

Abstract
Inorganic Arsenic (iAs) is one of the largest toxic exposures to impact humanity worldwide. Exposure to iAs during pregnancy may disrupt the proper remodeling of the epigenome of F1 developing offspring and potentially their F2 grand-offspring via disruption of fetal primordial germ cells (PGCs). There is a limited understanding between the correlation of disease phenotype and methylation profile within offspring of both generations and whether it persists to adulthood. Our study aims to understand the intergenerational effects of in utero iAs exposure on the epigenetic profile and onset of disease phenotypes within F1 and F2 adult offspring, despite the life-long absence of direct arsenic exposure within these generations. We exposed F0 female mice (C57BL6/J) to the following doses of iAs in drinking water 2 weeks before pregnancy until the birth of the F1 offspring: 1 ppb, 10 ppb, 245 ppb, and 2300 ppb. We found sex- and dose-specific changes in weight and body composition that persist from early time to adulthood within both generations. Fasting blood glucose challenge suggests iAs exposure causes dysregulation of glucose metabolism, revealing generational, exposure, and sex specific differences. Toward understanding the mechanism, genome-wide DNA methylation data highlights exposure-specific patterns in liver, finding dysregulation within genes associated with cancer, T2D, and obesity. We also identified regions containing persistently differentially methylated CpG sites between F1 and F2 generations. Our results indicate the F1 developing embryos and their PGCs, which will result in F2 progeny, retain epigenetic damage established during the prenatal period and are associated with adult metabolic dysfunction.
Keywords: DNA Methylation, DOHaD, Arsenic, Intergenerational, Mouse, Germ Cell
1. Introduction
Arsenic (iAs) exposure via drinking water is one of the largest global exposures affecting prenatal health, and has a growing body of evidence that suggests gestational iAs exposure influences intergenerational epigenetic inheritance(National Research Council (U.S.). Subcommittee on Arsenic in Drinking Water. 1999; Hossain et al. 2017; Nohara et al. 2011; Nohara, Suzuki, and Okamura 2020). Data from mother-infant pairs show prenatal iAs exposure alters DNA methylation(Xie et al. 2007; Zhao et al. 2002; Tsang et al. 2012) and birth outcomes(Marie et al. 2018; Laine et al. 2015; Gilbert-Diamond et al. 2016; Fei et al. 2013) in newborns. Studies using rodent models find prenatal iAs exposure causes type 2 diabetes (T2D)(Young, Cai, and States 2018; Liu et al. 2014), obesity(Rodriguez et al. 2016b; C et al. 2019), and differential methylation(Martin, Stýblo, and Fry 2017; Rojas et al. 2015; Bailey et al. 2013) in adult mice. However, most rodent studies use doses of iAs that are considered carcinogenic(“TOXICOLOGICAL PROFILE FOR ARSENIC | Enhanced Reader,” n.d.) thus the role of methylation and disease may be skewed, and methylation data reported from epidemiological studies are confounded by co-exposures. Due to its prevalence as a health threat in the United States and around the world, it is important to characterize the multigenerational epigenetic damage caused by iAs exposure and its long-term health effects in adult offspring. However, there are no studies to date investigating the intergenerational impact of maternal iAs exposure inherited through the female germline.
Epigenetic inheritance occurs by escape from complete epigenetic reprogramming. Epigenetic reprogramming is critical for regulation of gene expression and tissue differentiation in the developing embryo, and imperative for sex differentiation within primordial germ cells (PGCs)(Feng, Jacobsen, and Reik 2010; Cedar and Bergman 2012). Reprogramming occurs during early and late embryogenesis, where DNA methylation is erased then re-established in two waves of demethylation/re-methylation. The first reprogramming event takes place within the primordial germ cells (F2, PGCs that result in the F2 progeny) of F1 offspring, and the second in the somatic F1 post-fertilization pluripotent stem cells(Sasaki and Matsui 2008). Since the epigenome is vulnerable during development, disruption of epigenetic reprogramming by environmental exposures impacts both the developing zygote (F1 generation) and the PGCs (F2 generation), and is known to cause the onset of disease in subsequent generations(Painter et al. 2008; Skinner and Guerrero-Bosagna 2014; Titus-Ernstoff et al. 2008). When the F1 PGCs develop into oocytes that will give rise to the F2 progeny, become fertilized, and undergo the waves of somatic demethylation/re-methylation as the developing F2 zygote during pregnancy, it is unclear if grand-maternal epigenetic damage caused by in utero iAs exposure will persist in adult F2s, despite the reprogramming event. Thus, we designed an exposure paradigm to 1) identify the intergenerational epigenetic scarring caused by in utero iAs exposure and 2) assess the contribution to the onset of disease in adulthood using a mouse model.
The epigenetic damage of iAs exposure during pregnancy is hypothesized to be associated with iAs metabolism and the 1-carbon pathway. iAs metabolism acts as an inhibitor of DNA methylation due to competition for DNA methylation precursors. Biotransformation of iAs utilizes the one carbon metabolism pathway, the same metabolic pathway that generates methyl groups for the synthesis of DNA. The universal methyl donor, S-aenosyl-l-methione (SAM), is utilized by iAs-3-methyltransferase (AS3MT) to generate metabolites monomethylarsonic acid (MMA) and dimethylarsonic acid (DMA) for urinary excretion(Spratlen et al. 2017). SAM is also used by DNMTs to add methyl groups to convert unmodified cytosines to 5-methylcytosine (5-mC)(Thomas, Waters, and Styblo 2004). Since both AS3MT and DNMT1 compete for SAM, evidence suggests utilization of SAM is responsible for the lower global methylation changes seen in exposed individuals(Nohara et al. 2011; Zhao et al. 2002; Xie et al. 2007). Thus, in utero iAs exposure has the potential to disrupt normal methylation patterning during epigenetic reprogramming.
We hypothesized the disruption of DNA methylation by in utero iAs exposure would target both the developing fetus (F1) and primordial germ cell (F2) epigenomes leading to the onset of adulthood diseases. We exposed F0 females to human relevant doses of iAs throughout pregnancy that represented the WHO limit (10 ppb)(National Research Council (U.S.). Subcommittee on Arsenic in Drinking Water. 1999), the highest average global exposure (245 ppb) (Argos 2015), and the known prenatal carcinogenic exposure in mice (2300 ppb)(Waalkes et al. 2004). Our gestational exposure window was chosen to restrict the impact of iAs to development and causally link early exposure to later-in-life metabolic diseases. We assess the effects of maternal in utero iAs exposure on eight, total body fat, behavior, and glucose tolerance at multiple time points into adulthood in both sexes of the F1 and F2 generations. We found arsenic causes physiological metabolic changes in a sex-, dose-, and generation-specific manner. Further, we found differentially methylated CpGs (DMCs) and differentially methylated regions (DMRs) that were unique or persisted between sex, dose, and generations, identifying DMCs within genes associated T2D or obesity.
2. Materials and Methods
Mouse model, exposure, and breeding paradigm
F0 C57BL/6j mice (n=16, 8 weeks of age) were purchased from Jackson Laboratory (Bar Harbor, Maine). Mice were maintained on a 12:12 light/dark cycle and had ad libitum access to the standard lab diet Envigo 2018 (18% protein) chow and tap water (tested at 1ppb iAs) until the beginning of the exposure. All animals were individually housed until breeding. Tap water from the University of Minnesota was treated with iAsIII (Millipore Sigma) at the following doses: 10 ppb, 245 ppb, and 2300 ppb. Water quality and arsenic concentrations were tested at Tri-City/William Lloyd Analytical Lab (Bloomington, MN). Female mice from the F1 generation (n=4 per treatment), were used to produce the F2 generation. Mice used for glucose tolerance were selected at random for the initial 8 week measurement and remained enrolled for glucose tolerance at the 25 and 38 week time points throughout the study. The following quantity of mice survived the 40 weeks without health confounding factors, and were used for behavior testing and weekly body weight; (F1 1 ppb: male n= 10, female n=7; F1 10 ppb: male n=9, female n = 4; F1 245 ppb: male n= 8, female n=4; F1 2300 ppb: male n=7, female n=7; F2 1 ppb: male n=10, female n=8; F2 10 ppb: male n=10, female n=12; F2 245 ppb: male n=10, female n=11; F2 2300: male n=6, female n=5). Total mice, litter size, and sex distribution are found in Table 1. Mice were treated humanely and in accordance with the Guidelines for the Care and Use of Laboratory Animals (Institute of Laboratory Animal Resources, 1996). The study protocol was approved by the University of Minnesota Institutional Animal Care and Use Committee (IACUC).
Table 1. F1 and F2 Litter Size.
Reported litter size for F1 and F2 generation.
| Total | Total | ||||||
|---|---|---|---|---|---|---|---|
| 1 ppb Breeder 1 | 1 | 2 | 3 | 1 ppb Breeder 1 | 2 | 1 | 3 |
| 1 ppb Breeder 2 | 2 | 3 | 5 | 1 ppb Breeder 2 | 3 | 4 | 7 |
| 1 ppb Breeder 3 | 4 | 2 | 6 | 1 ppb Breeder 3 | 5 | 3 | 8 |
| 1 ppb Breeder 4 | 3 | 4 | 7 | 1 ppb Breeder 4 | - | - | - |
| 10 ppb Breeder 1 | 4 | 3 | 7 | 10 ppb Breeder 1 | 2 | 4 | 6 |
| 10 ppb Breeder 2 | 4 | 3 | 7 | 10 ppb Breeder 2 | 4 | 5 | 9 |
| 10 ppb Breeder 3 | 1 | 2 | 3 | 10 ppb Breeder 3 | 4 | 3 | 7 |
| 245 ppb Breeder 1 | 4 | 3 | 7 | 245 ppb Breeder 1 | 1 | 5 | 6 |
| 245 ppb Breeder 2 | 3 | 3 | 6 | 245 ppb Breeder 2 | 4 | 4 | 8 |
| 245 ppb Breeder 3 | 0 | 3 | 3 | 245 ppb Breeder 3 | 5 | 2 | 7 |
| 2300 ppb Breeder 1 | 4 | 2 | 6 | 2300 ppb Breeder 1 | 2 | 4 | 6 |
| 2300 ppb Breeder 2 | 2 | 6 | 8 | 2300 ppb Breeder 2 | 4 | 1 | 5 |
| 2300 ppb Breeder 3 | 1 | 3 | 4 | 2300 ppb Breeder 3 | - | - | - |
Glucose Tolerance Testing
At ages 8, 25, and 38 weeks, mice (n=4 per treatment) underwent a 6 hour fast before receiving a 100 μL glucose bolus by oral gavage. Blood glucose was monitored before glucose administration (0 minutes) and recorded every 15 minutes for 1.5 hours by Accu-Check Blood Glucose Meter (Roche Diabetes Care, Inc.). Statistical significance was determined by a 2-way ANOVA (p <0.05), and Area Under the Curve (AUC) was calculated using the AUC function in Prism 9.
Behavior Testing
To assess anxiety and compulsive behavior, we performed marble burying in a standard marble burying condition. A set of 20 glass marbles were evenly spaced in a rat cage on sawdust bedding. For the duration of 20 minutes, mice explored and buried marbles undisturbed. Marbles that were at least two-thirds covered by corn cob bedding at the end of the testing period were counted as buried. The number of marbles buried was scored by a single skilled observer; inter-group differences were assessed by one-way ANOVA (p-value < 0.05).
To assess spatial working memory and cognitive integrity, we performed a Y-maze behavior test(“Purpose and Cognition: The Determiners of Animal Learning.” n.d.; Hughes 2004). Mice were placed in one arm of a standard Y-maze (MazeEngineers, Boston, MA), consisting of a high walled chamber with three arms connected at 120°. Mice were video recorded exploring the maze undisturbed for 10 minutes. After each test, the maze was cleaned with 70% ethanol and then water to eliminate confounding scents for the subsequent trials. The total number of maze arm entries and spontaneous alterations (SA) were scored by a treatment-blinded skilled observer. Spontaneous alternation percentage was calculated as the number of spontaneous alterations ÷ (number of entries −2) × 100. One spontaneous alternation was counted when three consecutive entries into unique arms (e.g., A, B, C) were recorded on video. Arm entries were recorded when the test mouse was positioned with all four feet inside the maze arm. The total number of arm entries was also recorded as a measure of exploration. Results were determined significant by a one-way ANOVA (p-value < 0.05).
