

Abstract
Rationale and Objective:
Clonal hematopoiesis of indeterminate potential (CHIP), defined by the age-related ontogenesis of expanded leukemogenic mutations indicative of a genetically distinct clonal leukocyte population, is associated with risk of hematologic malignancy and cardiovascular disease. In experimental models, recapitulation of CHIP promotes kidney interstitial fibrosis with direct tissue infiltration of donor macrophages. We tested the hypothesis that CHIP is associated with kidney function decline in the general population.
Study Design:
Cohort Study.
Setting and Participants:
12,004 individuals from three community-based cohorts in the TOPMed Consortium.
Exposure:
CHIP status from blood DNA-derived whole genome sequences.
Outcome:
Risk of 30% decline in estimated glomerular filtration rate (eGFR) and percent eGFR decline per year during follow-up.
Analytical approach:
Cox proportional hazards models for 30% eGFR decline endpoint and generalized estimating equations for annualized relative change in eGFR with meta-analysis. Study-specific estimates were combined using fixed-effect meta-analysis.
Results:
Median baseline eGFR was 84 ml/min/1.73m2. Prevalence of CHIP was 6.6%, 9.0% and 12.2% in persons 50–60, 60–70 and >70 years old, respectively. Over a median follow-up of 8 years, 205 kidney function decline events occurred among 1,002 CHIP carriers (2.1 events per 100-person-years), and 2,041 kidney function decline events in persons without CHIP (1.7 events per 100-person-years). In meta-analysis, CHIP was associated with kidney function decline (17% higher risk; 95%CI: 1% to 36% higher; p-value=0.036). Differences were not observed between those with baseline eGFR above or below 60 ml/min/1.73m2, age above or below 60 years, or with or without diabetes.
Limitations:
Small number of participants with moderate-to-advanced kidney disease and restricted set of CHIP driver mutations.
Conclusions:
We report an association between CHIP and kidney function decline in three general population cohorts without known kidney disease. Further studies are needed to investigate this novel condition and its potential impact among individuals with overt kidney disease.
INTRODUCTION
Clonal hematopoiesis of indeterminate potential (CHIP) is an age-related disorder among asymptomatic adults defined by the ontogenesis of leukemogenic mutations in blood DNA indicative of a genetically distinct clonal leukocyte population.1,2 CHIP is caused by somatic mutations within a restricted set of cancer driver genes in hematopoietic stem cells (primarily DNMT3A, TET2, ASXL1, JAK2, and TP53).1 The resulting progeny propagate through the hematopoietic system to produce a resilient clonal population with a selective survival advantage. By definition, CHIP is not overtly malignant; however, clonal leukocytes harboring CHIP mutations increase the risk of future hematologic malignancy presumably through a second genetic hit.3 In addition to cancer, CHIP is also associated with cardiovascular diseases, including myocardial infarction, stroke, and heart failure.4–8 In murine models, Tet2-deficient hematopoietic stem cells accelerate atherogenesis and promote cardiac and kidney interstitial fibrosis with direct tissue infiltration of donor macrophages.4,9–11
Kidney function declines progressively with aging. The rate of decline is variable across individuals and is associated with multiple risk factors including hypertension, diabetes, and inflammatory markers.12,13 The pathologic hallmark of progressive chronic kidney disease (CKD) is tubulointerstitial fibrosis, which is characterized by the accumulation of inflammatory infiltrates and fibroblasts within the kidney interstitium and permanent loss of tubular epithelial cells.
Given mechanistic links connecting CHIP with both atherosclerosis and tubulointerstitial fibrosis, we hypothesized that CHIP would be associated with greater kidney function decline compared to those without CHIP in the general population. To test this hypothesis, we ascertained CHIP status of 12,004 individuals from three community-based cohort studies, and we delineated associations with the decline in the estimated glomerular filtration rate (eGFR) over follow-up.
METHODS
Study Populations
The NHLBI Trans-Omics for Precision Medicine (TOPMed) program was designed to facilitate research in precision medicine by integrating whole-genome genetic sequencing and molecular data across established epidemiology studies.14 For the current analysis, we studied participants from three community-based cohort studies in the freeze 8 release of TOPMed. Included studies were the Atherosclerosis Risk in Communities Study (ARIC, n=6,575)15, the Cardiovascular Health Study (CHS, n=1,701)16, and the Multi-Ethnic Study of Atherosclerosis (MESA, n=3,728)17. We selected participants who were ≥50 years old due to the rarity of CHIP in younger persons; additional inclusion criteria were valid ascertainment of CHIP status from the TOPMed genomic data and at least two longitudinal measurements of serum creatinine to assess changes in kidney function. Participants provided written consent per each study’s IRB approved protocol.
