

Abstract
The sEH has been identified as an attractive target for anti-inflammatory drug design in recent years. Picomolar level compound G1 against sEH was obtained by introducing the hydrophilic group homopiperazine and hydrophobic fragment propionyl onto the structure of lead compound A. G1 showed good microsomal stability, moderate plasma protein binding rate, good oral bioavailability and well tolerated in rats. G1 has significant analgesic effects on CFA-induced AIA mice, ameliorated the pancreatic injury in acute pancreatitis induced by L-arginine and reversed pancreatic injury, edema and neutrophil infiltration, increased survival time of C57BL/6 mice in an LPS-induced sepsis model, the expression levels of sEH, COX-2, NOS-2, VCAM, IL-6, MCP-5, and TNF-α were measured by Western blot or ELISA, with varying degrees of decrease. These results suggested G1 is a drug candidate worthy of further evaluation for the treatment of inflammation-induced diseases such as arthritis, acute pancreatitis and sepsis.
INTRODUCTION
Inflammation is a complex biological protective response to harmful stimuli such as pathogens, damaged cells, irritants etc, it involves immune cells, blood vessels and molecular mediators. But, excessive inflammatory response can cause physical damage, just as the severe COVID-19 patients may die from cytokine storm-induced sepsis and subsequent multiple organ failure(cytokine storm is also called inflammatory storm). Chronic inflammation is associated with many diseases, such as AD, arthritis, cancer etc.
Acute pancreatitis (AP) is an inflammatory disease related to the injury and necrosis of pancreatic exocrine tissue. It has an acute onset, severe condition and poor prognosis, with an incidence rate of 10 / 3400001, 2. The prevalence of AP has been increasing in recent years, with a mortality rate of up to 30 %3. The disease can cause varying degrees of damage to the lung, kidney, liver, heart, and other important substantive organs, and in severe cases, systemic inflammatory response syndrome (SIRS), multiple organ dysfunction syndrome (MODS), and even death can occur in the early stage of the disease, for which there is no specific treatment method4–6. According to research, inflammatory cytokines play an important role in AP progression, and AP progression is driven by an inflammatory cascade initiated by Toll-like receptors (TLRs)-nuclear factor B (NF-κB) activation and cytokine production by glandular follicle cells7. More recently, it has been demonstrated that mice with EPHX2 knocked out systemically exhibit attenuated arginine-induced AP, which is due to the potent anti-inflammatory properties of EET and a reduction in endoplasmic reticulum (ER) stress, implying that sEH inhibitors can be used to treat AP8, 9.
Rheumatoid arthritis (RA) is a chronic inflammatory disease with an unknown etiology that causes multiple, symmetrical joint swelling and pain, abnormal synovial proliferation, cellular invasion, and progressive destruction of cartilage and bone, resulting in further joint destruction, stiffness, deformity, and disability10. Inflammation causes pain in many RA patients. Pain is a complex signaling process caused by harmful substances being damaged and the release of inflammatory mediators such as cytokines, ions, bradykinin, prostaglandins, and leukotrienes, which act directly on pain receptors and drive action potentials to produce the sensation of pain11. The current clinical use of nonsteroidal anti-inflammatory drugs, glucocorticoids, and other anti-rheumatic drugs and traditional therapies can reduce symptoms and slow the progression of rheumatoid arthritis, but long-term use of these drugs can result in a variety of adverse reactions, including liver and kidney damage, gastrointestinal irritant injury, and so on12, 13. As a result, a new treatment for RA with no side effects is desperately needed. Pain is caused by biological or chemical inflammation, and the prostaglandin metabolite PGE2 is an important inflammatory mediator and pain-causing substance14. Many studies have shown that inhibiting sEH activity with small molecules lowers prostaglandin-2 (PGE2) levels, thereby inhibiting inflammation and exerting analgesic effects15–17. They are more effective than nonsteroidal anti-inflammatory drugs (NSAIDs) and even improve NSAIDs’ anti-inflammatory effects. sEH inhibitors changed the levels of not only EET and diol metabolites, but also some other metabolites in the arachidonic acid cascade of cyclooxygenase (COX) and lipoxygenase (LOX) metabolic pathways15. sEH inhibitors have been shown to have therapeutic and protective effects in acute lung injury18–20, various renal diseases21, the heart22, 23, and the blood-brain barrier24. Because sEH inhibitors are not addictive in terms of analgesia, they merit further development and investigation25.
Sepsis is a clinical syndrome characterized by a dysregulated inflammatory response (also known as cytokine storm or inflammatory storm) to infections (including bacteria, fungi, viruses, and parasites) and severe trauma26, 27. The COVID-19 pandemic has now become a global crisis, more lethal than any other infectious disease in history. It has had a significant physical and psychological impact on a large portion of the global population, as well as destroyed the global business and social order28. According to current research, the pathogenesis of COVID-19 is primarily related to immunopathology29. The rapid and massive production of cytokines such as TNF-α, IL-1, IL-6, IL-12, MCP-5, and IL-8 in body fluids following infection is known as cytokine storm, and it is a major cause of acute respiratory distress syndrome and multi-organ failure30, 31. Corticosteroids are commonly used in clinic to treat cytokine storm, but they have an immunosuppressive effect and, to some extent, delay the body’s clearance of viruses and other pathogens, thereby exacerbating the disease32, 33. sEH inhibitors inhibit the production of inflammatory factors and cytokines via the endoplasmic reticulum (ER) stress pathway, allowing the endoplasmic reticulum stress response to evolve toward maintaining the balance of the inflammatory response in vivo, rather than a single cytokine34. They show good application prospects in the treatment of sepsis.
Three sEH inhibitors have entered the clinic to date (Figure 1): AR-9281 (for hypertension and insulin resistance)35, GSK-2256294 (for chronic obstructive pulmonary disease)36, and EC-5026 (for neuropathic and inflammatory chronic pain)37, but none of them are currently approved for marketing. Because sEH play an important role in the development and progression of inflammation and pain, we designed and synthesized a series of sEH inhibitors and then tested their sEH inhibitory effects. The result showed that most compounds had good inhibiting activities, especially compound G1 with picomolar level inhibitory activity. The safety, metabolic stability, pharmacokinetic and therapeutic effects of compound G1 against rheumatoid arthritis, acute pancreatitis and sepsis were further evaluated. The results showed that compound G1 is a candidate for the treatment of rheumatoid arthritis, acute pancreatitis and sepsis and sEH inhibitors have a wide range of clinical application prospects.
Figure 1.

Chemical structures of sEH inhibitors that moved into clinical trials.
RESULTS AND DISCUSSION
Rational Design of the sEH Inhibitors.
Based on previous explorations made in our laboratory on sEH inhibitors, we chose compound A as the lead compound (Scheme 1)38, 39. The memantine part and the introduction of a fluorine atom at the 2-position of the benzene ring retard the metabolism of compound A, and it has been demonstrated that the fluorine atom breaks the structural symmetry, lowers the melting point, and increases the solubility of the compound38, 39. The protein code of sEH used for docking is 3WKE, and the eutectic molecule is t-AUCB. The structure of t-AUCB is very similar to that of compound A, both of which have urea segment and carboxylic acid segment, so compound A was selected as the basis for docking. The molecular docking revealed that compound A formed key hydrogen bonds with Try 383 and Try 466, and that the carboxyl group was exposed to the solvent with a large cavity, allowing the introduction of polar functional groups to improve the compound’s activity, physicochemical properties, and drug-like properties (Figure 2). Firstly, we introduced the R fragment with small amine and substituted amino group onto the 3- or 4-position of piperidine group to get two series of compounds A1-A5 and B1-B4, and found that the activity of the substituent at the 3-position of piperidine was better than that of 4-position, but the inhibitory activity of the substituent at position 4 against rat sEH (MsEH) is better than that against human sEH (HsEH). Then cyclohexylamine was used instead of piperidine, and the R segment of polar small molecule amine or alcohol was introduced to obtain C series compounds. To increase compound flexibility, we reduced the carbonyl group to methylene to generate compounds D1-D10, and their effects against sEH were improved when compared to the B series compounds. However, when we reduced the A series compounds to get compounds E1-E4, the activities were not increased. This result indicated that the substituent on the 3 position of piperidine was optimal, so we introduced the hydrophilic group homopiperazine on the 3 position of piperidine and integrated the R fragments of EC5026 and TPPU to obtain compounds F1-F3, while reducing the carbonyl group of the F series compounds to obtain G series compounds. The evaluation of these compounds against sEH revealed that compound G1 demonstrated strong inhibitory effects against HsEH and MsEH, with IC50 values of 0.05 and 0.14 nM, respectively. The result evoked our curiosity in the homopiperazine fragment. We changed the order of piperidine and homopiperazine and introduced R fragments of small molecule acids and amines to obtain compounds H1-H4, and then reduce the carbonyl group of H series compounds and introduce the R fragments of small alcohol to obtain I series compounds, but bioactivity results showed that no compound had better activity than G1, so compound G1 was chosen for further investigation.
Scheme 1.

Design strategy of target compounds based on lead compound A
Figure 2.

Docked pose of compound A in blue bound to sEH (PDB ID: 3WKE).
Chemistry.
The technique used to synthesize compounds A1-A5 was shown on Scheme 2. Intermediate 1 was obtained by commercially available 3-fluoro-4-nitrobenzoic acid with SOCl2 in the presence of methanol. A reduction of intermediate 1 was catalyzed with 5 % Pd-C under H2 atmosphere to provide intermediate 2. In the presence of triphosgene and triethylamine, the amino group of memantine was transformed to isocyanate, and then condensed with intermediate 2 to obtain intermediate 3. Intermediate 4 was produced immediately after hydrolysis of intermediate 3 in the presence of sodium hydroxide, while intermediate 5 was prepared by condensing intermediate 4 with piperidine-4-carboxylic acid methyl ester under HATU and DIPEA conditions. The hydrolysis of intermediate 5 gave the key intermediate 6. Intermediate 6 was condensed with diethylamine, ethanolamine, isopropanolamine, 2-(piperazin-1-yl-ethan)-1-ol or 2-aminopropan-1,3-diol in the presence of HATU and DIPEA to give the target compounds A1-A5, respectively.
Scheme 2.

Synthetic route to compounds A1-A5. Reactions and conditions: (i) SOCl2, MeOH, rt; (ii) H2 (g), 5 % Pd-C, EtOH, 60 °C, 12 h; (iii) (1) memantine, triphosgene, Et3N, DCM, −78 °C; (2) Et3N, DCM, 0 °C, 2 h; (vi) NaOH, THF/H2O, 60 °C, 1 h; (v)methyl piperidine-4-carboxylate, HATU, DIPEA, DCM, rt 2 h; (vi) NaOH, THF / H2O, 60 °C, 1 h; (vii) amines, HATU, DIPEA, DCM, rt.
Compounds B1-B4 were synthesized from intermediate 4 using the technique indicated in Scheme 3. Intermediate 7 was formed by condensing intermediate 4 with ethyl piperidine-3-carboxylate in the presence of HATU and DIPEA. Intermediate A was formed by hydrolyzing intermediate 7 with sodium hydroxide. Condensation of key intermediate A with diethylamine, ethanolamine, iso-propanolamine, or 2-(piperazin-1-yl-ethan)-1-ol yielded compounds B1-B4 respectively.
Scheme 3.

Synthetic route to compounds B1-B4. Reactions and conditions: (i) ethyl piperidine-3-carboxylate, HATU, DIPEA, DCM, rt; (ii) NaOH, THF / H2O, 60 °C, 1 h; (iii) amines, HATU, DIPEA, DCM, rt.
The synthesis of compounds C1-C8 was similar to the preparation of compounds A1-A5 depicted on Scheme 2, intermediate 4 was reacted with methyl 4-aminocyclohexane-1-carboxylate in the presence of HATU and DIPEA to give intermediate 8, and the hydrolysis of intermediate 8 under sodium hydroxide gave intermediate 9. Similarly intermediate 9 was condensed with tert-butyl 4-(2-aminoethyl)piperazine-1-carboxylate, 2-(piperazin-1-yl-ethan)-1-ol, ethanolamine, iso-propanolamine, diethanolamine, ammonia, 2-aminopropan-1,3-diol, 2-amino-2-(hydroxymethyl)propan-1,3-diol to give compounds C1-C8, respectively (Scheme 4).
Scheme 4.

Synthetic route to compounds C1-C8. Reactions and conditions: (i) methyl 4-aminocyclohexane-1-carboxylate, HATU, DIPEA, DCM, rt, (ii) NaOH, THF / H2O, 60 °C, 1 h; (iii) amines, HATU, DIPEA, DCM, rt, 2h.
Compounds D1-D9 were prepared via the procedure depicted in Scheme 5 from commercially available 3-fluoro-4-nitrobenzoic acid. Intermediate 10 was obtained by reducing 3-fluoro-4-nitrobenzoic acid under borane. Then intermediate 10 underwent nucleophilic substitution reaction with phosphorus tribromide to give intermediate 11. Intermediate 11 underwent nucleophilic substitution reaction with ethyl piperidine-3-carboxylate at the presence of K2CO3 and KI to give intermediate 12. Reduction of 12 catalyzed with 5 % Pd-C under H2 atmosphere provided 13. In the presence of triphosgene and triethylamine, the amino group of memantine was transformed to isocyanate, and then condensed with intermediate 13 to obtain intermediate 14. Immediately afterwards compound 15 was hydrolyzed in the presence of sodium hydroxide to give the key intermediate D. Intermediate D was condensed with ammonia, dimethylamine, diethylamine, iso-propanolamine, diethanolamine, ethanolamine, 2-aminopropan-1,3-diol, 2-amino-2-(hydroxymethyl)propan-1,3-diol or 2-(piperazin-1-yl-ethan)-1-ol in the presence of HATU and DIPEA to obtain the target compounds D1-D9, respectively.
Scheme 5.

Synthetic route to compounds D1-D10. Reactions and conditions: (i) Borane-tetrahydrofuran complex, THF, 0 °C, 2h; (ii) phosphorus tribromide, DCM, rt, 8 h; (iii) ethyl piperidine-3-carboxylate, K2CO3, ACN, 50 °C, 4h; (iv) H2 (g), 5 % Pd-C, EtOH, 60 °C, 12 h; (v) (1) memantine, triphosgene, Et3N, DCM, −78 °C; (2) Et3N, DCM, 0 °C, 2 h; (vi) NaOH, THF / H2O, 60 °C, 1 h; (vii) amines, HATU, DIPEA, DCM, rt, 2h.
The synthesis of compounds E1-E5 was similar to the preparation of compounds A1-A5 depicted on Scheme 2. Intermediate 11 reacted with piperidine-4-carboxylic acid methyl ester in the presence of K2CO3 and KI to obtain intermediate 15, followed immediately by reduction, urea formation and hydrolysis to obtain key intermediate 18. Similarly intermediate 18 was condensed with ammonia, iso-propanolamine, ammonia, 2-aminopropane-1,3-diol, 2-amino-2-(hydroxymethyl)propane-1,3-diol or diethanolamine in the presence of HATU and DIPEA to give the target compounds E1-E5, respectively (Scheme 6).
Scheme 6.

Synthetic route to compounds E1-E5. Reactions and conditions: (i) ethyl piperidine-3-carboxylate, K2CO3, ACN, 50 °C, 4h; (ii) H2 (g), 5 % Pd-C, EtOH, 60 °C, 12 h; (iii) (1) memantine, triphosgene, Et3N, DCM, −78 °C; (2) Et3N, DCM, 0 °C, 2 h; (iv) NaOH, THF / H2O, 60 °C, 1 h; (v) amines, HATU, DIPEA, DCM, rt, 2h.
Compound A and tert-butyl 1,4-diazepane-1-carboxylate in the presence of the condensing agent HATU and the organic base DIPEA to give intermediate 19. Intermediate 19 was deprotected under trifluoroacetic acid to give intermediate 20. The condensation of key intermediate 20 with acetic acid, propionic acid or 2-methylbutyric acid gave compounds F1-F3 respectively (Scheme 7).
Scheme 7.

Synthetic route to compounds F1-F3. Reactions and conditions: (i) tert-butyl 1,4-diazepane-1-carboxylate, HATU, DIPEA, DCM, rt, 2h. (ii) TFA, DCM, rt, 2 h; (iii) acids, HATU, DIPEA, DCM, rt, 2h.
Compound D was reacted with tert-butyl 1,4-diazepane-1-carboxylate and deprotected Boc group to give intermediate 22. Then the target compounds were obtained by condensation with propionic acid, 2-methylbutyric acid, acrylic acid, methacrylic acid or cyclopropanecarboxylic acid in the presence of HATU and DIPEA, respectively (Scheme 8).
Scheme 8.

Synthetic route to compounds F1-F3. Reactions and conditions: (i) tert-butyl 1,4-diazepane-1-carboxylate, HATU, DIPEA, DCM, rt, 2h. (ii) TFA, DCM, rt, 2 h; (iii) acids, HATU, DIPEA, DCM, rt, 2h.
Compounds H1-H4 were obtained as shown on Scheme 9. By acylating tert-butyl 1,4-diazepane-1-carboxylate with 3-fluoro-4-nitrobenzoic acid to give compound 23. Reduction of 23 under H2 atmosphere in presence of 5 % Pd-C gave 24. In the presence of triphosgene and triethylamine, the amino group of memantine was transformed to isocyanate, and then condensed with compound 24 to obtain compound 25. Intermediate 26 was obtained by deprotecting Boc in the presence of trifluoroacetic acid, and then condensed with 2-(1-(tert-butoxycarbonyl)piperidin-4-yl)acetic acid in the presence of HATU and DIPEA to give intermediate 27. The key intermediate 28 was obtained by deprotecting Boc in the presence of trifluoroacetic acid. Intermediate 28 was condensed with propionic acid, 2-methylbutyric acid and acrylic acid in the presence of HATU and DIPEA to give the target compounds H1-H3, respectively. Intermediate 28 underwent nucleophilic substitution with 2-chloro-N,N-dimethylpropan-1-amine in the presence of K2CO3 and KI to give compound H4(Scheme 9).
Scheme 9.

Synthetic route to compounds H1-H4. Reactions and conditions: (i) tert-butyl 1,4-diazepane-1-carboxylate, HATU, DIPEA, DCM, rt, 2h; (ii) H2 (g), 5 % Pd-C, EtOH, 60 °C, 12 h; (iii) (1) memantine, triphosgene, Et3N, DCM, −78 °C, 2h; (iv) TFA, DCM, rt, 2 h; (v) tert-butyl 1,4-diazepane-1-carboxylate, HATU, DIPEA, DCM, rt, 2h; (vi) TFA, DCM, rt, 2 h; (vii) acids, HATU, DIPEA, DCM, rt, 2h; (viii) 2-chloro-N,N-dimethylpropan-1-amine, K2CO3, ACN, 50 °C, 4h.
Intermediate 11 was condensed with tert-butyl 1,4-diazepane-1-carboxylate to give the key intermediate 29, Intermediates 34 was obtained from intermediate 29 through reduction, urea-forming, removal of Boc group, acylation, and removal of Boc group. Intermediates 34 were condensed with acetic acid, propionic acid or 2-methylbutyric acid in the presence of HATU and DIPEA to give the target compounds I1-I3 respectively. In the presence of triphosgene and triethylamine, the amino group of intermediate 34 was transformed to isocyanate, and then condensed with methoxyammonium chloride to obtain compound I4. Intermediate 34 in the presence of K2CO3 and KI with 2-chloroethane-1-ol, 3-chloropropane-1,2-diol, 2-chloro-N,N-dimethylethane-1-amine or 2-chloro-N,N-diethylethane-1-amine underwent nucleophilic substitution to give compounds I5-I8 respectively (Scheme 10).
Scheme 10.

Synthetic route to compounds I1-I8. Reactions and conditions: (i) tert-butyl 1,4-diazepane-1-carboxylate, HATU, DIPEA, DCM, rt, 2h; (ii) H2 (g), 5 % Pd-C, EtOH, 60 °C, 12 h; (iii) (1) memantine, triphosgene, Et3N, DCM, −78 °C, 2h; (iv) TFA, DCM, rt, 2 h; (v) tert-butyl 1,4-diazepane-1-carboxylate, HATU, DIPEA, DCM, rt, 2h; (vi) TFA, DCM, rt, 2 h; (vii) acids, HATU, DIPEA, DCM, rt, 2h; (viii) chlorides, K2CO3, ACN, 50 °C, 4h.
Molecular docking.
The possible binding mode of compound G1 with the sEH protein active pockets (PDB ID:3WKE) was docked with Discovery Studio 2016 software, as shown in Figure 3. The compound G1 urea group formed three hydrogen bonds with the catalytic triplet consisting of residues Asp 335 and Tyr 383 of the backbone, one more hydrogen bond than the lead compound A. It also filled the cavity around the carboxyl group and bound more tightly to the protein, which explains the much higher activity of compound G1 than that of compound A.
Figure 3.

Molecular simulation of compounds G1 bound to sEH (PDB ID: 3WKE)
Structure activity relationships of series derivatives.
Compounds with memantine core are promising sEH inhibitor in our previous research38. The activities of target compounds against HsEH and MsEH were evaluated according to the method reported in literature40 with the lead compounds A and t-TUCB as control. The results were shown on Table 1–Table 5
Table 1.
IC50 values of A1-A5 and B1-B4 against Human sEH (HsEH) and Murine sEH (MsEH)a.

|
||||||
|---|---|---|---|---|---|---|
| Compd. | R | Subst. | HsEH (nM) | MsEH (nM) | cLogPb | LLEc |
| A1 |

|
4 | 2.2 | 0.53 | 3.70 | 4.954 |
| A2 |

|
4 | 3.43 | 0.26 | 2.262 | 6.203 |
| A3 |

|
4 | 4.5 | 0.18 | 2.639 | 5.708 |
| A4 |

|
4 | 12.5 | 0.32 | 2.639 | 6.264 |
| A5 |

|
4 | 7.1 | 0.55 | 2.639 | 5.510 |
| B1 |

|
3 | 0.35 | 0.38 | 3.839 | 5.617 |
| B2 |

|
3 | 0.35 | 0.15 | 2.397 | 7.069 |
| B3 |

|
3 | 0.28 | 0.23 | 2.774 | 6.779 |
| B4 |

|
3 | 0.84 | 0.15 | 2.560 | 6.516 |
| A | - | 3 | 4.6 | 1.3 | 3.358 | 4.979 |
| t-TUCB | - | 0.28 | 2.8 | 5.489 | 4.064 | |
IC50 values were recorded on recombinant human sEH (HsEH) and murine sEH (MsEH) using PHOME as substrate at 50 mM concentration.
The cLogP values were predicted by BIOVIA Discovery Studio 2016.
Ligand lipophilicity efficiency (LLE) = pIC50-cLogP.
Table 5.
IC50 values of H1-H4 and I1-I8 against Human sEH(HsEH) and Murine sEH (MsEH)a.

|
||||||
|---|---|---|---|---|---|---|
| Compd. | X | R | HsEH (nM) | MsEH (nM) | cLogPb | LLEc |
| H1 |

|

|
0.9 | 0.39 | 3.198 | 5.848 |
| H2 |

|

|
1.0 | 0.35 | 4.117 | 4.883 |
| H3 |

|

|
1.0 | 0.13 | 3.470 | 5.530 |
| H4 |

|

|
19.6 | 0.8 | 3.788 | 3.920 |
| I1 |

|

|
0.49 | 0.11 | 3.191 | 6.119 |
| I2 |

|

|
0.33 | 0.07 | 3.858 | 5.623 |
| I3 |

|

|
0.77 | 0.16 | 4.777 | 4.337 |
| I4 |

|

|
1.1 | 0.41 | 3.834 | 5.125 |
| I5 |

|

|
4.69 | 0.61 | 3.392 | 4.937 |
| I6 |

|

|
58.1 | 1.25 | 2.881 | 5.986 |
| I7 |

|

|
2.59 | 0.67 | 4.07 | 4.517 |
| I8 |

|

|
2.1 | 1.37 | 4.768 | 3.910 |
| A | - | 4. | 1.3 | 3.358 | 4.979 | |
| t-TUCB | - | 0.28 | 2.8 | 5.489 | 4.064 | |
IC50 values were recorded on recombinant human sEH (HsEH) and murine sEH (MsEH) using PHOME as substrate at 50 mM concentration.
The cLogP values were predicted by BIOVIA Discovery Studio 2016.
Ligand lipophilicity efficiency (LLE) = pIC50-cLogP.
The activity of carboxylic acid derivative such as amide at position 4 of piperidine was first investigated, and N,N-diethylamide derivative compound A1 showed potent effects (IC50: HsEH = 2.2 nM, MsEH = 0.53 nM). The activities of compound A1 against HsEH and MsEH were more than twice that of the lead compound A, but they still need to be improved compared with t-TUCB (Table 1). To increase the water solubility and activity of the compounds, 2-aminoethan-1-ol, 1-aminopropan-2-ol, 2-(piperazin-1-yl)ethan-1-ol, or 2-aminopropane-1,3-diol were reacted to yield compounds A2-A5 to provide additional polar groups respectively. The structure-activity relationship revealed that small molecule amine alcohols significantly improved MsEH activity, with A3 having the best IC50: MsEH = 0.18 nM. Next we changed the position of amide onto the third position of piperidine from the fourth position of piperdine and obtained compound B1 (IC50: HsEH=0.35 nM, MsEH = 0.38 nM). The activity against HsEH is equivalent to that of t-TUCB, and it is 7 times that of t-TUCB against MsEH. When we introduced additional polar groups on the structure of compound A by the similar way to obtain compounds B2-B3, their activities against HsEH remained powerful effects; while 2-(piperazin-1-yl) ethan-1-ol was introduced, its activity against HsEH slightly decreased maybe because the steric hindrance was a little big. The result that compound B3 (IC50: HsEH = 0.28 nM, MsEH = 0.23 nM) is more active than t-TUCB is encouraging. According to the preliminary structure-activity relationship, the 3 position of piperidine has priority over the 4 position, the introduction of polar groups is beneficial.
We speculate that the poor activity of piperidine at position 4 may be due to the fact that N is on the ring, the rigidity of the compound is too strong, and the bond is not easy to rotate. Therefore, we replaced piperidine with cyclohexylamine, and explored the structure-activity relationship. When the introduction of cyclic tert- butyl 4-(2-aminoethyl) piperazine-1-carboxylate or 2-(piperazin-1-yl) ethan-1-ol provided compounds C1 and C2, their activities against HsEH obviously decreased. A series of small molecular amine fragments were introduced to obtain compounds C3-C8. Among them, C8 (IC50: HsEH = 0.35 nM, MsEH = 0.12 nM) has the best activities (Table 2). The preliminary structure-activity relationship showed that the activities of B series compounds were better than these of C series compounds in general, and the activity of piperidine fragments was better than that of cyclohexylamine, and the activity of compound C8 with the moiety of 2-amino-2 - (hydroxymethyl) propane-1,3-diol was the best.
Table 2.
IC50 values of C1-C8 against Human sEH(HsEH) and Murine sEH (MsEH)a.

