

Abstract
The positive relationship between increased levels of circulating triglycerides (TGs) and cardiovascular events has been observed for decades. Driven by genetic cohort studies, inhibitors of apolipoprotein (APO) C3 and angiopoietin like protein (ANGPTL) 3 that reduce circulating TGs are poised to enter clinical practice. We will review the biology of how inhibition of these two proteins affects circulating lipoproteins as well as the current state of clinical development of monoclonal antibodies, anti-sense oligonucleotides, and silencing RNAs targeting APOC3 and ANGPTL3.
Graphical Abstract
INTRODUCTION:
In this review, we will provide an update on the status of two exciting therapeutic approaches, presently in clinical trials, for the treatment of dyslipidemia: Inhibition of apolipoprotein (APO) C3 and angiopoietin-like protein (ANGPTL) 3. We begin with an historical overview of the term, dyslipidemia, and a brief definition of its lipid phenotype. The next section integrates the functions of APOC3 and ANGPTL3 with the physiology of the lipoproteins comprising dyslipidemia. Finally, we review the remarkable progression of clinical trials with inhibitors of APOC3 and ANGPTL3. We will not present any molecular or biochemical background for APOC3 or ANGPTL3 other than what is necessary to integrate their functions with abnormalities in lipoproteins metabolism. Additionally, we decided not to include ANGPTL4 or ANGPTL8 in this review, despite ongoing studies of both of these ANGPTL family members as well as the very significant interactions between ANGPTL8 and both ANGPTL3 and ANGPTL4. There are several recent reviews on this topic (1), (2), (3).
1. DEFINITION OF DYSLIPIDEMIA:
This term first emerged in the mid to late 1980s, and was used in the title of the New England Journal of Medicine report of the Helsinki Heart Study results in 1987 (4). After that publication, “dyslipidemia” increasingly appeared as a way to describe the multi-lipoprotein phenotype increased circulating levels of very low density lipoprotein (VLDL) triglyceride (TG), as well as alterations in the plasma levels and compositions of low density lipoproteins (LDL) and high density lipoproteins (HDL) in people with diabetes mellitus, renal disease, and atherosclerotic cardiovascular disease (ASCVD) (5), (6). In ATP III, published in 2001, dyslipidemia was linked to insulin resistance and the metabolic syndrome Although the concentrations of each of the lipoproteins defining dyslipidemia are determined to significant degrees by any individual’s genetic make-up, it is fair to state that the central pathophysiologic drivers of dyslipidemia are insulin resistance and increased assembly and secretion of VLDL (7). Relevant to this review, there are significant pre-clinical and clinical data supporting roles for APOC3 and/or ANGPTL3 in the metabolism of each of the lipoproteins that defines dyslipidemia.
2. LIPOPROTEIN METABOLISM IN DYSLIPIDEMIA AND THE ROLES OF APOC3 AND ANGPTL3:
Assembly and secretion of VLDL:
The rate of VLDL secretion is a key determinant of plasma TG levels, the generation of TG-rich lipoproteins (TRL) remnants, and the production of LDL. VLDL secretion is the culmination of several steps, including co-translational and post-translational addition of neutral lipids, mainly TG but also cholesterol ester (CE), to a nascent particle comprised of phospholipid and APOB100. These steps are followed by the transport of the mature lipoprotein (25–70 nm diameter) from the endoplasmic reticulum to the Golgi body in specialized, large COPII vesicles. The complex intracellular itinerary of VLDL requires quality control at several points and the involvement of numerous proteins (8). Although abnormalities in several of these proteins can significantly reduce VLDL secretion in mice, loss of function variants of APOB, microsomal TG transfer protein, and secretion associated Ras related GTPase 1B also cause decreased VLDL assembly and secretion in humans. In contrast, the increased secretion of APOB100 and TG that is foundational for dyslipidemia is secondary to the effects of insulin resistance and hepatic steatosis, which in most people results from the interaction of polygenic predispositions and environmental factors.
Does APOC3 play a role in VLDL secretion?
Studies in McA-Rh7777 cells demonstrated that overexpression of either wild-type APOC3 or a mutant APOC3 with defective lipid binding capacity resulted in increased or decreased secretion of large TG-rich VLDL, respectively (9), (10). These data were supported in part by studies with an APOC3 transgenic mouse that assembled and secreted larger, but not increased numbers of VLDL particles (11). In both the cell and mouse overexpression studies, increased APOC3 could have reduced intrahepatic lipolysis or re-uptake of newly secreted particles by reducing access to lipoprotein surface TG or displacing other apoproteins such as APOC2 and APOE. In contrast, three in vivo lipoprotein kinetic studies in humans with either complete (12) or partial loss of function (LOF) (13), (14) variants in APOC3 demonstrated normal rates of secretion of VLDL TG and APOB100. The human data were consistent with studies in mice showing no effect of an APOC3 antisense oligonucleotide (ASO) on VLDL secretion (15), (16). It is also important to note that many of the factors regulating chylomicron assembly and secretion, including insulin resistance, are the same as those affecting VLDL secretion (17). However, in the most recent of the three human studies (14), there was also no effect of partial LOF variants in APOC3 on APOB48 secretion. Overall, these data indicate that in vivo, in humans and in mice, APOC3 does not have a significant role in the assembly or secretion of either VLDL or chylomicron.
Does ANGPTL3 play a role in VLDL secretion?
In the first published study, which utilized the KKSan model of complete LOF of ANGPTL3, VLDL TG secretion was not different from wild-type KK mice; APOB secretion was not determined (18). More than a decade later, inactivation of ANGPTL3 by a monoclonal antibody was associated with decreased secretion of TG but not APOB100 (19), and knockdown of ANGPTL3 by an ASO generated similar results (20). However, silencing ANGPTL3 with siRNA or CRISPR/Cas9 in HUH7 or HepG2 cells resulted in marked reductions in secretion of APOB100; there were no parallel measures of TG secretion in this study (21). Determination of the in vivo kinetics of APOB100 metabolism in a family with ANGPTL3 LOF showed a dose-response reduction in VLDL APOB100 production comparing two compound heterozygotes, three heterozygotes, and two normal family members; VLDL TG secretion was also reduced in the compound heterozygous subjects (22). In a very recent study of four patients with homozygous familial hypercholesterolemia (HoFH), evinacumab, a monoclonal antibody to ANGPTL3 reduced VLDL secretion rates in two of the subjects but did not affect this parameter in the other two subjects (23). Overall, because of different models and different approaches to reducing ANGPTL3 in preclinical studies, together with very limited human data, it is too soon to draw a conclusion about ANGPTL3 and VLDL secretion.
Lipolysis of VLDL TG and TRL-remnants:
Although increased VLDL secretion is a critical factor in the genesis of dyslipidemia, the ranges of APOB100 and TG secretion, about 3-fold and 6-fold, respectively, cannot explain why plasma TG levels span a range from 50–2000 mg/dl or greater in the population. The simple answer is that the plasma concentration of any molecule derives from its absolute rate of entry into plasma and the efficiency of its removal (fractional clearance rate or FCR). The latter, in the case of TRLs, is determined by lipolysis of TG in VLDL and chylomicrons, as well as removal of VLDL and chylomicron remnant particles, and the remaining TG they carry, directly from the circulation.
TG removal from VLDL and chylomicrons involves the actions of post-heparin lipases, a family of enzymes that includes lipoprotein lipase (LPL), hepatic TG lipase (HL), and endothelial lipase (EL) (24). These enzymes differ in their optimal substrates, with LPL most active in larger TRLs, whereas HL and EL prefer smaller VLDL particles, remnants and intermediate density lipoproteins (IDL), and HDL. LPL is essential for the initial conversion of TRL TGs into fatty acids, and its complete loss leads to fasting hyperchylomicronemia with severe hypertriglyceridemia. Normally, lipolysis of nascent chylomicrons in non-hepatic tissues removes approximately 50–70% of the core TG and converts chylomicrons into smaller chylomicron remnant particles. Lipolysis of VLDL generates smaller particles, referred to as VLDL remnants or IDL. Although essentially all chylomicron remnants are taken up by the liver, VLDL remnants and IDL can either be removed by the liver or be converted to LDL, likely through the actions of HL. Human studies with VLDL labeled with radioactive iodine or stable isotopes of amino acids indicate that the percent of VLDL converted to LDL ranges from 25–75%; the determinants of this wide-range of conversion and, conversely, of hepatic removal of VLDL remnants and IDL, is not fully characterized, but HL (25) and EL (26) are involved.