Ascertainment of CHIP
We obtained CHIP genotypes from TOPMed blood DNA-derived whole genome sequences generated using GATK MuTect2, as previously described.2,18 Several quality control steps were applied to identify and remove sequencing artifacts and germline mutations from the call set.2 At present, a universal standard for CHIP driver variants definition does not exist. However, many groups, including ours, employ the criteria defined by Jaiswal et al in their seminal work describing CHIP.4 Samples were assigned CHIP carriers status were identified based on the presence a leukemogenic driver mutation at variant allele frequency (VAF) > 2% in 74 pre-specified genes known to promote clonal expansion of hematopoietic stem cells (see Supplemental Table 1). For 23 of these genes, only truncating (frameshifting/nonsense/splicing) variants are permitted; for another 26, only select missense variants are permitted (mostly gain-of-function); and for the remaining 25 genes, truncating and select missense variants are permitted. These gene-specific criteria were curated to minimize the number of germline and passenger variants as well as sequencing artefacts. Median VAF for CHIP carriers was 16%. To test for associations between clonal hematopoiesis due to specific mutations and kidney disease progression, in secondary analyses we categorized CHIP driver gene mutations into those in DNMT3A (the most common driver gene) and those not in DNMT3A.
Ascertainment of kidney function decline and microalbuminuria
We estimated the eGFR at each study visit from serum creatinine concentrations, age, and sex using the 3-variable 2021 CKD-EPI equation.19 We defined kidney function decline as a ≥30% decrease in eGFR from the baseline value.20 This definition was selected to balance a clinically meaningful change in kidney function with sufficient numbers of events for analysis. Moreover, relative changes in eGFR are less dependent on the baseline value than absolute changes. As secondary outcomes, we assessed a ≥40% decline in eGFR, the percent eGFR decline per year over follow-up and incident eGFR <60 ml/min/1.73m2.21 We defined microalbuminuria by a spot urine albumin to creatinine ratio ≥30 mg/g.22–25
Statistical Analysis
We tabulated baseline characteristics within each study according to CHIP status. We estimated cross-sectional associations of CHIP with baseline eGFR and microalbuminuria using linear, binary, and multinomial logistic regression with age adjustment and internal standardization. We constructed Cox proportional hazards models to delineate associations of CHIP status at baseline with the first occurrence of a ≥30% decline in eGFR from the baseline value in each study cohort. Participants were censored due to death, loss to follow-up, or the end of the study data collection period, whichever came first. Regression models were adjusted for age, age-squared, sex, baseline eGFR, self-reported race, and diabetes. Models in CHS and MESA additionally adjusted for log-transformed urine albumin to creatinine ratio (uACR). Urine albumin to creatinine ratio was not measured concomitantly with CHIP in the ARIC study. Study-specific hazard ratios were combined using fixed-effect meta-analysis. For secondary analyses of annualized relative change in log(eGFR), we used a mixed effects model with random intercept with adjustment for age, age-squared, sex, self-reported race, diabetes and log-transformed uACR. We tested for multiplicative interactions by age, baseline eGFR and baseline diabetes using a Wald test on the product term. Analyses were conducted using Stata 17 (College Station, TX).
RESULTS
The mean age was 57 ±4 years in the ARIC cohort, 72 ±5 years in CHS, and 64 ±8 years in MESA. The median baseline eGFR among all cohorts was 84 ±6 ml/min/1.73m2; 7.3% of participants had a baseline eGFR <60 ml/min/1.73m2. The prevalence of CHIP across all study cohorts was 6.6% in persons 50–60 years old, 9.0% in persons 60–70 years old, and 12.2% in persons greater than 70 years of age. The prevalence of CHIP was similar among men and women and in participants with and without diabetes (Table 1).
Table 1.
Baseline Characteristics of Study Populations
| Atherosclerosis Risk in Communities Study (n=6,575) | Cardiovascular Health Study (n=1,701) | Multi-Ethnic Study of Atherosclerosis (n=3,728) | ||||
|---|---|---|---|---|---|---|
| CHIP status | Yes | No | Yes | No | Yes | No |
| N | 584 | 5,991 | 240 | 1,461 | 178 | 3550 |
| Age, years | 58 (4) | 58 (4) | 74 (5) | 73 (5) | 68 (8) | 64 (8) |
| Male Sex, n(%) | 295 (50) | 2,789 (46) | 113 (47) | 626 (43) | 83 (46) | 1728 (48) |
| Race/ethnicity, n(%) | ||||||
| White | 489 (84) | 4,895 (82) | 205 (85) | 1,223 (84) | 85 (48) | 1462 (42) |
| Black | 95 (16) | 1095 (19) | 35 (15) | 238 (16) | 44 (24) | 851 (24) |
| Chinese | - | - | - | - | 14 (8) | 473 (14) |
| Hispanic | - | - | - | - | 35 (20) | 764 (22) |
| Diabetes, n(%) | 64 (10) | 650 (11) | 33 (14) | 222 (15) | 22 (12) | 428 (12) |
| BMI, kg/m2 | 28 (6) | 28 (6) | 26 (4) | 26 (4) | 28 (6) | 28 (6) |
| eGFR, ml/min/1.73m2 | 98 (14) | 100 (12) | 69 (16) | 71 (16) | 76 (14) | 80 (14) |
| Urine albumin-to-creatinine ratio, mg/g | - | - | 9 (6, 24) | 9 (5, 21) | 6 (3, 12) | 6 (3, 12) |
| Microalbuminuria, n(%) | 49 (10) | 437 (8) | 35 (15) | 223 (15) | 27 (16) | 293 (8) |
After age adjustment, the percentage of participants with a baseline eGFR <45, 45–59, and ≥60 ml/min/1.73m2 was similar by CHIP status (Figure 1, left panel). The presence of CHIP was not associated with baseline prevalent CKD after adjustment for age and age-squared (13% higher odds, 95% CI 10% lower to 42% higher; p-value = 0.27). Age-adjusted rates of microalbuminuria were 12.5% in participants with CHIP and 10.6% in participants without CHIP (Figure 1, right panel). The presence of CHIP was associated with a 17% higher urine albumin to creatinine ratio after age-adjustment (95% CI 3% to 33% higher; p-value = 0.02).