|
|||||
|---|---|---|---|---|---|
| Compd. | R | HsEH (nM) | MsEH (nM) | cLogPb | LLEc |
| C1 |

|
183 | 2.0 | 4.466 | 2.272 |
| C2 |

|
16 | 0.53 | 4.141 | 3.655 |
| C3 |

|
0.8 | 0.18 | 2.997 | 6.100 |
| C4 |

|
1.54 | 0.44 | 3.374 | 5.438 |
| C5 |

|
2.94 | 0.23 | 2.663 | 5.869 |
| C6 |

|
0.84 | 0.35 | 3.330 | 5.746 |
| C7 |

|
0.91 | 0.55 | 2.486 | 6.555 |
| C8 |

|
0.35 | 0.12 | 1.803 | 7.653 |
| A | - | 4.6 | 1.3 | 3.358 | 4.979 |
| t-TUCB | - | 0.28 | 2.8 | 5.489 | 4.064 |
IC50 values were recorded on recombinant human sEH (HsEH) and murine sEH (MsEH) using PHOME as substrate at 50 mM concentration.
The cLogP values were predicted by BIOVIA Discovery Studio 2016.
Ligand lipophilicity efficiency (LLE) = pIC50-cLogP.
In order to improve the potency, we also reduced the carbonyl group on the benzene ring of B series compounds to increase the flexibility and rotation of the compounds. Excitingly, the activities of compound D (IC50: HsEH = 2.3 nM, MsEH = 0.61 nM) were twice that of compound A. We continued to introduce small molecules of amines to obtain amide compounds D1-D4. Among them, the activities of compound D1 (IC50: HsEH = 0.14 nM, MsEH = 0.087 nM) were superior to t-TUCB, especially the activity against MsEH. When a series of small molecule aminol fragments were introduced, compounds D5-D10 were obtained. The results showed that the activities of both HsEH and MsEH remained almost similar activities compared to the B series compounds. Similarly for the carbonyl reduction of the A series compounds, a series of compounds E1-E5 were synthesized, with compound E1 (IC50: HsEH = 1.54 nM, MsEH = 0.058 nM) showing the best activities (Table 3). The structure-activity relationship showed that the activity of 3- substituted piperidine is better than that of 4- substituted piperidine, and short-chain small molecular amine with additional polar group was the best substituent.
Table 3.
IC50 values of D1-D9 and E1-E5 against Human sEH (HsEH) and Murine sEH (MsEH)a.

|
||||||
|---|---|---|---|---|---|---|
| Compd. | R | Subst. | HsEH (nM) | MsEH (nM) | cLogPb | LLEc |
| D | -OH | 3 | 2.3 | 0.61 | 0.982 | 7.656 |
| D1 |

|
3 | 0.14 | 0.087 | 3.390 | 6.464 |
| D2 |

|
3 | 0.7 | 0.12 | 3.801 | 5.354 |
| D3 |

|
3 | 0.35 | 0.56 | 4.499 | 4.957 |
| D4 |

|
3 | 0.28 | 0.58 | 3.434 | 6.119 |
| D5 |

|
3 | 0.28 | 0.29 | 2.723 | 6.879 |
| D6 |

|
3 | 0.63 | 0.18 | 3.056 | 6.145 |
| D7 |

|
3 | 0.28 | 0.35 | 2.545 | 7.008 |
| D8 |

|
3 | 0.14 | 0.32 | 1.862 | 7.992 |
| D9 |

|
3 | 0.98 | 0.55 | 3.219 | 5.790 |
| E1 |

|
4 | 1.54 | 0.058 | 3.255 | 5.557 |
| E2 |

|
4 | 6.0 | 0.47 | 4.955 | 3.267 |
| E3 |

|
4 | 31.6 | 2.25 | 2.410 | 5.090 |
| E4 |

|
4 | 6.51 | 0.12 | 1.727 | 6.459 |
| E5 |

|
4 | 6.72 | 0.55 | 2.588 | 5.585 |
| A | - | - | 4.6 | 1.3 | 3.358 | 4.979 |
| t-TUCB | - | - | 0.28 | 2.8 | 5.489 | 4.064 |
IC50 values were recorded on recombinant human sEH (HsEH) and murine sEH (MsEH) using PHOME as substrate at 50 mM concentration.
The cLogP values were predicted by BIOVIA Discovery Studio 2016.
Ligand lipophilicity efficiency (LLE) = pIC50-cLogP.
Next, we further modified the amide fragment at 3 position on the piperidine with the hydrophilic group homopiperazine instead of the small molecule amine and introduced a small molecular acyl fragment on the other N atom of homopiperazine according to the design concept of EC-5026 and TPPU. Firstly, acetyl, propionyl or 2-methylbutyryl group were introduced to obtain compound F1-F3. The activities of compound F1-F3 were several times higher than that of lead compound A (Table 4). Then we reduced the carbonyl group on the benzene ring to obtain compound G1-G2. To our delight, compound G1 showed very strong activities (IC50: HsEH = 0.05 nM, MsEH = 0.14 nM). The activity of G1 against HsEH is 5.6 times that of t-TUCB, and the activity of G1 against MsEH is 20 times that of t-TUCB. Meanwhile, the activity against HsEH of compound G1 is 2.8 times that against MsEH. In order to increase the binding ability with sEH, we tried to use α,β-unsaturated double bond to form covalent bonds with sEH, and introduce acryl or 2-methylacryl group to obtain compounds G3 and G4. Unfortunately, they didn’t work and didn’t show high effects. The structure-activity relationship showed that the activity was improved when the carbonyl group was reduced to methylene, and the propionyl group was the best substituent.
Table 4.
IC50 values of F1-F3 and G1-G5 against Human sEH (HsEH) and Murine sEH (MsEH)a.

|
||||||
|---|---|---|---|---|---|---|
| Compd. | X | R | HsEH (nM) | MsEH (nM) | cLogPb | LLEc |
| F1 |

|

|
0.6 | 0.26 | 2.421 | 6.801 |
| F2 |

|

|
0.56 | 0.58 | 3.088 | 6.164 |
| F3 |

|

|
1.4 | 0.9 | 4.007 | 4.847 |
| G1 |

|

|
0.05 | 0.14 | 3.748 | 7.553 |
| G2 |

|
|
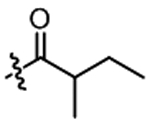
0.25 | 0.62 | 4.616 | 4.986 |
| G3 |

|

|
0.42 | 1.1 | 3.78 | 5.598 |
| G4 |

|

|
0.9 | 1.3 | 4.225 | 4.821 |
| G5 |

|

|
0.42 | 0.8 | 3.839 | 4.498 |
| A | - | 4.6 | 1.3 | 3.358 | 4.979 | |
| t-TUCB | - | 0.28 | 2.8 | 5.489 | 4.064 | |
IC50 values were recorded on recombinant human sEH (HsEH) and murine sEH (MsEH) using PHOME as substrate at 50 mM concentration.
The cLogP values were predicted by BIOVIA Discovery Studio 2016.
Ligand lipophilicity efficiency (LLE) = pIC50-cLogP.
Due to the introduction of homopiperazine and the propionyl group, the activity of compound G1 was significantly improved. This result stimulated our interest in homopiperazine. Based on the structure of compound G1, we tried to exchange the position of homopiperazine with piperidine, and substituted piperidine with piperidine acetic acid to synthesize a series of compounds H1-H4. Unfortunately, compared with compound G1, theirs activities decreased significantly. Then, after carbonyl reduction, acetyl, propionyl, 2-methylbutyryl or methoxycarbonyl were introduced to obtain compounds I1-I4, respectively. Compared with H1-H4, the activities of compounds I1-I4 have obviously increased. Compound I2 (IC50: HsEH = 0.35 nM, MsEH = 0.12 nM) has the best activities. Nextly small molecule amines and alcohols were introduced to obtain compounds I5-I8. But the activities of compounds I5-I8 decreased significantly (Table 5). Structure-activity relationship showed that the activity of the compound with reduction of carbonyl group was improved, and propionyl is the best small molecular substituent. The compound G1 (IC50: HsEH = 0.05 nM, MsEH = 0.12 nM) is the best compound, and has a high LLE value (7.553).
Microsomal stability.
The microsomal stability of compound G1 was evaluated in human and rat and mouse liver microsomes, respectively, which are widely used to determine the extent of possible primary metabolic clearance in the liver (Table 6). Compound G1 showed satisfactory microsomal stability in vitro with half-lives (t1/2) of 3.70 h and 3.15 h in human and rat liver microsomes, respectively. Preliminarily, the good metabolic stability of compound G1 suggests that it has value for further investigation.
Table 6.
Mean of concentrations of G1 in microsomal buffer at different time (μg / mL).
| Time(h) | 0 | 10 | 30 | 60 | t1/2(h) |
|---|---|---|---|---|---|
| Human | 100 | 81.12 | 74.76 | 68.78 | 3.7 |
| Rat | 100 | 75.04 | 66.63 | 62.01 | 3.15 |
Percent plasma protein binding.
Percent plasma protein binding (% PPB) was measured in SD rats by a classical equilibrium dialysis device. Compound G1 showed a moderate % PPB rate (72.57 %). Since the therapeutic effect of drugs depends on the concentration of free drugs, % PPB of medium intensity (72.57 %) may contribute to the therapeutic effect in vivo, thus contributing to its analgesic effect.
Pharmacokinetic study in vivo.
To determine the metabolic stability of compound G1 in vivo, the pharmacokinetic profile was therefore determined in Sprague-Dawley (SD) rats by oral (po) single dose of 50 mg / kg and intravenous (iv) single dose of 10 mg / kg, and the pharmacokinetic parameters are shown in Table 7. After oral administration, blood levels were detected within 8 h. The maximum concentration reached after 0.25 h (2.92 nM); the area under the curve (AUC0–8h) was 6.39 (nM∙h); the plasma t1/2 for G1 was 5.55 h. The maximum concentration (3.77 nM) was reached 1.8 min after intravenous injection of compound G1; the area under the curve (AUC0–8 h) was 6.13 nM∙h; the plasma t1/2 of G1 was 5.72 h. G1 had a moderate bioavailability of 20.85 % laying a good foundation for the next in vivo activity exploration.
Table 7.
Pharmacokinetics of G1 in rats following intravenous and oral administration (n = 3).
| Parameter | G1 (n = 3) |
G1 (n = 3) |
|---|---|---|
| iv (10 mg / kg) | po (50 mg / kg) | |
| Tmax(h) | 0.03 | 0.25 |
| Cmax(nM) | 3.77 | 2.92 |
| t1/2(h) | 5.72 | 5.55 |
| CL(L / h / kg) | 0.29 | 1.70 |
| VZ(L / kg) | 2.39 | 13.12 |
| AUC(0–8) (nM∙h) | 6.13 | 6.39 |
| AUC(0−∞) (nM∙h) | 11.47 | 10.15 |
| F (%) | 20.85 |
Safety of compound G1.
To further evaluate whether compound G1 has potential toxicological effects and adverse events, we conducted acute toxicity tests on mice. After seven consecutive days of oral administration of high dose of G1 (200 mg / kg), the survival status and body weight of mice were examined and no adverse events were observed, indicating that compound G1 was well tolerated and no toxicity was observed with continuous oral administration of high dose (200 mg / kg) (Figure 4).
Figure 4.

Effect of compound G1 on body weight of mice. G1 was prepared in 0.5 % CMC-Na solution and administered orally at a dose of 200 mg / kg for seven consecutive days.
Study of compound G1 against CFA-induced arthritis model in mice.
The mouse model of adjuvant-induced arthritis (AIA) induced by CFA has similar characteristics to human rheumatoid arthritis (RA) in terms of pathogenesis, joint pain, bone destruction and synovial hyperplasia, and is now widely used in the study of human RA pathogenesis and treatment mechanisms41, 42. To investigate if compound G1 is efficacious in Kunming mice with AIA, mice were randomly divided into 3 groups: the model group (CFA), the G1 group (10 mg / kg, ip), and the celecoxib group (10 mg / kg, ip). The health status of the animals was assessed by observing changes in their body weight during the experiment (Figure 5A), and the results revealed that no increase in body weight occurred in the mice of the three groups at 24 h of CFA administration, indicating that CFA-induced RA caused discomfort in the mice, but an increase in body weight was found in the G1 and celecoxib groups at 24 h of drug treatment while no weight gain was observed in the model group. Simultaneously, the pain valves of AIA mice were evaluated using the hot plate technique 10 minutes, 45 minutes, 120 minutes, and 240 minutes after injection (Figure 5B), and a pain valve enhancement rate curve was generated (Figure 5C). When compared to the basic pain valve, all three groups showed a decrease in pain threshold after 24 hours of modeling with CFA. Compound G1 began to work after 10 minutes of administration, peaked at 2 hours, and remained unchanged for 4 hours. Celecoxib did not significantly raise the pain threshold at 2 h and did not have a significant effect until 4 h. Compound G1 had a faster onset of analgesia than celecoxib and was more effective in terms of the pain threshold improvement rate curve. Finally, the thickness and breadth of mouse’s paws were compared before and after modeling. Compound G1 significantly reduced the swelling rate of mouse paw thickness and was superior to celecoxib, as shown in Figure 5D, and compound G1 was also superior to celecoxib in terms of swelling rate of mouse paw width (Figure 5E). In conclusion, compound G1 has significant analgesic and anti-inflammatory effects on CFA-induced AIA mice and is better than celecoxib.
Figure 5.
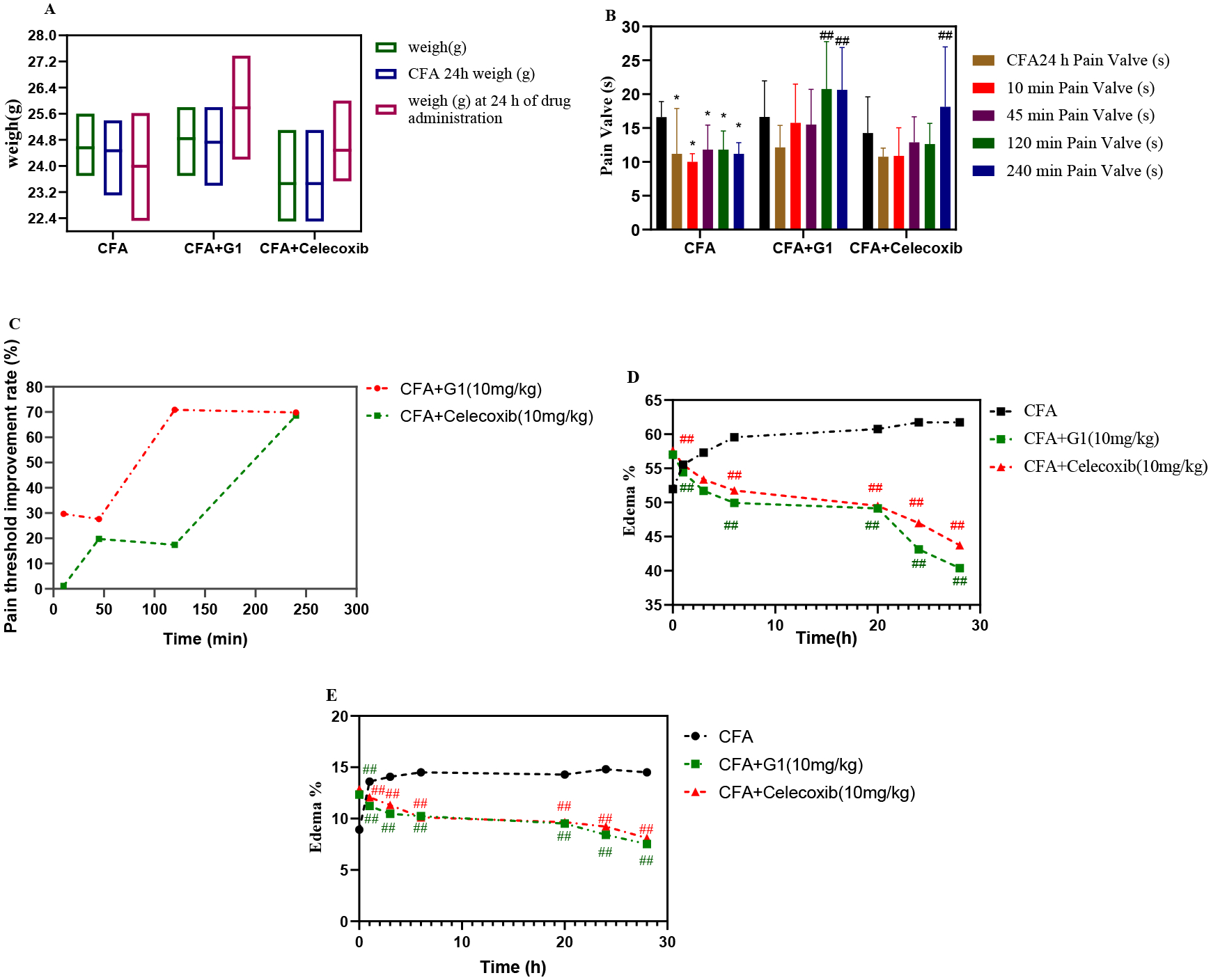
Evaluation of compound G1 for the treatment of (CFA)-induced arthritis in a mouse model (AIA); (A) Body weight changes of mice in CFA group, CFA + G1 group and CFA + Celecoxib group; (B) Changes in pain thresholds of mice in CFA group, CFA + G1 group and CFA + Celecoxib group; (C) Curves of pain threshold improvement in mice in CFA + G1 group and CFA + Celecoxib group;(D) Curves of paw thickness swelling rate of mice in CFA group, CFA + G1 group and CFA + Celecoxib group; (E) Curves of paw width swelling rate of mice in CFA group, CFA + G1 group and CFA + Celecoxib group; G1 (10 mg / kg, ip) and Celecoxib (10 mg / kg, ip) were administered to mice 24 h and 44 h after induction of CFA by AAI (25 μL / 25g). (n = 8 per group); Significance: ##P < 0.01 and #P < 0.05 compared with the Mod group; **P < 0.01 and *P < 0.05 compared with the Con group.
Study of compound G1 against L-arginine-induced acute pancreatitis model in mice.
AP is a potentially life-threatening gastrointestinal disease whose incidence has been increasing over the past decades. There are no effective therapeutic drugs in clinic and drug for the treatment of AP are urgent. Given the association of sEH with AP pathogenesis, the sEH inhibitor G1 at a dose of 3 mg / kg and 8 mg / kg was evaluated in the L-arginine-induced acute pancreatitis model in mice and compared with celecoxib (5 mg / kg). First, histological analysis of the pancreas was performed to determine whether G1 treatment reduced the severity of L-arginine-induced pancreatitis. Pathological changes were studied on H&E-stained pancreatic sections (Figure 6A).
Figure 6.

Results of the histologic analysis of pancreas from mice treated with control, Model, Celecoxib and compound G1. (A) Representative H&E-stained sections of the pancreas from the in vivo efficacy study. Arrows indicate inflammatory cells and edema. Bold arrows indicate intracellular vacuoles (the red arrows on the image represent edema and inflammatory cells). (B-E) Histologic scoring of pancreatic tissues. (B), edema. (C), inflammatory cells (mononuclear and polymorphonuclear). (D), parenchymal atrophy. (E), total scoring (edema, mononuclear and polymorphonuclear and parenchymal atrophy). (n = 4 per group); Significance: *p < 0.05, and **p < 0.01vs control. #p < 0.05 and ##p < 0.01vs Mod.
As expected, the L-arginine model group (6.25 ± 0.96) showed pancreatic injury representative of AP, including edema (Figure 6B), inflammatory cell infiltration (Figure 6C), and parenchymal atrophy (Figure 6D). In contrast, both compounds G1 8 mg / kg (3.50 ± 0.58) and G1 3 mg / kg (4.25 ± 1.5) improved L-arginine-induced pancreatic injury in AP. The effect of compound G1 (8 mg / kg) was better than the effect of 3 mg / kg, and compound G1 at the dose of 8 mg / kg (3.50 ± 0.58) exhibited more effective reversal of pancreatic injury, edema and neutrophil infiltration than equimolar concentrations of celecoxib 5 mg / kg (3.75 ± 0.96) (Figure 6E).
Study of compound G1 against LPS-induced sepsis model.
The potential therapeutic efficacy of compound G1 against severe sepsis was investigated. Compound G1 (5 mg / kg, ip) was administrated four hours after the sepsis model was induced by a high dosage (30 mg / kg, ip) of LPS. As a positive control group, dexamethasone (5 mg/kg, ip) was also given. G1 (5 mg / kg, ip) and dexamethasone (5 mg / kg, ip) were given for the second time 12 hours later. The number of mice that survived per hour was recorded, and the specific data are shown in Figure 7A. Because of the higher LPS dose, all of the mice in the dexamethasone group died after 28 hours. The mouse in compound G1 had a 37.5 % survival rate at this point, and when the period was extended to 35 hours, all of the mice in the solvent group died, while the animals in the G1 group had a 12.5 % survival rate. The final results showed that compound G1 outperformed dexamethasone in terms of extending the life of sick mice. Certain inflammatory related components such as sEH, COX-2, NOS-2, VCAM, IL-6, MCP-5, and TNF-α in blood were examined using Western blot or ELISA. At first, the expression of sEH and COX-2 were detected, and as expected, compound G1 significantly reduced the expression of sEH compared to the model group, and the similar result was observed in the dexamethasone group. However, COX-2 expression levels in compound G1 group and dexamethasone group were higher than in the control group. NOS-2 expression and excessive NO production are linked to endotoxic hypotension43. Compound G1 reduced NOS-2 expression, implying that sEH inhibitors can inhibit NOS-2 induction, reducing NO production and endotoxin-induced hypotension. Compared to the solvent group, the LPS group and compound G1 group had little change in the determination of vascular cell adhesion molecule (VCAM) (Figure 7B). IL-6 is important in the development of acute inflammatory responses, including endothelial and lymphocyte activation, as well as fever induction44. We detected a significant reduction in IL-6 levels in both compound G1 and dexamethasone groups by ELISA, and our findings suggest that compound G1 reduces IL-6 production, which is consistent with the antipyretic effect of EETs and the action of cyclooxygenase inhibitors45, 46. The chemokine MCP-5 is a potent monocyte chemoattractant47. Although the effect of compound G1 lowering MCP-5 was lower than dexamethasone, it demonstrated that the lymphocytes of mice in the LPS group were inhibited but still responded in the presence of sEH inhibitor. TNF-α, initially produced by resident monocytes and macrophages, is considered an early innate inflammatory mediator. Among its many physiological effects is the induction of vascular cell adhesion molecule-1 on endothelial cells, a critical factor for the transmigration of leukocytes from vascular compartments into inflamed tissues. Compound G1 and the dexamethasone considerably lowered TNF-α levels, inhibiting further inflammation and having a therapeutic effect. (Figure 7C).
Figure 7.

