

Abstract
Neisseria gonorrhoeae is an urgent public health threat due to the emergence of antibiotic resistance. As most isolates in the US are susceptible to at least one antibiotic, rapid molecular antimicrobial susceptibility tests (ASTs) would offer the opportunity to tailor antibiotic therapy, thereby expanding treatment options. With genome sequence and antibiotic resistance phenotype data for nearly 20,000 clinical N. gonorrhoeae isolates now available, there is an opportunity to use statistical methods to develop sequence-based diagnostics that predict antibiotic susceptibility from genotype. N. gonorrhoeae therefore provides a useful example illustrating how to apply machine learning models to aid in the design of sequence-based ASTs. We present an overview of this framework, which begins with establishing the assay technology, the performance criteria, the population in which the diagnostic will be used, and the clinical goals, and extends to the choices that must be made to arrive at a set of features with the desired properties for predicting susceptibility phenotype from genotype. While we focus on the example of N. gonorrhoeae, the framework generalizes to other organisms for which large-scale genotype and antibiotic resistance data can be combined to aid in diagnostics development.
Keywords: Neisseria gonorrhoeae, antimicrobial resistance, machine learning, diagnostics
Graphical Abstract
Rapid molecular antimicrobial susceptibility tests (ASTs) offer the opportunity to tailor antibiotic therapy for antibiotic-resistant Neisseria gonorrhoeae. Using genome sequence and antibiotic resistance data from clinical N. gonorrhoeae isolates, there is an opportunity to use statistical methods to develop sequence-based diagnostics that predict antibiotic susceptibility from genotype. We present an overview of how to apply machine learning models to aid in the design of sequence-based ASTs for N. gonorrhoeae, and this framework should be generalizable to other organisms.
INTRODUCTION
The need for new strategies to control antibiotic-resistant Neisseria gonorrhoeae
Antimicrobial resistance (AMR) is a formidable and persistent challenge requiring continuing innovation in health care and public health systems, with one estimate attributing 1.27 million deaths globally in 2019 to infection with AMR pathogens1. Without interventions, the trajectory of AMR points to a worsening crisis that threatens the foundations of modern medical practice. There is a pressing need for new tools to aid in clinical and public health efforts to curb the spread of resistance.
One of the most urgent AMR pathogens is Neisseria gonorrhoeae, the cause of the sexually transmitted infection gonorrhea2. N. gonorrhoeae can infect and transmit among mucosal tissues, including the urethra, cervix, rectum, and pharynx, and can cause invasive disease, including pelvic inflammatory disease and disseminated gonococcal infection with septic arthritis, tenosynovitis, and endocarditis3. Severe sequelae from pelvic inflammatory disease include ectopic pregnancy and infertility, and infection of the conjunctiva can lead to blindness3. The incidence has nearly doubled in the US since a historic low in 2009, and rates are similarly increasing globally, with the UK seeing a rise of 26% from 2018 to 2019 and Australia seeing a rise of 63% from 2012 to 20164–6.
Infection with N. gonorrhoeae is most commonly diagnosed by nucleic acid amplification test (NAAT), with culture and antibiotic susceptibility testing now relegated to only a small fraction of clinical microbiology laboratories7. NAATs are faster, only requiring several hours, and less labor intensive than culturing. However, while highly sensitive and specific for gonorrhea diagnosis, the genetic loci queried by NAATs do not provide information on antibiotic susceptibilities8. Consequently, almost all antibiotic treatment of gonorrhea is empiric, informed by surveillance data, and health care providers are encouraged to remain vigilant for treatment failure7.
N. gonorrhoeae has developed resistance to each of the first-line antibiotics used to treat gonorrhea9,10. Only a single antibiotic, the third-generation cephalosporin ceftriaxone, remains as the recommended treatment for gonococcal infections in the US7. The carbapenem class antibiotic ertapenem has been used to treat cases of ceftriaxone failure11 but, given the prevalence of gonorrhea, the widespread use of an antibiotic of last resort for many types of infection would be expected to result in bystander selection12 and have consequences for AMR across multiple pathogens13. Two drugs, zoliflodacin and gepotidacin, are in phase 3 trials for treatment of uncomplicated urogenital gonorrhea; however, in phase 2 trials they showed limited success in treating pharyngeal gonorrhea14,15, raising questions about how they will best be used if approved.
The increase in incidence and appearance of strains resistant to all approved antibiotics, however, belie substantial geographic and demographic variation in N. gonorrhoeae antibiotic susceptibility16–22. Both surveillance data from clinical microbiology laboratories and genomic epidemiology have demonstrated that resistance spreads locally and internationally17,23–25, leading to complex population dynamics that are a function of patterns of antibiotic use and contact networks, among other factors26–28.
Given this overall context, N. gonorrhoeae is paradigmatic of the AMR challenges posed by other bacterial pathogens. The increasing levels of resistance, the critical need for new treatment options, and varying patterns of resistance by location capture characteristics seen for other sexually transmitted pathogens (e.g., Shigella29), for enteric pathogens (e.g., Salmonella typhi30 and Enterococcus31,32), and for respiratory pathogens (e.g., Mycobacterium tuberculosis33), among others.
Genetic basis of AMR in N. gonorrhoeae
Much work has been done to determine the genetic modulators of AMR in N. gonorrhoeae34–48, with a focus on the antibiotics for which AST phenotypes are available—thus, most data are for antibiotics in recent use (ceftriaxone, cefixime, azithromycin, ciprofloxacin) as well as others for which surveillance programs often collect data (penicillin, tetracycline)16,17,23,25,49–51,51–72. Most of the loci that account for a large extent of resistance in circulating populations in areas of high surveillance have been well characterized66,73, with several recent additions to the list of genetic modulators being described,18,74–77 including some of increasing prevalence, such as the mosaic mtr78,79 (where mosaicism refers to interspecies recombination between N. gonorrhoeae and another Neisseria species).
How much of observed resistance in N. gonorrhoeae can be explained by known resistance determinants? Linear models offer one approach to answer this question18,52,80. However, the implementation of these models does not include interaction terms, and thus assumes that each of the determinants acts independently. As new resistance modulators are discovered, characterized, and incorporated into these models18,74–76,78,79 the correlation between predicted minimum inhibitory concentration (MIC) and observed MIC has steadily improved. For ciprofloxacin, the correlation as represented by R2 is very high; in the rare cases of mismatch between predicted and observed MIC, the mismatch is likely due to error in phenotyping or specimen labeling. For other drugs, like azithromycin and ceftriaxone, R2 is lower, reflecting that our understanding of the genetic basis of resistance is incomplete, either implicating unappreciated interactions among determinants or undescribed resistance determinants75. Moreover, the introduction of the blaTEM and tetM plasmids into N. gonorrhoeae in the 1970s and 1980s40,81 are reminders that new mechanisms of resistance can emerge, and that our assessment of our ability to explain observed resistance is necessarily restricted to a static and retrospective collection of isolates.
Rapid diagnostics that predict antimicrobial susceptibility
A strategy for managing AMR that is garnering much attention is the development and use of rapid diagnostics that can predict drug susceptibilities and help clinicians select optimal therapy. By broadening choices beyond empiric therapy, mathematical models suggest that diversification of selective pressures may slow the emergence and spread of resistance and prolong the clinically useful lifespan of antibiotics82–84.
While numerous strategies have been proposed for new AMR diagnostics, a longstanding effort has been to translate understanding of the genetic basis of resistance into sequence-based diagnostics85. These strategies leverage DNA amplification technologies that offer high specificity and sensitivity for pathogen presence and for resistance markers and that have substantially shorter turn-around times than culture-based methods. Such diagnostics have been developed and deployed for a number of pathogens, including detection of rifampin resistance-conferring mutations in rpoB in M. tuberculosis86, detection of mecA to identify methicillin-resistant Staphylococcus aureus87, and, recently, the development of a test for N. gonorrhoeae that reports on ciprofloxacin susceptibility by examining gyrA codon 9188.
The vast amount of pathogen genome sequencing being performed and the links to phenotypic antibiotic susceptibility testing has prompted a surge of interest in using statistical methods to predict AST from nucleotide sequencing52,72,80,89–92 (Table 1). While the applications of machine learning tools are being explored in many bacterial pathogens93–95, this strategy has urgency and relevance in N. gonorrhoeae given the potential clinical and public health benefits of rapid AST diagnostics.