Figure 1. Age-adjusted associations of CHIP with baseline eGFR and microalbuminuria categories.
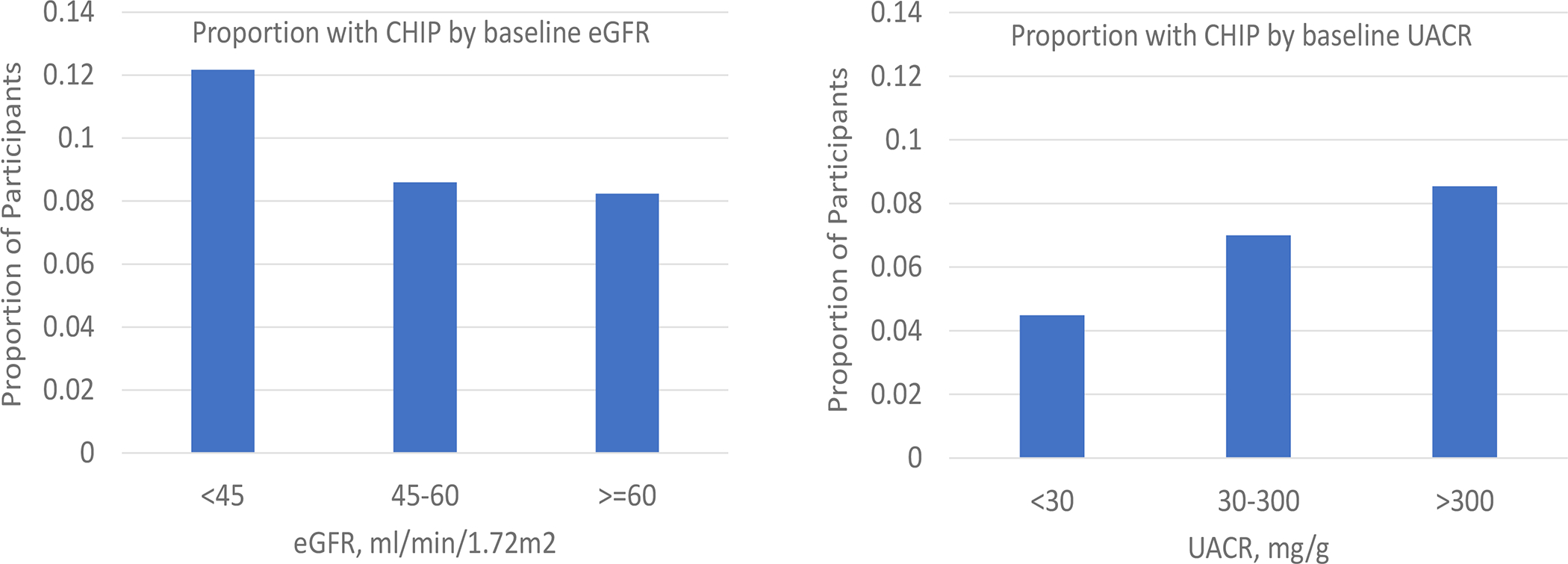
Y-axis depicts the age-adjusted (age and age-squared) proportions of participants with CHIP within each category of baseline eGFR <45, 45–59, and ≥60 ml/min/1.73m2 and within each category of baseline albumin-to-creatinine ratio <30, 30–300 and >300mg/g. Urine albumin-to-creatinine ratio was not concomitantly measured with CHIP in the ARIC Study and was not included in the estimates. Age-adjusted estimates were determined using Poisson regression. CHIP = Clonal hematopoiesis of Indeterminate Potential, eGFR = estimated glomerular filtration rate, uACR = urine albumin-to-creatinine ratio.
Median follow-up was 9 years in ARIC, 7 years in CHS, and 9 years in MESA (Table 2). The median number of eGFR measurements in these studies ranged from 2–4. There were 205 kidney function decline events over follow-up in persons with CHIP (2.1 events per 100-person years), and 2,041 kidney function decline events in persons without CHIP (1.7 events per 100-person years). In meta-analysis, the presence of CHIP at baseline was associated with a 17% greater risk of kidney function decline (Table 2; 95% CI 1% to 36% greater), after adjustment for baseline age, age-squared, sex, eGFR, uACR and diabetes (Figure 1). After adjustment for the same covariates, CHIP was not associated with the outcome of a 40% decline in eGFR (Supplemental Table 2), nor with incident eGFR<60 ml/min/1.73m2 (Supplemental Table 3). The least-squares mean slope of eGFR decline was −1.14 ml/min/1.73m2 per year in persons with CHIP and −1.15 ml/min/1.73m2 per year in persons without CHIP (Table 3). After adjustment, there was no significant difference in the slope of eGFR by CHIP status.