Body Composition
After weaning (3 weeks of age), F1 and F2 mice were weighed weekly for 40 weeks. Final body weight was recorded at the time of euthanasia. Wet weight was recorded for adipose deposits on the right abdomen and the left kidney. To determine differences in body composition, ratios of bodyweight to adipose mass (body weight/fat pad mass) and bodyweight to kidney mass (body weight/kidney weight) were calculated and compared using a one-way ANOVA (p-value < 0.05).
DNA Extractions and RRBS Data Analysis
Reduced Represented Bisulfite Sequencing (RRBS) Genomic DNA from F1 and F2 liver were isolated using the Zymo Quick-DNA Mini Prep Kit (Zymo Research Corp.). Upon submission, the gDNA samples were quantified using a fluorometric PicoGreen assay and analyzed for quality using the Nanodrop. All sampled passed the Input DNA Requirements outlines in Tecan’s Ovation RRBS Methyl-Seq System (Publication Number: M01394v7). gDNA samples were converted to sequencing libraries using Tecan’s Ovation RRBS Methyl-Seq with TrueMethyl oxBS System following manufacturers protocols (Part # 9522-A01). In summary, extracted gDNA was digested by MspI and then ligated to indexed sequencing adapters. Libraries were bisulfite converted (oxBS module was not used) and amplified. Final library size distribution was validated using capillary electrophoresis and quantified using fluorimetry (PicoGreen). Libraries were pooled and sequenced on one lane of an S4 2×150 paired-end run on the Illumina NovaSeq 6000 System.
R (R Core Team (2021). R: A language and environment for statistical computing. R Foundation for Statistical Computing, Vienna, Austria. https://www.R-project.org/.) and Linux command line tools were used for RRBS analysis. FastQC (version11.3) assessed overall sequencing quality, and TrimGalore(F. Krueger 2015) (version 4.5) with default settings trimmed low-quality bases, adapter sequences, and end-repair bases from 3’ ends. Bismark(Felix Krueger and Andrews 2011) (version 19.0) aligned and called methylation. Reads were aligned to mm10 mouse reference genome using Bowtie2(Langmead and Salzberg 2012) (version 2.3.4) using default parameters. Methylation calls were reported for all nucleotides with the minimum read depth of 5. Paired raw fastq.gz reads were uploaded to the Sequence Read Arcive and can be accesed under Bioproject Number (PRJNA881737). The DSS package in R (version 2.32.0) detected differential methylation in liver for F1 and F2 females versus sex- and generation- matched 1ppb controls (n = 3 per group) for sites with a minimum coverage of 10 reads. The DMLtest and callDML functions were used to detect differentially methylated CpGs (DMCs). Smoothing was set to FALSE in DMLtest as is recommended for sparse data. Differentially methylated regions (DMRs) were identified with callDMR. The p-value threshold was set to FDR < 0.001 for both tests with delta > 0.01 required to call DMCs. A delta value was not enforced for DMRs; all CpGs within a potential DMR need not exhibit the same direction of change, meaning a delta threshold could lead to false negatives. The Annotatr R package was used to annotate DMCs and DMRs to CpG islands, genes, and intergenic regions in the mm10 genome(Cavalcante and Sartor 2016). The functions plot_annotation and plot_categorical functions were used to generate figures.
3. Results
3.1. Maternal iAs exposure alters wean weights, adult weight gain, and final body weight ratios in of F1 offspring
To understand the effects of in utero iAs exposure on the developing embryo and primordial germ cells, we exposed F0 females to iAs treated water for 2 weeks prior to mating (Figure 1). To ensure intergenerational effects were passed through the maternal line, unexposed male mice were placed in female cages to generate the F1 population. Male mice were separated from females after 48 hours to avoid confounding paternal exposure. Treated water was removed within 24 hours after the F1 offspring were born. F1 mice were weaned at 3 weeks of age and weekly weights were recorded until 40 weeks of age. F1 female mice (n=4 per treatment, 8 weeks of age) were bred with unexposed, unrelated C57BL/6J male mice (8 weeks of age) to produce the F2 generation. F2 mice were weaned at 3 weeks of age. None of the F1 or F2 mice were directly exposed to iAs drinking water during their lifetime.
Figure 1. Breeding and exposure paradigm.
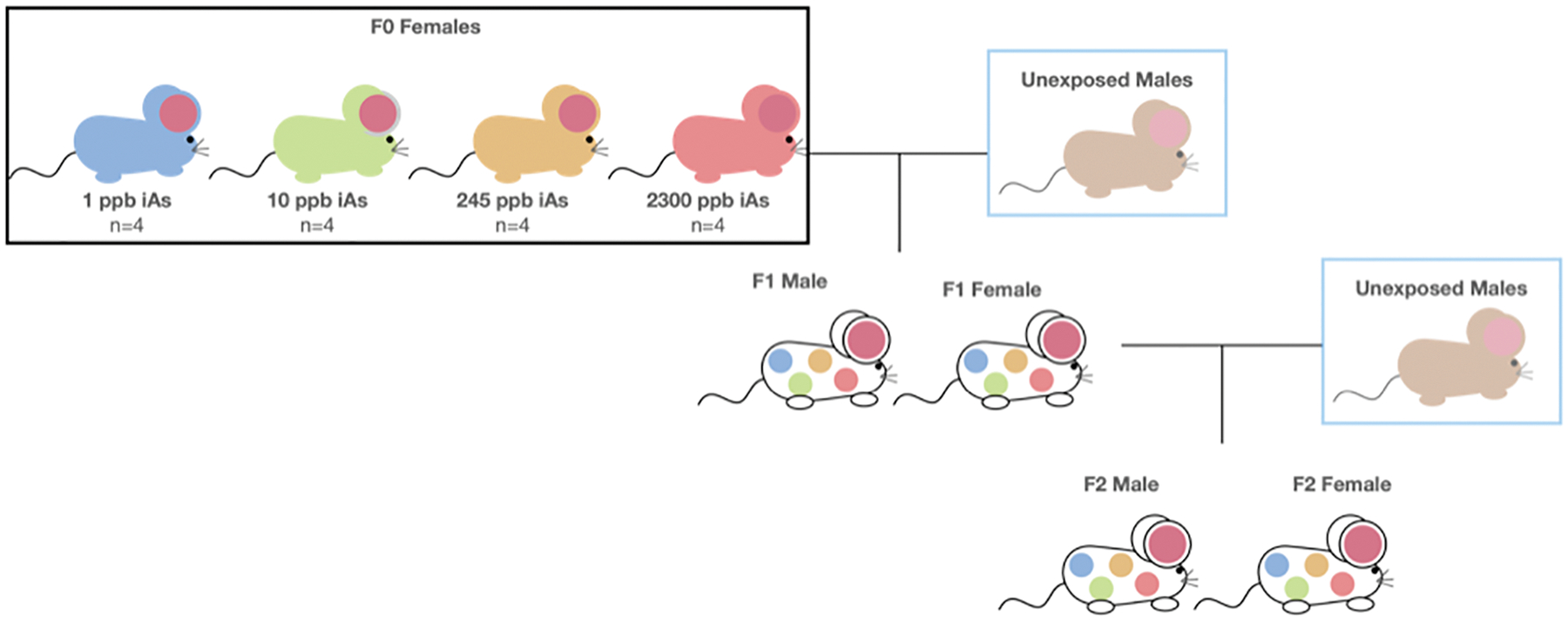
F0 females (n=4 per treatment) were exposed to iAs treated water two weeks prior to breeding and throughout pregnancy. F0 females were bred with unexposed males to produce the F1 generation. F1 females were mated with unexposed males to produce the F2 generation. F1 and F2 mice did not consume iAs treated water.
Male and female F1 mice born to mothers exposed to 10 ppb and 2300 ppb had lower wean weights compared to control, where there were no significant differences in body weight to fat pad mass raio (Figure 2.A–E). Over a period of 40 weeks, male F1 offspring from 10 ppb and 2300 ppb dams had lower final body weight compared to the control average (Figure 2.C). Despite low final body weight, F1 male mice had no significant difference in weight gain for the 40 weeks, or body weight to fat pad mass ratio (Figure 2.D, Figure 2.E). Thus, maternal exposure to iAs at three ranges alters wean weight and final body weight in a sex-specific manner.
Figure 2. F1 Body weight and composition.
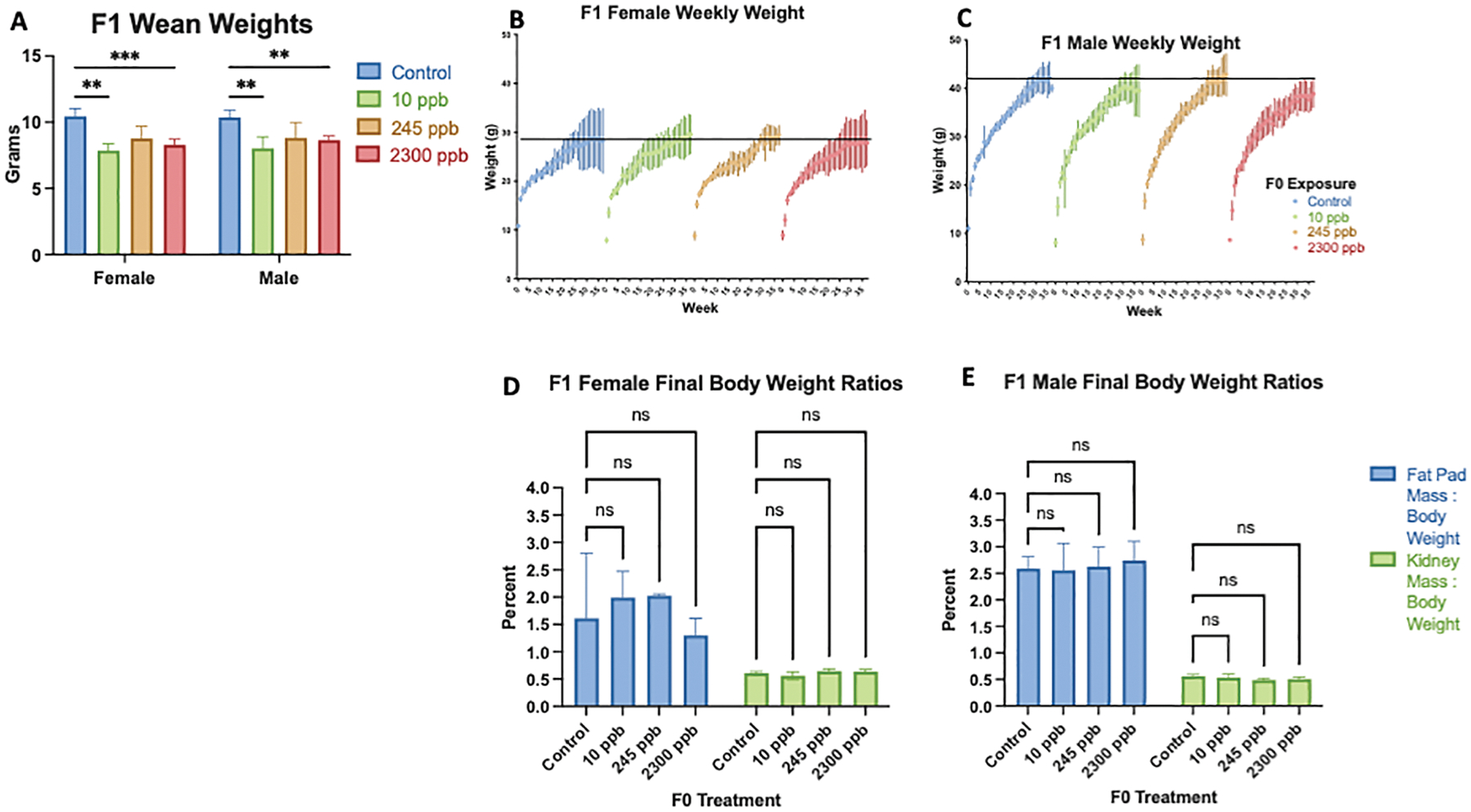
A) Wean weight of in utero exposed F1 mice were compared to F1 control mice. B) F1 Female weekly weight over a 40 week time period C) F1 Male weekly weight over a 40 week time period. Fat pad mass to body weight ratio and kidney mass to body weight ratio percentages at 40 weeks of age. C) Final body composition of F1 female mice. D) Final body composition of F1 male mice. All data were compared with control, presented as mean ±SEM and determined significant by a one-way ANOVA ***p<0.0001; standard error of mean. (F1 1 ppb: male n= 10, female n=7; F1 10 ppb: male n=9, female n = 4; F1 245 ppb: male n= 8, female n = 4; F1 2300 ppb: male n=7, female n=7)
3.2. F1 Females, but not males, show delayed glucose response similar to type II diabetes
Blood glucose was measured at three lifetime points (8, 25, and 38 weeks) to understand if in utero iAs exposure causes type II diabetes (T2D) associated phenotypes in F1 offspring (Figure 3.A and Figure 3.B). Throughout the study, F1 female offspring of 10 ppb dams exhibited consistent (i.e. multi-timepoint) dysregulated metabolism of glucose during the 25- and 38-week oral glucose challenges (Figure 3.A). F1 male mice exhibited no differences in glucose metabolism (Figure 3.B). No significant differences were identified in Area Under the Curve (AUC) analysis. Our data indicate a low exposure of 10 ppb iAs during pregnancy contributes to irregular metabolism of glucose in late life F1 females.