Table 1:
WGS AMR prediction models in N. gonorrhoeae.
| Year | First author/s | Antibiotics | # isolates | Preprocessing | Genotype encoding | Phenotype encoding | Feature selection | Model | Evaluation schema | |
|---|---|---|---|---|---|---|---|---|---|---|
| Validation | Test | |||||||||
| 2016 | Demzcuk54 | AZI | 246 | Known alleles | MIC | Reverse feature elimination using R2 | Linear regression | Leave-one-out cross validation | ||
| Grad95 | CFX, CRO, ESC, AZI | 1,102 | Categorical | Manual | Presence/absence | |||||
| 2017 | Eyre52 | AZI, CFX, CIP, TET, PEN | 681 | Known alleles | MIC | Reverse forward feature elimination using AIC | Linear regression | Leave-one-out cross validation | ||
| 2019 | Drouin91 | ~200 | k-mer | Categorical | Built-in | Set covering machine | Ten-fold cross validation | |||
| Hicks88 | CIP & AZI | 4358 | Down sampling minority class | k-mer of known alleles | Categorical & MIC | Removal of unique k-mer | Set covering machine & random forest | Five-fold cross validation | Stratified by categorical phenotype and dataset | |
| Deng96 | CFX | 415 | Known alleles | Categorical | Manual multivariate supervised selection | Presence/absence | ||||
| 2020 | Břinda90 | CRO, CFX, AZI, CIP | 1,102 | Reads | Categorical | Nearest-neighbors | ||||
| Demczuk79 | AZI, CRO, CFX, CIP, TET, PEN | 2,806 | Known alleles | MIC | Reverse forward feature elimination using AIC | Linear regression | Test dataset CA (n = 1,095) and international (n = 431) | |||
| 2021 | Lin97 | CRO | 3,821 | Known alleles | Categorical | Manual multivariate supervised selection | Presence/absence | |||
| Sánchez-Busó66 | AZI, CRO, CFX, CIP, TET, PEN | 3,987 | Known alleles | Categorical | Presence/absence | External test set | ||||
| Shaskolskiy98 | CRO | 5,631 | Known alleles | MIC | Reverse feature elimination using AIC | Poisson regression | External test set, Russian (n = 448) and WHO (n = 14) | |||
| 2022 | Mortimer & Zhang89 | TET, PEN | PEN (n = 6935), TET (n = 5727) | k-mer | MIC | Conditional GWAS | Presence/absence | External test set CDC (n = 1479) | ||
| Yasir99 | CFX, CIP, AZI | 3,787 | 8,290 unitigs |
MIC | Univariate supervised | 35 ML models | Ten-fold validation | 20/80 test train split | ||
AZI: azithromycin, CXF: cefixime, CRO: ceftriaxone, CIP: ciprofloxacin, TET: tetracycline, PEN: penicil.
How can machine learning help move from the large number of genome sequences and associated antibiotic resistance phenotypes to diagnostics? In the case of genome sequencing as a diagnostic, how much of the 2.2 Mb genome of N. gonorrhoeae needs to be evaluated to inform clinical decisions? And for diagnostics that evaluate only some of the genome, which loci should be included in a diagnostic to achieve optimal performance?
Below, we highlight the decisions and considerations that are part of developing and interpreting machine learning approaches for predicting AMR phenotype from pathogen genotype, with an emphasis on applications to the development of sequence-based assays for gonorrhea to inform clinical decisions in a dynamic and evolving pathogen population (Figure 1).
Figure 1.
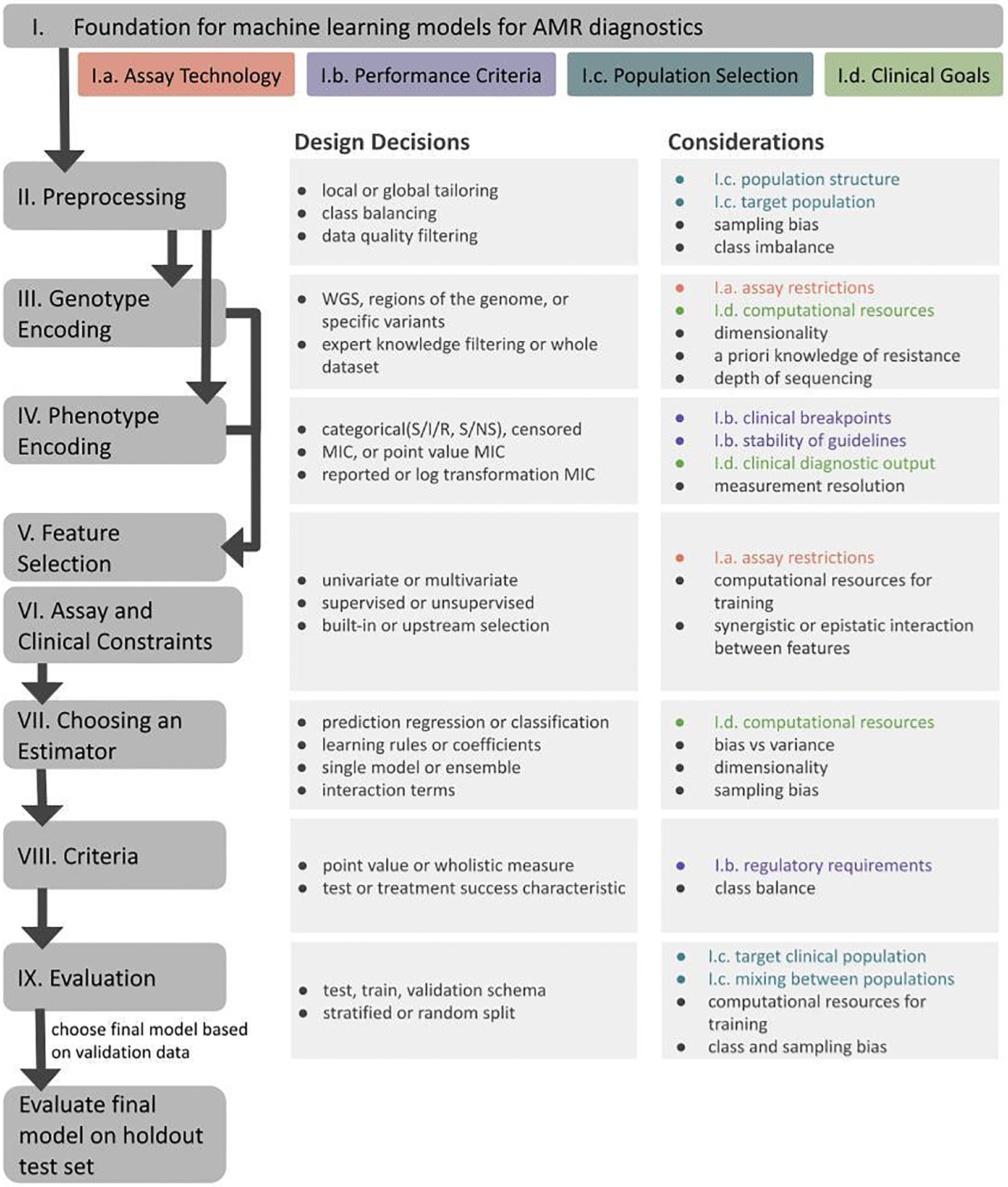
Framework for designing machine learning models for antimicrobial susceptibility diagnostics. Rounded boxes represent sections in the text. Black lines represent the order of decisions. Rectangular boxes to the right of each rounded box represent the key design decisions at each step. The rightmost column contains the considerations for each choice. Colors are coded to align with subsections of the first section listed at the top of the figure, with grey bullets representing computational and machine learning considerations.
FRAMEWORK FOR MACHINE LEARNING MODELS FOR ANTIMICROBIAL SUSCEPTIBILITY DIAGNOSTICS
Setting the foundation for machine learning models for AMR diagnostics
Assay technology
A diagnostic combines an assay and framework for interpretation to inform treatment decisions. In the absence of point-of-care pathogen genome sequencing, three existing and developing methods offer near-term promise for point-of-care sequence-based diagnostics: multiplexed polymerase chain reaction (PCR), loop-mediated isothermal amplification (LAMP) and SHERLOCK, a CRISPR-based lateral flow diagnostic85,96,97. The performance of a diagnostic relies on the assay’s accuracy to detect its targets as well as the ability of the algorithm to predict susceptibility. The choice of assay impacts which and how many markers can be detected and the clinical resources required. These parameters are key factors in overall feasibility of the diagnostic and present tradeoffs between accessibility across resource settings and accuracy.
Performance criteria
Regulatory standards for antimicrobial susceptibility diagnostics depend on the country and regulatory body. In the United States, the Food and Drug Administration (FDA) has criteria for short-term incubation cycle AST systems, but not specifically for sequence-based molecular diagnostics98. They consider three error types: very major, major, and minor. For approval, the rate of major error in susceptible isolates must be lower than 3%, the 95% confidence interval upper bound of very major error must be lower than 7.5%, and the essential/categorical agreement must be above 90%.