Table 2.
Associations of Clonal Hematopoiesis of Indeterminate Potential with 30% Decline in eGFR.
| CHIP | Number of participants | Number of eGFRs (IQR) | Median follow-up time, years (IQR) | Number of events | Incidence per 100 person-years | Unadjusted HR (95% CI) | Adjusted* HR (95%CI) | |
|---|---|---|---|---|---|---|---|---|
| ARIC | Yes | 584 | 3 (3,4) | 9 (8,22) | 143 | 2.20 | 1.23 (1.04,1.46) | 1.20 (1.01, 1.43) |
| No | 5,991 | 3 (3,4) | 9 (8,23) | 1,392 | 1.94 | reference | reference | |
| CHS | Yes | 240 | 3 (2,3) | 7 (3,7) | 40 | 2.14 | 1.48 (1.02, 2.13) | 1.38 (0.92, 2.06) |
| No | 1,461 | 3 (2,3) | 7 (4,7) | 216 | 1.53 | reference | reference | |
| MESA | Yes | 178 | 4 (4, 4) | 9 (9, 10) | 22 | 1.44 | 0.99 (0.64, 1.52) | 0.85 (0.55, 1.30) |
| No | 3,550 | 4 (4, 4) | 9 (9, 10) | 433 | 1.39 | reference | reference | |
| Meta-Analysis | Yes | 1,002 | 3 (3,4) | 8 (7, 14) | 205 | 2.05 | 1.24 (1.07, 1.43) | 1.17 (1.01, 1.36) |
| No | 11,002 | 3 (3,4) | 8 (7, 15) | 2,041 | 1.71 | reference | reference | |
| p = 0.004 | p = 0.036 |
ARIC = Atherosclerosis Risk in Communities Study; CHS = Cardiovascular Health Study; MESA = Multi-Ethnic Study of Atherosclerosis; CHIP = Clonal hematopoiesis of indeterminate potential; eGFR = estimated glomerular filtration rate.
Adjusted for age, age-squared, sex, baseline eGFR, diabetes, and log-urine albumin to creatinine ratio (where available at baseline).
Table 3.
Associations of Clonal Hematopoiesis of Indeterminate Potential with Longitudinal Change in eGFR.
| Cohort | CHIP | Number of participants | Number of eGFRs | Follow up time, years median (IQR) | Percent eGFR decline per year | Adjusted* difference (95% CI), %/year | Absolute eGFR decline per year | Adjusted* difference (95% CI), ml/min/1.73m2/year |
|---|---|---|---|---|---|---|---|---|
| ARIC | Yes | 584 | 3 (3,4) | 9 (8,22) | −2.30 (3.08) | −0.25 (−0.51, 0.01) | −1.87 (2.30) | −0.17 (−0.34, −0.01) |
| No | 5,991 | 3 (3,4) | 9 (8,23) | −2.02 (2.80) | reference | −1.69 (2.06) | reference | |
| CHS | Yes | 240 | 3 (2,3) | 7 (3,7) | −0.98 (4.60) | −0.13 (−0.60, 0.35) | −0.57 (2.67) | −0.09 (−0.39, 0.20) |
| No | 1,461 | 3 (2,3) | 7 (4,8) | −0.85 (3.89) | reference | −0.47 (2.29) | reference | |
| MESA | Yes | 178 | 4 (4, 4) | 9 (9, 10) | −1.60 (2.60) | 0.36 (−0.01, 0.74) | −1.03 (1.44) | 0.22 (0.01, 0.44) |
| No | 3,550 | 4 (4, 4) | 9 (9, 10) | −1.71 (2.44) | reference | −1.16 (1.49) | reference | |
| Meta-Analysis | Yes | 1,002 | 3 (3,4) | 9 (7, 16) | −1.75 (1.82) | −0.02 (−0.40, 0.36) | −1.14 (1.11) | −0.02 (−0.26, 0.23) |
| No | 11,002 | 3 (3,4) | 9 (8, 17) | −1.66 (1.66) | reference | −1.15 (1.07) | reference | |
| p = 0.934 | p = 0.906 |
ARIC = Atherosclerosis Risk in Communities Study; CHS = Cardiovascular Health Study; MESA = Multi-Ethnic Study of Atherosclerosis; CHIP = Clonal hematopoiesis of indeterminate potential; eGFR = estimated glomerular filtration rate.
Adjusted for age, age-squared, sex, baseline eGFR, diabetes, and log-urine albumin to creatinine ratio (where available at baseline).
The size of the association between CHIP and kidney function decline was similar among DNMT3A and non-DNMT3A gene driver mutations (Figure 2, Supplemental Tables 4,5). Associations were also similar among participants with an eGFR<60 versus > 60 min/1.73m2 at baseline, and those aged<60 versus >60 years at baseline (Figure 2, Supplemental Tables 6–9). These distinctions were not statistically significant (p-values for interactions > 0.5).
