

Abstract
The pore-forming BCL-2 family proteins are effectors of mitochondrial poration in apoptosis initiation. Two atypical effectors—BOK and truncated BID (tBID)—join the canonical effectors BAK and BAX. Gene knockout revealed developmental phenotypes in the absence the effectors, supporting their roles in vivo. During apoptosis effectors are activated and change shape from dormant monomers to dynamic oligomers that associate with and permeabilize mitochondria. BID is activated by proteolysis, BOK accumulates on inhibition of its degradation by the E3 ligase gp78, while BAK and BAX undergo direct activation by BH3-only initiators, autoactivation, and crossactivation. Except tBID, effector oligomers on the mitochondria appear as arcs and rings in super-resolution microscopy images. The BH3-in-groove dimers of BAK and BAX, the tBID monomers, and uncharacterized BOK species are the putative building blocks of apoptotic pores. Effectors interact with lipids and bilayers but the mechanism of membrane poration remains elusive. I discuss effector-mediated mitochondrial poration.
Keywords: mitochondrial apoptosis, mitochondrial poration, pore-forming effector BCL-2 family proteins, effector activation, effector dimerization, effector oligomerization, arc and ring oligomerization
Graphical Abstract
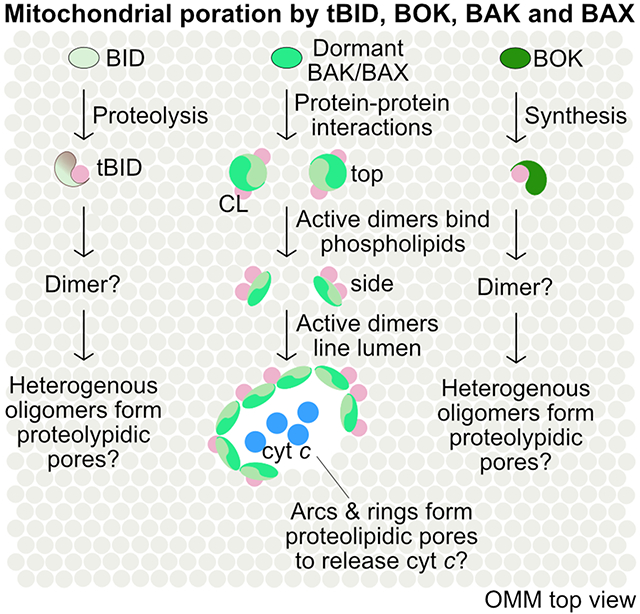
The pore-forming, effector BCL-2 family proteins BAK, BAX, BOK, and tBID initiate apoptosis through mitochondrial outer membrane permeabilization (MOMP). They are activated by upstream death stimuli leading to their proteolysis, protein-protein and protein-lipid interactions, and synthesis, ultimately adopting elusive oligomeric conformations of apoptotic pores via active putative monomers and dimers.
Introduction
Mitochondrial poration by the BCL-2 family proteins initiates apoptosis through the release of caspase activators and has been one of the most exciting, highly debated and poorly understood processes in apoptosis research [1-11]. This process is dysregulated in diseases including cancer and has been the main target of drugs designed to initiate apoptosis in tumors [12-17]. Mitochondrial poration is executed by the effector BCL-2 family proteins through an elusive mechanism in part because we have not been able to isolate and investigate the apoptotic pores, which have a heterogenous proteolipic nature [1,18]. The canonical BCL-2 family effectors, BAK and BAX, are the most extensively studied pore-forming proteins [2]. During apoptosis BAK and BAX are regulated through specific protein-protein interactions with the upstream pro-apoptotic BH3-only initiators, which activate the effectors directly and indirectly, and with the downstream pro-survival guardians, which recognize and sequester the activated effectors and the BH3-only initiators (Figure 1) [19]. In contrast, BOK porates mitochondria largely independent of the canonical BCL-2 proteins, its pro-apoptotic function being downregulated through degradation by the gp78 E3 ligase associated with the ER protein degradation system, as well as by sequestration in complex with the IP3 receptors [2,10]. On the other hand, BID has been historically studied extensively as a potent direct agonist of BAK and BAX and an antagonist of the BCL-2 pro-survival proteins [20], and although others have likened it to a BAX-like BH3 protein [21], only recently its pore-forming potential was definitively established [22]. In this problems and paradigms essay I present the evidence supporting the function of BCL-2 effectors reassessing the dogma of how the mitochondrial outer membrane is permeabilized mechanistically. The mitochondrial inner membrane is also permeabilized during apoptosis, but it comes secondary to the outer membrane permeabilization and apoptosis initiation and will not be discussed in any detail here because we know very little about it although it may have broad implications in inflammatory signaling via mitochondrial DNA sensing if and when apoptotic caspases are inhibited [23-26].
Figure 1.

Canonical and non-canonical effector BCL-2 family proteins execute mitochondrial poration.
BH, BCL-2 homology region; TM, transmembrane region. We note that some BH3-only initiators may have C-terminal TMs [41] or outer mitochondrial membrane targeting sequences that do not insert as TM helices (e.g. BIM [142]) and therefore we did not annotate them as TM-containing.
Innovative genetic models at the forefront of delineating hidden effector functions
The genetic mouse models of BCL-2 family proteins have been instrumental in elucidating and validating their functions. Analyses of the developmental phenotypes of knockout mice and of the apoptotic sensitivity of cultured cells isolated from these mice have been the gold standard in the field. However, the redundancy of the BCL-2 family proteins has made it challenging to genetically interrogate their roles in vivo. Single knockout (KO) of pro-death BCL-2 family genes have not revealed many developmental phenotypes (reviewed in [3,9,10]). Nonetheless, careful analyses of double, triple, and quadruple knockout mice in Andreas Strasser lab revealed the critical importance of the canonical effector BAK and BAX in embryogenesis (Table 1). The BAK BAX DKO mice had the most severe phenotypes among any pro-death BCL-2 family DKO mice [27,28]. Combined the deletions of three effectors in BAK BAX BOK TKO mice [28] and four effectors in BAK BAX BOK BID QKO mice [29] marginally albeit significantly exacerbated the already severe phenotypes of BAK BAX DKO mice [28]. Most DKO, TKO, and QKO mice did not survive to weaning but a few of them remarkably survived to adulthood, which suggests that mitochondrial apoptosis is not strictly required for the normal development of most organs. It remains unclear what compensatory mechanisms, such as proliferation, differentiation or cell death are responsible for balancing the lack of mitochondrial apoptosis under these conditions but neither extrinsic apoptosis, necroptosis, pyroptosis, or autophagy were upregulated in the DKO and TKO mice [28]. Moreover, not even inactivation of extrinsic and intrinsic apoptosis and necroptosis in Caspase-8 MLKL BOK BAK BAX quintuple knockout mice prevented some of the mice from surviving healthy into adulthood [30]. Anatomical examination of these mice revealed multiple midline defects visible at E18.5 including spina bifida, exencephaly, omphalocele, and multiple skeletal abnormalities (Table 1). Surprisingly, the QKO mice did not exhibit cleft abnormalities which were observed in many of the DKO and TKO mice. Many of the newborn mice died from an inability to suckle due to cleft palate (DKO and TKO), or aortic arch defects of which the most severe kind would prevent blood oxygenation on birth (this is a fully penetrant phenotype in QKOs). Noteworthy, and possibly further exacerbating the developmental cardiovascular defects, BID has recently been implicated in the homeostatic mitochondrial cristae remodeling and the BID KO mice exhibited abnormal left ventricular mitochondrial cristae making these mice more susceptible to acute cardiac stress induced damage by Epinephrine [31]. The QKO fetuses also exhibited extra tissue compared to TKO and DKO fetuses in multiple regions of the head, forebrain, nasal cavity, and tongue, anomalies in the eye, and kidney development defects, and abnormalities in urogenital tract development of female fetuses [29].
Table 1.
Summary of phenotypes of key gene knockout in mice and cell lines.
| Knockout mice | BAK/BAX DKO |
BAK/BAX/BOK TKO |
BAK/BAX/BOK/BID QKO |
|---|---|---|---|
| Offsprings E18.5-E19 | 42/54/216 p=0.0695 | 39/58/232 p=0.0157 | 26/29.25/117 p=0.7 |
| Offsprings weaning | 10/56 /223 p<0.0001 | 20/194/776 p<0.0001 | 15/144/576 p<0.0001 |
| Offsprings adulthood | 4/69/277 p<0.0001 | 6/173/692 p<0.0001 | 10/124/498 p<0.0001 |
| Midline defects | |||
| Cleft palate | ✓ | ✓✓✓ | X |
| Spina bifida | ✓ | ✓ | ✓ |
| Exencephaly/omphalocele | ✓ | ✓✓ | ✓✓ |
| Skeletal abnormalities | ✓ | ✓✓ | ✓✓✓ |
| Aortic arch defects | ✓ | ✓✓ | ✓✓✓ |
| Abnormal tissue growth | ✓ | ✓ | ✓✓ |
| Cell lines | BAK/BAK DKO MEF |
BCL2allKO (AKO) HCT116 |
|
| Resistance | |||
| DNA damage | ✓✓✓ | ✓✓✓ | |
| Radiation | ✓✓✓ | ✓✓✓ | |
| Chemotherapy | ✓✓✓ | ✓✓✓ | |
| Nutrient withdrawal | ✓✓✓ | ✓✓✓ | |
| Growth factor withdrawal | ✓✓✓ | ✓✓✓ |
Mouse offspring phenotypes were adapted from references [28,29]. The number of offsprings and p values are represented as observed/expected/total and corresponding p value. One or more check marks indicate phenotype occurrence and exacerbated phenotype, respectively, and the cross mark indicates lack of phenotype.
Some of the abnormalities observed in the BAK BAX DKO mice were also phenocopied in knockouts of their upstream BCL-2 family activators. The canonical effectors are potently activated through direct activation by the BH3-only proteins including BID, BIM, PUMA, NOXA and others (described later; Figure 1). Supporting the essential regulation by these BH3-only proteins, the BID BIM PUMA TKO mice and the BID BIM PUMA NOXA QKO mice were born at a lower Mendelian ratio than expected, exhibited persistence of interdigital webs and imperforated vaginas and were resistant to γ-irradiation in many organs and certain immune cell types [32,33], overall mimicking some of the phenotypes of BAK BAX DKO mice [27,28].
For more than two decades mouse embryonic fibroblasts (MEFs) generated from the BAK BAX DKO mice have served as the gold standard in functional investigation of BCL-2 family mediated mitochondrial apoptosis [27,34]. These cells are resistant to mitochondrial apoptosis inducers including DNA damaging agents, radiation, chemotherapeutics and withdrawal of nutrients and growth factors (Table 1). For mechanistic studies WT BAK or BAX and their mutants have been reintroduced into BAK BAX DKO MEFs to investigate their contributions in apoptosis initiation through mitochondrial poration. Nonetheless, these cells have limitations when assessing effector regulation because they express most of the BCL-2 family protein repertoire making it impossible to pinpoint exactly how effector mutations may affect their interactome and which proteins participate in the direct regulation of effectors. To circumvent this problem, an innovative tour-de-force genetic engineering milestone was achieved in the Xu Luo lab who developed a derivative of the HCT116 human colon tumor cell line in which 17 BCL-2 family genes where inactivated through a combination of TALEN, CRISPR and nickase genomic targeting (Table 1) [35]. The generation of the BCL2allKO HCT116 cell line (AKO) is commendable as it has become the new gold standard in the field for investigations of the apoptotic role of BCL-2 proteins on a clean genetic background; examples of its use follow.
