

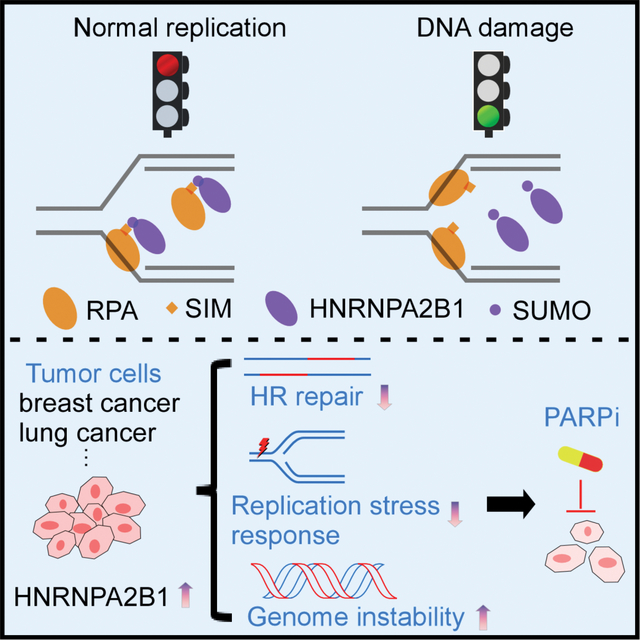
Summary
Replication Protein A (RPA) is a major regulator of eukaryotic DNA metabolism involved in multiple essential cellular processes. Maintaining appropriate RPA dynamics is crucial for cells to prevent RPA exhaustion, which can lead to replication fork breakage and replication catastrophe. However, how cells regulate RPA availability during unperturbed replication and in response to stress has not been well elucidated. Here, we show that HNRNPA2B1SUMO functions as an endogenous inhibitor of RPA during normal replication. HNRNPA2B1SUMO associates with RPA through recognizing the SUMO-interacting motif (SIM) of RPA to inhibit RPA accumulation at replication forks and impede local ATR activation. Declining HNRNPA2SUMO induced by DNA damage will release nuclear soluble RPA to localize to chromatin and enable ATR activation. Furthermore, we characterize that HNRNPA2B1 hinders homologous recombination (HR) repair via limiting RPA availability, thus conferring sensitivity to PARP inhibitors. These findings establish HNRNPA2B1 as a critical player in RPA-dependent surveillance networks.
In brief
In this study, Zhu et al. show that HNRNPA2B1SUMO associates with RPA to inhibit RPA accumulation at replication forks during normal replication. Declining HNRNPA2SUMO induced by DNA damage will release nuclear soluble RPA to localize to chromatin and enable ATR activation.
Graphical Abstract:
Introduction
Heterogeneous nuclear ribonucleoprotein A2/B1 (HNRNPA2B1) is one of the most abundant RNA-binding proteins (RBPs) that belongs to the HNRNP protein family1,2. HNRNPA2B1 is expressed as two alternatively spliced isoforms, HNRNPA2 and HNRNPB1. The shorter isoform HNRNPA2 lacks 12 amino acids at its N terminus and is the major isoform in most tissues3,4. HNRNPA2B1 contributes to multiple aspects of RNA metabolism5–8. Overexpression of HNRNPA2B1 has been described in many human cancers9–11. Previous studies have linked HNRNPA2B1’s function in mRNA processing to cancer cell migration, epithelial-mesenchymal transition and aerobic glycolysis10,12,13. In addition to prominent functions in RNA biology, HNRNPA2B1 also functions in telomere maintenance and infection by DNA viruses14,15. However, the potential role of HNRNPA2B1 in the regulation of DNA replication stress and DNA damage response (DDR) has not yet been definitively established.
