

Summary:
Genetic models suggested that SMARCA5 was required for DNA templated events including transcription, DNA replication and DNA repair. We engineered a degron tag into the endogenous alleles of SMARCA5, a catalytic component of the imitation switch (ISWI) complexes, in three different human cell lines to define the effects of rapid degradation of this key regulator. Degradation of SMARCA5 was associated with a rapid increase in global nucleosome repeat length, which may allow greater chromatin compaction. However, there were few changes in nascent transcription within the first 6hr of degradation. Nevertheless, we demonstrated a requirement for SMARCA5 to control nucleosome repeat length at G1/S and during the S phase. SMARCA5 co-localized with CTCF and H2A.Z, and we found a rapid loss of CTCF DNA binding and disruption of nucleosomal phasing around CTCF binding sites. This spatiotemporal analysis indicates that SMARCA5 is continuously required for maintaining nucleosomal spacing.
Keywords: SMARCA5, PRO-seq, MNase-seq, ATAC-seq, nucleosome repeat length, CTCF, H2A.Z
In Brief
Bomber et al. utilize degron tagging of the endogenous human chromatin remodeling enzyme SMARCA5, coupled with a multi-omics approach, to define the requirements for SMARCA5-mediated nucleosome sliding. SMARCA5 co-localized with H2A.Z and CTCF, and was required for CTCF DNA binding, nucleosomal phasing at these sites, and to maintain nucleosome repeat length.
Graphical Abstract
Introduction
The basic unit of chromatin is the nucleosome, which consists of approximately 147 bp of DNA wrapped around a histone octamer core1,2. This allows for both the efficient packaging of all the genetic material within the nucleus and mediates the accessibility of the DNA to other regulatory proteins, including those controlling transcription, DNA replication and DNA repair. Neighboring nucleosomes are typically separated by 20–50 bp of linker DNA, which is more easily accessible to regulatory proteins and can also be bound by structural proteins, such as histone H1, to promote the formation of higher order chromatin structures3–5. Chromatin remodeling enzymes hydrolyze ATP and slide or evict nucleosomes to modify chromatin structure and accessibility, as well as cooperate with histone chaperones to facilitate histone variant exchange6–8. Therefore, through complex mechanisms governing chromatin dynamics, these critical enzyme complexes have the ability to regulate all DNA-templated cellular processes.
Smarca5 (SNF2H) along with Smarca1 (SNF2L) encode the two ATPase components of the imitation switch (ISWI) chromatin remodeling complex and participate in multiple distinct ISWI subcomplexes. SNF2L associates with BPTF and RbAp46/48 in the NURF complex, while SNF2H is a component of CERC2-containing remodeling complex (CERF), ATP-utilizing chromatin assembly factor (ACF), chromatin accessibility complex (CHRAC), nucleolar remodeling complex (NoRC), Williams Syndrome Transcription Factor (WSTF)-ISWI chromatin remodeling complex (WICH), and remodeling and spacing factor (RSF)9–17. ISWI complexes have been implicated in the control of multiple processes including DNA replication, DNA repair, and transcription. RNAi mediated knockdown of SMARCA5 impaired DNA replication and S-phase progression, and the ISWI complexes in particular were important for replication through heterochromatin18–21. Similarly, the WICH-ISWI complex associated with PCNA and localized to sites of DNA replication22. In addition, multiple ISWI complexes have been implicated in the DNA damage response and double strand break repair22–25. Finally, the RSF-ISWI complex is required for the initiation of transcription from chromatinized DNA templates in vitro26, suggesting that SMARCA5-containing complexes may directly regulate gene expression. In fact, defects in transcriptional control were proposed to underlie a number of phenotypes observed in Smarca5-deficient mouse and zebrafish models27,28.
Unlike the related SWI/SNF chromatin remodeling complexes, the ISWI complex cannot evict nucleosomes, rather it slides them to maintain appropriately spaced nucleosome arrays25,29–33. Deletion of Smarca5 from mouse embryonic stem cells revealed that it was critical for the maintenance of appropriate nucleosome repeat length (NRL)34. As the length of the core-associated DNA is static (147 bp), changes in nucleosome repeat length reflect a lengthening or shortening of the linker DNA. Interestingly, NRL correlates with specific genomic features. For example, the gene body of highly expressed genes tends to have a relatively short nucleosome repeat length, which may prevent nucleosomal packing, while heterochromatin is associated with a longer nucleosome repeat length, which may allow nucleosomal stacking and compaction3. In addition, nucleosome positioning can be variable at some genomic features, but well defined at others. For instance, chromatin insulators bound by CTCF and transcription start sites (TSS) of active genes exhibited more rigid nucleosome phasing patterns3,34,35. It is likely that these unique nucleosomal patterns have both structural and regulatory consequences.
In addition to changes in nucleosome repeat length and consistent with previous studies, Smarca5 deletion in mouse embryonic stem cells resulted in a loss of CTCF binding and disruption of topology associated domain (TAD) structure, suggesting that Smarca5 is required for CTCF binding and function34. SMARCA5 also associates with CTCF and RAD21 in co-immunoprecipitation assays, further connecting SMARCA5 with CTCF function36–38. However, due to the static nature of the knockout studies, the exact mechanism by which Smarca5 altered CTCF binding could not be addressed. These results suggested that CTCF could direct SMARCA5 to slide nucleosomes to organize chromatin at its DNA binding sites. In contrast, a degron tagged version of the only ISWI member in Drosophila S2 cells caused changes in nucleosome positioning only around “housekeeping” genes whose expression was only modestly affected in the first 6hr after degradation39. These results may reflect fundamental differences between the Drosophila cell line and mammalian cells. For example, there is a profound difference in the requirement of CTCF in Drosophila versus vertebrates, as deletion of CTCF in Drosophila had only modest effects on genome architecture40.
While germline deletion of Smarca5 caused embryonic lethality, tissue-specific deletion also suggested a profound role in normal tissue development and differentiation, which was associated with large changes in gene expression, as well as proliferation defects41–43. These genetic deletion models helped define the role of Smarca5 in biology and development, yet they cannot effectively distinguish direct and indirect chromatin effects, and thus have not allowed a mechanistic examination of Smarca5 function in cells. In contrast, in vitro biochemical reconstitution assays have provided details on how these chromatin remodeling enzymes bind to DNA and hydrolyze ATP to move nucleosomes32,44. In fact, single-molecule analysis of SMARCA5 nucleosomal sliding indicated that a 1–2 base pair step (or sub-step) occurs within 10–30 seconds, highlighting the dynamic nature of nucleosomal positioning32,44. These in vivo versus in vitro analyses highlight the challenge of defining the contribution of nucleosome remodeling complexes to gene expression, DNA replication and cell cycle progression.
Here, we used CRISPR-Cas9D10A and homology-directed DNA repair to modify the endogenous allele of SMARCA5 to make it sensitive to a small molecule proteolysis targeting chimera (PROTAC), dTAG-47, to begin to close this gap45,46. We modified the endogenous SMARCA5 loci in acute myeloid leukemia cells (Kasumi-1), erythroleukemia cells (HEL), and a diffuse large B-cell lymphoma cell line (OCI-LY1). By using a small bi-functional molecule to rapidly degrade SMARCA5 we were able to show that SMARCA5 had only modest effects on nascent transcription or cytoplasmic pools of mRNA within the first 6hr after degradation. Conversely, ATAC-seq and MNase-seq identified rapid changes in nucleosome repeat length throughout the genome. Moreover, these effects were observed in synchronized cell cultures at the G1/S phase and in the mid to late S phase of the cell cycle. This was particularly acute near CTCF DNA binding sites, where CTCF was rapidly lost from the genome. These results suggest that nucleosome positioning is dynamic throughout the cell cycle and might suggest that SMARCA5 plays a role as a counterweight to other nucleosome sliding events in the cell.
Results
Engineering an endogenous SMARCA5-FKBP12F36V allele for inducible degradation
Chromatin-remodeling enzymes are the subject of intense drug discovery efforts, as they could be therapeutic targets in cancer, including acute leukemia42,47, and other chronic disease syndromes48,49. Small molecule inhibitors of chromatin remodeling enzymes are also invaluable tools for the discovery of the mechanism of action of these enzymes, as they allow rapid enzyme inhibition. Because a selective small molecule inhibitor was not readily available for SMARCA5, we used CRISPR-Cas9D10A nickase to induce two ssDNA breaks at the end of the coding region of SMARCA5 and homology-directed repair to insert FKBP12F36V-FLAG to generate a C-terminal fusion protein in the t(8;21)-containing acute myeloid leukemia (AML) cell line Kasumi-1, the human erythroleukemia cell line (HEL), and a diffuse large B-cell lymphoma cell line (OCI-LY1) (Figure 1A)50. This approach yielded an endogenous SMARCA5 protein that was rapidly degraded upon treatment with the PROTAC, dTAG-47 (Figure 1A)45,50. We performed RNA-seq analysis on the parental and CRISPR-edited Kasumi-1 cells to ensure that the addition of the degron tag alone did not dramatically alter gene expression. We observed few changes in gene expression between parental and SMARCA5-FKBP12F36V-FLAG cell lines with a Pearson correlation coefficient of the log2 (FPKM) between these cells of 0.9965 (Figure S1A).
Figure 1. Selective degradation of endogenously tagged SMARCA5 leads to decreased cell growth.
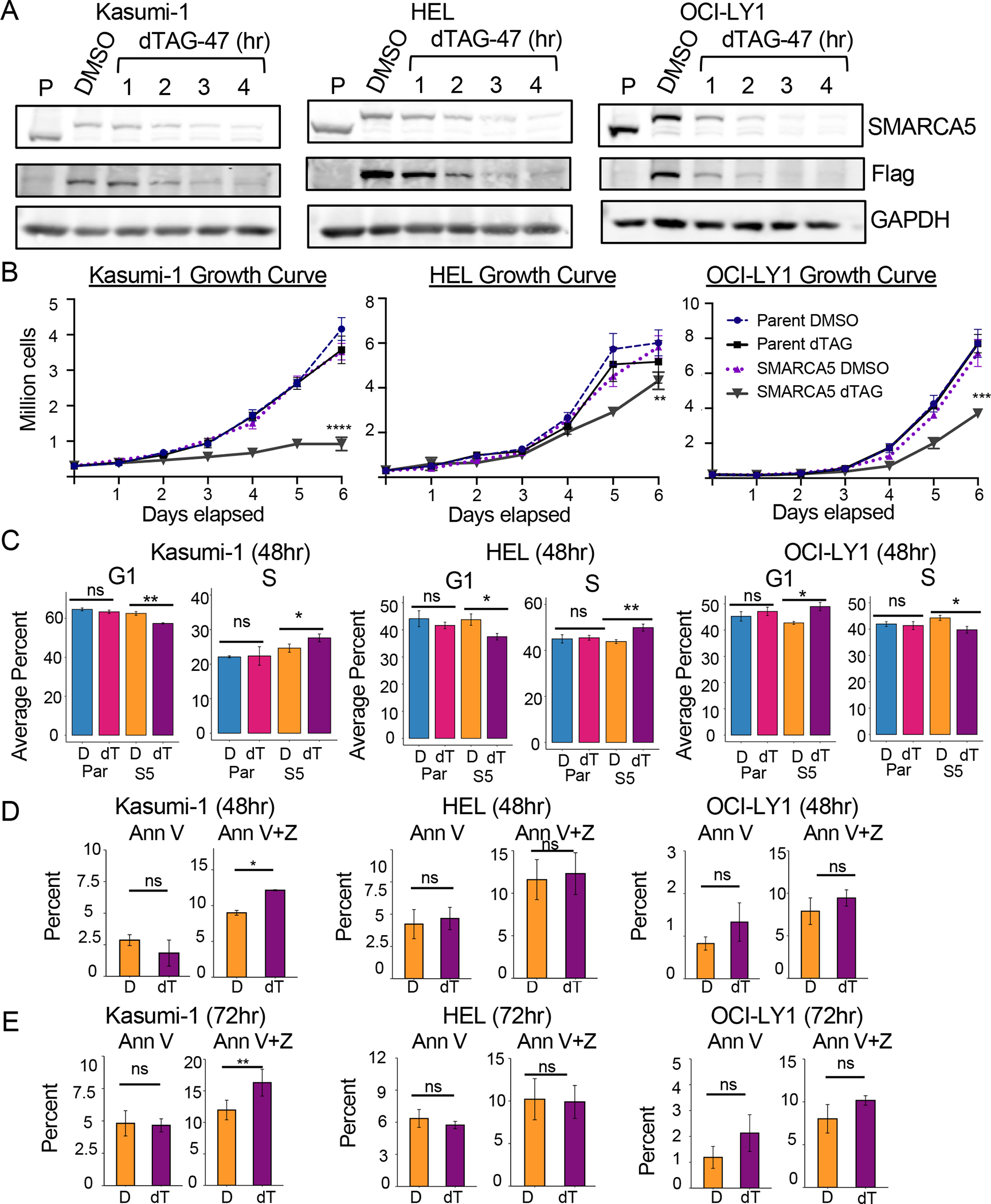
(A) Degradation time course showing rapid loss of SMARCA5 in Kasumi-1, HEL, and OCI-LY1 cells (Parental and SMARCA5FKBP12F36V-Flag) after 500nM dTAG-47 treatment. Western blot of SMARCA5, Flag, and loading control, GAPDH. (B) Cell growth curve of Kasumi-1, HEL, and OCI-LY1 Parental versus SMARCA5-FKBP12F36V-FLAG cells over a 6-day period after dTAG-47 treatment. Cell viability was determined using Trypan Blue exclusion (n=3). (C) Bar graphs of flow cytometry analysis of BrdU incorporation versus propidium iodide (PI) staining of Kasumi-1, HEL, and OCI-LY1 Parental versus SMARCA5-FKBP12F36V-FLAG-expressing cells after 48h dTAG-47 treatment (n=3). Two-way repeat measure ANOVA: **** = p<0.0001; *** = p<0.0007; ** = p<0.0014. (D, E) Bar graphs of flow cytometry analysis of Annexin V staining vs Zombie-NIR staining of Kasumi-1 (n=5), HEL (n=5), and OCI-LY1 (n=3) SMARCA5-FKBP12F36V-FLAG-expressing cells after 48h (D) or 72h (E) dTAG-47 treatment. D, DMSO; dT, dTAG-47; P-values (Welch two-tailed t test) are indicated by ns = p > 0.05, * = p <0.05, ** p< 0.001. Error bars show standard deviation.