Prophylactic and therapeutic treatment of G1 prevents death from LPS administration in mice. G1 (5 mg / kg, ip) and Dexamethasome (5 mg / kg, ip) were administered to mice 4 h and 12 h after induction of ALI by a lethal dose of LPS (30 mg / kg, ip). The mortality of the mice was monitored per hour, (A) the percent survival rate was expressed as a Kaplan-Meier survival curve (n = 12–14 per group); (B) The expression of inflammatory factors of COX-2, sEH, NOS-2 and VCAM in mouse plasma was detected by Western blot; (C) Measurement of plasma IL-6, MCP-5 and TNF-α inflammatory factors in mice by ELISA. Significance: ##P < 0.01 and #P < 0.05 compared with the LPS group; **P < 0.01 and *P < 0.05 compared with the blank group.
CONCLUSIONS
As an important factor of controlling CYP metabolic pathway of arachidonic acid, sEH has been identified as a suitable target for several inflammatory diseases. In past decades, a lot of sEH inhibitors have been reported, three out of them, AR9281, GSK2256294 and EC5026, have entered clinical trials. In this work, based on a rational structure-based drug design, the picomolar level compound G1 against sEH (IC50: HsEH = 0.05 nM, MsEH = 0.14 nM) was obtained by introducing the hydrophilic group homopiperazine and hydrophobic fragment propionyl of TPPU onto the structure of lead compound A. Compound G1 showed good microsomal stability (Human t1/2 = 3.70 h, Rat t1/2 = 3.15 h), and has moderate plasma protein binding rate(72.57 %) and good oral bioavailability in rats (F % = 20.85 %). All the results pave the foundation for the assessment of in vivo efficacy. In a mouse model of arthritis (AIA) induced by Freund’s complete adjuvant (CFA), compound G1 significantly increased the pain valve (70.10 %), reduced foot swelling (thickness: 21.4 %, width: 6.98 %) and outperformed celecoxib (thickness: 17.99 %, width: 6.4 %). Compound G1 of 8 mg / kg dose ameliorated L-arginine-induced acute pancreatic injury (score: 3.50 ± 0.58 vs 6.25 ± 0.96) and reversed pancreatic injury, edema and neutrophil infiltration. Compound G1 increased the survival time of animals in the LPS-induced sepsis model and outperformed dexamethasone, while the results of Western blot or ELISA showed that the expression and levels of relevant inflammatory factors were decreased to different degrees. In preliminary safety evaluation experiment, compound G1 was administered orally at a high dose of 200 mg / kg for seven consecutive days, no adverse events were observed. It showed that compound G1 has high safety. These results suggests that compound G1 is a drug candidate worthy of further evaluation for the treatment of inflammation-induced diseases such as arthritis, acute pancreatitis and sepsis.
EXPERIMENTAL SECTION
Chemistry.
All reactions were carried out with magnetic stirring and in dried glassware. The reactions were monitored by thin-layer chromatography (TLC: HG/T2354–92, GF254), and compounds were visualized on TLC with UV light. All chemicals and solvents were purchased from commercial sources and used without purification treatment. Analytical thin-layer chromatography was carried out on 0.20 mm silica gel plates (Haiyang, Qingdao, Shandong, China) with the QF-254 UV indicator. Column chromatography was conducted using Haiyang silica gel 60 (300–400 mesh). Melting points were determined with an X-4 apparatus and were uncorrected. The nuclear magnetic resonance (NMR) spectra were recorded on a Bruker 400 MHz spectrometer in CDCl3 or DMSO-d6 using tetramethylsilane (TMS) as an internal standard. Peak multiplicity of NMR signals were as follows: s, singlet; brs, broad singlet; d, doublet; t, triplet; q, quartet; m, multiplet. Chemical shift (δ): ppm relative to Me4Si (internal standard). Coupling constant: J (Hz). High-resolution mass spectra (HRMS) of all target compounds were performed by a Waters Q-TOF Premier spectrometer with acetonitrile and water as solvents. Electrospray ionization mass spectrometry (ESI-MS) analyses were recorded in an Agilent 1100 Series MSD Trap SL (Santa Clara, CA, USA). All final compounds are >95% pure by HPLC analysis.
Methyl 3-fluoro-4-nitrobenzoate(1).
The 3-fluoro-4-nitrobenzoic acid (3.0 g, 16.2 mmol) was dissolved in MeOH (20 mL), and 98% sulfuric acid (0.86 mL, 16.2 mmol) was added. The mixture was heated to reflux 12 h. The reaction solution was cooled to room temperature and concentrated in vacuum. The resulting crude residue was dissolved in a sat. NaHCO3(aq) solution and extracted with DCM (20 mL × 2). The combined organic layers were washed with brine, dried over MgSO4, and concentrated to afford the 1 (3.06 g, 95%) as a colorless oily substance, 1H NMR (400 MHz, CDCl3): δ (ppm) 8.13–8.09 (m, 1H), 7.98–7.94 (m, 2H), 3.99 (s, 3H).
Methyl 4-amino-3-fluorobenzoate (2).
To a solution of methyl 3-fluoro-4-nitrobenzoate (3.06 g, 15.40 mmol) in anhydrous EtOH (100 mL) was added 5 % Pd/C (10% w/w). The reaction mixture was stirred with H2 at 50 °C for 12 h. After the reaction mixture was cooled to rt, the reaction mixture was filtered through a celite pad and concentrated in vacuum to give a white solid 2 (2.34 g, 90%), ESI-MS: m/z 170.04 [M+H]+.
Methyl 4-(3-((1r,3R,5S,7r)-3,5-dimethyladamantan-1-yl)ureido)-3-fluorobenzoate (3).
A solution of memantine (2.98 g, 16.60 mmol) and Et3N (5.60 g, 55.36 mol) in DCM (30 mL) was added dropwise over 1 h to a thick slurry of triphosgene (2.46 g, 8.3 mmol) in DCM (30 mL) in a cold trap while maintaining the temperature below −65 °C. The cold bath was removed, and the mixture was warmed to rt and stirred for 30 min. The reaction was cooled to −78 °C, and a slurry of methyl 4-amino-3-fluorobenzoate (2.34 g, 13.84 mmol) and Et3N (5.60 g, 55.36 mol) in DCM (30 mL) was added in small portions over 0.5 h while maintaining the temperature below −65 °C. The reaction was warmed to rt and stirred overnight. The reaction mixture was washed with 10% aq HCl (4 × 50 mL). The organic layer was dried over MgSO4 and concentrated under reduced pressure to give a crude compound 3 as a viscous yellow oil (4.14 g) which was used in the next step without further purification, 1H NMR (400 MHz, CDCl3): δ (ppm) 8.26 (t, J=8.32 Hz, 1H), 7.76 (d, J=8.72 Hz, 1H), 7.69–7.66 (m, 1H), 6.84 (s, 1H), 4.93 (s, 1H), 3.88 (s, 3H), 2.16 (s, 1H), 1.85 (s, 2H), 1.66 s, 3H), 1.40–1.29 (m, 4H), 1.16 (s, 2H), 0.85 (s, 6H).
4-(3-((1r,3R,5S,7r)-3,5-Dimethyladamantan-1-yl)ureido)-3-fluorobenzoic acid (4).
To a solution of methyl 4-(3-((1r,3R,5S,7r)-3,5-dimethyladamantan-1-yl)ureido)-3-fluorobenzoate 3 (4.14 g, 11.06 mmol), EtOH (30 mL) and water (5 mL) were added NaOH (2.2 g, 55.3 mmol). The resulting mixture was heated to 70 °C for 0.5h. After the reaction was completed, the mixture was cooled to the rt, and the solvent was evaporated in vacuum. The residues were poured into water (40 mL), and adjusted pH to 1 using concentrated hydrochloric acid at 0 °C, which was extracted with EtOAc (30 mL × 2). Then, the combined organic layers were respectively washed with water (30 mL × 2) and brine (40 mL). The organic layer was separated, dried with anhydrous MgSO4, filtered, and concentrated in vacuum produce light yellow oil (3.39 g), which was used without further purification, ESI-MS: m/z 361.2 [M+H]+.
Methyl-1-(4-(3-((1r,3R,5S,7r)-3,5-dimethyladamantan-1-yl)ureido)-3-fluorobenzoyl)piperidine-4-carboxylate (5).
In a round-bottom flask, the intermediate obtained from the previous step (3.39 g, 6.99 mmol) was dissolved in anhydrous DCM (20 mL). Methyl piperidine-4-carboxylate (1.2 g, 8.39 mmol), HATU (3.19 g, 8.39 mmol), and DIPEA (2.17 g, 16.78 mmol) were added sequentially. The reaction mixture was stirred at room temperature for 2 h. After the reaction was completed, the residues were poured into water (10 mL), which was extracted with DCM (20 mL × 2). Then, the combined organic layers were respectively washed 10% aq HCl (20 mL × 2), 10% aq Na2CO3 (20 mL × 2) and brine (20 mL). The organic layer was separated, dried with anhydrous MgSO4, filtered, and concentrated in vacuum produce light yellow oil (2.71 g), which was used without further purification, 1H NMR (400 MHz, CDCl3): δ (ppm) 8.14 (t, J = 8.12 Hz, 1H), 7.28 (d, J = 2.92 Hz, 1H), 7.07–7.03 (m, 2H), 5.59 (s, 1H), 4.15 (q, J = 7.12 Hz), 3.06 (t, 2H, J = 10.44 Hz), 2.61–2.54 (m, 1H), 2.13–2.11 (m, 1H), 1.94 (s, 2H), 1.81–1.80 (m, 2H), 1.72 (s, 2H), 1.64–1.57 (m, 4H), 1.37–1.34 (m, 2H), 1.28–1.24 (m, 6H), 1.17–1.10 (m, 2H), 0.83 (s, 6H)..
1-(4-(3-((1r,3R,5S,7r)-3,5-Dimethyladamantan-1-yl)ureido)-3-fluorobenzoyl)piperidine-4-carboxylic acid (6).
According to the synthetic process of compound 4, compound 6 as a light yellow oil (0.40 g, 29%) was obtained by taking methyl1-(4-(3-((1r,3R,5S,7r)-3,5-dimethyl adamantan-1-yl)ureido)-3-fluorobenzoyl)piperidine-4-carboxylate 5 (2.71 g, 5.58 mmol) as starting material, compound 6, ESI-MS: m/z 472.26 [M+H]+, 494.24 [M+Na]+.
1-(4-(3-((1r,3R,5S,7r)-3,5-Dimethyladamantan-1-yl)ureido)-3-fluorobenzoyl)-N,N-diethylpiperidine-4-carboxamide (A1).
According to the synthetic process of compound 5, compound A1 as a white solid (0.20 g, 60%) was obtained by taking 1-(4-(3-((1r,3R,5S,7r)-3,5-dimethyladamantan-1-yl)ureido)-3-fluorobenzoyl)piperidine-4-carboxylic acid 6 (0.3 g, 0.64 mmol) and diethylamine (55 mg, 0.76 mmol) as starting material, compound A1, m.p. 113–114°C. 1H NMR (400 MHz, CDCl3): δ (ppm) 8.15 (t, J = 8.5 Hz, 1H), 7.33 (brs, 1H), 7.07–7.05 (m, 2H), 5.65 (brs, 1H), 4.61 (brs, 1H), 3.93 (brs, 1H), 3.41–3.33 (m, 4H), 3.03–2.95 (m, 2H), 2.75–2.69 (m, 1H), 2.14–2.13 (m, 1H), 1.82 (s, 2H), 1.72 (brs, 3H), 1.64 (s, 5H), 1.39–1.36 (m, 2H), 1.29–1.23 (m, 6H), 1.16–1.09 (m, 5H), 0.82 (s, 6H). 13C NMR (100 MHz, CDCl3): δ (ppm) 173.03, 169.72, 154.14, 130.26, 130.17, 127.87, 123.27, 120.48, 113.85, 52.82, 50.04, 47.98, 42.72, 41.98, 40.56, 40.47, 38.49, 32.39, 30.17, 30.13, 28.82, 15.06, 13.10. HRMS (ESI) calcd for C30H44FN4O3 [M+H]+: 527.3319, found 527.3405.
1-(4-(3-((1r,3R,5S,7r)-3,5-Dimethyladamantan-1-yl)ureido)-3-fluorobenzoyl)-N-(2-hydroxyethyl)piperidine-4-carboxamide (A2).
According to the synthetic process of compound 5, compound A2 as a white solid (0.21 g, 65%) was obtained by taking 1-(4-(3-((1r,3R,5S,7r)-3,5-dimethyladamantan-1-yl)ureido)-3-fluorobenzoyl)piperidine-4-carboxylic acid 6 (0.3 g, 0.64 mmol) and and ethanolamine (46 mg, 0.76 mmol) as starting material, compound A2, m.p. 125–126°C. 1H NMR (400 MHz, DMSO-d6): δ (ppm) 8.30 (s, 1H), 8.20 (t, J = 8.32 Hz, 1H), 7.82 (t, J = 5.20 Hz, 1H), 7.20 (d, J = 11.6 Hz, 1H), 7.09 (d, J = 8.44 Hz, 1H), 6.58 (s, 1H), 4.65 (t, J = 5.48 Hz, 1H), 4.33 (s, 1H), 3.72 (s, 1H), 3.40–3.37 (m, 1H), 3.10 (q, J = 5.84 Hz, 2H), 2.91 (s, 2H), 2.42–2.37 (m, 1H), 2.09 (s, 1H), 1.76 (s, 2H), 1.68 (s, 2H), 1.58 (s, 4H), 1.50–1.48 (m, 2H), 1.35–1.32 (m, 2H), 1.27–1.24 (m, 2H), 1.12 (s, 2H), 0.83 s, 6H). 13C NMR (100 MHz, DMSO-d6): δ 174.32, 168.27, 153.81, 149.77, 123.80, 119.30, 114.38, 114.17, 60.33, 60.33, 52.13, 52.13, 50.71, 47.99, 47.99, 42.76, 42.76, 42.19, 41.85, 32.38, 32.38, 32.38, 32.38, 30.52,30.52, 30.52, 30.03, 30.03, 29.04. HRMS (ESI) calcd for C28H40FN4O4 [M+H]+: 515.2955, found 515.3042.
1-(4-(3-((1r,3R,5S,7r)-3,5-Dimethyladamantan-1-yl)ureido)-3-fluorobenzoyl)-N-(2-hydroxypropyl)piperidine-4-carboxamide (A3).
According to the synthetic process of compound 5, compound A3 as a white solid (0.21 g, 63%) was obtained by taking 1-(4-(3-((1r,3R,5S,7r)-3,5-dimethyladamantan-1-yl)ureido)-3-fluorobenzoyl)piperidine-4-carboxylic acid 6 (0.3 g, 0.64 mmol) and and iso-propanolamine (57 mg, 0.76 mmol) as starting material, compound A3, m.p. 115–116°C. 1H NMR (400 MHz, DMSO-d6): δ (ppm) 8.30 (s, 1H), 8.19 (t, J = 8.36 Hz, 1H), 7.63 (d, J = 7.76 Hz, 1H), 7.20 (dd, J = 11.68, J = 1.68 Hz), 7.09 (d, 1H, J = 8.44 Hz), 6.58 (s, 1H), 4.52 (s, 1H), 3.43 (s, 1H), 2.89 (s, 2H), 2.36–2.30 (m, 1H), 2.09 (s, 1H), 1.78–1.76 (m, 2H), 1.72–1.71 (m, 2H), 1.58 (s, 4H), 1.49–1.46 (m, 2H), 1.35–1.32 (m, 2H), 1.27–1.32 (m, 4H), 1.17–1.15 (m, 4H), 1.21 (s, 3H), 0.83 (s, 6H). 13C NMR (100 MHz, DMSO-d6): δ 173.42, 168.28, 153.81, 152.18, 149.77, 123.81, 119.30, 114.38, 114.17, 68.63, 52.13, 50.71, 47.99, 47.99, 47.42, 42.76, 42.76, 42.24, 42.24, 34.40, 34.40, 32.38,32.38, 32.38, 30.73, 30.52, 30.52, 30.52, 30.04, 29.00. HRMS (ESI) calcd for C29H42FN4O4 [M+H]+: 529.3112, found 529.3196.
1-((1r,3R,5S,7r)-3,5-Dimethyladamantan-1-yl)-3-(2-fluoro-4-(4-(4-(2-hydroxyethyl)piperazine-1-carbonyl)piperidine-1-carbonyl)phenyl)urea (A4).
According to the synthetic process of compound 5, compound A4 as a white solid (0.22 g, 60%) was obtained by taking 1-(4-(3-((1r,3R,5S,7r)-3,5-dimethyladamantan-1-yl)ureido)-3-fluorobenzoyl)piperidine-4-carboxylic acid 6 (0.3 g, 0.64 mmol) and and 2-(piperazin-1-yl)ethan-1-ol (98 mg, 0.76 mmol) as starting material, compound A4, m.p. 128–129°C. 1H NMR (400 MHz, DMSO-d6): δ (ppm) 8.29 (s, 1H), 8.19 (t, J = 8.24 Hz, 1H), 7.20 (d, J = 11.72 Hz, 1H), 7.08 (d, J = 8.32 Hz, 1H), 6.58 (s, 1H), 4.44 (s, 1H), 3.50–3.49 (m, 4H), 3.43 (s, 2H), 2.92 (s, 2H), 2.40–2.37 (m, 4H), 2.33 (s, 2H), 2.09 (s, 1H), 1.76 (s, 2H), 1.58 (s, 6H), 1.49–1.46 (m, 3H), 1.35–1.32 (m, 2H), 1.27–1.24 (m, 3H), 1.12 (s, 2H), 1.05 (s, 2H), 0.83 (s, 6H). 13C NMR (100 MHz, DMSO-d6): δ 172.43, 168.26, 153.82, 152.19, 149.78, 123.82, 119.28, 114.42, 114.21, 60.54, 58.93, 58.93, 54.20, 53.43, 52.13, 50.71, 47.99, 47.99, 46.15, 45.28, 42.76, 42.76, 41.60, 37.41, 32.39, 32.39, 32.39, 30.52, 30.52, 30.52, 30.03, 28.88. HRMS (ESI) calcd for C32H47FN5O4 [M+H]+: 584.3534, found 584.3618.
1-(4-(3-((1r,3R,5S,7r)-3,5-Dimethyladamantan-1-yl)ureido)-3-fluorobenzoyl)-N-(1-hydroxypropan-2-yl)piperidine-4-carboxamide (A5).
According to the synthetic process of compound 5, compound A5 as a white solid (0.18 g, 54 %) was obtained by taking 1-(4-(3-((1r,3R,5S,7r)-3,5-dimethyladamantan-1-yl)ureido)-3-fluorobenzoyl) piperidine-4-carboxylic acid 6 (0.3 g, 0.64 mmol) and and 2-aminopropan-1-ol (57 mg, 0.76 mmol) as starting material, compound A5, m.p. 116–117°C. 1H NMR (400 MHz, DMSO-d6): δ (ppm) 8.36 (s, 1H), 8.20 (t, J = 8.32 Hz, 1H), 7.63 (d, J = 8.00 Hz, 1H), 7.21 (d, J = 11.68 Hz, 1H), 7.09 (d, J = 8.68 Hz, 1H), 6.67 (s, 1H), 4.62 (t, J = 5.44 Hz, 2H), 3.71–3.66 (m, 1H), 3.40–3.39 (m, 2H), 3.08–3.06 (m, 4H), 2.10 (s, 1H), 1.76 (s, 1H), 1.68 (s, 1H), 1.59 (s, 3H), 1.50–1.48 (m, 2H), 1.41–1.35 (m, 1H), 1.32 (s, 1H), 1.28–1.24 (m, 3H), 1.21–1.18 (m, 5H), 1.12 (s, 2H), 0.83 (s, 6H). 13C NMR (100 MHz, DMSO-d6): δ 174.24, 168.29, 153.86, 152.19, 149.79, 123.80, 119.35, 114.38, 114.17, 60.59, 60.59, 53.16, 52.13, 50.72, 47.99, 47.99, 45.87, 45.87, 42.77, 42.19, 32.38, 32.38, 32.38, 30.53, 30.53, 30.53, 30.03, 29.03, 8.94. HRMS (ESI) calcd for C29H42FN4O5 [M+H]+: 545.3061, found 545.3148.
Ethyl-1-(4-(3-((1r,3R,5S,7r)-3,5-dimethyladamantan-1-yl)ureido)-3-fluorobenzoyl) piperidine-3-carboxylate (7).
According to the synthetic process of compound 5, compound 7 as a light yellow oil (3.33 g) was obtained by taking 4-(3-((1r,3R,5S,7r)-3,5-dimethyladamantan-1-yl)ureido)-3-fluorobenzoic acid 4 (3 g, 8.33 mmol) and ethyl piperidine-3-carboxylate (1.57g, 10.0 mmol) as starting material, compound 7, ESI-MS: m/z 500.4 [M+H]+, 522.4 [M+Na]+.
1-(4-(3-((1r,3R,5S,7r)-3,5-Dimethyladamantan-1-yl)ureido)-3-fluorobenzoyl)piperidine-3-carboxylic acid (A).
According to the synthetic process of compound 4, compound A as a white solid (2.82 g, 90%) was obtained by taking ethyl 1-(4-(3-((1r,3R,5S,7r)-3,5-dimethyladamantan-1-yl)ureido)-3-fluorobenzoyl)piperidine-3-carboxylate 7 (3.33 g, 6.66 mmol) as starting material, compound A, m.p.154–155 °C. 1H NMR (400 MHz, CDCl3): δ (ppm) 8.11 (t, J = 7.76 Hz, 1H), 7.72–7.63 (m, 1H), 7.03–7.00 (m, 2H), 4.48 (s, 1H), 3.93–3.69 (m, 2H), 3.39–3.33 (m, 2H), 2.50 (s, 1H), 2.12–2.03 (m, 2H), 1.83 (s, 3H), 1.67–1.59 (m, 4H), 1.37–1.26 (m, 4H), 1.17–1.10 (m, 2H), 0.83 (s, 6H). 13C NMR (100 MHz, DMSO-d6): δ 182.7, 167.8, 167.5, 156.9, 144.4, 130.3, 130.2, 129.8, 127.9, 52.3, 51.5, 50.8, 48.6, 47.3, 46.9, 42.9, 40.9, 32.9, 32.3, 31.9, 30.6, 30.1. HRMS (ESI) calcd for C26H35FN3O4 [M+H]+: 472.2533, found 472.2615.
1-(4-(3-((1r,3R,5S,7S)-3,5-Dimethyladamantan-1-yl)ureido)-3-fluorobenzoyl)-N,N-diethylpiperidine-3-carboxamide (B1).
According to the synthetic process of compound 5, compound B1 as a white solid (0.19 g, 58%) was obtained by taking 1-(4-(3-((1r,3R,5S,7r)-3,5-dimethyladamantan-1-yl)ureido)-3-fluorobenzoyl)piperidine-3-carboxylic acid A (0.3 g, 0.64 mmol) and diethylamine (55 mg, 0.76 mmol) as starting material, compound B1, m.p. 137–138°C. 1H NMR (400 MHz, CDCl3): δ (ppm) 8.20 (t, J = 8.0 Hz, 1H), 7.57–7.54 (m, 1H), 7.07–6.99 (m, 2H), 5.63 (brs, 1H), 4.63 (brs, 1H), 3.81 (s, 1H), 3.48–3.35 (m, 4H), 3.18–2.99 (m, 2H), 2.73–2.56 (m, 1H), 2.13 (s, 1H), 1.91 (s, 2H), 1.84 (s, 3H), 1.63 (s, 4H), 1.38–1.35 (m, 2H), 1.29–1.24 (m, 4H), 1.18–1.10 (m, 7H), 0.83 (s, 6H). 13C NMR (100 MHz, CDCl3): δ (ppm) 172.26, 169.89, 154.13, 136.38, 130.30, 127.80, 123.48, 120.02, 113.97, 52.72, 50.62, 48.01, 42.72, 42.02, 40.61, 40.38, 32.37, 30.13, 14.95, 13.06. HRMS (ESI) calcd for C30H44FN4O3 [M+H]+: 527.3319, found 527.3404.
1-(4-(3-((1r,3R,5S,7S)-3,5-dimethyladamantan-1-yl)ureido)-3-fluorobenzoyl)-N-(2-hydroxyethyl)piperidine-3-carboxamide (B2).
According to the synthetic process of compound 5, compound B2 as a white solid (0.20 g, 61.90%) was obtained by taking 1-(4-(3-((1r,3R,5S,7r)-3,5-Dimethyladamantan-1-yl)ureido)-3-fluorobenzoyl) piperidine-3-carboxylic acid A (0.3 g, 0.64 mmol) and ethanolamine (46 mg, 0.76 mmol) as starting material, compound B2, m.p. 128–129°C. 1H NMR (400 MHz, DMSO-d6): δ (ppm) 8.30 (s, 1H), 8.20 (t, J = 8.36 Hz, 1H), 7.91 (s, 1H), 7.20 (dd, J = 11.68 Hz, J = 1.56 Hz,), 7.08 (d, J = 8.44 Hz, 1H), 6.58 (s, 1H), 4.66 (s, 1H), 4.34 (s, 1H), 3.62 (s, 1H), 3.36 (s, 2H), 3.09 (s, 2H), 2.89 (s, 2H), 2.36–2.30 (m, 1H), 2.09 (s, 1H), 1.87–1.84 (m, 1H), 1.76 (s, 2H), 1.62–1.55 (m, 5H), 1,35–1.32 (m, 3H), 1.27–1.24 (m, 3H), 1.12 (s, 2H), 0.83 (s, 6H). 13C NMR (100 MHz, DMSO-d6): δ173.03, 168.33, 153.81, 152.15, 149.75, 123.84, 119.24, 114.44, 114.24, 60.25, 60.25, 52.14, 50.71, 47.99, 47.99, 42.76, 42.76, 41.81, 32.39, 32.39, 32.39, 30.52, 30.52,30.52, 30.04, 30.04, 28.19. HRMS (ESI) calcd for C28H40FN4O4 [M+H]+: 515.2955, found 515.3041.