In addition to the amount of active LPL protein, other factors modulate TRL lipolysis (24) (Figure 1). Foremost, are several apoproteins that circulate on the surface of the lipoproteins: these apoproteins can either augment or inhibit LPL activity. APOC2 is the obligate activator of LPL and total deficiency of APOC2 leads to severe hypertriglyceridemia, including fasting chylomicronemia; the phenotype is essentially the same as complete deficiency of LPL. Recent studies with APOC2 mimetics suggest that higher APOC2 levels augment TG clearance (27). APOA5 also activates lipolysis and complete LOF variants in the APOA5 gene have been associated with severe hypertriglyceridemia. The exact mechanism whereby APOA5 activates lipolysis has not yet been definitively determined, but it may facilitate the attachment of TRLs to the luminal endothelial surface in proximity to LPL. Another possibility that is relevant to this review, is that APOA5 can inhibit the formation of the LPL inhibitory ANGPTL3/8 complex (28). Lipase maturation factor 1 is an intracellular chaperone necessary for LPL secretion from adipose and muscle cells. Glycosylphosphatidylinositol anchored HDL binding protein 1 is required for the transport of secreted LPL across the capillary endothelial cells and the binding of LPL to the luminal surface of those cells.
Figure 1. Regulation of lipolysis:
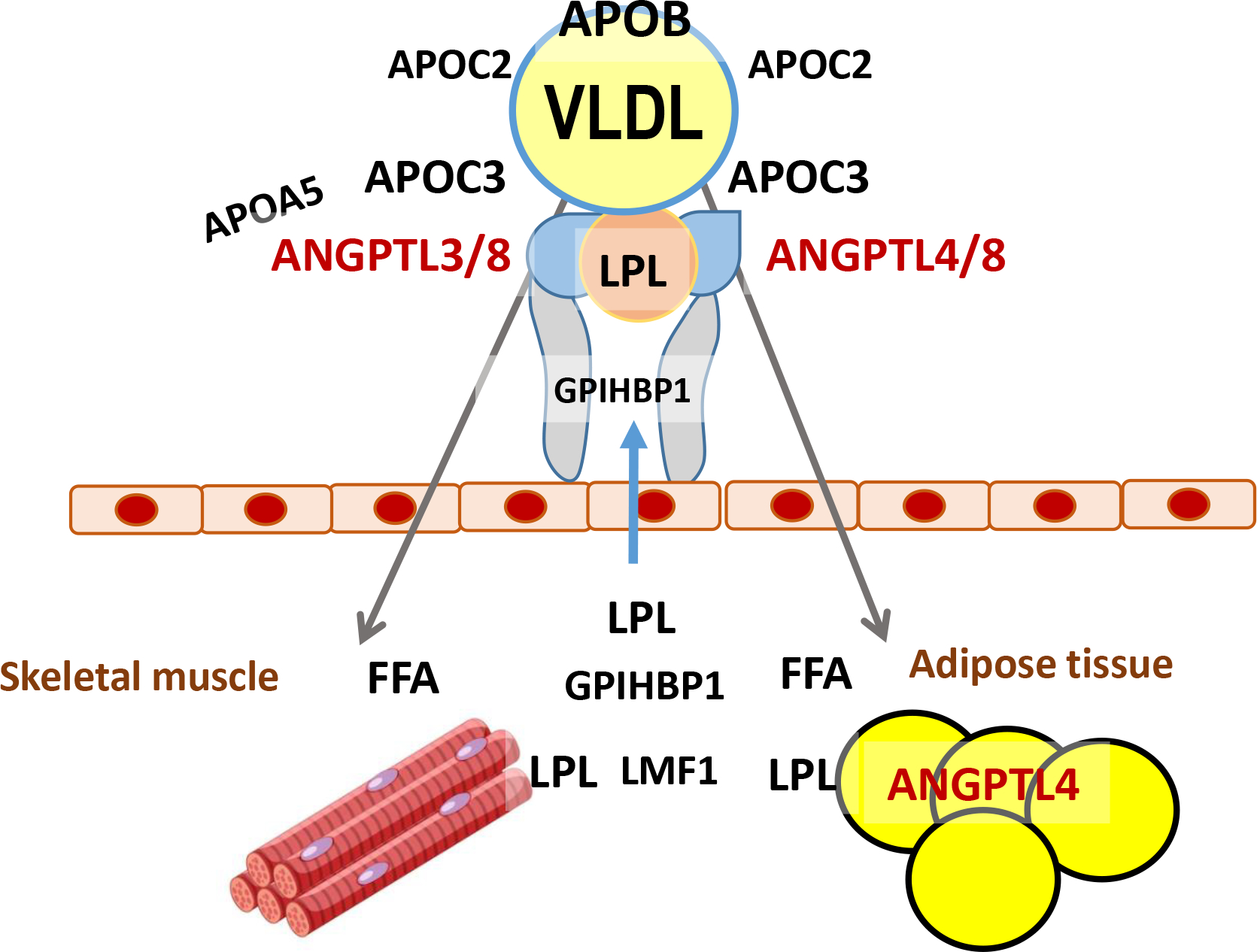
VLDL (and chylomicrons) lose much of their core lipids due to lipolysis in the circulation. The initial step in this process is mediated by lipoprotein lipase (LPL). LPL is synthesized in myocytes and adipocytes and requires the presence of lipase maturation factor 1 (LMF1) for secretion. LPL actions require its transport to the luminal endothelial cell surface in association with glycosylphosphotidylinositol anchored HDL binding protein, which also maintains LPL in an active form on the endothelial cells. LPL activity only occurs in the presence of a co-factor APOC2. LPL is inhibited by APOC3 and the complexes ANGPTL3/8 and ANGPTL4/8. APOA5 may prevent LPL inactivation by ANGPTL3/8.
Does APOC3 play a role in LPL-mediated lipolysis of TG in VLDL and chylomicrons?
Soon after its isolation and characterization, APOC3 was identified as an inhibitor of LPL (29). Two decades later, studies of the APOC3 transgenic mouse described earlier included several in vivo and in vitro experiments that led the authors to conclude that excess APOC3 caused hypertriglyceridemia not by inhibiting LPL-mediated lipolysis of VLDL TG, but by displacing APOE from TRLs, thereby reducing tissue uptake of those lipoproteins (11). Soon after, studies in an APOC3 knockout mouse supported increased tissue uptake of APOC3-depleted TRLs, rather than more efficient lipolysis of their TG, as the basis of the hypotriglyceridemic phenotype in these mice (30). In contrast to the studies in mice, all three in vivo lipoprotein kinetic studies in humans with complete or partial LOF variants in APOC3 (12), (13), (14) demonstrated markedly increased FCRs for VLDL TG and APOB100; there was no significant increase in the direct removal of VLDL particles, indicating that the major effect of lower APOC3 was increased LPL-mediated lipolysis of VLDL TG. The marked increase in the FCR of chylomicron TG and APOB48 in the third study is further support for this conclusion (14). Overall, while mouse data support a major role for APOC3 as an inhibitor of the removal of remnants of VLDL and chylomicrons by the liver, human studies support a major role for APOC3 as an inhibitor of LPL-mediated lipolysis, with minimal effect of LOF of APOC3 on hepatic remnant removal except when LPL activity is significantly decreased or completely absent.
Does ANGPTL3 play a role in LPL-mediated lipolysis of VLDL and chylomicrons?
A LOF mutation of ANGPTL3 was first associated with reduced circulating TG levels in diabetic mice (31). This same group then went on to show that LOF of ANGPTL3 reduced TG levels due to an increase in the FCR of VLDL and not a reduction in TG secretion (18). Moreover, they established that ANGPTL3 inhibited LPL, but not HL, using in vitro assays. Targeted deletions of ANGPTL3 in mice resulted in low levels of plasma TG, similar rates of TG secretion in vivo, and increased activity of LPL in post-heparin plasma compared to wild-type mice (32). A monoclonal antibody to ANGPTL3, which blocked inhibition of LPL activity by ANGPTL3, lowered plasma TG in normo- and dyslipidemic mice (33). An ASO targeting ANGPTL3 significantly increased clearance of a radiolabeled TG emulsion without affecting clearance of radiolabeled VLDL proteins, suggesting the major effect of reduced ANGPTL3 was increased LPL-mediated lipolysis. In this same study, ANGPTL3 ASO treated mice had significantly increased LPL but not HL activity (20). Importantly, increased LPL mass and activity were present in post-heparin plasma of humans homozygous for the S17K LOF mutation in ANGPTL3 who also had low levels of plasma TG; heterozygous carriers of the mutation had normal LPL mass and activity. (34) (35). In the family described earlier with LOF mutations of ANGPTL3, VLDL TG FCRs were increased 2–3 fold in the two compound heterozygous and three simple heterozygous members compared to unaffected family members and two unrelated control subjects (22).