Both germline and Vav-Cre-mediated Smarca5 deletion was embryonic lethal in mice43,51. Therefore, we first tested the effect of SMARCA5 degradation on cell growth and viability (Figure 1B). In Kasumi-1 cells, cell growth slowed 2–3 days after the addition of dTAG-47, while SMARCA5 degradation in HEL and OCI-LY1 cell lines did not cause a decrease in growth until day 5 (Figure 1B). The decreased cell growth in Kasumi-1 cells was associated with an increase in CD11b expression, which is indicative of myeloid differentiation (Figure S1B). There were also subtle changes in BrdU incorporation, suggesting that SMARCA5 may have contributed to efficient transit through the S phase in the Kasumi-1 and HEL cell lines (Figures 1C and S1C), which was previously observed after SMARCA5 knockdown52,53. The OCI-LY1 cell line showed a small decrease in the percentage of cells in the S phase with an accumulation in G1. Degradation of SMARCA5 in Kasumi-1 cells led to a statistically significant increase in Zombie NIR/AnnexinV double positive cells at 48hr and 72hr following dTAG-47 treatment, while HEL and OCI-LY1 cells did not display increased cell death upon SMARCA5 degradation (Figure 1D, 1E and S1D), consistent with the more dramatic effect on cell growth in Kasumi-1 cells (Figure 1B).
Genomic localization of SMARCA5
When engineering the SMARCA5-FKBP12F36V allele, we incorporated a FLAG epitope tag to simplify the analysis of endogenous SMARCA5. We used anti-FLAG in cleavage under targets and release using nuclease (CUT&RUN) analysis in the presence or absence of dTAG-47-mediated degradation to localize SMARCA5 throughout the genome54. We identified 9,650 SMARCA5 peaks in Kasumi-1, 12,048 in HEL, and 8,546 in OCI-LY1 that were reduced by at least 1.5-fold following dTAG-47 treatment (Figure 2A). These changed peaks were enriched in promoters, intronic, and intergenic regions (Figure S2), and many of these sites overlapped between cell lines (Figure 2B). For example, the region around SERTAD1 shows multiple conserved SMARCA5 (FLAG) peaks (Figure 2C). The consensus binding site for CTCF and the related BORIS were commonly enriched under the changed peaks within each cell line (Figure 2D). These results are consistent with ChIP-seq data from mESCs demonstrating frequent colocalization of CTCF and SMARCA538.
Figure 2. Genomic localization of endogenously tagged SMARCA5-FKBP12F36V-FLAG.
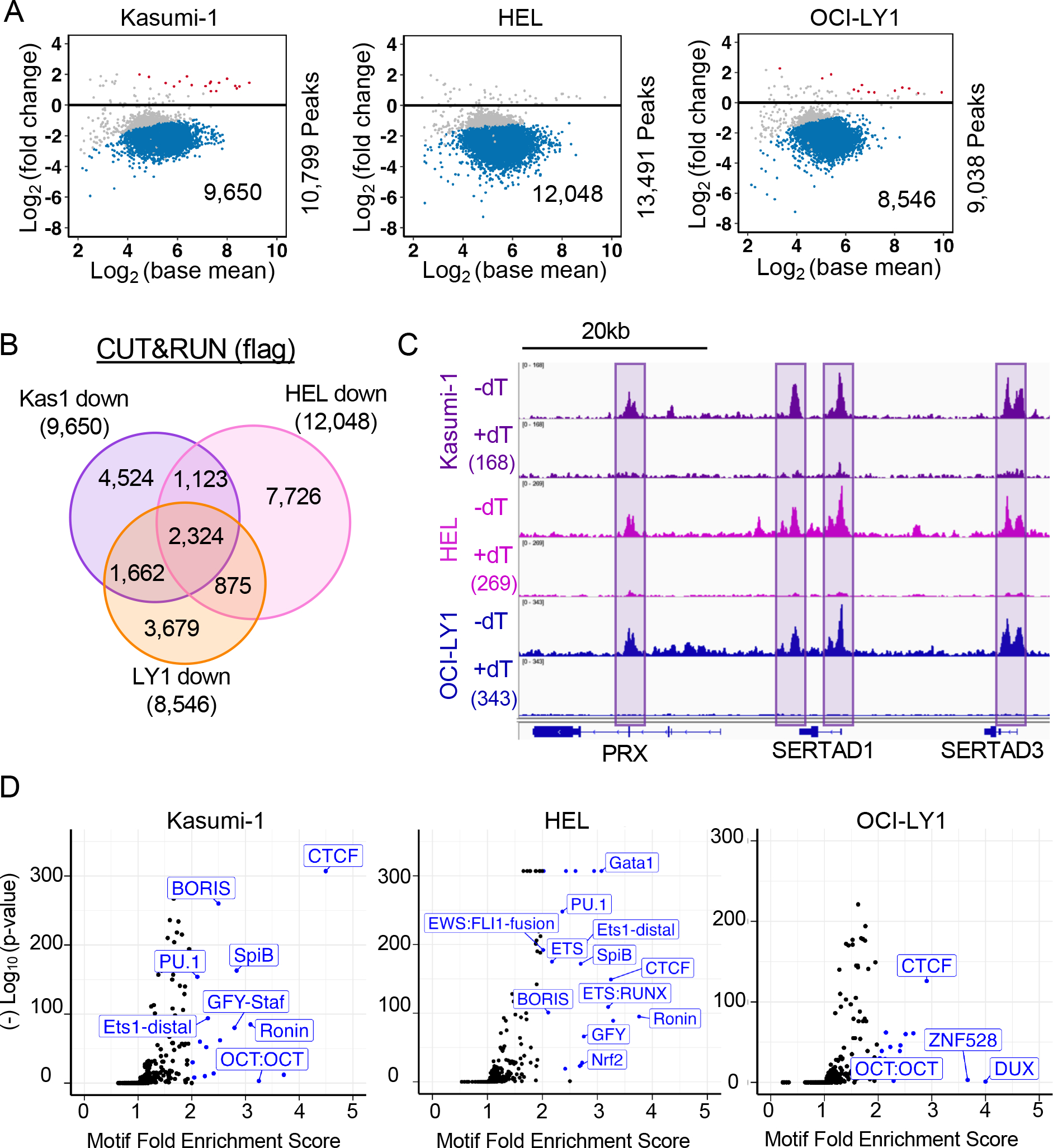
(A) MA plots of the changes in peaks upon degradation of SMARCA5-FKBP12F36V-FLAG. The blue dots represent peaks down-regulated at least 1.5-fold. (B) Venn diagram showing the overlap between the SMARCA5 CUT&RUN decreased peaks from each of the three cell lines. (C) IGV browser tracks showing an example of peaks conserved in all three cell lines. (D) Volcano plots of motif enrichment scores (percent enrichment/percent background) of the significantly down-regulated peaks versus the Log10 (p-value). Blue dots represent motifs enriched by 2-fold.
SMARCA5 loss affects CTCF localization
Smarca5 deletion in mouse ES cells resulted in a loss of CTCF binding34. Therefore, we performed CUT&RUN to assess the ability of CTCF to bind DNA during a time course of SMARCA5 degradation (Figure 3A–C). Remarkably, the intensity of all the CTCF binding was reduced following SMARCA5 degradation, with the observed decrease beginning at 2hr. While the number of peaks significantly reduced within 2hr of dTAG-47 treatment varied among the three cell lines, all of the peaks were trending lower and by 6hr there was a dramatic and significant loss of CTCF DNA binding (Figure 3A–C). Although these cell types represent different hematopoietic lineages, there was a great deal of conservation among the CTCF sites that were lost at 6hr (Figure 3D).
Figure 3. SMARCA5 is required for CTCF DNA binding.
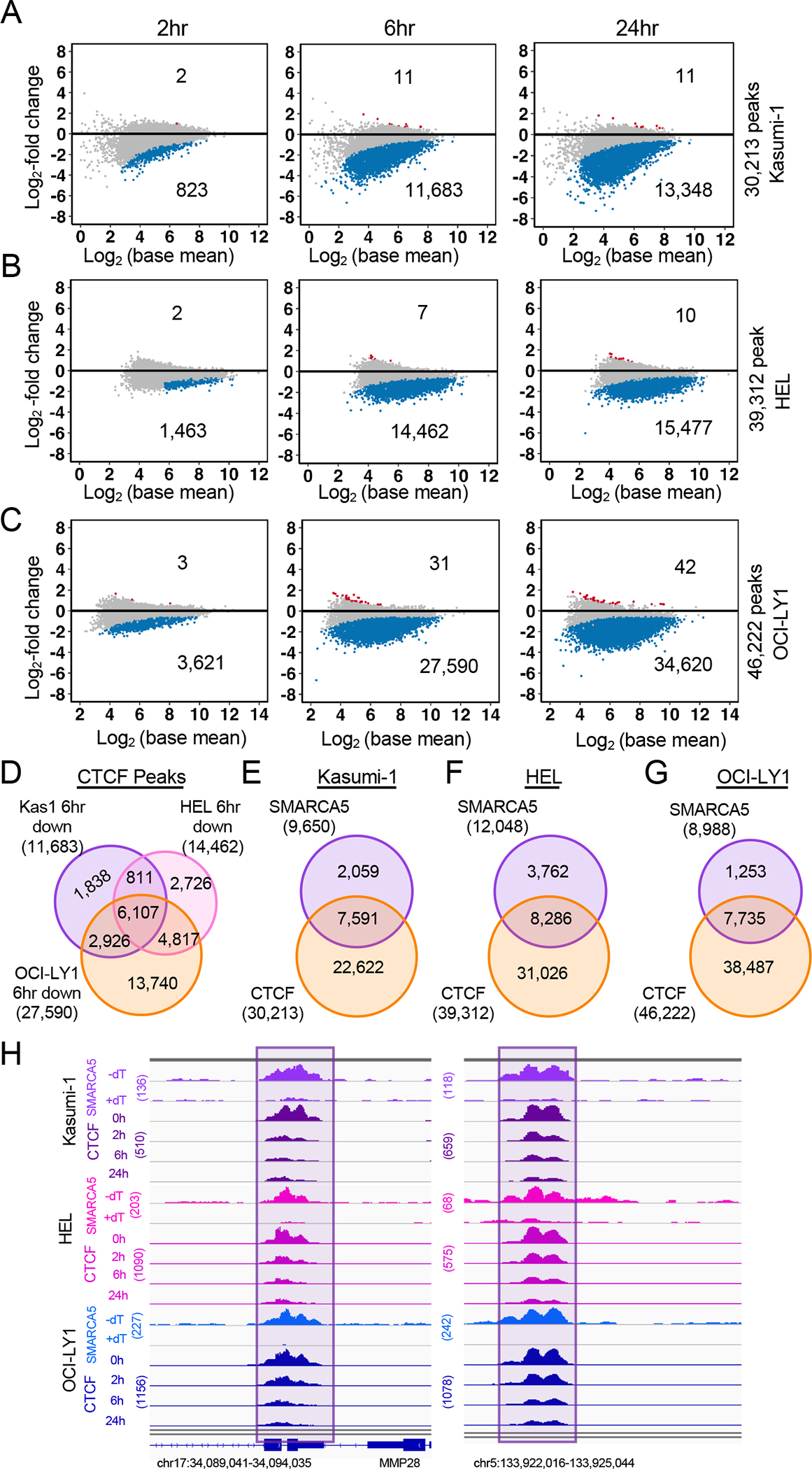
(A-C) MA plots of differential analysis of CTCF CUT&RUN peaks at the indicated time points in (A) Kasumi-1 (n=2), (B) HEL (n=2), (C) OCI-LY1 (n=2) cells expressing SMARCA5-FKBP12F36V-FLAG. The blue dots represent the down-regulated CTCF peaks. The red dots represent the up-regulated CTCF peaks. (D) Venn diagram showing the CTCF sites that were lost 6hr after degradation of SMARCA5-FKBP12F36V-FLAG from Kasumi-1, HEL, and OCI-LY1 cells. (E-G) Venn diagrams of the SMARCA5 CUT&RUN peaks and CTCF CUT&RUN peaks in (E) Kasumi-1, (F) HEL, (G) OCI-LY1 cells expressing SMARCA5-FKBP12F36V-FLAG. (H) IGV browser tracks of SMARCA5 CUT&RUN and CTCF CUT&RUN at the indicated time after dTAG-47 treatment in Kasumi-1 (purple), HEL (pink), and OCI-LY1 (blue) SMARCA5-FKBP12F36V-FLAG cell lines.
Given that it appeared that all CTCF binding sites were changing over the time course of SMARCA5 degradation, we overlapped all CTCF peaks with the SMARCA5 peaks, and found a 70–80% overlap of CTCF with SMARCA5 peaks (Figure 3E–G). For example, in the region around MMP28, there were both SMARCA5 and CTCF peaks that were lost upon degradation of SMARCA5 in all three cell lines (Figure 3H). Thus, it appears that degradation of SMARCA5 caused a rapid loss of CTCF DNA binding.
While the loss of CTCF DNA binding was closely associated with the degradation of SMARCA5, to ensure that other DNA binding proteins were not affected, we performed CUT&RUN assays to detect RUNX1 (Figure S3A) and the t(8;21) fusion protein, AML1-ETO (Figure S3E). Only 5 RUNX1 sites changed within 24hr of SMARCA5 degradation (Figure S3A), while 7,031 AML1-ETO sites were significantly down-regulated (Figure S3B). The larger number of AML1-ETO binding sites that were lost was intriguing, so we used motif analysis and found that these sites were not enriched for a RUNX1-binding motif, but contained a CTCF motif (Figure S3C). Not surprisingly, these down-regulated AML1-ETO sites did not overlap with the previously published AML1-ETO regulated enhancer peaks (Figure S3D)55. Indeed, many of these AML1-ETO binding sites were bound by CTCF (Figure S3E), suggesting that the loss of these weak AML1-ETO binding sites could be secondary to the loss of CTCF.
SMARCA5 maintains accessible chromatin around CTCF motifs
Given the ability of SMARCA5 to slide nucleosomes along the DNA56,57, we used ATAC-seq to identify areas of the genome that were affected by degradation of SMARCA558. We performed a time course analysis at 0, 2, 6, and 24hr after the addition of dTAG-47 to the Kasumi-1 cell line to assess changes in transposase accessibility over time. This analysis identified few areas of the genome that were affected within the first two hours, but there were substantial changes within 6hr with 8,513 peaks lost and 6,347 gained, and by 24hr there were 12,669 peaks lost and 10,766 gained (Figures 4A, S4A, and S4B). We also performed ATAC-seq after 6hr of dTAG-47 treatment in the HEL and OCI-LY1 cell lines containing the SMARCA5-FKBP12F36V fusion and also found large changes in accessibility (Figure 4B and 4C). The down-regulated peaks in each of the three cell lines showed significant overlap with 888 peaks found in all three cell lines and roughly 50% overlap when comparing just two of the cell lines at a time (Figure 4D). On the other hand, the up-regulated peaks showed only 24 overlapping peaks between all three cell lines and 5% overlap when comparing just two cell lines (Figure 4E).
Figure 4. Loss of SMARCA5 alters chromatin accessibility.