1-(4-(3-((1r,3R,5S,7S)-3,5-Dimethyladamantan-1-yl)ureido)-3-fluorobenzoyl)-N-(2-hydroxypropyl)piperidine-3-carboxamide (B3).
According to the synthetic process of compound 5, compound B3 as a white solid (0.22 g, 66%) was obtained by taking 1-(4-(3-((1r,3R,5S,7r)-3,5-dimethyladamantan-1-yl)ureido)-3-fluorobenzoyl)piperidine-3-carboxylic acid A (0.3 g, 0.64 mmol) and iso-propanolamine (57 mg, 0.76 mmol) as starting material, compound B3, m.p.141–142 °C. 1H NMR (400 MHz, CDCl3): δ (ppm) 8.20 (s, 1H), 7.61 (s, 1H), 7.05–7.03 (m, 2H), 5.85 (br, 1H), 4.05 (br, 1H), 3.86 (brs, 1H), 3.55 (brs, 4H), 3.38–3.35 (m, 1H), 3.29–3.20 (m, 1H), 3.2–2.95 (m, 1H), 2.81 (brs, 1H), 2.48 (brs, 1H), 2.13 (brs, 1H), 2.06–2.02 (m, 1H), 1.88–1.84 (m, 1H), 1.81 (s, 2H), 1.62 (s, 5H), 1.44–1.43 (m, 1H), 137–1.34 (m, 2H), 1.30–1.24 (m, 3H), 1.44–1.11 (m, 5H), 0.84 (m, 6H). 13C NMR (100 MHz, CDCl3): δ (ppm) 173.03, 169.72, 154.14, 130.26, 130.17, 127.87, 123.27, 120.48, 113.85, 52.82, 50.04, 47.98, 42.72, 41.98, 40.56, 40.47, 38.49, 32.39, 30.17, 30.13, 28.82, 15.06, 13.10. HRMS (ESI) calcd for C29H42FN4O4 [M+H]+: 529.3112, found 529.3198.
1-((1r,3R,5S,7r)-3,5-Dimethyladamantan-1-yl)-3-(2-fluoro-4-(3-(4-(2-hydroxyethyl) piperazine-1-carbonyl)piperidine-1-carbonyl)phenyl)urea (B4).
According to the synthetic process of compound 5, compound B4 as a white solid (0.22 g, 54.5%) was obtained by taking 1-(4-(3-((1r,3R,5S,7r)-3,5-dimethyladamantan-1-yl)ureido)-3-fluorobenzoyl)piperidine-3-carboxylic acid A (0.3 g, 0.64 mmol) and 2-(piperazin-1-yl)ethan-1-ol (98 mg, 0.76 mmol) as starting material, compound B4, m.p. 141–145°C. 1H NMR (400 MHz, DMSO-d6): δ (ppm) 8.30 (s, 1H), 8.20 (t, J = 8.36 Hz, 1H), 7.24 (d, J=11.80 Hz, 1H), 7.11 (d, J= 8.52 Hz, 1H), 6.58 (s, 1H), 4.43 (s, 1H), 4.32 (s, 1H), 3.49 (s, 6H), 3.05 (s, 1H), 2.79 (s, 2H), 2.38 (s, 5H), 2.09 (s, 1H), 1.82–1.76 (m, 3H), 1.58 (s, 8H), 1.35–1.32 (m, 2H), 1.27–1.24 (m, 3H), 1.18–1.12 (m, 3H), 0.83 (s, 6H). 13C NMR (100 MHz, DMSO-d6): δ 168.44, 153.81, 152.16, 149.76, 123.75, 119.34, 114.31, 114.11, 60.44, 58.79, 54.04, 53.29, 52.13, 52.13, 50.71, 47.98, 47.98, 45.19, 42.76, 42.76, 41.39, 38.29, 32.38, 32.38, 32.38, 32.38, 30.52, 30.52, 30.52, 30.04, 30.04, 27.98. HRMS (ESI) calcd for C32H47FN5O4 [M+H]+: 584.3534, found 584.3619.
Methyl-4-(4-(3-((1r,3R,5S,7r)-3,5-dimethyladamantan-1-yl)ureido)-3-fluorobenzamido)cyclohexane-1-carboxylate (8).
According to the synthetic process of compound 5, compound 8 as a light yellow oil (3.54 g) was obtained by taking 4-(3-((1r,3R,5S,7r)-3,5-dimethyladamantan-1-yl)ureido)-3-fluorobenzoic acid 4 (3 g, 8.33 mmol) and methyl 4-aminocyclohexane-1-carboxylate (1.57g, 10.0 mmol) as starting material, compound 8, 1H NMR (400 MHz, DMSO-d6): δ (ppm) 8.35 (s, 1H), 8.23 (t, J = 8.52 Hz, 1H), 8.11 (d, J = 7.76 Hz, 1H), 7.66–7.59 (m, 2H), 6.60 (s, 1H), 3.76–3.68 (m, 1H), 3.61 (s, 3H), 2.11–2.10 (m, 1H), 1.97–1.94 (m, 2H), 1.89–1.86 (m, 2H), 1.76 (s, 2H), 1.59 (s, 4H), 1.45–1.41 (m, 2H), 1.36–1.33 (m, 3H), 1.28–1.24 (m, 4H), 1.13 (s, 2H), 0.84 (s, 6H).
4-(4-(3-((1r,3R,5S,7r)-3,5-Dimethyladamantan-1-yl)ureido)-3-fluorobenzamido) cyclohexane-1-carboxylic acid (9).
According to the synthetic process of compound 4, compound 9 as a light yellow oil (3.10 g) was obtained by taking methyl-4-(4-(3-((1r,3R,5S,7r)-3,5-dimethyladamantan-1-yl)ureido)-3-fluorobenzamido)cyclohexane-1-carboxylate 8 (3.54 g, 7.09 mmol) as starting material, compound 9, which was used without further purification, 1H NMR (400 MHz, DMSO-d6): δ (ppm) 8.35 (s, 1H), 8.23 (t, J = 8.52 Hz, 1H), 8.11 (d, J = 7.76 Hz, 1H), 7.67–7.59 (m, 2H), 6.60 (s, 1H), 3.76–3.68 (m, 1H), 3.61 (s, 3H), 2.16–2.07 (m, 1H), 1.96–1.94 (m, 2H), 1.88–1.86 (m, 2H), 1.76 (s, 2H), 1.59 (s, 4H), 1.41–1.33 (m, 5H), 1.28–1.24 (m, 4H), 1.12 (s, 2H), 0.83 (s, 6H).
Tert-butyl-4-(2-(4-(4-(3-((1r,3R,5S,7r)-3,5-dimethyladamantan-1-yl)ureido)-3-fluorobenzamido)cyclohexane-1-carboxamido)ethyl)piperazine-1-carboxylate (C1).
According to the synthetic process of compound 5, compound C1 as a white solid (0.26 g, 60.0%) was obtained by taking 4-(4-(3-((1r,3R,5S,7r)-3,5-dimethyladamantan-1-yl)ureido)-3-fluorobenzamido)cyclohexane-1-carboxylic acid 9 (0.3 g, 0.64 mmol) and tert-butyl 4-(2-aminoethyl)piperazine-1-carboxylate (0.17 g, 0.76 mmol) as starting material, compound C1, m.p. 131–132°C. 1H NMR (400 MHz, DMSO-d6) δ 8.36 (s, 1H), 8.23 (t, J = 8.52 Hz, 1H), 8.09 (d, J = 7.88 Hz, 1H), 7.68–7.59 (m, 3H), 6.62 (s, 1H), 3.75–3.66 (m, 1H), 3.29 (s, 3H), 3.16 (s, 2H), 2.33 (s, 5H), 2.10–2.06 (m, 2H), 1.87–1.85 (m, 2H), 1.76 (s, 4H), 1.59 (s, 4H), 1.47–1.45 (m, 1H), 1.39 (s, 10H), 1.34–1.32 (m, 4H), 1.28 (s, 2H), 1.25–1.23 (m, 2H), 1.12 (s, 2H), 0.83 (s, 6H). 13C NMR (100 MHz, DMSO-d6): δ 175.21, 164.27, 154.25, 153.69, 152.09, 149.70, 124.04, 118.47, 114.22, 114.01, 79.21, 57.44, 52.91, 52.16, 52.16, 50.70, 48.38, 47.98, 47.98, 46.12, 43.72, 43.72, 42.76, 42.76, 36.45, 32.39, 32.39, 31.98, 31.98, 30.51, 30.51, 30.04, 30.04, 28.83, 28.83, 28.52, 28.52, 28.52. HRMS (ESI) calcd for C38H58FN6O5 [M+H]+: 697.4374, found 697.4460.
4-(3-((1r,3R,5S,7r)-3,5-Dimethyladamantan-1-yl)ureido)-3-fluoro-N-(4-(4-(2-hydroxyethyl)piperidine-1-carbonyl)cyclohexyl)benzamide (C2).
According to the synthetic process of compound 5, compound C2 as a white solid (0.22 g, 58 %) was obtained by taking 4-(4-(3-((1r,3R,5S,7r)-3,5-dimethyladamantan-1-yl)ureido)-3-fluorobenzamido)cyclohexane-1-carboxylic acid 9 (0.3 g, 0.64 mmol) and 2-(piperidin-4-yl)ethan-1-ol (98 mg, 0.76 mmol) as starting material, compound C2, m.p. 126–127 °C. 1H NMR (400 MHz, DMSO-d6) δ 8.35 (s, 1H), 8.23 (t, J = 8.4 Hz, 1H), 8.13 (d, J = 7.56 Hz, 1H), 7.66–7.60 (m, 2H), 6.61 (s, 1H), 4.44 (s, 1H), 3.71 (s, 1H), 3.53–3.44 (m, 6H), 2.42–2.41 (m, 4H), 2.35 (s, 2H), 2.10 (s, 1H), 1.87–1.85 (m, 2H), 1.76–1.69 (m, 4H), 1.59 (s, 4H), 1.46 (s, 1H), 1.43 (s, 1H), 1.40 (s, 1H), 1.35 (s, 1H), 1.32 (s, 2H), 1.28–1.23 (m, 4H), 1.12 (s, 2H), 0.83 (s, 6H). 13C NMR (100 MHz, DMSO-d6): δ 173.30, 164.38, 153.69, 152.07, 149.68, 124.15, 118.46, 114.26, 114.05, 60.54, 58.90, 54.26, 53.45, 52.16, 52.16, 50.70, 48.70, 47.98, 47.98, 45.29, 42.75, 42.75, 41.50, 38.71, 32.39, 32.39, 31.83, 31.83, 30.51, 30.51, 30.39, 30.39, 28.68. HRMS (ESI) calcd for C34H50FN4O4 [M+H]+: 597.3738, found 597.3775.
4-(3-((1r,3R,5S,7r)-3,5-Dimethyladamantan-1-yl)ureido)-3-fluoro-N-(4-((2-hydroxyethyl)carbamoyl)cyclohexyl)benzamide (C3).
According to the synthetic process of compound 5, compound C3 as a white solid (0.20 g, 61 %) was obtained by taking 4-(4-(3-((1r,3R,5S,7r)-3,5-dimethyladamantan-1-yl)ureido)-3-fluorobenzamido) cyclohexane-1-carboxylic acid 9 (0.3 g, 0.64 mmol) and ethanolamine (46 mg, 0.76 mmol) as starting material, compound C3, m.p. 117–118 °C. 1H NMR (400 MHz, DMSO-d6) δ 8.38 (s, 1H), 8.23 (t, J = 8.40 Hz, 1H), 8.09 (d, J = 7.8 Hz, 1H), 7.73 (t, J = 5.32 Hz, 1H), 7.66–7.59 (m, 2H), 6.64 (s, 1H), 4.64 (s, 1H), 3.71–3.69 (m, 1H), 3.24 (s, 1H), 3.12–3.08 (m, 2H), 3.01–3.00 (m, 1H), 2.09 (s, 2H), 1.87–1.84 (m, 2H), 1.76 (s, 4H), 1.59 (s, 4H), 1.47–1.41 (m, 2H), 1.37–1.32 (m, 4H), 1.28–1.25 (m, 2H), 1.12 (s, 2H), 0.83 (s, 6H). 13C NMR (100 MHz, DMSO-d6): δ 175.40, 164.27, 153.71, 152.09, 149.70, 124.07, 118.50, 114.21, 114.01, 60.44, 60.44, 52.16, 52.16, 50.70, 48.36, 47.98, 47.98, 46.01, 43.70, 42.75, 41.80, 32.39, 32.39, 32.00, 30.51, 30.51, 30.04, 28.86, 28.86. HRMS (ESI) calcd for C29H42FN4O4 [M+H]+: 529.3112, found 529.3198.
4-(3-((1r,3R,5S,7r)-3,5-Dimethyladamantan-1-yl)ureido)-3-fluoro-N-(4-((2-hydroxypropyl)carbamoyl)cyclohexyl)benzamide (C4).
According to the synthetic process of compound 5, compound C4 as a white solid (0.20 g, 59 %) was obtained by taking 4-(4-(3-((1r,3R,5S,7r)-3,5-dimethyladamantan-1-yl)ureido)-3-fluorobenzamido) cyclohexane-1-carboxylic acid 9 (0.3 g, 0.64 mmol) and iso-propanolamine (57 mg, 0.76 mmol) as starting material, compound C4, m.p. 123–124 °C. 1H NMR (400 MHz, DMSO-d6) δ 8.35 (d, J = 2.96 Hz, 1H), 8.23 (t, J = 8.52 Hz, 1H), 8.09 (d, J = 7.92 Hz, 1H), 7.69–7.59 (m, 3H), 6.60 (s, 1H), 4.63 (s, 1H), 3.74–3.67 (m, 1H), 3.64–3.59 (m, 1H), 2.98 (t, J = 5.88 Hz, 2H), 2.13–2.09 (m, 2H), 1.87–1.84 (m, 2H), 1.76 (s, 3H), 1.59 (s, 3H), 1.48–1.42 (m, 2H), 1.35–1.34 (m, 1H), 1.32 (s, 2H), 1.28 (s, 2H), 1.23 (s, 2H), 1.17 (s, 1H), 1.12 (s, 2H), 1.00 (d, J = 6.2 Hz, 3H), 0.83 (s, 6H). 13C NMR (100 MHz, DMSO-d6): δ 175.40, 164.27, 153.69, 152.10, 149.71, 124.04, 118.50, 114.22, 114.01, 65.75, 65.75, 52.17, 52.17, 50.71, 48.38, 47.99, 47.99, 46.67, 43.68, 42.76, 32.39, 32.39, 32.00, 30.51, 30.51, 30.05, 28.93, 28.84, 21.51. HRMS (ESI) calcd for C30H44FN4O4 [M+H]+: 543.3268, found 543.3353.
N-(4-(Bis(2-hydroxyethyl)carbamoyl)cyclohexyl)-4-(3-((1r,3R,5S,7r)-3,5-dimethyladamantan-1-yl)ureido)-3-fluorobenzamide (C5).
According to the synthetic process of compound 5, compound C5 as a white solid (0.22 g, 62 %) was obtained by taking 4-(4-(3-((1r,3R,5S,7r)-3,5-dimethyladamantan-1-yl)ureido)-3-fluorobenzamido) cyclohexane-1-carboxylic acid 9 (0.3 g, 0.64 mmol) and 2,2’-azanediylbis(ethan-1-ol) (80 mg, 0.76 mmol) as starting material, compound C5, m.p. 104–105 °C. 1H NMR (400 MHz, DMSO-d6) δ 8.38 (d, J = 2.72 Hz, 1H), 8.23 (t, J = 8.52 Hz, 1H), 8.11 (d, J = 7.68 Hz, 1H), 7.66–7.59 (m, 2H), 6.63 (s, 1H), 4.88 (s, 1H), 4.67 (s, 1H), 3.71–3.69 (m, 1H), 3.52–3.51 (m, 2H), 3.44–3.43 (m, 4H),3.34–3.32 (m, 1H),2.61–2.55 (m, 1H), 2.10 (s, 1H), 1.88–1.85 (m, 2H), 1.76–1.70 (m, 4H), 1.59 (s, 4H), 1.48–1.44 (m, 2H), 1.41–1.38 (m, 2H), 1.35–1.32 (m, 3H), 1.28–1.25 (m, 2H), 1.12 (s, 2H), 0.83 (s, 6H). 13C NMR (100 MHz, DMSO-d6): δ 175.61, 164.37, 153.70, 152.09, 149.69, 124.09, 118.51, 114.24, 114.03, 59.81, 59.40, 52.16, 50.89, 50.70, 48.76, 48.66, 47.98, 47.98, 42.76, 39.12, 32.39, 32.39, 32.39, 31.89, 30.51, 30.51, 30.51, 30.04, 28.91, 28.91. HRMS (ESI) calcd for C31H46FN4O5 [M+H]+: 573.3374, found 573.3458.
N-(4-Carbamoylcyclohexyl)-4-(3-((1r,3R,5S,7r)-3,5-dimethyladamantan-1-yl)ureido)-3-fluorobenzamide (C6).
According to the synthetic process of compound 5, compound C6 as a white solid (0.17 g, 62 %) was obtained by taking 4-(4-(3-((1r,3R,5S,7r)-3,5-dimethyladamantan-1-yl)ureido)-3-fluorobenzamido)cyclohexane-1-carboxylic acid 9 (0.3 g, 0.64 mmol) and ammonium hydroxide (88 mg, 0.76 mmol) as starting material, compound C6, m.p. 107–108°C. 1H NMR (400 MHz, DMSO-d6) δ 8.53 (s, 1H), 8.22 (t, J = 8.12 Hz, 1H), 8.15 (d, J = 7.64 Hz, 1H), 7.68–7.60 (m, 2H), 7.26 (s, 1H), 6.88 (s, 1H), 6.70 (s, 1H), 3.69 (s, 1H), 3.05 (q, J = 6.84 Hz, 4H), 2.69 (s, 1H), 2.09 (s, 1H), 1.77 (s, 3H), 1.59 (s, 3H), 1.45–1.35 (m, 4H), 1.32 (s, 2H), 1.27 (s, 2H), 1.12 (s, 2H), 0.83 (s, 6H). 13C NMR (100 MHz, DMSO-d6): δ 177.47, 164.29, 153.82, 152.11, 149.71, 124.07, 118.57, 114.21, 114.02, 52.15, 50.72, 48.41, 47.99, 45.76, 45.76, 43.46, 42.77, 38.71, 32.38, 32.38, 32.00, 30.53, 30.53, 30.03, 28.79, 8.89, 8.89. HRMS (ESI) calcd for C27H38FN4O3 [M+H]+: 485.2850, found 485.3297.
N-(4-((1,3-Dihydroxypropan-2-yl)carbamoyl)cyclohexyl)-4-(3-((1r,3R,5S,7r)-3,5-dimethyladamantan-1-yl)ureido)-3-fluorobenzamide (C7).
According to the synthetic process of compound 5, compound C7 as a white solid (0.20 g, 56 %) was obtained by taking 4-(4-(3-((1r,3R,5S,7r)-3,5-dimethyladamantan-1-yl)ureido)-3-fluorobenzamido) cyclohexane-1-carboxylic acid 9 (0.3 g, 0.64 mmol) and 2-aminopropane-1,3-diol (69 mg, 0.76 mmol) as starting material, compound C7, m.p. 110–111 °C. 1H NMR (400 MHz, DMSO-d6) δ 8.38 (s, 1H), 8.22 (t, J = 8.48 Hz, 1H), 8.09 (d, J = 7.88 Hz, 1H), 7.65–7.59 (m, 2H), 7.42 (d, J = 8.08 Hz, 1H), 6.64 (s, 1H), 4.62 (s, 2H), 3.71–3.66 (m, 2H), 3.39–3.38 (m, 4H), 2.10 (s, 1H), 1.86–1.84 (m, 2H), 1.76 (s, 3H), 1.59 (s, 4H), 1.47–1.41 (m, 2H),1.37–1.32 (m, 4H), 1.28 (s, 2H), 1.25 (s, 2H), 1.12 (s, 2H), 0.83 (s, 6H). 13C NMR (100 MHz, DMSO-d6): δ 175.33, 164.23, 153.72, 152.10, 149.71, 124.05, 118.52, 114.21, 114.01, 60.67, 60.67, 53.04, 52.16, 52.16, 50.70, 48.37, 47.98, 47.98, 43.66, 42.76, 32.39, 32.39, 32.39, 32.00, 30.52, 30.52, 30.04, 28.92, 28.92. HRMS (ESI) calcd for C30H44FN4O5 [M+H]+: 559.3217, found 559.3304.
N-(4-((1,3-Dihydroxy-2-(hydroxymethyl)propan-2-yl)carbamoyl)cyclohexyl)-4-(3-((1r,3R,5S,7r)-3,5-dimethyladamantan-1-yl)ureido)-3-fluorobenzamide (C8).
According to the synthetic process of compound 5, compound C8 as a white solid (0.22 g, 60 %) was obtained by taking 4-(4-(3-((1r,3R,5S,7r)-3,5-dimethyladamantan-1-yl)ureido)-3-fluorobenzamido)cyclohexane-1-carboxylic acid 9 (0.3 g, 0.64 mmol) and 2-amino-2-(hydroxymethyl)propane-1,3-diol (92 mg, 0.76 mmol) as starting material, compound C8, m.p. 121–122 °C. 1H NMR (400 MHz, DMSO-d6) δ 8.53 (s, 1H), 8.22 (t, J = 8.40 Hz, 1H), 8.14 (d, J = 7.80 Hz, 1H), 7.67–7.60 (m, 2H), 7.29 (s, 1H), 6.89 (s, 1H), 4.88 (s, 3H), 3.70 (s, 1H), 3.52 (s, 5H), 3.17 (d, J = 5.16 Hz, 1H), 2.26 (s, 1H), 2.09 (s, 1H), 1.85–1.77 (m, 6H), 1.59 (s, 3H), 1.46–1.40 (m, 2H),1.36–1.32 (m, 4H), 1.27–1.25 (m, 3H), 1.12 (s, 2H), 0.83 (s, 6H). 13C NMR (100 MHz, DMSO-d6): δ 177.09, 164.28, 153.84, 152.11, 149.72, 124.08, 118.59, 114.21, 114.00, 62.62, 61.27, 61.27, 52.15, 52.15, 50.73, 49.04, 48.35, 47.99, 47.99, 43.86, 42.77, 38.71, 32.38, 32.38, 32.38, 31.89, 30.53, 30.53, 30.53, 30.03, 28.98. HRMS (ESI) calcd for C31H46FN4O6 [M+H]+: 589.3323, found 589.3411.
(3-Fluoro-4-nitrophenyl)methanol (10).
To a solution of 3-fluoro-4-nitrobenzoic acid (10 g, 54.05 mmol), THF (60 mL) was added BH3 (2.2 g, 55.3 mmol) slowly dropwise at 0 °C. Transfer to room temperature and continue the reaction for 12h. After the reaction was completed, the reaction solution was lowered to 0°C and MeOH (100 mL) was slowly added. The reaction solution is concentrated under vacuum to produce a light yellow oil 10 (7.86 g), which is used without further purification, ESI-MS: m/z 194.1[M+H]+.
4-(Bromomethyl)-2-fluoro-1-nitrobenzene (11).
To a solution of (3-fluoro-4-nitrophenyl)methanol 10 (7.86 g, 45.96 mmol), DCM (50 mL) was added PBr3 (14.93 g, 55.15 mmol) slowly dropwise at 0 °C. Transfer to room temperature and continue the reaction for 12h. After the reaction was completed, the reaction mixture was washed with sat. NaHCO3 (3 × 30 mL). The combined organic layer was dried over MgSO4 and concentrated under reduced pressure to give a crude compound 11 as a viscous yellow oil (8.57 g) which was used in the next step without further purification, ESI-MS: m/z 233.2[M+H]+.
Ethyl-1-(3-fluoro-4-nitrobenzyl)piperidine-3-carboxylate (12).
To a solution of 4-(bromomethyl)-2-fluoro-1-nitrobenzene 11 (8.57 g, 36.77 mmol), acetonitrile (40 mL) were added ethyl piperidine-3-carboxylate (6.93 g, 44.12 mmol), K2CO3 (7.6 g, 55.2 mmol), KI (1.2 g, 7.35 mmol). The resulting mixture was heated to 70 °C for 8 h. After the reaction was completed, the mixture was cooled to the rt, and the solvent was evaporated in vacuum. The residues were poured into water (30 mL), which was extracted with EtOAc (40 mL × 2). Then, the combined organic layers were respectively washed with water (30 mL × 2) and brine (30 mL). The organic layer was separated, dried with anhydrous MgSO4, filtered, and concentrated in vacuum produce light yellow oil 12 (7.98 g), which was used without further purification, ESI-MS: m/z 311.1 [M+H]+.
Ethyl-1-(4-amino-3-fluorobenzyl)piperidine-3-carboxylate (13).
According to the synthetic process of compound 2, compound 13 as a light yellow oil (6.48 g) was obtained by taking ethyl 1-(3-fluoro-4-nitrobenzyl)piperidine-3-carboxylate 12 (7.98 g, 25.73 mmol) as starting material, compound 13, which was used without further purification, ESI-MS: m/z 281.1 [M+H]+.
Ethyl-1-(4-(3-((1r,3R,5S,7r)-3,5-dimethyladamantan-1-yl)ureido)-3-fluorobenzyl) piperidine-3-carboxylate (14).
According to the synthetic process of compound 3, compound 14 as a light yellow oil (7.86 g) was obtained by taking ethyl 1-(4-amino-3-fluorobenzyl)piperidine-3-carboxylate 13 (6.48 g, 23.13 mmol) as starting material, compound 14, which was used without further purification, ESI-MS: m/z 486.33 [M+H]+.
1-(4-(3-((1r,3R,5S,7r)-3,5-Dimethyladamantan-1-yl)ureido)-3-fluorobenzyl) piperidine-3-carboxylic acid (D).
According to the synthetic process of compound 4, compound D as a white solid (5.92 g, 70%) was obtained by taking ethyl-1-(4-(3-((1r,3R,5S,7r)-3,5-dimethyladamantan-1-yl)ureido)-3-fluorobenzyl)piperidine-3-carboxylate 14 (7.86 g, 16.20 mmol) as starting material, compound D, m.p. 211–212 °C. 1H NMR (400 MHz, DMSO-d6): δ (ppm) 11.06 (s, 1H), 8.38 (s, 1H), 8.20 (t, J = 8.42 Hz, 1H), 7.52 (d, J = 12.08 Hz, 1H), 7.22 (d, J = 8.24 Hz, 1H), 6.72 (s, 1H), 4.20 (s, 2H), 3.35–3.24 (m, 4H), 2.96–2.94 (m, 3H), 2.09–1.76 (m, 6H), 1.61–1.55 (m, 3H), 1.35–1.24 (m, 4H), 1.11 (s, 2H), 0.83 (s, 6H). 13C NMR (100 MHz, DMSO-d6): δ 173.7, 154.1, 152.6, 129.7, 127.4, 119.8, 117.6, 59.2, 52.8, 52.4, 52.1, 51.5, 50.8, 48.5, 48.2, 48.1, 46.3, 42.9, 42.8, 41.9, 32.4, 30.5, 30.1, 25.7, 22.2. HRMS (ESI) calcd for C26H37FN3O3 [M+H]+: 458.2741, found 458.2823.
1-(4-(3-((1r,3R,5S,7r)-3,5-Dimethyladamantan-1-yl)ureido)-3-fluorobenzyl)piperidine −3-carboxamide (D1).
According to the synthetic process of compound 5, compound D1 as a white solid (0.18 g, 60 %) was obtained by taking 1-(4-(3-((1r,3R,5S,7r)-3,5-dimethyladamantan-1-yl)ureido)-3-fluorobenzyl)piperidine-3-carboxylic acid D (0.3 g, 0.66 mmol) and aqueous ammonia (92 mg, 0.79 mmol) as starting material, compound D1, m.p. 87–88°C. 1H NMR (400 MHz, CDCl3): δ (ppm) 8.27–8.17 (m, 1H), 7.81–7.56 (m, 1H), 7.04–7.00 (m, 2H), 3.83–3.46 (m, 8H), 2.43–2.35 (m, 2H), 2.12–2.11 (m, 1H), 2.01 (s, 1H), 1.79 (s, 2H), 1.62–1.61 (m, 5H), 1.37–1.33 (m, 2H), 1.29–1.26 (m, 3H), 1.18–1.10 (m, 5H), 0.83 (s, 6H). 13C NMR (100 MHz, DMSO-d6): δ (ppm) 173.7, 170.9, 154.1, 153.9, 130.2, 128.1, 122.7, 120.0, 113.4, 52.7, 52.6, 50.8, 50.6, 48.0, 46.9, 46.1, 42.7, 40.6, 40.6, 32.4, 30.2, 30.1, 29.7, 28.3, 26.8, 26.2, 9.6, 9.5. HRMS (ESI) calcd for C26H38FN4O2 [M+H]+: 457.2901, found 457.2982.
1-(4-(3-((1r,3R,5S,7r)-3,5-Dimethyladamantan-1-yl)ureido)-3-fluorobenzyl)-N,N-dimethylpiperidine-3-carboxamide (D2).