Importantly, ANGPTL3 activity is augmented by its association with another member of this gene family ANGPTL8 and this complex acts mainly by inhibiting LPL in oxidative tissues such as skeletal and cardiac muscle (1), (2), (3). The mechanism whereby ANGPTL3/8 inhibits LPL may be either by unfolding LPL and destabilizing it or by causing the release of LPL from glycosylphosphotidylinositol anchored HDL binding protein 1 and the capillary endothelium in an inactive form (36). ANGPTL3 also inhibits EL, so not surprisingly decreased levels of ANGPTL3 are associated with reductions in both TRLs and HDL. Overall, in vitro data and results in mouse and human models of ANGPTL3 LOF convincingly demonstrate its function as an important inhibitor of LPL, particularly when ANGPTL3 is in a complex with ANGPTL8
Metabolism of TRL Remnants:
At some point during the lipolysis of VLDL TG, the particle becomes smaller and denser. Some of these remnants can still be isolated as VLDL, i.e. with a density of <1.006g/ml, and some become IDL, with a density of 1.006–1.019g/ml. Additionally, while losing TG, some of these particles become enriched in CE via the action of cholesterol ester transfer protein (CETP), which catalyzes the exchange of TG in VLDL with CE in HDL and LDL. As a result, the content of CE in remnants, in molecules per particle, can be 2–3 times greater than the CE content of LDL. Finally, some of these particles can be cleared from the circulation, mainly into the liver, by one or more pathways, while others become LDL (37) (Figure 2). It should be noted that mice lack CETP, and therefore the percent of VLDL that is converted to LDL may differ from that demonstrated in humans.
Figure 2. Triglyceride-rich lipoprotein metabolism.
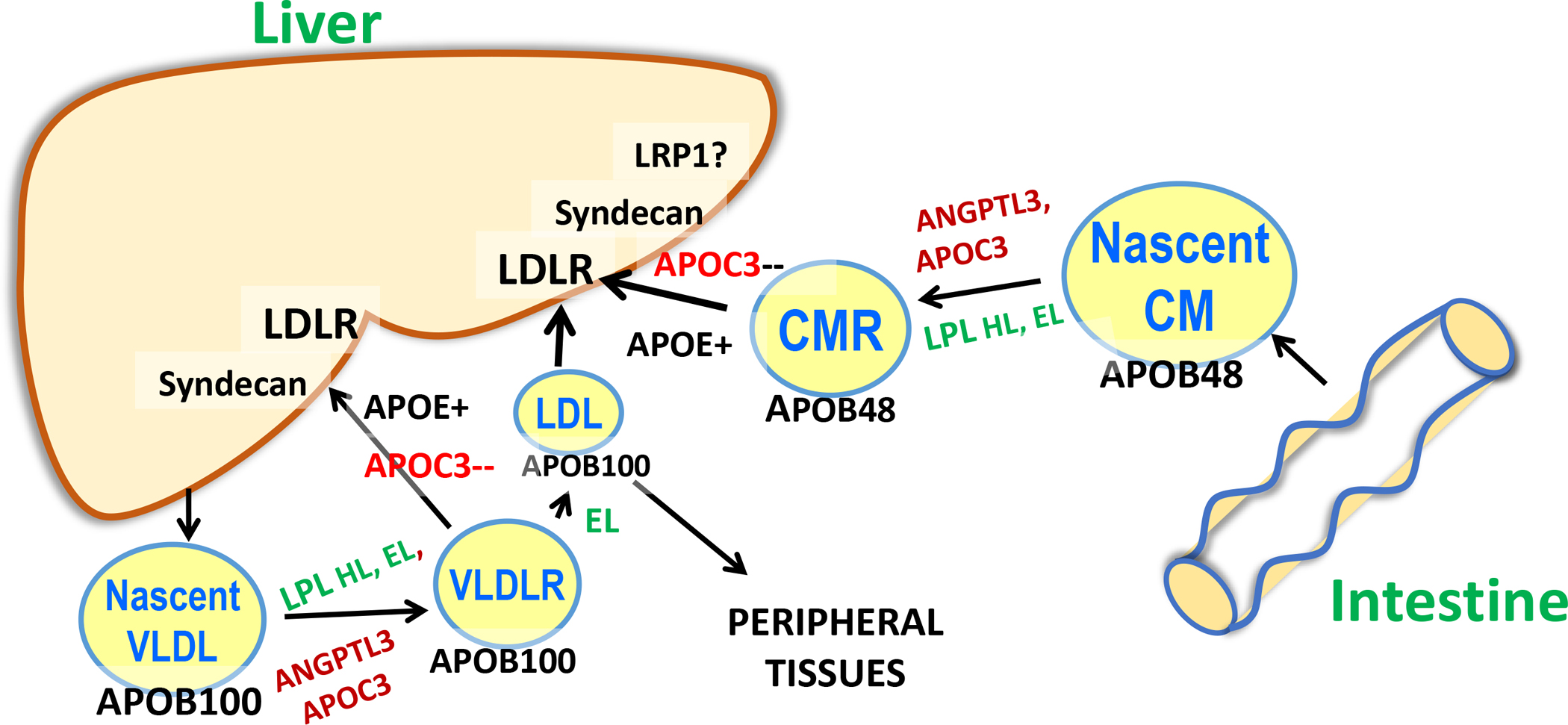
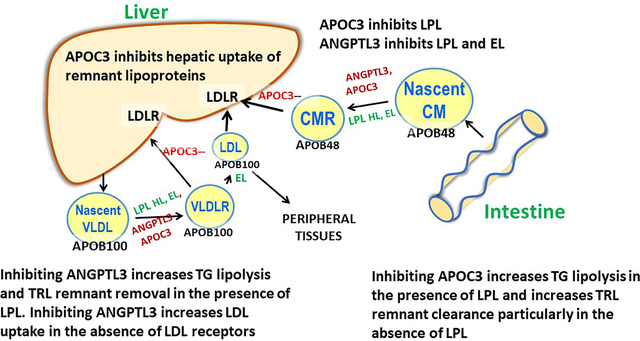
Although the initial lipolysis of TG in chylomicrons (CMs) and VLDL requires LPL, thereafter their metabolism diverges, as chylomicrons become smaller remnants (CMRs) that are removed by the liver via LDLR, syndecan, and possibly LRP1. In contrast, an average of 50% of circulating VLDL are converted to LDL rather than being removed by the liver. These two TG-rich lipoproteins also differ in that VLDL have a large form of APOB, APOB100, whereas the APOB in chylomicrons is only 48% of the total protein. Endothelial lipase (EL) is more active when ANGPTL3 is inactivated, and this enzyme along with hepatic lipase (HL) may remove lipid from smaller VLDL and CMRs. In addition, EL may play a role in generating remnants that can be taken up by the liver in the absence of LDL receptors.
Several receptors clear APOB lipoproteins by the liver: the LDL receptor (LDLR), LDLR related protein 1 (LRP1), and scavenger receptor B1 (38). In addition, syndecan, a heparin sulfate proteoglycan, captures circulating lipoproteins and modulates liver uptake of APOB lipoproteins (16).
Our vague characterization of TRL remnants is appropriate based on the difficulty in isolating pure remnants from plasma of normal and dyslipidemic individuals. This methodologic problem has led to a proliferation of publications with several different measurement methods. Most of what we know about TRL remnants is based on lipoproteins that accumulate in mouse models lacking APOE, and in humans with dysbetalipoproteinemia, a rare form of dyslipidemia characterized by homozygosity for APOE2, an isoform of APOE defective in its capacity to bind to LDLRs. Importantly, TRL remnants are at least as atherogenic, if not more so, than LDL, although they are typically present in much lower concentrations than LDL (37), (39).
Does APOC3 play a role in hepatic uptake of VLDL and chylomicron remnants?