Population selection
Resistance mechanisms for a given antibiotic can vary regionally. In some areas, resistance may be mediated by a single gene, whereas other regions may have more diverse genotypes of resistance. For example, azithromycin resistance can be through A2058G, A2059G, and C2611T mutations in the 23S rRNA35,41,77, variants in the mtr operon76,78,79, and mutations in rplD75 . Resistance prevalence can vary, too, with ciprofloxacin resistance at or near 100% in most of Asia, compared to less than 30% in the US and less than 70% in Europe. Similarly, resistance prevalence can vary by sexual behavior16,69.
Given this variation across populations, it might be tempting to reduce assay complexity by tailoring it to strains in a specific population instead of incorporating all genotypes circulating globally. However, tailoring to a single population might make the diagnostic less resilient to imported resistance or de novo acquisitions of resistance mechanisms that appear in other regions. Another challenge of local tailoring is that surveillance data for N. gonorrhoeae has been primarily collected in a small subset of countries66, so our ability to tailor a diagnostic to other endemic settings is limited in the absence of additional surveillance.
Clinical goals
The design of a diagnostic is informed by its intended influence on clinical treatment algorithms. If the assumption is that an infection will be susceptible to the empiric therapy, then the diagnostic should report on the presence of resistance markers that are highly sensitive for resistance. Conversely, if the provider assumes that the infection will be resistant to a specific antibiotic, then the goal is to identify markers that are highly sensitive for susceptibility. These two goals do not require accurate prediction of MIC across the full range of MICs; instead, their performance depends on the ability to predict categorical non-susceptibility or susceptibility, respectively.
A diagnostic may not be feasible in a clinical setting, due to lack of computational, laboratory, and/or monetary resources. Therefore, the design of a diagnostic should weigh the tradeoff between performance gained via assay or model complexity and feasibility across clinical settings.
While many sequence-based resistance diagnostics have focused on a single antibiotic, diagnostics can in principle report on multiple antibiotics. Balancing which antibiotics, given the extent of overlapping genetic modulators of resistance, and the multiplex capacity of the diagnostics, is another goal for machine learning strategies.
Preprocessing: Adjusting for biases in genotypic and phenotypic data
Machine learning models that predict resistance phenotype from genotype rely on datasets that provide both. Regional gonococcal surveillance programs (such as GISP, Euro GASP, WHO-GASP, ASGAP) and smaller epidemiological studies have reported genome sequences and AMR profiles from over 18,000 clinical isolates of N. gonorrhoeae16,17,23,49,52–55,57–70,99,100; many of these have been curated and aggregated in databases such as Pathogenwatch66. But the sampling strategies differ among surveillance programs, as can the quality of the sequencing or phenotypic data.
In an ideal machine learning setting, the training data would be a representative sample of the population in which we are trying to implement the model. The population level bias in this data lies in which isolates are sampled. Clinical isolates may be collected, sequenced, and undergo resistance phenotyping in the context of surveillance, investigation of a specific outbreak, clone or lineage, or clinical care.
Surveillance programs collect isolates to understand resistance prevalence and inform treatment guidelines. However, these programs have their own sampling biases: the limited set of sentinel surveillance clinics, the patient population using those clinics, the anatomical sites sampled, and the type of susceptibility testing and sequencing. For example, the CDC’s GISP program is designed to sample men with urethritis; recent expansions of the sampling strategy, including SURRGE and enhanced GISP aim to broaden the sampling to include women and more anatomical sites101. Because of the population structure of N. gonorrhoeae and differences in lineage prevalence by geography and demography, these differences in the structure and approach of surveillance programs need to be considered when deciding which data is appropriate for training a model for a given clinical population. Similarly, samples derived from analyses of an outbreak, clone, or lineage or as part of clinical care may be biased in over-representing specimens with a particular pattern of antibiotic resistance.
As with any effort to make inferences from the genomes of collections of bacteria, each individual isolate cannot be considered independent and must be considered within the context of the underlying genetic relatedness among them. A robust model relies on a diversity of data across the phylogeny, but models can also be targeted to specific patient populations and their associated bacterial lineages, depending on the goals.
Aggregation of data across sources requires confidence that the data from each source surpasses quality standards and that the reported data are comparable, despite any differences in how they were generated. From a sequencing perspective, the method, that is, long read versus short read, may change the resolution of the genomic data. The threshold of quality to include in the model needs to be selected. Alternatively, one could limit the data to a single study or set of studies52,80; however, this reduces the amount of data and might reinforce errors arising from the impact of the local population structure.
A similar set of considerations pertains to antibiotic resistance phenotypic data, where these data may be acquired by agar dilution, Etest, or disk diffusion. These tests have different outputs and error profiles that must be reconciled if combined. Agar dilution tests for resistance via a 2-fold dilution series, and with measurement error of one doubling dilution; results of 0.5 μg/mL and 4 μg/mL have vastly different expected error, [-0.25,0.5] and [-2,4], respectively. In contrast, Etests offer a finer scale assessment of MIC and have a distinct error profile. The non-uniformity of error may be resolved by transforming the MICs (e.g., log transformation), but doing so itself has implications for the machine learning algorithms and interpretation of the results.
In some cases, left- or right-censored intervals of MICs may be reported (for example, reporting that an MIC is <0.06 μg/mL or >8μg /mL, respectively). This labor- and cost-savings approach may be used when clinical or surveillance purposes do not require an exact MIC but only whether the level of resistance for a given isolate is above or below a threshold. These breakpoints can vary regionally. For instance, in the Australian gonococcal surveillance program the epidemiologically relevant breakpoint MICs for tetracycline are 8 μg/mL and 16 μg/mL, which differ from the CLSI clinical breakpoints of 0.25 μg/mL and 2 μg/mL69,102. Depending on the level of resolution of phenotype, whether that be point MIC via Etest, censored MIC via agar dilutions, or phenotype as determined by breakpoints, isolates will need to be discarded to have a uniform dependent variable.
From a machine learning perspective, the combination of sampling methods, underlying resistance prevalence, and interpretive breakpoints result in class imbalance. An overrepresentation of resistant isolates might lead to overfitting of a regression model on high MICs, resulting in prediction of all isolates as resistant, in an extreme case, or to poorer prediction for low MIC isolates. One strategy to address class imbalance is by resampling the data or synthetically generating data to equalize the classes. Resampling can be done by either oversampling the minority class or undersampling the majority class (resulting in lost data). Resampling should be done after splitting the data into training and testing sets, so that training isolates are not included in the test data. Synthetic methods rely on the generation of new samples via interpolation of the data, that is, using isolates from the training data to generate novel genomes and corresponding resistance profiles in the minority class. However, this requires strong assumptions of how certain alleles may interact to contribute to resistance and may reinforce underlying biases in the data. Generating and testing the synthetic samples experimentally would address concerns about their validity but can be labor- and time-intensive at large scales. Alternatively, class imbalance can be addressed later in the training process, either by using balanced performance metrics in training and supervised feature selection or by feeding the prediction model the sample weights.
Genotype encoding
Once the dataset is chosen and cleaned up, the next step is to determine how to encode genome sequences into features. Different scales of genetic data can be fed into a machine learning prediction model, including the whole genome sequence (WGS)91,92, sequences of regions known to modulate resistance89, and specific variants17,52,80,90,103. Each approach has potential drawbacks: whereas using WGS data could result in overfitting to noncausal mutations that appear in resistant lineages, using a restricted set of genes and variants known to contribute to resistance could exclude uncharacterized resistance modulators and reduce assay performance.
The goal of genetic encoding is to input genetic features and convert them into a numerical matrix. This is commonly done via one-hot encoding, which expands categorical features into multiple columns (for example, expanding a single variant into the columns that account for the variant and the alternative alleles). Ordinal encoding can be an alternative to one-hot, by representing all substitutions as a different number. However, this may lead the algorithm to believe non-ordered data has numerical values. Therefore, it is best for this method to be reserved for alleles with innate numerical relation, like copy number variation. While these two encoders are often sufficient for this context, this is not an exhaustive list of encoders104.
A second consideration for models that use unfiltered genotypic data is bacterial population structure: each clinical isolate used in the model is not genetically independent of the others, thus requiring a decision on how to adjust weighting based on genetic relatedness. The field has seen it as an opportunity, creating “neighbor typing” approaches using WGS91, and as a problem to be solved, designing methods that control for population structure to tease out causal links between genetic variation and resistance75,105,106. With an eye towards assay complexity, neighbor typing would require sequencing at point of care and an up-to-date database of circulating strains. Since N. gonorrhoeae is highly recombinogenic107,108, such an approach would require particular attention to characterizing novel combinations of genotypes that arose via recombination.