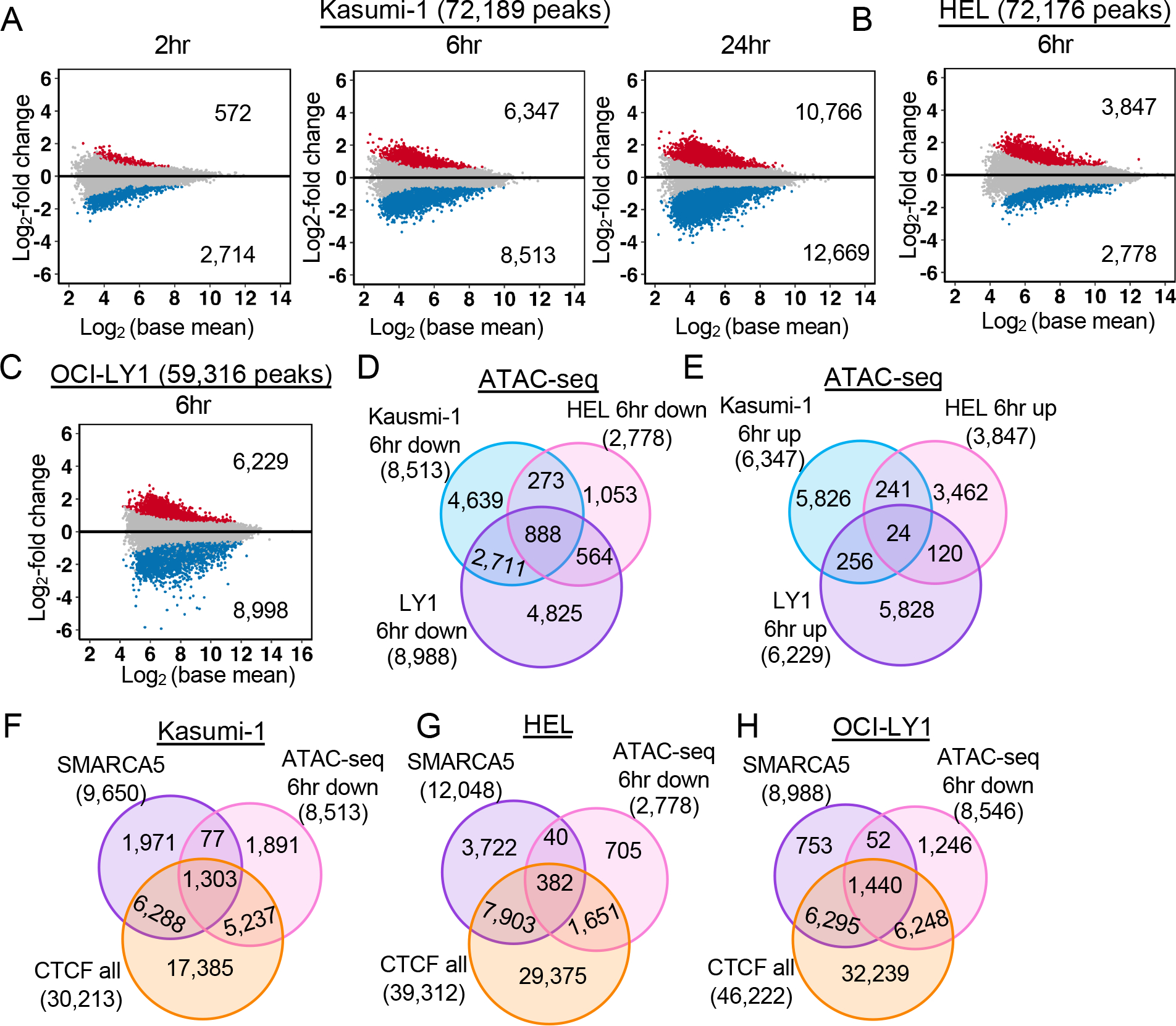
(A-C) MA plots of differential analysis of ATAC-seq peaks at the indicated time points of dTAG-47-mediated degradation of SMARCA5-FKBP12F36V-FLAG in (A) Kasumi-1 (n=4), (B) HEL (n=2), (C) OCI-LY1 (n=2). The blue dots represent peaks down-regulated and red dots up-regulated by at least 1.5-fold. (D-E) Venn diagrams showing that some down-regulated (D) and up-regulated (E) ATAC-seq peaks from A, B, and C are conserved in Kasumi-1, HEL, and OCI-LY1 cells. (F-H) Venn diagrams showing the overlap of the SMARCA5 CUT&RUN peaks, CTCF CUT&RUN peaks, and ATAC-seq 6hr after degradation of SMARCA5-FKBP12F36V-FLAG in Kasumi-1 (F), HEL (G), and OCI-LY1 (H) cells.
Next, we asked if the accessibility changes were associated with the genomic binding sites of SMARCA5 or with the CTCF binding sites. The ATAC-seq peaks that were lost showed a significant overlap with CTCF binding sites in all three cell lines (Figure 4F–H). Interestingly, there was a better overlap between SMARCA5 DNA association and loss of accessibility in the Kasumi-1 and OCI-LY1 cells than in HEL cells (Figure 4F–H). Nevertheless, motif enrichment analysis of the sequences identified by the down-regulated ATAC-seq peaks found that the CTCF/BORIS motif was almost 15-fold enriched in comparison to any other motif in all three cell lines tested (Figure S4C). Moreover, when considering the 888 ATAC-seq peaks that were found in all three cell lines (Figure 4D), the enrichment for CTCF motifs was 20-fold (not shown). In contrast, there was little enrichment among the up-regulated peaks.
SMARCA5 only modestly affects RNA polymerase dynamics
Next, we sought to determine whether the changes in chromatin accessibility observed upon SMARCA5 degradation were associated with changes in gene expression. Having a small molecule that rapidly degraded SMARCA5-FKBP12F36V allowed us to assess gene expression changes in the first hours after SMARCA5 degradation to determine if it directly regulates transcription. Given that SMARCA5 has been linked to the control of RNA polymerase elongation59, we performed precision nuclear run-on sequencing (PRO-seq)60 at 0, 2, 6, and 24hr after the addition of dTAG-47 to Kasumi-1 cells containing the SMARCA5-FKBP12F36V fusion protein (Figure 5 and S5). This analysis identified very few genes that exhibited reduced or increased gene body (GB) transcription with 50 genes up and 63 genes down at least two-fold at 6hr after degrading SMARCA5 (Figure 5A). Moreover, there was very little overlap of the transcriptional changes over time with only 19 genes exhibiting increased gene body transcription and 23 genes exhibiting reduced gene body transcription at all 3 time points (Figure 5B). Roughly a third of these 6hr changed genes could be associated with a SMARCA5 binding site within +/− 25 KB of the TSS (Figure 5C). Interestingly, many more of these 6hr changed genes were associated with a nearby CTCF binding site (50 genes within 500 bp of the TSS; Figure 5D). While a similar number of changes were detected by RNA-seq analysis at 6hr after degrading SMARCA5, the changes in nascent transcription only affected a small number of mRNAs even at 24hr after degradation of SMARCA5 (40 down; 18 up; Figure 5E, 5F).
Figure 5: Degradation of SMARCA5 causes modest transcriptional changes.
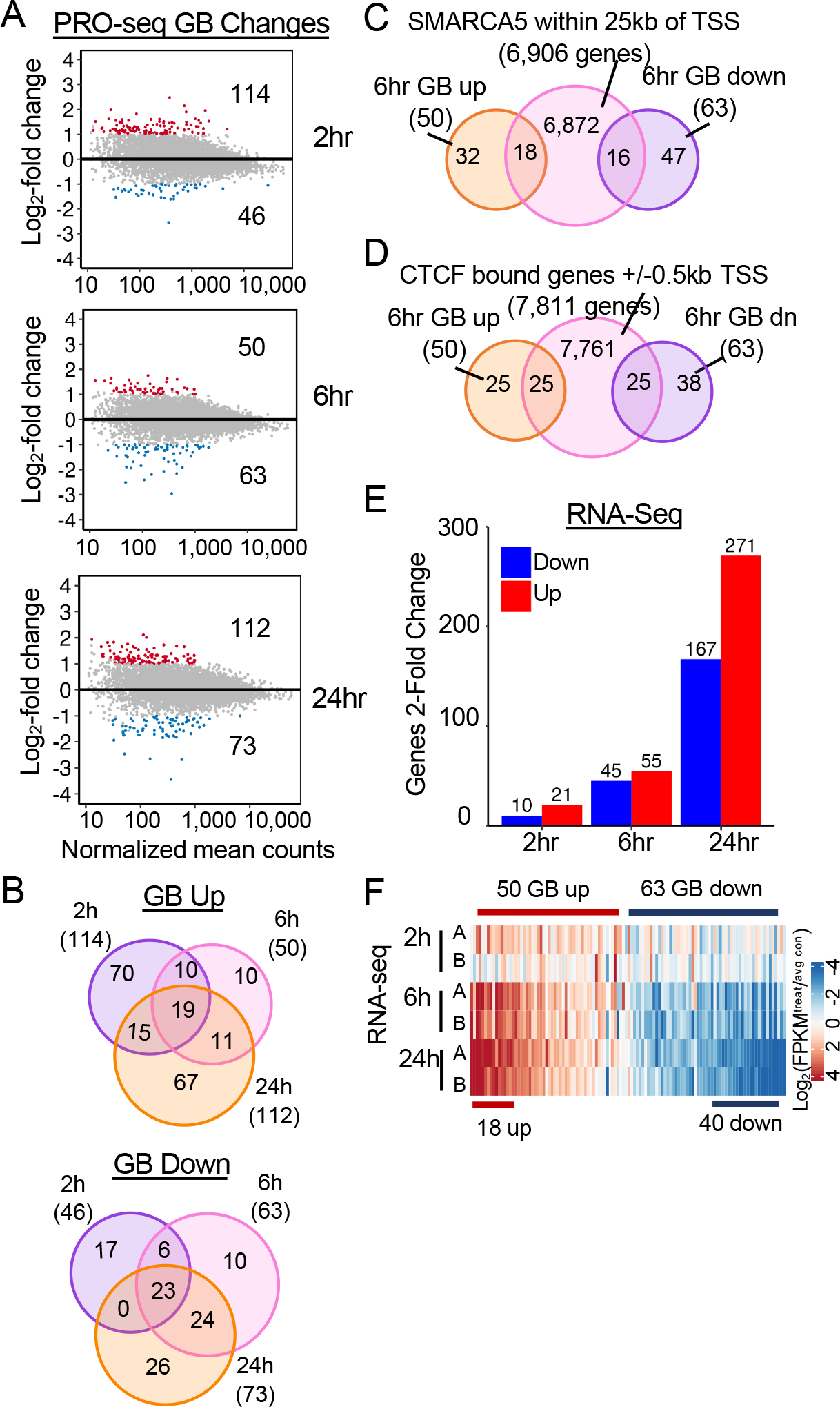
(A) MA plots of the log2 fold change vs the normalized mean counts of the gene body. The colored dots are the significant (adjusted p-value < 0.05 and fold change > 2; red = Up, blue = Down) gene body changes. (B) Venn diagram showing the overlap between changes within the gene body of up- or down-regulated genes at 2, 6, 24hr. (C) Venn diagram showing the overlap between gene body changes of up- or down-regulated genes at 6hrs after SMARCA5 degradation intersected with SMARCA5 CUT&RUN peaks annotated to +/−25kb of the TSS of the nearest neighbor gene. (D) Venn diagram showing the overlap between genes up- or down-regulated 6hr after SMARCA5 degradation intersected with CTCF CUT&RUN peaks annotated to +/−500bp of the TSS of the nearest neighbor gene. (E) Bar graph of the changes in mRNA pools measured by RNA-seq at 2h, 6h and, 24h after dTAG-47 treatment to degrade SMARCA5 in Kasumi-1 cells. (F) Heatmap of log2 (FPKM/avgFPKM) of each RNA-seq replicate 2h, 6h, and 24h after SMARCA5 degradation for genes regulated in the PRO-seq analysis at 6h.
PRO-seq also provides information about RNA polymerase pausing around the TSSs of expressed genes. We quantified the promoter-proximal (pp) RNA polymerase density and found that rapid degradation of SMARCA5 resulted in only modest effects (Figure S5A–B) and only 26 of these genes showed a loss of expression in our RNA-seq datasets (Figure S5C). Interestingly, 53 of these pp down genes contained a CTCF CUT&RUN binding site, within 500bp of the transcription start site and 46 of these pp down genes were also bound SMARCA5 (Figure S5D–E), suggesting that these effects could be secondary to the loss of CTCF DNA binding.
SMARCA5 loss affects chromatin architecture
Given that SMARCA5 can position nucleosomes, we further probed the ATAC-seq data to examine nucleosome repeat length as a measure of nucleosome compaction within the three different cell lines. The algorithm uses a local regression fit model with two smoothing parameters (only one shown, red; Figure 6A) to calculate the average nucleosome repeat length throughout the region of interest34. This analysis detected a lengthening in the nucleosome repeat length within 6hr of dTAG-47 treatment that was even further increased by 24hr in Kasumi-1 cells (Figures 6A and 6B). Analysis of the HEL and OCI-LY1 ATAC-seq data also showed a significant increase in global nucleosome repeat length 6hr after dTAG-47 treatment (Figures 6C–6F). While our ATAC-seq sequencing depth was not sufficient to measure nucleosome repeat length around individual features within the genome, it still appeared that SMARCA5 was regulating global nucleosome repeat length.
Figure 6: SMARCA5 regulates nucleosome repeat length independent of the cell cycle.
