

Abstract
Chronic pain caused by injury or disease of the nervous system (neuropathic pain) has been linked to persistent electrical hyperactivity of the sensory neurons (nociceptors) specialized to detect damaging stimuli and/or inflammation. This pain and hyperactivity are considered maladaptive because both can persist long after injured tissues have healed and inflammation has resolved. While the assumption of maladaptiveness is appropriate in many diseases, accumulating evidence from diverse species, including humans, challenges the assumption that neuropathic pain and persistent nociceptor hyperactivity are always maladaptive. We review studies indicating that persistent nociceptor hyperactivity has undergone evolutionary selection in widespread, albeit selected, animal groups as a physiological response that can increase survival long after bodily injury, using both highly conserved and divergent underlying mechanisms.
Keywords: Aplysia, cephalopod, chronic pain, human, primary afferent neuron, spontaneous activity
Chronic neuropathic pain and associated nociceptor hyperactivity are nearly always assumed to be maladaptive
In evolutionary medicine, it is recognized that some disease symptoms commonly assumed to be maladaptive, such as diarrhea and fever, are in fact evolutionary adaptations promoting survival and reproductive success – notwithstanding the distress they cause [1]. One malady almost universally considered maladaptive is chronic neuropathic pain (see Glossary) [2,3], which contrasts with acute pain that has clear protective functions. In humans and rodents, many examples of neuropathic pain have been linked to persistent electrical hyperactivity in primary nociceptors (Figure 1, Box 1) (reviewed in 2,4,5]). Because nociceptor activation evokes conscious pain in humans [6,7], and because nociceptor activity is sufficient and sometimes necessary for behaviors in rodents that indicate pain [8–11], continuing hyperactivity in nociceptors is recognized as a major source of persistent mammalian pain. Importantly, nociceptor hyperactivity is often generated “spontaneously” at “ectopic” sites near axonal injury or in nociceptor cell bodies rather than in the peripheral terminals where activation normally occurs (Figure 1). Moreover, both nociceptor activity and the associated pain sometimes persist after healing of an initiating injury. These properties are often taken as evidence of maladaptive pathology. Here we present evidence from diverse taxa indicating that persistent nociceptor hyperactivity can be an evolutionarily adaptive trait. While our focus is on nociceptor hyperactivity, alterations of various cell types in the central nervous system (central sensitization) are also important for persistent pain-like behavior, at least in mammals [2,12] and probably in insects (see below).
Figure 1. Nociceptor hyperactivity.
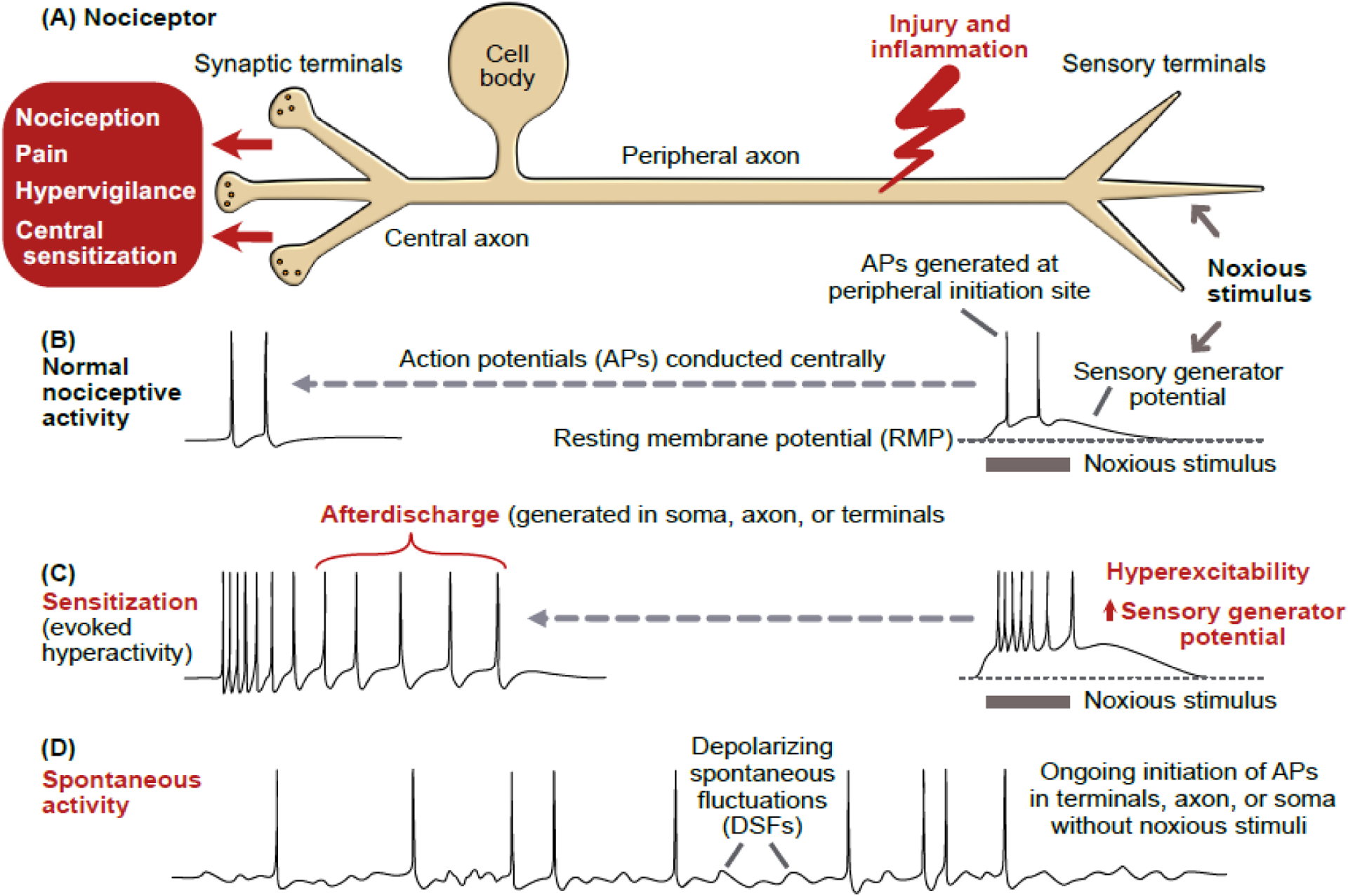
(A) Schematic of a primary nociceptor in vertebrates, gastropod molluscs, and leeches. The cell body is centrally located and distant from peripheral terminals where survivable injury and inflammation are most likely. In vertebrates, the cell body is in a ganglion near the central nervous system (CNS). In molluscs and leeches, nociceptor cell bodies are in ganglia within the CNS. (B) Normal nociceptive activity initiated by noxious stimulation of peripheral terminals produces a depolarizing sensory generator potential that may reach threshold for action potentials (APs). These APs are conducted to the CNS, sometimes evoking pain. (C) Illustration of sensitization of nociceptive activity evoked by the same noxious stimulus as in panel B, with the evoked hyperactivity following injury caused by a larger sensory generator potential, increased terminal excitability, and afterdischarge triggered by the evoked APs, which sometimes evoke pain, hypervigilance, and central sensitization. Experimentally, injection of pulses or ramps of depolarizing current (usually into the cell body) is often used to reveal hyperexcitability (see Box 1). (D) Spontaneous activity after injury, defined as ongoing activity generated in the absence of concurrent extrinsic stimulation of a neuron (the spontaneous activity is sometimes generated by depolarizing spontaneous fluctuations, DSFs) [100]. The term spontaneous activity is often used more loosely in referring to any nociceptor activity in the absence of evident ongoing noxious stimulation, including cases where intrinsic (cell autonomous) hyperexcitability and unobserved inflammatory signals combine to drive ongoing hyperactivity. Spontaneous activity and/or afterdischarge have been found in peripheral terminals, injured axons, and cell bodies of mammals and molluscs.
Box 1. Hyperactivity and hyperexcitability are not the same.
The terms hyperexcitability and hyperactivity are often used interchangeably but hyperactivity is arguably more important physiologically and evolutionarily. In studies of neuronal plasticity involving alterations of AP function, the most common experimental indicator of an altered electrophysiological state is modified electrical excitability – either hyperexcitability or hypoexcitability. Hyperexcitability is demonstrated when electrical stimulation more readily triggers an AP. Experimental delivery of depolarizing current, either in a series of increasingly depolarizing current steps or in response to a smooth depolarizing ramp of current, yields two indicators of hyperexcitability: generation of an AP by stimulation with less stimulating current (sometimes referred to as a decrease in rheobase) and/or AP generation at a more negative membrane potential. Under natural conditions, hyperexcitability would make it more likely that a physiological stimulus such as a sensory generator potential (Figure 1) or synaptic potential would reach AP threshold. For most neurons, the frequency and pattern of APs appear to be the critical physiological endpoints of excitability because they determine a neuron’s immediate effect on its postsynaptic targets and can control activity-dependent synaptic plasticity.
Hyperexcitability usually promotes hyperactivity, but many other intrinsic and extrinsic states of a neuron and its inputs will determine whether a neuron is hyperactive and what its level and pattern of activity are. As shown with examples in Figure 2, numerous drivers may contribute to hyperactivity in mammalian nociceptors after bodily injury, including intrinsic hyperexcitability, intrinsic hypersensitivity to extrinsic excitatory chemical signals, increased exposure to extrinsic excitatory signals, decreased exposure to extrinsic inhibitory signals, and hyposensitivity to inhibitory signals.
Evolutionary selection could change any or all of the intrinsic and extrinsic drivers of persistent nociceptor hyperactivity to make some species more or less likely to experience persistent pain-related states driven by continuing activity of nociceptors. The accessibility of nociceptors in diverse animal taxa enables direct inquiry into potentially adaptive functions of persistent nociceptor hyperactivity, as well as into similarities and differences in the underlying mechanisms.
Persistent nociceptor hyperactivity can have many sources
In mammals, scores of extrinsic injury- and inflammation-related signals are known (Figure 2A). Many can directly excite and/or sensitize nociceptors, and some of these signals may be persistently released in chronic pain conditions [13–15]. Extrinsic signals detected by nociceptors may be released by dying cells (damage-associated molecular patterns, DAMPs), released from (or detected by direct contact with) microbes (pathogen-associated molecular patterns, PAMPs), and released by numerous cell types, including cells in the peripheral and central nervous systems and the innate and adaptive immune systems (releasing amino acids, biogenic amines, purines, leukotrienes, cytokines, growth factors, and other signals) [16–25]. Furthermore, various cell types, including immune cells, release signals such as endogenous opioids that inhibit nociceptor activity [26]. In principle, continuing disinhibition by reduced release of inhibitory signals could promote persistent hyperactivity (Fig. 2A). In invertebrates, far less is known about the extrinsic signals that promote persistent nociceptor hyperactivity and the cells releasing them [27], although sensitizing signals coming from immune cells have been implicated in the marine snail Aplysia californica [28,29]. A fascinating question that exceeds the scope of our review concerns the evolution of immune systems and inflammatory mediators [e.g., 30,31–33]. Thus far, little is known about the important likelihood of coevolution of nociceptors and immune cells [34].