This choice has both algorithmic and assay considerations. First, sequencing data has high dimensionality, such that the number of features may be larger than the number of isolates. Thus, the complexity of the sequence data may need to be reduced in some way before feeding it into a ML algorithm. Second, the features are the basis of the model output, and as such the features must align with the capabilities and limitations of the target assay chosen.
Phenotype encoding
As discussed above, susceptibility information can be measured and reported differently across laboratories. For use in a ML model this data must be standardized into either numerical or categorical phenotypes. We have focused on phenotypes from the clinical microbiology laboratory—MIC—and from clinical guidelines, with MIC breakpoints defining susceptibility and resistance, such as those established by EUCAST and by CLSI, which aim to reflect the clinical outcomes of treatment success and failure102,109. Between susceptible and resistant breakpoints exists the non-susceptible or dose-dependent susceptible category. N. gonorrhoeae has defined breakpoints for penicillin, tetracycline, and ciprofloxacin, but breakpoints for azithromycin, cefixime, and ceftriaxone are limited to alert values. Breakpoints can also change, as treatment failure data continues to be collected. For instance, cefixime treatment failures have been reported at 0.12 MIC, despite “reduced susceptibility” breakpoints of CLSI ≥ 0.25 and EUCAST ≥ 0.125102,109.
Treatment failure is subject to factors beyond the pathogen, such as infection site, patient characteristics, and medication dosing. Whereas for treatment of uncomplicated gonorrhea the CDC recommends ceftriaxone 500 mg in a single dose, the British Association for Sexual Health and HIV recommends ceftriaxone 1 g as a single dose; both recent dose increases were in response to rising ceftriaxone MICs110. While training a classifier model on categorical phenotype can achieve better performance than training the model on MIC89, shifting and heterogenious treatment practices and circulating lineages point to the need for either regular updating of the model with changing interpretive criteria or training models on MIC.
Dichotomizing a continuous variable discards statistical power by making the model unaware of the magnitude of error, so it is considered bad practice111. Antibiotics with class imbalance may have few isolates that are resistant, but many isolates that have a broad range on MICs within the susceptible range. In this case, training on MIC may allow the model to leverage variation within the majority class to better predict the minority class. Even without a class imbalance, the model trained on MIC may better extrapolate phenotype for novel genotypes. For instance, a novel genotype containing molecular determinants that caused reduced susceptibility independently, within the training dataset, in combination may cause resistance. However, a classifier may predict this novel genotype as susceptible since all features are only seen in the susceptible class.
Feature selection
While sequence-based diagnostics allow for the entire genome to be queried, locus-based diagnostics, such as PCR, target a limited number of alleles. This raises the question, how are genetic features selected for inclusion in sequence-based AMR-predicting diagnostics? Feature selection can be manual, based on expert knowledge of causal variants, or automatic. If automatic, then there are several additional choices. First, within machine learning approaches, selection can be supervised or unsupervised. Supervised selection incorporates the phenotype, as selected in the above section (the dependent variable), and is thus most appropriate for the task here; unsupervised selection is best used for tasks where the categories that distinguish isolates are unknown. The next choice is whether to treat the features as univariate (features treated individually) or multivariate (features treated in combination) in their prediction of resistance phenotype. Supervised multivariate selection is computationally expensive, leading many algorithms to rely on greedy approaches that do not guarantee a globally optimal set of features (algorithms that do guarantee global optimality require conditions unlikely to be met by current datasets or without substantial preprocessing112).
Feature selection can also be done upstream of model training, especially in datasets with high dimensionality, or it can be built into the model’s fitting procedure. Built-in feature selection is often accomplished by adding a penalty for the size of the feature set, thereby allowing the input of the entire feature set. However, algorithms with built-in feature selection often do not allow for assessment of performance differences by set size, making it unclear if the penalty term adequately weighs the tradeoff between error and the number of features. In contrast, a separate feature selection algorithm enables explicit characterization of this tradeoff.
Multivariate feature selection algorithms rely on stepwise removal (e.g., reverse feature elimination), addition (e.g., forward selection), or a combination of removal and addition of features to maximize the objective for the diagnostic. One limitation with these methods is that they assume that the optimal feature set is a subset (for elimination) or superset (for addition) of the current set of features, which might be false when features have synergistic predictive power. While all heuristics will lead to a local maximum, certain selection algorithms might lead to a more optimal solution. Stepwise approaches can be augmented to take a breadth-first approach, where multiple optimal paths are searched.
Univariate feature selection methods evaluate and drop features individually. In an unsupervised setting, this is commonly done by removing rare alleles in the training population, while supervised selection may remove genetic features below a threshold predictive value or correlation with the outcome variable. The benefit of univariate approaches is that they are computationally cheap and can be run on large feature sets. A drawback is that they do not account for epistatic interactions. In contrast, multivariate approaches may become too computationally burdensome as the feature set grows and require greediness to run in a reasonable time. Machine learning pipelines can incorporate a combination of these approaches starting with filter-based univariate methods and then can move towards more complex methods, maximizing performance within a feasible runtime.
Incorporating clinical and assay constraints in feature selection
The type of diagnostic assay constrains which genetic features can be detected individually and in combination. For instance, if the assay was WGS, we could query most or all genetic features, depending on whether the sequence is in draft or finished form. Genetic locus–based assays, in contrast, focus on one or more specific regions of the genome to detect sequence variation at those loci. The genetic variation within the targeted locus and the association of that variation with the phenotype of interest then inform the complexity needed for a sequence-based assay.
When categorical resistance is associated with a single nucleotide polymorphism or an insertion/deletion, and there is limited other allelic variation, design of an assay to detect the variant is most straightforward. For example, to distinguish between ciprofloxacin susceptibility and resistance in N. gonorrhoeae, querying codon 91 in gyrA is sufficient. As allelic diversity increases and as the association between sequence variation and the phenotype of interest increases—such as with multiple mutations within a gene contributing additively to antibiotic resistance—the challenges associated with sequence-based diagnostics can also increase. For example, mosaicism in penA is associated with resistance to penicillin and reduced susceptibility to cephalosporins. But since there are multiple mosaic alleles in the N. gonorrhoeae population, such that one specific mutation does not reliably distinguish mosaic from non-mosaic alleles, multiple features would be needed to query this locus.
Next the feature selection algorithm needs measurable heuristics to evaluate the ability of a marker or set of markers to robustly and reliably detect the target feature. For PCR, this could be minimizing the estimated primer free energy (dimerization) score and the range of annealing temperatures across all markers. These heuristics could then be aggregated into a proxy for the diagnostic’s ability to return an accurate and interpretable result. If a marker were specific to resistance, but the assay had low accuracy of detection for this marker, it might not be a good marker to include in the feature set.
Choosing an estimator
Modeling the relationship between genotypic features and phenotype requires selection of the statistical method to relate the two. In the case of AMR, we expect the molecular determinants to contribute to resistance both individually and through interactions with each other. In this context we also need to think about interpretability of the model, for two reasons: first, so that the assumptions in the model can be evaluated experimentally, and second because employing a complex model in a diagnostic could require computational resources not available in all health care settings.
Models can be differentiated by what and how they learn. For linking genotype to resistance phenotype, algorithms can predict MIC using regression methods or categorical (e.g., susceptible, intermediate, resistant) phenotype using classification methods. Some models learn rules to make predictions. Such rules allow the algorithm to cluster the data into phenotypically similar subpopulations, with examples including decision trees and nearest neighbor algorithms. Others learn coefficients that quantify the contributions of each feature or sets of features onto the response variable, with examples including regression methods (i.e., linear, logistic, polynomial), and neural networks.
A well-fitting model minimizes the tradeoff between bias, the number of assumptions made by the model, and variance, the variability of the estimator given another training set. In the context of predicting AMR, a low bias model (for example, decision trees) can learn epistatic/nonlinear interactions between genetic determinants of resistance, but these models have high variance because they tend to overfit to population structure, while low variance models (e.g., linear/logistic regression) rely on assumptions about how molecular determinants contribute to resistance, which may lack the complexity of molecularly mediated resistance.