BID, the prototypical BH3-only initiator is also an atypical effector
BID was originally discovered as a BCL-2 and BAX interacting pro-apoptotic ligand exhibiting homology with the BCL-2 family only in the BH3 region, which mediates the interactions with its binding partners [36]. Shortly thereafter, BID was characterized as the nexus between the extrinsic apoptotic pathway, mediated by death receptor (DR) signaling (e.g. Fas, TNF, TRAIL), and the mitochondrial pathway [37,38] (Figure 2A). On DR ligation activation of the initiator Caspase-8 promotes BID cleavage in the loop proceeding the BH3 region. Compared to uncleaved BID, cleaved BID targeted mitochondria readily via the C-terminal domain, interacted with BCL-xL with stronger affinity, induced the release of cytochrome (cyt) c from purified mitochondria at ~100× lower concentration, and promoted much better the hallmarks of mitochondrial apoptosis. Mutagenesis in the BH3 region attenuated cyt c release but not mitochondrial targeting [37,38]. It was intriguing at that time that complete cyt c release from purified HL-60 mitochondria was induced by subnanomolar doses of cleaved BID at ~500× lower concentration than required for release by BAX in that system, presumably by activating endogenous BAK [38]. Although the NMR structures of BID revealed a folded globular domain similar to folded BCL-2 proteins [39,40] (Figure 2B), the field had assumed that BID acts primarily as a BH3-only initiator (described later). Phylogenetically, BID is closely related to the folded BCL-2 effectors and guardians aligning with the BH3-only initiators, which are intrinsically disordered proteins, only in the BH3 region [41]. It could be speculated that BID diverged from a common folded BCL-2 homolog who lost the BH1, BH2, and BH4 motifs during evolution, whereas the BH3-only initiators probably arose from convergent evolution suggesting multiple origins of BH3-only proteins.
Figure 2.

BID is a direct activator and non-canonical effector
A. Proteolytic cleavage activates BID.
B. Cartoon representation of apo BID structure.
C. Biochemical properties of BID constructs that revealed its role as direct activator and effector. ND, not determined.
D. Cartoon representation of LPPG micelle-associated tBID structure.
E. The tBID conformations at mitochondria are not fully defined but may act as monomer in the initiation of poration and lining the pore lumen.
Access to the AKO cell line has enabled researchers to directly ask if BID is an effector? This question was definitively answered in a recent comprehensive study spearheaded in the Ana Garcia-Saez lab [22]. The authors transiently expressed several BCL-2 family proteins in AKO cells revealing their potency in triggering apoptosis based on analysis of known apoptotic hallmarks including release of SMAC and cyt c from mitochondria, changes in nuclear morphology, PARP cleavage, disruption of plasma membrane integrity, and caspase activation. Evaluation of the effector-mediated apoptotic response in AKO cells revealed that BAK and BAX were most potent (~90% cell death), tBID exhibited intermediate potency (~65% cell death) while BID was weakly active (~25% cell death), whereas BIM, BCL-xL, or empty vector did not induce apoptosis (~10% baseline cell death). Co-expression of tBID + BCL-xL (a potent inhibitor of tBID) largely inhibited apoptosis of AKO cells, similar to tBID expression in BAK BAX DKO HCT116 cells which have a full repertoire of pro-survival BCL-2 proteins. Additionally, the combination of BH3 mimetics ABT-737 + S63845, which inhibits all pro-survival BCL-2 proteins but A1, enabled apoptosis in BAK BAX DKO HCT116 cells ectopically expressing tBID. Moreover, TRAIL-induced endogenous BID cleavage to tBID in these cells synergized with ABT-737 + S63845 in apoptosis induction supporting a role of tBID as effector in a genetic background lacking the canonical effectors yet otherwise containing a complete BCL-2 family repertoire, albeit neutralized with the BH3 mimetics. As control these stimuli did not induce apoptosis when BID was silenced or knocked out in these cells. Furthermore, tBID also synergized with ABT-737 + S63845 in mediating the release of mitochondrial DNA in BAK BAX DKO U2OS cells, as previously shown for BAX and BAK [23,24], by enabling mitochondrial inner membrane permeabilization. The authors demonstrated the effector role of tBID independent of BAK and BAX in two pathological circumstances: 1) during Shigella infection when BID is cleaved by calpain to tBID [42], which released mitochondrial SMAC in WT (or BAK BAX DKO) HeLa cells surprisingly with undetectable BAK and BAX activation; it remains unclear why BAK and/or BAX are not activated by tBID but one could speculate that they are post-translationally modified and somehow inactivated; and 2) in TRAIL-sensitive and BH3 mimetics-insensitive Nalm6 pre-B acute lymphoblasic leukemia cells lacking BAX and expressing the inactive BAK mutant R127H, TRAIL-induced BID cleavage to tBID promoted robust apoptosis in the absence of detectable BAK activation.
Mechanistically, tBID-mediated apoptosis, surprisingly, did not require the BH3 region helix α3 as mutations in this region known to block apoptosis mediated by direct activation of the canonical effectors had minimal effect on apoptosis compared to WT tBID (G94E, Figures 2B-2D). Instead, missense mutations in the central helix α6 found in cancer patients in the TCGA database showed an attenuated ability of tBID to induce apoptosis in AKO cells compared to WT tBID, although these mutants were similarly able to induce apoptosis in BID KO MEFs confirming that the WT BH3 region in the cancer mutants and WT tBID promotes apoptosis through BAK and BAX similarly. Remarkably a double mutant in helix α6, K157A/K158A substantially incapacitated tBID-induced apoptosis in AKO cells further supporting the role of helix α6 in mitochondrial permeabilization by tBID (Figures 2B-2D). Based on this study, it is now possible to uncouple through mutagenesis the effector role of tBID from its role as a BH3-only initiator, raising the possibility of further testing the two functions in vivo independently through knock-in strategies. Importantly, although the WT BH3 region is not required for apoptosis initiation by tBID, its tight binding to pro-survival BCL-2 proteins blocks the effector function of tBID and therefore BH3 mimetics are necessary to relieve this block. On the other hand, upstream upregulation of additional BH3-only proteins through standard inducers of the intrinsic pathway (e.g. DNA damage) could also antagonize the pro-survival BCL-2 proteins and thus tBID could contribute additively to mitochondrial poration as effector alongside BAK and BAX during circumstances where tBID is in fact produced. One such scenario involves death receptor-mediated apoptosis during which Caspase-8, but not executioner caspases, cleave BID to tBID [43]. tBID can also be generated through BID cleavage by granzyme B during cytotoxic T lymphocyte-mediated cell death [44,45] and by calpains [46,47] (Figure 2A-2B).
How does BID directly mediate mitochondrial poration?
Super-resolution stimulated emission depletion (STED) confocal microscopy revealed tBID as largely homogenously distributed in the mitochondria of AKO cells [22] rather than forming large supramolecular assemblies produced by BAX [23,24,48,49], BAK [50], and BOK [51] (described later, Figures 3, 4). However, the resolution of this technique precludes definitive conclusions on the extent of tBID oligomerization at the mitochondria or if tBID oligomers are made up of the putative dimer or monomer building blocks. Therefore, the interpretation is that tBID permeabilizes the outer mitochondrial membranes by concentration-dependent accumulated crowding rather than forming the proteolipidic pores with arc-like and ring-like features proposed for the other effectors (described later).
Figure 3.

BOK is a non-canonical effector activated through protein synthesis and stabilization
A. The ERAD gp78 is a key E3 ligase that degrades BOK in conjunction with VCP and the proteasome, while the IP3 receptors (IP3R) act as a sink together downregulating BOK-mediated apoptosis.
B. BOK structural analyses have revealed protein dynamics and intrinsic instability as a driver to BOK autoactivation in mitochondrial poration in the absence of regulation by BCL-2 family proteins.
C. How BOK oligomerizes to porate mitochondria is unclear but arcs and rings have been observed in super-resolution microscopy.
Figure 4.

The canonical effectors BAK and BAX dimerize to porate mitochondria.
A. BAK and BAX are activated through cooperation between direct activation by BH3-only initiators, autoactivation, and crossactivation. Representative structures identify key residues implicated in BAK and BAX activation as described in the text. Mutations in the BAK BH3 (e.g. D83A) or BAK activation groove (e.g. R127A) impaired the ability of BAK to autoactivate but it did not block direct activation by BID BH3 or the small molecule SJ572946. These mutations are expected to affect BH3-in-groove dimerization downstream of activation. Mutation R109D in BAX activation groove turned BAX into a potent activator but apparently blocked its ability to permeabilize mitochondria.
B. BH3-in-groove BAK dimers may initiate poration by binding to lipids and inducing membrane curvature.
C. BH3-in-groove BAX dimers may line the lumen of the apoptotic pores.
D. Super-resolution microscopy indicated BAK and BAX oligomerization via arcs and rings intermediates during apoptosis.
Early studies in the context of BAK-expressing mitochondria have indicated that tBID BH3 region is not required for mitochondrial targeting, as constructs comprising helices α4-α8 targeted mitochondria as readily as tBID; the BH3 region was required for cyt c release through physical interaction with BAK inducing conformational changes and BAK oligomerization; this study set the stage for tBID’s role as a BH3-only initiator (Figure 2C) [52]. tBID could bind cardiolipin (CL), phosphatidylethanolamine, and phosphatidic acid in liposomes whereas full-length BID was largely resistant to binding these lipids, and the α4-α6 region, named cardiolipid binding domain (CBD) was identified as the most minimal region for the interaction with lipids [53]. Interestingly, CBD, which does not contain the BH3 region, is almost as potent as tBID in mediating cyt c release from isolated HeLa mitochondria and both have been shown to promote leakage of liposomes [54] as well as induce lipid mixing in liposomes (Figure 2C) [55]. Interestingly, both tBID and CBD were similarly potent in releasing 10 kDa fluorescent dextran (FD) from liposomes pretreated with octylglucoside activated BAX, which partially permeabilized those liposomes, but only tBID could promote release of the 70 kDa FD, whereas CBD + BAX did not release FDs [56]. Importantly, BIM could not promote FD release in this system, suggesting that tBID and CBD cooperate with BAX to form large membrane openings through BH3-dependent and BH3-independent mechanisms [56]. These observations suggest that tBID and CBD may act on opened bilayers to possibly open them further (Figure 2C).
