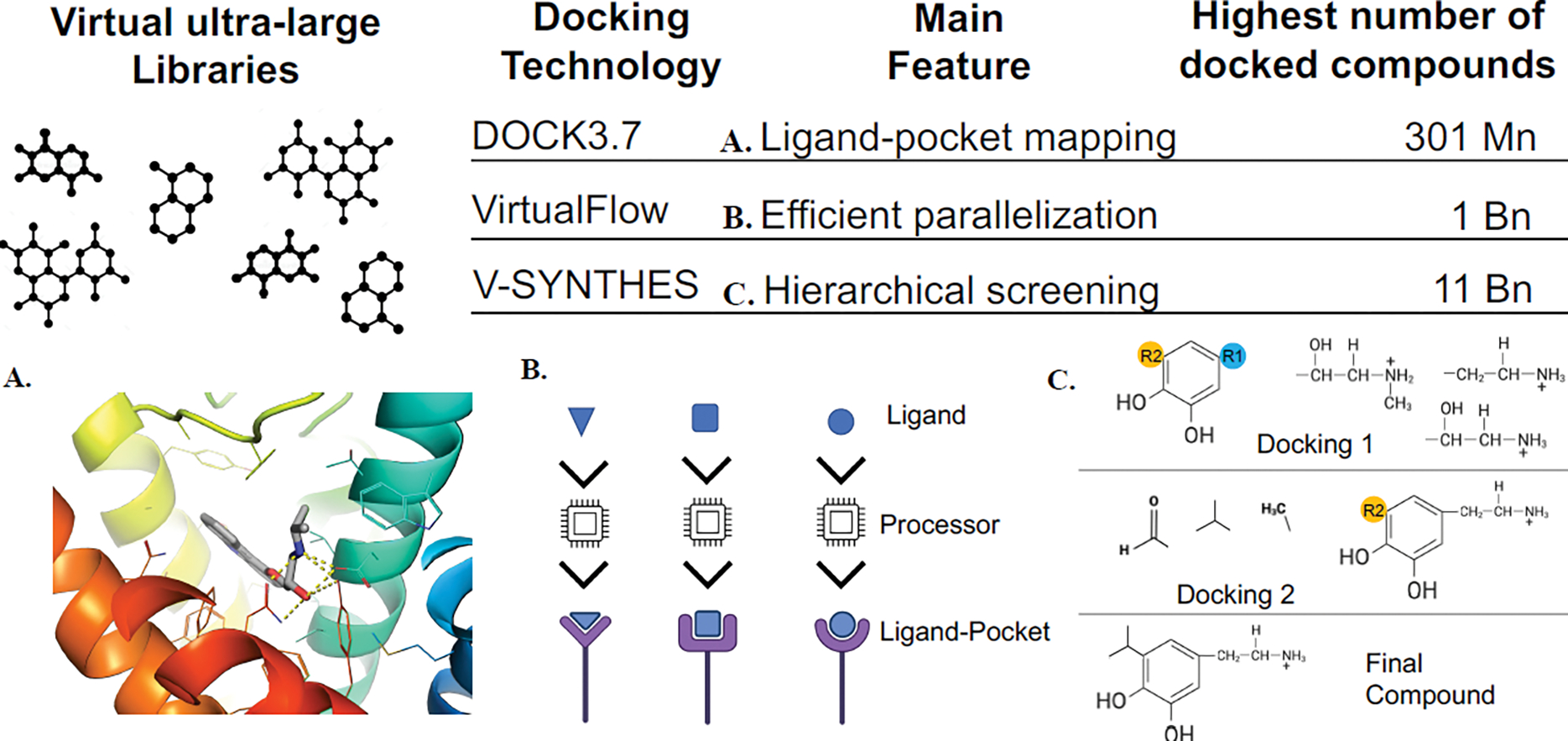
Ultra-large screening technologies. A. The ligand-pocket interactions of one or more known ligands are mapped to restrict the space of solutions during docking. B. An efficient parallelization mechanism allows multiple ligands to be screened simultaneously. C. Only one of the two reactive groups is replaced, and the resulting compound docked. Most promising fragments are then redocked upon substitution of the second reactive group.
Outstanding questions.