The Replication Protein A (RPA) heterotrimeric complex, comprised of the RPA70 (RPA1), RPA32 (RPA2) and RPA14 subunits, plays critical roles in almost all aspects of DNA metabolism16. In eukaryotic cells RPA is the major single-stranded DNA (ssDNA)-binding protein via its oligonucleotide-binding fold domains17. Obstacles for DNA replication forks often generate increasing amounts of ssDNA which are rapidly coated by RPA, thus forming a nucleoprotein structure that recruits the ATR-ATRIP kinase complex and its regulators18. RPA is also involved in homologous recombination (HR) repair. When a DNA double-strand break (DSB) occurs in a cell, the binding of RPA to the resected DNA end allows for the formation of RAD51 nucleoprotein filament to catalyze DNA strand exchange for high-fidelity repair of DSBs by HR19. Moreover, RPA exhaustion induced by ATR inhibition during replication stress will generate unprotected ssDNA and the ensuing DNA breakage20, suggesting that keeping appropriate RPA dynamics is crucial to protect cells against replication catastrophe. Yet how cells regulate RPA availability during DNA replication under unperturbed and stressed conditions is largely unknown. HNRNPA2B1 was reported to displace RPA from telomeric ssDNA in a cell cycle-dependent manner, thus contributing to the prevention of persistent ATR activation at telomeres21. It remains elusive whether the dynamics of RPA in the contexts of DNA replication stress and DDR are also regulated by HNRNPA2B1.
Post-translational modification by the small ubiquitin-like modifier (SUMO), referred to as SUMOylation, is an important event for substrate protein regulation22. SUMOylation is regulated by the dedicated enzymes of SUMO-activating enzyme (E1, SAE1), SUMO-conjugating enzyme (E2, UBC9) and SUMO ligase (E3)23. SUMO conjugates can be removed by Sentrin (SUMO)-specific proteases (SENPs)24. The three ubiquitous SUMO isoforms (SUMO1, SUMO2 and SUMO3) can be covalently conjugated in the form of a single moiety (SUMO1) or in the form of polymeric chains (SUMO2 and SUMO3)25. Mature SUMO2 and SUMO3 are ~95% identical in sequence and thus referred as SUMO2/326. Additionally, a large number of proteins have been described to non-covalently associate with SUMO modules through SUMO-interacting motifs (SIMs) implicated in a range of cellular processes27,28.
In this study, we characterized HNRNPA2B1SUMO as an endogenous inhibitor of RPA through its recognition of the SIM motif in RPA. Our findings elucidate how HNRNPA2B1SUMO participates in replication and the genotoxic stress responses via regulating RPA dynamics.
Results
HNRNPA2 is SUMOylated at K108 by PIAS2
A previous study described that HNRNPA2B1 is SUMOylated in exosomes based on an observed shift in molecular weight and anti-SUMO1 signal8. SUMOylated HNRNPA2B1 plays a role in sorting miRNA into exosome, but whether SUMOylation of HNRNPA2B1 has a nuclear function or not is unclear. Given that SUMOylation plays a pivotal role in controlling the function of HNRNP protein family29–31, it deserves deeper examination. To investigate whether HNRNPA2B1 is a bona fide SUMOylation substrate, FLAG-tagged SUMO1 or SUMO2/3 were transiently transfected into cells. HNRNPA2B1 is commonly detected as two alternatively spliced isoforms: HNRNPA2 and HNRNPB1. However, our Western blot analysis of HNRNPA2B1 from inputs of U2OS cells (Figure 1A) and HEK293T cells (Figure S1A) showed two obvious higher molecular weight bands (~10 kDa more) in comparison with the main HNRNPA2B1 bands (the expected normal size of HNRNPA2B1 is 37 kDa), indicating that HNRNPA2B1 might be post-translationally modified in cells. We observed that HNRNPA2B1 could be strongly detected by SUMO1 immunoprecipitation but not by the highly related proteins SUMO2/3 (Figures 1A and S1A), as shown by two specific shifted bands of SUMOylated HNRNPA2 (HNRNPA2SUMO) and HNRNPB1 (HNRNPB1SUMO).
Figure 1. HNRNPA2 is SUMOylated by PIAS2 at K108.