The structure of tBID has been probed in solution, detergent micelles, and bilayers by electron paramagnetic resonance (EPR), NMR, and circular dichroism (CD) spectroscopies. These biophysical studies of tBID in the absence of micelles or membranes have suggested that although it maintains α-helical secondary structure detected by CD spectroscopy and is monomeric according to size exclusion chromatography (SEC), it exhibits features of dynamically disordered proteins by NMR indicating that the protein adopts multiple conformations and undergoes dynamic conformational exchange in the intermediate range of the microsecond to millisecond NMR time scale [57]. Moreover, tBID adopts a micelle or bilayer-associated α-helical conformation with none of the helices adopting a transmembrane conformation, inserted perpendicular to the bilayer, rather being parallel to and embedded in the bilayer (Figure 2D) [58-60]. In isolation, helices α6 and α7 of BID have been proposed to act as hairpins and insert perpendicular to the bilayer (Figure 2C) [61]. All tBID helices maintain secondary structure in micelles but they no longer contact one another as in the apo full-length BID structure suggesting that the protein restructures its interhelical hydrophobic core by partitioning it within the acyl chain phase of the micelle [60]. tBID readily associated with detergent micelles, liposomes, or mitochondria remaining monomeric and it dissociated from the N-terminal fragment in the context of cleaved BID (also known as N/C BID), but the N-terminal fragment did not interact with these media according to biochemical and EPR investigations [62]. In contrast, others have detected tBID dimers in liposomes of mitochondrial lipid composition in fluorescence resonance energy transfer studies with tBID labeled with fluorophores, but that investigation has not disrupted dimerization by mutagenesis to provide mechanistic insights [63]. At the mitochondria, tBID interaction with the membrane is enabled by Mtch2, an outer mitochondrial membrane insertase of helical proteins [64], which facilitated the conformational changes of tBID to the open extended conformation embedded in membranes [65,66], although the exact mechanism is not yet defined. In contrast, mitochondrial-associated hexokinases 1 and 2 promoted tBID retrotranslocation, or recycling from the mitochondria into the cytosol, inhibiting death receptor apoptosis in a process that requires interactions with the tBID BH3 region, but the exact mechanism of retrotranslocation is unclear [67]. Remarkably, tBID has also been shown to form lipoparticles (nanodiscs) with an average diameter of 11.5-nm (at 20:1 lipid:protein ratio) exhibiting a broad size distribution according to SEC analysis, and with a composition of 56% α-helix and 6% β-strand based on CD spectroscopy and with only 30−40% of the amide signals out of 136-residues visible in 1H/15N 2D NMR suggesting intrinsic disorder in the NMR visible regions as well as slow tumbling of the tBID remainder in the lipoprotein assembly [68]. It is possible that tBID interacts with opened bilayers by lining the apoptotic pore lumen like the apoptotic dimers of BAK and BAX (described below). Overall, monomeric tBID likely destabilizes membranes by insertion in an open loosely associated helical conformation that can further organize with additional nearby monomers to generate and line the pore lumen forming heterogeneous proteolipidic pores that remain to be characterized structurally (Figure 2E, Table 2).
Table 2.
Similarities, differences, and missing aspects of mitochondrial poration by BCL-2 family effectors.
| Effectors | BAK | BAX | BOK | tBID |
|---|---|---|---|---|
| Activators | BH3-only initiators Sphingosine metabolites BAK |
BH3-only initiators Sphingosine metabolites BAK p53 DRP-1 |
Synthesis Heating Degradation inhibition |
Caspase-8 Granzyme B Calpains Mtch2 |
| Inhibitors | Guardians MARCHF5 |
Guardians | Gp78/VCP/proteasome IP3R MCL-1 |
Guardians |
| Lipid preference | CL | CL | CL | CL |
| Building block | BH3-in-groove dimers | BH3-in-groove dimers | ? | Monomers |
| SRM/AFM structures | Lines, arcs, rings | Lines, arcs, rings | Lines, arcs, rings | ? |
| Pore initiation | Dimer | Dimer? | ? | Monomer |
| Pore lumen lining | Yes, dimer | Yes, dimer | ? | Yes, monomer? |
| Pore structure | ? | ? | ? | ? |
SRM, super resolution microscopy; AFM, atomic force microscopy.
The emergence of BOK as a non-canonical effector
BOK is the least studied effector originally identified in a yeast two-hybrid screen using MCL-1 as bait and through mining the GeneBank database for cDNAs homologous to BCL-2 proteins [69,70]. BOK interacts poorly with other BCL-2 proteins and its overexpression induces apoptosis [69-71]. BOK has been characterized as one of the ligands of the IP3 receptor interacting network and has been shown to regulate the ER–mitochondria contact sites and possibly Ca2+ signaling between the two organelles [72-75]. The role of BOK in apoptosis has been difficult to pinpoint because it is not activated in conventional ways, for instance by activating the mitochondrial pathway with intrinsic apoptotic inducers dependent on BAK or BAX mediated apoptosis [10,51]. Instead, endogenous BOK is activated by inhibition of the machinery that regulates its degradation, the gp78 E3 ligase of the ER-associated degradation system (gp78, VCP, proteasome), which degrades proteins from inside the ER but also cytosolic proteins (Figure 3A) [76]. Interfering with this system pharmacologically or genetically stabilized BOK which targeted and permeabilized the mitochondria unassisted by other BCL-2 family proteins and in the absence of BAK and BAX [76]. Proteasome inhibition induced potent BOK-mediated apoptosis in BAK BAX DKO MEFs stably expressing BOK using a Tet-On strategy that promotes BOK accumulation over time [76,77]. Similarly, when transiently overexpressed in AKO cells, BAK BAX DKO cells, or BAK BAX DKO cells expressing BCL-2, BOK induced apoptosis supporting its effector role independent of BAK and BAX or of inhibition by BCL-2 [51]. Purified FL-BOK is weakly active compared to BOK-ΔTM, which exhibits activity comparable to that of BH3 ligand-activated BAK and BAX in mitochondria and liposome permeabilization assays [51,76,77]. Mild heating, previously shown to activate BAX and BAK [78], triggered significant activation of FL-BOK to promote similar extent of permeabilization observed with BOK-ΔTM, suggesting that the TM region is inhibitory [51]. It is unclear how FL-BOK is intrinsically inhibited but it likely adopts a similar structure to that of BAX with the C-terminal TM region bound to the hydrophobic groove which keeps BAX cytosolic and dormant (see Figure 4A) [79]. Structural investigations of BOK-ΔTM have revealed conformational heterogeneity between helices α2 and α4 in the hydrophobic groove exhibiting a short helix α3 [77,80] as well as conformational dynamics in this region and the α1 helix detected by NMR [77], overall explaining BOK’s independence of BH3 ligands for activation and that its intrinsic instability is a key feature enabling autoactivation at the mitochondria (Figure 3B). BOK is not inhibited by pro-survival BCL-2 proteins which bind poorly BOK’s BH3 region but MCL-1’s TM region has been identified as a negative regulator of BOK by direct interaction with BOK’s TM, presumably in the lipid bilayer (Figure 1) [81].
BOK mediated mitochondrial poration
Upon inhibition of its degradation or through overexpression, BOK spontaneously targets and permeabilizes the mitochondria. Supporting the essential role of BOK’s TM in mitochondrial targeting, when transiently expressed in AKO cells BOK-ΔTM was largely cytosolic exhibiting no apoptotic activity whereas FL-BOK exhibited significant albeit half of the activity of BAX achieved on transient expression in these cells [51]. FL-BOK localized at the mitochondria and ER, and their contact sites. In contrast, the chimeric BOK protein with the BCL-xL TM replacing that of BOK, BOK-TMxL, was more potent in triggering apoptosis presumably by targeting the mitochondria better than FL-BOK. The lipid composition determines the efficiency of membrane permeabilization by BOK with maximal activity observed in vesicles rich in cardiolipin (phosphatidylcholine (PC):CL 8:2) compared to intermediate and low activity supported in vesicles with lipid composition resembling mitochondria or 100% PC, respectively [51]. At the mitochondria, BOK was visualized by STED microscopy and underwent oligomerization into ring-like structures, as described for BAK and BAX (Figure 3C, see below). Like BAK and BAX, BOK exhibited concentration-dependent oligomerization in supported lipid bilayers (SLBs) prepared from mixtures of PC:CL 8:2 liposomes and fluorescently labeled BOK-ΔTM. In SLBs BOK-ΔTM species range between 1 to 8 monomers with monomers and dimers predominating at high and low concentration, while higher order oligomers at the high concentration. Surprisingly, BOK did not exhibit exclusively an even number of monomers in the oligomers as described previously for BAX [82], but this could be also a consequence of different incubation times that may lead predominantly to dimer formation for BAX. It is unclear if BOK is similar to tBID, exhibiting activity as monomer, BAK and BAX, relying on active dimers, or whether it adopts a yet to be define multimeric active conformation. Like BAK and BAX, BOK forms a large single pore in PC:CL 8:2 liposomes visualized by negative staining EM; the pore lacks proteinaceous features produced by the gasdermin D pores in similar liposomes. Unlike the gasdermin D pores, which have been resolved at high resolution [83], we currently lack structures of the BOK pores or any of their active intermediates (Table 2).
BAK and BAX are directly activated by BH3-only initiators, autoactivated and crossactivated
BAK and BAX are dormant in non-apoptotic healthy cells being uniquely turned on by direct interactions with certain BH3-only initiators activated during apoptosis. Dormant BAK is constitutively targeted to the mitochondrial outer membrane via the TM with the folded soluble core facing the cytosol [84,85], while dormant BAX is localized in the cytosol with the TM region stably bound to the activation groove [79]. The BH3-only initiators BIM, PUMA, NOXA and possibly others are activated by upstream intrinsic cellular stress (e.g. DNA damage, ER stress, cytokine withdrawal), while tBID is generated extrinsically through BID proteolysis (described above), and together have been characterized as promiscuous BAK and BAX activators [19,33,52,86-90]. BH3 activators engage the activation groove of effectors (delimited by helices α2-α5 and α8) [91-98] and potentially a non-canonical trigger site located on the opposite face to the activation groove (defined by helices α1 and α6 and the α1-α2 loop in BAX) [99-102] (Figure 4A). Additionally, as active effectors undergo conformational changes in the presence of the outer mitochondrial membrane, they transiently expose their BH3 regions, which in the case of BAK act as potent effector activators in trans triggering nearby dormant BAK and BAX activation to amplify the activation signaling (Figure 4A) [97,103,104]. The structural basis of BAK direct activation and autoactivation in trans have revealed a common mechanism whereby BH3 ligand binding to the activation groove destabilizes electrostatic contacts at the bottom of the groove thereby releasing the inhibitory helix α1 [97]. The BH3 activating ligands use hydrophobic residues to bind six pockets in the activation groove of BAK (numbered 0-5) and form hydrogen bonds with residues on the periphery of the groove most notably mediated by R127 side chain (Figure 4A) [97]. Similar mechanisms also govern BAX activation as recently proposed [18], although activated BAX with exposed BH3 readily forms BH3-in-groove dimers rather than acting as activator in trans as shown in a recent study investigating effector autoactivation in isolated mitochondria controlled by effector activating antibodies [104]. It is conceivable that the role of BAX as effector activator could be more significant in cells, but these studies are nontrivial with the available tools.
The process of direct activation and autoactivation can be uncoupled through mutagenesis as demonstrated recently for BAK [97]. Mutants of BH3 residues that engage hydrophobic pockets 3 and 4 to autoactivate BAK in trans are resistant to autoactivation but they can be directly activated by BH3 ligands, albeit requiring a higher concentration (2-4×) compared to wildtype (WT) BAK to permeabilize membranes. Even mutants that affected direct activation and autoactivation because of their location in the activation groove could be activated by BH3 ligands engineered to bind more potently to this groove. Remarkably, even a BAK mutant (R127A), which does not bind the prototypical BID BH3 ligand, could be activated in vitro by a recently described novel small molecule BAK activator, SJ572946 [105]. In AKO cells R127A BAK is so severely defective, with signs of misfolding and degradation, that only the combination of SJ572946 + BID could activate it [105]. Importantly, all activation impaired BAK mutants can be expressed stably in AKO cells whereas WT BAK could not be expressed [97,105], as documented in a recent study also for WT BAX [67]. However, we identified a BAK mutant, V74A, that behaves like the wildtype BAK for all intents and purposes, and which can be stably expressed in AKO cells providing an unprecedented opportunity to investigate cellular BAK alone in the absence of the BCL-2 family repertoire. This mutant was used to investigate autoactivation and direct activation by tBID and small molecules [97,105]. To our knowledge a similar mutant has not been characterized for BAX yet.