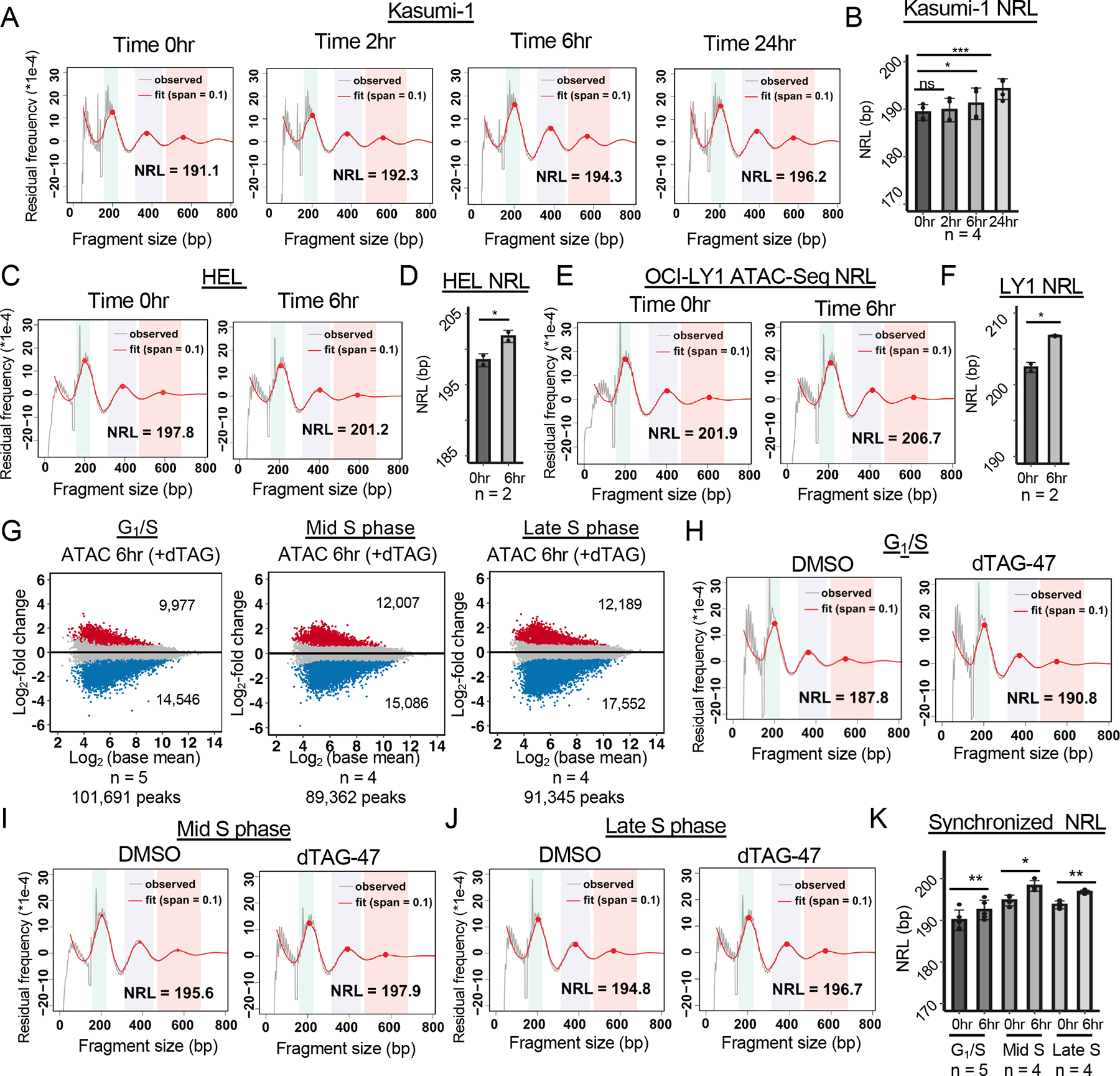
(A) Nucleosome phasing graph showing changes in the nucleosome repeat length (NRL) over the time course of degradation of SMARCA5-FKBP12F36V-FLAG. Red line shows the “fit” of the curve versus the observed ATAC-seq signal of one replicate (grey line) using the entire genome. (B) Bar graph of the NRL of all replicates (n=4) at each of the indicated time points. (C) Nucleosome phasing graph showing changes in the NRL at 6hr after degradation of SMARCA5-FKBP12F36V-FLAG in HEL cells using the entire genome. (D) Bar graph of the NRL of all replicates (n=2) at 6hr after degradation of SMARCA5 in HEL cells. (E) Nucleosome phasing graph showing changes in the NRL at 6hr after degradation of SMARCA5-FKBP12F36V-FLAG in OCI-LY1 cells using the entire genome. (F) Bar graph of the NRL of all replicates (n=2) at 6hr after degradation of SMARCA5 in OCI-LY1 cells. (G) MA plots showing changes in accessible peaks 6hr after degrading SMARCA5 at the G1/S, mid S and late S phases of the cell cycle. The blue and red dots represent the changed ATAC peaks with an at least 1.5-fold change. (H-J) Nucleosome phasing graphs showing changes in the nucleosome repeat length in Kasumi-1 cells synchronized at G1/S (H), Mid S phase (6hr after release from double thymidine block; I), or late S phase (12hr after release from block; J). (K) Bar graph of the NRL of individual replicates (dots) at each of the indicated phases of the cell cycle. For B, D, F and K, the significance was calculated with a two sampled paired t-test: ****=p<0.0001, ***=p<0.001, **=p<0.01, *=p<0.05; error bars represent standard deviation.
A major advantage of the dTAG system is the ability to probe the action of SMARCA5 during the cell cycle. We used a double thymidine block to synchronize Kasumi-1 cells at the G1/S boundary prior to the degradation of SMARCA5-FKBP12F36V and used BrdU incorporation to track the progression of cells through the S phase (Figures S6A–S6C). At the initiation of the time course, the vast majority of the cells were at G1/S and did not incorporate BrdU. However, within the first 2hr after removal of the thymidine block, the cells had re-entered the S phase, indicating that SMARCA5 was not required to initiate DNA replication. Within the first 6hr after release from the block, more than 60% of the cells had incorporated BrdU (Figure S6A–S6C) and within 12hr the control cells showed a roughly 50% reduction in the percentage of cells in the S phase, as these cells traversed and then exited the S phase. In contrast, Kasumi-1 cells lacking SMARCA5 were retained in the late S phase 12hr after release into the S phase and displayed a significant lag in returning to the G1 phase (Figure S6A–S6C). These data indicate that SMARCA5 may contribute to the passage of the late S phase in Kasumi-1 cells.
Using this information, we then performed ATAC-seq on synchronized Kasumi-1 cells with or without a 6hr degradation of SMARCA5-FKBP12F36V at G1/S, mid S (6hr after release), or late S (12hr after release). First, we identified accessible peaks and performed differential analysis to define the effect of synchronization on general accessibility. This analysis showed more dramatic changes in chromatin accessibility with a greater loss of peaks than gain of peaks in each of the cell cycle phases in comparison to untreated cells (Figure 6G). The up- and down-regulated peaks also showed a high degree of overlap between these phases of the cell cycle (40–60%; Figure S6D–S6E), and a high degree of overlap with binding of CTCF at each phase of the cell cycle (Figure S6F–H). This suggests that loss of CTCF was likely a key component of the loss of accessibility upon degradation of SMARCA5 at these different stages of the cell cycle.
Next, we analyzed the nucleosome repeat length of these populations of cells. We noted that nucleosome repeat length in the control cells changed as cells progressed from G1/S into S phase with a longer distance between nucleosomes at mid and late S phase after release from the block (0hr time points, Figure 6H–J). Even though the starting point for assessing the effects of SMARCA5 degradation was greater during the S phase, upon degradation of SMARCA5, we found a small but reproducible increase of the average length between nucleosomes at all three stages of the cell cycle assessed (Figure 6H–K). Together, these results suggest that SMARCA5 regulated chromatin accessibility independent of the cell cycle, and that SMARCA5 was required for nucleosomal spacing and compaction in both late G1 and the S phase of the cell cycle.
While ATAC-seq is a relatively rapid procedure, to deepen our analysis of chromatin structure and to examine different genomic features upon inactivation of SMARCA5, we performed micrococcal nuclease coupled with sequencing (MNase-seq) in Kasumi-1 cells34,56,61–63. Like the ATAC-seq analysis, MNase-Seq detected a subtle lengthening in the nucleosome repeat length within two hours after the addition of dTAG-47 that was more distinct by 6hr and continued to expand at the 24hr time point (Figure 7A and 7B). Using the MNase-seq datasets, we were also able to measure the nucleosome repeat length around other sites throughout the genome. We assessed the nucleosome repeat length around the TSS of all expressed genes and at CTCF sites. We found no significant change in nucleosome repeat length over time around the start sites of expressed genes (Figure S7A and S7B), which is consistent with the small effects that were found on transcription. Furthermore, there was no change in nucleosome positioning when we plotted the normalized nucleosome occupancy −0.5kb and + 1kb from the TSSs of expressed genes (Figure S7C).
Figure 7: SMARCA5 maintains nucleosome structure around CTCF motifs in Kasumi-1 cells.
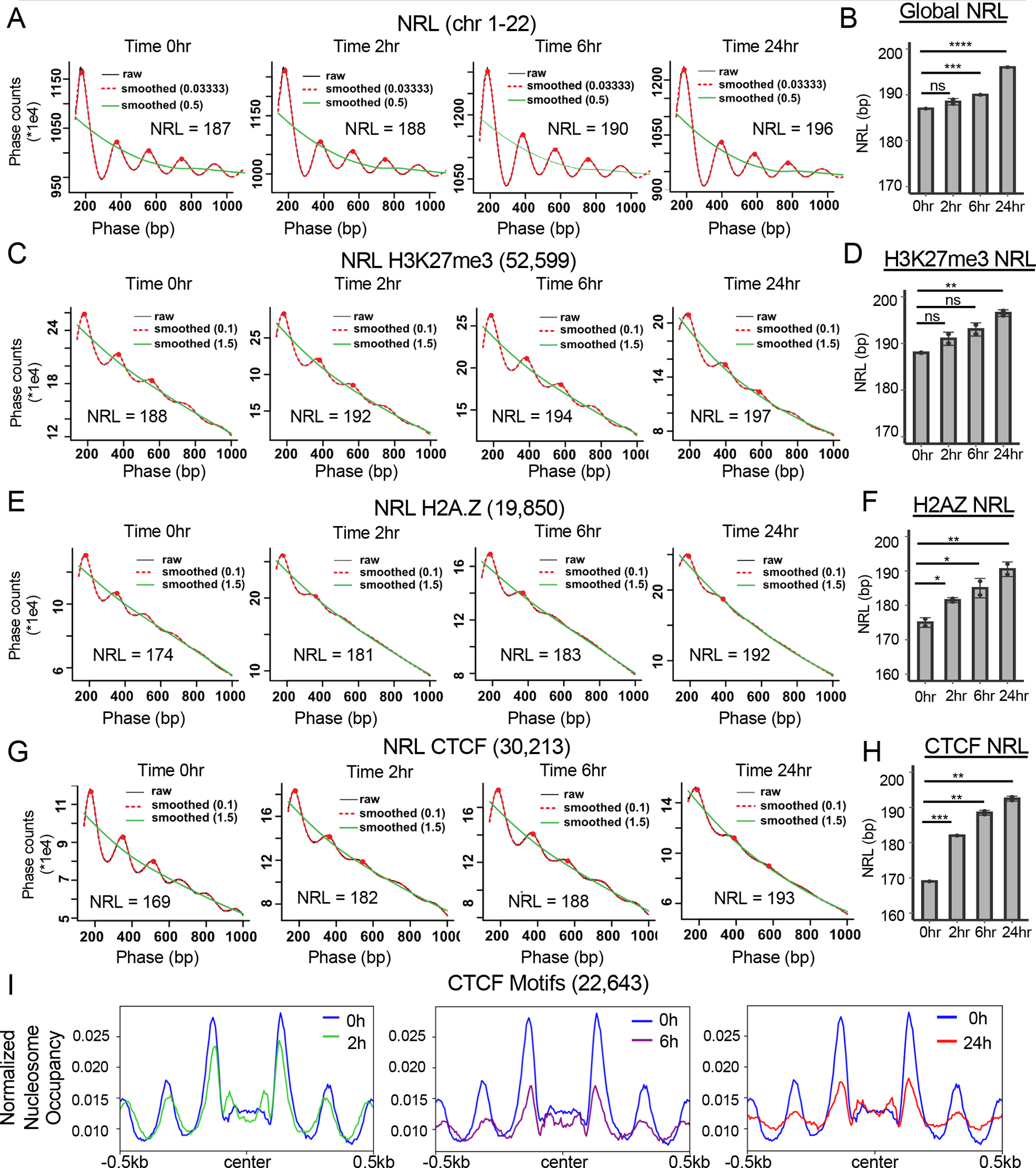
(A) “Phasogram” of the nucleosome repeat length (NRL) during the time course of SMARCA5 degradation (red line, span = 0.1, and green line, span = 1.5) derived from the MNase-seq signal of a representative replicate (grey line) using the entire genome. (B) Bar graph quantification from biological replicates of the NRL calculated from the MNase-seq signal at each of the indicated time points. (C) “Phasogram” of the NRL of SMARCA5 degradation at indicated times around H3K27me3 peaks +/−1kb. (D) Bar graph quantification of NRL at H3K27me3 peaks from biological replicates calculated using the MNase-seq signal at each of the indicated time points. (E) “Phasogram” of the NRL during the time course of SMARCA5 degradation around H2A.Z CUT&RUN peaks +/−1kb. (F) Bar graph quantification of the NRL at H2A.Z peaks of biological replicates calculated from the MNase-seq signal at each of the indicated time points. (G) “Phasogram” of the NRL at the indicated times within 1kb of the peak center of CTCF CUT&RUN peak motifs. H. Bar graph quantification of the NRL of biological replicates within 1kb of CTCF motifs at the indicated time points. I. Histograms of nucleosome occupancy using MNase-seq data plotted around all CTCF motif centers (31,015) from the CTCF CUT&RUN data. The signal was plotted +/− 500bp from the motif center and the changes after SMARCA5 degradation at the indicated time points are shown. For B, D, F, and H, the significance was calculated with Welch’s two sample t-test: ****=p<0.0001, ***=p<0.001, **=p<0.01, *=p<0.05; error bars represent standard deviation.
Next, we used CUT&RUN to assess the location of H3K27me3 to mark closed chromatin and to assess the nucleosome repeat length around H3K27me3-marked sites. Although using subsets of the genome reduced the robustness of the phasing due to the inclusion of fewer reads in the analysis, it appeared that the nucleosome repeat length around H3K27me3 was longer than the global nucleosome repeat length at the beginning of the time course, which is consistent with a more closed chromatin conformation (Figure 7C and 7D). The nucleosome repeat length still increased in H3K27me3-marked regions upon degradation of SMARCA5 (Figure 7C and 7D).
H2A.Z marks open chromatin and has also been linked to SMARCA5 function, as H2A.Z was better at stimulating the ATPase activity of SMARCA5 than H2A63. Moreover, H2A.Z also co-localized to CTCF binding sites64. Therefore, we used CUT&RUN to map H2A.Z genomic localization and assessed the co-localization of SMARCA5 and CTCF (Figure S7D). It appeared that all essentially all of the H2A.Z peaks were associated with at least a low amount of SMARCA5 that was lost upon degradation, and roughly 70% of the called SMARCA5 peaks co-localized with both H2A.Z and CTCF (Figure S7E). The majority of these SMARCA5 binding sites were found within the more intense peaks found in clusters 1 and 2 after k-means clustering of the H2A.Z signal (Figure S7D and data not shown). Likewise, essentially all of the H2A.Z peaks were associated with CTCF, and CTCF was rapidly lost from these H2A.Z peaks upon degradation of SMARCA5 with a larger decrease associated with clusters 1 and 2 (Figure S7D and S7F). The proportion of down-regulated CTCF binding sites that overlapped with an H2A.Z site remained consistent throughout the time course of SMARCA5 degradation (25–30%, Figure S7F).