Figure 3. Blood Glucose Metabolism in F1 Female and Male Mice.
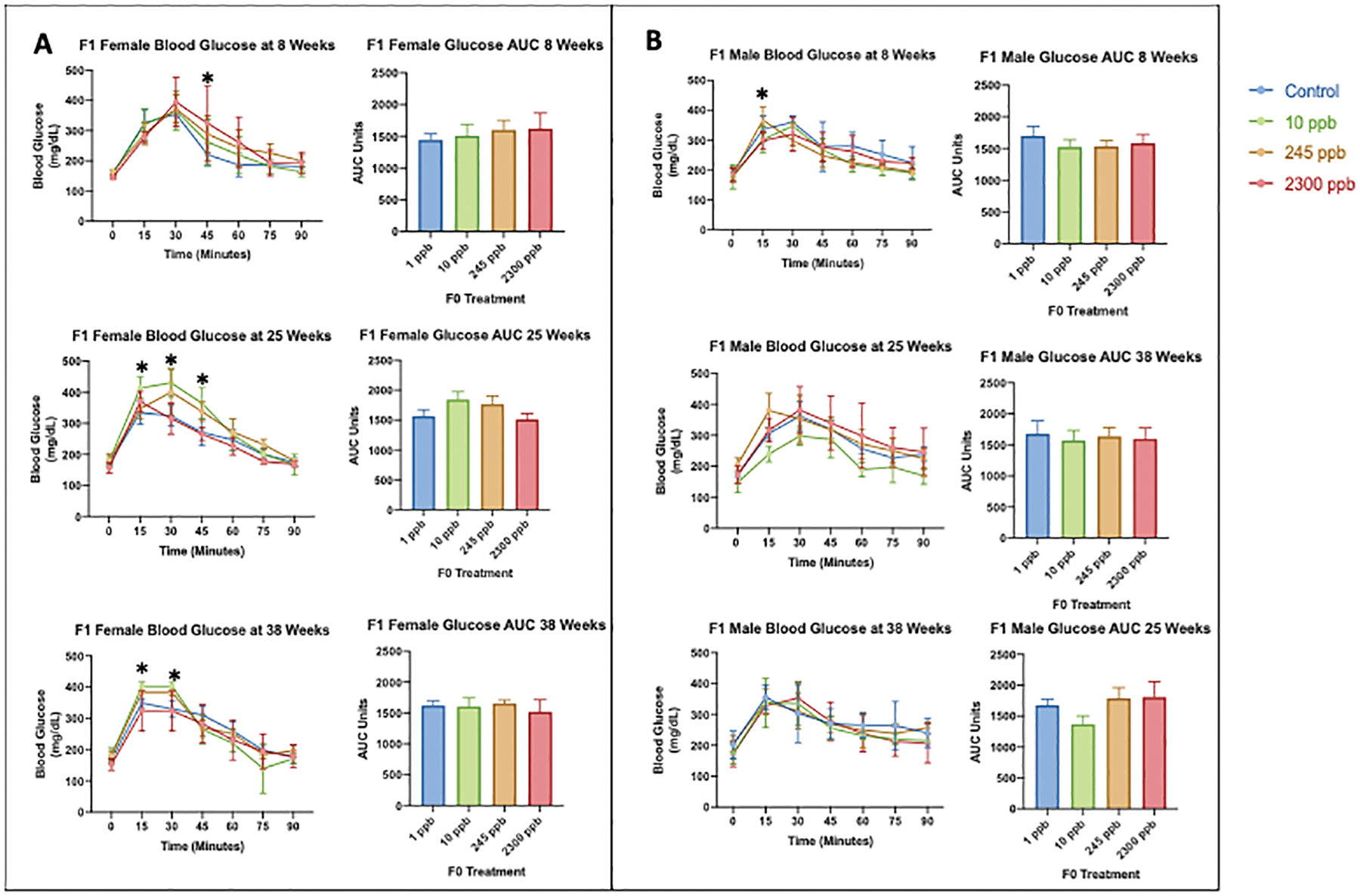
At ages 8, 25, and 38 weeks, the glucose tolerance test was preformed after a 6 hour fasting period followed by an oral administration of glucose. Tail vein blood glucose concentrations were measured at 0, 15, 30, 45, 60, 75, and 90 minutes A) F1 female mouse blood glucose data with Area Under the Curve analysis B) F1 male blood glucose data with Area Under the Curve Analysis. All exposed data were compared to the control values to determine significant differences. Statistical significance was determined using a two-way ANOVA *p<0.05; Standard Error of Mean (SEM) (n=4 per treatment, per sex)
3.3. Grandmaternal iAs exposure alters life-long body composition in male and female F2 offspring
Wean weights, weekly body weight, and final body composition in F2 offpsring were altered by grandmaternal exposure (Figure 4.A–E). Wean weights in F2 females were normal, whereas F2 10 ppb male mice had significantly higher wean weight compared to the F2 control (Figure 4.B and Figure 4.C). F2 females had no significant differences in body weight through 40 weeks of age (Figure 4.B). Remarkably, F2 male mice in all treatment groups had significantly lower body weight compared to control starting at week 28 that persisted through 40 weeks of age (Figure 4.C). Similar results were seen in body mass to fat pad mass ratios (Figure 4.D and Figure 4.E). No significant differences were seen in F2 female final body composition (Figure 4.D). Consistent with low body weight, F2 males in the 10 ppb and 2300 ppb iAs exposure groups had significantly lower body mass to fat pad mass ratio (Figure 4.E).
Figure 4. F2 Body weight and composition.
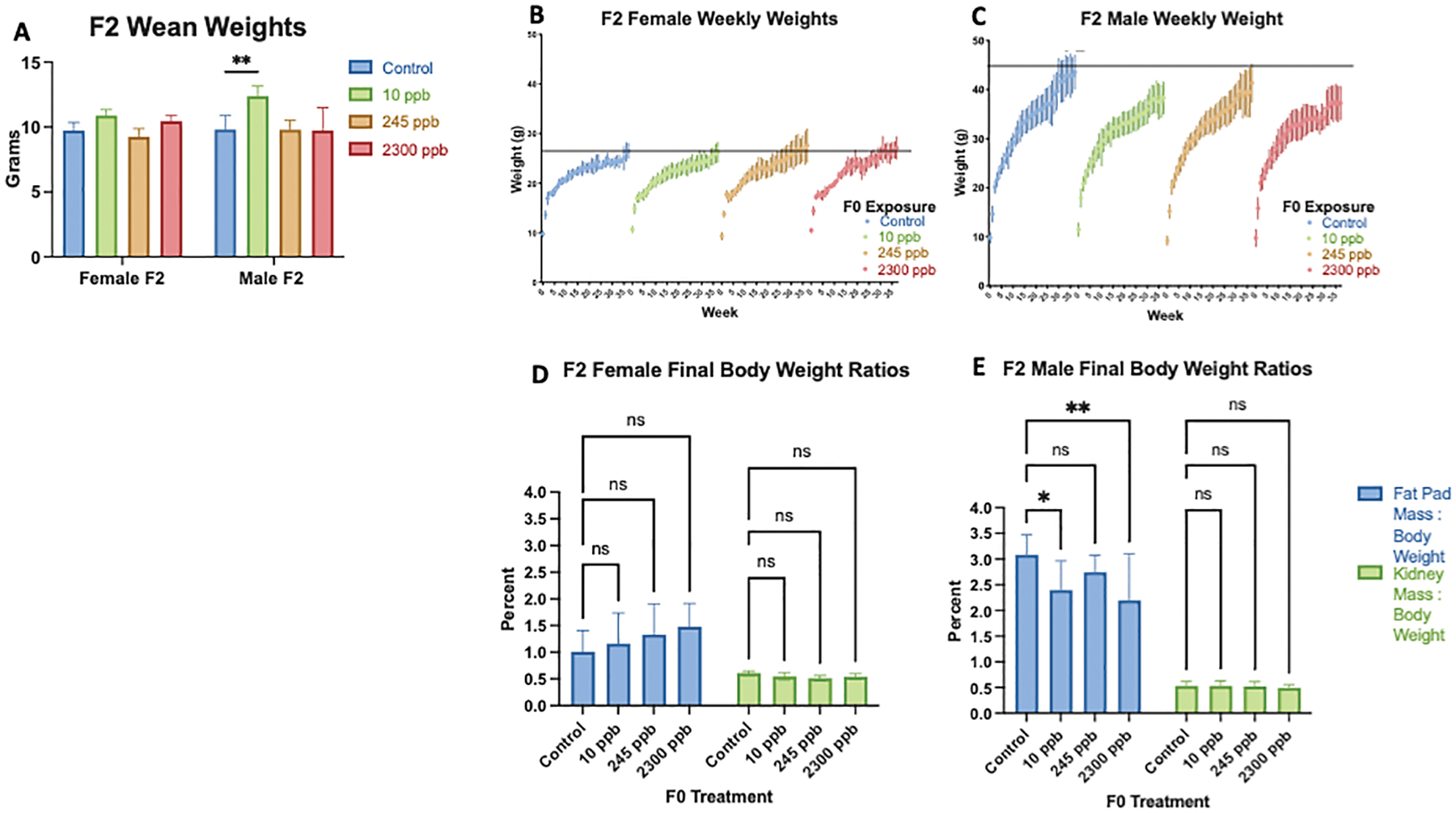
A) Wean weight of in utero exposed F2 mice were compared to F2 control mice. B) F2 Female weekly weight over a 40 week time period C) F2 Male weekly weight over a 40 week time period. Fat pad mass to body weight ratio and kidney mass to body weight ratio percentages at 40 weeks of age. C) Final body composition of F2 female mice. D) Final body composition of F2 male mice. All data were compared to control, presented as mean ±SEM, and were calculated as significantly different by a on–way ANOVA *p<0.05. **p<0.01; standard error of mean. (F2 1 ppb: male n=10, female n=8; F2 10 ppb: male n=10, female n=12; F2 245 ppb: male n=10, female n=11; F2 2300: male n=6, female n=5).
3.4. Dysregulated glucose metabolism in adult F2 male and F2 female mice
We measured blood glucose at three lifetime points (8, 25, and 38 weeks) within the F2 exposure groups to understand the effects of grandmaternal exposure on adult F2 diabetic phenotypes (Figure 5.A and Figure 5.B). The F0 exposure of 245 ppb causes a T2D phenotype in F2 female mice starting at an early age (3 weeks) that is persistent through adulthood (38 weeks) (Figure 5.A). In addition, F2 male mice developed dysregulated glucose metabolism only in late life (38 weeks) (Figure 5.B). We identified no significant changes in AUC analysis. These results indicate primordial germ cells are damaged by grandmaternal iAs exposure and result in adult metabolic disruption that is dose-specific.