Nonetheless, the mutant R109D BAX is reported to auto-trigger and to uncouple BH3-in-groove dimerization and autoactivation in trans, being very potent in activating BAK [104], i.e. to mediate crossactivation. Importantly, R109D BAX induced permeabilization of BAK KO mitochondria similar to WT BAX in “autoactivation mode” at higher doses (200 nM), yet only the mutant but not WT BAX can activate BAK in WT mitochondria [104]. On the other hand, BAK appears to be very potent in feed-forward activation of BAK and BAX, leading to signal amplification [104]. Combined the mutagenesis and structural studies provide compelling evidence supporting the mechanisms of BAK and BAX activation through combination of direct activation, autoactivation in trans, and crossactivation (Figure 4A) [97,104,105].
Mechanisms of BAK and BAX activation beyond the BH3-only initiators
Under certain conditions, documented in cultured cells, BAK and BAX are found sequestered by the pro-survival BCL-2 proteins, known as mode 2 binding, whereas mode 1 refers to the sequestration of BH3-only protein by the pro-survival BCL-2 proteins (Figure 1) [87,106]. The entire pool of effectors in each cell need not be found in mode 2, and a mixture of dormant and mode 2 effectors is usually observed as detected by proteolytic sensitivity which can distinguish between the two effector conformations [87,107]. Exactly how the conversion of the effectors to mode 2 conformation occurs is unclear but the effectors must present an open conformation with the BH3 region exposed for sequestration by the pro-survival BCL-2 proteins. Derepression of pro-survival BCL-2 proteins with inhibitors known as BH3 mimetics can induce robust apoptosis when effectors are present in mode 2. Remarkably, even when the eight known BH3-only proteins, BAD, BID, BIM, BMF, BIK, HRK, PUMA, and NOXA, were genetically inactivated in HCT116 (OctaKO HCT116) cells the BH3 mimetics induced robust apoptosis, which lead to the idea that the effectors are activated by the mitochondrial outer membrane [35,108]. It is documented that BAK and BAX are activated by sphingosine-1-phosphate and hexadecenal which cooperate with tBID, respectively [109,110], although the role of these lipids has not been reassessed in the AKO cells. Moreover, BAK and BAX are known to prefer CL in targeting membranes for efficient permeabilization [111,112]. One could speculate that in OctaKO HCT116 cells the effectors constitutively exhibit mode 2 complexes which are easily derepressed with BH3 mimetics. The effectors could be activated by proteins other than ‘canonical’ BH3-only initiators including p53 [113] and DRP1 [114], yet to be characterized proteins [115], lipids [11,110,116], uncharacterized metabolites, or may be sequestered in mode 2 via the inhibition of the E3 ubiquitin ligase MARCHF5/MITOL/RNF15, which prevents BAK’s mode 2 complex formation by an unknown mechanism [117]. Taken together these observations suggest that we know relatively little about the regulation of BAK and BAX outside the BCL-2 family, including matters of their synthesis, turnover, and even retrotranslocation [118,119], besides a known interacting partner of BAK, the outer mitochondrial porin VDAC2, which has been implicated in BAK and BAX-mediate mitochondrial poration and BAX retrotranslocation in poorly understood ways [120-123]. Future investigations of genetically edited cell lines, such as the OctaKO and AKO HCT116 cells, will likely reveal new effector biology in these areas.
BAK and BAX dimerize for efficient mitochondrial poration
During apoptosis BAK and BAX undergo extensive conformational changes upon activation leading to major opening of the globular core. In the process three elements dissociate including the N-terminal helix α1 which becomes disordered [85,124], the α6-α8 helical latch which may exhibit an extended helical conformation [125], and the α2-α5 helical core which organizes into symmetric BH3-in-groove dimers with another nearby open effector (Figures 4B-4D) [98,126]. The α2-α5 BH3-in-groove dimers present an extensive hydrophobic face opposite from the intertwined hydrophilic activation grooves occupied by the amphipathic BH3 helices, thereby generating a massive amphipathic structure thought to be the building block of apoptotic pores [98,125,126]. The α2-α5 core spontaneously dimerizes on expression in E. coli [98,125-128]. The isolated BAK α2-α5 core dimer binds a variety of lipids that can be exchanged in the presence of detergents forming trimers of dimers (hexamers) or dimers of dimers (tetramers), both of which exhibit the hydrophobic faces buried at the center and interacting with the acyl chains of phospholipids, and with the polar head groups bound to hydrophilic residues on the periphery [127]. The phospholipid acyl chains are partially resolved in the crystal structure suggesting heterogeneity in their binding modes (Figure 4B). The α2-α5 core dimer of BAX was captured bound on the periphery of bicelles by NMR and structure-guided mutants of the lipid-binding interface inhibited membrane permeabilization [128], supporting the long-standing hypothesis of capping the bilayer pore lumen by these dimers proposed from double electron-electron resonance (DEER) EPR investigations (Figure 4C) [129]. The symmetric BH3-in-groove dimers have been validated biochemically and biophysically [130-133]. The apoptotic pores are believed to be heterogeneously made up of oligomers of dimers [134] that steadily grow during apoptosis, with the BAK pores growing slower than the BAX pores possibly through arc-like and ring-like intermediates (Figure 4D) [50]. Moreover, BAK and BAX co-assemble into the same supra-molecular structures [50]. The latch and the TM of effectors represent the points of contact and heterogeneity of dimer oligomers as indicated by crosslinking and DEER EPR investigations [129,133,135,136]. Nonetheless, the organization of the BAK and BAX apoptotic oligomers or the pores has not yet been elucidated structurally (Table 2). It is unclear how the apoptotic pores are initiated to grow but the recent structure of the α2-α8 dimer of BAK induced in C12E9 detergent micelles has suggested that the purified dimer is very potent in permeabilization of liposomes and mitochondria supporting a role of this dimer in initiating the permeabilization (Figure 4B) [125].
Conclusions and open questions
Evaluation of apoptotic effectors reveals that they all undergo changes on activation from a globular closed conformation to an open membrane associated conformation. The progress in the field over the past five years has been remarkable with BID and BOK being rigorously and formally validated as effectors (Table 2). Intriguingly, even the pro-survival BCL-2 family proteins have been implicated in pore formation when proteolytically processed. For instance caspase-3 proteolysis of BCL-2 [137] and BCL-xL [138] in their helix α1-α2 loop during apoptosis produces tBID-like prodeath domains which amplify the prodeath signaling while mitigating the prosurvival response. Additionally, separase proteolytic processing of the N-terminus of helix α1 of MCL-1 during mitotic catastrophe promotes MCL-1 dimerization and mitochondrial poration in the absence of BAK and BAX [139]. These studies have ramification for our understanding the differences between prosurvival and prodeath folded BCL-2 proteins: the intact prosurvival BCL-2 family proteins are stable and do not undergo opening and conformational changes on binding BH3-only initiators as demonstrated for BAK and BAX; on the other hand, truncated prosurvival BCL-2 family proteins are highly destabilized possibly adopting tBID-like pore-forming conformations. Future structural studies will elucidate the prodeath conformations of the truncated prosurvival BCL-2 family proteins. How membrane phospholipids, of which CL appears to be a key ingredient, or their metabolites contribute to efficient effector activation and membrane permeabilization remains incompletely defined. It is unknown if tBID or BOK form apoptotic homodimers as characterized for BAK and BAX, or how they oligomerize. Super-resolution microscopy has suggested that unlike BAK, BAX, and BOK, which aggregate into arcs and rings on the mitochondrial membrane, and which have been associated with lipid removal from support bilayers in atomic force microscopy studies, tBID exhibits homogenous mitochondrial distribution without gross oligomeric features. Apoptotic marker released from the mitochondria precede the detection of effector aggregates revealing a limitation of the live microscopy studies and suggesting that the early apoptotic pores could be more dynamic and less well defined than the arcs and rings that form later. Moreover, except for tBID, which was characterized in detergent micelles as an open α-helical monomer without a hydrophobic core, it is unknown how the open active effector monomers (un)fold or whether open monomers permeabilize membranes. Interestingly, BAX has been shown to permeabilize the membranes of nanodiscs as monomers in cryo-EM studies, but the resolution was too low to reveal structural insights [140,141]. Over the next five years I anticipate that the field will seek to reveal the structural bases of the pore initiation and the growth of effector oligomers into apoptotic pores. Additionally, the field will investigate the effector interactome mechanistically and structurally beyond the BCL-2 family proteins. Our mechanistic understanding will fuel the discovery and design of effector chemical probes with novel mechanisms of action and inform therapeutic design for targeting pathologies with dysregulated apoptosis.
Funding
This work was supported by NIGMS R01GM129470 to TM.
Abbreviations:
- AKO
BCL2allKO HCT116 cells with 17 BCL2 gene knock-out
- cyt c
cytochrome c
- CL
cardiolipin
- CBD
cardiolipid binding domain
- PC
phosphatidylcholine
- DKO
double knock-out
- MEF
mouse embryonic fibroblast
- QKO
quadruple knock-out
- TKO
triple knock-out
- STED
Super-resolution stimulated emission depletion
- DEER
double electron-electron resonance
- EPR
electron paramagnetic resonance
- CD
circular dichroism
- SEC
size exclusion chromatography
- SLBs
supported lipid bilayers
Data availability statement
No data was generated in this work.