Figure 2. Associations of CHIP with 30% decline in eGFR among subgroups of interest.
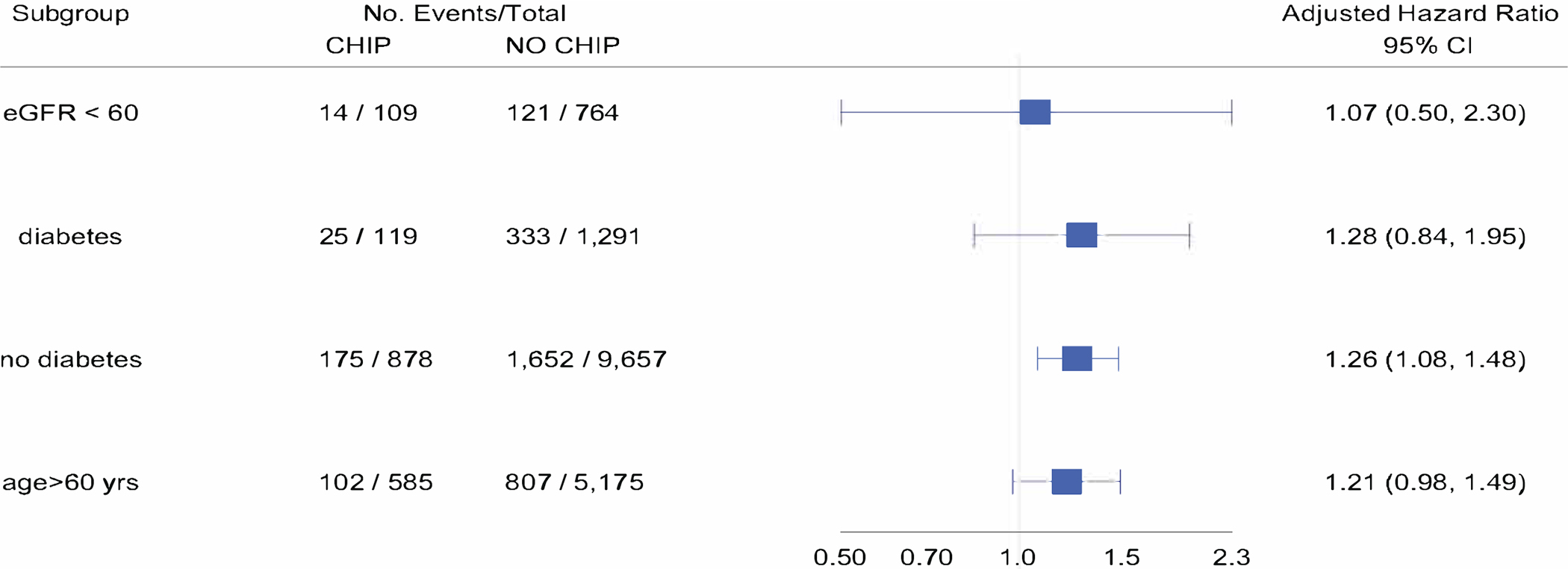
Forest plot of adjusted hazard ratio for association of CHIP with 30% decline in eGFR (meta-analysis of ARIC, CHS and MESA) among 1) individuals with eGFR<60ml/min/1.73m2 at baseline, 2) diabetes at baseline, 3) no diabetes at baseline and age >60 years at baseline. Adjusted for age, age-squared, sex, baseline eGFR, diabetes, and log-urine albumin to creatinine ratio (where available at baseline).CHIP = Clonal hematopoiesis of Indeterminate Potential, eGFR = estimated glomerular filtration rate.
DISCUSSION
In this study, we assessed the association between clonal hematopoiesis and kidney function decline and found CHIP was associated with 17% higher risk of eGFR decline among adults with relatively intact kidney function. We observed non-significant results for outcomes of a 40% eGFR decline, incident eGFR<60ml/min/1.73m2 and continuous slope of decline. Heterogeneity in the results was seen across the studies with numerically positive associations in the ARIC and CHS cohorts and null results in MESA. Taken together, these findings provide evidence for a link between CHIP and kidney function decline in the general population. However, wide confidence limits, low CHIP prevalence, and inherent methodologic limitations of these cohorts for assessing longitudinal changes in kidney function leave residual uncertainty regarding the potential kidney consequences of CHIP and support further studies of this question. Three previous studies have examined the association of CHIP and kidney function in humans, with conflicting results. Among 190,487 participants in the UK Biobank, individuals with myeloid clonal hematopoiesis had lower eGFR – as estimated from cystatin but not from creatinine.26 Among those with CKD, CHIP was associated with higher odds of cardiovascular events and death.26 In a small study of patients with advanced CKD, Vlasschaert et al reported a high prevalence of CHIP (25%) and that those with the condition had a 2.2-fold greater risk of kidney failure within 5 years.27 In contrast, a recent study of individuals with diabetic kidney disease found no association of CHIP with incident or progressive decline in kidney function.28
Evidence from animal models also suggests that clonal hematopoiesis could plausibly contribute to kidney function decline. In hypercholesterolemic mice, transplantation of hematopoietic stem cells genetically deficient for Tet2, one of the most commonly altered genes in CHIP, leads to accelerated age-related glomerulosclerosis.4 Transplantation of Tet2-deficient hematopoietic stem cells results in rapid infiltration of Tet2-deficient donor cells into the kidney interstitium, replacement of resident macrophages, and accelerated interstitial fibrosis.11 Moreover, transplantation of Tet2-deficient hematopoietic stem cells leads to the expansion of atherosclerotic plaque size and stimulation of pro-inflammatory cytokines; these experimental results are corroborated by associations of CHIP with incident cardiovascular events in human populations.4,5
It is possible that CHIP accelerates kidney function decline in persons with established kidney disease but has smaller effects on kidney function in the absence of pre-existing disease or injury. We did not detect significant differences in the size of the association between CHIP and eGFR decline among participants with a baseline eGFR <60 versus ≥60 ml/min/1.73m2.