Figure 2. Sources of nociceptor hyperactivity following injury.
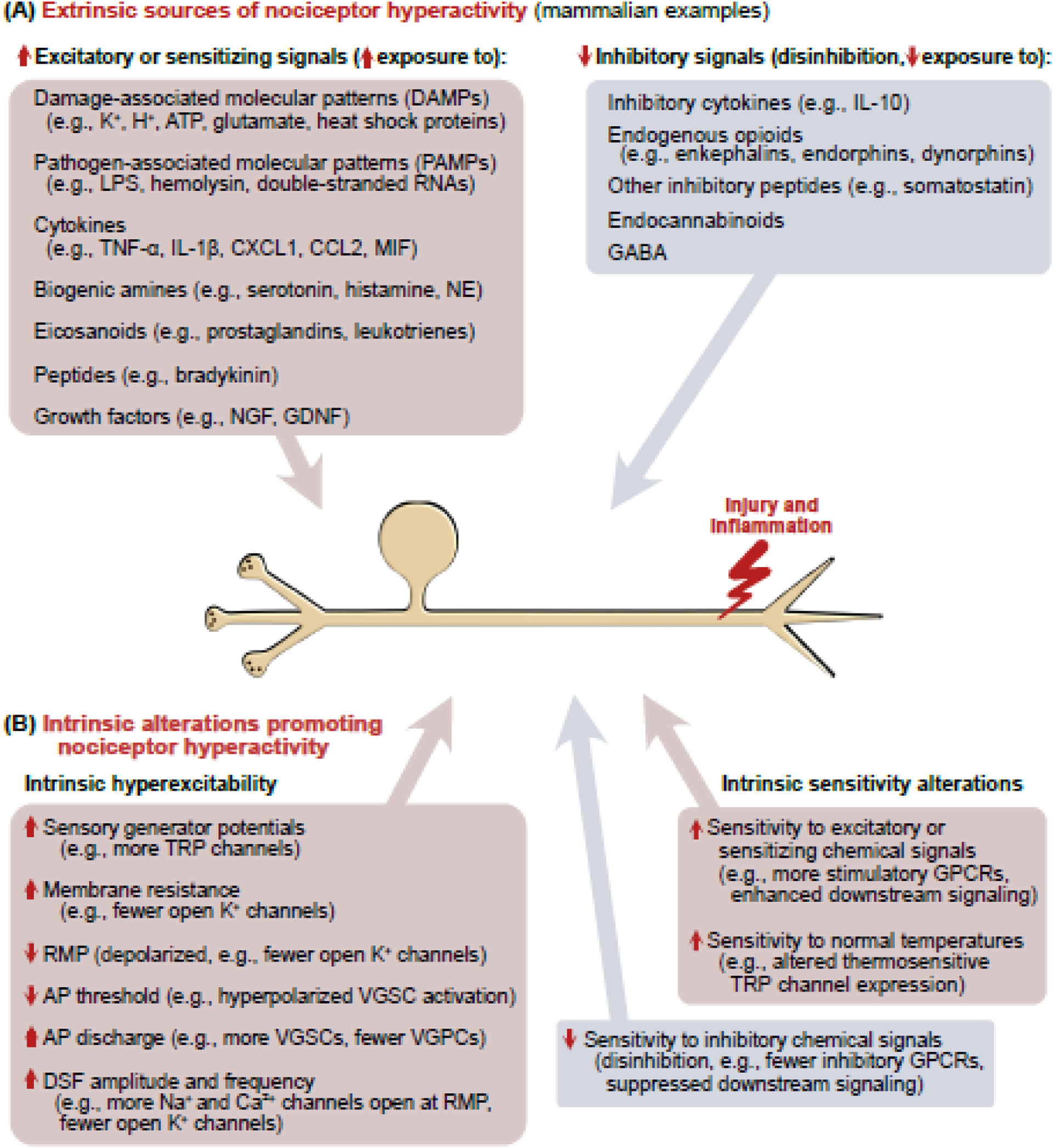
(A) Left: Some of the many extrinsic chemical signals implicated in mammals to excite and/or sensitize nociceptors. Many of these have multiple sources (e.g., glutamate and ATP are DAMPs released from ruptured cells but also are secreted exocytotically from various cell types including neurons). Right: Hyperactivity may also be promoted by reducing exposure to inhibitory chemical signals (disinhibition). (B) In principle, nociceptor hyperactivity can be promoted by intrinsic hyperexcitability via any or all the listed electrophysiological alterations [100,104]. Hyperactivity may also be promoted by increasing a nociceptor’s intrinsic sensitivity to excitatory or sensitizing sensory stimuli (including normal body temperature) or chemical signals, and by decreasing intrinsic sensitivity to inhibitory chemical signals. Abbreviations: AP, action potential; ATP, adenosine triphosphate, DSF, depolarizing spontaneous fluctuation; GABA, gamma-aminobutyric acid; GDNF, glial-derived neurotrophic factor; GPCR, G protein-coupled receptor; IL-1β, interleukin-1β; IL-10, interleukin 10; LPS, lipopolysaccharide; MIF, macrophage migration inhibitory factor; NGF, nerve growth factor; RMP, resting membrane potential; TNFα, tumor necrosis factor α; TRP, transient receptor potential; VGPC, voltage-gated potassium channel; VGSC, voltage-gated sodium channel.
Apart from extrinsic signals detected by nociceptors, intrinsic, cell-autonomous sources of persistent hyperactivity after bodily injury are also prominent in mammalian nociceptors (Figure 2B) and in some invertebrate nociceptors. Intrinsic alterations in identified nociceptors are readily compared across taxa, a major aim of this article. Intrinsic alterations include biophysical changes producing hyperexcitability, as well as alterations making a nociceptor either more sensitive to excitatory or sensitizing extrinsic signals, less sensitive to inhibitory signals, or more sensitive to normal body temperature. Importantly, intrinsic and extrinsic signaling often work cooperatively. Indeed, because nociceptors are normally electrically silent and not hyperexcitable, hyperactivity elicited by injury outside the nociceptor necessarily involves extrinsic triggers. These can have complex, long-lasting effects. For example, transient exposure to serotonin in Aplysia or rats triggers an intrinsic hyperactive state that lasts minutes or hours after the signal is removed, but prolonged or repeated exposure to serotonin or other inflammatory signals may sustain hyperactivity for longer periods, or induce an enduring, memory-like hyperactive state [35–39].
Primary nociceptors and evolution
Because nociceptors detect injury, and injury affecting survival and reproduction can occur in any animal, cells with this function should be widespread. Indeed, nociceptors have been identified in diverse animals, including roundworms, fruit flies, leeches, snails, squid, fish, birds, rodents, cats, pigs, monkeys, and humans [40,41]. Nociceptors thus provide an opportunity for broad comparative studies into evolutionary selection pressures for nociceptive plasticity and the roles of shared and divergent mechanisms. Adding to the evolutionary interest of nociceptors, adaptive cellular responses to injury or the threat of injury probably antedated the earliest animals, putatively shaping core mechanisms later used for basic neural function as well as nociception [42–44]. Nociceptors probably evolved very early, but may have also evolved independently in different lineages in response to common injury-related selection pressures.
Taxa in which persistent nociceptor hyperactivity may not be functionally important
Insight into the adaptive roles of a physiological trait can be gained by comparing the animals and ecological conditions in which the trait is prominent with those in which it appears to be weak or absent. We begin with nociceptors exhibiting little evidence of persistent hyperactivity after bodily injury.
Nematodes
Caenorhabditis elegans has polymodal nociceptors that repeatedly respond to noxious mechanical or heat stimuli and receive neuromodulatory input to prevent habituation to noxious stimuli [45]. Unusually, C. elegans neurons primarily signal by graded depolarizing responses rather than action potentials (APs, see Figure 1) [46]. In principle, persistent depolarization without APs might maintain nociceptive sensitization, but neither persistent depolarization nor acute or persistent AP generation has been reported in nematode nociceptors after injury. While a lack of observed nociceptor hyperactivity in this group (or any other taxon) does not prove it is absent, the short lifespan of C. elegans (about 20 days) and its tiny body size might be factors that make persistent nociceptor alterations relatively inconsequential (Table 1), despite the existence of predators of C. elegans [47].
Table 1.
Summary of evidence for functionally important, persistent nociceptor hyperactivity and complementary neural and immune alterations after injury in different taxa
| Phylum (genus or species) | Injury-induced nociceptor hyperactivity? | Survival link? | Required cell signaling in nociceptors | Complementary alterations in other cell types | Selected references |
|---|---|---|---|---|---|
| Nematoda (C. elegans) | Not observed | No | Probably none | Unknown | 45, 46 |
| Arthropoda (Drosophila - fly) | Unclear | Unknown | Unclear | Sensitization of central interneurons, central disinhibition | 50–54 |
| Mollusca (Aplysia - snail) | Hyperexcitability, afterdischarge | Unknown but plausible | cAMP, PKA, CREB, ERK, MNK, eIF4E | Sensitization of central interneurons, central disinhibition | 35, 55, 66–74, 106, 109 |
| Mollusca (Doryteuthis – squid) | Hyperexcitability, afterdischarge, spontaneous activity | Direct evidence | Unknown | Unknown | 75, 86, 87 |
| Chordata (Mus or Rattus - laboratory mice or rats) | Hyperexcitability, afterdischarge, spontaneous activity | Indirect evidence | cAMP, PKA, CREB, ERK, MNK, eIF4E | Sensitization of central interneurons, central disinhibition, glial and immune cell activation | 2–5, 11, 12, 17, 25, 26, 52, 77, 78, 81, 82, 88, 93, 100–103, 107, 108, 110–112, 114–118 |
| Chordata (Heterocephalus naked mole rat) | Not observed | No | Probably none | Unknown | 59, 60, 63–65, |
| Chordata (Homo sapiens) | Hyperexcitability, spontaneous activity | Unknown but plausible | Unknown | Probable sensitization of central interneurons, central disinhibition, glial activation, direct evidence for immune cell activation in DRG | 2, 7, 13, 24, 79, 80, 83, 84, 104 |
cAMP, cyclic adenosine monophosphate; CREB, cAMP response element-binding protein; DRG, dorsal root ganglia; eIF4E, eukaryotic translation initiation factor 4E; ERK, extracellular-signal-regulated kinase; MNK, mitogen-activated protein kinase-interacting kinase; PKA, protein kinase A.