References
- 1.Flores-Romero H, Ros U, & Garcia-Saez AJ (2020). Pore formation in regulated cell death. The EMBO Journal, 39(23). 10.15252/embj.2020105753 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Moldoveanu T, & Czabotar PE (2020). BAX, BAK, and BOK: A coming of age for the BCL-2 family effector proteins. Cold Spring Harbor Perspectives in Biology, 12(4). 10.1101/cshperspect.a036319 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Chipuk JE, Moldoveanu T, Llambi F, Parsons MJ, & Green DR (2010). The BCL-2 Family Reunion. In Molecular Cell (Vol. 37, Issue 3, pp. 299–310). 10.1016/j.molcel.2010.01.025 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Czabotar PE, Lessene G, Strasser A, & Adams JM (2014). Control of apoptosis by the BCL-2 protein family: Implications for physiology and therapy. In Nature Reviews Molecular Cell Biology (Vol. 15, Issue 1, pp. 49–63). 10.1038/nrm3722 [DOI] [PubMed] [Google Scholar]
- 5.Kale J, Osterlund EJ, & Andrews DW (2018). BCL-2 family proteins: Changing partners in the dance towards death. In Cell Death and Differentiation (Vol. 25, Issue 1, pp. 65–80). Nature Publishing Group. 10.1038/cdd.2017.186 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Uren RT, Iyer S, & Kluck RM (2017). Pore formation by dimeric Bak and Bax: An unusual pore? In Philosophical Transactions of the Royal Society B: Biological Sciences (Vol. 372, Issue 1726). Royal Society Publishing. 10.1098/rstb.2016.0218 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Moldoveanu T, Follis AV, Kriwacki RW, & Green DR (2014). Many players in BCL-2 family affairs. In Trends in Biochemical Sciences (Vol. 39, Issue 3, pp. 101–111). 10.1016/j.tibs.2013.12.006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Wolf P, Schoeniger A, & Edlich F (2022). Pro-apoptotic complexes of BAX and BAK on the outer mitochondrial membrane. In Biochimica et Biophysica Acta - Molecular Cell Research (Vol. 1869, Issue 10). Elsevier B.V. 10.1016/j.bbamcr.2022.119317 [DOI] [PubMed] [Google Scholar]
- 9.Voss AK, & Strasser A (2020). The essentials of developmental apoptosis. In F1000Research (Vol. 9). F1000 Research Ltd. 10.12688/f1000research.21571.1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Naim S, & Kaufmann T (2020). The Multifaceted Roles of the BCL-2 Family Member BOK. In Frontiers in Cell and Developmental Biology (Vol. 8). Frontiers Media S.A. 10.3389/fcell.2020.574338 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Luo X, O’neill KL, & Huang K (2020). The third model of Bax/Bak activation: a Bcl-2 family feud finally resolved? F1000Research, 9, 935–949. 10.12688/f1000research.25607.1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Ashkenazi A, Fairbrother WJ, Leverson JD, & Souers AJ (2017). From basic apoptosis discoveries to advanced selective BCL-2 family inhibitors. In Nature Reviews Drug Discovery (Vol. 16, Issue 4, pp. 273–284). Nature Publishing Group. 10.1038/nrd.2016.253 [DOI] [PubMed] [Google Scholar]
- 13.Pogmore JP, Uehling D, & Andrews DW (2021). Pharmacological Targeting of Executioner Proteins: Controlling Life and Death. Journal of Medicinal Chemistry, 64(9), 5276–5290. 10.1021/acs.jmedchem.0c02200 [DOI] [PubMed] [Google Scholar]
- 14.Walensky LD (2019). Targeting BAX to drug death directly. In Nature Chemical Biology (Vol. 15, Issue 7, pp. 657–665). Nature Research. 10.1038/s41589-019-0306-6 [DOI] [PubMed] [Google Scholar]
- 15.Diepstraten ST, Anderson MA, Czabotar PE, Lessene G, Strasser A, & Kelly GL (2022). The manipulation of apoptosis for cancer therapy using BH3-mimetic drugs. In Nature Reviews Cancer (Vol. 22, Issue 1, pp. 45–64). Nature Research. 10.1038/s41568-021-00407-4 [DOI] [PubMed] [Google Scholar]
- 16.Leverson JD, Boghaert ER, Ackler SL, Catron ND, Chen J, Dayton BD, Ding H, Enschede SH, Fairbrother WJ, S Huang DC, Hymowitz SG, Jin S, Lin Khaw S, Kovar PJ, Lam LT, Lee J, Maecker HL, Marsh KC, Mason KD, … Souers AJ (2013). ABT-199, a potent and selective BCL-2 inhibitor, achieves antitumor activity while sparing platelets. Nature Medicine. 10.1038/nm.3048 [DOI] [PubMed] [Google Scholar]
- 17.Spitz AZ, & Gavathiotis E (2022). Physiological and pharmacological modulation of BAX. In Trends in Pharmacological Sciences (Vol. 43, Issue 3, pp. 206–220). Elsevier Ltd. 10.1016/j.tips.2021.11.001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Sekar G, Ojoawo A, & Moldoveanu T (2022). Protein-protein and protein-lipid interactions of pore-forming BCL-2 family proteins in apoptosis initiation. Biochemical Society Transactions, 50, 1091–1103. 10.1042/BST20220323 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Singh R, Letai A, & Sarosiek K (2019). Regulation of apoptosis in health and disease: the balancing act of BCL-2 family proteins. Nature Reviews Molecular Cell Biology, 20, 175–193. 10.1038/s41580-018-0089-8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Shamas-Din A, Brahmbhatt H, Leber B, & Andrews DW (2011). BH3-only proteins: Orchestrators of apoptosis. In Biochimica et Biophysica Acta - Molecular Cell Research (Vol. 1813, Issue 4, pp. 508–520). 10.1016/j.bbamcr.2010.11.024 [DOI] [PubMed] [Google Scholar]
- 21.Billen LP, Shamas-Din A, & Andrews DW (2008). Bid: A Bax-like BH3 protein. In Oncogene (Vol. 27, pp. S93–S104). 10.1038/onc.2009.47 [DOI] [PubMed] [Google Scholar]
- 22.Flores-Romero H, Hohorst L, John M, Albert M, King LE, Beckmann L, Szabo T, Hertlein V, Luo X, Villunger A, Frenzel LP, Kashkar H, & Garcia-Saez AJ (2022). BCL-2-family protein tBID can act as a BAX-like effector of apoptosis. The EMBO Journal, 41(2). 10.15252/embj.2021108690 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Mcarthur K, Whitehead LW, Heddleston JM, Li L, Padman BS, Oorschot V, Geoghegan ND, Chappaz S, Davidson S, Chin HS, Lane RM, Dramicanin M, Saunders TL, Lessene R, Osellame LD, Chew T-L, Dewson G, Lazarou M, Ramm G, … Kile BT (2018). BAK/BAX macropores facilitate mitochondrial herniation and mtDNA efflux during apoptosis. Science, 359, 1–12. 10.1126/science.aao6047 [DOI] [PubMed] [Google Scholar]
- 24.Riley JS, Quarato G, Cloix C, Lopez J, O’prey J, Pearson M, Chapman J, Sesaki H, Carlin LM, Passos JF, Wheeler AP, Oberst A, Ryan KM, & Tait SW (2018). Mitochondrial inner membrane permeabilisation enables mtDNA release during apoptosis. The EMBO Journal, 37, 99238. 10.15252/embj.201899238 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Marchi S, Guilbaud E, G Tait SW, Yamazaki T, & Galluzzi L (2022). Mitochondrial control of inflammation. Nature Reviews Immunology. 10.1038/s41577-022-00760-x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Vringer E, & Tait SWG (2022). Mitochondria and cell death-associated inflammation. Cell Death Differ. 10.1038/s41418-022-01094-w [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Lindsten T, Ross AJ, King A, Zong W-X, Rathmell JC, Shiels HA, Ulrich E, Waymire KG, Mahar P, Frauwirth K, Chen Y, Wei M, Eng VM, Adelman DM, Simon MC, Ma A, Golden JA, Evan G, Korsmeyer SJ, … Thompson CB (2000). The Combined Functions of Proapoptotic Bcl-2 Family Members Bak and Bax Are Essential for Normal Development of Multiple Tissues. In Molecular Cell (Vol. 6). 10.1016/s1097-2765(00)00136-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Ke FFS, Vanyai HK, Cowan AD, Delbridge ARD, Whitehead L, Grabow S, Czabotar PE, Voss AK, & Strasser A (2018). Embryogenesis and Adult Life in the Absence of Intrinsic Apoptosis Effectors BAX, BAK, and BOK. Cell, 173(5), 1217–1230.e17. 10.1016/j.cell.2018.04.036 [DOI] [PubMed] [Google Scholar]
- 29.Ke FS, Holloway S, Uren RT, Wong AW, Little MH, Kluck RM, Voss AK, & Strasser A (2022). The BCL −2 family member BID plays a role during embryonic development in addition to its BH3 -only protein function by acting in parallel to BAX, BAK and BOK. The EMBO Journal, 41(15). 10.15252/embj.2021110300 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Ke FFS, Brinkmann K, Voss AK, & Strasser A (2022). Some mice lacking intrinsic, as well as death receptor induced apoptosis and necroptosis, can survive to adulthood. In Cell Death and Disease (Vol. 13, Issue 4). Springer Nature. 10.1038/s41419-022-04731-x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Salisbury-Ruf CT, Bertram CC, Vergeade A, Lark DS, Shi Q, Heberling ML, Fortune NL, Donald Okoye G, Gray Jerome W, Wells QS, Fessel J, Moslehi J, Chen H, Jackson Roberts LI, Boutaud O, Gamazon ER, & Zinkel SS (2018). Bid maintains mitochondrial cristae structure and function and protects against cardiac disease in an integrative genomics study. ELife, 7. 10.7554/eLife.40907.001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Ren D, Tu H-C, Kim H, Wang GX, Bean GR, Takeuchi O, Jeffers JR, Zambetti GP, J-D Hsieh J, & H-Y Cheng E (2010). BID, BIM, and PUMA Are Essential for Activation of the BAX- and BAK-Dependent Cell Death Program. Science, 330(6009), 1390–1393. 10.1126/science.1190217 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Chen H-C, Kanai M, Inoue-Yamauchi A, Tu H-C, Huang Y, Ren D, Kim H, Takeda S, Reyna DE, Chan PM, Ganesan YT, Liao C-P, Gavathiotis E, Hsieh JJ, & Cheng EH (2015). An interconnected hierarchical model of cell death regulation by the BCL-2 family. Nature Cell Biology, 17, 1270–1281. 10.1038/ncb3236 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Wei MC, Zong W-X, Y Cheng EH, Lindsten T, Panoutsakopoulou V, Ross AJ, Roth KA, MacGregor GR, Thompson CB, & Korsmeyer SJ (2001). Proapoptotic BAX and BAK: A Requisite Gateway to Mitochondrial Dysfunction and Death. Science, 292(5517), 727–730. https://www.science.org [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.O’neill KL, Huang K, Zhang J, Chen Y, & Luo X (2016). Inactivation of prosurvival Bcl-2 proteins activates Bax/Bak through the outer mitochondrial membrane. Genes and Development, 30(8), 973–988. 10.1101/gad.276725.115 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Wang K, Yin X-M, Chao DT, Milliman CL, & Korsmeyer SJ (1996). BID: a novel BH3 domain-only death agonist. Genes and Development, 10, 2859–2869. [DOI] [PubMed] [Google Scholar]
- 37.Li H, Zhu H, Xu C, & Yuan J (1998). Cleavage of BID by Caspase 8 Mediates the Mitochondrial Damage in the Fas Pathway of Apoptosis. Cell, 94, 491–501. [DOI] [PubMed] [Google Scholar]