The nucleosome repeat length was shorter at sites occupied by H2A.Z than the global nucleosome repeat length, consistent with a more open configuration of H2A.Z-containing nucleosomes (Figure 7E, 0hr). While the phasing was less distinct at these areas of open chromatin, there was a large increase in nucleosome repeat length over the 24hr time course of SMARCA5 degradation at these regions (Figure 7E and 7F). Similarly, the nucleosome repeat length around CTCF motifs showed a rather short nucleosome repeat length that increased dramatically upon degradation of SMARCA5 (Figure 7G and 7H). Because of the apparent large changes in chromatin structure around CTCF sites upon SMARCA5 inactivation, we plotted the normalized nucleosome occupancy centered on the CTCF DNA binding motif under the CTCF peaks (Figure 7I). We found that the nucleosome phasing around the CTCF motifs was beginning to erode even at the 2hr time point and the phasing was significantly affected within the first 6hr of SMARCA5 degradation (Figure 7I). These data suggest that SMARCA5 is required to maintain the regular nucleosome spacing in concert with CTCF and H2A.Z, and that SMARCA5 acts across the genome and at different stages of the cell cycle as a nucleosome ruler to prevent undue chromatin compaction.
Discussion
SMARCA5 is an ATP-dependent helicase that is able to slide nucleosomes and order nucleosome arrays in vitro to act as a “ruler”57. Genetic studies using gene deletion or siRNA inhibition have implicated SMARCA5 as being required for the control of gene expression, DNA replication and repair, chromatin accessibility, as well as nucleosome spacing and even higher order chromatin looping15,25,34,37,39,51,59,65. However, these steady-state analyses performed a few days to weeks after inactivation of SMARCA5 were unable to differentiate direct versus indirect effects. By engineering the endogenous alleles of SMARCA5 for rapid degradation in mammalian cells, we established that SMARCA5 is continuously required to maintain nucleosomal spacing throughout the genome. This implies that nucleosomes are constantly moving or being moved, and that SMARCA5 was required to push these nucleosomes together to maintain more relaxed or accessible chromatin. These effects were independent of the phase of the cell cycle or whether the chromatin was more open or closed and suggests that chromatin is highly dynamic. This is in agreement with recent findings that individual loci are far more dynamic than previously envisioned66.
Genetic models have established CTCF as a central factor in SMARCA5-mediated chromatin regulation34,37,67. CTCF is required to establish chromatin loops and transcription activation domains or TADs, which are a key feature of chromosome organization34,66,68–70. However, because these genetic models reached a steady state in which nearly all CTCF DNA binding was lost34, these genetic approaches did not have the ability to link SMARCA5 to the loss of CTCF. Our time course approach showed that rapid degradation of SMARCA5 led to a relatively quick decline in all CTCF DNA binding with about a third of the CTCF sites being significantly reduced within the first 6hr of dTAG-47 addition (Figure 3A–3C). In addition, our CUT&RUN analysis indicated that roughly 70% of the SMARCA5 peaks overlapped with a CTCF and a H2A.Z peak (Figure S7E) and 70–80% of the ATAC-seq peaks that decreased overlapped with a CTCF binding site (Figure 4F–4H). Interestingly, the ATAC-seq peaks that were gained were not linked to CTCF or another DNA binding factor, indicating that these sites were gained stochastically. Thus, we hypothesize that inactivation of SMARCA5 allowed nucleosomes, likely containing H2A.Z to be positioned over the CTCF binding sites causing a rapid loss of CTCF binding and eventually a loss of DNA looping as observed in mESCs34. This is consistent with a rapid loss of nucleosomal phasing around CTCF binding sites (Figure 7I).
The use of a degron tag allowed us to synchronize the cells prior to SMARCA5 degradation, which indicated that SMARCA5 contributed to maintaining chromatin accessibility at the G1/S phase boundary, mid S phase and late S/G2 phase (Figure 6). Thus, SMARCA5 may contribute to maintaining nucleosomal phasing around CTCF and H2A.Z sites during multiple phases of the cell cycle. Indeed, even during the S phase, the ATAC-seq peaks that were lost were strongly associated with CTCF binding motifs (data not shown). These data argue that the impairment that we observed in Kasumi-1 cells late in the S phase is unlikely to be due to an effect in the G1 or G2/M phases of the cell cycle and point to a more direct role for SMARCA5 in the S phase27,28,30,34,37,42,47. Interestingly, Kasumi-1 cells have lost an allele of RAD2171, which raises the possibility that disruptions of nucleosome repeat length or CTCF binding could be synthetic lethal with cohesin mutations, which are prevalent in various types of leukemia and solid cancers. Given that the defect in S phase transit and the impaired growth were more evident in the t(8;21) cell line (Kasumi-1), than in the erythroleukemia cell line HEL or DLBCL cell line OCI-LY1 (Figure 1), it is possible that SMARCA5 could be a therapeutic target in t(8;21) AML or other types of cancer containing cohesin mutations72–75.
Limitations
While we have performed deep genomic analysis after degradation of endogenous SMARCA5 in three different cell lines that represent different cell lineages, they are all hematopoietic cell types. This raises the question as to whether SMARCA5 inactivation would have the same impact in other types of tissues. This is especially important given that SMARCA1 is not expressed in these cells, such that inactivation of SMARCA5 eliminated all ISWI functions. It is also notable that Kasumi-1 cells are haploinsufficient for the cohesin RAD2171. Given that Kasumi-1 cells showed a more significant growth defect and possible delay late in the S phase, it is possible that the alterations in accessibility around CTCF sites were amplified by the cohesion-low state.
STAR Methods
Resource Availability
Lead contact
Further information and requests for resources or reagents should be directed and filled by Scott Hiebert (scott.hiebert@vanderbilt.edu).
Materials availability
All the materials generated in this study are accessible upon request.
Data and code availability
All genomic datasets have been deposited at GEO and are publicly available at GEO: GSE160470. The raw western blot images were deposited to Mendeley Data and are publicly as of the date of publication. doi:10.17632/cf5vbhy67d.1.
The code and software used in this paper is described in detail on github: https://github.com/monnieb92/SMARCA5_paper (DOI: 10.5281/zenodo.7438719).
Any additional information required to reanalyze the data reported in this paper is available from the lead contact upon request.
Experimental model and Subject details
Cell culture
The Kasumi-1 cell line was purchased from ATCC and grown in RPMI supplemented with 20% fetal bovine serum, 1% L-glutamine, and 1% penicillin and streptomycin. The HEL cell line was grown in RPMI supplemented with 10% fetal plex, 1% L-glutamine, and 1% penicillin and streptomycin. The OCI-LY1 cell line was grown in IMDM supplemented with 10% fetalplex, 1% L-glutamine, and 1% penicillin and streptomycin. Drosophila S2 cells were grown in Schneider media supplemented with 10% fetal bovine serum, 1% penicillin and streptomycin.
dTAG-47
The chemical dTAG-47 was synthesized by the Vanderbilt University Molecular Design and Synthesis Center as previously described50, and reconstituted at 5mM in DMSO and stored at −20°C.
METHOD DETAILS
Gibson Cloning
The homology directed DNA repair (HDR) plasmids were created using Gibson cloning (NEB, E5510S) to assemble the 5’ and 3’ SMARCA5 homology arms (amplified by PCR from genomic DNA) and the FKBP12 plasmid containing either mCherry or blue fluorescent protein (pAW62.YY1.FKBP.knock-in.mCherry/BFP; #104370/104371 AddGene). The homology arms were amplified with Phusion (NEB, M0530S). The PCR reactions were performed per NEB manufacture protocol and PCR products were gel purified (Promega, A9282) before Gibson Assembly. Site directed mutagenesis (Agilent, 210518) was performed on final BFP and mCherry plasmids per the manufacturer protocol.
Western blot
Cells were washed with cold phosphate buffered saline (PBS), lysed with a buffer containing 50mM Tris-HCl pH 7.4, 150mM NaCl, 0.1% SDS, 0.5% DOC, 1% NP40, sonicated briefly on ice, and the lysates were clarified by centrifugation. Immunoblotting was performed with anti-SMARCA5 (1:1000 abcam, 72499), anti-GAPDH (1:1000 SantaCruz, sc-365062), anti-FLAG (1:500 Sigma M2Flag, F1804) in blocking buffer. The membrane was visualize using fluorescently tagged secondary antibodies from LICOR Biosciences and visualized with the Odyssey scanner.
CRISPR knock-in and electroporation
Cells were seeded at 6 × 105/ml the day before electroporation. The gRNAs complexes were mixed at a ratio of 1.5uL of 100uM crRNA_C (IDT, Alt-R® CRISPR-Cas9 crRNA, 2 nmol): 5’ GATGGCGCACCTGATGGTCG 3’, 1.5uL of 100uM crRNA_D (IDT, Alt-R® CRISPR-Cas9 crRNA, 2 nmol): 5’ GGTGAAGACTGAAAGGGACAA 3’, 1.5uL of 200uM tracrRNA (IDT, 1072532), and 1.5uL of R buffer (Neon Kit), and heated at 95°C for 5 minutes and then allowed to cool to 20° C. Ribonucleoprotein (RNP) complexes were formed with 10ug of gRNA complex and 15 ug of Cas9D10A-Nickase (IDT, 1081062) by incubation at 20° C for 25 minutes. The Neon System was used to electroporate using a 100uL tip (ThermoScientific, MPK100025) with the following conditions: 1350V, 35ms, 1 pulse (Kasumi-1); 1300V, 20ms, 1 pulse (HEL); 1700V, 20ms, 1 pulse (OCI-LY1). 1 × 106 cells were resuspended in 100uL of R buffer (ThermoScientific, MPK100025) and added to the RNP reaction with 4 ug of mCherry and 4ug of BFP HDR plasmids prior to electroporation. Electroporated cells were placed in antibiotic free media overnight at 37°C 5% CO2 and the media replaced 24 hours after electroporation.
Cell proliferation
Cells were seeded at 3 × 105/mL, then treated with 500nM dTAG-47 or DMSO. Cell viability was assessed using Trypan Blue (Gibco, 15250-061) exclusion and counted on a hemocytometer every day for six days. The cells were re-seeded at 3 × 105/mL, then treated with 500nM dTAG-47 or DMSO on the third day.
Cell cycle analysis
Cells were seeded at 3 × 105/mL (Kasumi-1) or 2 × 105/mL (HEL and OCI-LY1) and treated with 500nM dTAG-47 at indicated time points. BrdU (20mM) was diluted 1:1000 into cell cultures for 1.5 hours at 37°C 5% CO2, the cells were washed with PBS, and then fixed overnight with 5 mL of 70% EtOH. Cells were collected using centrifugation, resuspended in 1mL 2N HCl supplemented with 0.5mg/mL Pepsin (Sigma, P7012-1G) and after 30 min. at 37°C, the solution was neutralized with 3mL of 0.1M Sodium Tetraborate pH 8.5. The cells were pelleted and washed in 0.5% BSA in PBS, then permeabilized with 1mL of 0.5% BSA/0.5% Tween-20 in PBS. Cells were stained with 15uL of FITC-anti-BrdU (BD, 556025) in 85uL of 0.5% BSA PBS and incubated for 45 minutes at 20°C in the dark. Cells were washed with 1mL of 0.5% BSA/0.5% Tween-20 in PBS, collected using centrifugation and resuspended in 400uL of PBS with Proprium Iodide (PI) (BD, 51-66211E) supplemented with RNAse A (Sigma, CAS 9001-99-4) before flow cytometry in which cells were acquired at 400 events per second.
Synchronized cells
Parental Kasumi-1 and Kasumi-1- SMARCA5FKBP12F36V-expressing cells were seeded at 4 × 105/mL then treated for 18hr at 37°C 5% CO2 with thymidine (Sigma, T9250-10G) at a final concentration of 2mM. The cells were washed with pre-warmed PBS then placed in media for 9hrs at 37°C 5% CO2. The cells were treated with a second thymidine block for 18hrs at 37°C 5% CO2. Before releasing the cells from the double thymidine block, they were treated for 2hrs with DMSO or 500nM dTAG-47. Cells were released from G1/S by washing with pre-warmed PBS and incubating with fresh media. Cells were then collected and analyzed at the indicated time points (0hr, 2hr, 4hr, 6hr, 8hr, 12hr, 24hr) following the cell cycle analysis methods section mentioned above.
Flow cytometry analysis
Cells were treated for the indicated times with 500nM dTAG-47 and 1 million cells were harvested and stained with FITC-AnnexinV (BD 556419), Zombie-NIR (BioLegend, 423106) following the manufacturer’s protocol (substituting Zombie-NIR for propidium iodide in the 100uL 1x binding buffer at 1:2000). Cells were treated for the indicated times with 500nM dTAG-47 and 1 million cells were harvested and stained with APC-CD11b (BD 550019).
CUT&RUN analysis
Cells at 5 ×105/mL were treated at indicated time points with 500nM dTAG-47. Cells were washed, attached to concanavalin A beads, permeabilized with digitonin and incubated overnight with anti-Flag (1:200, Sigma, F1804), anti-CTCF (1:100, Millipore, 07-729), anti-RUNX1 (1:100, SantaCruz, sc-365644), anti-H3K27me3 (1:100,CST, 9733), anti-H2AZ (1:100, ab4174), or anti-ETOZnf (1:100, made in house) at 4°C overnight. The next day cells were washed 3 times with PBS, then incubated with anti-mouse secondary antibody (1:200, abcam, ab46540) or anti-rabbit secondary antibody (1:200, Invitrogen, 31238) for 1.5 hours at 4°C as described54. Beads were washed and incubated with pA/G-MNAse (Epicypher, 15-1116) for 10 min at 20° C and the MNase activated with 1mM CaCl2 for 2 hours at 4° C, before adding the Stop Buffer. Libraries were created with the NEB Next Ultra II DNA Library Prep Kit (NEB, E7645S) per the manufacturer’s protocol, amplifying for 14 cycles. The samples were sequenced by the VANTAGE Sequencing Core on the NovaSeq 6000 instrument.
RNA-seq analysis
Kasumi-1-SMARCA5FKBP12F36V-expressing cells at 5 ×105/mL were treated with 500nM dTAG-47 at the indicated times in duplicate. RNA was extracted from 5 × 106 Kasumi-1 cells using 0.75mL Trizol (Invitrogen, 15596026) and incubated at 20°C for 10 minutes. The RNA was then further extracted by adding 0.15mL chloroform, vortexed on high for 1 minute, and centrifuged at 16,000 × G for 15 minutes to separate the aqueous phase. The RNA was precipitated using isopropanol. DNA was removed using DNAse I (Invitrogen, 18068-015) degradation and the RNA collected using ethanol precipitation. The RNA-seq libraries were created and sequenced by the VANTAGE Sequencing Core (Vanderbilt) on the NovaSeq6000 instrument.
PRO-seq analysis
Kasumi-1-SMARCA5FKBP12F36V-expressing cells at 5 ×105/mL were treated with 500nM dTAG-47 at the indicated times in duplicate. Nuclei was isolated and PRO-seq performed as previously described76,77. The samples were sequenced by VANTAGE Sequencing Core on the NextSeq instrument.