Insects
Larvae of the fruit fly Drosophila melanogaster show behavioral sensitization after tissue injury, including a lowering of threshold for heat- or mechanically induced defensive rolling following UV irradiation of the epidermis [48,49]. Behavioral sensitization was linked to various molecular changes in heat-sensitive nociceptors [50], and accompanied by an increase in the range of temperatures that evoked continuing generation of APs [51]. In rodents, “spontaneous” pain may be driven by continuing activity in heat-sensitive nociceptors resulting from a lowering of threshold into the range of normal body temperature after injury [52]. However, for heat allodynia in Drosophila, UV-induced hypersensitivity of defensive behavior was recently found to depend on the acquisition of heat sensitivity by central neurons rather than enhanced heat sensitivity in nociceptors [53]. Thus, if injury-induced nociceptor hyperactivity occurs (which has not been demonstrated physiologically), it appears to contribute little to the UV-induced behavioral sensitization. Results from adult Drosophila also point to alterations in the central nervous system (CNS) underlying injury-induced hypersensitivity of a defensive behavior, without obvious contributions from injured primary nociceptors [54]. Amputation of one leg lowered the threshold for jumping escape to heat stimulation for at least 3 weeks. Initial nociceptor activation was necessary for inducing permanent central alterations (excitotoxic loss of GABAergic inhibitory neurons, producing central sensitization) and behavioral sensitization to heat test stimuli. However, no nociceptor alterations were described other than loss of all nociceptors in the amputated limb [54]. The use of amputation highlights an unusual feature of Drosophila nociceptors that may be important for the evolution of injury-induced persistent nociceptor hyperactivity. Unlike in leeches, Aplysia, squid, and vertebrates, where nociceptor cell bodies occur within centralized ganglia, Drosophila’s nociceptor cell bodies are located just beneath the epidermis where they can easily be killed by superficial tissue injuries. This anatomical feature in insects should prevent persistent site-specific nociceptor sensitization after injury [55]. It is consistent with the lack of enhanced sensory activity recorded from nerves innervating a previously injured region in larvae of the moth Manduca sexta [56], and with central mechanisms implicated in generalized sensitization and hypervigilance in Drosophila [53,54].
Mole-rats
Most investigations of persistent nociceptor hyperactivity in mammals have used laboratory rats (specifically, Rattus norvegicus), and mice (Mus musculus), which show complex alterations of both behavior and nociceptor function after injury. Quite different nociceptive reactions have been found in naked mole-rats (Heterocephalus glaber), which show distinctive traits related to their subterranean habitat [57]. Two differences have been found between nociceptors of naked mole-rats and laboratory rodents that are consistent with limited injury-induced nociceptor hyperactivity in mole-rats.
First, mole-rats have far fewer cutaneous nociceptors. In most mammals, the skin is innervated by ~4 times as many unmyelinated C-fibers as myelinated A-fibers, and most cutaneous C-fibers are nociceptors [58]. In adult naked mole-rats, the ratio of cutaneous C- to A-fibers is ~1:1 [59,60]. Fewer nociceptors in a subterranean animal might represent an evolutionary response to reduce the energy costs of maintaining defensive neurons that are less useful in an environment where predation is reduced, as has been suggested for a central defensive neuron, the Mauthner cell, in a cave fish species [61,62].
Second, nociceptors in naked mole-rats exhibit adaptations that limit their ability to drive pain under inflammatory conditions. Presumed nociceptors in mammals are often identified by sensitivity to capsaicin, the pain-evoking ingredient of chili peppers, and by expression of the capsaicin receptor, transient receptor potential vanilloid 1 (TRPV1). In other mammals, TRPV1+ nociceptors are important for driving inflammatory pain. Unexpectedly, in naked mole-rats the synapses of TRPV1+ sensory neurons are distributed randomly throughout the dorsal spinal cord [59], rather than being concentrated in the superficial dorsal horn of the spinal cord that is critical for relaying nociception in other mammals. Another adaptation reduces acid sensitivity. Tissue injury and inflammation are accompanied by tissue acidosis, and most mammals utilize a plethora of acid-sensitive receptors and ion channels to activate nociceptors and drive inflammatory pain [63]. However, naked mole-rats live in large colonies in poorly ventilated burrows, and thus experience high CO2 levels. CO2 can produce carbonic acid, releasing protons that activate acid receptors of naked mole-rats and other rodents. Surprisingly, acid fails to evoke APs or consequent pain behavior in naked mole-rats. This is because a mutation in voltage-gated Na+ channel NaV1.7 enables protons to block the channel and prevent AP generation under acidic conditions [64].
A third adaptation is reduced function of the naked mole-rat tropomyosin receptor kinase A (TrkA) for nerve growth factor (NGF), which is released in mammals during injury and inflammation. This TrkA hypofunctionality results in an absence of NGF-induced hyperalgesia in naked mole-rats and might also contribute developmentally to reduced nociceptor number. These adaptations in the naked mole-rat and additional adaptations in related mole-rats [65] should reduce the likelihood of persistent nociceptor hyperactivity being induced by injury and inflammation (although this prediction has yet to be tested directly). Loss of nociceptor hyperactivity might have been promoted during evolution by the relative safety from predation afforded by subterranean environments.
Taxa in which persistent nociceptor hyperactivity is likely to be functionally important
All the species in which persistent nociceptor hyperactivity has been observed after nerve or tissue injury have probably been subject to high levels of predation in their recent evolutionary history. In each case, persistent hyperactivity is generated near a site of injury and sometimes also in nociceptor cell bodies distant from the body surface (Figure 1) (Table 1).
Gastropod molluscs
Aplysia californica, lacks a shell and is one of the larger invertebrates, reaching weights of ~1 kg in its 2-year lifespan. Identified Aplysia nociceptors are activated preferentially by pinching or crushing stimuli and are unresponsive to chemical or thermal stimuli. Hyperactivity lasting days to months after experimental injury (nerve crush or wounding that transects major nerve branches) is expressed as reduced AP threshold and afterdischarge (Figure 1C) generated in nociceptor peripheral terminals [66], the injured axon [67], and the cell body [68]. The function of nociceptor cell body hyperactivity in Aplysia is, via afterdischarge, to amplify the number of APs reaching central synaptic terminals, thereby enhancing defensive behavioral responses [69]. Many invertebrates, including Aplysia and leeches, lack image-forming eyes and depend upon chemical and mechanical senses to detect predatory threats. In Aplysia, nociceptive sensitization after injury reduces nociceptor mechanical threshold into the innocuous range [70], which may promote escape at the first contact with slowly moving predatory gastropods [71]. Aplysia also exhibit short-term central sensitization expressed in identified interneurons [72,73], and in vitro evidence is consistent with long-term central sensitization that may increase serotonin release onto nociceptors [74].
Cephalopod molluscs
Nociceptors that respond to intense mechanical stimuli and fail to respond to heat have also been identified in the squid Doryteuthis pealeii. Activity recorded in fin-innervating nerves showed that nociceptors are activated by forces that can damage the fin, and, following noxious activation, the nociceptor mechanical activation threshold decreased dramatically (~50 fold) [75]. Two findings in this study were unexpected. First, injury produced either experimentally to the fin in vivo or more naturally to various body regions (caused by attacks from other squid) produced spontaneous activity (Figure 1D) lasting at least 24 hours after injury. This remains the only reported invertebrate example (to our knowledge) of injury-induced spontaneous activity in primary afferent neurons. Second, after injury to one side of the animal, both nociceptor sensitization to mechanical stimulation and spontaneous activity were found in nociceptors innervating the contralateral fin. This generalized hyperactivity contrasts with site-specific hyperactivity in mammalian and gastropod nociceptors that is usually restricted to neurons with innervation close to injured regions.
Mammals
Most studies of persistent nociceptor hyperactivity in mammals have utilized rat or mouse neuropathic pain models [reviewed by 2,4,5]. Hyperactivity can manifest in nociceptors as increased generation of APs evoked by extrinsic signals or generated by intrinsic alterations (spontaneous activity) (Figures 1C, 1D, 2A, 2B, Box 1). Cell body hyperactivity is typically assumed to be an uncommon and less important parallel to hyperactivity generated in peripheral terminals. The more severe an injury, however, the more likely peripheral nerve branches are to be crushed or cut, disconnecting affected nociceptors from their normal sources of excitation. Peripherally disconnected nociceptors may still contribute ongoing information about injury because sensory receptors and ion channels accumulate at the tips of cut axons, which along with other alterations, can impart sensitivity, hyperexcitability, and spontaneous activity to axons at the injury site [76–78]. Ongoing pain in humans from peripheral injury or neuropathy [79] can be eliminated rapidly by blocking peripheral nerve conduction with a non-selective voltage-gated sodium channel (NaV) inhibitor, lidocaine, which also can partially ameliorate pain from amputation [80]. In addition, persistent spontaneous activity may be generated in the cell body [81,82]. For example, lidocaine delivered to human dorsal root ganglia (DRG) innervating an amputated limb at concentrations that block local AP generation, without blocking conduction through the ganglia, rapidly and reversibly relieved phantom limb pain [83] (Figure 3, Table 1), providing strong evidence for the clinical importance of spontaneous activity generated in sensory neuron cell bodies. Spontaneous activity also occurs in dissociated DRG neurons from cancer patients with neuropathic pain who have had DRGs removed during thoracic vertebrectomy [84].
Figure 3. Persistent nociceptor hyperactivity can be adaptive.