Decision trees are more commonly used in an ensemble format to make up for their high variance. Ensembles rely on multiple trees that are trained either in sequence or independently, referred to as boosting (with Adaboost as one example) and bagging (with Random Forest as an example), respectively, on subsets of the data. Both types of ensembles can result in 100+ trees, requiring a clinical setting to have the computational resources to run a prediction. Due to the method of training, bagging might miss rare lineages, while boosting tends to prioritize them. As a replacement for ensembles, set covering machines and INGOT-DR learn logical operators as opposed to hierarchal rules like decision trees and have performed similarly to random forest implementations89,92,113. Linear regression models tend to be highly interpretable but assume linear and independent contributions of features to phenotype. Linear mixed models, an extension of linear regression, require fewer assumptions and can incorporate interaction terms. As the models become more complex, that is, neural networks, more coefficients need to be trained, leading towards a high dimensionality problem requiring larger datasets. We note that while we mention multiple algorithms commonly used in this context, this does not represent an exhaustive list. We recommend the Nature review A guide to machine learning for biologists as a broader resource for machine learning algorithms and methods in a biological context104.
Performance criteria
Formal guidelines for the accuracy of molecular tests have not yet been established, but there do exist such standards for phenotypic tests and clinical treatment success. In the design of machine learning models, evaluation may need to be performed to judge performance at preprocessing, feature selection, and model choice steps. For instance, if feature selection methods may not achieve above zero specificity at two features, then a more holistic measure that is not bounded by a threshold should be used to differentiate the performance among sets of two features.
There are common metrics used to evaluate a model’s ability to classify or predict a certain outcome. Broadly these can be separated into classification and regression metrics. Classification metrics include point-based values like sensitivity/recall, specificity, precision/positive predictive value, and negative predictive value. These metrics are calculated using interpretative thresholds or probability thresholds as reported by a classification model or tuning to error bounds. In the context of ASTs, a model might not be able to achieve FDA performance across all criteria, therefore understanding the tradeoff between the errors assists us in tuning the model to clinically relevant error.
The choice of metric impacts the number and set of features required for the task. For instance, if the goal is predicting N. gonorrhoeae susceptibility to ciprofloxacin, then a single marker, gyrA codon 91, achieves >0.99 sensitivity and specificity (as in the SpeeDx diagnostic)88. However, if the goal is to predict the MIC within one doubling dilution in 90% of isolates, then eight genetic features are needed80. What is the tradeoff between the number of loci in the diagnostic and performance? Common classification performance metrics like Akaike information criterion (AIC) and Bayesian information criterion (BIC) include a cost term for the number of features to indicate overfitting on the training data, though it is worth noting that this penalty term is not aligned with either clinical or assay constraints. These differences underscore important questions about the goals of diagnostics: Should diagnostics only be measured on classification success? On treatment outcomes?
Another consideration is whether the metrics represent a test characteristic or confidence in the test. Categorical measures, like sensitivity and specificity, measure accuracy of prediction for each class in isolation. Positive predictive value (PPV) and negative predictive value are measures in the confidence of a positive or negative diagnosis, respectively. For rare diseases the PPV might be low, even as the test has high sensitivity and specificity. Since PPV varies based on disease burden in the population, it might be better used retrospectively to evaluate the test data and potential clinical efficacy, but we caution against its use in the training process since it might overfit class imbalances.
The receiver operator characteristic curve (ROC) and the precision recall curve (PR) together can give us a more holistic understanding of the model’s ability to discriminate between the classes. The ROC curve quantifies the tradeoff between specificity and sensitivity. The PR curve represents the predicted clinical efficacy (positive predictive value) at varying sensitivities. Both curves can be summarized by the area under them, where an area of 0.5 indicates that classifier is no better than random chance and a maximum area of one meaning the classifier has perfect performance.
Evaluating in silico performance in the context of population dynamics
The goal of evaluation is to test the model’s ability to perform on unseen data, and more broadly to evaluate the diagnostic’s predicted clinical efficacy. Machine learning approaches start by reserving 10–20% of data for testing. The remainder of the data is split into training and validation sets. Various approaches to data splitting have been used for N. gonorrhoeae, including leave-one-out cross-validation52, k-fold cross-validation89, and/or split by dataset80,89. Data can be split into these folds or into the test set at random or stratified to maintain a proportion of a given characteristic in each split, where characteristics can include year, study, and sexual behavior. Once parameters are chosen, the full validation/training dataset is used to train the model, which in turn uses the test data to assess the model’s predictions. Data can be nonrandomly chosen to better understand the transferability of a diagnostic designed for one population to another clinical population89.
The continued evolution of the target bacterial population after the development and implementation of a diagnostic will reflect selective pressures from the diagnostic itself114–116, antibiotic treatment,9 and adaptation to other host and environment factors18. To evaluate an AST in the context of an evolving population, one approach is to split the training and test data by time, thereby giving a sense of how well the model would perform given the fluctuating pathways to resistance. But no matter the performance on historical collections of isolates, sequence-based diagnostics should be monitored for possible diagnostic escape, as has been seen in N. gonorrhoeae, Chlamydia trachomatis, Plasmodium falciparum, and methicillin-resistant Staphylococcus aureus87,114,117,118. Escape could be achieved through mutations around a probe’s target that reduce binding and thus the diagnostic’s sensitivity, through dropping the probe’s target altogether, or through developing novel resistance mutations or novel combinations of mutations (a particular concern for N. gonorrhoeae, given that it is highly recombinogenic). This continued evolution in the pathogen population means that routine surveillance will be a critical part of regularly assessing sequence-based diagnostics that predict resistance phenotype from genotype89,119,120.
CONCLUSION
The combination of large-scale genomic data and antimicrobial susceptibility phenotypes provides an opportunity for machine learning methods to design and develop sequence-based diagnostics that predict antimicrobial susceptibility and that can improve clinical outcomes by rapidly enabling tailored therapy. To achieve these goals successfully and sustainably, the application of machine learning models requires a rigorous framework that considers factors including the datasets employed in training the models and their relationship to the populations in which the models will be used, the targeted performance metrics, and the constraints of the diagnostic platform itself. These issues are particularly salient when considering the use of such diagnostics in low resource settings, where surveillance data and access to first-line therapies may be limited. For N. gonorrhoeae, these methods hold promise, though their application will require transparency and commitment to the continued surveillance that will be necessary to monitor and maintain their usefulness over time and across populations.
Box 1: Evaluation metrics for predicting antibiotic susceptibility.
Accuracy: The probability of a correct prediction of a class given a classification model.
Balanced accuracy: A measure of accuracy useful when one class in a classification model appears much more frequently than the other, such as when resistant isolates are much more common than susceptible isolates.
Sensitivity/recall: The proportion of susceptible isolates predicted to be susceptible.
Major error*: The portion of susceptible isolates that are predicted to be resistant. The FDA requires this to be below 3% for AST in vitro systems.
Specificity: The proportion of non-susceptible isolates predicted to be non-susceptible.
Very major error*: proportion of resistant isolates predicted susceptible. FDA requires the 95% confidence interval of VME to be below 7.5% for AST in vitro systems.
Minor error*: proportion of the total isolates that are either intermediate but predicted susceptible/resistant or susceptible/resistant isolates predicted intermediate. FDA recommends this to be below 10% for AST in vitro systems but allows greater error if major and very major error meet required values.
Positive predictive value: The probability that isolates predicted to be susceptible are truly susceptible.
Negative predictive value: The probability that isolates predicted to be non-susceptible are truly non-susceptible.
Treatment efficacy: The proportion of cases given a treatment that are successfully treated. The World Health Organization recommends this to be above 95% for treatment of gonorrhea.
Precision recall curve: The tradeoff between precision and recall with varying susceptibility breakpoints on predicted values.
Receiver operator characteristic: The tradeoff between sensitivity and specificity at varying susceptibility breakpoints on predicted values. This metric may be misleading in cases of large class imbalance.
Root mean squared error: The transformed measure of absolute error.
R2: The proportion of MIC variation captured by the model and features.
Essential agreement*: The proportion of the diagnostic’s reported MICs that are within one doubling dilution of the actual MIC. The FDA requires this to be above 90% for AST in vitro systems.
Bayesian information criterion/Akaike information criterion: The log likelihood of the data, penalized by the number of features.
*FDA criteria
ACKNOWLEDGMENTS
S.L.M. and Y.H.G. were supported in part by CDC contract #200-2016-91779, and T.D.M. and Y.H.G. in part by the NIH National Institute of Allergy and Infectious Diseases (F32AI145157 and R01 AI132606). Y.H.G. is also supported by the Smith Family Foundation Odyssey Award.