Figure 5. Blood Glucose Metabolism in F2 Female and Male Mice.
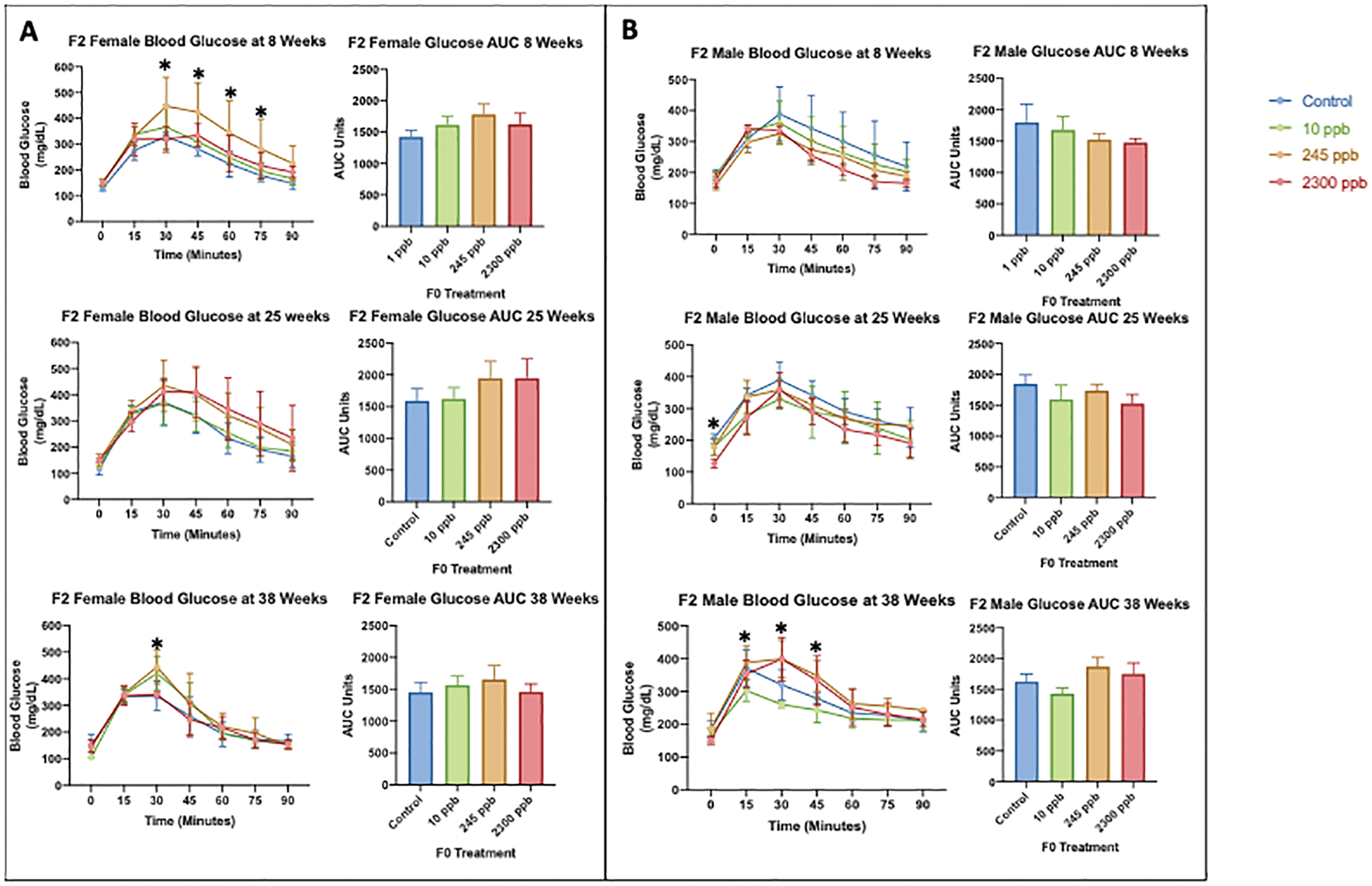
At ages 8, 25, and 38 weeks, the glucose tolerance test was preformed after a 6 hour fasting period followed by an oral administration of glucose. Tail vein blood glucose concentrations were measured at 0, 15, 30, 45, 60, 75, and 90 minutes A) F2 female mouse blood glucose data and Area Under the Curve analysis B) F2 male blood glucose data and Area Under the Curve Analysis. All exposed data were compared to the control values to determine significant differences. Statistical significance was calculated by a
3.5. Maternal iAs exposure does not influence anxiety or memory behaviors in F1 or F2 offspring
We performed marble burying to determine whether gestational iAs exposure induced anxiety and a Y-maze assessment for spatial memory within adult F1 and F2 mice (Figure 6.A and Figure 6.B). We identified no significant differences between exposure groups and control for either generation in both the marble burying and spontaneous alteration behavior in the Y-maze. These results suggest that in utero iAs exposure does not cause anxiety or impair spatial memory in F1 and F2 adult mice.
Figure 6. F1 Male and Female Behavior Data.
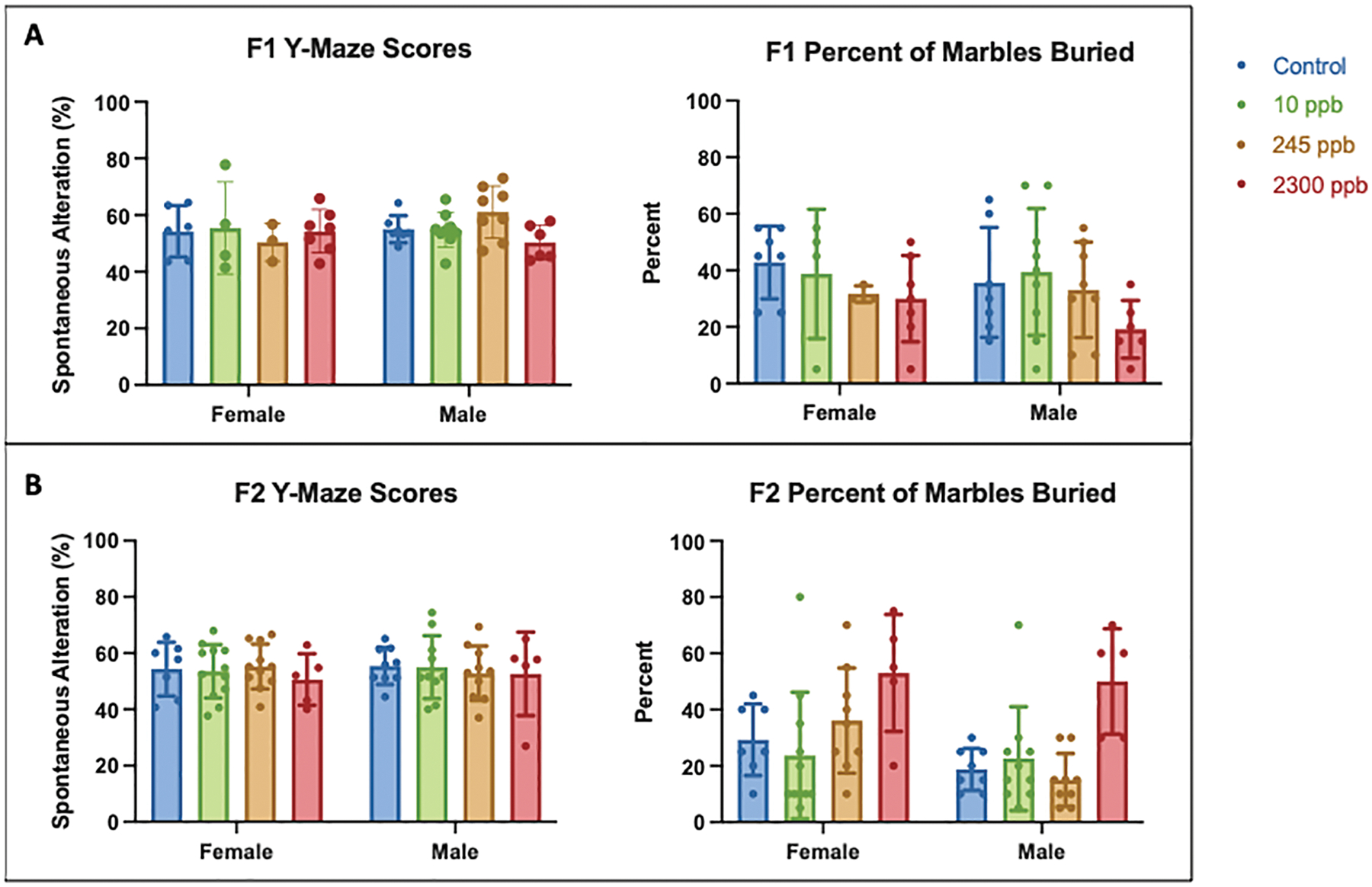
A) F1 γ-maze scores and percent of marbles buried. B) F2 γ-maze scores and percent of marbles buried. Control values were compared to exposed data to determine significance. (F1 1 ppb: male n= 10, female n=7; F1 10 ppb: male n=9, female n = 4; F1 245 ppb: male n= 8, female n=4; F1 2300 ppb: male n=7, female n=7; F2 1 ppb: male n=10, female n=8; F2 10 ppb: male n=10, female n=12; F2 245 ppb: male n=10, female n=11; F2 2300: male n=6, female n=5).
3.6. Preferential Hypermethylation in DMCs and DMRs of F1 Generation
To identify a potential mechanism for epigenetic damage, we assessed differentially methylated CpGs (DMCs) and differentially methylated regions (DMRs) in the liver of 10 ppb and 245 ppb F1 mice (Table 2) since these dose levels are most relevant to human exposures. In the F1 animals of both sexes, we found an excess of hypermethylated CpG sites at both doses. Specifically, in F1 females, we found 259 (66%) hypermethylated and 132 (34%) hypomethylated DMCs within the 10-ppb group, and 157 (61%) hypermethylated and 99 (39%) hypomethylated DMCs within the 245-ppb group. Only 4 (80%) DMRs were hypermethylated and 1 DMR hypomethylated in the 10-ppb group, whereas no DMRs were identified in the 245-ppb group. In F1 males, we identified 299 (65%) hypermethylated and 196 (42%) hypomethylated DMCs within the 10-ppb group, and 265 (64%) hypermethylated and 151 (36%) hypomethylated DMCS in the 245-ppb group. Only 3 (43%) hypermethylated and 4 (57%) hypomethylated DMRs were identified in the 10 ppb iAs group, whereas 9 (82%) hypermethylated and 2 (18%) hypomethylated DMRs were identified in the 245-ppb group. Our results show F1 males have more dysregulated methylation in DMCs and DMRs compared to F1 females, regardless of the exposure group.
Table 2. Differential Methylation Across Generation, Sex, and Dose in Liver by RRBS Data.