- 38.Luo X, Budihorjo I, Zou H, Slaughter C, & Wang X (1998). Bid, a Bcl2 Interacting Protein, Mediates Cytochrome c Release from Mitochondria in Response to Activation of Cell Surface Death Receptors. Cell, 94, 481–490. [DOI] [PubMed] [Google Scholar]
- 39.Chou JJ, Li H, Salvesen GS, Yuan J, & Wagner G (1999). Solution Structure of BID, an Intracellular Amplifier of Apoptotic Signaling. Cell, 96, 615–624. [DOI] [PubMed] [Google Scholar]
- 40.Mcdonnell JM, Fushman D, Milliman CL, Korsmeyer SJ, & Cowburn D (1999). Solution Structure of the Proapoptotic Molecule BID: A Structural Basis for Apoptotic Agonists and Antagonists. Cell, 96, 625–634. [DOI] [PubMed] [Google Scholar]
- 41.Aouacheria A, Rech de Laval V, Combet C, & Hardwick JM (2013). Evolution of Bcl-2 homology motifs: homology versus homoplasy. Trends in Cell Biology, 23(3), 103–111. 10.1016/J.TCB.2012.10.010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Andree M, Seeger JM, Schüll S, Coutelle O, Wagner-Stippich D, Wiegmann K, Wunderlich CM, Brinkmann K, Broxtermann P, Witt A, Fritsch M, Martinelli P, Bielig H, Lamkemeyer T, Rugarli EI, Kaufmann T, Sterner-Kock A, Wunderlich FT, Villunger A, … Kashkar H (2014). BID-dependent release of mitochondrial SMAC dampens XIAP-mediated immunity against Shigella. The EMBO Journal, 33(19), 2171–2187. 10.15252/embj.201387244 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Huang K, Zhang J, O’Neill KL, Gurumurthy CB, Quadros RM, Tu Y, & Luo X (2016). Cleavage by caspase 8 and mitochondrial membrane association activate the BH3-only protein bid during TRAIL-induced apoptosis. Journal of Biological Chemistry, 291(22), 11843–11851. 10.1074/jbc.M115.711051 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Barry M, Heibein JA, Pinkoski MJ, Lee S-F, Moyer RW, Green DR, & Bleackley ARC (2000). Granzyme B Short-Circuits the Need for Caspase 8 Activity during Granule-Mediated Cytotoxic T-Lymphocyte Killing by Directly Cleaving Bid. Molecular and Cellular Biology, 20(11), 3781–3794. https://journals.asm.org/journal/mcb [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Sutton VR, Sedelies K, Dewson G, Christensen ME, Bird PI, Johnstone RW, Kluck RM, Trapani JA, & Waterhouse NJ (2012). Granzyme B triggers a prolonged pressure to die in Bcl-2 overexpressing cells, defining a window of opportunity for effective treatment with ABT-737. Cell Death and Disease, 3(7). 10.1038/cddis.2012.73 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Chen M, He H, Zhan S, Krajewski S, Reed JC, & Gottlieb RA (2001). Bid is Cleaved by Calpain to an Active Fragment in Vitro and during Myocardial Ischemia/Reperfusion. Journal of Biological Chemistry, 276(33), 30724–30728. 10.1074/jbc.M103701200 [DOI] [PubMed] [Google Scholar]
- 47.Mandic A, Viktorsson K, Strandberg L, Heiden T, Hansson J, Linder S, & Shoshan MC (2002). Calpain-Mediated Bid Cleavage and Calpain-Independent Bak Modulation: Two Separate Pathways in Cisplatin-Induced Apoptosis. Molecular and Cellular Biology, 22(9), 3003–3013. 10.1128/mcb.22.9.3003-3013.2002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Große L, Wurm CA, Brüser C, Neumann D, Jans DC, & Jakobs S (2016). Bax assembles into large ring-like structures remodeling the mitochondrial outer membrane in apoptosis. The EMBO Journal, 35(4), 402–413. 10.15252/embj.201592789 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Salvador-Gallego R, Mund M, Cosentino K, Schneider J, Unsay J, Schraermeyer U, Engelhardt J, Ries J, & García-Sáez AJ (2016). Bax assembly into rings and arcs in apoptotic mitochondria is linked to membrane pores. The EMBO Journal, 35(4), 389–401. 10.15252/embj.201593384 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Cosentino K, Hertlein V, Jenner A, Dellmann T, Gojkovic M, Peña-Blanco A, Dadsena S, Wajngarten N, Danial JSH, Thevathasan JV, Mund M, Ries J, & Garcia-Saez AJ (2022). The interplay between BAX and BAK tunes apoptotic pore growth to control mitochondrial-DNA-mediated inflammation. Molecular Cell, 82(5), 933–949.e9. 10.1016/j.molcel.2022.01.008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Shalaby R, Diwan A, Flores-Romero H, Hertlein V, & Garcia-Saez AJ (2022). Visualization of BOK pores independent of BAX and BAK reveals a similar mechanism with differing regulation. Cell Death & Differentiation. 10.1038/s41418-022-01078-w [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Wei MC, Lindsten T, Mootha VK, Weiler S, Gross A, Ashiya M, Thompson CB, & Korsmeyer SJ (2000). tBID, a membrane-targeted death ligand, oligomerizes BAK to release cytochrome c. Genes and Development, 14, 2060–2071. 10.1101/gad.14.16.2060 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Lutter M, Fang M, Luo X, Nishijima M, Xie X-S, & Wang X (2000). Cardiolipin provides specificity for targeting of tBid to mitochondria. Nature Cell Biology, 2, 754–756. [DOI] [PubMed] [Google Scholar]
- 54.Liu J, Weiss A, Durrant D, Chi N-W, Lee RM, & City L (2004). The cardiolipin-binding domain of Bid affects mitochondrial respiration and enhances cytochrome c release. Apoptosis, 9, 533–541. [DOI] [PubMed] [Google Scholar]
- 55.Epand RF, Martinou JC, Fornallaz-Mulhauser M, Hughes DW, & Epand RM (2002). The apoptotic protein tBid promotes leakage by altering membrane curvature. Journal of Biological Chemistry, 277(36), 32632–32639. 10.1074/jbc.M202396200 [DOI] [PubMed] [Google Scholar]
- 56.Terrones O, Antonsson B, Yamaguchi H, Wang HG, Liu J, Lee RM, Herrmann A, & Basañez G (2004). Lipidic pore formation by the concerted action of proapoptotic BAX and tBID. Journal of Biological Chemistry, 279(29), 30081–30091. 10.1074/jbc.M313420200 [DOI] [PubMed] [Google Scholar]
- 57.Yao Y, Bobkov AA, Plesniak LA, & Marassi FM (2009). Mapping the interaction of pro-apoptotic tBID with pro-survival BCL-XL. Biochemistry, 48(36), 8704–8711. 10.1021/bi901171n [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Kyoung JO, Barbuto S, Meyer N, Kim RS, Collier MJ, & Korsmeyer SJ (2005). Conformational changes in BID, a pro-apoptotic BCL-2 family member, upon membrane binding: A site-directed spin labeling study. Journal of Biological Chemistry, 280(1), 753–767. 10.1074/jbc.M405428200 [DOI] [PubMed] [Google Scholar]
- 59.Gong XM, Choi J, Franzin CM, Zhai D, Reed JC, & Marassi FM (2004). Conformation of membrane-associated proapoptotic tBid. Journal of Biological Chemistry, 279(28), 28954–28960. 10.1074/jbc.M403490200 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Wang Y, & Tjandra N (2013). Structural insights of tBid, the caspase-8-activated bid, and its BH 3 domain. Journal of Biological Chemistry, 288(50), 35840–35851. 10.1074/jbc.M113.503680 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.García-Sáez AJ, Mingarro I, Pérez-Payá E, & Salgado J (2004). Membrane-insertion fragments of Bcl-xL, Bax, and Bid. Biochemistry, 43(34), 10930–10943. 10.1021/bi036044c [DOI] [PubMed] [Google Scholar]
- 62.Bleicken S, García-Sáez AJ, Conte E, & Bordignon E (2012). Dynamic interaction of cBid with detergents, liposomes and mitochondria. PLoS ONE, 7(4). 10.1371/journal.pone.0035910 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Shivakumar S, Kurylowicz M, Hirmiz N, Manan Y, Friaa O, Shamas-Din A, Masoudian P, Leber B, Andrews DW, & Fradin C (2014). The proapoptotic protein tBid forms both superficially bound and membrane-inserted oligomers. Biophysical Journal, 106(10), 2085–2095. 10.1016/j.bpj.2014.03.049 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Guna A, Stevens TA, Inglis AJ, Replogle JM, Esantsi TK, Muthukumar G, L Shaffer KC, Wang ML, Pogson AN, Jones JJ, Lomenick B, Chou T-F, Weissman JS, & Voorhees RM (2022). MTCH2 is a mitochondrial outer membrane protein insertase. Science, 378, 317–322. https://www.science.org [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Zaltsman Y, Shachnai L, Yivgi-Ohana N, Schwarz M, Maryanovich M, Houtkooper RH, Vaz FM, de Leonardis F, Fiermonte G, Palmieri F, Gillissen B, Daniel PT, Jimenez E, Walsh S, Koehler CM, Roy SS, Walter L, Hajnóczky G, & Gross A (2010). MTCH2/MIMP is a major facilitator of tBID recruitment to mitochondria. Nature Cell Biology, 12(6), 553–562. 10.1038/ncb2057 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Shamas-Din A, Bindner S, Zhu W, Zaltsman Y, Campbell C, Gross A, Leber B, Andrews DW, & Fradin C (2013). TBid undergoes multiple conformational changes at the membrane required for bax activation. Journal of Biological Chemistry, 288(30), 22111–22127. 10.1074/jbc.M113.482109 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Lauterwasser J, Fimm-Todt F, Oelgeklaus A, Schreiner A, Funk K, Falquez-Medina H, Klesse R, Jahreis G, Zerbes RM, O’neill K, van der Laan M, Luo X, & Edlich F (2021). Hexokinases inhibit death receptor-dependent apoptosis on the mitochondria. PNAS, 118(33), 1–10. 10.1073/pnas.2021175118/-/DCSupplemental [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Ekanayake V, Nisan D, Ryzhov P, Yao Y, & Marassi FM (2018). Lipoprotein Particle Formation by Proapoptotic tBid. Biophysical Journal, 115(3), 533–542. 10.1016/j.bpj.2018.06.021 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Hsu SY, Kaipia A, Mcgee E, Lomeli M, & W Hsueh AJ (1997). Bok is a pro-apoptotic Bcl-2 protein with restricted expression in reproductive tissues and heterodimerizes with selective anti-apoptotic Bcl-2 family members (apoptosisprogrammed cell deathovarytestisuterus) (Vol. 94). www.pnas.org. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Inohara N, Ekhterae D, Garcia I, Carrio R, Merino J, Merry A, Chen S, & Nú G (1998). Mtd, a Novel Bcl-2 Family Member Activates Apoptosis in the Absence of Heterodimerization with Bcl-2 and Bcl-XL. The Journal of Biological Chemistry, 273(15), 8705–8710. http://www.jbc.org [DOI] [PubMed] [Google Scholar]
- 71.Einsele-Scholz S, Malmsheimer S, Bertram K, Stehle D, Johänning J, Manz M, Daniel PT, Gillissen BF, Schulze-Osthoff K, & Essmann F (2016). Bok is a genuine multi-BH-domain protein that triggers apoptosis in the absence of Bax and Bak. Journal of Cell Science, 129(15), 3054. 10.1242/jcs.193946 [DOI] [PubMed] [Google Scholar]