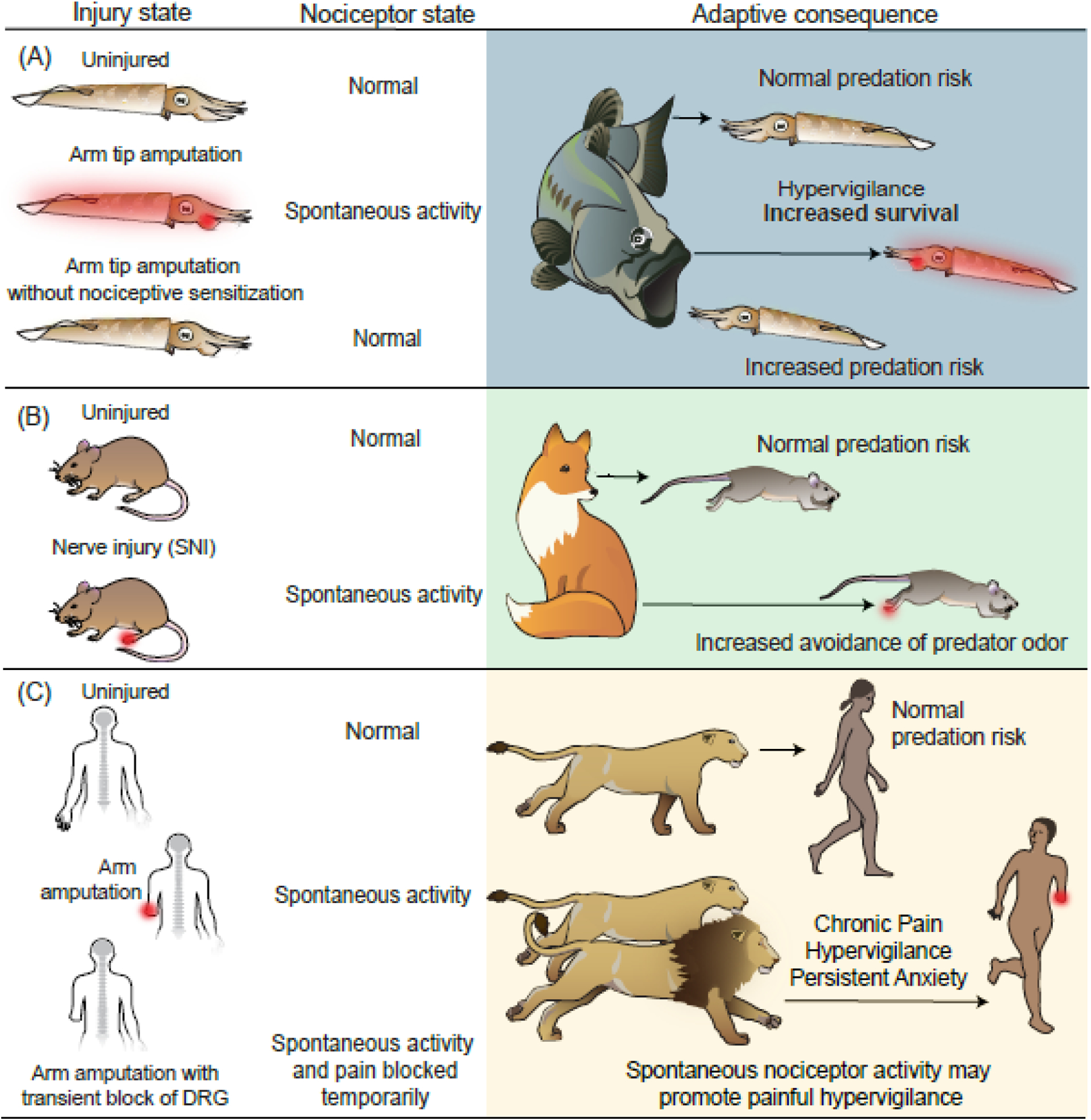
(A) Squid survival is enhanced by nociceptor activity after injury. Peripheral tissue injury causes long-lasting nociceptor hyperactivity expressed over much of the body surface [75] (red). In staged 30-minute encounters with fish predators [87], ~75% of uninjured subjects survived. Nearly half of squid previously receiving a minor arm amputation survived. Although selectively targeted by the fish, injured squid began their escape behavior farther from approaching fish (indicated by arrow length) than uninjured squid [87]. Squid that were injured without nociceptive sensitization and persistent nociceptor hyperactivity (prevented by transient block of nociceptor activity during amputation) waited longer to begin escape, with only ~20% surviving, indicating the survival benefit of persistent nociceptor hyperactivity after injury [86]. (B) Enhanced avoidance of a predator cue by mice with neuropathic pain. Mice with a spared nerve injury model exhibit nociceptor hyperactivity generated in the hindpaw (red) [138] and DRG [25,139] (not shown). When exposed to fox urine, injured mice chose a route that took them farther from the predator odor [88]. This suggests that injury-induced nociceptor hyperactivity promotes hypervigilance and increased risk aversion. (C) Speculative benefit of chronic nociceptor hyperactivity in ancestral humans at high risk for predation after injury (e.g., amputation injury in the illustration). The top two rows parallel the arguments for squid illustrated in panel A. The bottom row illustrates a situation of transient blockade of DRG APs in amputees that blocks ongoing pain [83]. If spontaneous activity in human nociceptors persistently drives hypervigilance as well as pain (as suggested by anxiety being a comorbidity of pain [90]), this hyperactivity should be protective for injured individuals who are more likely to be targeted by predators.
An experimental advantage of cell-body generation of hyperactivity in mammalian (and gastropod) nociceptors is that the mechanisms can be investigated more directly than is possible in other neuronal compartments. Although dissociation produces highly abnormal test conditions, spontaneous activity induced in vivo is surprisingly resistant to dissociation of rodent DRG neurons. In a DRG compression model, similar spontaneous activity was recorded from a DRG neuron in vivo before and after complete isolation [85], and in a spinal cord injury model, remarkable parallels were found in the pattern and discharge frequency of spontaneous activity generated in cell bodies of probable nociceptors in vivo and after dissociation [82].
Evidence that pain-like behavior associated with nociceptor hyperactivity is adaptive in cephalopod molluscs and mice
As mentioned, injury in squid induces long-lasting nociceptor spontaneous activity, which was suggested to drive a state of generalized hypervigilance [75,86]. The adaptiveness of this state was tested by examining the effects on survival of squid in staged interactions with natural predators (Figure 3A) [87]. A relatively minor injury, distal amputation of one of a squid’s eight arms, produced immediate defensive behavior and sensitization of responses to tactile and visual stimuli, but no lasting effects on motor function [86]. When exposed to predatory sea bass (Centropristis striata), squid with amputation injury were more likely to be attacked than uninjured squid, but they also began their escape behavior sooner than uninjured squid [87]. Briefly blocking nociceptor activation/sensitization during amputation by transient delivery of an anesthetic dose of MgCl2 resulted in injured squid later failing to show early responses to approaching sea bass and being less likely to escape. This result demonstrates how nociceptive sensitization enhances evolutionary fitness by increasing survival during predatory encounters and, when combined with electrophysiological observations, it suggests that persistent nociceptor spontaneous activity can drive a hypervigilant state that protects animals from attack during periods of increased vulnerability (Table 1).
Analogous results [88] were found in mice experiencing chronic pain in a spared nerve injury model (Figure 3B). When neuropathic mice had to choose between long and short routes to a food reward, they were more likely than uninjured mice to avoid the short route if it exposed them to the smell of fox urine, suggesting that continuing awareness of their injury (perhaps driven by nociceptor spontaneous activity, [25]) influenced their behavioral choices during elevated predatory risk[88]. Persistent injury-induced hypervigilance in animals appears functionally similar to anxiety in humans [89], and anxiety or anxiety-related behaviors are a common comorbidity of chronic pain in humans and rodents [90].
Are the hypervigilant states driven by nociceptor hyperactivity in cephalopods associated with motivational properties resembling those associated with affective pain in mammals? This question has not yet been addressed experimentally in squid, to our knowledge, but a study in Octopus bocki [91] using the noxious stimulus acetic acid showed that nociceptive states with pain-like aversive features occur in at least one cephalopod species. Specifically, octopuses remembered a chamber in which an arm was injected with acetic acid, and subsequently avoided it (referred to as conditioned place aversion). When acetic acid injection was followed by lidocaine injection into the same site immediately prior to placement in a different chamber, octopuses later chose to spend time in the chamber associated with lidocaine’s block of ongoing nociceptive activity (i.e., conditioned place preference). Thus, the octopus’s voluntary behavior revealed a pain-like link between flexible cognitive processing (including complex associative learning, memory, and decision making) and an aversive state induced by noxious stimulation. Although recordings could not be made from primary afferent neurons in the octopus arm, downstream electrical activity recorded in a central pathway from the injected arm to the brain continued for at least 30 minutes after acetic acid injection. The immediate block of this activity by lidocaine delivered to the acetic acid injection site is consistent with ongoing nociceptor activity generated at an injury site driving pain-like motivational and cognitive processes that enable conditioned place aversion and place preference in octopuses. These findings [91] parallel observations in rodents where spontaneous nociceptor activity is associated with a tonic aversive state [11,92].
Indirect evidence consistent with persistent nociceptor hyperactivity being adaptive in mammals
Several lines of evidence are consistent with the adaptiveness of spontaneous activity generated persistently in the cell bodies of mammalian nociceptors after injuries severe enough to increase an individual’s vulnerability to attack [93]. First, mammalian primary sensory neuron cell bodies are likely to survive serious peripheral injury because they are protected by their central location far from their receptive fields (Figure 1A), and by the bone and dura covering sensory ganglia. Second, the cell bodies are uniquely placed to integrate many injury signals (Figure 2) that may induce or persistently maintain hyperactivity [93–95]. Such signals may be intrinsic to injured neurons and conveyed electrically or by axonal transport from sites of peripheral damage. In addition, numerous extrinsic signal molecules may be conveyed to nociceptor cell bodies in the cerebrospinal fluid and the blood (because of the absence of an effective vascular permeability barrier in the ganglia). Peripheral or central tissue injury may also lead to local release of extrinsic signals near nociceptor cell bodies (from satellite glial cells and resident or infiltrating immune cells, [95–97]) and near sensory neuron presynaptic terminals within the spinal dorsal horn (from postsynaptic neurons and spinal glia [93,94]). While these anatomic and signaling features have many biological explanations, a functional consequence is that they enable nociceptor cell bodies to survive peripheral injury, to integrate disparate sources of information about injury severity in tissues that may have been disconnected from the CNS, and through persistent spontaneous activity to inform the CNS about continuing peripheral dysfunction.
A third line of evidence comes from the complex, complementary physiological mechanisms found to drive hyperactivity in the nociceptor cell body, providing some evidence of “functional design” by natural selection [98,99]. Specifically, in terms of membrane potential, there are three possible alterations that can result in AP generation in the absence of excitation from fast synaptic potentials (which mammalian nociceptors do not receive) or sensory receptor potentials (Figure 1B, which are unlikely to be generated in the cell body): depolarization of resting membrane potential (RMP) to reach AP threshold, hyperpolarization of AP threshold to reach RMP, and enhancement of depolarizing spontaneous fluctuations of RMP (DSFs, Figure 1D) to bridge the gap between RMP and AP threshold. Recordings from dissociated rodent nociceptors in a variety of different persistent pain models showed that all three properties are altered in these neurons compared to control conditions to promote spontaneous activity [100–103]. Spontaneous activity and all three underlying alterations were also found in probable nociceptors isolated from human cancer patients suffering from neuropathic pain [84,104]. Importantly, these complementary alterations occurred almost exclusively in nociceptors taken from ganglia innervating dermatomes reported by the patients to have ongoing pain.
RMP, action potential threshold, and DSFs in nociceptors each involve numerous ion channels, and the interactions between membrane potential, channel activity, and associated intracellular signaling are highly complex. Although spontaneous activity promoting neuropathic pain may sometimes be a purely pathological side effect of various insults to the body and nervous system, the intricate, functionally cohesive electrophysiological alterations that underpin spontaneous activity suggest that some damaging conditions may recruit a persistent, specialized state that was selected during evolution to ensure that nociceptor hyperactivity continues after injury severe enough to produce lasting physical impairment detectable by predators and competitors. Continuing pain would promote protective behavior (including hypervigilance, sometimes manifested in mammals as anxiety [90]) during increased vulnerability (Table 1), which could persist chronically for severe injuries such as amputation (Figure 3A, C).