Elegant studies in the 1980s by Richard Havel and colleagues, using perfused rat livers, demonstrated that chylomicron and VLDL remnant uptake was regulated by the proportions of non-APOB apoproteins, particularly the APOC proteins and APOE, present on the surface (40). In a later study, this same group demonstrated that hepatic uptake of rat chylomicron and VLDL remnants was inhibited by addition of APOC1, APOC2, APOC3-1, and APOC3-2 (41). As described in the sub-section on the role of APOC3 on lipolysis, more recent results from an APOC3 transgenic mouse and an APOC3 knockout mouse (11),(30) supported the results of the liver perfusion experiments. However, as noted above, in vivo studies of lipoprotein kinetics in humans indicated that the dominant effect of LOF of APOC3 was on the lipolytic pathway (12), (13), (14). On the other hand, reductions of APOC3 using an ASO in individuals completely lacking LPL protein or activity resulted in marked reductions of plasma TG levels (42), (43). These striking results indicated that ASO-mediated knockdown of APOC3 in people without any LPL resulted in LPL-independent clearance of TRLs, presumably by the liver. The latter results are consistent with exhaustive studies in various mouse knockout models demonstrating that treatment with APOC3 ASO lowered TG in the absence of each of the following alone: LPL, syndecan 1, LDLR, or LRP1. However, if both LDLR and LRP1 were reduced, APOC3 ASO treatment did not lower TG concentrations (16). Overall, data from studies in rodents and cultured hepatocytes, together with the APOC3 ASO studies in humans lacking LPL activity, support a role for APOC3 in remnant removal by the liver. However, because lipolysis is the major pathway for TG removal, the augmentation of LPL activity resulting from administration of an APOC3 ASO is likely the dominant process leading to reduced plasma TG levels in humans with active LPL.
Does ANGPTL3 play a role in hepatic uptake of VLDL and chylomicron remnants?
Individuals homozygous for ANGPTL3 LOF have significantly reduced plasma levels of TG, LDL-C, and APOB (22) (35). The reduction in TG is compatible with the loss of an inhibitor of LPL. Reductions in APOB and LDL-C levels, however, suggest that ANGPTL3 LOF increases clearance of TRL remnants or LDL particles, or both. In mice, inhibition of ANGPTL3 reduces LDL levels and this may result from the remodeling of TRLs by activation of EL, which is normally inhibited by ANGPTL3 (44), (45). It appears from those studies that removal of remnants and LDL, when ANGPTL3 is inhibited, occurs by both LDLR dependent and independent pathways. Clear support for an LDLR independent pathway derives from studies where LDL levels were reduced by targeting ANGPTL3 with a monoclonal antibody or an ASO in LDLR receptor deficient mice (19), (20), as well as studies in humans with HoFH (23), (46). Overall, studies in mice and humans indicate that ANGPTL3 is involved in hepatic uptake of TRL remnants via an EL-mediated pathway that is at least partially independent of LDLRs.
Metabolism of LDL and HDL in dyslipidemia:
As noted above, rates of conversion of VLDL and their remnants to LDL range from about 25 to 75%, with lower conversion rates seen in individuals with higher plasma levels of TRLs. Although only 50% of VLDL are, on average, converted to LDL, there are typically 10-fold greater LDL than VLDL particles. This is because the FCR of LDL is, on average, about 1/20 that of the FCR of VLDL. The LDL FCR is normal in moderate hypertriglyceridemia, but can be increased in individuals with severe hypertriglyceridemia.
Increased levels of TRLs and their remnants not only impact LDL particle numbers, but are also major determinants of LDL size and composition. Increased VLDL secretion, inefficient lipolysis of TRL TG, and/or reduced hepatic uptake of TRL remnants will generate TG-rich LDL via CETP mediated exchange. When the TG in LDL are lipolyzed by LPL (47) and probably HL and EL, the overall LDL size distribution will shift toward smaller particles.
The mechanisms for the well-known inverse correlation of HDL and TG levels are the same as those determining the size distribution and cholesterol content of LDL. When levels of TRL are elevated, CETP transfer of TRL TG for HDL CE reduces HDL-C and creates substrates for HL and EL that in turn reduce the overall HDL size and lipid content. HDL are cleared both in the liver and kidney, and smaller HDL are more rapidly cleared from the circulation (48), (49). In addition, deficient LPL activity results in reduced generation of lipids and proteins from TRLs that normally contribute to HDL size and number (47). Thus, even in rodents that are CETP deficient, defective lipolysis due to LPL deficiency leads to reduced HDL levels (16).
Do APOC3 and ANGPTL3 play roles in LDL and HDL metabolism?
Overall, it is very likely that ANGPTL3 and APOC3 are important determinants of LDL and HDL particle number, size, and CE content via their effects on LPL-mediated lipolysis and TRL-remnant removal. Reduced levels of plasma APOC3 will increase the size and CE-content of LDL, and depending on the initial levels of TRLs and LDL, possibly increase LDL particle number by increasing conversion of VLDL to LDL. Inhibition of APOC3 will increase the size and CE-content of HDL; these changes appear to reduce the FCR of APOA1, increasing the level of this apoprotein. The effects of ANGPTL3 inhibition on LDL will have some of the same effects as inhibition of APOC3 related to lowering of TRL levels. However, reducing ANGPTL3 levels, likely via dis-inhibition of EL, can increase hepatic uptake of TRL remnants and, importantly LDL. This appears to account for the significant reduction of LDL-C in patients with isolated elevations of LDL and HoFH.
3. CLINICAL DEVELOPMENT OF INHIBITORS OF APOC3 AND ANGPTL3:
Rationale for targeting APOC3:
Human genetic findings played a critical role in the development of methods to reduce APOC3 as an approach to the treatment of hypertriglyceridemia and potentially ASCVD. A genome-wide association study in 2008 showed that members of the Old Order Amish population who were heterozygous carriers of the null mutation, R19X, in the APOC3 gene had lower serum TG levels and reduced subclinical atherosclerosis compared to non-carriers (50). In 2014, LOF variants in the APOC3 gene were shown to be associated with 40% lower plasma TG levels and a 40% lower risk of CVD (51), (52). Finally, stable isotope kinetic studies in a small group of Old Order Amish demonstrated markedly increased FCRs of VLDL APOB100 and TG in the heterozygous carriers of the APOC3 mutation compared to their unaffected siblings (13), and this finding was very recently confirmed in another study of Caucasian subjects (14). All of these data generated significant enthusiasm for APOC3, leading to the development of novel molecules targeting APOC3 for treatment of both severe hypertriglyceridemia to prevent pancreatitis and moderate hypertriglyceridemia to prevent ASCVD.
Clinical Development of APOC3 inhibitors:
It is less than 10 years since publication of the Phase I clinical trial results with an APOC3 ASO (ISIS 304801) demonstrated excellent efficacy for APOC3 and TG lowering in normal participants (15). The clinical development of ISIS 304801, a 2’–O-(2-methoxyethyl–modified) ASO, which became known as volanesorsen, proceeded rapidly (we will use the latter name from this point on although it was not used in the early trials). In a small but critical study published in 2014, three patients with Familial Chylomicronemia Syndrome (FCS) who were either homozygotes or compound heterozygotes for LOF mutations in LPL, had marked reductions in plasma APOC3 and TG while receiving volanesorsen (42).
Also in 2014, 57 non-FCS patients participated in a Phase 2 monotherapy trial of 100, 200, and 300 mg volanesorsen versus placebo administered once per week for 13 weeks (NCT01529424) (53). This cohort was not on any TG-lowering medications and at baseline, had a mean TG level of close to 600 mg/dl, a mean HDL-C of 32 mg/dl, and a mean LDL-C of 88 mg/dl. There were dose-dependent decreases in TG, with a fall of 79% in the 300 mg group compared to an increase of 20% in the placebo group. There were significant increases in both HDL-C and LDL-C, and plasma APOB levels were unchanged. Twenty-eight additional patients who were on stable treatment with fibrate, were studied in parallel. They had mean baseline TG levels of 376 mg/dl and essentially the same percent reduction in TG (53). A small study in patients with type 2 diabetes mellitus, published in 2016, demonstrated similar efficacy of volanesorsen for lowering APOC3 and TG concentrations. In this study, there was a suggestion of improvement in insulin sensitivity and HbA1c levels (54).