Can modern computational methods predict multiple conformational states at high accuracy?
Can predicted structures enrich ultra-large library screening of active compounds or experimental information is still needed to refine the binding pocket?
Can state-annotated conformations enrich a hit discovery campaign of molecules with the desired pharmacological properties?
How far are we from an automatic pipeline going from a protein sequence directly to the identification of active compounds?
Highlights.
The availability of ultra-large virtual libraries in combination with efficient docking algorithms has improved hit discovery in a way that is now possible to detect a significant number of active molecules dissimilar to known binding ligands, opening new therapeutic opportunities.
GPCRs are highly desirable drug targets due to their ability of regulating cellular response. The limited availability of experimentally determined GPCR conformations prevents the extensive use of structure-based virtual screenings for hit discovery.
The recent development of computational workflows able to predict multiple or user-defined GPCR conformations at high accuracy opens the door to virtual screening against predicted functional states, with multiple potential prospects for drug discovery.
Acknowledgments
This work was supported by the Deutsche Forschungsgemeinschaft (DFG, German Research Foundation) through CRC 1423, project number 421152132, subproject Z04. This work was also supported by the NIH (R01 GM080403, R01 A046138, R01 HL122010, and R01 GM129261).
Glossary
- AlphaFold2 (AF2):
AlphaFold2 is a deep machine learning algorithm designed to predict the 3D structure of proteins. It has been widely hailed as a major breakthrough technology in the field
- Chemical libraries:
in drug discovery, collections of molecules representing a fraction of the chemical space that can be synthesized and can potentially have drug-like properties
- Chemotypes:
chemical structure moieties that are common to a group of molecules
- DNA-encoded libraries (DELs):
collections of molecules covalently bound to distinct DNA fragments acting as amplifiable identification barcodes
- Evolutionary couplings:
protein residue pairs carrying a relevant structural or functional role. They occur when a random mutation is followed by a mutation in a different residue that compensates the loss of function. Analysis of mutational covariance can provide information on residue-residue interactions occurring in a protein structure and dynamics
- G-protein-coupled receptors (GPCRs):
are the largest and most diverse group of membrane receptors in eukaryotes. They convert extracellular signals into intracellular responses. Their ability to regulate biological processes upon binding of small molecules has made them a common drug target
- Hit compounds:
molecules that upon screening result active against a drug target, where active means having a high-enough binding affinity to cause the expected effect in biochemical or cellular assays
- Metastable:
that has a longer lifetime than higher energy states but shorter than global energetic minima states
- Microswitches:
specific amino acid residues within the receptor that are thought to play a role in controlling its activity by switching between different conformations in response to the binding of a ligand
- Molecular dynamics (MD):
a computational method able to provide an atomistic view of protein dynamics by simulating the evolution of a system over time based on its forces expressed as force fields
- Multiple sequence alignment (MSA):
a number of evolutionarily related protein sequences aligned according to their similarities in amino acid composition to achieve maximal matching
- Pan-assay interference compounds (PAINS):
molecules that often interfere with drug screening assays by returning false positives hits in vitro whereas they have no biological activity in vivo
- REadily AccessibLe (REAL) molecules:
molecules that can be synthesized through prevalidated chemical reactions of reactants acting as building blocks
- Structure-based drug discovery (SBDD):
is a process for designing, searching and developing new drugs based on the detailed three-dimensional structure of target proteins
- Tanimoto coefficient:
a metric to measure the similarity of two sets of elements in a range from 0 to 1. Often used for measuring chemical compounds similarity
- Ultra-large libraries (ULLs):
virtual collections of chemicals spanning the range from hundreds of millions to billions of molecules and beyond