ATAC-seq analysis
Cells were seeded at 5 ×105/mL then treated with 500nM dTAG-47 at the indicated times in duplicate for the analysis of asynchronous cells. Kasumi-1-SMARCA5FKBP12F36V-expressing cells were also synchronized with a double thymidine block as described above. Before releasing the cells from the double thymidine block, they were treated for 6hr with DMSO or 500nM dTAG-47. The nuclei were isolated and incubated with Tn5 following the protocol as previously described78, adding in 1% Drosophila S2 nuclei. The sequencing libraries were created as previously described58. The nuclei from either asynchronous cells (Kasumi-1, OCI-LY1, HEL) or synchronous cells (Kasumi-1) were isolated and incubated with Tn5 following the Active motif protocol (Active Motif, 53150) adding in 1% Drosophila S2 nuclei. The sequencing libraries were created and SPRI clean-up was performed with the Active Motif kit per protocol. Samples were sequenced by the VANTAGE Sequencing Core on the NovaSeq6000 instrument.
MNase-seq analysis
Kasumi-1-SMARCA5FKBP12F36V-expressing cells at 5 ×105/mL were treated with 500nM dTAG-47 at the indicated times in duplicate. 10 million cells were lysed with cell lysis buffer (10mM Tris-HCl pH 7.4, 300mM sucrose, 3mM CaCl2, 2M MgCl2, 1% Triton-X100, 100mM DTT), spun down, placed in reaction buffer (50mM Tris HCl pH8, 5mM CaCl2) and 2 million cell equivalents were incubated with 0.1U/uL MNase (Sigma N3755) for 4 minutes at 20°C79. The sequencing libraries were created from gel extracted mono-nucleosomes using the NEB Ultra II DNA Prep Kit (NEB E7645L) with 7 cycles of PCR. Samples were sequenced by the VANTAGE Sequencing Core on the NovaSeq6000 instrument.
QUANTIFICATION AND STATISTICAL ANALYSIS
Raw sequencing data were trimmed and aligned as described in github (https://github.com/monnieb92/SMARCA5_paper), using trimmomatic, bowtie2, and samtools80–82. Bam files were analyzed using MACS2 for peak calling, bedTools to remove blacklist peaks, and Diffbind was used to create counts tables. DESeq2 was used for the default normalization83–87. Due to global changes in SMARCA5 and CTCF peaks, these analyses could not use DESeq2 for normalization, so normalization was done using total read counts. CUT&RUN data was visualized using deepTools88 and HOMER was used to annotate peaks and perform motif analysis89. RNA-seq analysis was performed as using trimmomatic v0.3280, tophat v2.0.1190, and cuffdiff v2.2.191 as described in github (https://github.com/monnieb92/SMARCA5_paper). The heat maps were clustered using the hierarchical ward.D2 method92.
PRO-seq data were analyzed as previously described using the Nascent RNA Sequencing Analysis (NRSA) platform, which yielded differential gene body and promoter proximal pausing count tables93,94. The count tables for gene body counts and promoter proximal counts from 0h (n = 5), 2h (n = 3), 6h (n = 4), and 24h (n = 2) from NRSA were concatenated and filtered for expressed genes within at least one time point (gene body density >= 0.004 (4 reads/kb) and promoter proximal density > 0.01(0.5 reads/bp in 50 bp bin)). Gene body counts tables were then analyzed and normalized with batch effect using DESeq283, and promoter proximal counts tables were analyzed using normalization factor from gene body counts along with providing batch effect as described in the github (https://github.com/monnieb92/SMARCA5_paper).
ATAC-seq data were trimmed and analyzed as described in github (https://github.com/monnieb92/SMARCA5_paper), using trimmomatic, bowtie2, and samtools 80–82. Data were further analyzed as described in github (https://github.com/monnieb92/SMARCA5_paper) using Genrich (v0.6.1, available at https://github.com/jsh58/Genrich, parameters: -q 0.05 -j -r -E) for peak calling, Diffbind for creating a counts table, and DESeq2 with batch effect for differential peaks83–85. CUT&RUN data were visualized using deepTools88. HOMER was used to annotate peaks89. Nucleosome repeat length was determined using a previously published loess model34.
MNase-seq data was trimmed and analyzed as described in github (https://github.com/monnieb92/SMARCA5_paper), using trimmomatic, “bowtie2—very-sensitive”, and samtools80–82. MNase-seq data were further analyzed using “deepTools – alignmentseive” to isolate fragment sizes between 130 and 210 bp as well as remove “blacklist peaks” from ENCODE hg19 blacklist peaks, then isolated genomic region of interest +/−2kB with bedTools86, and the data visualized via metaplots using “deepTools—MNase”88. A previously published loess model algorithm was used to determine nucleosome repeat length34,95.
Supplementary Material
Key resources table.
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
| Antibodies | ||
| α-SNF2H | Abcam | CAT#ab72499, RRID:AB_1270821 |
| Monoclonal ANTI-FLAG® M2 antibody | Sigma-Aldrich | CAT#F1804, RRID:AB_262044 |
| α-GAPDH Antibody (G9) | Santa Cruz | CAT#sc-365062; RRID: AB_10847862 |
| α-RUNX1 | Santa Cruz | CAT#sc-365644; RRID: AB_10843207 |
| α-H2A.Z | Abcam | CAT#ab4174, RRID: AB_304345 |
| BD Pharmingen™ FITC Mouse α- BrdU Set | BD Biosciences | CAT#556028 |
| Zombie-NIR | BioLegend | CAT#423106 |
| α-CTCF | Millipore | CAT #07-729, RRID: AB_441965 |
| Rabbit α -Mouse IgG H&L | Abcam | CAT#ab46540 |
| Donkey α -Rabbit IgG (H+L) Cross-Adsorbed Secondary Antibody | Invitrogen | CAT#31238 |
| α-ETOZnf | Made in house | N/A |
| α-H3K27me3 | Cell Signaling Technology | CAT#9733l RRID: AB_2616029 |
| α-CD11b/MAC-1 Clone ICRF44 APC | Fisher Scientific | CAT#BDB550019; RRID: AB_398456 |
| Bacterial and virus strains | ||
| Chemicals, peptides, and recombinant proteins | ||
| RPMI | Fisher Scientific | CAT#MT15040CV |
| IMDM | Fisher Scientific | CAT#12440053 |
| Fetal Plex | Gemini Bio-Products | CAT#100-602 |
| Fetal Bovine Serum | R&D Systems | CAT#S11150 |
| Corning Penicillin/Streptomycin | Fisher Scientific | CAT#MT30002CI |
| Corning L-glutamine Solution | Fisher Scientific | CAT#12440053 |
| dTAG-47 | Vanderbilt Chemical Synthesis Core | Custom dTAG-47 Synthesis |
| DMSO | Millipore Sigma | CAT#D8418; CAS: 67-68-5 |
| Trizol | ThermoFisher | CAT#15596026; CAS: 108-95-2, 1762-95-4, 593-84-0 |
| Phenol:Chloroform:Isoamyl Alcohol | Millipore Sigma | CAT#P3803; CAS: 108-95-2, 67-66-3, 123-51-3 |
| Biotin-11-CTP | PerkinElmer | CAT#NEL542001EA |
| Digitonin | Millipore Sigma | CAT#300410, CAS: 11024-24-1 |
| BioMag Plus Concanavalin A Beads | Bangs Laboratories, Inc | CAT#BP531 |
| Sarkosyl | Fisher Scientific | CAT#IB07080; CAS: 7631-98-3 |
| Glycine | Fisher Scientific | CAT#808831; CAS: 56-40-6 |
| RNAse A | Sigma-Aldrich | CAS: 9001-99-4 |
| Nuclease micrococcal from Staphylococcus aureus | Sigma-Aldrich | CAT#N3755 |
| Pepsin | Sigma-Aldrich | CAT#P7012-1G |
| SUPERase•In™ RNase Inhibito | Invitrogen | CAT#AM2694 |
| DNAse I | Invitrogen | CAT#18068-015 |
| Propium Iodide | BD Biosciences | CAT#51-66211E |
| Critical commercial assays | ||
| QuickChange Lightning Site-Directed Mutagenesis kit | Agilent | CAT#210518 |
| High Capacity cDNA Reverse Transcription Kit | ThermoFisher | CAT#4368814 |
| NEBNext Ultra II DNA Library Prep Kit | NEB | CAT#E7645L |
| Gibson Assembly Cloning kit | NEB | CAT#E5510S |
| QIAGEN QIAfilter Maxi Kit | QIAGEN | CAT#12263 |
| Wizard® SV Gel and PCR Clean-Up System | Promega | CAT#A9282 |
| ATAC-Seq Kit | Active Motif | CAT#53150 |
| Deposited data | ||
| GEO | GSE160470 | |
| Github | DOI: 10.5281/zenodo.7438719 | |
| Mendeley Data | https://data.mendeley.com/datasets/cf5vbhy67d | |
| Kasumi-1 | ATCC | CAT#CRL-2724; RRID: CVCL_0589 |
| OCI-LY1 | Gift from B. Hilda Ye (Einstein) | N/A |
| HEL | ATCC | CAT#TIB-180 |
| Drosophila S2 | Gift from Emily Hodge’s Lab (Vanderbilt) | N/A |
| Experimental models: Organisms/strains | ||
| Kasumi-1 | ATCC | CAT#CRL-2724; RRID: CVCL_0589 |
| Kasumi-1-SMARCA5-FKBP12F36V | This paper | N/A |
| HEL | ATCC | CAT#TIB-180 |
| HEL -SMARCA5-FKBP12F36V | This paper | N/A |
| OCI-LY1 | ATCC | |
| OCI-LY1-SMARCA5-FKBP12F36V | This paper | N/A |
| Oligonucleotides | ||
| Alt-R® CRISPR-Cas9 tracrRNA | Integrated DNA Technologies | CAT#1072534 |
| crRNA_C SMARCA5 (SNF2h): 5’ GATGGCGCACCTGATGGTCG 3’ | Integrated DNA Technologies | CAT#Alt-R® CRISPR-Cas9 crRNA |
| crRNA_D SMARCA5 (SNF2h): 5’ GGTGAAGACTGAAAGGGACAA 3’, | Integrated DNA Technologies | CAT#Alt-R® CRISPR-Cas9 crRNA |
| 5’ tgggttgatgtctggtttttccttgaCTGTTAGTCTCTAAGCTTTGATTATTTCAG 3’ (Forward-5hgDNA-SMARCA5) | Integrated DNA Technologies | Custom DNA Oligo |
| 5’ gatggtttccacctgcactcctccggatccTAGTTTCAGCTTCTTTTTTCTTCCTCGACC 3’ (Reverse-5hgDNA-SMARCA5) | Integrated DNA Technologies | Custom DNA Oligo |
| 5’ ccaccggcggcatggacgagctgtacaagtaaATATGTTTTTGTTTCATAATCACTAACTTTAAACCAG 3’ (Forward-BFP-3UTRgDNA) | Integrated DNA Technologies | Custom DNA Oligo |
| 5’ accggcggcatggacgagctgtacaagtaaATATGTTTTTGTTTCATAATCACTAACTTTAAACC 3’ (Forward-mCherry-3UTRgDNA) | Integrated DNA Technologies | Custom DNA Oligo |
| 5’ tcgagctcggtacccggggatccTGGAATTATAGTCAGTCTTGCTAACAC 3’ (Reverse-3UTRgDNA) | Integrated DNA Technologies | Custom DNA Oligo |
| 5’ GTCTCATACCTATTACTTTGTAGTAGTTGT 3’ (Forward-Fragment 1),’ | Integrated DNA Technologies | Custom DNA Oligo |
| 5 tcaaggaaaaaccagacatcaacccacttg 3’ (Reverse-Fragment 1) | Integrated DNA Technologies | Custom DNA Oligo |
| 5’ GGTCGAGGAAGAAAAAAGAAGCTGAAACTAggatccggaggagtgcaggtggaaaccatc 3’ (Forward-Fragment 2) | Integrated DNA Technologies | Custom DNA Oligo |
| 5’ AGTTAGTGATTATGAAACAAAAACATATttacttgtacagctcgtccatgccgccggtgg 3’ (Reverse-mCherry-Fragment 2) | Integrated DNA Technologies | Custom DNA Oligo |
| 5’ AAAGTTAGTGATTATGAAACAAAAACATATttaattaagcttgtgccccagtttgctagg 3’ (Reverse-BFP-Fragment 2) | Integrated DNA Technologies | Custom DNA Oligo |
| 5’AATGGATGGCGCACCTGACGGCCGAGGAAGAAAAAAGAAGCTGA 3’ (Forward-SDM) | Integrated DNA Technologies | Custom DNA Oligo |
| 5’ TCAGCTTCTTTTTTCTTCCTCGGCCGTCAGGTGCGCCATCCATT 3’ (Reverse-SDM) | Integrated DNA Technologies | Custom DNA Oligo |
| Recombinant DNA | ||
| pAW62.YY1.FKBP.knock-in.mCherry | AddGene | CAT#104370 |
| pAW62.YY1.FKBP.knock-in.BFP | AddGene | CAT#104371 |
| pAW62.SMARCA5.FKBP.knock-in.BFP | This paper | |
| pAW62.SMARCA5.FKBP.knock-in.mCherry | This paper | |
| Software and algorithms | ||
| Bowtie2 (v. 2.2.2) | Langmead and Salzberg, 201281 | N/A |
| MACS2 peak caller (narrowPeak; q-0.01 or q-0.001, broadPeak q-0.01; v. 2.2.6) | Feng et al., 201284,87 | N/A |
| Genrich peak caller (-q 0.05 -j -r -E ; v. 0.6) | https://github.com/jsh58/Genrich | N/A |
| DiffBind | Ross-Innes et al. 201285 | N/A |
| DESeq2 | Love et al., 201483 | N/A |
| HOMER | Heinz et al., 201089 | N/A |
| deepTOOLS (v. 3.4.1) | Ramírez et al., 201688 | N/A |
| FASTX toolkit (v. 0.0.13) | http://hannonlab.cshl.edu | N/A |
| TopHat (v. 2.0.11) | Trapnell et al., 201291 | N/A |
| Cuffdiff (v. 2.1.1) | Trapnell et al., 201291 | N/A |
| Nascent RNA Sequencing Anaylsis (NRSA) | Wang et al., 201893 | N/A |
| Bedtools (v. 2.27.1) | Quinlan et al. 201086 | N/A |
| Samtools (v. 1.10) | Li et al., 200982 | N/A |
| Trimmomatic-0.32 | Bolger et al., 201480 | N/A |
| Other | ||
| Alt-R® S.p. Cas9 D10A Nickase V3 | Integrated DNA Technologies | CAT#1081062 |
| CUTANA pAG-MNase for ChIC/CUT&RUN Workflows | Epicypher | CAT#15-1016 |
| Trypan Blue | Gibco | CAT#15250-061 |
Highlights.