APPROACH, published in 2019, was a phase 3, 52-week randomized trial of 66 patients with FCS (41 homozygous LOF mutations in LPL, 11 compound heterozygous LOF mutations for any of the genes required for LPL activity, and 14 with the same phenotype and very low LPL activity (NCT02211209) (43). Baseline mean plasma TG levels were 2209 mg/dl. Volanesorsen, 300 mg/week reduced plasma TG levels by 77%. In the volanesorsen group, injection-site reactions and thrombocytopenia with platelet levels of <100,000/μL were seen in 60% and 45% of the patients, respectively. Pancreatitis incidence was adjudicated in an exploratory analysis: over 52 weeks, there were 4 episodes in 3 patients on placebo and 1 episode in the volanesorsen group (43).
Also in 2019, a modification of volanesorsen with a tri-antennary N-acetyl galactosamine (GalNac) molecule at the 5’ end of the nucleic acid sequence (AKCEA-APOCIII-LRX), was introduced in a Phase 1/2a trial (NCT02900027) (55). The hepatic GalNac-targeting of the ASO allowed for much lower dosing, leading to many fewer adverse events, including injection site reactions and systemic flu-like symptoms. AKCEA-APOCIII-LRX reduced TG in multiple dose cohorts 50 to 66% from baseline at doses of 15 or 30 mg weekly or 60 mg every 4 weeks. There are several clinical trials ongoing with this modified APOC3 ASO, now called Olezarsen.
COMPASS, published in 2021, was a phase 3 trial of 113 patients with multifactorial severe hypertriglyceridemia (plasma TG ≥500 mg/dl) or FCS (7 subjects) and a mean baseline TG of 1261 mg/dl (56). They were randomized to receive 300 mg/week of volanesorsen or placebo for 26 weeks (NCT02300233). Volanesorsen reduced plasma TG levels by 71% versus no change in the placebo group. HDL-C levels increased 61% from a baseline of 25 mg/dl. LDL-C increased 96% from a baseline of 64 mg/dl in the volanesorsen group, whereas serum APOB concentration did not change significantly. In the volanesorsen group, 24% of the patients exhibited injection-site reactions and one patient exhibited thrombocytopenia with platelet level of <50,000/μL. There were 5 adjudicated cases of acute pancreatitis in 3 of the 38 patients in the placebo group and none in the 75 patients treated with volanesorsen.
Other methods to reduce APOC3 are in clinical trials, including the use of a liver directed silencing RNA. In a Phase 1 study presented at AHA in 2021, ARO-APOC3 reduced TG as much as 90% in 4 subjects with FCS and 25 subjects with multifactorial chylomicronemia syndrome. Data from an interim analysis of the ARO-APOC3 Phase 2 trial in patients with a median TG levels of 650–700 mg/dl presented at the recent AHA 2022 meeting, demonstrated reductions in plasma TG from 78–86% across 3 doses over 16 weeks after administration of the RNAi at day 0 and week 12. Non-HDL-C levels fell 35–45%, HDL-C increased 75–99%, and LDL-C increased 13–22%.
Rationale for targeting ANGPTL3:
Interest in developing inhibitors of ANGPTL3, derives from both preclinical and clinical data described earlier in this review. Cohort studies further increased the enthusiasm for ANGPTL3 as a target. In 2017, a case-control study of 58,000 individuals in a healthcare system demonstrated that carriers of 13 different ANGPTL3 LOF variants had markedly lower plasma ANGPTL3 levels than noncarriers, and this reduction in ANGPTL3 was associated with reduced plasma concentrations of TG (27%), LDL-C (9%), and HDL-C (4%), as well as a 41% reduction in rate of ASCVD (57). In the same year, a study of 180,000 subjects demonstrated reductions of 17% in plasma TG and 12% in plasma LDL-C in association with a 34% reduction in CAD in carriers of heterozygous ANGPTL3 LOF variants (58). Importantly, unlike LOF mutations in APOB and MTP associated with hypobetalipoproteinemia, patients with ANGPTL3 deficiency did not have hepatic steatosis (59).
Clinical Development of ANGPTL3 inhibitors:
In contrast to the ASO-focused clinical development of APOC3 inhibitors, three approaches have been used to knock down ANGPTL3: a monoclonal antibody, an ASO, and a siRNA. In 2019, detailed results of a phase 1, single ascending dose (SAD) clinical trial of a humanized monoclonal antibody, evinacumab, were reported (NCT02107872) (60). This study, which was conducted in 83 healthy individuals with TG levels between 150 and 450 mg/dl and an LDL of 100 mg/dl or greater, was also described briefly in reference 57 (NCT01749878). Evinacumab was administered once to each subject at 5, 10, or 20 mg/kg intravenously (IV) or 75, 150, or 250 mg subcutaneously (SC). Plasma TG concentrations were reduced at all doses, with a maximum fall of 77% after 10 mg/kg IV of evinacumab. In a companion multiple ascending dose (MAD) study, 56 subjects received IV or SC doses, similar to those in the SAD study, for 8 weeks, with a maximum reduction in TG of 83% on the 20 mg/kg IV dose. Maximal reductions in TG levels with SC dosing were in the range of 50 %. In the SAD study, LDL-C levels were lowered by 15–20% by the various IV and SC doses. In the MAD study, LDL-C concentrations fell by 20–25%. These intriguing clinical data led to a bifurcated development program for evinacumab that focused on severe hypertriglyceridemia and familial hypercholesterolemia.
Further development of evinacumab included studies of additional individuals in the SAD clinical trial described above who had more severe hypertriglyceridemia (NCT01748678; NCT02107872). In this trial, 7 subjects with “moderate” hypertriglyceridemia (TG between 450 to 1500 mg/dl) had a placebo-corrected peak median reduction in TG of approximately 60%. An additional 9 subjects with “severe” hypertriglyceridemia (TG greater than 1000 mg/dl and sequence variants in APOA5, APOC2, and LPL, including 4 subjects with FCS) had a very heterogeneous response to treatment, with reductions in TG ranging from 0.9 to 93.2% (61). This paper, published in 2021, concluded that the heterogeneity in responsiveness may have been caused by “variability of disease-causing sequences in the LPL pathway.”
The program to develop evinacumab as a single therapy to lower both TG and LDL was expanded based on findings that LDL-C fell 20–25% in subjects with TG levels between 150 and 450 mg/dl in the first published phase 1 trial (NCT01748678; NCT02107872). Likely based on studies in which evinacumab lowered LDL-C levels in mice lacking LDLRs, further development targeted HoFH. In a proof of concept, open-label study published in 2017, 9 patients with HoFH (3 with null alleles) and LDL-C levels at baseline of 376 mg/dl despite aggressive multi-drug therapy, had evinacumab added to their regimen (NCT02265952). After 4 weeks, LDL-C was reduced by 49% with a range of 25–90%. APOB concentrations fell by 46% (62).
A larger study, the ELIPSE HoFH trial (NCT03399786) with 65 subjects, was published in 2020 and confirmed that evinacumab could reduce LDL-C 47%. Fifteen subjects with null-null variants received evinacumab and had a fall in LDL-C of 43%; making evinacumab the only LDL-reducing therapy that does not require the presence of LDLRs. Additionally, APOB levels were reduced by 41% whereas Lp(a) levels were unaffected (46). Evinacumab has also been shown to reduce LDL-C in subjects with refractory hypercholesterolemia despite multi-drug treatment (NCT03175367). Patients were eligible if they had an LDL-C greater than 70 mg/dl with clinical ASCVD or 100 mg/dl without clinical ASCVD and were receiving treatment with a PCSK9 inhibitor and a maximally tolerated dose of a statin with or without ezetimibe (63). As a result of these studies, evinacumab has been approved by the FDA for the treatment of HoFH.
Complementing the monoclonal antibody approach, both ASO and siRNA therapeutics are in development to knockdown ANGPTL3. Several studies have been reported with the ASO vupanorsen (ISIS 703802; AKCEA-ANGPTL3-LRx; IONIS-ANGPTL3-LRx), which is a GalNAc-conjugated ASO that targets ANGPTL3 mRNA. In a phase 1 trial published in 2017, 44 healthy participants (plasma TG >90 mg/dl) were enrolled in a SAD protocol (12 subjects) with 20, 40 or 60 mg as a single dose, or a MAD protocol (32 subjects) with 10, 20, 40, or 60 mg per week for 6 weeks) of vupanorsen versus placebo (NCT02709850). In the MAD study, TG levels fell 63%, LDL-C levels were reduced 33%, HDL-C concentrations fell 27%, and APOB levels were reduced 26% at the highest dose. Similar results were observed 15 days after dosing in the SAD protocol (20).