- Ultra-large library screening (ULLS):
computational screening of virtual ultra-large libraries
Footnotes
Declaration of interests
No interests are declared.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Lin A et al. (2018) Mapping of the Available Chemical Space versus the Chemical Universe of Lead-Like Compounds. ChemMedChem 13, 540–554 [DOI] [PubMed] [Google Scholar]
- 2.Ahn S et al. (2017) Allosteric “beta-blocker” isolated from a DNA-encoded small molecule library. Proc. Natl. Acad. Sci. U. S. A. 114, 1708–1713 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Goodnow RA et al. DNA-encoded chemistry: Enabling the deeper sampling of chemical space., Nature Reviews Drug Discovery, 16. 09-Dec-(2017), Nature Publishing Group, 131–147 [DOI] [PubMed] [Google Scholar]
- 4.Sunkari YK et al. (2022) High-power screening (HPS) empowered by DNA-encoded libraries. Trends Pharmacol. Sci. 43, 4–15 [DOI] [PubMed] [Google Scholar]
- 5.Lerner RA and Neri D (2020) Reflections on DNA-encoded chemical libraries. Biochem. Biophys. Res. Commun. 527, 757–759 [DOI] [PubMed] [Google Scholar]
- 6.Gironda-Martínez A et al. (2021) DNA-Encoded Chemical Libraries: A Comprehensive Review with Succesful Stories and Future Challenges. ACS Pharmacol. Transl. Sci. 4, 1265–1279 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Walters WP (2019) Virtual Chemical Libraries. J. Med. Chem. 62, 1116–1124 [DOI] [PubMed] [Google Scholar]
- 8.Sterling T and Irwin JJ (2015) ZINC 15 - Ligand Discovery for Everyone. J. Chem. Inf. Model. 55, 2324–2337 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Irwin JJ et al. (2020) ZINC20 - A Free Ultralarge-Scale Chemical Database for Ligand Discovery. J. Chem. Inf. Model. 60, 6065–6073 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.REAL Space (Enamine, 2022); https://enamine.net/library-synthesis/real-compounds/real-space-navigator.
- 11.Grygorenko OO et al. (2020) Generating Multibillion Chemical Space of Readily Accessible Screening Compounds. iScience 23, 101681. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Warr WA et al. (2022) Exploration of Ultralarge Compound Collections for Drug Discovery. J. Chem. Inf. Model. 62, 2021–2034 [DOI] [PubMed] [Google Scholar]
- 13.Gentile F et al. Artificial intelligence–enabled virtual screening of ultra-large chemical libraries with deep docking., Nature Protocols, 17. 01-Mar-(2022), Nature Research, 672–697 [DOI] [PubMed] [Google Scholar]
- 14.Coley CW (2021) Defining and Exploring Chemical Spaces. Trends Chem. 3, 133–145 [Google Scholar]
- 15.Yang D et al. G protein-coupled receptors: structure- and function-based drug discovery., Signal Transduction and Targeted Therapy, 6. 08-Jan-(2021), Nature Publishing Group, 1–27 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Congreve M et al. (2020) Impact of GPCR Structures on Drug Discovery. Cell 181, 81–91 [DOI] [PubMed] [Google Scholar]
- 17.Kooistra AJ et al. (2021) GPCRdb in 2021: Integrating GPCR sequence, structure and function. Nucleic Acids Res. 49, D335–D343 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Sadybekov AA et al. Structure-based virtual screening of ultra-large library yields potent antagonists for a lipid gpcr., Biomolecules, 10. (2020), 1–15 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Stein RM et al. (2020) Virtual discovery of melatonin receptor ligands to modulate circadian rhythms. Nature 579, 609–614 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Lyu J et al. (2019) Ultra-large library docking for discovering new chemotypes. Nature 566, 224–229 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Sadybekov AVAA et al. (2022) Synthon-based ligand discovery in virtual libraries of over 11 billion compounds. Nature 601, 452–459 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Fink EA et al. (2022) Structure-based discovery of nonopioid analgesics acting through the α2A-adrenergic receptor. Science 377, eabn7065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Kaplan AL et al. (2022) Bespoke library docking for 5-HT2A receptor agonists with antidepressant activity. Nat. 2022 DOI: 10.1038/s41586-022-05258-z [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Jumper J et al. (2021) Highly accurate protein structure prediction with AlphaFold. Nature 596, 583–589 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Baek M et al. (2021) Accurate prediction of protein structures and interactions using a three-track neural network. Science (80-.). 