REFERENCES
- 1.Murray CJ et al. Global burden of bacterial antimicrobial resistance in 2019: a systematic analysis. The Lancet 399, 629–655 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Centers for Disease Control and Prevention (U.S.). Antibiotic resistance threats in the United States, 2019. https://stacks.cdc.gov/view/cdc/82532 (2019) doi: 10.15620/cdc:82532. [DOI] [Google Scholar]
- 3.Bennett John E, Dolin Raphael, & Blaser Martin J.. Neisseria gonorrhoeae (Gonorrhea). in Mandell, Douglas, and Bennett’s Principles and Practice of Infectious Diseases (Elsevier, 2020). [Google Scholar]
- 4.Australian Government Department of Health. Diagnoses of gonorrhoea in Australia. Australian Government Department of Health; https://www.health.gov.au/resources/pregnancy-care-guidelines/part-g-targeted-maternal-health-tests/gonorrhoea (2018). [Google Scholar]
- 5.Centers for Disease Control and Prevention. National Overview - Sexually Transmitted Disease Surveillance, 2019. Sexually Transmitted Disease Surveillance 2019 https://www.cdc.gov/std/statistics/2019/overview.htm (2021). [Google Scholar]
- 6.Wise J Gonorrhoea cases in England hit highest level since records began. BMJ 370, m3425 (2020). [DOI] [PubMed] [Google Scholar]
- 7.Centers for Disease Control and Prevention. Gonococcal Infections Among Adolescents and Adults - STI Treatment Guidelines. Sexually Transmitted Infections Treatment Guidelines, 2021 https://www.cdc.gov/std/treatment-guidelines/gonorrhea-adults.htm (2022). [Google Scholar]
- 8.Gaydos CA & Melendez JH Point-by-Point Progress: Gonorrhea Point of Care Tests. Expert Rev. Mol. Diagn 20, 803–813 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Unemo M & Shafer WM Antimicrobial resistance in Neisseria gonorrhoeae in the 21st century: past, evolution, and future. Clin. Microbiol. Rev 27, 587–613 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Goire N et al. Molecular approaches to enhance surveillance of gonococcal antimicrobial resistance. Nat. Rev. Microbiol 12, 223–229 (2014). [DOI] [PubMed] [Google Scholar]
- 11.Eyre DW et al. Detection in the United Kingdom of the Neisseria gonorrhoeae FC428 clone, with ceftriaxone resistance and intermediate resistance to azithromycin, October to December 2018. Euro Surveill. 24, (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Tedijanto C, Olesen SW, Grad YH & Lipsitch M Estimating the proportion of bystander selection for antibiotic resistance among potentially pathogenic bacterial flora. Proc. Natl. Acad. Sci 115, E11988–E11995 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.MacFadden DR, Lipsitch M, Olesen SW & Grad Y Multidrug-resistant Neisseria gonorrhoeae: implications for future treatment strategies. Lancet Infect. Dis 18, 599 (2018). [DOI] [PubMed] [Google Scholar]
- 14.Taylor SN et al. Single-Dose Zoliflodacin (ETX0914) for Treatment of Urogenital Gonorrhea. N. Engl. J. Med 379, 1835–1845 (2018). [DOI] [PubMed] [Google Scholar]
- 15.Taylor SN et al. Gepotidacin for the Treatment of Uncomplicated Urogenital Gonorrhea: A Phase 2, Randomized, Dose-Ranging, Single-Oral Dose Evaluation. Clin. Infect. Dis 67, 504–512 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Mortimer TD et al. The Distribution and Spread of Susceptible and Resistant Neisseria gonorrhoeae Across Demographic Groups in a Major Metropolitan Center. Clin. Infect. Dis 73, e3146–e3155 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Grad YH et al. Genomic epidemiology of Neisseria gonorrhoeae with reduced susceptibility to cefixime in the USA: a retrospective observational study. Lancet Infect. Dis 14, 220–226 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Ma K et al. Adaptation to the cervical environment is associated with increased antibiotic susceptibility in Neisseria gonorrhoeae. Nat. Commun 11, 4126 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Clifton S et al. Prevalence of and factors associated with MDR Neisseria gonorrhoeae in England and Wales between 2004 and 2015: analysis of annual cross-sectional surveillance surveys. J. Antimicrob. Chemother 73, 923–932 (2018). [DOI] [PubMed] [Google Scholar]
- 20.Barbaric J et al. Surveillance and epidemiology of syphilis, gonorrhoea and chlamydia in the non-European Union countries of the World Health Organization European Region, 2015 to 2020. Euro Surveill. 27, (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Sánchez-Busó L et al. Europe-wide expansion and eradication of multidrug-resistant Neisseria gonorrhoeae lineages: a genomic surveillance study. Lancet Microbe 3, e452–e463 (2022). [DOI] [PubMed] [Google Scholar]
- 22.Machado H. de M. et al. National surveillance of Neisseria gonorrhoeae antimicrobial susceptibility and epidemiological data of gonorrhoea patients across Brazil, 2018–20. JAC-Antimicrob. Resist 4, dlac076 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Fifer H et al. Sustained transmission of high-level azithromycin-resistant Neisseria gonorrhoeae in England: an observational study. Lancet Infect. Dis 18, 573–581 (2018). [DOI] [PubMed] [Google Scholar]
- 24.Lahra MM et al. Cooperative Recognition of Internationally Disseminated Ceftriaxone-Resistant Neisseria gonorrhoeae Strain. Emerg. Infect. Dis 24, 735–740 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.De Silva D et al. Whole-genome sequencing to determine transmission of Neisseria gonorrhoeae: an observational study. Lancet Infect. Dis 16, 1295–1303 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Fingerhuth SM, Bonhoeffer S, Low N & Althaus CL Antibiotic-Resistant Neisseria gonorrhoeae Spread Faster with More Treatment, Not More Sexual Partners. PLOS Pathog. 12, e1005611 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Sun DS et al. Analysis of multiple bacterial species and antibiotic classes reveals large variation in the association between seasonal antibiotic use and resistance. PLOS Biol. 20, e3001579 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Olesen SW et al. Azithromycin Susceptibility Among Neisseria gonorrhoeae Isolates and Seasonal Macrolide Use. J. Infect. Dis 219, 619–623 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Baker KS et al. Intercontinental dissemination of azithromycin-resistant shigellosis through sexual transmission: a cross-sectional study. Lancet Infect. Dis 15, 913–921 (2015). [DOI] [PubMed] [Google Scholar]
- 30.da Silva KE et al. The international and intercontinental spread and expansion of antimicrobial-resistant Salmonella Typhi: a genomic epidemiology study. Lancet Microbe S2666–5247(22)00093–3 (2022) doi: 10.1016/S2666-5247(22)00093-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Gilmore MS et al. Genes Contributing to the Unique Biology and Intrinsic Antibiotic Resistance of Enterococcus faecalis. mBio 11, e02962–20 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.van Hal SJ et al. The global dissemination of hospital clones of Enterococcus faecium. Genome Med. 13, 52 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Manson AL et al. Genomic analysis of globally diverse Mycobacterium tuberculosis strains provides insights into the emergence and spread of multidrug resistance. Nat. Genet 49, 395–402 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Belland RJ, Morrison SG, Ison C & Huang WM Neisseria gonorrhoeae acquires mutations in analogous regions of gyrA and parC in fluoroquinolone-resistant isolates. Mol. Microbiol 14, 371–380 (1994). [DOI] [PubMed] [Google Scholar]
- 35.Galarza PG et al. New Mutation in 23S rRNA Gene Associated with High Level of Azithromycin Resistance in Neisseria gonorrhoeae. Antimicrob. Agents Chemother 54, 1652–1653 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Gill MJ et al. Gonococcal Resistance to β-Lactams and Tetracycline Involves Mutation in Loop 3 of the Porin Encoded at the penB Locus. Antimicrob. Agents Chemother 42, 2799–2803 (1998). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Hu M, Nandi S, Davies C & Nicholas RA High-level chromosomally mediated tetracycline resistance in Neisseria gonorrhoeae results from a point mutation in the rpsJ gene encoding ribosomal protein S10 in combination with the mtrR and penB resistance determinants. Antimicrob. Agents Chemother 49, 4327–4334 (2005). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Ito M et al. Emergence and spread of Neisseria gonorrhoeae clinical isolates harboring mosaic-like structure of penicillin-binding protein 2 in Central Japan. Antimicrob. Agents Chemother 49, 137–143 (2005). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Lindbäck E, Rahman M, Jalal S & Wretlind B Mutations in gyrA, gyrB, parC, and parE in quinolone-resistant strains of Neisseria gonorrhoeae. APMIS Acta Pathol. Microbiol. Immunol. Scand 110, 651–657 (2002). [DOI] [PubMed] [Google Scholar]