However, the number of participants with CKD, particularly moderate-advanced disease, was too small to reliably address this question, motivating future studies in kidney disease populations.
It is also possible that the study lacked sensitivity for detecting a true association between CHIP and kidney function decline. The included cohorts included relatively few serum creatinine measurements over time, reducing precision and increasing the possibility for differential censoring. Most serum creatinine measurements were within the normal range, in which GFR estimating equations are least precise. The number of participants with CHIP who experienced a 30% decline in eGFR was relatively small, contributing to the wide confidence limits. Larger declines in eGFR are clinically meaningful and used as outcomes in randomized trials; however, such declines were rare in this study population.
In summary, we detected an association between CHIP and kidney function decline in three general population cohorts without known kidney disease. Further studies are needed to investigate this novel condition and its potential impact on kidney disease.
Supplementary Material
Table S1. Leukemogenic driver mutations used for CHIP variant calling
Table S2. Associations of Clonal Hematopoiesis with 40% Decline in eGFR
Table S3. Associations of Clonal Hematopoiesis with Incident eGFR < 60 ml/min/1.73m2
Table S4. Associations of DNMT3A clonal hematopoiesis with 30% decline in eGFR and longitudinal change in eGFR
Table S5. Associations of non-DNMT3A clonal hematopoiesis with 30% decline in eGFR and longitudinal change in eGFR
Table S6. Associations of Clonal Hematopoiesis with 30% decline in eGFR and longitudinal change in eGFR among individuals with eGFR<60 at baseline
Table S7. Associations of Clonal Hematopoiesis with 30% decline in eGFR and longitudinal change in eGFR among individuals with diabetes at baseline
Table S8. Associations of Clonal Hematopoiesis with 30% decline in eGFR and longitudinal change in eGFR among individuals without diabetes at baseline
Table S9. Associations of Clonal Hematopoiesis with 30% decline in eGFR and longitudinal change in eGFR among individuals aged > 60 years at baseline
Support:
Whole genome sequencing (WGS) for the Trans-Omics in Precision Medicine (TOPMed) program was supported by the National Heart, Lung and Blood Institute (NHLBI). Genome sequencing for “NHLBI TOPMed: Whole Genome Sequencing and Related Phenotypes in the Framingham Heart Study” (phs000974.v1.p1) was performed at the Broad Institute Genomics Platform (3R01HL092577-06S1, 3U54HG003067-12S2). Genome sequencing for “NHLBI TOPMed: the Atherosclerosis Risk in Communities Study” (phs001211.v1.p1) was performed at the Broad Institute Genomics Platform (3U54HG003273-12S2, HHSN268201500015C, 3R01HL092577-06S1). Genome sequencing for “NHLBI TOPMed: the Multi-Ethnic Study of Atherosclerosis” (phs001416.v1.p1) was performed at the Broad Institute Genomics Platform (HHSN268201600034I, 3U54HG003067-13S1). Genome sequencing for “NHLBI TOPMed: the Cardiovascular Health Study” (phs001368.v1.p1) was performed at the Broad Institute Genomics Platform (HHSN268201600034I) and Baylor College of Medicine Human Genome Sequencing Center (HHSN268201600033I). Centralized read mapping and genotype calling, along with variant quality metrics and filtering were provided by the TOPMed Informatics Research Center (3R01HL-117626-02S1; contract HHSN268201800002I). Phenotype harmonization, data management, sample-identity QC, and general study coordination were provided by the TOPMed Data Coordinating Center (3R01HL-120393-02S1; contract HHSN268201800001I). We gratefully acknowledge the studies and participants who provided biological samples and data for TOPMed. PN is supported by grants from the National, Heart, Lung, and Blood Institute (R01HL142711, R01HL148050, R01HL151283, R01HL148565, R01HL135242, R01HL151152), National Institute of Diabetes and Digestive and Kidney Diseases (R01DK125782), Fondation Leducq (TNE-18CVD04), and Massachusetts General Hospital (Paul and Phyllis Fireman Endowed Chair in Vascular Medicine).