According to the synthetic process of compound 5, compound D2 as a white solid (0.18 g, 58 %) was obtained by taking 1-(4-(3-((1r,3R,5S,7r)-3,5-dimethyladamantan-1-yl)ureido)-3-fluorobenzyl)piperidine-3-carboxylic acid D (0.3 g, 0.66 mmol) and dimethylamine (36 mg, 0.79 mmol) as starting material, compound D2, m.p. 111–112°C. 1H NMR (400 MHz, CDCl3): δ (ppm) 7.93 (t, J = 8.92Hz, 1H),7.13–7.12 (m, 1H), 6.91–6.85 (m, 2H), 5.53 (s, 1H), 3.39–3.27 (m, 2H), 2.94 (s, 3H), 2.83 (s, 3H), 2.79–2.72 (m, 3H), 2.07–2.02 (m, 2H), 1.89–1.85 (m, 1H), 1.76–1.70 (m, 3H), 1.62–1.54 (m, 6H), 1.42–1.39 (m, 1H), 1.30–1.27 (m, 2H), 1.21–1.18 (m, 2H), 1.10–1.02 (m, 2H), 0.75 (s, 6H). 13C NMR (100 MHz, DMSO-d6): δ (ppm) 174.5, 154.5, 153.4, 151.1, 124.9, 120.9, 115.2, 115.1, 65.6, 62.5, 55.6, 53.6, 52.7, 50.7, 48.1, 42.8, 40.7, 39.6, 37.2, 35.6, 32.4, 30.5, 30.2, 30.2, 29.7, 27.3, 24.9, 13.7. HRMS (ESI) calcd for C28H42FN4O2 [M+H]+: 485.3214, found 485.3297.
1-(4-(3-((1r,3R,5S,7r)-3,5-Dimethyladamantan-1-yl)ureido)-3-fluorobenzyl)-N,N-diethylpiperidine-3-carboxamide (D3).
According to the synthetic process of compound 5, compound D3 as a white solid (0.20 g, 61 %) was obtained by taking 1-(4-(3-((1r,3R,5S,7r)-3,5-dimethyladamantan-1-yl)ureido)-3-fluorobenzyl)piperidine-3-carboxylic acid D (0.3 g, 0.66 mmol) and diethylamine (58 mg, 0.79 mmol) as starting material, compound D3, m.p. 159–160°C. 1H NMR (400 MHz, DMSO-d6): δ (ppm) 8.27–8.22 (m, 2H), 7.42–7.14 (m, 2H), 6.55 (s, 1H), 4.31–4.24 (m, 1H), 3.27–3.17 (m, 6H), 2.91 (s, 3H), 2.09 (s, 1H), 1.75 (s, 5H), 1.57 (s, 4H), 1.41 (s, 2H), 1.40–1.25 (m, 5H), 1.12–1.09 (m, 5H), 1.00–0.98 (m, 3H), 0.83 (s, 6H).13C NMR (100 MHz, DMSO-d6): δ (ppm) 172.5, 153.8, 152.0, 150.4, 129.9, 127.5, 119.8, 117.3, 59.3, 54.0, 52.2, 52.1, 52.1, 52.0, 50.7, 48.0, 42.7, 42.3, 41.8, 32.5, 32.3, 30.5, 30.0, 28.9, 26.8, 23.0, 22.9. HRMS (ESI) calcd for C30H46FN4O2 [M+H]+: 513.3527, found 513.3611.
1-(4-(3-((1r,3R,5S,7r)-3,5-Dimethyladamantan-1-yl)ureido)-3-fluorobenzyl)-N-(2-hydroxypropyl)piperidine-3-carboxamide (D4).
According to the synthetic process of compound 5, compound D4 as a white solid (0.20 g, 61 %) was obtained by taking 1-(4-(3-((1r,3R,5S,7r)-3,5-dimethyladamantan-1-yl)ureido)-3-fluorobenzyl)piperidine-3-carboxylic acid D (0.3 g, 0.66 mmol) and iso-propanolamine (59 mg, 0.79 mmol) as starting material, compound D4, m.p. 91–92°C. 1H NMR (400 MHz, CDCl3): δ (ppm) 7.98 (t, J = 7.36Hz, 1H), 7.01–6.94 (m, 3H), 5.29 (s, 1H), 3.91–3.85 (m, 1H), 3.50–3.26 (m, 4H), 3.10–3.07 (m, 3H), 2.81 (s, 2H), 2.51 (s, 1H), 2.30 (s, 2H), 2.14 (s, 1H), 1.84 (s, 3H), 1.15–1.58 (m, 7H), 1.27–1.22 (m, 4H), 1.16–1.14 (m, 4H), 0.84 (s, 6H).13C NMR (100 MHz, DMSO-d6): δ (ppm) 176.5, 154.3, 153.6, 151.2, 125.3, 121.3, 115.6, 115.4, 67.4, 67.3, 62.3, 54.2, 53.9, 52.9, 50.6, 48.1, 47.3, 47.0, 46.9, 42.7, 40.7, 32.4, 30.2, 30.1, 26.7, 22.5, 20.9, 8.8, 8.6. HRMS (ESI) calcd for C29H44FN4O3 [M+H]+: 515.3319, found 515.3404.
1-(4-(3-((1r,3R,5S,7r)-3,5-Dimethyladamantan-1-yl)ureido)-3-fluorobenzyl)-N,N-bis(2-hydroxyethyl)piperidine-3-carboxamide (D5).
According to the synthetic process of compound 5, compound D5 as a white solid (0.21 g, 59 %) was obtained by taking 1-(4-(3-((1r,3R,5S,7r)-3,5-dimethyladamantan-1-yl)ureido)-3-fluorobenzyl)piperidine-3-carboxylic acid D (0.3 g, 0.66 mmol) and diethanolamine (83 mg, 0.79 mmol) as starting material, compound D5, m.p. 123–124°C. 1H NMR (400 MHz, CDCl3): δ (ppm) 8.08 (t, J = 8.36Hz, 1H),6.95–6.90 (m, 2H), 5.67 (s, 1H), 3.73–3.62 (m, 2H), 3.45–3.43 (m, 1H), 3.14 (q, J = 8.00Hz, 2H), 2.86 (s, 2H), 2.45 (s, 1H), 2.14 (s, 2H), 1.98–1.97 (m, 3H), 1.89–1.83 (m, 3H), 1.65 (s, 4H), 1.57 (s, 2H), 1.42–1.33 (m, 5H), 1.30–1.24 (m, 5H), 1.15–1.14 (m, 2H), 0.75 (s, 6H). 13C NMR (100 MHz, DMSO-d6): δ (ppm) 174.8, 154.5, 154.4, 153.5, 151.1, 125.3, 120.9, 115.1, 125.3, 120.9, 115.1, 69.7, 62.5, 60.4, 54.3, 54.1, 52.8, 50.6, 48.1, 47.3, 42.7, 40.7, 33.8, 32.4, 30.9, 30.2, 26.7, 22.2, 21.1, 14.2, 8.9. HRMS (ESI) calcd for C30H46FN4O4 [M+H]+: 545.3425, found 545.3147.
1-(4-(3-((1r,3R,5S,7r)-3,5-Dimethyladamantan-1-yl)ureido)-3-fluorobenzyl)-N-(2-hydroxyethyl)piperidine-3-carboxamide (D6).
According to the synthetic process of compound 5, compound D6 as a white solid (0.20 g, 62 %) was obtained by taking 1-(4-(3-((1r,3R,5S,7r)-3,5-dimethyladamantan-1-yl)ureido)-3-fluorobenzyl)piperidine-3-carboxylic acid D (0.3 g, 0.66 mmol) and ethanolamine (48 mg, 0.79 mmol) as starting material, compound D6, m.p. 94–95°C. 1H NMR (400 MHz, CDCl3): δ (ppm) 8.00 (t, J = 8.33 Hz, 1H), 7.08 (s, 1H), 7.00–6.95 (m, 2H), 5.46 (s, 1H), 3.64–3.62 (m, 2H), 3.51–3.45 (m, 1H), 3.37–3.35 (m, 3H), 3.16–3.09 (m, 2H), 2.79 (s, 1H), 2.49 (s, 1H), 2.29 (s, 2H), 2.14 (s, 1H), 1.83 (s, 2H), 1.65–1.62 (m, 6H), 1.42–1.26 (m, 8H), 1.14 (s, 4H), 0.84 (s, 6H). 13C NMR (100 MHz, DMSO-d6): δ (ppm) 176.4, 154.3, 151.2, 127.3, 125.3, 121.1, 115.5, 115.3, 62.3, 62.1, 54.1, 52.9, 50.6, 48.1, 47.2, 42.7, 42.2, 40.7, 32.4, 30.2, 30.1, 26.7, 22.6, 22.5, 14.1, 8.9. HRMS (ESI) calcd for C28H42FN4O3 [M+H]+: 501.3163, found 501.3246.
N-(1,3-Dihydroxypropan-2-yl)-1-(4-(3-((1r,3R,5S,7r)-3,5-dimethyladamantan-1-yl)ureido)-3-fluorobenzyl)piperidine-3-carboxamide (D7).
According to the synthetic process of compound 5, compound D7 as a white solid (0.20 g, 59 %) was obtained by taking 1-(4-(3-((1r,3R,5S,7r)-3,5-dimethyladamantan-1-yl)ureido)-3-fluorobenzyl) piperidine-3-carboxylic acid D (0.3 g, 0.66 mmol) and 2-aminopropane-1,3-diol (72 mg, 0.79 mmol) as starting material, compound D7, m.p. 113–114°C. 1H NMR (400 MHz, CDCl3): δ (ppm) 7.87–7.86 (m, 1H), 7.45 (s, 1H), 7.04–7.01 (m, 1H), 6.85–6.83 (m, 1H), 5.88 (s, 1H), 3.87 (s, 1H), 3.67–3.30 (m, 7H), 3.01–2.70 (m, 2H), 2.43 (s, 1H), 2.03 (s, 1H), 1.73 (s, 3H), 1.55(s, 7H), 1.28–1.77 (m, 7H), 1.05 (s, 3H), 0.74 (s, 6H). 13C NMR (100 MHz, DMSO-d6): δ (ppm) 166.5, 150.0, 148.9, 146.5, 122.8, 120.7, 116.0, 110.9, 57.2, 57.0, 55.7, 49.4, 47.9, 47.6, 45.9, 43.4, 41.9, 37.9, 35.8, 27.6, 27.2, 25.4, 24.9, 24.6, 22.1, 17.9, 17.6, 16.3, 9.4, 9.3. HRMS (ESI) calcd for C29H44FN4O4 [M+H]+: 531.3268, found 531.3340.
N-(1,3-Dihydroxy-2-(hydroxymethyl)propan-2-yl)-1-(4-(3-((1r,3R,5S,7r)-3,5-dimethyladamantan-1-yl)ureido)-3-fluorobenzyl)piperidine-3-carboxamide (D8).
According to the synthetic process of compound 5, compound D8 as a white solid (0.22 g, 61 %) was obtained by taking 1-(4-(3-((1r,3R,5S,7r)-3,5-dimethyladamantan-1-yl)ureido)-3-fluorobenzyl)piperidine-3-carboxylic acid D (0.3 g, 0.66 mmol) and 2-amino-2-(hydroxymethyl)propane-1,3-diol (96 mg, 0.79 mmol) as starting material, compound D8, m.p. 111–112°C. 1H NMR (400 MHz, CDCl3): δ (ppm) 7.88 (t, J = 8.16Hz, 1H), 7.16–7.09 (m, 2H), 6.96–6.94 (m, 1H), 5.50 (s, 1H), 4.63 (s, 2H), 3.67–3.60 (m, 4H), 3.46–3.35 (m, 2H), 3.12 (q, J = 7.28Hz, 2H), 2.81–2.78 (m, 2H), 2.50 (s, 1H), 2.29 (s, 1H), 2.13 (s, 1H), 1.81 (s, 3H), 1.63 (s, 6H), 1.37–1.26 (m, 7H), 1.17–1.10 (m, 2H), 0.84 (s, 6H). 13C NMR (100 MHz, DMSO-d6): δ (ppm) 175.4, 154.1, 152.5, 150.9, 127.7, 125.3, 119.9, 115.6, 62.6, 61.9, 61.1, 60.6, 55.6, 53.3, 52.0, 50.7, 48.1, 46.2, 42.7, 32.4, 32.4, 32.3, 30.6, 30.5, 30.0, 27.7, 23.8. HRMS (ESI) calcd for C30H46FN4O5 [M+H]+: 561.3374, found 561.3447.
1-((1r,3R,5S,7r)-3,5-Dimethyladamantan-1-yl)-3-(2-fluoro-4-((3-(4-(2-hydroxyethyl) piperazine-1-carbonyl)piperidin-1-yl)methyl)phenyl)urea (D9).
According to the synthetic process of compound 5, compound D9 as a white solid (0.21 g, 56 %) was obtained by taking 1-(4-(3-((1r,3R,5S,7r)-3,5-dimethyladamantan-1-yl)ureido)-3-fluorobenzyl)piperidine-3-carboxylic acid D (0.3 g, 0.66 mmol) and 2-(piperazin-1-yl-ethan)-1-ol (96 mg, 0.79 mmol) as starting material, compound D9, m.p. 116–117°C. 1H NMR (400 MHz, CDCl3): δ (ppm) 7.99 (t, J = 8.24 Hz, 1H), 7.01–6.98 (m, 2H), 6.80 (s, 1H), 5.12 (s, 1H), 3.65–3.62 (m, 3H), 3.49–3.47 (m, 5H), 3.65–3.62 (m, 3H), 3.49–3.47 (m, 5H), 2.89–2.78 (m, 4H), 2.55 (t, J = 5.32Hz, 2H), 2.50–2.40 (m, 4H), 2.15–2.14 (m, 3H), 1.84–1.73 (m, 4H), 1.66 (s, 4H), 1.36–1.33 (m, 2H), 1.31–1.25 (m, 4H), 1.16–1.15 (m, 2H), 0.84 (s, 6H). 13C NMR (100 MHz, DMSO-d6): δ (ppm) 172.6, 154.0, 153.5, 151.1, 125.1, 120.9, 115.4, 115.2, 62.3, 59.2, 57.8, 55.6, 53.7, 53.2, 52.9, 52.6, 50.6, 48.2, 46.8, 45.5, 42.7, 41.6, 40.7, 32.7, 30.2, 30.1, 29.7, 27.3, 24.7, 22.7, 9.2. HRMS (ESI) calcd for C32H49FN5O3 [M+H]+: 570.3741, found 570.3812.
Ethyl-1-(3-fluoro-4-nitrobenzyl)piperidine-4-carboxylate (15).
According to the synthetic process of compound 12, compound 15 as a light yellow oil (4.79 g) was obtained by taking 4-(bromomethyl)-2-fluoro-1-nitrobenzene 11 (5 g, 21.46 mmol) and ethyl piperidine-4-carboxylate (4.04 g, 25.76 mmol) as starting material, compound 15, which was used without further purification, ESI-MS: m/z 311.12 [M+H]+.
Ethyl-1-(4-amino-3-fluorobenzyl)piperidine-4-carboxylate (16).
According to the synthetic process of compound 2, compound 16 as a light yellow oil (3.94 g) was obtained by taking ethyl 1-(3-fluoro-4-nitrobenzyl)piperidine-4-carboxylate 15 (4.79 g, 15.44 mmol) as starting material, compound 16, which was used without further purification, 1H NMR (400 MHz, CDCl3): δ (ppm) 7.10 (dd, J = 11.28 Hz, J = 1.76 Hz, 1H), δ 7.03 (dd, J = 8.08 Hz, J = 1.4 Hz, 1H), 6.76 (t, J = 8.48 Hz, 1H), 4.28 (s, 0.5H), 4.13 (q, J = 7.08 Hz, 2H), 4.02 (s, 0.5H),3.51 (s, 2H), 3.19 (t, J = 10.8 Hz, 1H), 3.04 (td, J = 12.44 Hz, J = 1.96 Hz, 1H),2.52 (m, 1H), 2.17 (m, 1H), 1.74 (m, 2H), 1.53 (m,1H), 1.24 (t, J = 7.08 Hz, 4H).
Ethyl-1-(4-(3-((1r,3R,5S,7r)-3,5-dimethyladamantan-1-yl)ureido)-3-fluorobenzyl) piperidine-4-carboxylate (17).
According to the synthetic process of compound 3, compound 17 as a light yellow oil (4.64 g) was obtained by taking thyl-1-(4-amino-3-fluorobenzyl)piperidine-4-carboxylate (16) (3.94 g, 14.06 mmol) as starting material, compound 17, which was used without further purification, ESI-MS: m/z 486.3 [M+H]+, 508.32 [M+Na]+.
1-(4-(3-((1r,3R,5S,7r)-3,5-Dimethyladamantan-1-yl)ureido)-3-fluorobenzyl) piperidine-4-carboxylic acid (18).
According to the synthetic process of compound 4, compound 18 as a light yellow oil (3.85 g) was obtained by taking thyl 1-(4-(3-((1r,3R,5S,7r)-3,5-dimethyladamantan-1-yl)ureido)-3-fluorobenzyl)piperidine-4-carboxylate 17 (4.64 g, 9.56 mmol) as starting material, compound 18, which was used without further purification, ESI-MS: m/z 458.3 [M+H]+.
1-(4-(3-((1r,7r)-3,5-Dimethyladamantan-1-yl)ureido)-3-fluorobenzyl)piperidine-4-carboxamide (E1).
According to the synthetic process of compound 5, compound E1 as a white solid (0.20 g, 66 %) was obtained by taking 1-(4-(3-((1r,3R,5S,7r)-3,5-dimethyladamantan-1-yl)ureido)-3-fluorobenzyl)piperidine-4-carboxylic acid 18 (0.3 g, 0.66 mmol) and aqueous ammonia (92 mg, 0.79 mmol) as starting material, compound E1, m.p.117–118°C. 1H NMR (400 MHz, CDCl3): δ (ppm) 8.10 (s, 1H), 7.25–7.01 (m, 2H), 6.77 (s, 1H), 6.48 (s, 1H), 3.34 (s, 2H), 3.09 (s, 1H), 2.90 (s, 2H), 2.51 (s, 2H), 2.10 (s, 3H), 1.76–1.58 (m, 8H), 1.40–1.12 (m, 8H), 0.83 (s, 6H). 13C NMR (100 MHz, DMSO-d6): δ (ppm) 176.5, 161.4, 154.0, 152.4, 150.8, 125.8, 119.9, 115.9, 52.4, 52.0, 50.2, 48.2, 48.1, 46.2, 43.8, 42.9, 42.7, 42.2, 32.4, 32.3, 30.7, 30.5, 30.0, 28.7, 28.0. HRMS (ESI) calcd for C26H38FN4O2 [M+H]+: 457.2901, found 457.2971.
1-(4-(3-((1r,7r)-3,5-Dimethyladamantan-1-yl)ureido)-3-fluorobenzyl)-N-(2-hydroxypropyl)piperidine-4-carboxamide (E2).
According to the synthetic process of compound 5, compound E2 as a white solid (0.19 g, 57 %) was obtained by taking 1-(4-(3-((1r,3R,5S,7r)-3,5-dimethyladamantan-1-yl)ureido)-3-fluorobenzyl)piperidine-4-carboxylic acid 18 (0.3 g, 0.66 mmol) and iso-propanolamine (59 mg, 0.79 mmol) as starting material, compound E2, m.p. 99–100°C. 1H NMR (400 MHz, CDCl3): δ (ppm) 7.88 (t, J = 8.24Hz, 1H),7.02–6.93 (m, 2H), 6.84 (s, 1H), 6.28 (s, 1H), 5.12 (s, 1H), 3.92–3.88 (m, 1H), 3.41 (s, 3H), 3.14–2.96 (m, 3H), 2.88–2.86 (m, 2H), 2.14–1.96 (m, 4H), 1.83–1.72 (m, 5H), 1.68–1.62 (m, 4H), 1.38–1.26 (m, 4H), 1.18–1.15 (m, 4H), 0.86 (s, 6H). 13C NMR (100 MHz, DMSO-d6): δ (ppm) 176.5, 154.4, 151.5, 126.5, 124.8, 121.3, 115.3, 115.1, 67.4, 62.0, 52.9, 52.8, 50.6, 48.2, 46.9, 43.0, 42.7, 40.7, 32.4, 30.2, 30.1, 28.7, 20.9, 9.6. HRMS (ESI) calcd for C29H44FN4O3 [M+H]+: 515.3319, found 515.3394.
N-(1,3-Dihydroxypropan-2-yl)-1-(4-(3-((1r,7r)-3,5-dimethyladamantan-1-yl)ureido)-3-fluorobenzyl)piperidine-4-carboxamide (E3).
According to the synthetic process of compound 5, compound E3 as a white solid (0.19 g, 54 %) was obtained by taking 1-(4-(3-((1r,3R,5S,7r)-3,5-dimethyladamantan-1-yl)ureido)-3-fluorobenzyl)piperidine-4-carboxylic acid 18 (0.3 g, 0.66 mmol) and 2-aminopropane-1,3-diol (72 mg, 0.79 mmol) as starting material, compound E3, m.p. 150–151°C. 1H NMR (400 MHz, DMSO-d6): δ (ppm) 8.61–8.60 (m, 1H), 8.39 (dd, J = 8.36 Hz, J = 1.12 Hz, 1H), 8.36 (s, 1H), 8.12 (t, J = 8.44 Hz, 1H), 7.67 (d, J = 7.88 Hz, 1H), 7.41–7.38 (m, 2H), 7.15 (d, J = 8.64Hz, 1H), 6.75 (s, 1H), 4.66 (s, 2H), 3.95 (s, 2H), 3.68–3.64 (m, 1H), 3.39–3.38 (m, 6H), 2.09 (s, 1H), 1.80–1.76 (m, 5H), 1.61–1.54 (m, 4H), 1.36–1.27 (m, 4H), 1.11 (s, 3H), 0.83 (s, 6H). 13C NMR (100 MHz, DMSO-d6): δ (ppm) 173.4, 154.0, 152.0, 150.4, 129.1, 128.0, 119.7, 118.1, 61.4, 60.9, 58.9, 58.2, 54.8, 53.6, 51.0, 50.7, 49.0, 48.2, 42.7, 32.5, 32.2, 30.6, 29.5, 28.8, 26.2, 25.6, 22.8, 22.5. HRMS (ESI) calcd for C29H44FN4O4 [M+H]+: 531.3268, found 531.3337.
N-(1,3-Dihydroxy-2-(hydroxymethyl)propan-2-yl)-1-(4-(3-((1r,7r)-3,5-dimethyl adamantan-1-yl)ureido)-3-fluorobenzyl)piperidine-4-carboxamide (E4).
According to the synthetic process of compound 5, compound E4 as a white solid (0.20 g, 55 %) was obtained by taking 1-(4-(3-((1r,3R,5S,7r)-3,5-dimethyladamantan-1-yl)ureido)-3-fluorobenzyl)piperidine-4-carboxylic acid 18 (0.3 g, 0.66 mmol) and 2-amino-2-(hydroxymethyl)propane-1,3-diol (96 mg, 0.79 mmol) as starting material, compound E4, m.p. 117–118°C. 1H NMR (400 MHz, DMSO-d6): δ (ppm) 8.06–8.01 (m, 2H), 7.08–7.04 (m, 2H), 6.97–6.95 (m, 1H), 6.45 (s, 1H), 4.75 (t, J = 5.60Hz, 2H), 3.51–3.50 (m, 5H), 3.41–3.33 (m, 4H), 2.87–2.79 (m, 1H), 2.22 (s, 1H), 2.09–2.08 (m, 1H), 1.97–1.84 (m, 2H), 1.77–1.74 (m, 2H), 1.65–1.51 (m, 7H), 1.34–1.24 (m, 6H), 1.15–1.11 (m, 2H), 0.83 (s, 6H). 13C NMR (100 MHz, DMSO-d6): δ (ppm) 173.8, 154.1, 152.5, 150.9, 132.1, 125.2, 119.9, 115.4, 72.7, 61.9, 60.7, 60.6, 60.5, 55.7, 53.3, 52.9, 52.0, 50.7, 48.1, 46.2, 42.8, 42.7, 42.6, 32.3, 30.5, 30.0, 29.5, 27.7, 24.2, 22.5. HRMS (ESI) calcd for C30H46FN4O5 [M+H]+: 561.3374, found 561.3447.
1-(4-(3-((1r,7r)-3,5-Dimethyladamantan-1-yl)ureido)-3-fluorobenzyl)-N,N-bis(2-hydroxyethyl)piperidine-4-carboxamide (E5).
According to the synthetic process of compound 5, compound E4 as a white solid (0.22 g, 61 %) was obtained by taking 1-(4-(3-((1r,3R,5S,7r)-3,5-dimethyladamantan-1-yl)ureido)-3-fluorobenzyl)piperidine-4-carboxylic acid 18 (0.3 g, 0.66 mmol) and diethanolamine (83 mg, 0.79 mmol) as starting material, compound E5, m.p. 104–105°C. 1H NMR (400 MHz, CDCl3): δ (ppm) 7.88 (t, J = 8.44 Hz, 1H), 7.03–6.94 (m, 2H), 6.61 (s, 1H), 4.92 (s, 1H), 3.83–3.77 (m, 3H), 3.54–3.52 (m, 2H), 3.44 (s, 1H), 2.89–2.81 (m, 3H), 2.57 (s, 1H), 2.15 (s, 1H), 2.03–1.97 (m, 2H), 1.84 (s, 2H), 1.68–1.66 (m, 4H), 1.43 (s, 2H), 1.37–1.21 (m, 8H), 1.20–1.15 (m, 5H), 0.84 (s, 6H). 13C NMR (100 MHz, DMSO-d6): δ (ppm) 177.7, 154.0, 124.9, 121.1, 115.4, 115.2, 61.7, 60.8, 53.0, 52.6, 51.7, 50.6, 50.3, 48.2, 46.6, 42.7, 40.7, 38.8, 32.5, 31.6, 30.2, 30.1, 29.7, 28.7, 26.9, 22.6, 14.1, 10.1. HRMS (ESI) calcd for C30H46FN4O4 [M+H]+: 545.3425, found 545.3499.
Tert-butyl-4-(1-(4-(3-((1r,3R,5S,7r)-3,5-dimethyladamantan-1-yl)ureido)-3-fluorobenzoyl)piperidine-3-carbonyl)-1,4-diazepane-1-carboxylate (19).
According to the synthetic process of compound 5, compound 19 as a light yellow oil (3.33 g) was obtained by taking 1-(4-(3-((1r,3R,5S,7r)-3,5-dimethyladamantan-1-yl)ureido)-3-fluorobenzoyl)piperidine-3-carboxylic acid A (3 g, 6.37 mmol) and tert-butyl 1,4-diazepane-1-carboxylate (1.53 g, 7.64 mmol) as starting material, compound 19, which was used without further purification, 1H NMR (400 MHz, CDCl3): δ (ppm) 8.18–8.15 (m, 1H), 7.36–7.35 (m, 1H), 7.06 (s, 2H), 4.54 (s, 1H), 3.76–3.41 (m, 9H), 3.11–3.01 (m, 1H), 3.84–2.97 (m, 1H), 2.14 (t, J = 2.76 Hz, 1H), 1.91–1.72 (m, 7H), 1.64 (s, 4H), 1.45–1.36 (m, 1H), 1.33–1.23 (m, 4H), 1.19–1.15 (m, 2H), 0.85 (s, 6H).
1-(4-(3-(1,4-Diazepane-1-carbonyl)piperidine-1-carbonyl)-2-fluorophenyl)-3-((1r,3R,5S,7r)-3,5-dimethyladamantan-1-yl)urea (20).
To a solution of tert-butyl 4-(1-(4-(3-((1r,3R,5S,7r)-3,5-dimethyladamantan-1-yl)ureido)-3-fluorobenzoyl)piperidine-3-carbonyl)-1,4-diazepane-1-carboxylate 19 (3.33 g, 5.10 mmol), DCM (10 mL) were added TFA (5.82 g, 51 mmol) slowly dropwise at 0 °C. Transfer to room temperature and continue the reaction for 12h. After the reaction was completed, the reaction solution is concentrated under vacuum to produce a light yellow oil 20 (7.86 g), which is used without further purification, ESI-MS: m/z 554.44 [M+H]+.
1-(4-(3-(4-Acetyl-1,4-diazepane-1-carbonyl)piperidine-1-carbonyl)-2-fluorophenyl)-3-((1r,3R,5S,7r)-3,5-dimethyladamantan-1-yl)urea (F1).
According to the synthetic process of compound 5, compound F1 as a white solid (0.21 g, 66 %) was obtained by taking 1-(4-(3-(1,4-diazepane-1-carbonyl)piperidine-1-carbonyl)-2-fluorophenyl)-3-((1r,3R,5S,7r)-3,5-dimethyladamantan-1-yl)urea 20 (0.3 g, 0.56 mmol) and acetic acid (40 mg, 0.67 mmol) as starting material, compound F1, m.p. 136–137°C. 1H NMR (400 MHz, CDCl3): δ (ppm) 8.26–8.20 (m, 1H), 7.60 (s, 1H), 7.08 (s, 2H), 5.92 (s, 1H), 4.58 (s, 1H), 3.74–3.47 (m, 9H), 3.14–2.60 (m, 3H), 2.13–2.05 (m, 4H), 1.87–1.80 (m, 6H), 1.63 (s, 4H), 1.42–1.35 (m, 5H), 1.29–1.24 (m, 3H), 1.18–1.11(m, 2H), 0.83 (s, 6H). 13C NMR (100 MHz, DMSO-d6): δ (ppm) 175.8, 172.6, 168.5, 153.8, 151.7, 150.2, 130.3, 128.8, 123.8, 119.2, 52.1, 50.7, 48.2, 47.9, 46.7, 46.6, 46.2, 46.1, 45.1, 44.2, 42.7, 36.6, 36.5, 36.4, 32.4, 30.5, 30.0, 27.4, 27.3, 27.2, 18.1, 12.3, 12.2. HRMS (ESI) calcd for C33H47FN5O4 [M+H]+: 596.3534, found 596.3600.
1-((1r,3R,5S,7r)-3,5-Dimethyladamantan-1-yl)-3-(2-fluoro-4-(3-(4-propionyl-1,4-diazepane-1-carbonyl)piperidine-1-carbonyl)phenyl)urea (F2).