- 40.Morse SA, Johnson SR, Biddle JW & Roberts MC High-level tetracycline resistance in Neisseria gonorrhoeae is result of acquisition of streptococcal tetM determinant. Antimicrob. Agents Chemother 30, 664–670 (1986). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Ng L-K, Martin I, Liu G & Bryden L Mutation in 23S rRNA Associated with Macrolide Resistance in Neisseria gonorrhoeae. Antimicrob. Agents Chemother 46, 3020–3025 (2002). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Roberts MC et al. Erythromycin-resistant Neisseria gonorrhoeae and oral commensal Neisseria spp. carry known rRNA methylase genes. Antimicrob. Agents Chemother 43, 1367–1372 (1999). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Ropp PA, Hu M, Olesky M & Nicholas RA Mutations in ponA, the gene encoding penicillin-binding protein 1, and a novel locus, penC, are required for high-level chromosomally mediated penicillin resistance in Neisseria gonorrhoeae. Antimicrob. Agents Chemother 46, 769–777 (2002). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Sparling PF, Sarubbi FA & Blackman E Inheritance of low-level resistance to penicillin, tetracycline, and chloramphenicol in Neisseria gonorrhoeae. J. Bacteriol 124, 740–749 (1975). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Spratt BG Hybrid penicillin-binding proteins in penicillin-resistant strains of Neisseria gonorrhoeae. Nature 332, 173–176 (1988). [DOI] [PubMed] [Google Scholar]
- 46.Zhao S et al. Genetics of chromosomally mediated intermediate resistance to ceftriaxone and cefixime in Neisseria gonorrhoeae. Antimicrob. Agents Chemother 53, 3744–3751 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Hagman KE et al. Resistance of Neisseria gonorrhoeae to antimicrobial hydrophobic agents is modulated by the mtrRCDE efflux system. Microbiology 141, 611–622 (1995). [DOI] [PubMed] [Google Scholar]
- 48.Elwell LP, Roberts M, Mayer LW & Falkow S Plasmid-Mediated Beta-Lactamase Production in Neisseria gonorrhoeae. Antimicrob. Agents Chemother 11, 528–533 (1977). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Alfsnes K et al. Genomic epidemiology and population structure of Neisseria gonorrhoeae in Norway, 2016–2017. Microb. Genomics 6, e000359 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Buckley C et al. Use of whole genome sequencing to investigate an increase in Neisseria gonorrhoeae infection among women in urban areas of Australia. Sci. Rep 8, 1503 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Cehovin A et al. Identification of Novel Neisseria gonorrhoeae Lineages Harboring Resistance Plasmids in Coastal Kenya. J. Infect. Dis 218, 801–808 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Eyre David W. et al. WGS to predict antibiotic MICs for Neisseria gonorrhoeae. J. Antimicrob. Chemother 72, 1937–1947 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.de Korne-Elenbaas J, Bruisten SM, de Vries HJC & Van Dam AP Emergence of a Neisseria gonorrhoeae clone with reduced cephalosporin susceptibility between 2014 and 2019 in Amsterdam, The Netherlands, revealed by genomic population analysis. J. Antimicrob. Chemother 76, 1759–1768 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Demczuk W et al. Genomic Epidemiology and Molecular Resistance Mechanisms of Azithromycin-Resistant Neisseria gonorrhoeae in Canada from 1997 to 2014. J. Clin. Microbiol 54, 1304–1313 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Demczuk W et al. Whole-genome phylogenomic heterogeneity of Neisseria gonorrhoeae isolates with decreased cephalosporin susceptibility collected in Canada between 1989 and 2013. J. Clin. Microbiol 53, 191–200 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Didelot X et al. Genomic Analysis and Comparison of Two Gonorrhea Outbreaks. mBio 7, e00525–16 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Ezewudo MN et al. Population structure of Neisseria gonorrhoeae based on whole genome data and its relationship with antibiotic resistance. PeerJ 3, e806 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Gernert KM et al. Azithromycin susceptibility of Neisseria gonorrhoeae in the USA in 2017: a genomic analysis of surveillance data. Lancet Microbe 1, e154–e164 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Golparian D et al. Genomic epidemiology of Neisseria gonorrhoeae elucidating the gonococcal antimicrobial resistance and lineages/sublineages across Brazil, 2015–16. J. Antimicrob. Chemother 75, 3163–3172 (2020). [DOI] [PubMed] [Google Scholar]
- 60.Kwong JC et al. Whole-genome sequencing reveals transmission of gonococcal antibiotic resistance among men who have sex with men: an observational study. Sex. Transm. Infect 94, 151–157 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Lee RS et al. Genomic epidemiology and antimicrobial resistance of Neisseria gonorrhoeae in New Zealand. J. Antimicrob. Chemother 73, 353–364 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Parmar NR et al. Genomic Analysis Reveals Antibiotic-Susceptible Clones and Emerging Resistance in Neisseria gonorrhoeae in Saskatchewan, Canada. Antimicrob. Agents Chemother 64, e02514–19 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Peng J-P et al. A Whole-genome Sequencing Analysis of Neisseria gonorrhoeae Isolates in China: An Observational Study. EClinicalMedicine 7, 47–54 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Pinto M et al. Fifteen years of a nationwide culture collection of Neisseria gonorrhoeae antimicrobial resistance in Portugal. Eur. J. Clin. Microbiol. Infect. Dis 39, 1761–1770 (2020). [DOI] [PubMed] [Google Scholar]
- 65.Reimche JL et al. Genomic Analysis of the Predominant Strains and Antimicrobial Resistance Determinants Within 1479 Neisseria gonorrhoeae Isolates From the US Gonococcal Isolate Surveillance Project in 2018. Sex. Transm. Dis 48, S78 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Sánchez-Busó L et al. A community-driven resource for genomic epidemiology and antimicrobial resistance prediction of Neisseria gonorrhoeae at Pathogenwatch. Genome Med. 13, 61 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Thomas JC et al. Phylogenomic analysis reveals persistence of gonococcal strains with reduced-susceptibility to extended-spectrum cephalosporins and mosaic penA-34. Nat. Commun 12, 3801 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Town K et al. Genomic and Phenotypic Variability in Neisseria gonorrhoeae Antimicrobial Susceptibility, England. Emerg. Infect. Dis 26, 505–515 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Williamson DA et al. Bridging of Neisseria gonorrhoeae lineages across sexual networks in the HIV pre-exposure prophylaxis era. Nat. Commun 10, 3988 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Yahara K et al. Genomic surveillance of Neisseria gonorrhoeae to investigate the distribution and evolution of antimicrobial-resistance determinants and lineages. Microb. Genomics 4, (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Jamoralin MC et al. Genomic surveillance of Neisseria gonorrhoeae in the Philippines, 2013–2014. West. Pac. Surveill. Response J. WPSAR 12, 17–25 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Grad YH et al. Genomic Epidemiology of Gonococcal Resistance to Extended-Spectrum Cephalosporins, Macrolides, and Fluoroquinolones in the United States, 2000–2013. J. Infect. Dis 214, 1579–1587 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Mortimer TD & Grad YH Applications of genomics to slow the spread of multidrug-resistant Neisseria gonorrhoeae. Ann. N. Y. Acad. Sci 1435, 93–109 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Palace SG et al. RNA polymerase mutations cause cephalosporin resistance in clinical Neisseria gonorrhoeae isolates. eLife 9, e51407 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Ma KC et al. Increased power from conditional bacterial genome-wide association identifies macrolide resistance mutations in Neisseria gonorrhoeae. Nat. Commun 11, 5374 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Ma KC, Mortimer TD & Grad YH Efflux Pump Antibiotic Binding Site Mutations Are Associated with Azithromycin Nonsusceptibility in Clinical Neisseria gonorrhoeae Isolates. mBio 11, e01509–20 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Pham CD et al. Atypical Mutation in Neisseria gonorrhoeae 23S rRNA Associated with High-Level Azithromycin Resistance. Antimicrob. Agents Chemother 65, e00885–20 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Rouquette-Loughlin CE et al. Mechanistic Basis for Decreased Antimicrobial Susceptibility in a Clinical Isolate of Neisseria gonorrhoeae Possessing a Mosaic-Like mtr Efflux Pump Locus. mBio 9, e02281–18 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Wadsworth CB, Arnold BJ, Sater MRA & Grad YH Azithromycin Resistance through Interspecific Acquisition of an Epistasis-Dependent Efflux Pump Component and Transcriptional Regulator in Neisseria gonorrhoeae. mBio 9, e01419–18 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Demczuk W et al. Equations To Predict Antimicrobial MICs in Neisseria gonorrhoeae Using Molecular Antimicrobial Resistance Determinants. Antimicrob. Agents Chemother (2019) doi: 10.1128/AAC.02005-19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Ashford WinstonA., Golash RomanG. & Hemming ValG. Penicillinase-producing Neisseria gonorrhoeae. The Lancet 308, 657–658 (1976). [DOI] [PubMed] [Google Scholar]