In 2020, a phase 2a trial tested the efficacy and safety of vupanorsen, 40 or 80 mg every 4 weeks or 20 mg every week in 105 patients with plasma TG >150 mg/dl, type 2 diabetes, and hepatic steatosis (NCT03371355). The change from baseline to 6 months of treatment in plasma TG was the primary endpoint. The study cohort has a baseline median TG of 252 mg/dl, LDL-C of 101 mg/dl and HDL-C of 37 mg/dl. Vupanorsen reduced plasma TG levels by 36, 53, and 47% at dosages of 40 or 80 mg every 4 weeks or 20 mg every week SC, respectively. Remnant cholesterol levels were reduced by 38%, LDL-C by 7% HDL-C levels by 24%, and APOB levels by 9% at the 80 mg per 4 weeks dose. HbA1c was 8.2% and did not change with any of the doses. Hepatic fat fraction (HFF) was 17.6% at baseline and increased by 4.1% on the 80 mg every 4 weeks dose. On that same dose, there were small but significant increases in ALT and AST. Injection-site pruritus (14% of the participants) and injection-site erythema (12% of the participants) were the most common side effects. None of the participants exhibited a platelet level <100,000/mm3 (64).
Recently, 286 individuals with non-HDL-C of 100 mg/dl or greater and TG levels of 150–500 mg/dl on statins were enrolled in a phase 2b trial of vupanorsen versus placebo (NCT04516291). Participants received 1 of 7 dose regimens or placebo SC: every 2 or 4 weeks for 24 weeks. Vupanorsen reduced plasma lipids across all doses with a range of reductions in plasma non-HDL-C from 22.0% on 60 mg every 2 weeks to 27.7% on 80 mg every 2 weeks. TG levels were reduced between 41% on 120 mg every 4 weeks and 57% on 160 mg every 2 weeks. LDL levels fell between 8 and 16%, HDL-C fell between 12 and 35%, and APOB between 6 and 15% across the range of treatments, but without clear dose-responses. Injection site reactions and liver enzyme elevations were more common at higher doses. However, elevations of ALT or AST greater than 3 times the upper limit of normal occurred in 44% of the subjects on the highest dose used, 160 mg every 2 weeks. There were no elevations of bilirubin at any dose. HFF, which ranged from 5.7 to 9.9% at baseline, had a relative increase of 76% in the group receiving vupanorsen, 160 mg every 2 weeks (65).
Other methods to reduce ANGPTL3 are in clinical trials, including the use of a liver directed silencing RNA. At the recent AHA 2022 meeting, an interim analysis of the ARO-ANG3 Phase 2 trial in patients with mixed dyslipidemia was presented. Subjects with baseline median TG levels of about 230 mg/d, and mean LDL-C levels of 100–110 mg/dl, demonstrated reductions in plasma TG from 50–60% and LDL-C of 20–30% across 3 doses. Non-HDL-C levels fell 35–45%, HDL-C decreased between 17–30%, and APOB fell 13–20%. Importantly, and in contrast to the data described above for vupanorsen, ARO–ANG3 treatment was not associated with increased liver fat.
CONCLUSIONS
With novel and effective therapies for reducing circulating TRLs, what will be their roles in the clinic? Reduction in plasma APOC3 appears to be a more effective therapy for hypertriglyceridemia, particularly in monogenic FCS, whereas initial limited data suggests that reductions of plasma ANGPTL3 is not effective in these patients. However, unlike APOC3 inhibitors, ANGPTL3 inhibition effectively reduces LDL-C, particularly in individuals with modest hypertriglyceridemia or isolated hypercholesterolemia. The question for ANGPTL3 inhibition is whether another LDL-C reducing medication will fill a clinical need beyond its use in the very rare patients with HoFH, or will physicians choose this single medication instead of using a combination of LDL- and TG-lowering drugs, i.e. a statin and fibrate or fish oil. The recent failure of pemafibrate in the PROMINENT trial may make ANGPTL3 inhibitors more attractive as a combination agent. On the other hand, the successful REDUCE-IT trial with icosapent ethyl makes the latter a strong contender as the partner with statins. Over a quarter of the adult population has hypertriglyceridemia in the range of 150–400 mg/dl, and use of either of these new agents in this large population will require outcome trials demonstrating ASCVD benefit on top of conventional LDL reduction. Such data will not be available for several years, but we will all stay tuned until that time.
HIGHLIGHTS:
APOC3 is an inhibitor of LPL and hepatic uptake of TGRL
ANGPTL3 is an inhibitor of LPL and of EL
Both are attractive targets for treatment of dyslipidemia
Loss of function of APOC3 or ANPTL3 have been linked to reduced ASCVD
Clinical development of inhibitors of either of these proteins have had promising results
Support:
HNG is supported by NHLBI 5R35HL135833; IJG is supported by NHLBI HL45095
Nonstandard Abbreviations and Acronyms
- APO
apolipoprotein
- ANGPTL
angiopoietin-like protein
- VLDL
very low density lipoprotein
- LDL
low density lipoproteins
- HDL
high density lipoproteins
- ASCVD
atherosclerotic cardiovascular disease
- TRL
TG-rich lipoproteins
- HoFH
homozygous familial hypercholesterolemia
- FCR
fractional clearance rate
- LPL
lipoprotein lipase
- HL
hepatic TG lipase
- EL
endothelial lipase
- IDL
intermediate density lipoproteins
- CETP
cholesterol ester transfer protein
- LDLR
LDL receptor
- LRP1
LDLR related protein 1
- FCS
Familial Chylomicronemia Syndrome
- GalNac
tri-antennary N-acetyl galactosamine
Footnotes
Disclosures relevant to this manuscript:
HNG has consulted for Ionis and has a grants from Ionis as a site in two Multi-Center Trials.
IJG is on Advisory Boards for Ionis and Arrowhead.
Contributor Information
Henry N. Ginsberg, Department of Medicine, Vagelos College of Physicians and Surgeons, Columbia University. New York, NY 10032.
Ira J. Goldberg, Division of Endocrinology, Diabetes and Metabolism, New York University Grossman School of Medicine, New York, NY 10016.