Target-specific protein degradation of endogenous human SMARCA5
Degradation of SMARCA5 leads to a rapid loss in CTCF DNA binding
SMARCA5 regulates nucleosome repeat length independent of the cell cycle
CTCF and SMARCA5 co-localize at H2A.Z-containing sites
Acknowledgements
We especially thank Dr. Michael Stadler (Friedrich Miescher Institute for Biomedical Research, and the Swiss Institute of Bioinformatics, Basel, Switzerland) for sharing code and helpful guidance in using this code to calculate nucleosome repeat length. We also thank the members of the Hiebert lab for helpful discussions, reagents and advice. We thank the Flow Cytometry, Chemical Synthesis, and Genome Sciences Shared Resources for services and support. Core services were performed through Vanderbilt Digestive Disease Research grant (NIDDK P30DK58404) and the Vanderbilt-Ingram Cancer Center support grant (NCI P30CA68485). This work was supported by the T. J. Martell Foundation, the Robert J. Kleberg, Jr. and Helen C. Kleberg Foundation, National Institutes of Health grants (RO1-CA164605, R01-CA255446-01A1, T32-CA009582-33) and Department of Defense grant (W81XWH-20-1-0522). The project described was also supported by the National Center for Research Resources, Grant UL1 RR024975-01, and is now at the National Center for Advancing Translational Sciences, Grant 2 UL1 TR000445-06. The content is solely the responsibility of the authors and does not necessarily represent the official views of the NIH.
Inclusion and Diversity
We support inclusive, diverse, and equitable conduct of research. One or more of the authors of this paper self-identifies as a gender minority in their field of research. One or more of the authors of this paper self-identifies as a member of the LGBTQIA+ community. One or more of the authors of this paper self-identifies as living with a disability.
Footnotes
Declaration of Interests
The authors declare no competing interests. Although Scott Hiebert received research funding from Incyte Inc. through the Vanderbilt-Incyte Alliance, these funds did not support this work. Scott Hiebert is also a scientific advisor for the Edward P. Evans Foundation.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Luger K, Mäder AW, Richmond RK, Sargent DF, and Richmond TJ (1997). Crystal structure of the nucleosome core particle at 2.8 Å resolution. Nature 389, 251–260. 10.1038/38444. [DOI] [PubMed] [Google Scholar]
- 2.Kornberg RD (1977). Structure of chromatin. Annu Rev Biochem 46, 931–954. 10.1146/annurev.bi.46.070177.004435. [DOI] [PubMed] [Google Scholar]
- 3.Baldi S, Korber P, and Becker PB (2020). Beads on a string—nucleosome array arrangements and folding of the chromatin fiber. Nat. Struct. Mol. Biol. 27, 109–118. 10.1038/s41594-019-0368-x. [DOI] [PubMed] [Google Scholar]
- 4.Bakayev V v, and Georgiev GP (1976). Heterogeneity of Chromatin Subunits in Vitro and Location of Histone HI. Nucleic Acids Res 3, 477–492. 10.1093/nar/3.2.477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Thoma F, Koller T, and Klug A (1979). Involvement of histone H1 in the organization of the nucleosome and of the salt-dependent superstructures of chromatin. Journal of Cell Biology 83, 403–427. 10.1083/jcb.83.2.403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Hirschhorn JN, Brown SA, Clark CD, and Winston F (1992). Evidence that SNF2/SWI2 and SNF5 activate transcription in yeast by altering chromatin structure. Genes Dev 6, 2288–2298. 10.1101/gad.6.12a.2288. [DOI] [PubMed] [Google Scholar]
- 7.Schwartz BE, and Ahmad K (2005). Transcriptional activation triggers deposition and removal of the histone variant H3.3. Genes Dev 19, 804–814. 10.1101/gad.1259805. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Mizuguchi G, Shen X, Landry J, Wu W-H, Sen S, and Wu C (2004). ATP-Driven Exchange of Histone H2AZ Variant Catalyzed by SWR1 Chromatin Remodeling Complex. Science 303, 343–348. 10.1126/science.1090701. [DOI] [PubMed] [Google Scholar]
- 9.Hamiche A, Sandaltzopoulos R, Gdula DA, and Wu C (1999). ATP-Dependent Histone Octamer Sliding Mediated by the Chromatin Remodeling Complex NURF. Cell 97, 833–842. 10.1016/S0092-8674(00)80796-5. [DOI] [PubMed] [Google Scholar]
- 10.Barak O, Lazzaro MA, Lane WS, Speicher DW, Picketts DJ, and Shiekhattar R (2003). Isolation of human NURF: A regulator of Engrailed gene expression. EMBO Journal 22, 6089–6100. 10.1093/emboj/cdg582. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Kukimoto I, Elderkin S, Grimaldi M, Oelgeschläger T, and Varga-Weisz PD (2004). The Histone-Fold Protein Complex CHRAC-15/17 Enhances Nucleosome Sliding and Assembly Mediated by ACF. Mol Cell 13, 265–277. 10.1016/S1097-2765(03)00523-9. [DOI] [PubMed] [Google Scholar]
- 12.Yang JG, Madrid TS, Sevastopoulos E, and Narlikar GJ (2006). The chromatin-remodeling enzyme ACF is an ATP-dependent DNA length sensor that regulates nucleosome spacing. Nat Struct Mol Biol 13, 1078–1083. 10.1038/nsmb1170. [DOI] [PubMed] [Google Scholar]
- 13.Ito T, Bulger M, Pazin MJ, Kobayashi R, and Kadonaga JT (1997). ACF, an ISWI-Containing and ATP-Utilizing Chromatin Assembly and Remodeling Factor. Cell 90, 145–155. 10.1016/S0092-8674(00)80321-9. [DOI] [PubMed] [Google Scholar]
- 14.Loyola A, Huang J-Y, LeRoy G, Hu S, Wang Y-H, Donnelly RJ, Lane WS, Lee S-C, and Reinberg D (2003). Functional Analysis of the Subunits of the Chromatin Assembly Factor RSF. Mol Cell Biol 23, 6759–6768. 10.1128/mcb.23.19.6759-6768.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Cavellán E, Asp P, Percipalle P, and Farrants AKÖ (2006). The WSTF-SNF2h chromatin remodeling complex interacts with several nuclear proteins in transcription. Journal of Biological Chemistry 281, 16264–16271. 10.1074/jbc.M600233200. [DOI] [PubMed] [Google Scholar]
- 16.Strohner R, Nemeth A, Jansa P, Hofmann-Rohrer U, Santoro R, Längst G, and Grummt I (2001). NoRC--a novel member of mammalian ISWI-containing chromatin remodeling machines. EMBO J 20, 4892–4900. 10.1093/emboj/20.17.4892. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Zhou Y, Santoro R, and Grummt I (2002). The chromatin remodeling complex NoRC targets HDAC1 to the ribosomal gene promoter and represses RNA polymerase I transcription. EMBO J 21, 4632–4640. 10.1093/emboj/cdf460. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Bozhenok L, Wade PA, and Varga-Weisz P (2002). WSTF-ISWI chromatin remodeling complex targets heterochromatic replication foci. EMBO Journal 21, 2231–2241. 10.1093/emboj/21.9.2231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Santoro R, Li J, and Grummt I (2002). The nucleolar remodeling complex NoRC mediates heterochromatin formation and silencing of ribosomal gene transcription. Nat Genet 32, 393–396. 10.1038/ng1010. [DOI] [PubMed] [Google Scholar]
- 20.Collins N, Poot RA, Kukimoto I, García-Jiménez C, Dellaire G, and Varga-Weisz PD (2002). An ACF1-ISWI chromatin-remodeling complex is required for DNA replication through heterochromatin. Nat Genet 32, 627–632. 10.1038/ng1046. [DOI] [PubMed] [Google Scholar]
- 21.Guetg C, Lienemann P, Sirri V, Grummt I, Hernandez-Verdun D, Hottiger MO, Fussenegger M, and Santoro R (2010). The NoRC complex mediates the heterochromatin formation and stability of silent rRNA genes and centromeric repeats. EMBO Journal 29, 2135–2146. 10.1038/emboj.2010.17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Poot RA, Bozhenok L, van den Berg DLC, Steffensen S, Ferreira F, Grimaldi M, Gilbert N, Ferreira J, and Varga-Weisz PD (2004). The Williams syndrome transcription factor interacts with PCNA to target chromatic remodelling by ISWI to replication foci. Nat Cell Biol 6, 1236–1244. 10.1038/ncb1196. [DOI] [PubMed] [Google Scholar]
- 23.Min S, Jo S, Lee HS, Chae S, Lee JS, Ji JH, and Cho H (2014). ATM-dependent chromatin remodeler Rsf-1 facilitates DNA damage checkpoints and homologous recombination repair. Cell Cycle 13, 666–677. 10.4161/cc.27548. [DOI] [PubMed] [Google Scholar]
- 24.Varga-Weisz PD, Wilm M, Bonte E, Dumas K, Mann M, and Becker PB (1997). Chromatin-remodelling factor CHRAC contains the ATPases ISWI and topoisomerase II. Nature 388, 598–602. 10.1038/41587. [DOI] [PubMed] [Google Scholar]
- 25.Aydin ÖZ, Marteijn JA, Ribeiro-Silva C, Rodríguez López A, Wijgers N, Smeenk G, van Attikum H, Poot RA, Vermeulen W, and Lans H (2014). Human ISWI complexes are targeted by SMARCA5 ATPase and SLIDE domains to help resolve lesion-stalled transcription. Nucleic Acids Res 42, 8473–8485. 10.1093/nar/gku565. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.LeRoy G, Orphanides G, and Lane WS (1998). Requirement of RSF and FACT for transcription of chromatin templates in vitro. Science 282, 1900–1904. 10.1126/science.282.5395.1900. [DOI] [PubMed] [Google Scholar]
- 27.Ding Y, Li Y, Zhao Z, Cliff Zhang Q, and Liu F (2021). The chromatin-remodeling enzyme smarca5 regulates erythrocyte aggregation via keap1-nrf2 signaling. Elife 10. 10.7554/ELIFE.72557. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Ding Y, Wang W, Ma D, Liang G, Kang Z, Xue Y, Zhang Y, Wang L, Heng J, Zhang Y, et al. (2021). Smarca5-mediated epigenetic programming facilitates fetal HSPC development in vertebrates. Blood 137, 190. 10.1182/BLOOD.2020005219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Deindl S, Hwang WL, Hota SK, Blosser TR, Prasad P, Bartholomew B, and Zhuang X (2013). ISWI Remodelers Slide Nucleosomes with Coordinated Multi-Base-Pair Entry Steps and Single-Base-Pair Exit Steps. Cell 152, 442–452. 10.1016/j.cell.2012.12.040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Levendosky RF, and Bowman GD (2019). Asymmetry between the two acidic patches dictates the direction of nucleosome sliding by the ISWI chromatin remodeler. Elife 8, e45472.1–e45472.18. 10.7554/eLife.45472. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Grüne T, Brzeski J, Eberharter A, Clapier CR, Corona DFV, Becker PB, and Müller CW (2003). Crystal structure and functional analysis of a nucleosome recognition module of the remodeling factor ISWI. Mol Cell 12, 449–460. 10.1016/S1097-2765(03)00273-9. [DOI] [PubMed] [Google Scholar]
- 32.Gamarra N, Johnson SL, Trnka MJ, Burlingame AL, and Narlikar GJ (2018). The nucleosomal acidic patch relieves auto-inhibition by the ISWI remodeler SNF2h. Elife 7. 10.7554/ELIFE.35322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Dao HT, Dul BE, Dann GP, Liszczak GP, and Muir TW (2020). A basic motif anchoring ISWI to nucleosome acidic patch regulates nucleosome spacing. Nat Chem Biol 16, 134–142. 10.1038/s41589-019-0413-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Barisic D, Stadler MB, Iurlaro M, and Schübeler D (2019). Mammalian ISWI and SWI/SNF selectively mediate binding of distinct transcription factors. Nature 569, 136–140. 10.1038/s41586-019-1115-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Clarkson CT, Deeks EA, Samarista R, Mamayusupova H, Zhurkin VB, and Teif VB (2019). CTCF-dependent chromatin boundaries formed by asymmetric nucleosome arrays with decreased linker length. Nucleic Acids Res 47, 11181–11196. 10.1093/nar/gkz908. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Hakimi MA, Bochar DA, Schmiesing JA, Dong Y, Barak OG, Spelcher DW, Yokomori K, and Shiekhattar R (2002). A chromatin remodelling complex that loads cohesin onto human chromosomes. Nature 418, 994–998. 10.1038/nature01024. [DOI] [PubMed] [Google Scholar]
- 37.Dluhosova M, Curik N, Vargova J, Jonasova A, Zikmund T, and Stopka T (2014). Epigenetic control of SPI1 gene by CTCF and ISWI ATPase SMARCA5. PLoS One 9, e87448:1–11. 10.1371/journal.pone.0087448. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Song Y, Liang Z, Zhang J, Hu G, Wang J, Li Y, Guo R, Dong X, Babarinde IA, Ping W, et al. (2022). CTCF functions as an insulator for somatic genes and a chromatin remodeler for pluripotency genes during reprogramming. Cell Rep 39, 110626. 10.1016/J.CELREP.2022.110626. [DOI] [PubMed] [Google Scholar]
- 39.Hendy O, Serebreni L, Bergauer K, Muerdter F, Huber L, Nemčko F, and Stark A (2022). Developmental and housekeeping transcriptional programs in Drosophila require distinct chromatin remodelers. Mol Cell 82, 3598–3612.e7. 10.1016/J.MOLCEL.2022.08.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Kaushal A, Mohana G, Dorier J, Özdemir I, Omer A, Cousin P, Semenova A, Taschner M, Dergai O, Marzetta F, et al. (2021). CTCF loss has limited effects on global genome architecture in Drosophila despite critical regulatory functions. Nature Communications 2021 12:1 12, 1–16. 10.1038/s41467-021-21366-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Zikmund T, Kokavec J, Turkova T, Savvulidi F, Paszekova H, Vodenkova S, Sedlacek R, Skoultchi AI, and Stopka T (2019). ISWI ATPase Smarca5 Regulates Differentiation of Thymocytes Undergoing β-Selection. The Journal of Immunology 202, 3434–3446. 10.4049/jimmunol.1801684. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Stopka T, Zakova D, Fuchs O, Kubrova O, Blafkova J, Jelinek J, Necas E, and Zivny J (2000). Chromatin remodeling gene SMARCA5 is dysregulated in primitive hematopoietic cells of acute leukemia. Leukemia 14, 1247–1252. 10.1038/sj.leu.2401807. [DOI] [PubMed] [Google Scholar]