- 72.Schulman JJ, Wright FA, Kaufmann T, & Wojcikiewicz RJH (2013). The Bcl-2 protein family member bok binds to the coupling domain of inositol 1,4,5-trisphosphate receptors and protects them from proteolytic cleavage. Journal of Biological Chemistry, 288(35), 25340–25349. 10.1074/jbc.M113.496570 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Szczesniak LM, Bonzerato CG, Schulman JJ, Bah A, & Wojcikiewicz RJH (2021). Bok binds to a largely disordered loop in the coupling domain of type 1 inositol 1,4,5-trisphosphate receptor. Biochemical and Biophysical Research Communications, 553, 180–186. 10.1016/j.bbrc.2021.03.047 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Carpio MA, Means RE, Brill AL, Sainz A, Ehrlich BE, & Katz SG (2021). BOK controls apoptosis by Ca2+ transfer through ER-mitochondrial contact sites. Cell Reports, 34(10). 10.1016/j.celrep.2021.108827 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Schulman JJ, Szczesniak LM, Bunker EN, Nelson HA, Roe MW, Wagner LE, Yule DI, & Wojcikiewicz RJH (2019). Bok regulates mitochondrial fusion and morphology. Cell Death and Differentiation, 26(12), 2682–2694. 10.1038/s41418-019-0327-4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Llambi F, Wang YM, Victor B, Yang M, Schneider DM, Gingras S, Parsons MJ, Zheng JH, Brown SA, Pelletier S, Moldoveanu T, Chen T, & Green DR (2016). BOK Is a Non-canonical BCL-2 Family Effector of Apoptosis Regulated by ER-Associated Degradation. Cell, 165(2), 421–433. 10.1016/j.cell.2016.02.026 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Zheng JH, Grace CR, Guibao CD, McNamara DE, Llambi F, Wang YM, Chen T, & Moldoveanu T (2018). Intrinsic Instability of BOK Enables Membrane Permeabilization in Apoptosis. Cell Reports, 23(7), 2083–2094.e6. 10.1016/j.celrep.2018.04.060 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Pagliari LJ, Kuwana T, Bonzon C, Newmeyer DD, Tu S, Beere HM, & Green DR (2005). The multidomain proapoptotic molecules Bax and Bak are directly activated by heat. PNAS, 13, 17975–17980. 10.1073/pnas.0506712102 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Suzuki M, Youle RJ, & Tjandra N (2000). Structure of Bax: Coregulation of Dimer Formation and Intracellular Localization. Cell, 103, 645–654. [DOI] [PubMed] [Google Scholar]
- 80.Ke FFS, Vanyai HK, Cowan AD, Delbridge ARD, Whitehead L, Grabow S, Czabotar PE, Voss AK, & Strasser A (2018). Embryogenesis and Adult Life in the Absence of Intrinsic Apoptosis Effectors BAX, BAK, and BOK. Cell, 173(5), 1217–1230.e17. 10.1016/j.cell.2018.04.036 [DOI] [PubMed] [Google Scholar]
- 81.Lucendo E, Sancho M, Lolicato F, Javanainen M, Kulig W, Leiva D, Duart G, Andreu-Fernández V, Mingarro I, & Orzáez M (2020). Mcl-1 and Bok transmembrane domains: Unexpected players in the modulation of apoptosis. PNAS, 117(45), 27980–27988. 10.1073/pnas.2008885117/-/DCSupplemental [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Subburaj Y, Cosentino K, Axmann M, Pedrueza-Villalmanzo E, Hermann E, Bleicken S, Spatz J, & García-Sáez AJ (2015). Bax monomers form dimer units in the membrane that further self-assemble into multiple oligomeric species. Nature Communications, 6. 10.1038/ncomms9042 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Xia S, Zhang Z, Magupalli VG, Lorenzo Pablo J, Dong Y, Vora SM, Wang L, Fu T-M, Jacobson MP, Greka A, Lieberman J, Ruan J, Wu H, & Author JR ). (2021). Gasdermin D pore structure reveals preferential release of mature interleukin-1. Nature, 593(7860), 607–611. 10.1038/s41586-021-03478-3 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Moldoveanu T, Liu Q, Tocilj A, Watson M, Shore G, & Gehring K (2006). The X-Ray Structure of a BAK Homodimer Reveals an Inhibitory Zinc Binding Site. Molecular Cell, 24(5), 677–688. 10.1016/j.molcel.2006.10.014 [DOI] [PubMed] [Google Scholar]
- 85.Sperl LE, Rührnößl F, Schiller A, Haslbeck M, & Hagn F (2021). High-resolution analysis of the conformational transition of pro-apoptotic Bak at the lipid membrane. The EMBO Journal, 40(20). 10.15252/embj.2020107159 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Hockings C, Anwari K, Ninnis RL, Brouwer J, O’hely M, Evangelista M, Hinds MG, Lee EF, Fairlie WD, Dewson G, Kluck RM, Kluck W, & Hall E (2015). Bid chimeras indicate that most BH3-only proteins can directly activate Bak and Bax, and show no preference for Bak versus Bax. Citation: Cell Death and Disease, 6. 10.1038/cddis.2015.105 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Llambi F, Moldoveanu T, Tait SWG, Bouchier-Hayes L, Temirov J, McCormick LL, Dillon CP, & Green DR (2011). A Unified Model of Mammalian BCL-2 Protein Family Interactions at the Mitochondria. Molecular Cell, 44(4), 517–531. 10.1016/j.molcel.2011.10.001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Letai A, Bassik MC, Walensky LD, Sorcinelli MD, Weiler S, & Korsmeyer SJ (2002). Distinct BH3 domains either sensitize or activate mitochondrial apoptosis, serving as prototype cancer therapeutics. Cancer Cell, 2, 183–192. [DOI] [PubMed] [Google Scholar]
- 89.Kuwana T, Mackey MR, Perkins G, Ellisman MH, Latterich M, Schneiter R, Green DR, & Newmeyer DD (2002). Bid, Bax, and Lipids Cooperate to Form Supramolecular Openings in the Outer Mitochondrial Membrane. Cell, 111, 331–342. [DOI] [PubMed] [Google Scholar]
- 90.Kim H, Tu HC, Ren D, Takeuchi O, Jeffers JR, Zambetti GP, Hsieh JJD, & Cheng EHY (2009). Stepwise Activation of BAX and BAK by tBID, BIM, and PUMA Initiates Mitochondrial Apoptosis. Molecular Cell, 36(3), 487–499. 10.1016/j.molcel.2009.09.030 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Brouwer JM, Westphal D, Dewson G, Robin AY, Uren RT, Bartolo R, Thompson G. v., Colman PM, Kluck RM, & Czabotar PE (2014). Bak Core and Latch Domains Separate during Activation, and Freed Core Domains Form Symmetric Homodimers. Molecular Cell, 55(6), 938–946. 10.1016/j.molcel.2014.07.016 [DOI] [PubMed] [Google Scholar]
- 92.Brouwer JM, Lan P, Cowan AD, Bernardini JP, Birkinshaw RW, van Delft MF, Sleebs BE, Robin AY, Wardak A, Tan IK, Reljic B, Lee EF, Fairlie WD, Call MJ, Smith BJ, Dewson G, Lessene G, Colman PM, & Czabotar PE (2017). Conversion of Bim-BH3 from Activator to Inhibitor of Bak through Structure-Based Design. Molecular Cell, 68(4), 659–672.e9. 10.1016/j.molcel.2017.11.001 [DOI] [PubMed] [Google Scholar]
- 93.Moldoveanu T, Grace CR, Llambi F, Nourse A, Fitzgerald P, Gehring K, Kriwacki RW, & Green DR (2013). BID-induced structural changes in BAK promote apoptosis. Nature Structural and Molecular Biology, 20(5), 589–597. 10.1038/nsmb.2563 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Leshchiner ES, Braun CR, Bird GH, & Walensky LD (2013). Direct activation of full-length proapoptotic BAK. PNAS, 110(11). 10.1073/pnas.1214313110 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Yao S, Westphal D, Babon JJ, Thompson G. v., Robin AY, Adams JM, Colman PM, & Czabotar PE (2014). NMR studies of interactions between Bax and BH3 domain-containing peptides in the absence and presence of CHAPS. Archives of Biochemistry and Biophysics, 545, 33–43. 10.1016/j.abb.2014.01.003 [DOI] [PubMed] [Google Scholar]
- 96.Dai H, Smith A, Meng XW, Schneider PA, Pang YP, & Kaufmann SH (2011). Transient binding of an activator BH3 domain to the Bak BH3-binding groove initiates Bak oligomerization. Journal of Cell Biology, 194(1), 39–48. 10.1083/jcb.201102027 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Singh G, Guibao CD, Seetharaman J, Aggarwal A, Grace CR, Mcnamara DE, Vaithiyalingam S, Waddell MB, & Moldoveanu T (2022). Structural basis of BAK activation in mitochondrial apoptosis initiation. Nature Communications, 13(1). 10.1038/s41467-021-27851-y [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Czabotar PE, Westphal D, Dewson G, Ma S, Hockings C, Fairlie WD, Lee EF, Yao S, Robin AY, Smith BJ, Huang DCS, Kluck RM, Adams JM, & Colman PM (2013). Bax crystal structures reveal how BH3 domains activate Bax and nucleate its oligomerization to induce apoptosis. Cell, 152(3), 519–531. 10.1016/j.cell.2012.12.031 [DOI] [PubMed] [Google Scholar]
- 99.Gavathiotis E, Suzuki M, Davis ML, Pitter K, Bird GH, Katz SG, Tu H-C, Kim H, H-Y Cheng E, Tjandra N, & Walensky LD (2008). BAX activation is initiated at a novel interaction site. Nature, 455, 1076–1081. 10.1038/nature07396 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Dengler MA, Robin AY, Gibson L, Li MX, Sandow JJ, Iyer S, Webb AI, Westphal D, Dewson G, & Adams JM (2019). BAX Activation: Mutations Near Its Proposed Non-canonical BH3 Binding Site Reveal Allosteric Changes Controlling Mitochondrial Association. Cell Reports, 27(2), 359–373.e6. 10.1016/j.celrep.2019.03.040 [DOI] [PubMed] [Google Scholar]
- 101.Gavathiotis E, Reyna DE, Davis ML, Bird GH, & Walensky LD (2010). BH3-Triggered Structural Reorganization Drives the Activation of Proapoptotic BAX. Molecular Cell, 40(3), 481–492. 10.1016/j.molcel.2010.10.019 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Ye K, Meng WX, Sun H, Wu B, Chen M, Pang YP, Gao J, Wang H, Wang J, Kaufmann SH, & Dai H (2020). Characterization of an alternative BAK-binding site for BH3 peptides. Nature Communications, 11(1). 10.1038/s41467-020-17074-y [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Tan C, Dlugosz PJ, Peng J, Zhang Z, Lapolla SM, Plafker SM, Andrews DW, & Lin J (2006). Auto-activation of the apoptosis protein Bax increases mitochondrial membrane permeability and is inhibited by Bcl-2. The Journal of Biological Chemistry, 281(21), 14764–14775. 10.1074/JBC.M602374200 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Iyer S, Uren RT, Dengler MA, Shi MX, Uno E, Adams JM, Dewson G, & Kluck RM (2020). Robust autoactivation for apoptosis by BAK but not BAX highlights BAK as an important therapeutic target. Cell Death and Disease, 11(4). 10.1038/s41419-020-2463-7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Sekar G, Singh G, Qin X, Guibao CD, Schwam B, Inde Z, Grace CR, Zhang W, Slavish PJ, Lin W, Chen T, Lee RE, Rankovic Z, Sarosiek K, & Moldoveanu T (2022). Small molecule SJ572946 activates BAK to initiate apoptosis. IScience, 25(10). 10.1016/j.isci.2022.105064 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Hockings C, Alsop AE, Fennell SC, Lee EF, Fairlie WD, Dewson G, & Kluck RM (2018). Mcl-1 and Bcl-x L sequestration of Bak confers differential resistance to BH3-only proteins. Cell Death & Differentiation, 25, 719–732. 10.1038/s41418-017-0010-6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Singh G, & Moldoveanu T (2019). Methods to Probe Conformational Activation and Mitochondrial Activity of Proapoptotic BAK. Methods in Molecular Biology, 1877, 185–200. 10.1007/978-1-4939-8861-7_13 [DOI] [PubMed] [Google Scholar]