Liver tissue from F1 and F2 offspring at exposures 10 ppb and 245 ppb were assessed for differential DNA methylation as a result of in utero exposure. RRBS differential methylation was identified by DSS package in R studio. Statistical significance was determined by running a Wald Test in DSS, where data displayed have a p-value threshold set to FDR <0.001 (n= 3 per treatment, per sex).
| F1 Generation: DMC and DMR count in Female and Male Offspring | ||||||||
|---|---|---|---|---|---|---|---|---|
| Differentially Methylated CpGs (DMCs) | ||||||||
| 5mC | 10 ppb iAs | 245 ppb iAs | ||||||
| Female | Male | Female | Male | |||||
| Hypermethylated | 259 | 66% | 299 | 65% | 157 | 61% | 265 | 64% |
| Hypomethylated | 132 | 34% | 196 | 42% | 99 | 39% | 151 | 36% |
| Total | 391 | 462 | 256 | 416 | ||||
| Differentially Methylated Regions (DMRs) | ||||||||
| 5mC | 10 ppb iAs | 245 ppb iAs | ||||||
| Female | Male | Female | Male | |||||
| Hypermethylated | 4 | 80% | 3 | 43% | - | 0% | 9 | 82% |
| Hypomethylated | 1 | 20% | 4 | 57% | - | 0% | 2 | 18% |
| Total | 5 | 7 | - | 11 | ||||
| F2 Generation: DMC and DMR count in Female and Male Grand-Offspring | ||||||||
| Differentially Methylated CpGs (DMCs) | ||||||||
| 5mC | 10 ppb iAs | 245 ppb iAs | ||||||
| Female | Male | Female | Male | |||||
| Hypermethylated | 320 | 60% | 140 | 54% | 156 | 43% | 144 | 52% |
| Hypomethylated | 217 | 40% | 118 | 46% | 208 | 57% | 133 | 48% |
| Total | 537 | 258 | 364 | 277 | ||||
| Differentially Methylated Regions (DMRs) | ||||||||
| 5mC | 10 ppb iAs | 245 ppb iAs | ||||||
| Female | Male | Female | Male | |||||
| Hypermethylated | 9 | 56% | 1 | 100% | 1 | 20% | 1 | 100% |
| Hypomethylated | 7 | 44% | - | 4 | 80% | - | ||
| Total | 16 | 1 | 5 | 1 | ||||
The F2 mice sustained similar overall numbers of differentially methylated CpG sites and recapitulated the hypermethylation bias seen in F1 animals. Our F2 results indicate higher DMC and DMR content within F2 females compared to F2 males, a contrast to the F1 generation (Table 2). Females of the 10-ppb group had preferential DMC hypermethylation in the liver, where 320 (60%) hypermethylated and 217 (40%) hypomethylated DMCs were identified in the 10-ppb group. We identified 9 (56%) hypermethylated and 7 (44%) hypomethylated DMRs within the 10-ppb group. Females of the 245-ppb group had a slight bias towards hypomethylation, where 208 (57%) hypomethylated and 156 (43%) hypermethylated DMCs were identified. We found 1 (20%) hypermethylated and 4 (80%) hypomethylated DMRs in the 245 ppb F2 females. Within the 10-ppb F2 males, we identified 140 (54%) hypermethylated and 118 (46%) hypomethylated DMCs, detecting 1 hypermethylated DMR. F2 males in the 245-ppb group had 144 (52%) hypermethylated and 133 (48%) hypomethylated DMCs, and only 1 hypermethylated DMR. Our findings show female DMCs and DMRs within the liver are more reactive to prenatal exposure to iAs in both the 10 ppb and 245 ppb groups when compared to males.
3.7. Generational Intersect of DMCs and DMRs and Top DMC Containing Genes
The overlap of DMC-containing genes was assessed as no single-nucleotide positional DMCs were shared between generations. Overall, few DMC-containing genes were shared between F1 and F2, with most overlapping genes occurring between iAs doses within-generation (Figure 7). The top three greatest DMC containing gene overlap was identified within generations and sex (Male F110 ppb vs 245 ppb, Female F2 10 ppb vs 245 ppb, and F1 Female 10 ppb vs. 245 ppb). We identified generational DMCs, finding males exposed to 245 ppb in utero had the highest overlap (8 DMCs) compared to other generational comparisons.
Figure 7. Shared DMC Containing Genes Across Sex and Generation.
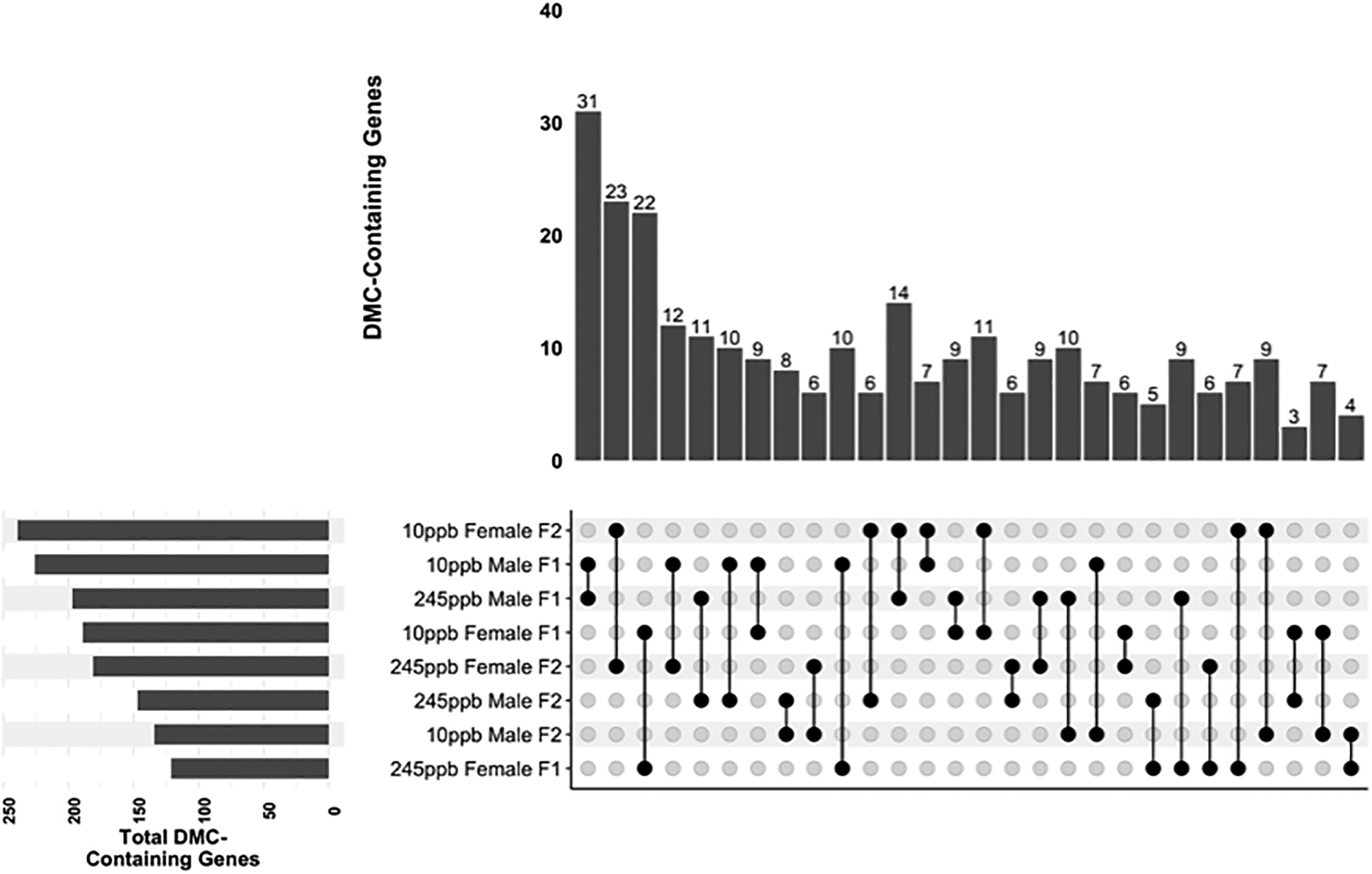
DMC containing genes were compared across sex and generation. Comparisons are shown from greatest gene overlap to least overlap, (n = 3 mice per sex, per treatment)
No DMR-containing genes were shared intergenerationally, although the genes Rlbp1 and Tcf4 both contained DMRs in 10ppb and 245ppb F1 males (Supplemental file “DMR_all_hits.xlsx”). The Rlbp1 DMRs were hypomethylated and the Tcf4 DMRs were hypermethylated in both doses relative to control. One hypermethylated intronic DMC was identified in Tcf4 in F2 females at the 245ppb dose but not in any other F2 animals (Supplementary file “DML_all_hits.xlsx”. No Rlbp1 DMCs were observed in the F2 generation. This data shows limited persistence of DMCs and DMRs through the F1 to F2 generation.
3.8. Differentially Methylated Genes and Targeted Genic Regions for Dysregulation
To evaluate the potential molecular consequences of iAs exposure, we identified genes containing DMCs and DMRs Identified genes were catalogued based on DMC content (Table 3) and based on the degree of differential methylation (Table 4). Several top DMC and DMR contain genes were associated with neurological function, retina or cornea function and anatomy, along with cell junction and membrane integrity. Genes identified with the greatest degree of differential methylation were associated with DNA helicase activity, cell adhesion, signal receptor binding, cell growth, muscle development, and nervous system development. In relation to the specific diseases connected to iAs exposure, we identified genes essential to liver function and associated diseases. For example, our data included genes associated with T2D and insulin resistance(Ali 2013) such as Pik3c2g(Saeed 2018) (F2 female 245 ppb), Ptprn2(Ouni et al. 2020) (F2 male 245 ppb), Adcy5(Sommese et al. 2018) (F1 male 10 ppb and F2 female 10 ppb), and Slc27a4(Rowlands et al. 2014) (F1 male 10 ppb). We also found dysregulated methylation within Slc43a2(Owaydhah et al. 2021) (F2 female 245 ppb) and Bicc1(Park et al. 2016) (F1 male 245 ppb), genes that are associated with placental development and embryogenesis. Within the generational overlap, we found several genes associated with T2D such as Irs2 (10 ppb females), Tcf12 (245 ppb males), Jazf1 (10 ppb females), Adcy5 (10 ppb males), and Slc27a (245 ppb females)(Ali 2013). We also identified genes associated with obesity, such as Adipor1 (F1 female 10 ppb), Fto (F1 male 10 ppb), Pparg (F2 male 245 ppb), and Adcy3 (F1 male 245 ppb) within our DMC list(Loos and Yeo 2021).
Table 3. Top Ten DMC Containing Genes.
Genes are categorized first by number of DMCs, then in alphabetical order. Statistical significance was determined by running a Wald Test in DSS, where data displayed have a p-value threshold set to FDR <0.001 (n= 3 per treatment, per sex).