According to the synthetic process of compound 5, compound F2 as a white solid (0.21 g, 63 %) was obtained by taking 1-(4-(3-(1,4-diazepane-1-carbonyl)piperidine-1-carbonyl)-2-fluorophenyl)-3-((1r,3R,5S,7r)-3,5-dimethyladamantan-1-yl)urea 20 (0.3 g, 0.56 mmol) and propionic acid (50 mg, 0.67 mmol) as starting material, compound F2, m.p. 96–98°C. 1H NMR (400 MHz, CDCl3): δ (ppm) 8.30 (s, 1H),8.16–8.13 (m, 1H), 7.59 (s, 1H), 6.99 (s, 2H), 5.88 (s, 1H), 4.48 (s, 1H), 3.71–3.60 (m, 7H), 3.07–3.04 (m, 3H), 2.69 (s, 2H), 2.25 (s, 3H), 2.05 (s, 1H), 1.74 (s, 6H), 1.56 (s, 4H), 1.22–1.18 (m, 3H), 1.06–1.01 (m, 5H), 0.76 (s, 6H). 13C NMR (100 MHz, DMSO-d6): δ (ppm) 173.9, 169.7, 168.4, 154.0, 152.2, 150.6, 130.4, 130.3, 120.1, 124.8, 54.7, 52.8, 50.6, 47.9, 47.4, 47.3, 42.8, 42.7, 40.6, 32.4, 30.2, 30.1, 29.7, 27.7, 26.5, 26.1, 25.9, 18.5, 17.1, 12.2, 9.6, 9.5, 9.4. HRMS (ESI) calcd for C34H49FN5O4 [M+H]+: 610.3690, found 610.3767.
1-((1r,3R,5S,7r)-3,5-Dimethyladamantan-1-yl)-3-(2-fluoro-4-(3-(4-(2-methylbutanoyl) −1,4-diazepane-1-carbonyl)piperidine-1-carbonyl)phenyl)urea (F3).
According to the synthetic process of compound 5, compound F3 as a white solid (0.23 g, 65 %) was obtained by taking 1-(4-(3-(1,4-diazepane-1-carbonyl)piperidine-1-carbonyl)-2-fluorophenyl)-3-((1r,3R,5S,7r)-3,5-dimethyladamantan-1-yl)urea 20 (0.3 g, 0.56 mmol) and 2-methylbutanoic acid (68 mg, 0.67 mmol) as starting material, compound F3, m.p. 149–150°C. 1H NMR (400 MHz, CDCl3): δ (ppm) 8.26–8.20 (m, 1H), 7.58 (s, 1H), 7.24–7.08 (m, 2H), 5.90 (s, 1H), 4.59 (s, 1H), 3.82–3.37 (m, 10H), 3.05–2.55 (m, 4H), 2.13–2.05(m, 2H), 1.80–1.62 (m, 11H), 1.37–1.24 (m, 6H), 1.14–1.06 (m, 5H), 0.86 (s, 2H), 0.83 (s, 6H). 13C NMR (100 MHz, DMSO-d6): δ (ppm) 175.8, 172.6, 168.5, 153.8, 121.7, 150.2, 130.4, 128.8, 123.8, 119.2, 52.1, 50.7, 48.2, 47.9, 47.8, 46.8, 46.7, 46.2, 46.1, 45.1, 44.2, 42.7, 36.7, 36.6, 36.5, 36.4, 32.4, 30.5, 30.0, 28.7, 27.4, 27.3, 27.2, 27.2, 18.0, 17.7. HRMS (ESI) calcd for C36H53FN5O4 [M+H]+: 638.4003, found 638.4076.
Tert-butyl-4-(1-(4-(3-((1r,3R,5S,7r)-3,5-dimethyladamantan-1-yl)ureido)-3-fluorobenzyl)piperidine-3-carbonyl)-1,4-diazepane-1-carboxylate (21).
According to the synthetic process of compound 5, compound 21 as a light yellow oil (3.27 g) was obtained by taking 1-(4-(3-((1r,3R,5S,7r)-3,5-dimethyladamantan-1-yl)ureido)-3-fluorobenzyl)piperidine-3-carboxylic acid D1 (3.0 g, 6.56 mmol) and tert-butyl 1,4-diazepane-1-carboxylate (1.57 g, 7.87 mmol) as starting material, compound 21, which was used without further purification, ESI-MS: m/z 640.53 [M+H]+.
1-(4-((3-(1,4-Diazepane-1-carbonyl)piperidin-1-yl)methyl)-2-fluorophenyl)-3-((1r,3R,5S,7r)-3,5-dimethyladamantan-1-yl)urea (22).
According to the synthetic process of compound 20, compound 22 as a light yellow oil (1.59 g) was obtained by taking tert-butyl-4-(1-(4-(3-((1r,3R,5S,7r)-3,5-dimethyladamantan-1-yl)ureido)-3-fluorobenzyl)piperidine-3-carbonyl)-1,4 diazepane-1-carboxylate 21 (3.27 g, 5.12 mmol) as starting material, compound 22, which was used without further purification, ESI-MS: m/z 540.41 [M+H]+.
1-((1r,3R,5S,7r)-3,5-Dimethyladamantan-1-yl)-3-(2-fluoro-4-((3-(4-propionyl-1,4-diazepane-1-carbonyl)piperidin-1-yl)methyl)phenyl)urea (G1).
According to the synthetic process of compound 5, compound G1 as a white solid (0.22 g, 68 %) was obtained by taking 1-(4-((3-(1,4-diazepane-1-carbonyl)piperidin-1-yl)methyl)-2-fluorophenyl)-3-((1r,3R,5S,7r)-3,5-dimethyladamantan-1-yl)urea 22 (0.3 g, 0.56 mmol) and acetic acid (40 mg, 0.67 mmol) as starting material, compound G1, m.p. 99–100°C. 1H NMR (400 MHz, CDCl3): δ (ppm) 8.02–7.97 (m, 1H), 7.02–7.69 (m, 2H), 6.85 (br, 1H), 5.20 (m, 1H), 3.84–3.73 (m, 1H), 3.69–3.58 (m, 2H), 3.56–3.49 (m, 4H), 3.47–3.33 (m, 4H), 2.90–2.75 (m, 3H), 2.37–2.38 (m, 2H), 2.15–2.14 (m, 2H), 1.98–1.92 (m, 1H), 1.84 (s, 4H), 1.74–1.69 (m, 2H), 1.66 (s, 4H), 1.64–1.59 (m, 1H), 1.55–1.46 (m, 1H), 1.40–1.37 (m, 2H), 1.33–1.24 (m, 3H), 1.20–1.13 (m, 3H), 1.12–1.08 (m, 2H), 0.85 (s, 6H). 13C NMR (100 MHz, CDCl3): δ (ppm) 174.19, 173.76, 173.59, 173.16, 154.02, 125.08, 120.93, 115.33, 115.14, 62.39, 56.09, 55.92, 53.42, 52.95, 50.66, 48.56, 47.47, 47.38, 46.92, 46.21, 44.53, 44.29, 42.74, 40.78, 39.47, 32.45, 30.11, 27.84, 27.67, 27.08, 26.54, 25.99, 24.75, 9.57, 9.41. HRMS (ESI) calcd forC34H51FN5O3 [M+H]+: 596.3898, found 596.4743.
1-((1r,3R,5S,7r)-3,5-Dimethyladamantan-1-yl)-3-(2-fluoro-4-((3-(4-(2-methylbutanoyl) −1,4-diazepane-1-carbonyl)piperidin-1-yl)methyl)phenyl)urea (G2).
According to the synthetic process of compound 5, compound G2 as a white solid (0.21 g, 60 %) was obtained by taking 1-(4-((3-(1,4-diazepane-1-carbonyl)piperidin-1-yl)methyl)-2-fluorophenyl)-3-((1r,3R,5S,7r)-3,5-dimethyladamantan-1-yl)urea 22 (0.3 g, 0.56 mmol) and 2-methylbutanoic acid (78 mg, 0.67 mmol) as starting material, compound G2, m.p. 108–110°C. 1H NMR (400 MHz, CDCl3): δ (ppm) 8.04–7.95 (m, 1H), 7.02–7.95 (m, 2H), 6.83 (br, 1H), 5.18 (m, 1H), 3.87–3.74 (m, 1H), 3.66–3.55 (m, 4H), 3.53–3.46 (m, 4H), 3.40–3.38 (m, 2H), 2.84–2.73 (m, 3H), 2.55–2.51 (m, 1H), 2.15 (s, 2H), 2.00–1.84 (m, 6H), 1.71–1.62 (m, 7H), 1.58–1.53 (m, 2H), 1.43–1.37 (m, 3H), 1.31 (s, 1H), 1.26–1.24 (m, 1H), 1.19–1.15 (m, 2H), 1.12–1.05 (m, 2H), 0.88–0.83 (m, 9H). 13C NMR (100 MHz, CDCl3): δ (ppm) 176.63, 176.48, 174.21, 174.12, 154.05, 125.02, 120.92, 115.30, 115.11, 62.45, 56.20, 56.10, 55.99, 53.51, 52.95, 50.66, 48.22, 42.75, 40.78, 37.34, 32.46, 30.23, 30.11, 28.04, 27.88, 27.68, 27.42, 27.35, 27.26, 27.13, 27.01, 24.81, 17.90, 17.46, 10.04. HRMS (ESI) calcd for C36H55FN5O3 [M+H]+: 624.4211, found 624.4286.
1-(4-((3-(4-acryloyl-1,4-Diazepane-1-carbonyl)piperidin-1-yl)methyl)-2-fluorophenyl) −3-((1r,3R,5S,7r)-3,5-dimethyladamantan-1-yl)urea (G3).
According to the synthetic process of compound 5, compound G3 as a white solid (0.21 g, 63 %) was obtained by taking 1-(4-((3-(1,4-diazepane-1-carbonyl)piperidin-1-yl)methyl)-2-fluorophenyl)-3-((1r,3R,5S,7r)-3,5-dimethyladamantan-1-yl)urea 22 (0.3 g, 0.56 mmol) and acrylic acid (55 mg, 0.67 mmol) as starting material, compound G3, m.p. 62–65°C. 1H NMR (400 MHz, DMSO-d6): δ (ppm) 8.93–8.85 (m, 2H), 8.17 (s, 2H), 7.20–7.09 (m, 2H), 6.52 (s, 1H), 6.51–5.59 (m, 1H), 3.65–3.59 (m, 8H), 3.53–3.51 (m, 6H), 2.09 (s, 1H), 1.75–1.60 (m, 6H), 1.58 (s, 5H), 1.34–1.32 (m, 5H), 1.28–1.23 (m, 3H), 0.86 (s, 6H). 13C NMR (100 MHz, DMSO-d6): δ (ppm) 168.5, 165.7, 165.3, 153.9, 150.5, 128.9, 128.3, 128.0, 119.8, 52.9, 52.7, 52.6, 52.2, 52.1, 50.7, 48.2, 44.8, 44.3, 42.7, 32.4, 30.5, 30.0, 27.7, 27.3, 25.6, 25.2, 24.8. HRMS (ESI) calcd for C34H49FN5O3 [M+H]+: 594.3741, found 594.3817.
1-((1r,3R,5S,7r)-3,5-Dimethyladamantan-1-yl)-3-(2-fluoro-4-((3-(4-methacryloyl-1,4-diazepane-1-carbonyl)piperidin-1-yl)methyl)phenyl)urea (G4).
According to the synthetic process of compound 5, compound G4 as a white solid (0.25 g, 65 %) was obtained by taking 1-(4-((3-(1,4-diazepane-1-carbonyl)piperidin-1-yl)methyl)-2-fluorophenyl)-3-((1r,3R,5S,7r)-3,5-dimethyladamantan-1-yl)urea 22 (0.3 g, 0.56 mmol) and methacrylic acid (65 mg, 0.67 mmol) as starting material, compound G4, m.p. 79–81°C. 1H NMR (400 MHz, DMSO-d6): δ (ppm) 7.99 (s, 1H), 7.19–7.02 (m, 2H), 5.15 (s, 1H), 4.96–4.93 (s, 1H), 3.60–3.49 (m, 7H), 3.13–3.11 (m, 4H), 2.15–2.08 (m, 1H), 1.93–1.90 (m, 2H), 1.84 (m, 2H), 1.77–1.76 (m, 2H), 1.66 (m, 3H), 1.31–1.25 (m, 14H), 1.17–1.15 (m, 2H), 0.85 (s, 6H), 0.80–0.77 (m, 2H). 13C NMR (100 MHz, DMSO-d6): δ (ppm) 171.9, 171.4, 153.9, 152.1, 150.5, 141.0, 119.8, 115.1, 114.5, 60.1, 52.1, 50.7, 49.4, 48.5, 48.0, 46.7, 46.4, 45.6, 44.9, 43.9, 43.3, 42.7, 32.5, 32.4, 32.2, 30.5, 30.0, 28.2, 28.0, 23.0, 20.7, 20.6, 19.1. HRMS (ESI) calcd for C35H51FN5O3 [M+H]+: 608.3898, found 608.3969.
1-(4-((3-(4-(Cyclopropanecarbonyl)-1,4-diazepane-1-carbonyl)piperidin-1-yl)methyl) −2-fluorophenyl)-3-((1r,3R,5S,7r)-3,5-dimethyladamantan-1-yl)urea (G5).
According to the synthetic process of compound 5, compound G5 as a white solid (0.20 g, 61 %) was obtained by taking 1-(4-((3-(1,4-diazepane-1-carbonyl)piperidin-1-yl)methyl)-2-fluorophenyl)-3-((1r,3R,5S,7r)-3,5-dimethyladamantan-1-yl)urea 22 (0.3 g, 0.56 mmol) and cyclopropanecarboxylic acid (65 mg, 0.67 mmol) as starting material, compound G5, m.p. 106–108°C. 1H NMR (400 MHz, DMSO-d6): δ (ppm) 8.00–7.96 (m, 1H), 7.05–6.98 (m, 2H), 6.74 (s, 1H), 5.04 (s, 1H), 3.76–3.48 (m, 12H), 2.87–2.78 (m, 3H), 2.16–2.14 (m, 1H), 1.93–1.90 (m, 1H), 1.83–1.73 (m, 4H), 1.72–1.66 (m, 8H), 1.40–1.37 (m, 2H), 1.28–1.26 (m, 2H), 1.20–1.12 (m, 2H), 0.96–0.91 (m, 2H), 0.85 (s, 6H), 0.77–0.75 (m, 2H). 13C NMR (100 MHz, DMSO-d6): δ (ppm) 169.6, 168.8, 168.4, 167.7, 149.5, 149.1, 120.5, 116.4, 110.7, 49.3, 48.7, 48.1, 45.9, 43.6, 43.1, 42.7, 42.5, 40.3, 39.7, 37.9, 37.4, 36.1, 28.0, 27.7, 25.6, 25.1, 23.7, 23.7, 23.0, 22.4, 21.9, 6.4, 4.4, 3.0, 2.8. HRMS (ESI) calcd for C35H51FN5O3 [M+H]+: 608.3898, found 608.3972.
Tert-butyl 4-(3-fluoro-4-nitrobenzoyl)-1,4-diazepane-1-carboxylate (23).
According to the synthetic process of compound 5, compound 23 as a light yellow oil (7.94 g) was obtained by taking 3-fluoro-4-nitrobenzoic acid (5 g, 27.03 mmol) and tert-butyl 1,4-diazepane-1-carboxylate (6.49 g, 32.44 mmol) as starting material, compound 23, which was used without further purification, ESI-MS: m/z 390.16 [M+Na]+.
Tert-butyl 4-(4-amino-3-fluorobenzoyl)-1,4-diazepane-1-carboxylate (24).
According to the synthetic process of compound 2, compound 24 as a light yellow oil (6.20 g) was obtained by taking tert-butyl 4-(3-fluoro-4-nitrobenzoyl)-1,4-diazepane-1-carboxylate 23 (7.94 g, 21.63 mmol) as starting material, compound 24, which was used without further purification, ESI-MS: m/z 360.1 [M+Na]+.
Tert-butyl-4-(4-(3-((1r,3R,5S,7r)-3,5-dimethyladamantan-1-yl)ureido)-3-fluorobenzoyl) −1,4-diazepane-1-carboxylate (25).
According to the synthetic process of compound 3, compound 25 as a light yellow oil (6.78 g) was obtained by taking tert-butyl 4-(4-amino-3-fluorobenzoyl)-1,4-diazepane-1-carboxylate 24 (6.20 g, 18.40 mmol) as starting material, compound 25, which was used without further purification, 1H NMR (400 MHz, CDCl3): δ (ppm) 8.13 (t, J = 7.8 Hz, 1H), 7.09–6.95 (m, 2H), 6.88–6.78 (m, 2H), 3.76–3.43 (m, 10H), 2.15 (t, J = 2.9 Hz, 1H), 1.96 (s, 1H), 1.83–1.82 (m, 2H), 1.64 (s, 5H), 1.47 (s, 7H), 1.39 (s, 2H), 1.31–1.26 (m, 2H), 1.19–1.12 (m, 2H), 0.85 (s, 6H).
1-(4-(1,4-Diazepane-1-carbonyl)-2-fluorophenyl)-3-((1r,3R,5S,7r)-3,5-dimethy ladamantan-1-yl)urea (26).
According to the synthetic process of compound 20, compound 26 as a light yellow oil (4.98 g) was obtained by taking tert-butyl-4-(4-(3-((1r,3R,5S,7r)-3,5-dimethyladamantan-1-yl)ureido)-3-fluorobenzoyl)-1,4-diazepane-1-carboxylate 25 (6.78 g, 12.51 mmol) as starting material, compound 26, which was used without further purification, 1H NMR (400 MHz, CDCl3): δ (ppm) 8.25–8.21 (m, 1H), 7.80–7.61 (m, 1H), 7.05–7.01 (m, 2H), 5.99 (s, 1H), 3.82–3.47 (m, 7H), 2.71–2.01 (m, 5H), 1.79 (s, 2H), 1.12–1.61 (m, 4H), 1.37–1.24 (m, 5H), 1.17–1.09 (m, 2H), 0.83 (s, 6H).
Tert-butyl-4-(2-(4-(4-(3-((1r,3R,5S,7r)-3,5-dimethyladamantan-1-yl)ureido)-3-fluorobenzoyl)-1,4-diazepan-1-yl)-2-oxoethyl)piperidine-1-carboxylate (27).
According to the synthetic process of compound 5, compound 27 as a light yellow oil (4.98 g) was obtained by taking 1-(4-(1,4-diazepane-1-carbonyl)-2-fluorophenyl)-3-((1r,3R,5S,7r)-3,5-dimethyladamantan-1-yl)urea 26 (4.98 g, 11.27 mmol) and 2-(1-(tert-butoxycarbonyl)piperidin-4-yl)acetic acid (3.29 g, 13.52 mmol) as starting material, compound 27, which was used without further purification, 1H NMR (400 MHz, CDCl3): δ (ppm) 8.23–8.16 (m, 1H), 7.16–7.15 (m, 1H), 7.06–7.00 (m, 2H), 4.08 (t, J = 11.48 Hz, 2H), 3.81–3.45 (m, 10H), 2.75–2.67 (m, 2H), 2.30–2.24 (m, 3H), 2.15–2.14 (m, 1H), 2.04–2.00 (m, 3H), 1.82 (s, 2H), 1.72–1.64 (m, 7H), 1.45–1.44 (m, 11H), 1.39–1.25 (m, 5H), 1.16–1.11 (m, 4H), 0.85 (s, 6H).
1-((1r,3R,5S,7r)-3,5-Dimethyladamantan-1-yl)-3-(2-fluoro-4-(4-(2-(piperidin-4-yl) acetyl)-1,4-diazepane-1-carbonyl)phenyl)urea (28).
According to the synthetic process of compound 20, compound 28 as a light yellow oil (5.82 g) was obtained by taking ert-butyl-4-(2-(4-(4-(3-((1r,3R,5S,7r)-3,5-dimethyladamantan-1-yl)ureido)-3-fluorobenzoyl)-1,4-diazepan-1-yl)-2-oxoethyl)piperidine-1-carboxylate 27 (6.46 g, 9.69 mmol) as starting material, compound 28, which was used without further purification, ESI-MS: m/z 568.44 [M+H]+.
1-((1r,3R,5S,7r)-3,5-Dimethyladamantan-1-yl)-3-(2-fluoro-4-(4-(2-(1-propionyl piperidin-4-yl)acetyl)-1,4-diazepane-1-carbonyl)phenyl)urea (H1).
According to the synthetic process of compound 5, compound H1 as a white solid (0.20 g, 61 %) was obtained by taking 1-((1r,3R,5S,7r)-3,5-dimethyladamantan-1-yl)-3-(2-fluoro-4-(4-(2-(piperidin-4-yl)acetyl)-1,4-diazepane-1-carbonyl)phenyl)urea 28 (0.3 g, 0.53 mmol) and propionic acid (65 mg, 0.67 mmol) as starting material, compound H1, m.p. 101–102°C. 1H NMR (400 MHz, CDCl3): δ (ppm) 8.27–8.22 (m, 1H), 7.68–7.55 (m, 1H), 7.04–7.02 (m, 2H), 4.60 (s, 1H), 3.72–3.46 (m, 9H), 3.02 (s, 1H), 2.58 (s, 1H), 2.37–2.25 (m, 4H), 2.13–1.99 (m, 4H), 1.88 (s, 4H), 1.63 (s, 5H), 1.38–1.26 (m, 4H), 1.14–1.11 (m, 7H), 0.84 (s, 6H). 13C NMR (100 MHz, DMSO-d6): δ (ppm) 169.9, 169.8, 168.1, 153.0, 129.4, 129.3, 127.1, 121.7, 119.1, 112.5, 59.4, 52.8, 51.8, 49.6, 47.1, 45.6, 41.7, 41.0, 40.8, 39.6, 32.0, 31.9, 31.7, 31.7, 31.4, 30.9, 29.2, 29.1, 20.4, 20.0, 17.6, 16.4, 13.2, 10.9. HRMS (ESI) calcd for C35H51FN5O4 [M+H]+: 624.3847, found 624.3921.
1-((1r,3R,5S,7r)-3,5-Dimethyladamantan-1-yl)-3-(2-fluoro-4-(4-(2-(1-(2-methyl butanoyl)piperidin-4-yl)acetyl)-1,4-diazepane-1-carbonyl)phenyl)urea (H2).
According to the synthetic process of compound 5, compound H2 as a white solid (0.20 g, 60 %) was obtained by taking 1-((1r,3R,5S,7r)-3,5-dimethyladamantan-1-yl)-3-(2-fluoro-4-(4-(2-(piperidin-4-yl)acetyl)-1,4-diazepane-1-carbonyl)phenyl)urea 28 (0.3 g, 0.53 mmol) and 2-methylbutanoic acid (68 mg, 0.67 mmol) as starting material, compound H2, m.p. 106–107°C. 1H NMR (400 MHz, CDCl3): δ (ppm) 8.26–8.21 (m, 1H), 7.57–7.42 (m, 1H), 7.05–7.00 (m, 2H), 4.63 (s, 1H), 3.73–3.46 (m, 9H), 3.02 (s, 1H), 2.66–2.59 (m, 2H), 2.30–2.27 (m, 2H), 2.14 (s, 2H), 1.99 (s, 1H), 1.82 (s, 4H), 1.71–1.64 (m, 5H), 1.40–1.36 (m, 3H), 1.30–1.19 (m, 3H), 1.19–1.10 (m, 3H), 1.10–1.07 (m, 4H), 0.90–0.87 (m, 3H), 0.83 (s, 6H). 13C NMR (100 MHz, DMSO-d6): δ (ppm) 175.1, 171.2, 171.0, 170.8, 153.9, 130.2, 128.3, 122.8, 120.1, 133.6, 60.4, 52.8, 50.7, 48.1, 45.7, 42.7, 42.11, 40.7, 39.6, 39.1, 36.9, 33.3, 33.2, 32.4, 30.2, 30.1, 27.1, 21.1, 17.3, 14.2, 10.7. HRMS (ESI) calcd for C37H55FN5O4 [M+H]+: 652.4160, found 652.4235.
1-((1r,3R,5S,7r)-3,5-Dimethyladamantan-1-yl)-3-(4-(4-(2-(1-methacryloylpiperidin-4-yl)acetyl)-1,4-diazepane-1-carbonyl)phenyl)urea (H3).
According to the synthetic process of compound 5, compound H3 as a white solid (0.18 g, 56 %) was obtained by taking 1-((1r,3R,5S,7r)-3,5-dimethyladamantan-1-yl)-3-(2-fluoro-4-(4-(2-(piperidin-4-yl)acetyl)-1,4-diazepane-1-carbonyl)phenyl)urea 28 (0.3 g, 0.53 mmol) and methacrylic acid (58 mg, 0.67 mmol) as starting material, compound H3, m.p. 95–96°C. 1H NMR (400 MHz, CDCl3): δ (ppm) 8.27–8.22 (m, 1H), 7.68–7.56 (m, 1H), 7.04–7.02 (m, 2H), 4.60 (s, 1H), 3.72–3.46 (m, 9H), 3.62 (s, 1H), 2.58 (s, 1H), 2.37–2.25 (m, 4H), 2.13–1.99 (m, 4H), 1.81 (s, 4H),1.63 (s, 5H), 1.38–1.24 (m, 4H), 1.14–1.11 (m, 7H), 0.84 (s, 6H). 13C NMR (100 MHz, DMSO-d6): δ (ppm) 171.2, 171.0, 170.9, 154.1, 130.4, 130.3, 128.1, 122.7, 120.1, 113.5, 60.4, 52.7, 50.6, 48.1, 46.4, 45.7, 43.9, 42.7, 41.9, 40.6, 39.6, 39.1, 33.2, 33.1, 32.4, 32.0, 30.2, 30.1, 29.7, 29.3, 26.6, 21.1, 14.2, 9.7. HRMS (ESI) calcd for C36H52N5O4 [M+H]+: 618.3941, found 618.3425.
1-((1r,3R,5S,7r)-3,5-Dimethyladamantan-1-yl)-3-(4-(4-(2-(1-(1-(dimethylamino) propan-2-yl)piperidin-4-yl)acetyl)-1,4-diazepane-1-carbonyl)-2-fluorophenyl)urea (H4).
According to the synthetic process of compound 12, compound H4 as a white solid (0.16 g, 46 %) was obtained by taking 1-((1r,3R,5S,7r)-3,5-dimethyladamantan-1-yl)-3-(2-fluoro-4-(4-(2-(piperidin-4-yl)acetyl)-1,4-diazepane-1-carbonyl)phenyl) urea 28 (0.3 g, 0.53 mmol) and 2-chloro-N,N-dimethylpropan-1-amine (81 mg, 0.67 mmol) as starting material, compound H4, m.p. 112–113°C. 1H NMR (400 MHz, CDCl3): δ (ppm) 8.28–8.20 (m, 1H), 7.76–7.57 (m, 1H), 7.05–7.02 (m, 2H), 5.97–5.84 (m, 1H), 3.81–3.40 (m, 9H), 2.88–2.80 (m, 3H), 2.47–2.43 (m, 1H), 2.31–2.25 (m, 7H), 2.25–2.23 (m, 2H), 2.13–2.12 (m, 2H), 2.04–1.91 (m, 3H), 1.80 (s, 2H), 1.67–1.62 (m, 6H), 1.37–1.26 (m, 7H), 1.81–1.11 (m, 2H), 1.01–0.99 (m, 4H), 0.84 (s, 6H). 13C NMR (100 MHz, DMSO-d6): δ (ppm) 174.3, 168.3, 153.9, 151.8, 150.2, 130.3, 123.8, 119.4, 114.2, 73.6, 61.5, 60.6, 58.2, 53.2, 52.1, 50.7, 47.9, 46.6, 45.9, 42.7, 42.2, 32.4, 32.3, 30.5, 30.0, 29.4, 29.1, 19.2. HRMS (ESI) calcd for C37H58FN6O3 [M+H]+: 653.4476, found 653.4549.
Tert-butyl 4-(3-fluoro-4-nitrobenzyl)-1,4-diazepane-1-carboxylate (29).
According to the synthetic process of compound 12, compound 29 as a light yellow oil (6.29 g) was obtained by taking 4-(bromomethyl)-2-fluoro-1-nitrobenzene (5.0 g, 21.46 mmol) and tert-butyl 1,4-diazepane-1-carboxylate (5.15 g, 25.76 mmol) as starting material, compound 29, which was used without further purification, ESI-MS: m/z 376.17 [M+Na]+.
Tert-butyl 4-(4-amino-3-fluorobenzyl)-1,4-diazepane-1-carboxylate (30).
According to the synthetic process of compound 2, compound 30 as a light yellow oil (5.24 g) was obtained by taking tert-butyl 4-(3-fluoro-4-nitrobenzyl)-1,4-diazepane-1-carboxylate 29 (6.29 g, 17.82 mmol) as starting material, compound 30, which was used without further purification, ESI-MS: m/z 324.20 [M+H]+.
Tert-butyl-4-(4-(3-((1r,3R,5S,7r)-3,5-dimethyladamantan-1-yl)ureido)-3-fluorobenzyl) −1,4-diazepane-1-carboxylate (31).
According to the synthetic process of compound 3, compound 31 as a light yellow oil (5.82 g) was obtained by taking used tert-butyl-4-(4-amino-3-fluorobenzyl)-1,4-diazepane-1-carboxylate 30 (5.24 g, 16.22 mmol) as starting material, compound 31, which was used without further purification, ESI-MS: m/z 529.3 [M+H]+.
1-(4-((1,4-Diazepan-1-yl)methyl)-2-fluorophenyl)-3-((1r,3R,5S,7r)-3,5-dimethyl adamantan-1-yl)urea (32).
According to the synthetic process of compound 20, compound 32 as a light yellow oil (4.15 g) was obtained by taking used tert-butyl-4-(4-(3-((1r,3R,5S,7r)-3,5-dimethyladamantan-1-yl)ureido)-3-fluorobenzyl)-1,4-diazepane-1-carboxylate 31 (5.82 g, 11.0 mmol) as starting material, compound 32, which was used without further purification, ESI-MS: m/z 529.45 [M+H]+.