References
- 1.Zhang R, Zhang K. An updated ANGPTL3-4-8 model as a mechanism of triglyceride partitioning between fat and oxidative tissues. Prog Lipid Res. 2022;85:101140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.DiDonna NM, Chen YQ, Konrad RJ. Angiopoietin-like proteins and postprandial partitioning of fatty acids. Current opinion in lipidology. 2022;33:39–46. [DOI] [PubMed] [Google Scholar]
- 3.Kersten S New insights into angiopoietin-like proteins in lipid metabolism and cardiovascular disease risk. Current opinion in lipidology. 2019;30:205–11. [DOI] [PubMed] [Google Scholar]
- 4.Frick MH, Elo O, Haapa K, Heinonen OP, Heinsalmi P, Helo P, et al. Helsinki Heart Study: primary-prevention trial with gemfibrozil in middle-aged men with dyslipidemia. Safety of treatment, changes in risk factors, and incidence of coronary heart disease. The New England journal of medicine. 1987;317:1237–45. [DOI] [PubMed] [Google Scholar]
- 5.Summary of the second report of the National Cholesterol Education Program (NCEP) Expert Panel on Detection, Evaluation, and Treatment of High Blood Cholesterol in Adults (Adult Treatment Panel II). Jama. 1993;269:3015–23. [PubMed] [Google Scholar]
- 6.Executive Summary of The Third Report of The National Cholesterol Education Program (NCEP) Expert Panel on Detection, Evaluation, And Treatment of High Blood Cholesterol In Adults (Adult Treatment Panel III). Jama. 2001;285:2486–97. [DOI] [PubMed] [Google Scholar]
- 7.Choi SH, Ginsberg HN. Increased very low density lipoprotein (VLDL) secretion, hepatic steatosis, and insulin resistance. Trends Endocrinol Metab. 2011;22:353–63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Ginsberg HN. ApoB SURFs a Ride from the ER to the Golgi. Cell Metab. 2021;33:231–3. [DOI] [PubMed] [Google Scholar]
- 9.Sundaram M, Zhong S, Bou Khalil M, Links PH, Zhao Y, Iqbal J, et al. Expression of apolipoprotein C-III in McA-RH7777 cells enhances VLDL assembly and secretion under lipid-rich conditions. Journal of lipid research. 2010;51:150–61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Qin W, Sundaram M, Wang Y, Zhou H, Zhong S, Chang CC, et al. Missense mutation in APOC3 within the C-terminal lipid binding domain of human ApoC-III results in impaired assembly and secretion of triacylglycerol-rich very low density lipoproteins: evidence that ApoC-III plays a major role in the formation of lipid precursors within the microsomal lumen. J Biol Chem. 2011;286:27769–80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Aalto-Setala K, Fisher EA, Chen X, Chajek-Shaul T, Hayek T, Zechner R, et al. Mechanism of hypertriglyceridemia in human apolipoprotein (apo) CIII transgenic mice. Diminished very low density lipoprotein fractional catabolic rate associated with increased apo CIII and reduced apo E on the particles. J Clin Invest. 1992;90:1889–900. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Ginsberg HN, Le NA, Goldberg IJ, Gibson JC, Rubinstein A, Wang-Iverson P, et al. Apolipoprotein B metabolism in subjects with deficiency of apolipoproteins CIII and AI. Evidence that apolipoprotein CIII inhibits catabolism of triglyceride-rich lipoproteins by lipoprotein lipase in vivo. J Clin Invest. 1986;78:1287–95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Reyes-Soffer G, Sztalryd C, Horenstein RB, Holleran S, Matveyenko A, Thomas T, et al. Effects of APOC3 Heterozygous Deficiency on Plasma Lipid and Lipoprotein Metabolism. Arteriosclerosis, thrombosis, and vascular biology. 2019;39:63–72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Taskinen MR, Bjornson E, Matikainen N, Soderlund S, Ramo J, Ainola MM, et al. Postprandial metabolism of apolipoproteins B48, B100, C-III and E in humans with APOC3 loss-of-function mutations. JCI Insight. 2022. 7:e160607. doi: 10.1172/jci.insight.16060 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Graham MJ, Lee RG, Bell TA 3rd, Fu W, Mullick AE, Alexander VJ, et al. Antisense oligonucleotide inhibition of apolipoprotein C-III reduces plasma triglycerides in rodents, nonhuman primates, and humans. Circ Res. 2013;112:1479–90. [DOI] [PubMed] [Google Scholar]
- 16.Gordts PL, Nock R, Son NH, Ramms B, Lew I, Gonzales JC, et al. ApoC-III inhibits clearance of triglyceride-rich lipoproteins through LDL family receptors. J Clin Invest. 2016;126:2855–66. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Xiao C, Dash S, Morgantini C, Lewis GF. New and emerging regulators of intestinal lipoprotein secretion. Atherosclerosis. 2014;233:608–15. [DOI] [PubMed] [Google Scholar]
- 18.Shimizugawa T, Ono M, Shimamura M, Yoshida K, Ando Y, Koishi R, et al. ANGPTL3 decreases very low density lipoprotein triglyceride clearance by inhibition of lipoprotein lipase. J Biol Chem. 2002;277:33742–8. [DOI] [PubMed] [Google Scholar]
- 19.Wang Y, Gusarova V, Banfi S, Gromada J, Cohen JC, Hobbs HH. Inactivation of ANGPTL3 reduces hepatic VLDL-triglyceride secretion. Journal of lipid research. 2015;56:1296–307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Graham MJ, Lee RG, Brandt TA, Tai LJ, Fu W, Peralta R, et al. Cardiovascular and Metabolic Effects of ANGPTL3 Antisense Oligonucleotides. The New England journal of medicine. 2017;377:222–32. [DOI] [PubMed] [Google Scholar]
- 21.Xu YX, Redon V, Yu H, Querbes W, Pirruccello J, Liebow A, et al. Role of angiopoietin-like 3 (ANGPTL3) in regulating plasma level of low-density lipoprotein cholesterol. Atherosclerosis. 2018;268:196–206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Musunuru K, Pirruccello JP, Do R, Peloso GM, Guiducci C, Sougnez C, et al. Exome sequencing, ANGPTL3 mutations, and familial combined hypolipidemia. The New England journal of medicine. 2010;363:2220–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Reeskamp LF, Millar JS, Wu L, Jansen H, van Harskamp D, Schierbeek H, et al. ANGPTL3 Inhibition With Evinacumab Results in Faster Clearance of IDL and LDL apoB in Patients With Homozygous Familial Hypercholesterolemia-Brief Report. Arteriosclerosis, thrombosis, and vascular biology. 2021;41:1753–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Basu D, Goldberg IJ. Regulation of lipoprotein lipase-mediated lipolysis of triglycerides. Current opinion in lipidology. 2020;31:154–60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Goldberg IJ, Le NA, Paterniti JR Jr., Ginsberg HN, Lindgren FT, Brown WV. Lipoprotein metabolism during acute inhibition of hepatic triglyceride lipase in the cynomolgus monkey. J Clin Invest. 1982;70:1184–92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Khetarpal SA, Vitali C, Levin MG, Klarin D, Park J, Pampana A, et al. Endothelial lipase mediates efficient lipolysis of triglyceride-rich lipoproteins. PLoS Genet. 2021;17:e1009802. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Wolska A, Lo L, Sviridov DO, Pourmousa M, Pryor M, Ghosh SS, et al. A dual apolipoprotein C-II mimetic-apolipoprotein C-III antagonist peptide lowers plasma triglycerides. Sci Transl Med. 2020;12(528) :eaaw7905.d.aaw7905. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Chen YQ, Pottanat TG, Zhen EY, Siegel RW, Ehsani M, Qian YW, et al. ApoA5 lowers triglyceride levels via suppression of ANGPTL3/8-mediated LPL inhibition. Journal of lipid research. 2021;62:100068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Brown WV, Baginsky ML. Inhibition of lipoprotein lipase by an apoprotein of human very low density lipoprotein. Biochem Biophys Res Commun. 1972;46:375–82. [DOI] [PubMed] [Google Scholar]
- 30.Maeda N, Li H, Lee D, Oliver P, Quarfordt SH, Osada J. Targeted disruption of the apolipoprotein C-III gene in mice results in hypotriglyceridemia and protection from postprandial hypertriglyceridemia. J Biol Chem. 1994;269:23610–6. [PubMed] [Google Scholar]
- 31.Koishi R, Ando Y, Ono M, Shimamura M, Yasumo H, Fujiwara T, et al. Angptl3 regulates lipid metabolism in mice. Nat Genet. 2002;30:151–7. [DOI] [PubMed] [Google Scholar]
- 32.Fujimoto K, Koishi R, Shimizugawa T, Ando Y. Angptl3-null mice show low plasma lipid concentrations by enhanced lipoprotein lipase activity. Experimental animals. 2006;55:27–34. [DOI] [PubMed] [Google Scholar]