Shared signaling mechanisms support nociceptor hyperactivity in gastropod molluscs and rodents
One of the interesting evolutionary aspects of persistent nociceptor hyperactivity is that it is found in two distantly related animal phyla, chordates and molluscs, suggesting common functions and potentially ancient mechanisms. However, even within placental mammals, nociceptor physiology shows clear divergences, raising questions about whether fundamental hyperactivity mechanisms are shared across the enormous phylogenetic distances between invertebrates and mammals.
Indeed, nociceptors in rodents and Aplysia share fundamental mechanisms important for synaptic plasticity [105] and hyperactivity. In Aplysia, long-lasting hyperactivity in dissociated nociceptors induced by serotonin was found to require protein synthesis [106]. Persistent axonal hyperactivity induced in Aplysia by nerve injury or nerve depolarization using elevated extracellular K+ to mimic the depolarization produced by axotomy [67], or serotonin application to a nerve [35], also requires protein synthesis, which is controlled by the mechanistic target of rapamycin (mTOR) pathway. Detailed studies in mice showed that activity-dependent mRNA translation regulated by mitogen-activated protein kinase (MAPK)-interacting kinase (MNK) phosphorylation of eukaryotic translation initiation factor 4E (eIF4E) is necessary for nociceptor hyperexcitability after peripheral nerve injury or inflammatory signals [107,108]. Aplysia has MNK1 and eIF4E proteins [109], with conservation of the eIF4E serine residue that is phosphorylated by MNK1 and required in mice for nociceptor hyperactivity induced by inflammatory mediators [107]. A selective MNK inhibitor was shown to potently inhibit the axonal hyperexcitability induced by depolarization of an Aplysia nerve, as do general inhibitors of tyrosine receptor kinases (upstream activators of MNKs) and of extracellular receptor kinases (ERKs, which activate MNKs) [109] (Figure 4).
Figure 4. Shared signaling pathways driving nociceptor hyperactivity in Aplysia and rodents.
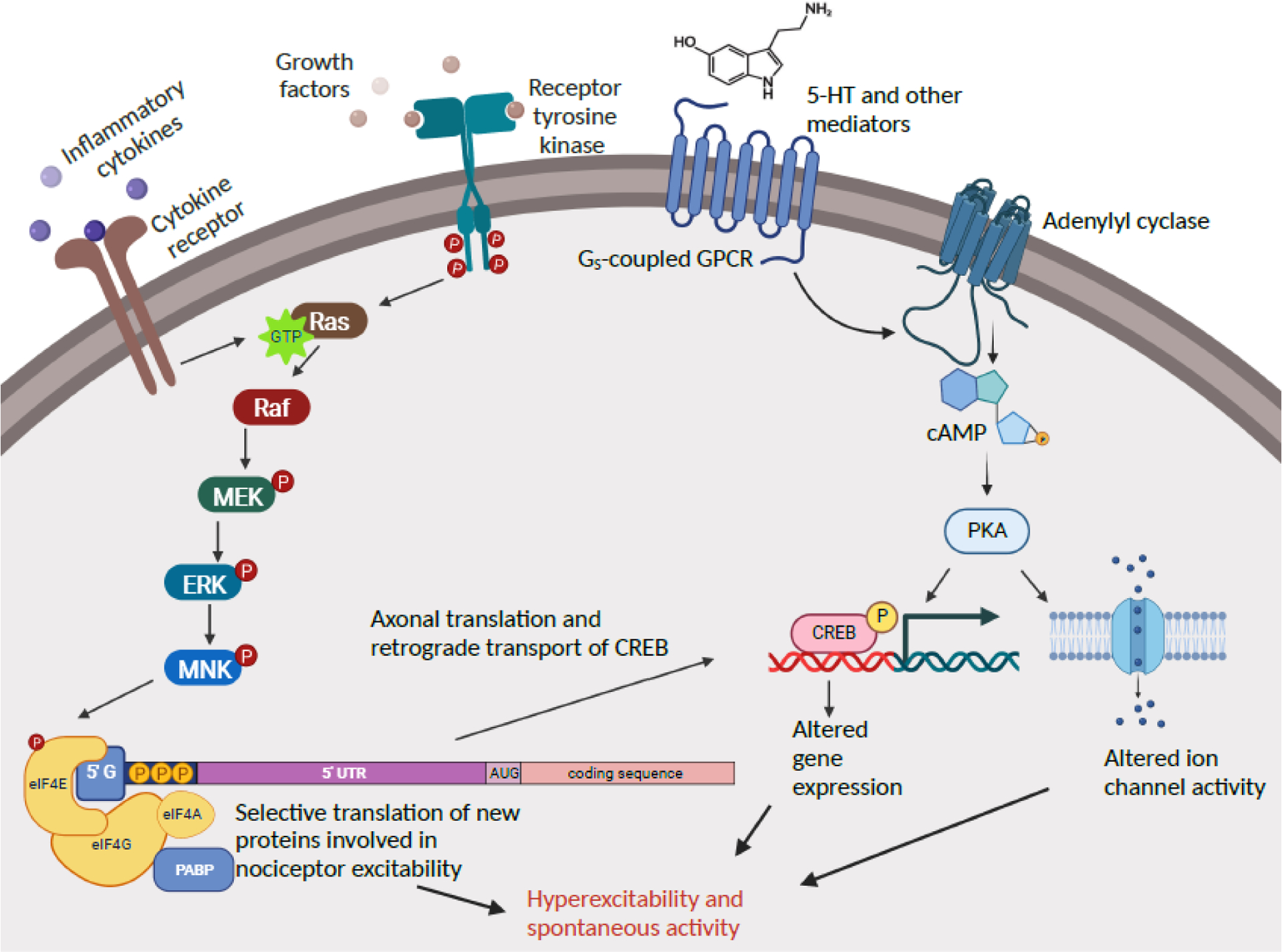
Two general cell signaling pathways have been implicated directly and indirectly in the maintenance and induction of persistent nociceptor hyperactivity both in a gastropod mollusc and in mammals: Ras-MNK signaling and cAMP-PKA signaling. These pathways are more complex than shown (e.g., cAMP has additional effectors implicated in nociceptor hyperactivity, and many protein kinases in addition to PKA can activate CREB), and each has additional interactions with the same and other cell signaling pathways. Only steps for which biochemical and/or pharmacological evidence is available are indicated. 5-HT, serotonin; 5’ UTR, 5’ untranslated region; cAMP, cyclic adenosine monophosphate; CREB, cAMP response element-binding protein; eIF4E, eukaryotic translation initiation factor 4E (correspondingly for eIF4G, eIF4A); ERK, extracellular-signal-regulated kinase; GPCR, G protein coupled receptor; Gs, stimulatory G protein; MEK, mitogen-activated protein kinase kinase; MNK, mitogen-activated protein kinase-interacting kinase; P, phosphorylation; PABP, poly-A-binding protein; PKA, protein kinase A; Raf, rapidly activated fibrosarcoma kinase; Ras, rat sarcoma GTPase. Created with BioRender.com.
A cell signal that plays prominent but complex roles in persistent hyperexcitability in Aplysia nociceptors is cyclic adenosine monophosphate (cAMP) and its downstream signals protein kinase A (PKA) and the transcription factor, cAMP response element-binding protein (CREB) [110–112] (Figure 4). Cyclic AMP has long been known to increase mammalian nociceptor excitability [113] and cAMP signaling is essential for maintaining rat nociceptor hyperactivity months after spinal cord injury, via protein kinase A [114], exchange protein activated by cAMP (EPAC) [101], and indirect positive feedback through depolarization and consequent C-Raf and ERK signaling, plus a reduction of adenylyl cyclase’s sensitivity to inhibition by opioids [115]. In addition, CREB activity is important for persistent nociceptor sensitization in rodents [116,117]. Thus, MNK-dependent protein synthesis, ERK signaling upstream of MNK [118], cAMP signaling, and CREB activity are associated with nociceptor hyperactivity in species (Table 1) whose last common ancestor lived ~600 million years ago.
Human nociceptors may be especially prone to hyperactivity
Sometime during the last 65 million years, after primates and rodents diverged from the common ancestor of placental mammals [119], notable modifications in nociceptors evolved, as reflected in several key differences between nociceptors in humans and rodents. Rodent nociceptors are typically divided into two distinct classes, peptidergic and non-peptidergic. Peptidergic nociceptors express TrkA receptors into adulthood, as well as neuropeptides such as calcitonin gene-related peptide (CGRP); they typically innervate deeper tissues and connect to neurons in lamina I of the spinal dorsal horn. Non-peptidergic nociceptors innervate the skin, do not express TrkA in adulthood, and connect to neurons in lamina II of the dorsal horn. The two classes have specializations for thermal and mechanical pain, respectively [120], although genetic tools are revealing finer functional subdivisions within each class. In contrast, all adult human nociceptors express TrkA and most express CGRP, suggesting that human nociceptors are generally peptidergic [121,122]. Unlike rodent nociceptors, among which ~50% express TRPV1, all human nociceptors express TRPV1. TRPV1 is readily sensitized by diverse inflammatory mediators such that its temperature activation threshold falls into the range of core body temperature [123], producing ongoing AP discharge (Figures 1D, 2B). In addition, a larger fraction of probable nociceptors in humans expresses one or more acid-sensing ion channels (ASIC1, 2, and 3) [124]. Thus, human nociceptors appear more “polymodal” than rodent nociceptors, and possibly than other primate nociceptors [122,125–127]. This should make them sensitive to a broader range of sensory stimuli and inflammatory mediators, thereby increasing extrinsically driven nociceptor hyperactivity and more readily inducing intrinsic hyperactivity (Figure 2).
Additional observations suggest that human nociceptors may be more intrinsically excitable than rodent nociceptors. First, dissociated human DRG neurons exhibit a greater density of tetrodotoxin (TTX)-sensitive and TTX-resistant NaV current than rodent nociceptors [128]. Second, RNA sequencing and functional experiments suggest that expression of the TTX-sensitive channel Nav1.7 is higher in human than rodent nociceptors [129]. Third, TTX-sensitive current is activated at more hyperpolarized potentials in human nociceptors [128]. Fourth, there is little or no use-dependent inactivation of TTX-resistant currents in human nociceptors [128]. All these differences should further promote spontaneous activity in human nociceptors, as observed in vivo [2,7] and in DRG neurons dissociated from neuropathic pain patients [84,104]. Interestingly, different densities and kinetics of human voltage-gated calcium (Cav) currents suggest that activity-dependent increases in intracellular Ca2+ may be smaller in human than rodent nociceptors [130]. If human evolution led to a high propensity for persistent nociceptor hyperactivity, then smaller Ca2+ responses might help protect against toxicity from excessive Ca2+ accumulation during continuing hyperactivity.