Tert-butyl-4-(2-(4-(4-(3-((1r,3R,5S,7r)-3,5-dimethyladamantan-1-yl)ureido)-3-fluorobenzyl)-1,4-diazepan-1-yl)-2-oxoethyl)piperidine-1-carboxylate (33).
According to the synthetic process of compound 5, compound 33 as a light yellow oil (4.94 g) was obtained by taking used 1-(4-((1,4-diazepan-1-yl)methyl)-2-fluorophenyl)-3-((1r,3R,5S,7r)-3,5-dimethyladamantan-1-yl)urea 32 (4.15 g, 9.70 mmol) and 2-(1-(tert-butoxycarbonyl)piperidin-4-yl)acetic acid (2.72 g, 11.64 mmol) as starting material, compound 33, which was used without further purification, ESI-MS: m/z 654.5 [M+H]+.
1-((1r,3R,5S,7r)-3,5-Dimethyladamantan-1-yl)-3-(2-fluoro-4-((4-(2-(piperidin-4-yl) acetyl)-1,4-diazepan-1-yl)methyl)phenyl)urea (34).
According to the synthetic process of compound 20, compound 34 as a light yellow oil (3.77 g) was obtained by taking used tert-butyl-4-(2-(4-(4-(3-((1r,3R,5S,7r)-3,5-dimethyladamantan-1-yl)ureido)-3-fluorobenzyl)-1,4-diazepan-1-yl)-2-oxoethyl)piperidine-1-carboxylate 33 (4.94 g, 7.57 mmol) as starting material, compound 34, which was used without further purification, ESI-MS: m/z 554.43 [M+H]+.
1-(4-((4-(2-(1-Acetylpiperidin-4-yl)acetyl)-1,4-diazepan-1-yl)methyl)-2-fluorophenyl)-3-((1r,3R,5S,7r)-3,5-dimethyladamantan-1-yl)urea (I1).
According to the synthetic process of compound 5, compound I1 as a white solid (0.20 g, 63 %) was obtained by taking 1-((1r,3R,5S,7r)-3,5-dimethyladamantan-1-yl)-3-(2-fluoro-4-((4-(2-(piperidin-4-yl)acetyl)-1,4-diazepan-1-yl)methyl)phenyl)urea 34 (0.3 g, 0.54 mmol) and acetic acid (39 mg, 0.65 mmol) as starting material, compound I1, m.p. 92–95°C. 1H NMR (400 MHz, CDCl3): δ (ppm) 8.05–8.00 (m, 1H), 7.11 (s, 1H), 7.03–6.99 (m, 2H), 6.95–6.93 (m, 1H), 5.47 (s, 1H), 4.63–4.57 (m, 1H), 3.81–3.78 (m, 1H), 3.72–3.65(m, 2H), 3.56–3.49 (m, 3H), 3.46–3.43 (m, 1H), 3.11–3.05 (m, 1H), 2.67 (s, 1H), 2.59–2.57 (m, 4H), 2.26–2.23 (m, 1H), 2.18 (s, 1H), 2.15–2.14 (m, 1H), 2.09 (s, 3H), 1.84 (s, 4H), 1.67 (s, 5H), 1.39–1.36 (m, 2H), 1.31–1.26 (m, 3H), 1.19–1.11 (m, 4H), 0.85 (s, 6H). 13C NMR (100 MHz, CDCl3): δ 171.12, 171.04, 169.01, 168.98, 154.25, 154.22, 151.15, 124.58, 120.72, 61.94, 61.58, 55.54, 55.34, 55.14, 53.68, 52.83, 52.81, 50.66, 48.19, 46.74, 46.70, 44.49, 42.75, 41.91, 41.86, 40.75, 39.51, 33.10, 32.80, 32.43, 31.98, 30.21, 30.14, 21.50. HRMS (ESI) calcd for C34H51FN5O3 [M+H]+: 596.3898, found 596.3972.
1-((1r,3R,5S,7r)-3,5-Dimethyladamantan-1-yl)-3-(2-fluoro-4-((4-(2-(1-propionyl piperidin-4-yl)acetyl)-1,4-diazepan-1-yl)methyl)phenyl)urea (I2).
According to the synthetic process of compound 5, compound I2 as a white solid (0.19 g, 59 %) was obtained by taking 1-((1r,3R,5S,7r)-3,5-dimethyladamantan-1-yl)-3-(2-fluoro-4-((4-(2-(piperidin-4-yl)acetyl)-1,4-diazepan-1-yl)methyl)phenyl)urea 34 (0.3 g, 0.54 mmol) and propionic acid (48 mg, 0.65 mmol) as starting material, compound I2, m.p. 86–89°C. 1H NMR (400 MHz, CDCl3): δ (ppm) 8.04–7.99 (m, 1H), 7.10–7.09 (s, 1H), 7.01–6.93 (m, 3H), 5.45–5.41 (m, 1H), 4.65–4.58 (m, 1H), 3.85–3.82 (d, J = 12.56 Hz, 1H), 3.71–3.43(m, 6H), 3.07–3.01 (m, 1H), 2.66 (s, 1H), 2.60–2.56 (m, 4H), 2.35 (q, J = 7.48 Hz, 2H), 2.26–2.14 (m, 4H), 1.85–1.84 (m, 4H), 1.67 (s, 5H), 1.39–1.36 (m, 2H), 1.30–1.27 (m, 3H), 1.16–1.12 (m, 6H), 0.85 (s, 6H). 13C NMR (100 MHz, CDCl3): δ 172.36, 172.32, 171.17, 171.10, 154.26, 154.23, 124.57, 120.86, 120.77, 61.93, 61.59, 55.55, 55.41, 55.07, 52.83, 52.81, 50.67, 48.18, 45.81, 45.76, 42.75, 40.75, 39.56, 39.54, 33.24, 33.21, 32.89, 32.43, 32.05, 30.22, 30.14, 26.65, 26.64, 9.72, 9.68. HRMS (ESI) calcd for C35H53FN5O3 [M+H]+: 610.4054, found 610.4096.
1-((1r,3R,5S,7r)-3,5-Dimethyladamantan-1-yl)-3-(2-fluoro-4-((4-(2-(1-(2-methyl butanoyl)piperidin-4-yl)acetyl)-1,4-diazepan-1-yl)methyl)phenyl)urea (I3).
According to the synthetic process of compound 5, compound I3 as a white solid (0.19 g, 57 %) was obtained by taking 1-((1r,3R,5S,7r)-3,5-dimethyladamantan-1-yl)-3-(2-fluoro-4-((4-(2-(piperidin-4-yl)acetyl)-1,4-diazepan-1-yl)methyl)phenyl)urea 34 (0.3 g, 0.54 mmol) and 2-methylbutanoic acid (66 mg, 0.65 mmol) as starting material, compound I3, m.p. 96–100°C. 1H NMR (400 MHz, CDCl3): δ (ppm) 7.99 (t, J = 8.36 Hz, 1H), 7.17 (d, J = 7.48 Hz, 1H), 7.06 (s, 1H), 6.99 (s, 1H), 6.96–6.93 (m, 1H), 5.50–5.48 (m, 1H), 4.68–4.61 (m, 1H), 3.97–3.83 (d, J = 12.76 Hz, 1H), 3.75–3.44 (m, 6H), 3.08–3.02 (m, 1H), 2.65–2.55 (m, 6H), 2.27–2.14 (m, 4H), 1.84 (s, 5H), 1.66 (s, 5H), 1.39–1.36 (m, 3H), 1.30–1.26 (m, 3H), 1.16–1.14 (m, 2H), 1.11–1.08 (m, 4H), 0.89–0.87 (m, 3H), 0.84 (s, 6H). 13C NMR (100 MHz, CDCl3): δ 175.04, 171.22, 171.14, 154.31, 154.31, 124.56, 121.00, 120.87, 114.94, 114.75, 61.91, 61.59, 55.53, 55.43, 52.81, 52.78, 50.68, 48.20, 47.17, 45.81, 45.35, 44.49, 42.76, 42.18, 40.74, 39.56, 36.95, 33.37, 33.27, 30.22, 30.13, 28.46, 27.51, 27.06, 17.39, 17.20, 12.01, 11.91. HRMS (ESI) calcd for C37H57FN5O3 [M+H]+: 638.4367, found 638.4437.
Methyl-4-(2-(4-(4-(3-((1r,3R,5S,7r)-3,5-dimethyladamantan-1-yl)ureido)-3-fluorobenzyl)-1,4-diazepan-1-yl)-2-oxoethyl)piperidine-1-carboxylate (I4).
According to the synthetic process of compound 5, compound I4 as a white solid (0.20 g, 60 %) was obtained by taking 1-((1r,3R,5S,7r)-3,5-dimethyladamantan-1-yl)-3-(2-fluoro-4-((4-(2-(piperidin-4-yl)acetyl)-1,4-diazepan-1-yl)methyl)phenyl)urea 34 (0.3 g, 0.54 mmol) and methyl carbonic acid (49 mg, 0.65 mmol) as starting material, compound I4, m.p. 121–122°C. 1H NMR (400 MHz, CDCl3): δ (ppm) 8.05–8.00 (m, 1H), 7.24–7.20 (m, 1H), 7.00–6.93 (m, 2H), 5.61–5.60 (m, 1H), 4.13–4.12 (m, 2H), 3.68 (s, 3H), 3.64–3.59 (m, 2H), 3.53–3.51 (m, 3H), 3.46 (s, 1H), 2.78 (s, 2H), 2.64–2.60 (m, 2H), 2.56–2.55 (m, 2H), 2.25–2.19 (m, 2H), 2.13–2.12 (m, 1H),1.83–1.78 (m, 4H), 1.74–1.72 (m, 2H), 1.65 (s, 4H), 1.38–1.35 (m, 2H), 1.29–1.26 (m, 3H), 1.15–1.14 (m, 3H), 0.85 (s, 6H). 13C NMR (100 MHz, DMSO-d6): 171.32, 171.26, 156.04, 154.13, 124.56, 120.86, 114.84, 61.85, 61.85, 55.63, 55.58, 55.02, 53.77, 52.88, 52.56, 50.64, 48.19, 48.19, 47.15, 44.50, 44.11, 42.73, 40.76, 39.70, 39.66, 33.04, 32.15, 32.15, 30.20, 30.12, 30.12, 27.48. HRMS (ESI) calcd for C34H51FN5O4 [M+H]+: 612.3847, found 612.3920.
1-((1r,3R,5S,7r)-3,5-Dimethyladamantan-1-yl)-3-(2-fluoro-4-((4-(2-(1-(2-hydroxyethyl)piperidin-4-yl)acetyl)-1,4-diazepan-1-yl)methyl)phenyl)urea (I5).
According to the synthetic process of compound 12, compound I5 as a white solid (0.13 g, 42 %) was obtained by taking 1-((1r,3R,5S,7r)-3,5-dimethyladamantan-1-yl)-3-(2-fluoro-4-((4-(2-(piperidin-4-yl)acetyl)-1,4-diazepan-1-yl)methyl)phenyl)urea 34 (0.3 g, 0.54 mmol) and 2-chloroethan-1-ol (52 mg, 0.65 mmol) as starting material, compound I5, m.p. 101–102°C. 1H NMR (400 MHz, DMSO-d6): δ (ppm) 8.14 (s, 1H), 8.04–8.00 (m, 1H), 7.08–7.04 (m, 1H Hz), 6.96–6.94 (m, 1H),6.57 (s, 1H), 5.34 (s, 1H), 4.48 (s, 1H), 3.52–3.46 (m, 10 H), 2.90–2.88 (m, 2H), 2.59 (s, 1H), 2.44 (s, 3H), 2.21–2.17 (m, 2H), 2.08–2.05 (m, 3H), 1.75 (s, 4H), 1.65–1.62 (m, 4H), 1.57 (s, 4H), 1.34–1.31 (m, 4H), 1.11 (s, 2H), 0.82 (s, 6H). 13C NMR (100 MHz, DMSO-d6): δ 170.97, 154.17, 153.00, 150.61, 124.67, 120.04, 115.09, 114.90, 63.05, 60.72, 60.62, 58.61, 56.03, 55.22, 54.52, 54.01, 53.79, 52.48, 51.99, 50.75, 48.09, 47.75, 46.73, 44.99, 44.39, 42.80, 32.66, 32.37, 31.90, 30.55, 30.05, 28.50, 27.42, 7.66. HRMS (ESI) calcd for C34H53FN5O3 [M+H]+: 598.4054, found 598.4125.
1-(4-((4-(2-(1-(2,3-Dihydroxypropyl)piperidin-4-yl)acetyl)-1,4-diazepan-1-yl)methyl)-2-fluorophenyl)-3-((1r,3R,5S,7r)-3,5-dimethyladamantan-1-yl)urea (I6).
According to the synthetic process of compound 12, compound I6 as a white solid (0.13 g, 38 %) was obtained by taking 1-((1r,3R,5S,7r)-3,5-dimethyladamantan-1-yl)-3-(2-fluoro-4-((4-(2-(piperidin-4-yl)acetyl)-1,4-diazepan-1-yl)methyl)phenyl)urea 34 (0.3 g, 0.54 mmol) and 3-chloropropane-1,2-diol (71 mg, 0.65 mmol) as starting material, compound I6, m.p. 104–105°C. 1H NMR (400 MHz, DMSO-d6): δ (ppm) 8.14 (s, 1H), 8.03–7.99 (m, 1H), 7.09–7.04 (m, 1H Hz), 6.98–6.94 (m, 1H),6.57 (s, 1H), 5.33 (s, 4H), 5.01 (s, 1H), 3.67 (s, 1H), 3.52–3.43 (m, 10 H), 2.99 (s, 3H), 2.60–2.59 (m, 2H), 2.21–2.19 (m, 4H), 2.09 (s, 1H), 1.75 (s, 5H), 1.68 (s, 5H), 1.57 (s, 3H), 1.34 (s, 1H), 1.31 (s, 1H), 1.11 (s, 2H), 0.82 (s, 6H). 13C NMR (100 MHz, DMSO-d6): δ 170.90, 154.18, 127.62, 124.68, 120.08, 65.23, 63.07, 56.03, 55.20, 54.51, 53.83, 52.50, 52.50, 50.76, 48.10, 48.10, 47.74, 46.71, 44.41, 42.80, 42.80, 32.38, 32.38, 30.55, 30.55, 30.05, 28.48, 27.39, 7.67, 7.67. HRMS (ESI) calcd for C35H55FN5O4 [M+H]+: 628.4160, found 628.4230.
1-((1r,3R,5S,7r)-3,5-Dimethyladamantan-1-yl)-3-(4-((4-(2-(1-(2-(dimethylamino) ethyl)piperidin-4-yl)acetyl)-1,4-diazepan-1-yl)methyl)-2-fluorophenyl)urea (I7).
According to the synthetic process of compound 12, compound I7 as a white solid (0.13 g, 39 %) was obtained by taking 1-((1r,3R,5S,7r)-3,5-dimethyladamantan-1-yl)-3-(2-fluoro-4-((4-(2-(piperidin-4-yl)acetyl)-1,4-diazepan-1-yl)methyl)phenyl)urea 34 (0.3 g, 0.54 mmol) and 2-chloro-N,N-dimethylethan-1-amine (69 mg, 0.65 mmol) as starting material, compound I7, m.p.108–109°C. 1H NMR (400 MHz, CDCl3): δ (ppm) 8.08–8.01 (m, 1H), 7.01–6.96 (m, 1H), 6.94–6.91 (m, 1H), 5.62 (s, 2H), 3.62–3.61 (m, 5H), 3.54 (s, 1H), 3.51–3.48 (m, 2H), 3.44–3.42 (m, 1H), 3.08–3.06 (m, 2H), 2.72 (s, 1H), 2.66 (s, 1H), 2.61 (s, 3H), 2.57–2.55 (m, 1H), 2.43 (s, 2H), 2.37 (s, 2H), 2.32 (s, 1H), 2.25–2.23 (m, 2H), 2.20–2.18 (m, 1H), 2.13 (s, 1H), 1.95 (s, 1H), 1.88 (s, 2H), 1.80–1.77 (m, 4H), 1.70 (s, 3H), 1.39 (s, 1H), 1.36 (s, 1H), 1.25 (s, 4H), 1.16 (s, 1H), 1.13 (s, 1H), 0.84 (s, 6H). 13C NMR (100 MHz, CDCl3): δ 171.33, 154.79, 154.73, 124.48, 120.86, 114.79, 114.61, 63.43, 62.07, 61.59, 53.95, 53.01, 53.01, 52.75, 50.74, 48.07, 47.26, 45.30, 45.15, 44.34, 42.83, 40.70, 39.37, 32.51, 32.51, 32.40, 31.62, 31.53, 30.20, 29.69, 28.69, 27.92, 8.13. HRMS (ESI) calcd for C36H58FN6O2 [M+H]+: 625.4527, found 625.4597.
1-(4-((4-(2-(1-(2-(Diethylamino)ethyl)piperidin-4-yl)acetyl)-1,4-diazepan-1-yl)methyl) −2-fluorophenyl)-3-((1r,3R,5S,7r)-3,5-dimethyladamantan-1-yl)urea (I8).
According to the synthetic process of compound 12, compound I8 as a white solid (0.11 g, 32 %) was obtained by taking 1-((1r,3R,5S,7r)-3,5-dimethyladamantan-1-yl)-3-(2-fluoro-4-((4-(2-(piperidin-4-yl)acetyl)-1,4-diazepan-1-yl)methyl)phenyl)urea 34 (0.3 g, 0.54 mmol) and 2-chloro-N,N-diethylethan-1-amine (88 mg, 0.65 mmol) as starting material, compound I8, m.p.111–112°C. 1H NMR (400 MHz, CDCl3): δ (ppm) 8.06–7.98 (m, 1H), 6.94–6.89 (m, 1H), 6.83–6.77 (m, 1H), 5.57 (s, 2H), 4.55 (s, 3H), 3.57–3.55 (m, 5H), 3.46–3.43(m, 3H), 3.35 (s, 1H), 3.10–3.07 (m, 1H), 3.01–2.98 (m, 2H), 2.95–2.93 (m, 2H), 2.86–2.84 (m, 2H), 2.80–2.78 (m, 2H), 2.63 (s, 1H), 2.55 (s, 2H), 2.51 (s, 1H), 2.23–2.15 (m, 2H), 2.05 (s, 1H), 1.91 (s, 1H), 1.82 (s, 1H), 1.74 (s, 3H), 1.65 (s, 2H), 1.32–1.29 (m, 2H), 1.24 (s, 1H), 1.22–1.21 (m, 3H), 1.18–1.15 (m, 5H), 1.09 (s, 1H), 1.05 (s, 1H), 0.77 (s, 6H). 13C NMR (100 MHz, CDCl3): δ 171.29,171.10, 154.99, 154.90, 124.63, 63.47, 61.60, 55.76, 55.44, 55.13, 54.78, 53.82, 53.04, 53.04, 52.69, 50.78, 48.31, 48.02, 48.02, 47.09, 44.09, 42.87, 40.68, 39.19, 32.38, 32.38, 32.07, 31.46, 31.19, 30.23,30.23, 29.68, 8.15. HRMS (ESI) calcd for C38H62FN6O2 [M+H]+: 653.4840, found 653.4910.
Biological activity assays in vitro.
All HsEH and MsEH IC50 values is the concentration of a compound that reduces the sEH activity by 50%. The IC50 values were determined using a fluorescent-based assay [(3-phenyloxiranyl) acetic acid cyano(6-methoxynaphthalen-2-yl)methyl ester (PHOME) as the substrate]31.The fluorescent assay was used with purified recombinant human, or mouse sEH proteins. The enzymes were incubated at 37 °C with the inhibitors ([I]final = 0.4 – 100,000 nM) for 10 min in 25 mM Tris HCl buffer (pH = 7.4) containing 0.1 mg/mL of BSA and 1% of DMSO. The substrate (PHOME) was then added ([S]final = 50 mM). Activity was assessed by measuring the formation of the fluorescent 6-methoxynaphthaldehyde product (lex = 330 nm, lem = 465 nm) on a SpectraMax M2 (molecular devices). Results were obtained by regression analysis from a linear region of the curve.
Microsomal stability.
Human and SD rat hepatic microsomes were purchased from Research Institute for Liver Diseases (Shanghai, China) Co., Ltd. The incubation mixture consisted of microsomal protein in PBS buffer (100 mM, pH = 7.4) and 100 μg / mL G1 in a final volume of 100 mL. The concentration of human hepatic microsomal protein was 0.5 mg/mL (0.53 mg / mL of SD rat). A 0.83 mg/mL NADPH solution was prepared and added to the PBS buffer. After the addition of the NADPH-generating system, the resulting mixture was incubated at 37 °C for 0, 10, 30, 60 min. The reaction was terminated by the addition of methanol 300 μL containing diphenhydramine (0.02 ng / mL). The mixture was vortexed for 1 min, centrifuged at 21,528 × g for 10 min at 4 °C. After centrifugation, 200 μL of the supernatant was transferred to 96-well plate, and 400 mL of purified water was added, and the solution was mixing with shaker at 500 rpm for 5 min, and the final concentration of compound G1 was analyzed by HPLC.
Pharmacokinetics study in vivo.
SPF-grade healthy Sprague-Dawley (3 females, 3 males, 8 weeks old), weighing 180 ± 20 g, were provided by Liaoning Changsheng Biotechnology Co., Ltd (Liaoning, China), license No. SCXK (Liao) 2015–0001. SD rats were housed under controlled environmental conditions at 22 – 24 °C, 50 – 60 % relative humidity, natural circadian rhythm, free access to water and food, and acclimatized for one week. The experiments were approved by the Animal Management and Use Committee of Shenyang Pharmaceutical University, and the experiments conformed to the relevant experimental animal research guidelines of the Ethics Committee. Six SD rats were randomly divided into two groups (half male and half female). Weigh before each dose to adjust the required volume. Three SD rats were given 10 mg / kg of compound G1 by tail vein injection, and the other three SD rats were given 50 mg / kg of compound G1 by gavage. Blood samples (iv groups) were collected at different times (0 min, 5 min, 15 min, 30 min, 1 h, 2 h, 4 h, 6 h, 8 h). Blood samples (po groups) were collected at different times (0 min, 10 min, 30 min, 1h, 2h, 4h, 6h, 8h). Blood was collected from the orbital vein on an Eppendorf tube (1.5 mL) containing EDTA and then centrifuged at 4800 rpm for 10 minutes. Each plasma sample (50 μL) was mixed with 150 μL tolbutamide in acetonitrile (5 ng / mL) and centrifuged at 13000 rpm for 10 min. Afterwards, 50 μL of supernatant was reconstituted with 200 μL of acetonitrile and the final concentration of compound G1 was analyzed by HPLC.
Percentage plasma protein binding (% PPB).
The detailed experimental procedure followed the procedures reported of our previously work31. The experimental assay was calibrated using warfarin of known % PPB: rat plasma (99.5 %).
In Vivo Efficacy.
All mice used for pharmacological experiments were C57BL/6 (males, body weight 20 – 22 g) and Kunming (female, weight 20–22 g) provided by Liaoning Changsheng Biotechnology Co., Ltd (Liaoning, China), License No. SCXK (Liao) 2015–0001. Mice were kept under controlled environmental conditions at 22 – 24 °C, 50 – 60 % relative humidity, natural circadian rhythm, free access to water and food, and acclimatized for one week. The experiments were approved by the Animal Management and Use Committee of Shenyang Pharmaceutical University, and the experiments conformed to the relevant experimental animal research guidelines of the Ethics Committee.
Evaluation of the efficacy of compound G1 in the AIA model.
Twenty-four Kunming mice (female, body weight 20 −22 g) were randomly divided into three groups, namely CFA, CFA + G1 and CFA + Celecoxib groups. The mice were fasted for 6 – 8 h and freely drinking water before the experiment. Each mouse was weighed and the basal pain threshold (s) was measured by XR1700 hot plate meter and the thickness and width of each mouse’s paw was measured by micrometer, and then CFA 25μL / 25 g was injected right in the middle of the paw, and the mice showed obvious redness and swelling after modeling, and the behavior was significantly reduced, indicating successful modeling. The mice were weighed again 24 h after modeling and the pain threshold (s) was measured. Compound G1 and celecoxib were dissolved in 10 % DMSO, 5 % Tween 80 and 85 % 0.9 % saline and injected intraperitoneally at the dose of G1 and celecoxib (10 mg / kg, ip), and the same volume of 0.9 % saline was given to the CFA group. The pain threshold (s) of each mouse was measured at 10 min, 45 min, 120 min, and 240 min after drug administration, and the increase in pain threshold (%) = (pain threshold measured after drug administration (s) - pain threshold after CFA modeling (s) / pain threshold after CFA modeling × 100 %, and the thickness and width of the paw of each mouse were also measured by micrometer at 1 h, 3 h, 6 h, and 20 h after drug administration. The same volume of 0.9 % saline was given to the CFA group. The thickness and width of the paw of each mouse were measured by micrometer at 24h and 28h. Edema (%) = (m / thickness or width of the paw of the mouse before CFA modeling) × 100 % (m = thickness or width of the paw measured after drug administration - thickness or width of the paw of the mouse before CFA modeling).
Evaluation of the efficacy of compound G1 in the AP model.
Twenty Kunming mice (females, body weight 20 – 22 g) were randomly divided into five groups: solvent group, model group (L-Arg), G1(3 mg / kg) group, G1(8 mg / kg) group, and celecoxib group. The mice were fasted for 6–8 h and freely drinking water before the experiment. Each mouse was weighed and injected intraperitoneally with 20 % L-arginine (2 g / kg, ip) except for the solvent group, which was re-injected with 20% L-arginine (2 g / kg, ip) after an interval of 1 h. After 14 h of modeling, G1(3 mg / kg, ip) group, G1(8 mg / kg, ip) and celecoxib (5 mg / kg, ip) were dissolved in 10 % DMSO, 5 % Tween 80 and 85 % 0.9 % saline. G1 and celecoxib (5 mg / kg, ip) were injected intraperitoneally, and the solvent group, and the modeling group (L-Arg) were injected intraperitoneally with an equal volume of 0.9 % saline. At 10 h of drug treatment, each group of mice was executed and the pancreatic tissue was removed and fixed in 10% formalin for the next step of sectioning. Basic sectioning process: embedding sections, dewaxing sections to water, staining, dehydration, transparency, sealing, slide scanning.