373, 871–876 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Tunyasuvunakool K et al. (2021) Highly accurate protein structure prediction for the human proteome. Nature 596, 590–596 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Varadi M et al. (2022) AlphaFold Protein Structure Database: Massively expanding the structural coverage of protein-sequence space with high-accuracy models. Nucleic Acids Res. 50, D439–D444 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Jumper J et al. (2021) Applying and improving AlphaFold at CASP14. Proteins Struct. Funct. Bioinforma. 89, 1711–1721 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Del Alamo D et al. (2022) Sampling alternative conformational states of transporters and receptors with AlphaFold2. Elife 11, 1–12 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Wayment-Steele HK et al. Prediction of multiple conformational states by combining sequence clustering with AlphaFold2. DOI: 10.1101/2022.10.17.512570 [DOI]
- 31.Sala D and Meiler J (2022) Biasing AlphaFold2 to predict GPCRs and Kinases with user-defined functional or structural properties. bioRxiv DOI: 10.1101/2022.12.11.519936 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Freidman NJ et al. (2022) Characterizing unexpected interactions of a glutamine transporter inhibitor with members of the SLC1A transporter family. J. Biol. Chem. 298, [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Rotem-Bamberger S et al. (2022) Structural insights into the role of the WW2 domain on tandem WW–PPxY motif interactions of oxidoreductase WWOX. J. Biol. Chem. 298, [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.del Alamo D et al. (2022) Integrated AlphaFold2 and DEER investigation of the conformational dynamics of a pH-dependent APC antiporter. Proc. Natl. Acad. Sci. U. S. A. 119, e2206129119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Stein RA et al. (2022) SPEACH_AF: Sampling protein ensembles and conformational heterogeneity with Alphafold2. PLOS Comput. Biol. 18, e1010483. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Heo L and Feig M (2022) Multi-state modeling of G-protein coupled receptors at experimental accuracy. Proteins Struct. Funct. Bioinforma. DOI: 10.1002/prot.26382 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Williams CJ et al. (2018) MolProbity: More and better reference data for improved all-atom structure validation. Protein Sci. 27, 293–315 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Roney JP and Ovchinnikov S (2022) State-of-the-Art Estimation of Protein Model Accuracy Using AlphaFold. Phys. Rev. Lett. 129, 238101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Fleetwood O et al. (2020) Energy Landscapes Reveal Agonist Control of G Protein-Coupled Receptor Activation via Microswitches. Biochemistry 59, 880–891 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Fleetwood O et al. (2021) Identification of ligand-specific g-protein coupled receptor states and prediction of downstream efficacy via data-driven modeling. Elife 10, 1–46 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Weis WI and Kobilka BK (2018) The Molecular Basis of G Protein–Coupled Receptor Activation. Annu. Rev. Biochem. 87, 897–919 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Saleh N et al. (2018) Multiple Binding Sites Contribute to the Mechanism of Mixed Agonistic and Positive Allosteric Modulators of the Cannabinoid CB1 Receptor. Angew. Chemie - Int. Ed. 57, 2580–2585 [DOI] [PubMed] [Google Scholar]
- 43.Suomivuori CM et al. (2020) Molecular mechanism of biased signaling in a prototypical G protein-coupled receptor. Science (80-.). 367, 881–887 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Lu S et al. (2021) Activation pathway of a G protein-coupled receptor uncovers conformational intermediates as targets for allosteric drug design. Nat. Commun. 12, 1–15 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Li Y et al. (2022) The full activation mechanism of the adenosine A1 receptor revealed by GaMD and Su-GaMD simulations. Proc. Natl. Acad. Sci. U. S. A. 119, e2203702119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Heuberger DM and Schuepbach RA (2019) Protease-activated receptors (PARs): Mechanisms of action and potential therapeutic modulators in PAR-driven inflammatory diseases. Thromb. J. 17, 4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Kilpatrick LE and Hill SJ (2021) Transactivation of G protein-coupled receptors (GPCRs) and receptor tyrosine kinases (RTKs): Recent insights using luminescence and fluorescence technologies. Curr. Opin. Endocr. Metab. Res. 16, 102–112 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Faron-Górecka A et al. (2019) Chapter 10 - Understanding GPCR dimerization. In G Protein-Coupled Receptors, Part B 149 (Shukla AKBT-M in C. B., ed), pp. 155–178, Academic Press; [DOI] [PubMed] [Google Scholar]