Could a high susceptibility to nociceptor-driven persistent pain in humans be evolutionarily adaptive? Although other species can display apparent empathy and helping behavior [131–133], humans are unique in the extent and flexibility of their helping behavior [134,135]. Thus, even early humans may have provided more help to injured family and group members than occurs in most species. Furthermore, humans are much more likely to communicate their pain to group members than are other species [136,137]. An intriguing possibility is that persistent nociceptor hyperactivity and other traits that maintain painful awareness of injury-induced vulnerability were subject to increased natural selection in hominins. The evolution of more flexible and complex hominin social behaviors may have enabled more effective and longer-lasting aid and protection for those needing help, with help-seeking being spurred by nociceptor-driven pain in humans weakened by injury [see 136].
Concluding remarks
Studies in both mammals and molluscs (Table 1) show that injuries sufficiently severe to damage nerves can induce persistent nociceptor hyperactivity. Findings in squid and mice suggest that persistently enhanced vulnerability resulting from impairment after injury can be lessened by behavioral alterations, including hypervigilance driven by prolonged nociceptor hyperactivity. The lack of clear evidence thus far for injury-induced persistent nociceptor hyperactivity in naked mole-rats, Drosophila, and C. elegans suggests either that a protective hypervigilance function has not been evolutionarily significant in some lineages (e.g., because of limited predation or short lifespans), or that alternative adaptations (e.g., central sensitization) were preferentially selected for this function (Table 1). In rodent and human nociceptors, synergistic electrophysiological alterations underpinning spontaneous activity are consistent with hyperactivity functioning to drive persistent hypervigilance. Current findings emphasize the need for further comparisons of species having and lacking persistent nociceptor hyperactivity (see Outstanding questions). Also needed is expanded investigation across taxa into common as well as divergent cellular and molecular mechanisms in neurons, glia, and immune cells driving persistent hyperactivity in nociceptors (and possibly in other types of primary somatosensory neurons). Finally, differences in sensitivity and excitability between nociceptors in humans and other mammals encourage further study into the possibility that, during recent hominin evolution, nociceptors increased their susceptibility to persistent hyperactivity, potentially increasing the propensity of humans to chronic pain.
Outstanding Questions.
What are the relevant biological needs shared by animals exhibiting persistent nociceptor hyperactivity? Do species that exhibit little inflammatory or neuropathic pain, such as naked mole-rats, lack persistent nociceptor hyperactivity after significant injuries?
To what extent are biophysical and molecular mechanisms (intrinsic and extrinsic, including persistent immune alterations) that drive persistent nociceptor hyperactivity conserved across vertebrate and invertebrate species, and among mammalian species?
Do types of primary afferent neurons other than nociceptors (e.g., pruriceptors and low-threshold mechanosensory neurons) exhibit persistent hyperactivity that might be adaptive in some conditions?
Are the apparently high excitability and sensitivity to extrinsic pain-related signals in human nociceptors linked to a high susceptibility of humans to persistent nociceptor hyperactivity and consequent pain?
Many clinical conditions in humans cause neuropathy, altered sensory function, and chronic pain (e.g., diabetes, chemotherapy, radiculopathy, postherpetic neuralgia). To what extent does pain in such conditions involve accidental recruitment of a persistent nociceptor hyperactivity state that evolved originally as an adaptation to severe peripheral injury versus purely pathological effects of neuropathy, such as metabolic dysfunction?
Highlights.
Injury to the nervous system can generate chronic pain driven by persistent hyperactivity of nociceptors, sensory neurons specialized to detect damaging stimuli.
Recent evidence argues against the near-universal assumption that nociceptor hyperactivity and resulting chronic pain are always maladaptive.
Widespread conservation of mechanisms of nociceptor function and hyperactivity are well documented, as well as distinct adaptations to divergent nociceptive needs.
Persistent nociceptor hyperactivity could have evolved to drive painful hypervigilance during persistent physical impairment, enhancing survival by decreasing subsequent risk of predatory or aggressive attack.
Differences between rodent and human nociceptors are consistent with the possibility that during relatively recent evolution nociceptors increased their susceptibility to persistent hyperactivity, potentially increasing the propensity of humans to chronic pain.
Acknowledgements
ETW was supported by NIH grants NS091759 and RO1NS111521. RJC was supported by an Allen Distinguished Investigator Award, a Paul G. Allen Frontiers Group advised grant of the Paul G. Allen Family Foundation, and NSF IOS 204-7331. GGN was supported by Australian National Health and Medical Research Council grants APP1026310, APP1028887, and APP1046090. TJP was supported by NIH grants NS111929 and NS102161. EStJS was supported by a grant from the Medical Research Council MR/W002426/1.
Declaration of interests
Edgar T. Walters declares no competing interests.
Robyn J. Crook declares no competing interests.
G. Gregory Neely declares no competing interests.
Theodore J. Price is a co-founder of 4E Therapeutics, Doloromics, PARMedics and NuvoNuro; he serves on the Board of Directors of 4E Therapeutics and Doloromics; he is an inventor on patents related to MNK inhibition for the treatment of pain.
Ewan St John Smith receives funding from AstraZeneca and GlaxoSmithKline.
Glossary
- Afterdischarge
APs generated in response to a preceding AP or burst of APs, prolonging the electrical activity.
- Depolarizing spontaneous fluctuation (DSF)
brief, intrinsically generated depolarizing fluctuations of RMP. In nociceptors, some DSFs may intermittently reach AP threshold, contributing to spontaneous activity under hyperactive conditions.
- Hyperactivity
state of increased discharge of APs resulting from drivers that are intrinsic and/or extrinsic to a neuron.
- Hyperexcitability
electrophysiological state in which the likelihood of generating at least one AP by depolarizing stimulation is increased. Hyperexcitability is electrical hypersensitivity, and represents one type of sensitization.
- Hypersensitivity
state of increased responsiveness to a biological signal (sensory, chemical, or electrical). The term is almost synonymous with sensitization, although in the pain field it tends to be used more descriptively and less mechanistically than sensitization.
- Neuropathic pain
pain produced by injury or disease of any part of the nervous system.
- Nociceptor
a primary sensory neuron that is selectively activated by stimuli causing actual or incipient tissue injury, or by signals reliably associated with tissue injury (e.g., inflammatory signals).
- Resting membrane potential (RMP)
electrical potential difference across the plasma membrane in the absence of APs, sensory potentials, or synaptic potentials. If DSFs occur, they are considered transient components of RMP.
- Sensitization (see also hypersensitivity)
a state in which a system (typically a neuron, neuronal pathway, or animal) exhibits increased responsiveness to biological signals.
- Spontaneous activity
in electrophysiology, the ongoing generation of APs by a cell without concurrent extrinsic sources of depolarization. Because it is often impractical to identify background excitatory or sensitizing contributions to ongoing AP discharge, ongoing discharge generated in neurons in the absence of observed sensory generator potentials or synaptic potentials is commonly referred to as spontaneous activity.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Williams GC and Nesse RM (1991) The dawn of Darwinian medicine. Q Rev Biol 66, 1–22 [DOI] [PubMed] [Google Scholar]
- 2.Finnerup NB et al. (2021) Neuropathic Pain: From Mechanisms to Treatment. Physiol Rev 101, 259–301 [DOI] [PubMed] [Google Scholar]
- 3.Sommer C et al. (2018) Inflammation in the pathophysiology of neuropathic pain. Pain 159, 595–602 [DOI] [PubMed] [Google Scholar]
- 4.Walters ET (2021) Nociceptors and chronic pain. In Oxford Research Encyclopedia of Neuroscience (Murray S. Oxford University Press [Google Scholar]
- 5.Roza C and Bernal L (2022) Electrophysiological characterization of ectopic spontaneous discharge in axotomized and intact fibers upon nerve transection: a role in spontaneous pain. Pflugers Arch 474, 387–396 [DOI] [PubMed] [Google Scholar]
- 6.Ochoa J and Torebjörk E (1989) Sensations evoked by intraneural microstimulation of C nociceptor fibres in human skin nerves. J Physiol 415, 583–599 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Serra J et al. (2012) Microneurographic identification of spontaneous activity in C-nociceptors in neuropathic pain states in humans and rats. Pain 153, 42–55 [DOI] [PubMed] [Google Scholar]
- 8.Cowie AM et al. (2018) Optogenetic Inhibition of CGRPα Sensory Neurons Reveals Their Distinct Roles in Neuropathic and Incisional Pain. J Neurosci 38, 5807–5825 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Beaudry H et al. (2017) Distinct behavioral responses evoked by selective optogenetic stimulation of the major TRPV1+ and MrgD+ subsets of C-fibers. Pain 158, 2329–2339 [DOI] [PubMed] [Google Scholar]
- 10.Daou I et al. (2016) Optogenetic Silencing of Nav1.8-Positive Afferents Alleviates Inflammatory and Neuropathic Pain. eNeuro 3, [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Yang Q et al. (2014) Persistent pain after spinal cord injury is maintained by primary afferent activity. J Neurosci 34, 10765–10769 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Woolf CJ (2011) Central sensitization: implications for the diagnosis and treatment of pain. Pain 152, S2–15 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Stein A et al. (2013) Pilot study: elevated circulating levels of the proinflammatory cytokine macrophage migration inhibitory factor in patients with chronic spinal cord injury. Arch Phys Med Rehabil 94, 1498–1507 [DOI] [PubMed] [Google Scholar]
- 14.Robinson SM et al. (2019) Systemic inflammation contributes to impairment of quality of life in chronic pancreatitis. Sci Rep 9, 7318. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Zhou WBS et al. (2021) Does Low Grade Systemic Inflammation Have a Role in Chronic Pain. Front Mol Neurosci 14, 785214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Abdo H et al. (2019) Specialized cutaneous Schwann cells initiate pain sensation. Science 365, 695–699 [DOI] [PubMed] [Google Scholar]