- 33.Gusarova V, Alexa CA, Wang Y, Rafique A, Kim JH, Buckler D, et al. ANGPTL3 blockade with a human monoclonal antibody reduces plasma lipids in dyslipidemic mice and monkeys. Journal of lipid research. 2015;56:1308–17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Robciuc MR, Maranghi M, Lahikainen A, Rader D, Bensadoun A, Öörni K, et al. Angptl3 deficiency is associated with increased insulin sensitivity, lipoprotein lipase activity, and decreased serum free fatty acids. Arteriosclerosis, thrombosis, and vascular biology. 2013;33:1706–13. [DOI] [PubMed] [Google Scholar]
- 35.Minicocci I, Montali A, Robciuc MR, Quagliarini F, Censi V, Labbadia G, et al. Mutations in the ANGPTL3 gene and familial combined hypolipidemia: a clinical and biochemical characterization. J Clin Endocrinol Metab. 2012;97:E1266–75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Ploug M. ANGPTL4: a new mode in the regulation of intravascular lipolysis. Current opinion in lipidology. 2022;33:112–9. [DOI] [PubMed] [Google Scholar]
- 37.Ginsberg HN, Packard CJ, Chapman MJ, Boren J, Aguilar-Salinas CA, Averna M, et al. Triglyceride-rich lipoproteins and their remnants: metabolic insights, role in atherosclerotic cardiovascular disease, and emerging therapeutic strategies-a consensus statement from the European Atherosclerosis Society. Eur Heart J. 2021;42:4791–806. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Out R, Kruijt JK, Rensen PC, Hildebrand RB, de Vos P, Van Eck M, et al. Scavenger receptor BI plays a role in facilitating chylomicron metabolism. J Biol Chem. 2004;279:18401–6. [DOI] [PubMed] [Google Scholar]
- 39.Salinas CAA, Chapman MJ. Remnant lipoproteins: are they equal to or more atherogenic than LDL? Current opinion in lipidology. 2020;31:132–9. [DOI] [PubMed] [Google Scholar]
- 40.Windler E, Chao Y, Havel RJ. Determinants of hepatic uptake of triglyceride-rich lipoproteins and their remnants in the rat. J Biol Chem. 1980;255:5475–80. [PubMed] [Google Scholar]
- 41.Windler E, Havel RJ. Inhibitory effects of C apolipoproteins from rats and humans on the uptake of triglyceride-rich lipoproteins and their remnants by the perfused rat liver. Journal of lipid research. 1985;26:556–65. [PubMed] [Google Scholar]
- 42.Gaudet D, Brisson D, Tremblay K, Alexander VJ, Singleton W, Hughes SG, et al. Targeting APOC3 in the familial chylomicronemia syndrome. The New England journal of medicine. 2014;371:2200–6. [DOI] [PubMed] [Google Scholar]
- 43.Witztum JL, Gaudet D, Freedman SD, Alexander VJ, Digenio A, Williams KR, et al. Volanesorsen and Triglyceride Levels in Familial Chylomicronemia Syndrome. The New England journal of medicine. 2019;381:531–42. [DOI] [PubMed] [Google Scholar]
- 44.Adam RC, Mintah IJ, Alexa-Braun CA, Shihanian LM, Lee JS, Banerjee P, et al. Angiopoietin-like protein 3 governs LDL-cholesterol levels through endothelial lipase-dependent VLDL clearance. Journal of lipid research. 2020;61:1271–86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Wu L, Soundarapandian MM, Castoreno AB, Millar JS, Rader DJ. LDL-Cholesterol Reduction by ANGPTL3 Inhibition in Mice Is Dependent on Endothelial Lipase. Circ Res. 2020;127:1112–4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Raal FJ, Rosenson RS, Reeskamp LF, Hovingh GK, Kastelein JJP, Rubba P, et al. Evinacumab for Homozygous Familial Hypercholesterolemia. The New England journal of medicine. 2020;383:711–20. [DOI] [PubMed] [Google Scholar]
- 47.Chung BH, Segrest JP, Franklin F. In vitro production of beta-very low density lipoproteins and small, dense low density lipoproteins in mildly hypertriglyceridemic plasma: role of activities of lecithin:cholester acyltransferase, cholesterylester transfer proteins and lipoprotein lipase. Atherosclerosis. 1998;141:209–25. [DOI] [PubMed] [Google Scholar]
- 48.Horowitz BS, Goldberg IJ, Merab J, Vanni TM, Ramakrishnan R, Ginsberg HN. Increased plasma and renal clearance of an exchangeable pool of apolipoprotein A-I in subjects with low levels of high density lipoprotein cholesterol. J Clin Invest. 1993;91:1743–52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Breslow JL, Eisenberg S, Brinton EA. Metabolic determinants of low HDL-C levels. Ann N Y Acad Sci. 1993;676:157–62. [DOI] [PubMed] [Google Scholar]
- 50.Pollin TI, Damcott CM, Shen H, Ott SH, Shelton J, Horenstein RB, et al. A null mutation in human APOC3 confers a favorable plasma lipid profile and apparent cardioprotection. Science. 2008;322:1702–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Jorgensen AB, Frikke-Schmidt R, Nordestgaard BG, Tybjaerg-Hansen A. Loss-of-function mutations in APOC3 and risk of ischemic vascular disease. The New England journal of medicine. 2014;371:32–41. [DOI] [PubMed] [Google Scholar]
- 52.Tg, Hdl Working Group of the Exome Sequencing Project NHL, Blood I, Crosby J, Peloso GM, Auer PL, et al. Loss-of-function mutations in APOC3, triglycerides, and coronary disease. The New England journal of medicine. 2014;371:22–31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Gaudet D, Alexander VJ, Baker BF, Brisson D, Tremblay K, Singleton W, et al. Antisense Inhibition of Apolipoprotein C-III in Patients with Hypertriglyceridemia. The New England journal of medicine. 2015;373:438–47. [DOI] [PubMed] [Google Scholar]
- 54.Digenio A, Dunbar RL, Alexander VJ, Hompesch M, Morrow L, Lee RG, et al. Antisense-Mediated Lowering of Plasma Apolipoprotein C-III by Volanesorsen Improves Dyslipidemia and Insulin Sensitivity in Type 2 Diabetes. Diabetes Care. 2016;39:1408–15. [DOI] [PubMed] [Google Scholar]
- 55.Alexander VJ, Xia S, Hurh E, Hughes SG, O’Dea L, Geary RS, et al. N-acetyl galactosamine-conjugated antisense drug to APOC3 mRNA, triglycerides and atherogenic lipoprotein levels. Eur Heart J. 2019;40:2785–96. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Gouni-Berthold I, Alexander VJ, Yang Q, Hurh E, Steinhagen-Thiessen E, Moriarty PM, et al. Efficacy and safety of volanesorsen in patients with multifactorial chylomicronaemia (COMPASS): a multicentre, double-blind, randomised, placebo-controlled, phase 3 trial. Lancet Diabetes Endocrinol. 2021;9:264–75. [DOI] [PubMed] [Google Scholar]
- 57.Dewey FE, Gusarova V, Dunbar RL, O’Dushlaine C, Schurmann C, Gottesman O, et al. Genetic and Pharmacologic Inactivation of ANGPTL3 and Cardiovascular Disease. The New England journal of medicine. 2017;377:211–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Stitziel NO, Khera AV, Wang X, Bierhals AJ, Vourakis AC, Sperry AE, et al. ANGPTL3 Deficiency and Protection Against Coronary Artery Disease. Journal of the American College of Cardiology. 2017;69:2054–63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Yue P, Tanoli T, Wilhelm O, Patterson B, Yablonskiy D, Schonfeld G. Absence of fatty liver in familial hypobetalipoproteinemia linked to chromosome 3p21. Metabolism. 2005;54:682–8. [DOI] [PubMed] [Google Scholar]
- 60.Ahmad Z, Pordy R, Rader DJ, Gaudet D, Ali S, Gonzaga-Jauregui C, et al. Inhibition of Angiopoietin-Like Protein 3 With Evinacumab in Subjects With High and Severe Hypertriglyceridemia. Journal of the American College of Cardiology. 2021;78:193–5. [DOI] [PubMed] [Google Scholar]
- 61.Ahmad Z, Banerjee P, Hamon S, Chan KC, Bouzelmat A, Sasiela WJ, et al. Inhibition of Angiopoietin-Like Protein 3 With a Monoclonal Antibody Reduces Triglycerides in Hypertriglyceridemia. Circulation. 2019;140:470–86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Gaudet D, Gipe DA, Pordy R, Ahmad Z, Cuchel M, Shah PK, et al. ANGPTL3 Inhibition in Homozygous Familial Hypercholesterolemia. The New England journal of medicine. 2017;377(3):296–7. [DOI] [PubMed] [Google Scholar]
- 63.Rosenson RS, Burgess LJ, Ebenbichler CF, Baum SJ, Stroes ESG, Ali S, et al. Evinacumab in Patients with Refractory Hypercholesterolemia. The New England journal of medicine. 2020;383:2307–19. [DOI] [PubMed] [Google Scholar]
- 64.Gaudet D, Karwatowska-Prokopczuk E, Baum SJ, Hurh E, Kingsbury J, Bartlett VJ, et al. Vupanorsen, an N-acetyl galactosamine-conjugated antisense drug to ANGPTL3 mRNA, lowers triglycerides and atherogenic lipoproteins in patients with diabetes, hepatic steatosis, and hypertriglyceridaemia. Eur Heart J. 2020;41:3936–45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Bergmark BA, Marston NA, Bramson CR, Curto M, Ramos V, Jevne A, et al. Effect of Vupanorsen on Non-High-Density Lipoprotein Cholesterol Levels in Statin-Treated Patients With Elevated Cholesterol: TRANSLATE-TIMI 70. Circulation. 2022;145:1377–86. [DOI] [PMC free article] [PubMed] [Google Scholar]