| Top Ten DMC Containing Genes: RRBS | |||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| F1 Female 10 ppb | F2 Female 10 ppb | F1 Female 245 ppb | F2 Female 245 ppb | ||||||||||||
| Gene Name | Number of DMCs | Mean Differential Methylation | SD | Gene Name | Number of DMCs | Mean Differential Methylation | SD | Gene Name | Number of DMCs | Mean Differential Methylation | SD | Gene Name | Number of DMCs | Mean Differential Methylation | SD |
| Lrp1 | 8 | 62.89 | 5.28 | Dok7 | 5 | −62.24 | 2.73 | Mad1l1 | 6 | 48.44 | 5.96 | Tmem116 | 5 | −55.60 | 7.31 |
| Nav2 | S | 68.59 | 7.53 | Eda | 4 | 47.12 | 0.10 | Nav2 | 4 | 69.01 | 8.64 | Slc43a2 | 3 | 55.32 | 0.85 |
| Dock9 | 4 | 70.26 | 10.16 | Fgf12 | 4 | −75.69 | 4.59 | Abcb10 | 2 | −59.89 | 2.22 | Cacna2d3 | 2 | 62.62 | 8.56 |
| St3gal3 | 4 | −66.09 | 7.30 | Sox5 | 4 | 76.07 | 5.24 | Dock9 | 2 | 72.59 | 15.58 | Dip2c | 2 | −68.37 | 7.87 |
| Tfr2 | 4 | −48.99 | 1.16 | Cacna1f | 3 | 47.52 | 5.61 | Gpx1 | 2 | 61.04 | 4.50 | Fam13c | 2 | 64.70 | 0.00 |
| Adgrg4 | 2 | −59.26 | 2.03 | Cdkl5 | 3 | 46.04 | 4.35 | Pde10a | 2 | 83.57 | 11.72 | Gipc3 | 2 | −61.91 | 0.73 |
| Afap1l2 | 2 | −66.62 | 0.19 | Entpd8 | 3 | 60.13 | 1.78 | Pelp1 | 2 | 71.91 | 7.09 | Igsf21 | 2 | −54.54 | 9.16 |
| Ak5 | 2 | −3.46 | 88.54 | Eya2 | 3 | 60.24 | 8.43 | Shroom3 | 2 | 60.72 | 0.00 | Kank1 | 2 | −2.66 | 71.40 |
| Cnksr2 | 2 | 76.22 | 0.62 | Srgap1 | 3 | −27.63 | 88.53 | Acacb | 1 | −67.30 | NA | Pik3c2g | 2 | −40.53 | 2.78 |
| Cpne8 | 2 | −77.88 | 0.20 | Ar | 2 | 45.33 | 1.44 | Acvr2b | 1 | 73.53 | NA | Slc7a5 | 2 | −69.36 | 6.40 |
| F1 Mate 10 ppb | F2 Male 10 ppb | F1 Male 245 ppb | F2 Male 245 ppb | ||||||||||||
| Gene Name | Number of DMCs | Mean Differential Methylation | SD | Gene Name | Number of DMCs | Mean Differential Methylation | SD | Gene Name | Number of DMCs | Mean Differential Methylation | SD | Gene Name | Number of DMCs | Mean Differential Methylation | SD |
| Tcf4 | 6 | 54.21 | 2.23 | Agxt | 2 | −60.46 | 5.95 | Acot7 | 6 | 57.15 | 2.33 | Katna1 | 4 | 73.97 | 8.40 |
| Rlbp1 | 4 | −68.24 | 4.62 | Btla | 2 | −76.68 | 0.00 | Tcf4 | 6 | 64.40 | 4.32 | Pou2f1 | 4 | 0.19 | 79.44 |
| Smarca2 | 4 | −73.41 | 3.83 | Clmn | 2 | 65.79 | 2.91 | Bicc1 | 4 | 56.25 | 2.37 | Abca4 | 3 | −80.70 | 1.48 |
| Atp8b4 | 3 | 48.99 | 5.33 | Pcdhb16 | 2 | 57.51 | 3.38 | Irag1 | 4 | 70.52 | 1.69 | Ddx46 | 3 | −69.67 | 0.00 |
| Cdk7 | 3 | 51.65 | 2.05 | Sptb | 2 | −66.01 | 2.31 | Cdk7 | 3 | 50.44 | 0.68 | Ptpm2 | 3 | −22.65 | 67.25 |
| Gzmk | 3 | 56.50 | 6.98 | Acot7 | 1 | 69.40 | NA | Erc2 | 3 | 58.84 | 9.66 | Spidr | 2 | 63.63 | 1.33 |
| Pkd1l3 | 3 | 44.99 | 2.86 | Acox1 | 1 | −71.26 | NA | Pick1 | 3 | 61.41 | 9.90 | Zbtb12 | 2 | −51.57 | 2.22 |
| Slc27a4 | 3 | 66.19 | 8.23 | Adamts14 | 1 | 46.30 | NA | Rlbp1 | 3 | −64.19 | 0.13 | Ady | 1 | 69.99 | NA |
| Acp7 | 2 | 63.06 | 2.36 | Aff3 | 1 | −64.38 | NA | Scpep1 | 3 | 66.22 | 15.71 | Acot7 | 1 | 70.18 | NA |
| Adcy5 | 2 | 60.53 | 1.18 | AI314278 | 1 | 68.79 | NA | Atp8b4 | 2 | 49.51 | 2.14 | Actr2 | 1 | −59.52 | NA |
Table 4. Degree of Differential Methylation in DMC Containing Genes.
DMC containing genes were categorized by highest degree of hypo- or hypermethylation. Statistical significance was determined by running a Wald Test in DSS, where data displayed have a p-value threshold set to FDR <0.001 (n = 3 mice per sex, per treatment)
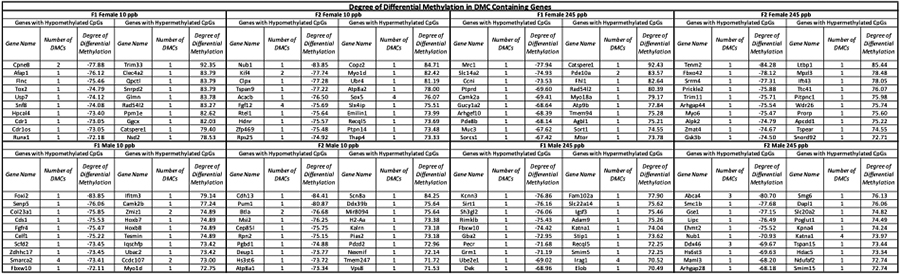
|
Using AnnotatR, we determined introns, 1 to 5 kb regions were the most sensitive while intergenic regions were most protected from iAs exposure by using the randomize regions function (Figure 8.A–C). This function determines if differential methylation happens by chance or if specific regions are more responsive to exposure compared to others. Across sex, dose, and generation, we found introns and 1 to 5 kb regions contained more DMCs compared to other genic regions. In addition, intergenic regions were less sensitive to iAs exposure, indicating intergenic regions may be protected from dysregulation caused by iAs exposure.
Figure 8. Differentially Methylated Loci localization.
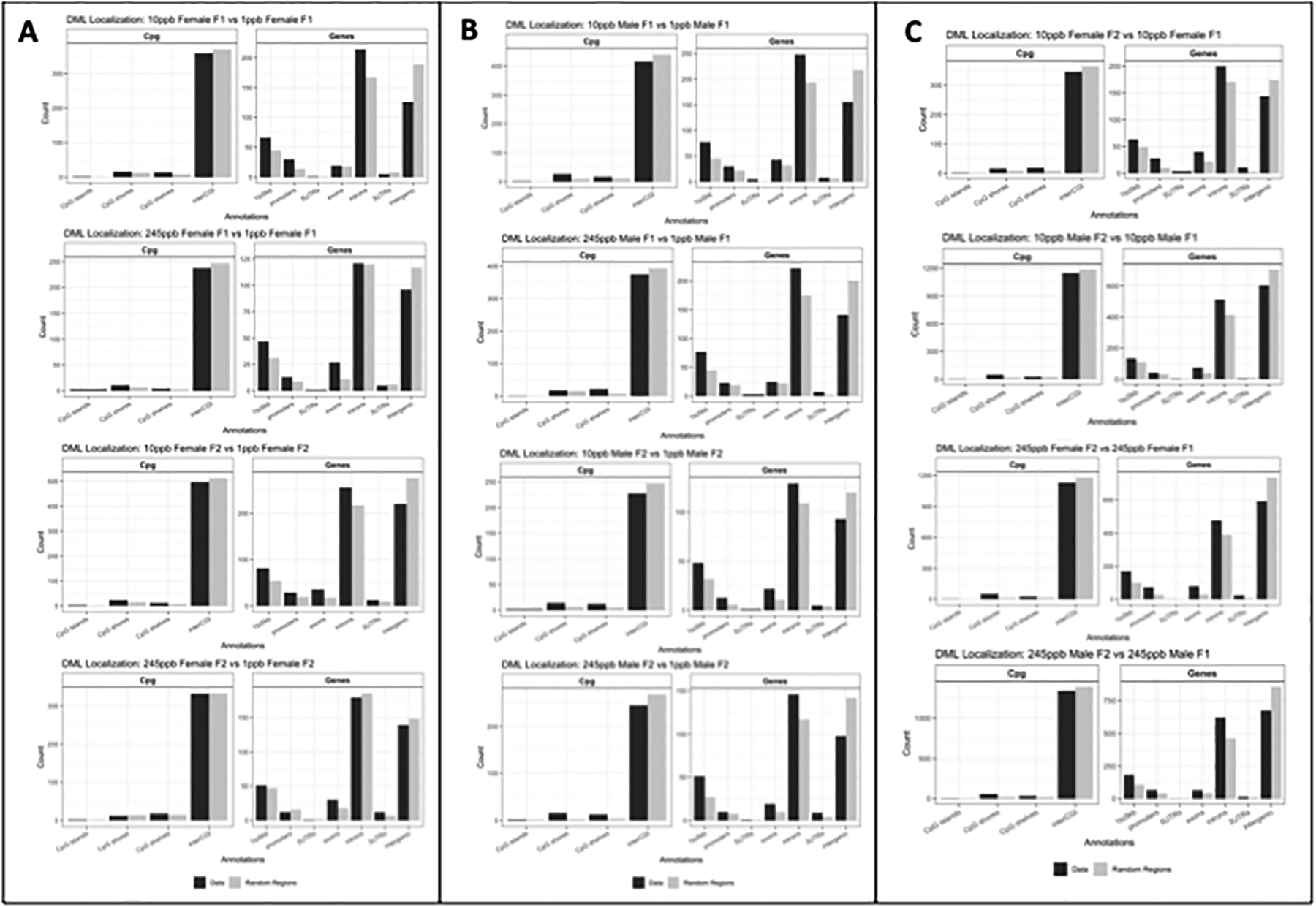
A) DML comparisons in Females within generations B) DML comparisons in males within generations C) DML comparisons within sex across generations. Statistical significance was determined by running a Wald Test in DSS, where data displayed have a p-value threshold set to FDR <0.001. (n= 3 per treatment per sex).
Discussion
There is increasing evidence suggesting the early life environment has an impact on multigenerational health. This study characterizes the intergenerational effects of iAs on offspring health, as inherited through the female germline. We provide evidence that iAs has sex-specific effects, such that dysregulated glucose metabolism affects both F1/F2 females and F2 males. More specifically, these effects were dose-specific with a late life onset. We show exposed F1 and F2 males have impaired growth as indicated by weekly body weight, where only a subset of the exposed F2 female lineage had surpassed the weight gain of F2 female control mice. We probed DNA methylation changes as a potential mechanism for intergenerational damage. We found DMCs and DMRs occur across sex and generations, identifying genes with high DMC/DMR content. Together, this data suggests in utero arsenic exposure alters metabolic phenotypes into adulthood of despite the absence of iAs exposure during adult lifetimes though with limited recapitulation of methylation changes across generations. The effects presented are sex-, dose-, and generation-specific indicating the prenatal environment influences the onset of disease that persists throughout generations; more studies exploring the intergenerational effects on epigenetic inheritance and the associated functionality are needed to link iAs exposure to changes in gene expression and protein function.
It has been well established that maternal in utero iAs exposure is associated with dysregulated glucose metabolism in the F1 populations(Young, Cai, and States 2018; Navas-Acien et al. 2019; Tinkelman et al. 2020). In a rat model, the maternal exposure of 500 ppb and 50 ppm throughout gestation and 2 months post-partum resulted in the onset of T2D phenotype in F1 offspring(Bonaventura et al. 2017). Mouse studies investigating in utero iAs of 100 ppb also identify diabetes related phenotypes such as increased plasma insulin, decreased pancreatic insulin production, or the onset of non-alcoholic fatty liver disease within offspring(Huang et al. 2018; Martin, Stýblo, and Fry 2017; Ditzel et al. 2016). Unlike our study using standard chow, these studies are coupled with nutritional arms (total western diet or supplementation with folate) and doses of iAs that exceed doses found in drinking water. Our exposure paradigm was designed to identify if iAs targeted the developing fetus and germ cells using human relevant exposures. Thus, our results indicate the dysregulated glucose metabolism phenotype in F1 and F2 generations are present at doses relevant to the human population.
Our findings on generational impacts on body weight and fat mass contributes to the growing body of in utero iAs exposure on offspring metabolism. Our data show maternal exposure causes sex- and dose-specific weight changes in F1 offspring. Additionally, grand maternal exposure alters the life-long body composition in both male and female F2 offspring. Previous studies using exposures ranging from 10 ppb – 42.5 ppm show maternal iAs exposure contributes to higher body weight in female offspring(Rodriguez et al. 2016a). Additionally, a study using a similar exposure of 250 ppb found no changes in body weight within the F1 offspring, early life stunted growth in F2 offspring that was restored in late life, and increased adiposity in F3 males(Gong et al. 2021). These studies focusing on rodent models are but a few indicating dose of iAs and sex have a role in adiposity. Further research is needed to identify the influence of iAs early- and late-life body composition and the implications on human metabolic health.