Evaluation of the efficacy of compound G1 in LPS-induced sepsis model.
Fifty C57BL/6 mice (males, body weight 20 – 22 g) were randomly divided into four groups: solvent group (12 mice), model group (LPS, 12 mice), G1 group (14 mice) and dexamethasone group (12 mice). The mice were fasted for 6–8 h and freely drinking water before the experiment. After each mouse was weighed, except for the solvent group Which each mouse was injected intraperitoneally with LPS (30 mg / kg), and 4 h later compound G1 and dexamethasone were dissolved with 10 % DMSO, 5 % Tween 80 and 85 % 0.9 % saline and injected intraperitoneally at the dose of G1 and dexamethasone (5 mg / kg), and the solvent group, and the LPS group were injected intraperitoneally with an equal volume of 0.9 % saline. Six mice from each group were executed at 12 h of drug treatment to obtain blood for Western blot and ELISA assays. Immediately afterwards, mice were injected intraperitoneally second time at the dose of compound G1 (5 mg / kg) and dexamethasone (5 mg / kg) and observed for death per hour.
Western blot. Total protein extraction and western blot were performed according to our previous study. The plasma of experimental animals was centrifuged at 5000rmp for 5min. The protein concentration determined with BCA Protein Assay Kit (Fdbiio science cat: FD2001). The proteins were mixed with loading buffer and denatured at 100 °C for 10 min. Then the lysates were separated using an SDS-PAGE gel, and the proteins were transferred onto 0.45-μm polyvinylidene difluoride (PVDF) membranes. The membranes were blocked with 5 % fat-free milk and were incubated overnight at 4 °C with mouse anti-COX-2 antibody (1: 2000, SANTA CRUZ Biothechnology, cat: sc-19999, USA), mouse anti-NOS-2 antibody (SANTA CRUZ Biothechnology, cat: sc-7271, USA), mouse anti-sEH antibody (1: 2000, SANTA CRUZ Biothechnology, cat: sc-166961, USA), mouse anti-VCAM-1 antibody (1: 2000, SANTA CRUZ Biothechnology, cat: sc13160, China), mouse anti-GAPDH antibody (1: 4000, Zhongshanjinqiao, cat: TA-08, China). Subsequently, the membranes were washed three times with TBST and were incubated for 1 h at room temperature with anti-mouse (COX-2, NOS2, sEH, VCAM-1, GAPDH, zhongshanjinqiao, cat: ZB-2305) horseradish peroxidase-conjugated secondary antibodies. Film was used to develop protein in darkroom.The expression of proteins in the plasma was respectively normalized to GAPDH as a loading control.
Enzyme-linked immunosorbent assay (ELISA).
The protein level of monocyte chemoattractant protein-5 (MCP-5), (Boster, Cat: EK1128, China), tumor necrosis factor α (TNF-α), (Multi Sciences, Cat: 70-EK282 / 4–96, China), interleukin-6 (IL-6), (Multi Sciences, Cat: EK206/3, China) in serum was measured using an ELISA kit according to the manufacturer’s instructions. The concentrations of the MCP-5, TNF-α, IL-6 were quantified by reference to a standard curve.
Molecular Docking.
The X-ray crystal structures of sEH (PDB ID: 3WKE) was retrieved from the Protein Data Bank. The protein structures were prepared using the Protein Preparation Wizard module. Hydrogen atoms were added, water molecules in the entire system were removed, followed by energy minimization and optimization by the Discovery Studio 2016 software. Grids of sEH was generated using receptor-ligand interactions, following the standard procedure recommended by Discovery Studio 2016 software. The small molecules was employed to prepare the compounds for molecular docking. To prepare ligand structures, hydrogens were added, and 3D geometries, ionization, and tautomeric states were generated. Finally, the ligand structures were minimized using the full minimization. The conformational ensembles were docked flexibly using libdock. Only poses with low energy conformations and good hydrogen-bond geometries were considered.
Supplementary Material
ACKNOWLEDGMENTS
This work was funded by the National Natural Science Foundation of China (No. 82273793), Key Research Project of Department of Education of Liaoning Province(LJKZZ20220108), the Liaoning Revitalization Talents Program (XLYC1908031, China), NIEHS RIVER Award R35ES030443, NINDS Counter Act Program U54NS127758, and NIEHS/Superfund Research Program P42 ES004699.
ABBREVIATIONS USED
- sEH
soluble epoxide hydrolase
- WB
Western Blot
- ELISA
enzyme linked immunosorbent assay
- COX-2
cyclooxygenase-2
- NOS-2
nitric oxide synthase-2
- VCAM
vascular cell adhesion molecule
- IL-6
interleukin-6
- MCP-5
monocyte chemotactic protein-5
- TNF-α
tumor necrosis factor-α
- NF-κB
nuclear factor B
- EET
epoxyeicosatrienoic acids
- EPHX2
epoxide hydrolase 2
- ER
endoplasmic reticulum
- HATU
2-(7-Azabenzotriazol-1-yl)-N,N,N’,N’-tetramethyluronium hexafluorophosphate
- DIPEA
N,N-diisopropylethylamine
- NADPH
nicotinamide adenine dinucleotide phosphate
- TFA
trifluoroacetic acid
Footnotes
Supporting Information
This material is available free of charge via the Internet at http://pubs.acs.org. Supplemental figures and tables; NMR, MS spectra; and HPLC purity traces for all final compounds demonstrating ≥ 95% purity (PDF),
Molecular formula strings (CSV)
compound G1 complex (PDB)
compound A complex (PDB)
The authors declare no competing financial interes
REFERENCES
- (1).Petrov MS; Yadav D Global epidemiology and holistic prevention of pancreatitis. Nat. Rev Gastro. Hepat 2019, 16, 175–184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (2).Xiao AY; Tan ML; Wu LM; Asrani VM; Windsor JA; Yadav D; Petrov MS Global incidence and mortality of pancreatic diseases: a systematic review, meta-analysis, and meta-regression of population-based cohort studies. Lancer. Gastroenterol 2016, 1, 45–55. [DOI] [PubMed] [Google Scholar]
- (3).Mei F; Yu J; Li M; Xiang M; Hong Y; Zhou Y; You Y; Xia H; Jin H; Wang W Magnesium isoglycyrrhizinate alleviates liver injury in obese rats with acute necrotizing pancreatitis. Pathol. Res Pract 2019, 215, 106–114. [DOI] [PubMed] [Google Scholar]
- (4).Yadav D; Lowenfels AB The epidemiology of pancreatitis and pancreatic cancer. Gastroenterology 2013, 144, 1252–1261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (5).Forsmark CE; Vege SS; Wilcox CM Acute pancreatitis. N. Engl. J. Med 2016, 375, 1972–1981. [DOI] [PubMed] [Google Scholar]
- (6).Krishna SG; Kamboj AK; Hart PA; Hinton A; Conwell DL The changing rpidemiology of acute pancreatitis hospitalizations: a decade of trends and the impact of chronic pancreatitis. Pancreas 2017, 46, 482–488. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (7).Kylanpaa L; Rakonczay Z; Oreilly DA The clinical course of acute pancreatitis and the inflammatory mediators that drive it. Int. J. Inflam 2012, 360685–360695. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (8).Bettaieb A; Chahed S; Bachaalany S; Griffey S; Hammock BD; Haj FG Soluble epoxide hydrolase pharmacological inhibition ameliorates experimental acute pancreatitis in mice. Mol. Pharmacol 2015, 88, 281–290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (9).Codony S; Pujol E; Pizarro J; Feixas F; Valverde E; Loza MI; Brea JM; Saez E; Oyarzabal J; Pineda-Lucena A; Perez B; Perez C; Rodriguez-Franco MI; Leiva R; Osuna S; Morisseau C; Hammock BD; Vazquez-Carrera M; Vazquez S 2-Oxaadamant-1-yl ureas as soluble epoxide hydrolase inhibitors: in vivo evaluation in a murine model of acute pancreatitis. J. Med. Chem 2020, 63, 9237–9257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (10).Zhu LP; Wang J; Wei TT; Gao J; He H; Chang XY; Yan TH Effects of naringenin on inflammation in complete freund′s adjuvant-induced arthritis by regulating bax / bcl-2 balance. Inflammation 2015, 38, 245–251. [DOI] [PubMed] [Google Scholar]
- (11).Woolf CJ; Ma Q Nociceptors-noxious stimulus detectors. Neuron 2007, 55, 353–364. [DOI] [PubMed] [Google Scholar]
- (12).Sultana F; Rasool MK A novel therapeutic approach targeting rheumatoid arthritis by combined administration of morin, a dietary flavanol and non-steroidal anti-inflammatory drug indomethacin with reference to pro-inflammatory cytokines, infl ammatory enzymes, RANKL and transcription factor. Chem-Biol. Interact 2015, 230, 58–70. [DOI] [PubMed] [Google Scholar]
- (13).Burmester GR; Stuhlmuller B; Keyszer G; Kinne RW Mononuclear phagocytes and rheumatoid synovitis. Mastermind or workhorse in arthritis?. Arthritis Rheum-Us, 1997, 40, 5–18. [DOI] [PubMed] [Google Scholar]
- (14).Schmelzer KR; Inceoglu B; Kubala L; Kim IH; Jinks S; Eiserich JP; Hammock BD Enhancement of antinociception by coadministration of nonsteroidal anti-inflammatory drugs and soluble epoxide hydrolase inhibitors. Proc. Natl. Acad. Sci. U.S.A 2006,103, 13646–13651. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (15).Schmelzer KR; Kubala L; Newman JW; Kim IH; Eiserich JP; Hammock BD Solubleepoxidehydrolaseisa therapeutictargetforacuteinflammation. Proc. Natl. Acad. Sci. U.S.A 2005, 102, 9772–9777. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (16).Inceoglu B; Jinks SL; Schmelzer KR; Waite T; Kim IH; Hammock BD Inhibition of soluble epoxide hydrolase reduces LPS-induced thermal hyperalgesia and mechanical allodynia in a rat model of inflammatory pain. Life. Sci 2006, 79, 2311–2319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (17).Fitzgerald GA; Coxibs and cardiovascular disease. N. Engl. J. Med 2004, 351, 1709–1711. [DOI] [PubMed] [Google Scholar]
- (18).Kiss L; Schuette H; Padberg W; Weissmann N; Mayer K; Gessler T; Voswinckel R; Seeger W; Grimminger F Epoxyeicosatrienoates are the dominant eicosanoids in human lungs upon microbial challenge. Eur. Respir. J 2010, 36, 1088–1098 [DOI] [PubMed] [Google Scholar]
- (19).Pfister SL; Gauthier KM; Campbell WB Vascular pharmacology of epoxyeicosatrienoic acids. Advances in Pharmacology 2010, 60, 27–59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (20).Ingraham RH; Gless RD Soluble epoxide hydrolase inhibitors and their potential for treatment of multiple pathologic conditions. Curr. med. chem 2011, 18, 587–603. [DOI] [PubMed] [Google Scholar]
- (21).Fang Q; Chen GZ; Wang Y; Wang DW Role of cytochrome P450 epoxygenase-dependent arachidonic acid metabolites in kidney physiology and diseases. Acta Physiologica Sinica, 2018, 70, 591–599. [PubMed] [Google Scholar]
- (22).Wu S; Chen W; Murphy E; Gabel S; Tomer KB; Foley J; Steenbergen C; Falck JR; Moomaw CR; Zeldin DC Molecular cloning, expression, and functional significance of a cytochrome P450 highly expressed in rat heart myocytes. J. Biol. Chem 1997, 272, 12551–12559. [DOI] [PubMed] [Google Scholar]
- (23).Seubert J; Yang B; Bradbury JA; Graves J; Degraff LM; Gabel S; Gooch R; Foley J; Newman J; Mao L; Rockman HA; Hammock BD; Murphy E; Zeldin DC Enhanced postischemic functional recovery in CYP2J2 transgenic hearts involves mitochondrial ATP-sensitive K+ channels and p42 / p44 MAPK pathway. Circ. Res 2004, 95, 506–514. [DOI] [PubMed] [Google Scholar]
- (24).Wu J; Zhao Y; Fan Z Soluble epoxide hydrolase inhibitor protects against blood-brain barrier dysfunction in a mouse model of type 2 diabetes via the AMPK/HO-1 pathway. Biochem Biophys Res Commun 2020, 524, 354–359. [DOI] [PubMed] [Google Scholar]
- (25).Hammock BD; McReynolds CB; Wagner K; Buckpitt A; Cortes-Puch I; Croston G; Lee KSS; Yang J; Schmidt WK; Hwang SH Movement to the clinic of soluble epoxide hydrolase inhibitor EC5026 as an analgesic for neuropathic pain and for use as a nonaddictive opioid alternative. J. Med. Chem 2021, 64, 1856–1872 [DOI] [PMC free article] [PubMed] [Google Scholar]
- (26).Cecconi M; Evans L; Levy M; Rhodes A Sepsis and septic shock. Lancet 2018, 392, 75–87. [DOI] [PubMed] [Google Scholar]
- (27).Rello J; Valenzuela-Sanchez F; Ruiz-Rodriguez M; Moyano S Sepsis: a review of advances in management. Adv. Ther 2017, 34, 2393–2411. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (28).Yang L; Xie X; Tu Z; Zhou Y; Yang L; Xie X; Tu Z; Zhou Y; Fu J; Xu D; Xu D The signal pathways and treatment of cytokine storm in COVID-19. Signal. Transduct. Tar 2021, 6, 255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (29).Vanderbeke L; Van Mol P; Van Herck Y; De Smet F; Humblet-Baron S; Martinod K; Antoranz A; Arijs I; Boeckx B; Bosisio FM; Casaer M; Dauwe D; De Wever W; Dooms C; Dreesen E; Emmaneel A; Filtjens J; Gouwy M; Gunst J; Hermans G; Jansen S; Lagrou K; Liston A; Lorent N; Meersseman P; Mercier T; Neyts J; Odent J; Panovska D; Penttila PA; Pollet E; Proost P; Qian J; Quintelier K; Raes J; Rex S; Saeys Y; Sprooten J; Tejpar S; Testelmans D; Thevissen K; Van Buyten T; Vandenhaute J; Van Gassen S; Velasquez Pereira LC; Vos R; Weynand B; Wilmer A; Yserbyt J; Garg AD; Matthys P; Wouters C; Lambrechts D; Wauters E; Wauters J Monocyte-driven atypical cytokine storm and aberrant neutrophil activation as key mediators of COVID-19 disease severity. Nat. Commun 2021, 12, 4117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (30).Conti P; Ronconi G; Caraffa Al.; Gallenga CE; Ross R; Frydas I; And Kritas SK Induction of pro-inflammatory cytokines (IL-1 and IL-6) and lung inflammation by coronavirus-19 (COVI-19 or SARS-CoV-2): anti-inflammatory strategies, J. Biol. Reg. Homeos. Ag 2020, 34 327–331. [DOI] [PubMed] [Google Scholar]
- (31).Mehta P; McAuley DF; Brown M; Sanchez E; Tattersall RS; Manson JJ COVID-19: consider cytokine storm syndromes and immunosuppression. Lancet 2020, 395, 1033–1034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (32).Calzetta L; Aiello M; Frizzelli A; Rogliani P; Chetta A Dexamethasone in patients hospitalized with COVID-19: whether, when and to whom. J. Clin. Med 2021, 10, 1607–1613. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (33).Tomazini BM; Maia IS; Cavalcanti AB; Berwanger O; Rosa RG; Veiga Vi. C.; Avezum A; Lopes RD; Bueno FR; Silva MVAO; Baldassare FP; Costa ELV; Moura RAB; Honorato MO; Costa AN; Damiani LP; Lisboa T; Kawano-Dourado L; Zampieri FG; Olivato GB; Righy C; Amendola CP; Roepke RML; Freitas DHM; Forte DN; Freitas FGR; Fernandes CCF; Melro LMG; Gedealvares FSJ; Morais DC; Machado FR; Azevedo LCP Effect of dexamethasone on days alive and ventilator-free in patients with moderate or severe acute respiratory distress syndrome and COVID-19 the codex ran-domized clinical trial. JAMA-J. Am. Med. Assoc 2020, 324 1307–1316. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (34).Inceoglu B; Bettaieb A; Trindade S, Carlos A; Lee KSS; Haj FG; Hammock BD Endoplasmic reticulum stress in the peripheral nervous system is a significant driver of neuropathic pain. Proc. Natl. Acad. Sci. U.S.A 2015, 112, 9082–9087. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (35).Chen D; Whitcomb R; MacIntyre E; Tran V; Do ZN; Sabry J; Patel DV; Anandan SK; Gless R; Webb HK Pharmacokinetics and pharmacodynamics of AR9281, an inhibitor of soluble epoxide hydrolase, in single-and multiple-dose studies in healthy human subjects. J. Clin. Pharmacol 2012, 52, 319–328. [DOI] [PubMed] [Google Scholar]
- (36).Lazaar AL; Yang L; Boardley RL; Goyal NS; Robertson J; Baldwin SJ; Newby DE; Wilkinson IB; Tal-Singer R; Mayer RJ; Cheriyan J Pharmaco-kintics, pharmacodynamics and adverse event profile of GSK2256294, a novel soluble epoxide hydrolase inhibitor. Br. J. Clin. Pharmacol 2016, 81, 971–979. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (37).Lee KSS; Ng JC; Yang J; Hwang SH; Morisseau C; Wagner K; Hammock BD Preparation and evaluation of soluble epoxide hydrolase inhibitors with improved physical properties and potencies for treating diabetic neuropathic pain, Bioorg. Med. Chem 2020, 28, 115735–115746. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (38).Du F; Sun W; Morisseau C; Hammock BD; Bao X; Liu Q; Wang C; Zhang T; Yang H; Zhou J; Xiao W; Liu Z; Chen G Discovery of memantyl urea derivatives as potent soluble epoxide hydrolase inhibitors against lipopolysaccharide-induced sepsis. Eur. J. Med. Chem 2021, 223, 113678–113692. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (39).Du F; Cao R; Chen L; Sun J.n; Shi Y; Fu Y; Hammock BD; Zheng Z; Liu Z; Chen G Structure-guided discovery of potent and oral soluble epoxide hydrolase inhibitors for the treatment of neuropathic pain. Acta. Pharm. Sin. B 2022, 12, 1377–138 [DOI] [PMC free article] [PubMed] [Google Scholar]
- (40).Wolf NM; Morisseau C; Jones PD; Hock B; Hammock BD Development of a high-throughput screen for soluble epoxide hydrolase inhibition. Anal Biochem 2006, 355, 71–80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (41).Gopal VB; Rojatkarb RS; Bodhankar L Anti-arthritic activity of methanol extract of cyathocline purpurea (whole plant) in Freund’s complete adjuvant-induced arthritis in rats. Biomed. Ag. Pathology 2014, 4: 197–206. [Google Scholar]
- (42).Andersen ML; Santos EHR; Seabra MV; da Silva AAB; Tufik Se. Evaluation of acute and chronic treatments with harpagophytum procumbens on freund’s adjuvant-induced arthritis in rats. J. Ethnopharmacol 2004, 91, 325–330. [DOI] [PubMed] [Google Scholar]
- (43).Thiemermann C Nitric oxide and septic shock. Gen. Pharmacol 1997, 29, 159–166. [DOI] [PubMed] [Google Scholar]
- (44).Machnicki M Lactoferrin regulates production of interleukin-6 and tumor necrosis factor alpha in mice. Postepy. Hig. Med. Dosw 1995, 49, 53–57. [PubMed] [Google Scholar]
- (45).Nakashima T; Yoshida Y; Miyata S; Kiyohara T Hypothalamic 11,12-epoxyeicosatrienoic acid attenuates fever induced by central interleukin-1β in the rat. Neurosci. Lett 2001, 310, 141–144. [DOI] [PubMed] [Google Scholar]
- (46).Kozak W; Kluger MJ; Kozak A; Wachulec M; Dokladny K Role of cytochrome P-450 in endogenous antipyresis. Am. J. Physiol 2000, 279, R455–R460. [DOI] [PubMed] [Google Scholar]
- (47).Kopydlowski KM; Salkowski CA; Cody MJ; van Rooijen N; Major J; Hamilton TA; Vogel SN Regulation of macrophage chemokine expression by lipopolysaccharide in vitro and in vivo. J. Immunol 1999, 163, 1537–1544. [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.