Footnotes
Financial Disclosure: PN reports investigator-initiated grants from Amgen, Apple, Boston Scientific, AstraZeneca, and Novartis, personal fees from Apple, AstraZeneca, Genentech, Novartis, Blackstone Life Sciences, and Foresite Labs, and spousal employment at Vertex, all unrelated to the present work. BP serves on the Steering Committee of the Yale Open Data Access Project funded by Johnson & Johnson. MBL reports speaker and advisory fees from Otsuka, Bayer, Sanofi Genzyme, and Reata. The remaining authors declare that they have no relevant financial interests.
Peer Review: Received January 26, 2022. Evaluated by 2 external peer reviewers, with direct editorial input from a Statistics/Methods Editor, an Associate Editor, and a Deputy Editor who served as Acting Editor-in-Chief. Accepted in revised form August 9, 2022. The involvement of an Acting Editor-in-Chief was to comply with AJKD’s procedures for potential conflicts of interest for editors, described in the Information for Authors & Journal Policies.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Data Sharing:
Individual whole-genome sequence data for TOPMed whole genomes, individual-level harmonized phenotypes, harmonized germline variant call sets, the CHIP somatic variant call sets, RNA-Seq and peripheral blood methylation data used in this analysis are available through restricted access via the dbGaP.
REFERENCES
- 1.Steensma DP, Bejar R, Jaiswal S, et al. Clonal hematopoiesis of indeterminate potential and its distinction from myelodysplastic syndromes. Blood. Jul 2015;126(1):9–16. doi: 10.1182/blood-2015-03-631747 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Bick AG, Weinstock JS, Nandakumar SK, et al. Inherited causes of clonal haematopoiesis in 97,691 whole genomes. Nature. 10 2020;586(7831):763–768. doi: 10.1038/s41586-020-2819-2 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Genovese G, Kähler AK, Handsaker RE, et al. Clonal hematopoiesis and blood-cancer risk inferred from blood DNA sequence. N Engl J Med. Dec 2014;371(26):2477–87. doi: 10.1056/NEJMoa1409405 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Jaiswal S, Natarajan P, Silver AJ, et al. Clonal Hematopoiesis and Risk of Atherosclerotic Cardiovascular Disease. N Engl J Med. 07 2017;377(2):111–121. doi: 10.1056/NEJMoa1701719 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Bick AG, Pirruccello JP, Griffin GK, et al. Genetic Interleukin 6 Signaling Deficiency Attenuates Cardiovascular Risk in Clonal Hematopoiesis. Circulation. 01 2020;141(2):124–131. doi: 10.1161/CIRCULATIONAHA.119.044362 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Mas-Peiro S, Hoffmann J, Fichtlscherer S, et al. Clonal haematopoiesis in patients with degenerative aortic valve stenosis undergoing transcatheter aortic valve implantation. Eur Heart J. 02 2020;41(8):933–939. doi: 10.1093/eurheartj/ehz591 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Dorsheimer L, Assmus B, Rasper T, et al. Association of Mutations Contributing to Clonal Hematopoiesis With Prognosis in Chronic Ischemic Heart Failure. JAMA Cardiol. 01 2019;4(1):25–33. doi: 10.1001/jamacardio.2018.3965 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Yu B, Roberts MB, Raffield LM, et al. Supplemental Association of Clonal Hematopoiesis With Incident Heart Failure. J Am Coll Cardiol. Jul 2021;78(1):42–52. doi: 10.1016/j.jacc.2021.04.085 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Fuster JJ, MacLauchlan S, Zuriaga MA, et al. Clonal hematopoiesis associated with TET2 deficiency accelerates atherosclerosis development in mice. Science. 02 2017;355(6327):842–847. doi: 10.1126/science.aag1381 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Sano S, Wang Y, Yura Y, et al. -Mediated Clonal Hematopoiesis Accelerates Pathological Remodeling in Murine Heart Failure. JACC Basic Transl Sci. Oct 2019;4(6):684–697. doi: 10.1016/j.jacbts.2019.05.013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Sano S, Oshima K, Wang Y, Katanasaka Y, Sano M, Walsh K. CRISPR-Mediated Gene Editing to Assess the Roles of Tet2 and Dnmt3a in Clonal Hematopoiesis and Cardiovascular Disease. Circ Res. 07 2018;123(3):335–341. doi: 10.1161/CIRCRESAHA.118.313225 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Qin X, Wang Y, Li Y, et al. Risk factors for renal function decline in adults with normal kidney function: a 7-year cohort study. J Epidemiol Community Health. Aug 2015;69(8):782–8. doi: 10.1136/jech-2014-204962 [DOI] [PubMed] [Google Scholar]
- 13.Young BA, Katz R, Boulware LE, et al. Risk Factors for Rapid Kidney Function Decline Among African Americans: The Jackson Heart Study (JHS). Am J Kidney Dis. 08 2016;68(2):229–239. doi: 10.1053/j.ajkd.2016.02.046 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Taliun D, Harris DN, Kessler MD, et al. Sequencing of 53,831 diverse genomes from the NHLBI TOPMed Program. Nature. 02 2021;590(7845):290–299. doi: 10.1038/s41586-021-03205-y [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.The Atherosclerosis Risk in Communities (ARIC) Study: design and objectives. The ARIC investigators. Am J Epidemiol. Apr 1989;129(4):687–702. [PubMed] [Google Scholar]