Our data show glucose metabolism and body composition are dysregulated by in utero iAs exposure, yet it is unclear if the onset of disease in our study is influenced by epigenetic alterations or other factors. The onset of T2D in adults chronically exposed to iAs may be a result of non-epigenetic mechanisms such as inhibition of a GLUT4, damage to ß-cells by ROS production, inhibition of glucose simulated insulin secretion, or increased stimulation of liver gluconeogenesis(Martin, Stýblo, and Fry 2017). Chronic iAs exposure is known to cause increased body weight and alterations in lipid metabolism(Castriota et al. 2018; C et al. 2019). Additionally, studies show the maternal metabolic milieu influences the onset of obesity in offspring ranging from adolescence and into adulthood(Tequeanes et al. 2009; Derraik et al. 2015). Despite the role of iAs in direct damage to tissues and the maternal metabolism, increasing evidence from mouse and human studies shows developmental iAs exposure alters DNA methylation in offspring with associations to metabolic phenotypes. In a longitudinal mother-child cohort in Bangladesh, differential methylation of CpG sites within in blood mononuclear cells were characterized, where 12 hypermethylated CpG sites were associated with prenatal iAs exposure(Gliga et al. 2018). Of the differentially methylated CpG sites, several were associated with proteins essential to insulin secretion by ß-cells in the pancreas. Similar to previously reported studies, our data finds differential methylation within genes associated with T2D(Ali 2013) and obesity(Loos and Yeo 2021) - Pik3c2g, Ptprn2, Adcy5, Slc27a4, Irs2, Tcf12, Jazf1, Adcy5, Slc27a, Adipor1, Fto, Pparg, and Adcy3. While in utero exposure is known to cause long-term health effects in developing fetuses and young children, often impacting the establishment of DNA methylation(Smeester and Fry 2018), further research is needed to explore the association between our DMCs and the onset of disease. It is clear the identification of differential methylation in offspring is associated with maternal iAs exposure, where differential methylation may be a plausible contributor to the onset of metabolic phenotypes. Although our DMC and DMR data provide insight on genes associated with disease, the exploration of differential methylation on gene expression is still necessary. The correlation between DMCs and DMRs with dysregulated gene expression would potentially link our data with the characterized disease phenotypes. Additionally, the exploration of the physiological state of the liver, such as morphology, lipid accumulation, or RNA expression, could provide context if differential methylation is an artifact of cell composition. While levels of iAs that exceed our dose regimen (up to 85 ppm iAs) are known to cause intergenerational carcinogenesis targeting the liver (Waalkes et al. 2007), we found no visible carcinomas or immediate morphological changes such as color across treatment groups. Further investigation of liver morphology, cell composition, and RNA transcription is needed to understand the effects of iAs on hepatotoxicity and metabolism.
Sex-, dose-, and generation-specific effects are common among prenatal environmental exposures. Previous studies have reported similar effects of paternal iAs exposure on glucose homeostasis through the F1 and F2 generations(Gong et al. 2021). Glucose intolerance and hepatic insulin resent was present in F1 females, not F1 males, without altering body weight. Despite low body wean weights, body weight was restored into adulthood within the F2 generation. In contrast to our study, Gong et. al explores the transmission of metabolic phenotypes through the paternal lineage (mating F1 male offspring), not the maternal lineage (mating F1 female offspring). These results indicate a paternal epigenetic influence on adult F1 and F2 offspring metabolic disease phenotypes. Another study exploring both maternal and paternal exposure on transgenerational health found altered DNA methylation and reproductive phenotypes(Nava-Rivera et al. 2021). Methylation in the ovary and testes was significantly lower in the F1 generation, remained unchanged in the F2 generation, and increased within the F3 generation. Sperm quality parameters were also lower in the F1 and F3 male offspring, not within the F2 offspring. Results from these two intergenerational studies are foundational for the understanding of iAs exposure and epigenetic reprogramming. Our shared DMC containing genes across sex and generation provides an exciting foundation for genes targeted by prenatal iAs exposure. Despite the absence of overlap between generations, it is possible CpG methylation within the detected DMCs was re-established during F2 embryogenesis. Our findings could indicate female PGCs may retain epigenetic damage from F0 exposure that persists regardless of epigenetic reprogramming. Our results join the few novel studies indicating intergenerational iAs exposure alters metabolic phenotypes and the epigenome in a sex-, dose-, and generation-specific manner.
Supplementary Material
Acknowledgements
The authors acknowledge the anonymous reviewers of this manuscript for their valuable input.
Funding
This work was supported by NIEHS F31ES030967 (Colwell), NIH Office of the Director T32OD010993 (Flack) NIH R21AG071908 (Faulk), Impetus Grant (Norn Foundation) (Faulk), USDA-NIFA MIN-16-129 (Faulk)
Footnotes
Conflicts of Interest
The authors declare that they have no conflicts of interest.
Citations
- Ali Omar. 2013. “Genetics of Type 2 Diabetes.” World Journal of Diabetes 4 (4): 114. 10.4239/WJD.V4.I4.114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Argos Maria. 2015. “Arsenic Exposure and Epigenetic Alterations: Recent Findings Based on the Illumina 450K DNA Methylation Array.” Current Environmental Health Reports 2 (2): 137. 10.1007/S40572-015-0052-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bailey Kathryn A., Wu Michael C., Ward William O., Smeester Lisa, Rager Julia E., García-Vargas Gonzalo, Del Razo Luz Maria, Drobná Zuzana, Stýblo Miroslav, and Fry Rebecca C.. 2013. “Arsenic and the Epigenome: Interindividual Differences in Arsenic Metabolism Related to Distinct Patterns of DNA Methylation.” Journal of Biochemical and Molecular Toxicology. 10.1002/jbt.21462. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bonaventura María Marta, Bourguignon Nadia Soledad, Bizzozzero Marianne, Rodriguez Diego, Ventura Clara, Cocca Claudia, Libertun Carlos, and Lux-Lantos Victoria Adela. 2017. “Arsenite in Drinking Water Produces Glucose Intolerance in Pregnant Rats and Their Female Offspring.” Food and Chemical Toxicology. 10.1016/j.fct.2016.12.025. [DOI] [PubMed] [Google Scholar]
- Rivas-Santiago C, González-Curiel I, Zarazua S, Murgu M, Ruiz Cardona A, Lazalde B, Lara-Ramírez EE, et al. 2019. “Lipid Metabolism Alterations in a Rat Model of Chronic and Intergenerational Exposure to Arsenic.” BioMed Research International 2019. 10.1155/2019/4978018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Castriota Felicia, Acevedo Johanna, Ferreccio Catterina, Smith Allan H., Liaw Jane, Smith Martyn T., and Steinmaus Craig. 2018. “Obesity and Increased Susceptibility to Arsenic-Related Type 2 Diabetes in Northern Chile.” Environmental Research. 10.1016/j.envres.2018.07.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cavalcante Raymond G, and Sartor Maureen A. 2016. “Annotatr: Associating Genomic Regions with Genomic Annotations.” BioRxiv, 1–9. 10.1101/039685. [DOI] [Google Scholar]
- Cedar Howard, and Bergman Yehudit. 2012. “Programming of DNA Methylation Patterns.” Annual Review of Biochemistry. 10.1146/annurev-biochem-052610-091920. [DOI] [PubMed] [Google Scholar]
- Derraik José G.B., Ahlsson Fredrik, Diderholm Barbro, and Lundgren Maria. 2015. “Obesity Rates in Two Generations of Swedish Women Entering Pregnancy and Associated Obesity Risk among Adult Daughters.” Scientific Reports 2015 5:1 5 (1): 1–5. 10.1038/srep16692. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ditzel Eric J., Nguyen Thu, Parker Patricia, and Camenisch Todd D.. 2016. “Effects of Arsenite Exposure during Fetal Development on Energy Metabolism and Susceptibility to Diet-Induced Fatty Liver Disease in Male Mice.” Environmental Health Perspectives 124 (2): 201. 10.1289/EHP.1409501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fei Dennis Liang, Koestler Devin C., Li Zhigang, Giambelli Camilla, Sanchez-Mejias Avencia, Gosse Julie A., Marsit Carmen J., Karagas Margaret R., and Robbins David J.. 2013. “Association between in Utero Arsenic Exposure, Placental Gene Expression, and Infant Birth Weight: A US Birth Cohort Study.” Environmental Health: A Global Access Science Source. 10.1186/1476-069X-12-58. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Feng Suhua, Jacobsen Steven E., and Reik Wolf. 2010. “Epigenetic Reprogramming in Plant and Animal Development.” Science. 10.1126/science.1190614. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gilbert-Diamond Diane, Emond Jennifer A., Baker Emily R., Korrick Susan A., and Karagas Margaret R.. 2016. “Relation between in Utero Arsenic Exposure and Birth Outcomes in a Cohort of Mothers and Their Newborns from New Hampshire.” Environmental Health Perspectives 124 (8): 1299–1307. 10.1289/EHP.1510065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gliga Anda R., Engström Karin, Kippler Maria, Skröder Helena, Ahmed Sultan, Vahter Marie, Raqib Rubhana, and Broberg Karin. 2018. “Prenatal Arsenic Exposure Is Associated with Increased Plasma IGFBP3 Concentrations in 9-Year-Old Children Partly via Changes in DNA Methylation.” Archives of Toxicology 92 (8): 2487. 10.1007/S00204-018-2239-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gong Yingyun, Xue Yanfeng, Li Xin, Zhang Zhao, Zhou Wenjun, Marcolongo Paola, Benedetti Angiolo, et al. 2021. “Inter- and Transgenerational Effects of Paternal Exposure to Inorganic Arsenic.” Advanced Science 8 (7). 10.1002/ADVS.202002715. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hossain Khaled, Suzuki Takehiro, Hasibuzzaman MM, Islam Md Shofikul, Rahman Atiqur, Paul Sudip Kumar, Tanu Tanzina, et al. 2017. “Chronic Exposure to Arsenic, LINE-1 Hypomethylation, and Blood Pressure: A Cross-Sectional Study in Bangladesh.” Environmental Health: A Global Access Science Source. 10.1186/s12940-017-0231-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang Madelyn C., Douillet Christelle, Dover Ellen N., and Stýblo Miroslav. 2018. “Prenatal Arsenic Exposure and Dietary Folate and Methylcobalamin Supplementation Alter the Metabolic Phenotype of C57BL/6J Mice in a Sex-Specific Manner.” Archives of Toxicology 92 (6): 1925–37. 10.1007/S00204-018-2206-Z/FIGURES/7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hughes Robert N. 2004. “The Value of Spontaneous Alternation Behavior (SAB) as a Test of Retention in Pharmacological Investigations of Memory.” Neuroscience and Biobehavioral Reviews 28 (5): 497–505. 10.1016/J.NEUBIOREV.2004.06.006. [DOI] [PubMed] [Google Scholar]
- Krueger F 2015. “Trim Galore!: A Wrapper Tool around Cutadapt and FastQC to Consistently Apply Quality and Adapter Trimming to FastQ Files.” Babraham Institute. 2015. 10.1002/maco.200603986. [DOI] [Google Scholar]