- 43.Stopka T, and Skoultchi AI (2003). The ISWI ATPase Snf2h is required for early mouse development. Proc Natl Acad Sci U S A 100, 14097–14102. 10.1073/pnas.2336105100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Sabantsev A, Levendosky RF, Zhuang X, Bowman GD, and Deindl S (2019). Direct observation of coordinated DNA movements on the nucleosome during chromatin remodelling. Nature Communications 2019 10:1 10, 1–12. 10.1038/s41467-019-09657-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Erb MA, Scott TG, Li BE, Xie H, Paulk J, Seo HS, Souza A, Roberts JM, Dastjerdi S, Buckley DL, et al. (2017). Transcription control by the ENL YEATS domain in acute leukaemia. Nature 543, 270–274. 10.1038/nature21688. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Layden HM, Eleuteri NA, Hiebert SW, and Stengel KR (2021). A protocol for rapid degradation of endogenous transcription factors in mammalian cells and identification of direct regulatory targets. STAR Protoc 2, 100530. 10.1016/J.XPRO.2021.100530. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Zikmund T, Paszekova H, Kokavec J, Kerbs P, Thakur S, Turkova T, Tauchmanova P, Greif PA, and Stopka T (2020). Loss of ISWI ATPase SMARCA5 (SNF2H) in acute myeloid leukemia cells inhibits proliferation and chromatid cohesion. Int J Mol Sci 21, 2073:1–13. 10.3390/ijms21062073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Roberts CWM, and Orkin SH (2004). The SWI/SNF complex - Chromatin and cancer. Nat Rev Cancer 4, 133–142. 10.1038/nrc1273. [DOI] [PubMed] [Google Scholar]
- 49.Erdel F, and Rippe K (2011). Chromatin remodelling in mammalian cells by ISWI-type complexes - Where, when and why? FEBS Journal 278, 3608–3618. 10.1111/j.1742-4658.2011.08282.x. [DOI] [PubMed] [Google Scholar]
- 50.Weintraub AS, Li CH, Zamudio A.v, Sigova AA, Hannett NM, Day DS, Abraham BJ, Cohen MA, Nabet B, Buckley DL, et al. (2017). YY1 Is a Structural Regulator of Enhancer-Promoter Loops. Cell 171, 1573–1588. 10.1016/j.cell.2017.11.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Kokavec J, Zikmund T, Savvulidi F, Kulvait V, Edelmann W, Skoultchi AI, and Stopka T (2017). The ISWI ATPase Smarca5 (Snf2h) Is Required for Proliferation and Differentiation of Hematopoietic Stem and Progenitor Cells. Stem Cells 35, 1614–1623. 10.1002/stem.2604. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Bhaskara S, Jacques V, Rusche JR, Olson EN, Cairns BR, and Chandrasekharan MB (2013). Histone deacetylases 1 and 2 maintain S-phase chromatin and DNA replication fork progression. Epigenetics Chromatin 6, 1–21. 10.1186/1756-8935-6-27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Poot RA, Dellaire G, Hülsmann BB, Grimaldi MA, Corona DFV, Becker PB, Bickmore WA, and Varga-Weisz PD (2000). HuCHRAC, a human ISWI chromatin remodelling complex contains hACF1 and two novel histone-fold proteins. EMBO Journal 19, 3377–3387. 10.1093/emboj/19.13.3377. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Skene PJ, Henikoff JG, and Henikoff S (2018). Targeted in situ genome-wide profiling with high efficiency for low cell numbers. Nat Protoc 13, 1006–1019. 10.1038/nprot.2018.015. [DOI] [PubMed] [Google Scholar]
- 55.Stengel KR, Ellis JD, Spielman CL, Bomber ML, and Hiebert SW (2021). Definition of a small core transcriptional circuit regulated by AML1-ETO. Mol Cell 81, 530–545. 10.1016/j.molcel.2020.12.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.He X, Fan HY, Garlick JD, and Kingston RE (2008). Diverse regulation of SNF2h chromatin remodeling by noncatalytic subunits. Biochemistry 47, 7025–7033. 10.1021/bi702304p. [DOI] [PubMed] [Google Scholar]
- 57.Aalfs JD, Narlikar GJ, and Kingston RE (2001). Functional Differences between the Human ATP-dependent Nucleosome Remodeling Proteins BRG1 and SNF2H. Journal of Biological Chemistry 276, 34270–34278. 10.1074/jbc.M104163200. [DOI] [PubMed] [Google Scholar]
- 58.Buenrostro JD, Giresi PG, Zaba LC, Chang HY, and Greenleaf WJ (2013). Transposition of native chromatin for fast and sensitive epigenomic profiling of open chromatin, DNA-binding proteins and nucleosome position. Nat Methods 10, 1213–1218. 10.1038/nmeth.2688. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Jimeno-González S, Ceballos-Chávez M, and Reyes JC (2015). A positioned +1 nucleosome enhances promoter-proximal pausing. Nucleic Acids Res 43, 3068–3078. 10.1093/nar/gkv149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Mahat DB, Kwak H, Booth GT, Jonkers IH, Danko CG, Patel RK, Waters CT, Munson K, Core LJ, and Lis JT (2016). Base-pair-resolution genome-wide mapping of active RNA polymerases using precision nuclear run-on (PRO-seq). Nat Protoc 11, 1455–1476. 10.1038/nprot.2016.086. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Tsukiyama T, Palmer J, Landel CC, Shiloach J, and Wu C (1999). Characterization of the imitation switch subfamily of ATP-dependent chromatin-remodeling factors in Saccharomyces cerevisiae. Genes Dev 13, 686–697. 10.1101/gad.13.6.686. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Whitehouse I, Stockdale C, Flaus A, Szczelkun MD, and Owen-Hughes T (2003). Evidence for DNA Translocation by the ISWI Chromatin-Remodeling Enzyme. Mol Cell Biol 23, 1935–1945. 10.1128/mcb.23.6.1935-1945.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Goldman JA, Garlick JD, and Kingston RE (2010). Chromatin remodeling by imitation switch (ISWI) class ATP-dependent remodelers is stimulated by histone variant H2A.Z. Journal of Biological Chemistry 285, 4645–4651. 10.1074/jbc.M109.072348. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Wen Z, Zhang L, Ruan H, and Li G (2020). Histone variant H2A.Z regulates nucleosome unwrapping and CTCF binding in mouse ES cells. Nucleic Acids Res 48, 5939–5952. 10.1093/NAR/GKAA360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Längst G, and Becker PB (2001). Nucleosome mobilization and positioning by ISWI-containing chromatin-remodeling factors. J Cell Sci 114, 2561–2568. 10.1242/jcs.114.14.2561. [DOI] [PubMed] [Google Scholar]
- 66.Gabriele M, Brandão HB, Grosse-Holz S, Jha A, Dailey GM, Cattoglio C, Hsieh THS, Mirny L, Zechner C, and Hansen AS (2022). Dynamics of CTCF- and cohesin-mediated chromatin looping revealed by live-cell imaging. Science 376, 476–501. 10.1126/science.abn6583. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Wiechens N, Singh V, Gkikopoulos T, Schofield P, Rocha S, and Owen-Hughes T (2016). The Chromatin Remodelling Enzymes SNF2H and SNF2L Position Nucleosomes adjacent to CTCF and Other Transcription Factors. PLoS Genet 12, e1005940:1–25. 10.1371/journal.pgen.1005940. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Luan J, Xiang G, Gómez-García PA, Tome JM, Zhang Z, Vermunt MW, Zhang H, Huang A, Keller CA, Giardine BM, et al. (2021). Distinct properties and functions of CTCF revealed by a rapidly inducible degron system. Cell Rep 34, 108783: 1–21. 10.1016/j.celrep.2021.108783. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Khoury A, Achinger-Kawecka J, Bert SA, Smith GC, French HJ, Luu PL, Peters TJ, Du Q, Parry AJ, Valdes-Mora F, et al. (2020). Constitutively bound CTCF sites maintain 3D chromatin architecture and long-range epigenetically regulated domains. Nat Commun 11, 2041–1723. 10.1038/s41467-019-13753-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Nora EP, Goloborodko A, Valton AL, Gibcus JH, Uebersohn A, Abdennur N, Dekker J, Mirny LA, and Bruneau BG (2017). Targeted Degradation of CTCF Decouples Local Insulation of Chromosome Domains from Genomic Compartmentalization. Cell 169, 930–944. 10.1016/j.cell.2017.05.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Ghandi M, Huang FW, Jané-Valbuena J, Kryukov G. v., Lo CC, McDonald ER, Barretina J, Gelfand ET, Bielski CM, Li H, et al. (2019). Next-generation characterization of the Cancer Cell Line Encyclopedia. Nature 569, 503–508. 10.1038/s41586-019-1186-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Negrini S, Gorgoulis VG, and Halazonetis TD (2010). Genomic instability an evolving hallmark of cancer. Nat Rev Mol Cell Biol 11, 220–228. 10.1038/nrm2858. [DOI] [PubMed] [Google Scholar]
- 73.Hanahan D, and Weinberg RA (2011). Hallmarks of Cancer: The Next Generation. Cell 144, 646–674. 10.1016/j.cell.2011.02.013. [DOI] [PubMed] [Google Scholar]
- 74.DepMap (2017). The Cancer Dependency Map Consortium. Nat Genet. 10.1038/ng.3984. [DOI] [Google Scholar]
- 75.Tsherniak A, Vazquez F, Montgomery PG, Weir BA, Kryukov G, Cowley GS, Gill S, Harrington WF, Pantel S, Krill-Burger JM, et al. (2017). Defining a Cancer Dependency Map. Cell 170, 564–576.e16. 10.1016/J.CELL.2017.06.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Sampathi S, Acharya P, Zhao Y, Wang J, Stengel KR, Liu Q, Savona MR, and Hiebert SW (2019). The CDK7 inhibitor THZ1 alters RNA polymerase dynamics at the 5’ and 3’ ends of genes. Nucleic Acids Res 47, 3921–3936. 10.1093/nar/gkz127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Kwak H, Fuda NJ, Core LJ, and Lis JT (2013). Precise maps of RNA polymerase reveal how promoters direct initiation and pausing. Science 339, 950–953. 10.1126/science.1229386. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Barnett KR, Decato BE, Scott TJ, Hansen TJ, Chen B, Attalla J, Smith AD, and Hodges E (2020). ATAC-Me Captures Prolonged DNA Methylation of Dynamic Chromatin Accessibility Loci during Cell Fate Transitions. Mol Cell 77, 1350–1364. 10.1016/j.molcel.2020.01.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Chereji RV, Bryson TD, and Henikoff S (2019). Quantitative MNase-seq accurately maps nucleosome occupancy levels. Genome Biol 20, 198:1–18. 10.1186/s13059-019-1815-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Bolger AM, Lohse M, and Usadel B (2014). Trimmomatic: a flexible trimmer for Illumina sequence data. Bioinformatics 30, 2114–2120. 10.1093/bioinformatics/btu170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Langmead B, and Salzberg SL (2012). Fast gapped-read alignment with Bowtie 2. Nat Methods 9, 357–359. 10.1038/nmeth.1923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Li H, Handsaker B, Wysoker A, Fennell T, Ruan J, Homer N, Marth G, Abecasis G, Durbin R, and Subgroup, 1000 Genome Project Data Processing (2009). The Sequence Alignment/Map format and SAMtools. Bioinformatics 25, 2078–2079. 10.1093/bioinformatics/btp352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Love MI, Huber W, and Anders S (2014). Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol 15, 550:1–21. 10.1186/s13059-014-0550-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Zhang Y, Liu T, Meyer CA, Eeckhoute J, Johnson DS, Bernstein BE, Nussbaum C, Myers RM, Brown M, Li W, et al. (2008). Model-based analysis of ChIP-Seq (MACS). Genome Biol 9, R137–R137.9. 10.1186/gb-2008-9-9-r137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Ross-Innes CS, Stark R, Teschendorff AE, Holmes KA, Ali HR, Dunning MJ, Brown GD, Gojis O, Ellis IO, Green AR, et al. (2012). Differential oestrogen receptor binding is associated with clinical outcome in breast cancer. Nature 481, 389–393. 10.1038/nature10730. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Quinlan AR, and Hall IM (2010). BEDTools: a flexible suite of utilities for comparing genomic features. Bioinformatics 26, 841–842. 10.1093/BIOINFORMATICS/BTQ033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Feng J, Liu T, Qin B, Zhang Y, and Shirley Liu X (2012). Identifying ChIP-seq enrichment using MACS. 10.1038/nprot.2012.101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Ramírez F, Ryan DP, Grüning B, Bhardwaj V, Kilpert F, Richter AS, Heyne S, Dündar F, and Manke T (2016). deepTools2: a next generation web server for deep-sequencing data analysis. Nucleic Acids Res 44, W160–W165. 10.1093/nar/gkw257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Heinz S, Benner C, Spann N, Bertolino E, Lin YC, Laslo P, Cheng JX, Murre C, Singh H, and Glass CK (2010). Simple Combinations of Lineage-Determining Transcription Factors Prime cis-Regulatory Elements Required for Macrophage and B Cell Identities. Mol Cell 38, 576–589. 10.1016/j.molcel.2010.05.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Trapnell C, Pachter L, and Salzberg SL (2009). TopHat: discovering splice junctions with RNA-Seq. Bioinformatics 25, 1105–1111. 10.1093/bioinformatics/btp120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Trapnell C, Roberts A, Goff L, Pertea G, Kim D, Kelley DR, Pimentel H, Salzberg SL, Rinn JL, and Pachter L (2012). Differential gene and transcript expression analysis of RNA-seq experiments with TopHat and cufflinks. Nat Protoc 7, 562–578. 10.1038/nprot.2012.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Murtagh F, and Legendre P (2014). Ward’s Hierarchical Agglomerative Clustering Method: Which Algorithms Implement Ward’s Criterion? J Classif 31, 274–295. 10.1007/s00357-014-9161-z. [DOI] [Google Scholar]
- 93.Wang J, Zhao Y, Zhou X, Hiebert SW, Liu Q, and Shyr Y (2018). Nascent RNA sequencing analysis provides insights into enhancer-mediated gene regulation. BMC Genomics 19, 633:1–18. 10.1186/s12864-018-5016-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Zhao Y, Liu Q, Acharya P, Stengel KR, Sheng Q, Zhou X, Kwak H, Fischer MA, Bradner JE, Strickland SA, et al. (2016). High-Resolution Mapping of RNA Polymerases Identifies Mechanisms of Sensitivity and Resistance to BET Inhibitors in t(8;21) AML. Cell Rep 16, 2003–2016. 10.1016/j.celrep.2016.07.032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Valouev A, Johnson SM, Boyd SD, Smith CL, Fire AZ, and Sidow A (2011). Determinants of nucleosome organization in primary human cells. Nature 474, 516–522. 10.1038/nature10002. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
All genomic datasets have been deposited at GEO and are publicly available at GEO: GSE160470. The raw western blot images were deposited to Mendeley Data and are publicly as of the date of publication. doi:10.17632/cf5vbhy67d.1.
The code and software used in this paper is described in detail on github: https://github.com/monnieb92/SMARCA5_paper (DOI: 10.5281/zenodo.7438719).
Any additional information required to reanalyze the data reported in this paper is available from the lead contact upon request.