- 16.Fried LP, Borhani NO, Enright P, et al. The Cardiovascular Health Study: design and rationale. Ann Epidemiol. 1991 1991;1(3):263–76. [DOI] [PubMed] [Google Scholar]
- 17.Bild DE, Bluemke DA, Burke GL, et al. Multi-ethnic study of atherosclerosis: objectives and design. Am J Epidemiol. Nov 2002;156(9):871–81. [DOI] [PubMed] [Google Scholar]
- 18.Cibulskis K, Lawrence MS, Carter SL, et al. Sensitive detection of somatic point mutations in impure and heterogeneous cancer samples. Nat Biotechnol. Mar 2013;31(3):213–9. doi: 10.1038/nbt.2514 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Inker LA, Eneanya ND, Coresh J, et al. New Creatinine- and Cystatin C-Based Equations to Estimate GFR without Race. N Engl J Med. Nov 4 2021;385(19):1737–1749. doi: 10.1056/NEJMoa2102953 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Coresh J, Turin TC, Matsushita K, et al. Decline in Estimated Glomerular Filtration Rate and Subsequent Risk of End-Stage Renal Disease and Mortality. JAMA. Jun 2014;doi: 10.1001/jama.2014.6634 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Levey AS, Inker LA, Matsushita K, et al. GFR decline as an end point for clinical trials in CKD: a scientific workshop sponsored by the National Kidney Foundation and the US Food and Drug Administration. Am J Kidney Dis. Dec 2014;64(6):821–35. doi: 10.1053/j.ajkd.2014.07.030 [DOI] [PubMed] [Google Scholar]
- 22.Kramer H, Jacobs DR, Bild D, et al. Urine albumin excretion and subclinical cardiovascular disease. The Multi-Ethnic Study of Atherosclerosis. Hypertension. Jul 2005;46(1):38–43. doi: 10.1161/01.HYP.0000171189.48911.18 [DOI] [PubMed] [Google Scholar]
- 23.de Boer IH, Katz R, Cao JJ, et al. Cystatin C, albuminuria, and mortality among older adults with diabetes. Diabetes Care. Oct 2009;32(10):1833–8. doi: 10.2337/dc09-0191 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Huang M, Matsushita K, Sang Y, Ballew SH, Astor BC, Coresh J. Association of kidney function and albuminuria with prevalent and incident hypertension: the Atherosclerosis Risk in Communities (ARIC) study. Am J Kidney Dis. Jan 2015;65(1):58–66. doi: 10.1053/j.ajkd.2014.06.025 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Arnlöv J, Evans JC, Meigs JB, et al. Low-grade albuminuria and incidence of cardiovascular disease events in nonhypertensive and nondiabetic individuals: the Framingham Heart Study. Circulation. Aug 2005;112(7):969–75. doi: 10.1161/CIRCULATIONAHA.105.538132 [DOI] [PubMed] [Google Scholar]
- 26.Dawoud AAZ, Gilbert RD, Tapper WJ, Cross NCP. Clonal myelopoiesis promotes adverse outcomes in chronic kidney disease. Leukemia. Feb 2022;36(2):507–515. doi: 10.1038/s41375-021-01382-3 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Vlasschaert C, McNaughton AJM, Chong M, et al. Association of Clonal Hematopoiesis of Indeterminate Potential with Worse Kidney Function and Anemia in Two Cohorts of Patients with Advanced Chronic Kidney Disease. J Am Soc Nephrol. May 2022;33(5):985–995. doi: 10.1681/ASN.2021060774 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Denicolo S, Vogi V, Keller F, et al. Clonal Hematopoiesis of Indeterminate Potential and Diabetic Kidney Disease: A Nested Case-Control Study. Kidney Int Rep. Apr 2022;7(4):876–888. doi: 10.1016/j.ekir.2022.01.1064 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Table S1. Leukemogenic driver mutations used for CHIP variant calling
Table S2. Associations of Clonal Hematopoiesis with 40% Decline in eGFR
Table S3. Associations of Clonal Hematopoiesis with Incident eGFR < 60 ml/min/1.73m2
Table S4. Associations of DNMT3A clonal hematopoiesis with 30% decline in eGFR and longitudinal change in eGFR
Table S5. Associations of non-DNMT3A clonal hematopoiesis with 30% decline in eGFR and longitudinal change in eGFR
Table S6. Associations of Clonal Hematopoiesis with 30% decline in eGFR and longitudinal change in eGFR among individuals with eGFR<60 at baseline
Table S7. Associations of Clonal Hematopoiesis with 30% decline in eGFR and longitudinal change in eGFR among individuals with diabetes at baseline
Table S8. Associations of Clonal Hematopoiesis with 30% decline in eGFR and longitudinal change in eGFR among individuals without diabetes at baseline
Table S9. Associations of Clonal Hematopoiesis with 30% decline in eGFR and longitudinal change in eGFR among individuals aged > 60 years at baseline
Data Availability Statement
Individual whole-genome sequence data for TOPMed whole genomes, individual-level harmonized phenotypes, harmonized germline variant call sets, the CHIP somatic variant call sets, RNA-Seq and peripheral blood methylation data used in this analysis are available through restricted access via the dbGaP.