- Krueger Felix, and Andrews Simon R.. 2011. “Bismark: A Flexible Aligner and Methylation Caller for Bisulfite-Seq Applications.” Bioinformatics. 10.1093/bioinformatics/btr167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Laine Jessica E., Bailey Kathryn A., Rubio-Andrade Marisela, Olshan Andrew F., Smeester Lisa, Drobná Zuzana, Herring Amy H., Stýblo Miroslav, García-Vargas Gonzalo G., and Fry Rebecca C.. 2015. “Maternal Arsenic Exposure, Arsenic Methylation Efficiency, and Birth Outcomes in the Biomarkers of Exposure to ARsenic (BEAR) Pregnancy Cohort in Mexico.” Environmental Health Perspectives. 10.1289/ehp.1307476. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Langmead Ben, and Salzberg Steven L.. 2012. “Fast Gapped-Read Alignment with Bowtie 2.” Nature Methods. 10.1038/nmeth.1923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu Su, Guo Xuechao, Wu Bing, Yu Haiyan, Zhang Xuxiang, and Li Mei. 2014. “Arsenic Induces Diabetic Effects through Beta-Cell Dysfunction and Increased Gluconeogenesis in Mice.” Scientific Reports. 10.1038/srep06894. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Loos Ruth J.F., and Yeo Giles S.H.. 2021. “The Genetics of Obesity: From Discovery to Biology.” Nature Reviews Genetics 2021 23:2 23 (2): 120–33. 10.1038/s41576-021-00414-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marie Cécile, Léger Stéphanie, Guttmann Aline, Rivière Olivier, Marchiset Nathalie, Lémery Didier, Vendittelli Françoise, and Sauvant-Rochat Marie Pierre. 2018. “Exposure to Arsenic in Tap Water and Gestational Diabetes: A French Semi-Ecological Study.” Environmental Research. 10.1016/j.envres.2017.11.016. [DOI] [PubMed] [Google Scholar]
- Martin Elizabeth M., Stýblo Miroslav, and Fry Rebecca C.. 2017. “Genetic and Epigenetic Mechanisms Underlying Arsenic-Associated Diabetes Mellitus: A Perspective of the Current Evidence.” Epigenomics. 10.2217/epi-2016-0097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- National Research Council (U.S.). Subcommittee on Arsenic in Drinking Water. 1999. Arsenic in Drinking Water. National Academy Press. http://www.who.int/news-room/fact-sheets/detail/arsenic. [Google Scholar]
- Nava-Rivera Lydia Enith, Betancourt-Martínez Nadia Denys, Lozoya-Martínez Rodrigo, Carranza-Rosales Pilar, Guzmán-Delgado Nancy Elena, Carranza-Torres Irma Edith, Delgado-Aguirre Hector, Zambrano-Ortíz José Omar, and Morán-Martínez Javier. 2021. “Transgenerational Effects in DNA Methylation, Genotoxicity and Reproductive Phenotype by Chronic Arsenic Exposure.” Scientific Reports 2021 11:1 11 (1): 1–16. 10.1038/s41598-021-87677-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Navas-Acien Ana, Spratlen Miranda J., Abuawad Ahlam, LoIacono Nancy J., Bozack Anne K., and Gamble Mary V.. 2019. “Early-Life Arsenic Exposure, Nutritional Status, and Adult Diabetes Risk.” Current Diabetes Reports 2019 19:12 19 (12): 1–8. 10.1007/S11892-019-1272-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nohara Keiko, Baba Takashi, Murai Hikari, Kobayashi Yayoi, Suzuki Takehiro, Tateishi Yukiyo, Matsumoto Michiyo, Nishimura Noriko, and Sano Tomoharu. 2011. “Global DNA Methylation in the Mouse Liver Is Affected by Methyl Deficiency and Arsenic in a Sex-Dependent Manner.” Archives of Toxicology. 10.1007/s00204-010-0611-z. [DOI] [PubMed] [Google Scholar]
- Nohara Keiko, Suzuki Takehiro, and Okamura Kazuyuki. 2020. “Gestational Arsenic Exposure and Paternal Intergenerational Epigenetic Inheritance.” Toxicology and Applied Pharmacology 409 (December): 115319. 10.1016/J.TAAP.2020.115319. [DOI] [PubMed] [Google Scholar]
- Ouni Meriem, Saussenthaler Sophie, Eichelmann Fabian, Jähnert Markus, Stadion Mandy, Wittenbecher Clemens, Rönn Tina, et al. 2020. “Epigenetic Changes in Islets of Langerhans Preceding the Onset of Diabetes.” Diabetes 69 (11): 2503–17. 10.2337/DB20-0204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Owaydhah Wejdan H., Ashton Nick, Verrey François, and Glazier Jocelyn D.. 2021. “Differential Expression of System L Amino Acid Transporter Subtypes in Rat Placenta and Yolk Sac.” Placenta 103 (January): 188–98. 10.1016/J.PLACENTA.2020.10.034. [DOI] [PubMed] [Google Scholar]
- Painter RC, Osmond C, Gluckman P, Hanson M, Phillips DIW, and Roseboom TJ. 2008. “Transgenerational Effects of Prenatal Exposure to the Dutch Famine on Neonatal Adiposity and Health in Later Life.” BJOG: An International Journal of Obstetrics and Gynaecology. 10.1111/j.1471-0528.2008.01822.x. [DOI] [PubMed] [Google Scholar]
- Park Sookhee, Blaser Susanne, Marchal Melissa A., Houston Douglas W., and Sheets Michael D.. 2016. “A Gradient of Maternal Bicaudal-C Controls Vertebrate Embryogenesis via Translational Repression of MRNAs Encoding Cell Fate Regulators.” Development (Cambridge, England) 143 (5): 864–71. 10.1242/DEV.131359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- “Purpose and Cognition: The Determiners of Animal Learning.” n.d. Accessed December 1, 2021. https://psycnet.apa.org/fulltext/1927-00608-001.pdf.
- Rodriguez Karina F., Ungewitter Erica K., Crespo-Mejias Yasmin, Liu Chang, Nicol Barbara, Kissling Grace E., and Yao Humphrey Hung Chang. 2016a. “Effects of in Utero Exposure to Arsenic during the Second Half of Gestation on Reproductive End Points and Metabolic Parameters in Female CD-1 Mice.” Environmental Health Perspectives. 10.1289/ehp.1509703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- ———. 2016b. “Effects of in Utero Exposure to Arsenic during the Second Half of Gestation on Reproductive End Points and Metabolic Parameters in Female CD-1 Mice.” Environmental Health Perspectives 124 (3): 336–43. 10.1289/EHP.1509703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rojas Daniel, Rager Julia E, Smeester Lisa, Bailey Kathryn A, Drobná Zuzana, Rubio-Andrade Marisela, Stýblo Miroslav, García-Vargas Gonzalo, and Fry Rebecca C. 2015. “Prenatal Arsenic Exposure and the Epigenome: Identifying Sites of 5-Methylcytosine Alterations That Predict Functional Changes in Gene Expression in Newborn Cord Blood and Subsequent Birth Outcomes.” Toxicological Sciences : An Official Journal of the Society of Toxicology 143 (1): 97–106. 10.1093/toxsci/kfu210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rowlands David S., Page Rachel A., Sukala William R., Giri Mamta, Ghimbovschi Svetlana D., Hayat Irum, Cheema Birinder S., et al. 2014. “Multi-Omic Integrated Networks Connect DNA Methylation and MiRNA with Skeletal Muscle Plasticity to Chronic Exercise in Type 2 Diabetic Obesity.” Physiological Genomics 46 (20): 747–65. 10.1152/PHYSIOLGENOMICS.00024.2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saeed Mohammad. 2018. “Locus and Gene-Based GWAS Meta-Analysis Identifies New Diabetic Nephropathy Genes.” Immunogenetics 70 (6): 347–53. 10.1007/S00251-017-1044-0/TABLES/2. [DOI] [PubMed] [Google Scholar]
- Sasaki Hiroyuki, and Matsui Yasuhisa. 2008. “Epigenetic Events in Mammalian Germ-Cell Development: Reprogramming and Beyond.” Nature Reviews Genetics. 10.1038/nrg2295. [DOI] [PubMed] [Google Scholar]
- Skinner Michael, and Guerrero-Bosagna Carlos. 2014. “Role of CpG Deserts in the Epigenetic Transgenerational Inheritance of Differential DNA Methylation Regions.” BMC Genomics 15 (1): 692. 10.1186/1471-2164-15-692. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smeester Lisa, and Fry Rebecca C.. 2018. “Long-Term Health Effects and Underlying Biological Mechanisms of Developmental Exposure to Arsenic.” Current Environmental Health Reports. 10.1007/s40572-018-0184-1. [DOI] [PubMed] [Google Scholar]
- Sommese Linda, Benincasa Giuditta, Lanza Michele, Sorriento Antonio, Schiano Concetta, Lucchese Roberta, Alfano Roberto, Nicoletti Giovanni Francesco, and Napoli Claudio. 2018. “Novel Epigenetic-Sensitive Clinical Challenges Both in Type 1 and Type 2 Diabetes.” Journal of Diabetes and Its Complications 32 (11): 1076–84. 10.1016/J.JDIACOMP.2018.08.012. [DOI] [PubMed] [Google Scholar]
- Spratlen Miranda Jones, Gamble Mary V., Grau-Perez Maria, Kuo Chin Chi, Best Lyle G., Yracheta Joseph, Francesconi Kevin, et al. 2017. “Arsenic Metabolism and One-Carbon Metabolism at Low-Moderate Arsenic Exposure: Evidence from the Strong Heart Study.” Food and Chemical Toxicology. 10.1016/j.fct.2017.05.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tequeanes Ana Lilia Lozada, Gigante Denise Petrucci, Assunção Maria Cecilia Formoso, Chica David Alejandro Gonzalez, and Horta Bernardo Lessa. 2009. “Maternal Anthropometry Is Associated with the Body Mass Index and Waist:Height Ratio of Offspring at 23 Years of Age.” The Journal of Nutrition 139 (4): 750–54. 10.3945/JN.108.100669. [DOI] [PubMed] [Google Scholar]
- Thomas David J., Waters Stephen B., and Styblo Miroslav. 2004. “Elucidating the Pathway for Arsenic Methylation.” Toxicology and Applied Pharmacology. 10.1016/j.taap.2003.10.020. [DOI] [PubMed] [Google Scholar]
- Tinkelman Naomi E., Miranda Jones Spratlen Arce Domingo-Relloso, Maria Tellez-Plaza Maria Grau-Perez, Francesconi Kevin A., Goessler Walter, et al. 2020. “Associations of Maternal Arsenic Exposure with Adult Fasting Glucose and Insulin Resistance in the Strong Heart Study and Strong Heart Family Study.” Environment International 137 (April): 105531. 10.1016/J.ENVINT.2020.105531. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Titus-Ernstoff Linda, Troisi Rebecca, Hatch Elizabeth E., Hyer Marianne, Wise Lauren A., Palmer Julie R., Kaufman Raymond, et al. 2008. “Offspring of Women Exposed in Utero to Diethylstilbestrol (DES): A Preliminary Report of Benign and Malignant Pathology in the Third Generation.” Epidemiology. 10.1097/EDE.0b013e318163152a. [DOI] [PubMed] [Google Scholar]
- “TOXICOLOGICAL PROFILE FOR ARSENIC | Enhanced Reader.” n.d.
- Tsang Verne, Fry Rebecca C., Niculescu Mihai D., Rager Julia E., Saunders Jesse, Paul David S., Zeisel Steven H., Waalkes Michael P., Stýblo Miroslav, and Drobná Zuzana. 2012. “The Epigenetic Effects of a High Prenatal Folate Intake in Male Mouse Fetuses Exposed in Utero to Arsenic.” Toxicology and Applied Pharmacology. 10.1016/j.taap.2012.08.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Waalkes Michael P., Liu Jie, Ward Jerrold M., and Diwan Bhalchandra A.. 2004. “Animal Models for Arsenic Carcinogenesis: Inorganic Arsenic Is a Transplacental Carcinogen in Mice.” Toxicology and Applied Pharmacology. 10.1016/j.taap.2003.10.028. [DOI] [PubMed] [Google Scholar]
- Xie Yaxiong, Liu Jie, Benbrahim-Tallaa Lamia, Ward Jerry M., Logsdon Daniel, Diwan Bhalchandra A., and Waalkes Michael P.. 2007. “Aberrant DNA Methylation and Gene Expression in Livers of Newborn Mice Transplacentally Exposed to a Hepatocarcinogenic Dose of Inorganic Arsenic.” Toxicology. 10.1016/j.tox.2007.03.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Young Jamie L., Cai Lu, and J. Christopher States. 2018. “Impact of Prenatal Arsenic Exposure on Chronic Adult Diseases.” Systems Biology in Reproductive Medicine, June, 1–15. 10.1080/19396368.2018.1480076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao CQ, Young MR, Diwan BA, Coogan TP, and Waalkes MP. 2002. “Association of Arsenic-Induced Malignant Transformation with DNA Hypomethylation and Aberrant Gene Expression.” Proceedings of the National Academy of Sciences. 10.1073/pnas.94.20.10907. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.