- 49.Yin R et al. (2022) Benchmarking AlphaFold for protein complex modeling reveals accuracy determinants. Protein Sci. 31, e4379. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Bryant P et al. (2022) Predicting the structure of large protein complexes using AlphaFold and Monte Carlo tree search. Nat. Commun. 13, 6028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Evans R et al. (2022) Protein complex prediction with AlphaFold-Multimer. bioRxiv DOI: 10.1101/2021.10.04.463034 [DOI] [Google Scholar]
- 52.Jones AJY et al. Structure and dynamics of GPCRs in lipid membranes: physical principles and experimental approaches., Molecules, 25. (2020) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Hilger D et al. (2018) Structure and dynamics of GPCR signaling complexes. Nat. Struct. Mol. Biol. 25, 4–12 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Sala D et al. (2022) Modeling of Protein Conformational Changes With Rosetta Guided by Limited Experimental Data. Structure DOI: 10.2139/ssrn.4041402 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Shteynberg DD et al. (2019) PTMProphet: Fast and Accurate Mass Modification Localization for the Trans-Proteomic Pipeline. J. Proteome Res. 18, 4262–4272 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Wingler LM et al. (2019) Angiotensin Analogs with Divergent Bias Stabilize Distinct Receptor Conformations. Cell 176, 468–478.e11 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Hekkelman ML et al. (2022) AlphaFill: enriching AlphaFold models with ligands and cofactors. Nat. Methods DOI: 10.1038/s41592-022-01685-y [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Luttens A et al. (2022) Ultralarge Virtual Screening Identifies SARS-CoV-2 Main Protease Inhibitors with Broad-Spectrum Activity against Coronaviruses. J. Am. Chem. Soc. 144, 2905–2920 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Jiang H et al. (2022) G protein-coupled receptor signaling: transducers and effectors. Am. J. Physiol. Physiol. 323, C731–C748 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Hauser AS et al. (2017) Trends in GPCR drug discovery: New agents, targets and indications. Nat. Rev. Drug Discov. 16, 829–842 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Hedderich JB et al. (2022) The pocketome of G-protein-coupled receptors reveals previously untargeted allosteric sites. Nat. Commun. 13, [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Lu H et al. (2020) Recent advances in the development of protein–protein interactions modulators: mechanisms and clinical trials. Signal Transduct. Target. Ther. 5, 213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Weis WI and Kobilka BK (2018) The Molecular Basis of G Protein-Coupled Receptor Activation. Annu. Rev. Biochem. 87, 897–919 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Zhang Y et al. (2022) Benchmarking Refined and Unrefined AlphaFold2 Structures for Hit Discovery. ChemRxiv at <https://chemrxiv.org/engage/chemrxiv/article-details/62b41f0c0bbbc117477285a4> [DOI] [PubMed] [Google Scholar]
- 65.Miller EB et al. (2021) Reliable and Accurate Solution to the Induced Fit Docking Problem for Protein–Ligand Binding. J. Chem. Theory Comput. 17, 2630–2639 [DOI] [PubMed] [Google Scholar]
- 66.Zhao Q et al. (2021) Enhanced Sampling Approach to the Induced-Fit Docking Problem in Protein–Ligand Binding: The Case of Mono-ADP-Ribosylation Hydrolase Inhibitors. J. Chem. Theory Comput. 17, 7899–7911 [DOI] [PubMed] [Google Scholar]
- 67.Liu X et al. (2019) Structural Insights into the Process of GPCR-G Protein Complex Formation. Cell 177, 1243–1251.e12 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Coleman RG et al. (2013) Ligand Pose and Orientational Sampling in Molecular Docking. PLoS One 8, e75992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Bender BJ et al. A practical guide to large-scale docking., Nature Protocols, 16. 24-Sep-(2021), Nature Publishing Group, 4799–4832 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Gorgulla C et al. (2020) An open-source drug discovery platform enables ultra-large virtual screens. Nature 580, 663–668 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Gaulton A et al. (2012) ChEMBL: A large-scale bioactivity database for drug discovery. Nucleic Acids Res. 40, D1100–D1107 [DOI] [PMC free article] [PubMed] [Google Scholar]