- 17.Donnelly CR et al. (2020) How Do Sensory Neurons Sense Danger Signals. Trends Neurosci 43, 822–838 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Lacagnina MJ et al. (2021) Autoimmune regulation of chronic pain. Pain Rep 6, e905. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Raoof R et al. (2021) Dorsal Root Ganglia Macrophages Maintain Osteoarthritis Pain. J Neurosci 41, 8249–8261 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Luo X et al. (2021) IL-23/IL-17A/TRPV1 axis produces mechanical pain via macrophage-sensory neuron crosstalk in female mice. Neuron 109, 2691–2706.e5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Szabo-Pardi TA et al. (2021) Sensory Neuron TLR4 mediates the development of nerve-injury induced mechanical hypersensitivity in female mice. Brain Behav Immun 97, 42–60 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Oggero S et al. (2022) Dorsal root ganglia CX3CR1 expressing monocytes/macrophages contribute to arthritis pain. Brain Behav Immun 106, 289–306 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Gangadharan V et al. (2022) Neuropathic pain caused by miswiring and abnormal end organ targeting. Nature 606, 137–145 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Ray PR et al. (2022) RNA profiling of human dorsal root ganglia reveals sex-differences in mechanisms promoting neuropathic pain. Brain awac266. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Zheng Q et al. (2022) Synchronized cluster firing, a distinct form of sensory neuron activation, drives spontaneous pain. Neuron 110, 209–220.e6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Plein LM and Rittner HL (2018) Opioids and the immune system - friend or foe. Br J Pharmacol 175, 2717–2725 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Marinesco S et al. (2004) Serotonergic modulation in aplysia. I. Distributed serotonergic network persistently activated by sensitizing stimuli. J Neurophysiol 92, 2468–2486 [DOI] [PubMed] [Google Scholar]
- 28.Clatworthy AL et al. (1994) Induction of a cellular defense reaction is accompanied by an increase in sensory neuron excitability in Aplysia. J Neurosci 14, 3263–3270 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Clatworthy AL and Grose E (1999) Immune-mediated alterations in nociceptive sensory function in Aplysia californica. J Exp Biol 202, 623–630 [DOI] [PubMed] [Google Scholar]
- 30.Tassia MG et al. (2017) Toll-like receptor pathway evolution in deuterostomes. Proc Natl Acad Sci U S A 114, 7055–7060 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Michelet C et al. (2019) Cross-Kingdom Analysis of Diversity, Evolutionary History, and Site Selection within the Eukaryotic Macrophage Migration Inhibitory Factor Superfamily. Genes (Basel) 10, E740. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Kubick N et al. (2021) Interleukins and Interleukin Receptors Evolutionary History and Origin in Relation to CD4+ T Cell Evolution. Genes (Basel) 12, 813. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Arch M et al. (2022) Drosophila melanogaster as a model to study innate immune memory. Front Microbiol 13, 991678. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Kraus A et al. (2021) Sensing the world and its dangers: An evolutionary perspective in neuroimmunology. Elife 10, e66706. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Weragoda RM and Walters ET (2007) Serotonin induces memory-like, rapamycin-sensitive hyperexcitability in sensory axons of aplysia that contributes to injury responses. J Neurophysiol 98, 1231–1239 [DOI] [PubMed] [Google Scholar]
- 36.Reichling DB and Levine JD (2009) Critical role of nociceptor plasticity in chronic pain. Trends Neurosci 32, 611–618 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Liu RY et al. (2011) The requirement for enhanced CREB1 expression in consolidation of long-term synaptic facilitation and long-term excitability in sensory neurons of Aplysia. J Neurosci 31, 6871–6879 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Price TJ and Inyang KE (2015) Commonalities between pain and memory mechanisms and their meaning for understanding chronic pain. Prog Mol Biol Transl Sci 131, 409–434 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Lopez ER et al. (2021) Serotonin enhances depolarizing spontaneous fluctuations, excitability, and ongoing activity in isolated rat DRG neurons via 5-HT4 receptors and cAMP-dependent mechanisms. Neuropharmacology 184, 108408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Smith ES and Lewin GR (2009) Nociceptors: a phylogenetic view. J Comp Physiol A Neuroethol Sens Neural Behav Physiol 195, 1089–1106 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Walters ET (2020) Evolutionary aspects of nociception and pain. In The Senses: A Comprehensive Reference, Vol 5, Pain (Fritzsch B et al. eds.), pp. 463–480, Elsevier, Academic Press [Google Scholar]
- 42.Monk T and Paulin MG (2014) Predation and the origin of neurones. Brain Behav Evol 84, 246–261 [DOI] [PubMed] [Google Scholar]
- 43.Brunet T and Arendt D (2016) From damage response to action potentials: early evolution of neural and contractile modules in stem eukaryotes. Philos Trans R Soc Lond B Biol Sci 371, 20150043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Moroz LL et al. (2021) Neural versus alternative integrative systems: molecular insights into origins of neurotransmitters. Philos Trans R Soc Lond B Biol Sci 376, 20190762. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Marques F et al. (2021) Signaling via the FLP-14/FRPR-19 neuropeptide pathway sustains nociceptive response to repeated noxious stimuli in C. elegans. PLoS Genet 17, e1009880. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Jiang J et al. (2022) C. elegans enteric motor neurons fire synchronized action potentials underlying the defecation motor program. Nat Commun 13, 2783. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Quach KT and Chalasani SH (2022) Flexible reprogramming of Pristionchus pacificus motivation for attacking Caenorhabditis elegans in predator-prey competition. Curr Biol 32, 1675–1688.e7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Babcock DT et al. (2009) Cytokine signaling mediates UV-induced nociceptive sensitization in Drosophila larvae. Curr Biol 19, 799–806 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Jang W et al. (2018) Duox mediates ultraviolet injury-induced nociceptive sensitization in Drosophila larvae. Mol Brain 11, 16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Follansbee TL et al. (2017) Drosophila nociceptive sensitization requires BMP signaling via the canonical SMAD pathway. J Neurosci 37, 8524–8533 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Im SH et al. (2015) Tachykinin acts upstream of autocrine Hedgehog signaling during nociceptive sensitization in Drosophila. Elife 4, e10735. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Banik RK and Brennan TJ (2009) Trpv1 mediates spontaneous firing and heat sensitization of cutaneous primary afferents after plantar incision. Pain 141, 41–51 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Gu P et al. (2022) Nociception and hypersensitivity involve distinct neurons and molecular transducers in Drosophila. Proc Natl Acad Sci U S A 119, e2113645119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Khuong TM et al. (2019) Nerve injury drives a heightened state of vigilance and neuropathic sensitization in Drosophila. Sci Adv 5, eaaw4099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Walters ET (1987) Multiple sensory neuronal correlates of site-specific sensitization in Aplysia. J Neurosci 7, 408–417 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Tabuena DR et al. (2017) Central neural alterations predominate in an insect model of nociceptive sensitization. J Comp Neurol 525, 1176–1191 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Buffenstein R et al. (2022) The naked truth: a comprehensive clarification and classification of current ‘myths’ in naked mole-rat biology. Biol Rev Camb Philos Soc 97, 115–140 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Schmalbruch H (1986) Fiber composition of the rat sciatic nerve. Anat Rec 215, 71–81 [DOI] [PubMed] [Google Scholar]
- 59.Park TJ et al. (2008) Selective inflammatory pain insensitivity in the African naked mole-rat (Heterocephalus glaber). PLoS Biol 6, e13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Omerbašić D et al. (2016) Hypofunctional TrkA Accounts for the Absence of Pain Sensitization in the African Naked Mole-Rat. Cell Rep 17, 748–758 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Gallman K et al. (2020) Differences in behavior between surface and cave Astyanax mexicanus may be mediated by changes in catecholamine signaling. J Comp Neurol 528, 2639–2653 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Tanvir Z et al. (2021) Evolutionary and homeostatic changes in morphology of visual dendrites of Mauthner cells in Astyanax blind cavefish. J Comp Neurol 529, 1779–1786 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Pattison LA et al. (2019) Evolution of acid nociception: ion channels and receptors for detecting acid. Philos Trans R Soc Lond B Biol Sci 374, 20190291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Smith ES et al. (2011) The molecular basis of acid insensitivity in the African naked mole-rat. Science 334, 1557–1560 [DOI] [PubMed] [Google Scholar]
- 65.Eigenbrod O et al. (2019) Rapid molecular evolution of pain insensitivity in multiple African rodents. Science 364, 852–859 [DOI] [PubMed] [Google Scholar]
- 66.Dulin MF et al. (1995) Recovery of function, peripheral sensitization and sensory neurone activation by novel pathways following axonal injury in Aplysia californica. J Exp Biol 198, 2055–2066 [DOI] [PubMed] [Google Scholar]
- 67.Weragoda RM et al. (2004) Memory-like alterations in Aplysia axons after nerve injury or localized depolarization. J Neurosci 24, 10393–10401 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Walters ET et al. (1991) Similar neuronal alterations induced by axonal injury and learning in Aplysia. Science 253, 797–799 [DOI] [PubMed] [Google Scholar]
- 69.Gasull X et al. (2005) Evidence that long-term hyperexcitability of the sensory neuron soma induced by nerve injury in Aplysia is adaptive. J Neurophysiol 94, 2218–2230 [DOI] [PubMed] [Google Scholar]
- 70.Billy AJ and Walters ET (1989) Long-term expansion and sensitization of mechanosensory receptive fields in Aplysia support an activity-dependent model of whole-cell sensory plasticity. J Neurosci 9, 1254–1262 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Pepino C et al. (2022) Sensitized by a sea slug: Site-specific short-term and general long-term sensitization in Aplysia following Navanax attack. Neurobiol Learn Mem 187, 107542. [DOI] [PubMed] [Google Scholar]
- 72.Frost WN et al. (1988) Parallel processing of short-term memory for sensitization in Aplysia. J Neurobiol 19, 297–334 [DOI] [PubMed] [Google Scholar]
- 73.Trudeau LE and Castellucci VF (1993) Sensitization of the gill and siphon withdrawal reflex of Aplysia: multiple sites of change in the neuronal network. J Neurophysiol 70, 1210–1220 [DOI] [PubMed] [Google Scholar]
- 74.Upreti C et al. (2019) Serotonin Induces Structural Plasticity of Both Extrinsic Modulating and Intrinsic Mediating Circuits In Vitro in Aplysia Californica. Cell Rep 28, 2955–2965.e3 [DOI] [PubMed] [Google Scholar]
- 75.Crook RJ et al. (2013) Squid have nociceptors that display widespread long-term sensitization and spontaneous activity after bodily injury. J Neurosci 33, 10021–10026 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Devor M et al. (1992) Eicosanoids, but not tachykinins, excite C-fiber endings in rat sciatic nerve-end neuromas. Neuroreport 3, 21–24 [DOI] [PubMed] [Google Scholar]
- 77.Bernal L et al. (2016) Spontaneous activity in C-fibres after partial damage to the saphenous nerve in mice: Effects of retigabine. Eur J Pain 20, 1335–1345 [DOI] [PubMed] [Google Scholar]
- 78.Yatziv SL and Devor M (2019) Suppression of neuropathic pain by selective silencing of dorsal root ganglion ectopia using nonblocking concentrations of lidocaine. Pain 160, 2105–2114 [DOI] [PubMed] [Google Scholar]
- 79.Haroutounian S et al. (2014) Primary afferent input critical for maintaining spontaneous pain in peripheral neuropathy. Pain 155, 1272–1279 [DOI] [PubMed] [Google Scholar]