- 108.Huang K, O’Neill KL, Li J, Zhou W, Han N, Pang X, Wu W, Struble L, Borgstahl G, Liu Z, Zhang L, & Luo X (2019). BH3-only proteins target BCL-xL/MCL-1, not BAX/BAK, to initiate apoptosis. Cell Research, 29(11), 942–952. 10.1038/s41422-019-0231-y [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Cohen DT, Wales TE, McHenry MW, Engen JR, & Walensky LD (2020). Site-Dependent Cysteine Lipidation Potentiates the Activation of Proapoptotic BAX. Cell Reports, 30(10), 3229–3239.e6. 10.1016/j.celrep.2020.02.057 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Chipuk JE, McStay GP, Bharti A, Kuwana T, Clarke CJ, Siskind LJ, Obeid LM, & Green DR (2012). Sphingolipid metabolism cooperates with BAK and BAX to promote the mitochondrial pathway of apoptosis. Cell, 148(5), 988–1000. 10.1016/j.cell.2012.01.038 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Lai YC, Li CC, Sung TC, Chang CW, Lan YJ, & Chiang YW (2019). The role of cardiolipin in promoting the membrane pore-forming activity of BAX oligomers. Biochimica et Biophysica Acta - Biomembranes, 1861(1), 268–280. 10.1016/j.bbamem.2018.06.014 [DOI] [PubMed] [Google Scholar]
- 112.Landeta O, Landajuela A, Gil D, Taneva S, DiPrimo C, Sot B, Valle M, Frolov VA, & Basañez G (2011). Reconstitution of proapoptotic BAK function in liposomes reveals a dual role for mitochondrial lipids in the BAK-driven membrane permeabilization process. Journal of Biological Chemistry, 286(10), 8213–8230. 10.1074/jbc.M110.165852 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Zheng JH, Viacava Follis A, Kriwacki RW, & Moldoveanu T (2016). Discoveries and controversies in BCL-2 protein-mediated apoptosis. FEBS Journal, 2690–2700. 10.1111/febs.13527 [DOI] [PubMed] [Google Scholar]
- 114.Jenner A, Peña-Blanco A, Salvador-Gallego R, Ugarte-Uribe B, Zollo C, Ganief T, Bierlmeier J, Mund M, Lee JE, Ries J, Schwarzer D, Macek B, & Garcia-Saez AJ (2022). DRP1 interacts directly with BAX to induce its activation and apoptosis. The EMBO Journal, 41(8). 10.15252/embj.2021108587 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Ruehl S, Guy CS, Li Z, Yang M, Moldoveanu T, & Green DR (2022). Anti-apoptotic BH3-only proteins inhibit Bak-dependent apoptosis. BioRxiv. 10.1101/2022.07.24.499430 [DOI] [Google Scholar]
- 116.Cohen DT, Wales TE, McHenry MW, Engen JR, & Walensky LD (2020). Site-Dependent Cysteine Lipidation Potentiates the Activation of Proapoptotic BAX. Cell Reports, 30(10), 3229–3239.e6. 10.1016/j.celrep.2020.02.057 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Huang AS, Chin HS, Reljic B, Djajawi TM, Tan IKL, Gong J-N, Stroud DA, Huang DCS, van Delft MF, & Dewson G (2022). Mitochondrial E3 ubiquitin ligase MARCHF5 controls BAK apoptotic activity independently of BH3-only proteins. Cell Death Differ. 10.1038/s41418-022-01067-z [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Todt F, Cakir Z, Reichenbach F, Emschermann F, Lauterwasser J, Kaiser A, Ichim G, Tait SW, Frank S, Langer HF, & Edlich F (2015). Differential retrotranslocation of mitochondrial Bax and Bak. The EMBO Journal, 34(1), 67–80. 10.15252/embj.201488806 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Edlich F, Banerjee S, Suzuki M, Cleland MM, Arnoult D, Wang C, Neutzner A, Tjandra N, & Youle RJ (2011). Bcl-xL retrotranslocates Bax from the mitochondria into the cytosol. Cell, 145(1), 104–116. 10.1016/j.cell.2011.02.034 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Lauterwasser J, Todt F, Zerbes RM, Nguyen TN, Craigen W, Lazarou M, van der Laan M, & Edlich F (2016). The porin VDAC2 is the mitochondrial platform for Bax retrotranslocation. Scientific Reports, 6. 10.1038/srep32994 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Cheng EH-Y, Sheiko T. v., Fisher JK, Craigen WJ, & Korsmeyer SJ (2003). VDAC2 Inhibits BAK Activation and Mitochondrial Apoptosis. Science, 301(5632), 513–517. 10.1126/science.1086462 [DOI] [PubMed] [Google Scholar]
- 122.Chin HS, Li MX, Tan IKL, Ninnis RL, Reljic B, Scicluna K, Dagley LF, Sandow JJ, Kelly GL, Samson AL, Chappaz S, Khaw SL, Chang C, Morokoff A, Brinkmann K, Webb A, Hockings C, Hall CM, Kueh AJ, … Dewson G (2018). VDAC2 enables BAX to mediate apoptosis and limit tumor development. Nature Communications, 9(1). 10.1038/s41467-018-07309-4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Yuan Z, Dewson G, Czabotar PE, & Birkinshaw RW (2021). VDAC2 and the BCL-2 family of proteins. Biochemical Society Transactions, 49(6), 2787–2795. 10.1042/BST20210753 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Sandow JJ, Tan IK, Huang AS, Masaldan S, Bernardini JP, Wardak AZ, Birkinshaw RW, Ninnis RL, Liu Z, Dalseno D, Lio D, Infusini G, Czabotar PE, Webb AI, & Dewson G (2021). Dynamic reconfiguration of pro-apoptotic BAK on membranes. The EMBO Journal, 40(20). 10.15252/embj.2020107237 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Birkinshaw RW, Iyer S, Lio D, Luo CS, Brouwer JM, Miller MS, Robin AY, Uren RT, Dewson G, Kluck RM, Colman PM, & Czabotar PE (2021). Structure of detergent-activated BAK dimers derived from the inert monomer. Molecular Cell, 81(10), 2123–2134.e5. 10.1016/j.molcel.2021.03.014 [DOI] [PubMed] [Google Scholar]
- 126.Brouwer JM, Westphal D, Dewson G, Robin AY, Uren RT, Bartolo R, Thompson G. v., Colman PM, Kluck RM, & Czabotar PE (2014). Bak Core and Latch Domains Separate during Activation, and Freed Core Domains Form Symmetric Homodimers. Molecular Cell, 55(6), 938–946. 10.1016/j.molcel.2014.07.016 [DOI] [PubMed] [Google Scholar]
- 127.Cowan AD, Smith NA, Sandow JJ, Kapp EA, Rustam YH, Murphy JM, Brouwer JM, Bernardini JP, Roy MJ, Wardak AZ, Tan IK, Webb AI, Gulbis JM, Smith BJ, Reid GE, Dewson G, Colman PM, & Czabotar PE (2020). BAK core dimers bind lipids and can be bridged by them. Nature Structural and Molecular Biology, 27(11), 1024–1031. 10.1038/s41594-020-0494-5 [DOI] [PubMed] [Google Scholar]
- 128.Lv F, Qi F, Zhang Z, Wen M, Kale J, Piai A, Du L, Wang S, Zhou L, Yang Y, Wu B, Liu Z, Rosario J, Pogmore J, Chou JJ, Andrews DW, Lin J, & OuYang B (2021). An amphipathic Bax core dimer forms part of the apoptotic pore wall in the mitochondrial␣membrane. The EMBO Journal, 40(14). 10.15252/embj.2020106438 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Bleicken S, Jeschke G, Stegmueller C, Salvador-Gallego R, García-Sáez AJ, & Bordignon E (2014). Structural Model of Active Bax at the Membrane. Molecular Cell, 56(4), 496–505. 10.1016/j.molcel.2014.09.022 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Dewson G, Kratina T, Sim HW, Puthalakath H, Adams JM, Colman PM, & Kluck RM (2008). To Trigger Apoptosis, Bak Exposes Its BH3 Domain and Homodimerizes via BH3:Groove Interactions. Molecular Cell, 30(3), 369–380. 10.1016/j.molcel.2008.04.005 [DOI] [PubMed] [Google Scholar]
- 131.Dewson G, Ma S, Frederick P, Hockings C, Tan I, Kratina T, & Kluck RM (2012). Bax dimerizes via a symmetric BH3:groove interface during apoptosis. Cell Death and Differentiation, 19(4), 661–670. 10.1038/cdd.2011.138 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132.Mandal T, Shin S, Aluvila S, Chen H-C, Grieve C, Choe J-Y, Cheng EH, Hustedt EJ, Kyoung, &, & Oh J (2016). Assembly of Bak homodimers into higher order homooligomers in the mitochondrial apoptotic pore OPEN. Scientific Reports, 6. 10.1038/srep30763 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133.Zhang Z, Subramaniam S, Kale J, Liao C, Huang B, Brahmbhatt H, Condon SG, Lapolla SM, Hays FA, Ding J, He F, Zhang XC, Li J, Senes A, Andrews DW, & Lin J (2016). BH3-in-groove dimerization initiates and helix 9 dimerization expands Bax pore assembly in membranes. The EMBO Journal, 35(2), 208–236. 10.15252/EMBJ.201591552 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134.Uren RT, Iyer S, Bartolo R, Shi MX, Brouwer JM, Alsop AE, Dewson G, & Kluck RM (2017). Disordered clusters of Bak dimers rupture mitochondria during apoptosis. ELife. 10.7554/eLife.19944.001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135.Iyer S, Bell F, Westphal D, Anwari K, Gulbis J, Smith BJ, Dewson G, & Kluck RM (2015). Bak apoptotic pores involve a flexible C-terminal region and juxtaposition of the C-terminal transmembrane domains. Cell Death and Differentiation, 22(10), 1665–1675. 10.1038/cdd.2015.15 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136.Dewson G, Kratina T, Czabotar P, Day CL, Adams JM, & Kluck RM (2009). Bak Activation for Apoptosis Involves Oligomerization of Dimers via Their α6 Helices. Molecular Cell, 36(4), 696–703. 10.1016/j.molcel.2009.11.008 [DOI] [PubMed] [Google Scholar]
- 137.Kirsch DG, Doseff A, Chau BN, Dae-Sik L, de Souza-Pinto NC, Hansford R, Kastan MB, Lazebnik YA, & Hardwick JM (1999). Caspase-3-dependent cleavage of Bcl-2 promotes release of cytochrome c. The Journal of Biological Chemistry, 274(30), 21155–21161. 10.1074/JBC.274.30.21155 [DOI] [PubMed] [Google Scholar]
- 138.Basañez G, Zhang J, Chau BN, Maksaev GI, Frolov VA, Brandt TA, Burch J, Hardwick JM, & Zimmerberg J (2001). Pro-apoptotic cleavage products of Bcl-xL form cytochrome c-conducting pores in pure lipid membranes. The Journal of Biological Chemistry, 276(33), 31083–31091. 10.1074/JBC.M103879200 [DOI] [PubMed] [Google Scholar]
- 139.Hellmuth S, & Stemmann O (2020). Separase-triggered apoptosis enforces minimal length of mitosis. Nature, 580(7804), 542–547. 10.1038/s41586-020-2187-y [DOI] [PubMed] [Google Scholar]
- 140.López CA, Swift MF, Xu XP, Hanein D, Volkmann N, & Gnanakaran S (2019). Biophysical Characterization of a Nanodisc with and without BAX: An Integrative Study Using Molecular Dynamics Simulations and Cryo-EM. Structure, 27(6), 988–999.e4. 10.1016/j.str.2019.03.013 [DOI] [PubMed] [Google Scholar]
- 141.Xu XP, Zhai D, Kim E, Swift M, Reed JC, Volkmann N, & Hanein D (2013). Three-dimensional structure of Bax-mediated pores in membrane bilayers. Cell Death and Disease, 4(6). 10.1038/cddis.2013.210 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142.Chi X, Nguyen D, Pemberton JM, Osterlund EJ, Liu Q, Brahmbhatt H, Zhang Z, Lin J, Leber B, & Andrews DW (2020). The carboxyl-terminal sequence of bim enables bax activation and killing of unprimed cells. ELife, 9. 10.7554/ELIFE.44525 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
No data was generated in this work.