- 82.Fingerhuth SM, Low N, Bonhoeffer S & Althaus CL Detection of antibiotic resistance is essential for gonorrhoea point-of-care testing: a mathematical modelling study. BMC Med. 15, 142 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Tuite AR et al. Impact of Rapid Susceptibility Testing and Antibiotic Selection Strategy on the Emergence and Spread of Antibiotic Resistance in Gonorrhea. J. Infect. Dis 216, 1141–1149 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Vegvari C et al. Using rapid point-of-care tests to inform antibiotic choice to mitigate drug resistance in gonorrhoea. Eurosurveillance 25, 1900210 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Donà V, Low N, Golparian D & Unemo M Recent advances in the development and use of molecular tests to predict antimicrobial resistance in Neisseria gonorrhoeae. Expert Rev. Mol. Diagn 17, 845–859 (2017). [DOI] [PubMed] [Google Scholar]
- 86.Marlowe EM et al. Evaluation of the Cepheid Xpert MTB/RIF Assay for Direct Detection of Mycobacterium tuberculosis Complex in Respiratory Specimens. J. Clin. Microbiol 49, 1621–1623 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Tenover FC et al. Updating Molecular Diagnostics for Detecting Methicillin-Susceptible and Methicillin-Resistant Staphylococcus aureus Isolates in Blood Culture Bottles. J. Clin. Microbiol 57, e01195–19 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Hadad R et al. Evaluation of the SpeeDx ResistancePlus® GC and SpeeDx GC 23S 2611 (beta) molecular assays for prediction of antimicrobial resistance/susceptibility to ciprofloxacin and azithromycin in Neisseria gonorrhoeae. J. Antimicrob. Chemother 76, 84–90 (2021). [DOI] [PubMed] [Google Scholar]
- 89.Hicks AL et al. Evaluation of parameters affecting performance and reliability of machine learning-based antibiotic susceptibility testing from whole genome sequencing data. PLOS Comput. Biol 15, e1007349 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Mortimer TD, Zhang JJ, Ma KC & Grad YH Loci for prediction of penicillin and tetracycline susceptibility in Neisseria gonorrhoeae: a genome-wide association study. Lancet Microbe 3, e376–e381 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Břinda K et al. Rapid inference of antibiotic resistance and susceptibility by genomic neighbour typing. Nat. Microbiol 5, 455–464 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Drouin A et al. Interpretable genotype-to-phenotype classifiers with performance guarantees. Sci. Rep 9, 4071 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Anahtar MN, Yang JH, Jason H Yang & Kanjilal, S. Applications of Machine Learning to the Problem of Antimicrobial Resistance: an Emerging Model for Translational Research. J. Clin. Microbiol 59, (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Lüftinger L et al. Learning From Limited Data: Towards Best Practice Techniques for Antimicrobial Resistance Prediction From Whole Genome Sequencing Data. Front. Cell. Infect. Microbiol 11, 610348–610348 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Kim JI et al. Machine Learning for Antimicrobial Resistance Prediction: Current Practice, Limitations, and Clinical Perspective. Clin. Microbiol. Rev 0, e00179–21 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Luo H et al. Development and application of Cas13a-based diagnostic assay for Neisseria gonorrhoeae detection and azithromycin resistance identification. J. Antimicrob. Chemother 77, 656–664 (2022). [DOI] [PubMed] [Google Scholar]
- 97.Ackerman CM et al. Massively multiplexed nucleic acid detection with Cas13. Nature 582, 277–282 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.U.S. Food and Drug Administration. Antimicrobial Susceptibility Test (AST) Systems - Class II Special Controls Guidance for Industry and FDA. FDA; (2018). [Google Scholar]
- 99.Lan PT et al. Genomic analysis and antimicrobial resistance of Neisseria gonorrhoeae isolates from Vietnam in 2011 and 2015–16. J. Antimicrob. Chemother 75, 1432–1438 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Harris SR et al. Public health surveillance of multidrug-resistant clones of Neisseria gonorrhoeae in Europe: a genomic survey. Lancet Infect. Dis 18, 758–768 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Centers for Disease Control and Prevention. CDC Pilots Drug-resistant Gonorrhea Testing. Centers for Disease Control and Prevention; https://www.cdc.gov/drugresistance/solutions-initiative/stories/pilot-expands-gonorrhea-testing.html (2022). [Google Scholar]
- 102.CLSI. Performance Standards for Antimicrobial Susceptibility Testing. 28th ed. CLSI supplement M100. Clinical & Laboratory Standards Institute; https://clsi.org/standards/products/microbiology/documents/m100/ (2018). [Google Scholar]
- 103.Shaskolskiy B et al. Prediction of ceftriaxone MIC in Neisseria gonorrhoeae using DNA microarray technology and regression analysis. J. Antimicrob. Chemother 76, 3151–3158 (2021). [DOI] [PubMed] [Google Scholar]
- 104.Greener JG, Kandathil SM, Moffat L & Jones DT A guide to machine learning for biologists. Nat. Rev. Mol. Cell Biol 23, 40–55 (2022). [DOI] [PubMed] [Google Scholar]
- 105.Lees JA, Galardini M, Bentley SD, Weiser JN & Corander J pyseer: a comprehensive tool for microbial pangenome-wide association studies. Bioinforma. Oxf. Engl 34, 4310–4312 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Earle SG et al. Identifying lineage effects when controlling for population structure improves power in bacterial association studies. Nat. Microbiol 1, 1–8 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Feil EJ et al. Recombination within natural populations of pathogenic bacteria: short-term empirical estimates and long-term phylogenetic consequences. Proc. Natl. Acad. Sci. U. S. A 98, 182–187 (2001). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Feil EJ & Spratt BG Recombination and the population structures of bacterial pathogens. Annu. Rev. Microbiol 55, 561–590 (2001). [DOI] [PubMed] [Google Scholar]
- 109.EUCAST. EUCAST: Clinical breakpoints and dosing of antibiotics. https://www.eucast.org/clinical_breakpoints/ (2022).
- 110.British Association for Sexual Health and HIV. BASHH Guidelines. British Association for Sexual Health and HIV; https://www.bashh.org/guidelines/. [Google Scholar]
- 111.Altman DG & Royston P The cost of dichotomising continuous variables. BMJ 332, 1080 (2006). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Reshetnikov KO et al. Feature selection and aggregation for antibiotic resistance GWAS in Mycobacterium tuberculosis: a comparative study. bioRxiv 2022.03.16.484601 (2022) doi: 10.1101/2022.03.16.484601. [DOI] [Google Scholar]
- 113.Zabeti H et al. INGOT-DR: an interpretable classifier for predicting drug resistance in M. tuberculosis. Algorithms Mol. Biol 16, 17 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Feleke SM et al. Plasmodium falciparum is evolving to escape malaria rapid diagnostic tests in Ethiopia. Nat. Microbiol 6, 1289–1299 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Unemo M et al. The Swedish new variant of Chlamydia trachomatis: genome sequence, morphology, cell tropism and phenotypic characterization. Microbiol. Read. Engl 156, 1394–1404 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Whiley DM et al. False-negative results using Neisseria gonorrhoeae porA pseudogene PCR - a clinical gonococcal isolate with an N. meningitidis porA sequence, Australia, March 2011. Euro Surveill. 16, 19874 (2011). [PubMed] [Google Scholar]
- 117.Ripa T & Nilsson P A variant of Chlamydia trachomatis with deletion in cryptic plasmid: implications for use of PCR diagnostic tests. Euro Surveill. 11, E061109.2 (2006). [DOI] [PubMed] [Google Scholar]
- 118.Guglielmino CJD, Appleton S, Vohra R & Jennison AV Identification of an Unusual 16S rRNA Mutation in Neisseria gonorrhoeae. J. Clin. Microbiol 57, e01337–19 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Hicks AL, Kissler SM, Lipsitch M & Grad YH Surveillance to maintain the sensitivity of genotype-based antibiotic resistance diagnostics. PLOS Biol. 17, (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Lin EY, Adamson PC, Ha S & Klausner JD Reliability of Genetic Alterations in Predicting Ceftriaxone Resistance in Neisseria gonorrhoeae Globally. Microbiol. Spectr 10, e02065–21. [DOI] [PMC free article] [PubMed] [Google Scholar]