- 80.Buch NS et al. (2019) The role of afferent input in postamputation pain: a randomized, double-blind, placebo-controlled crossover study. Pain [DOI] [PubMed] [Google Scholar]
- 81.Wall PD and Devor M (1983) Sensory afferent impulses originate from dorsal root ganglia as well as from the periphery in normal and nerve injured rats. Pain 17, 321–339 [DOI] [PubMed] [Google Scholar]
- 82.Bedi SS et al. (2010) Chronic spontaneous activity generated in the somata of primary nociceptors is associated with pain-related behavior after spinal cord injury. J Neurosci 30, 14870–14882 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Vaso A et al. (2014) Peripheral nervous system origin of phantom limb pain. Pain 155, 1384–1391 [DOI] [PubMed] [Google Scholar]
- 84.North RY et al. (2019) Electrophysiological and transcriptomic correlates of neuropathic pain in human dorsal root ganglion neurons. Brain 142, 1215–1226 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Ma C and LaMotte RH (2007) Multiple sites for generation of ectopic spontaneous activity in neurons of the chronically compressed dorsal root ganglion. J Neurosci 27, 14059–14068 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Crook RJ et al. (2011) Peripheral injury induces long-term sensitization of defensive responses to visual and tactile stimuli in the squid Loligo pealeii, Lesueur 1821. J Exp Biol 214, 3173–3185 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Crook RJ et al. (2014) Nociceptive sensitization reduces predation risk. Curr Biol 24, 1121–1125 [DOI] [PubMed] [Google Scholar]
- 88.Lister KC et al. (2020) Chronic pain produces hypervigilance to predator odor in mice. Curr Biol 30, R866–R867 [DOI] [PubMed] [Google Scholar]
- 89.Walters ET (1994) Injury-related behavior and neuronal plasticity: an evolutionary perspective on sensitization, hyperalgesia, and analgesia. Int Rev Neurobiol 36, 325–427 [DOI] [PubMed] [Google Scholar]
- 90.Kremer M et al. (2021) How to study anxiety and depression in rodent models of chronic pain. Eur J Neurosci 53, 236–270 [DOI] [PubMed] [Google Scholar]
- 91.Crook RJ (2021) Behavioral and neurophysiological evidence suggests affective pain experience in octopus. iScience 24, 102229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Okun A et al. (2011) Transient inflammation-induced ongoing pain is driven by TRPV1 sensitive afferents. Mol Pain 7, 4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Walters ET (2019) Adaptive mechanisms driving maladaptive pain: how chronic ongoing activity in primary nociceptors can enhance evolutionary fitness after severe injury. Philos Trans R Soc Lond B Biol Sci 374, 20190277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Walters ET (2012) Nociceptors as chronic drivers of pain and hyperreflexia after spinal cord injury: an adaptive-maladaptive hyperfunctional state hypothesis. Front Physiol 3, 309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Gazerani P (2021) Satellite Glial Cells in Pain Research: A Targeted Viewpoint of Potential and Future Directions. Front Pain Res (Lausanne) 2, 646068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Kim YS et al. (2016) Coupled Activation of Primary Sensory Neurons Contributes to Chronic Pain. Neuron 91, 1085–1096 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Malcangio M (2019) Role of the immune system in neuropathic pain. Scand J Pain 20, 33–37 [DOI] [PubMed] [Google Scholar]
- 98.Andrews PW et al. (2002) Adaptationism--how to carry out an exaptationist program. Behav Brain Sci 25, 489–504; discussion 504 [DOI] [PubMed] [Google Scholar]
- 99.Williams GC (1966) Adaptation and natural selection. Princeton University Press, Princeton, NJ: (1996 reprint edition). [Google Scholar]
- 100.Odem MA et al. (2018) Isolated nociceptors reveal multiple specializations for generating irregular ongoing activity associated with ongoing pain. Pain 159, 2347–2362 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Berkey SC et al. (2020) EPAC1 and EPAC2 promote nociceptor hyperactivity associated with chronic pain after spinal cord injury. Neurobiol Pain 7, 100040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Laumet G et al. (2020) Interleukin-10 resolves pain hypersensitivity induced by cisplatin by reversing sensory neuron hyperexcitability. Pain 161, 2344–2352 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Bavencoffe AG et al. (2022) Macrophage Migration Inhibitory Factor (MIF) Makes Complex Contributions to Pain-Related Hyperactivity of Nociceptors after Spinal Cord Injury. J Neurosci JN–RM [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.North RY et al. (2022) Electrophysiological alterations driving pain-associated spontaneous activity in human sensory neuron somata parallel alterations described in spontaneously active rodent nociceptors. J Pain S1526–5900(22)00047 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Orvis J et al. (2022) The evolution of synaptic and cognitive capacity: Insights from the nervous system transcriptome of Aplysia. Proc Natl Acad Sci U S A 119, e2122301119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Dale N et al. (1987) Serotonin produces long-term changes in the excitability of Aplysia sensory neurons in culture that depend on new protein synthesis. J Neurosci 7, 2232–2238 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Moy JK et al. (2017) The MNK-eIF4E Signaling Axis Contributes to Injury-Induced Nociceptive Plasticity and the Development of Chronic Pain. J Neurosci 37, 7481–7499 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Jeevakumar V et al. (2020) IL-6 induced upregulation of T-type Ca2+ currents and sensitization of DRG nociceptors is attenuated by MNK inhibition. J Neurophysiol 124, 274–283 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Mihail SM et al. (2019) MNK-eIF4E signalling is a highly conserved mechanism for sensory neuron axonal plasticity: evidence from Aplysia californica. Philos Trans R Soc Lond B Biol Sci 374, 20190289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Scholz KP and Byrne JH (1988) Intracellular injection of cAMP induces a long-term reduction of neuronal K+ currents. Science 240, 1664–1666 [DOI] [PubMed] [Google Scholar]
- 111.Liao X et al. (1999) Activation of protein kinase A contributes to the expression but not the induction of long-term hyperexcitability caused by axotomy of Aplysia sensory neurons. J Neurosci 19, 1247–1256 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Zhang Y et al. (2021) Quantitative description of the interactions among kinase cascades underlying long-term plasticity of Aplysia sensory neurons. Sci Rep 11, 14931. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Gold MS et al. (1998) Modulation of TTX-R INa by PKC and PKA and their role in PGE2-induced sensitization of rat sensory neurons in vitro. J Neurosci 18, 10345–10355 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Bavencoffe A et al. (2016) Persistent Electrical Activity in Primary Nociceptors after Spinal Cord Injury Is Maintained by Scaffolded Adenylyl Cyclase and Protein Kinase A and Is Associated with Altered Adenylyl Cyclase Regulation. J Neurosci 36, 1660–1668 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Garza Carbajal A et al. (2020) Depolarization-Dependent C-Raf Signaling Promotes Hyperexcitability and Reduces Opioid Sensitivity of Isolated Nociceptors after Spinal Cord Injury. J Neurosci 40, 6522–6535 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Melemedjian OK et al. (2014) Local translation and retrograde axonal transport of CREB regulates IL-6-induced nociceptive plasticity. Mol Pain 10, 45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Ferrari LF et al. (2015) Distinct terminal and cell body mechanisms in the nociceptor mediate hyperalgesic priming. J Neurosci 35, 6107–6116 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Yousuf MS et al. (2021) Pharmacological Manipulation of Translation as a Therapeutic Target for Chronic Pain. Pharmacol Rev 73, 59–88 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.O’Leary MA et al. (2013) The placental mammal ancestor and the post-K-Pg radiation of placentals. Science 339, 662–667 [DOI] [PubMed] [Google Scholar]
- 120.Cavanaugh DJ et al. (2009) Distinct subsets of unmyelinated primary sensory fibers mediate behavioral responses to noxious thermal and mechanical stimuli. Proc Natl Acad Sci U S A 106, 9075–9080 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Shiers SI et al. (2021) Convergence of peptidergic and non-peptidergic protein markers in the human dorsal root ganglion and spinal dorsal horn. J Comp Neurol 529, 2771–2788 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Tavares-Ferreira D et al. (2022) Spatial transcriptomics of dorsal root ganglia identifies molecular signatures of human nociceptors. Sci Transl Med 14, eabj8186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Julius D (2013) TRP channels and pain. Annu Rev Cell Dev Biol 29, 355–384 [DOI] [PubMed] [Google Scholar]
- 124.Papalampropoulou-Tsiridou M et al. (2022) Distribution of acid-sensing ion channel subunits in human sensory neurons contrasts with that in rodents. Brain Commun 4, fcac256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Middleton SJ et al. (2021) Studying human nociceptors: from fundamentals to clinic. Brain 144, 1312–1335 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Nguyen MQ et al. (2021) Single-nucleus transcriptomic analysis of human dorsal root ganglion neurons. Elife 10, e71752. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Yang L et al. (2022) Human and mouse trigeminal ganglia cell atlas implicates multiple cell types in migraine. Neuron 110, 1806–1821.e8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Zhang X et al. (2017) Voltage-gated Na+ currents in human dorsal root ganglion neurons. Elife 6, e23235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Wangzhou A et al. (2020) Pharmacological target-focused transcriptomic analysis of native vs cultured human and mouse dorsal root ganglia. Pain 161, 1497–1517 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Hartung JE et al. (2022) Voltage-gated calcium currents in human dorsal root ganglion neurons. Pain 163, e774–e785 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Mogil JS (2015) Social modulation of and by pain in humans and rodents. Pain 156 Suppl 1, S35–41 [DOI] [PubMed] [Google Scholar]
- 132.Bshary R and Raihani NJ (2017) Helping in humans and other animals: a fruitful interdisciplinary dialogue. Proc Biol Sci 284, 20170929. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133.Chen J (2018) Empathy for Distress in Humans and Rodents. Neurosci Bull 34, 216–236 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134.Burkart JM et al. (2014) The evolutionary origin of human hyper-cooperation. Nat Commun 5, 4747. [DOI] [PubMed] [Google Scholar]
- 135.Tomasello M (2014) The ultra-social animal. Eur J Soc Psychol 44, 187–194 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136.Finlay BL and Syal S (2014) The pain of altruism. Trends Cogn Sci 18, 615–617 [DOI] [PubMed] [Google Scholar]
- 137.Williams ACC (2019) Persistence of pain in humans and other mammals. Philos Trans R Soc Lond B Biol Sci 374, 20190276. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138.Smith AK et al. (2013) Mechanical sensitization of cutaneous sensory fibers in the spared nerve injury mouse model. Mol Pain 9, 61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139.Tappe-Theodor A et al. (2012) Gα(q/11) signaling tonically modulates nociceptor function and contributes to activity-dependent sensitization. Pain 153, 184–196 [DOI] [PubMed] [Google Scholar]