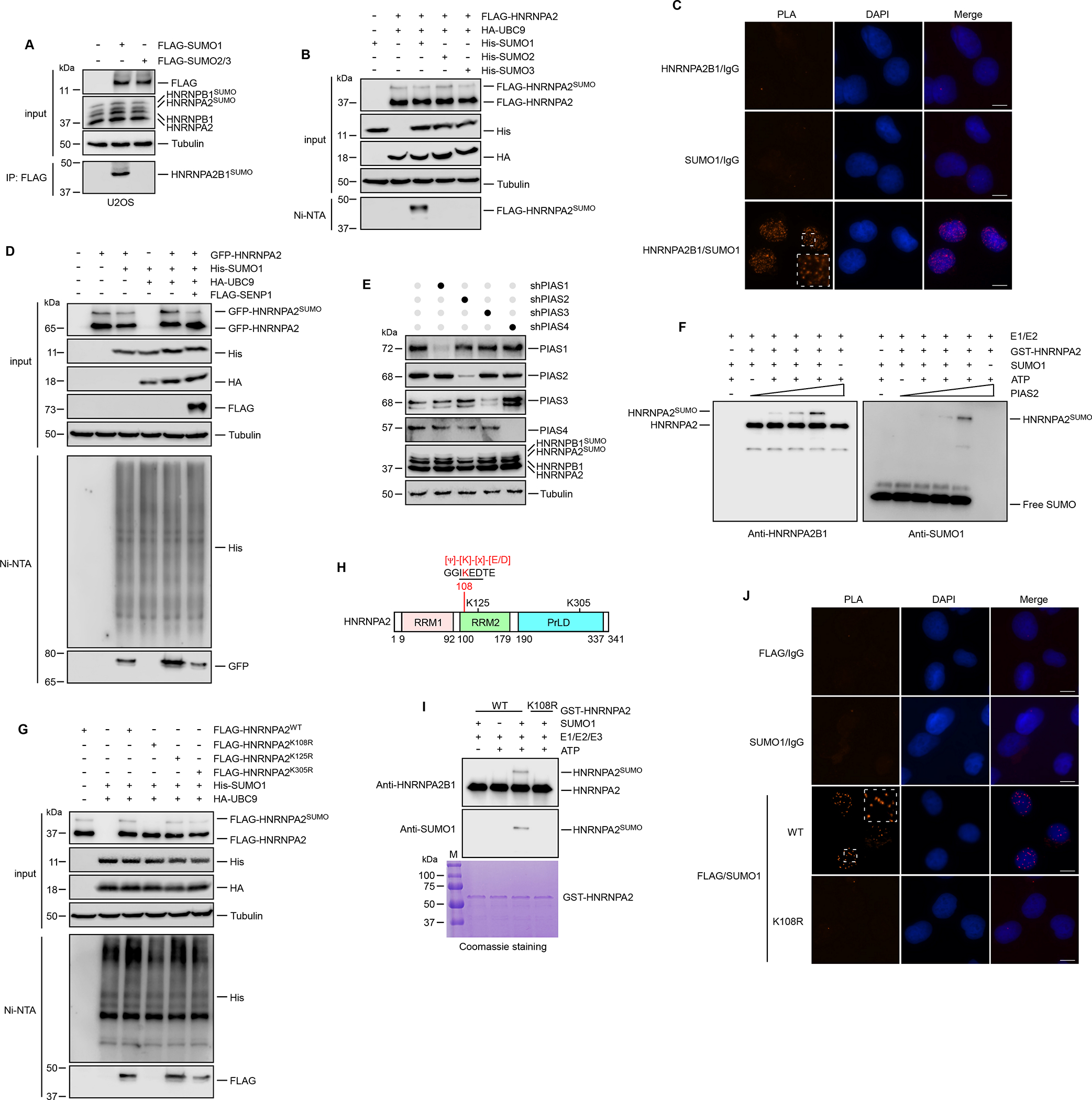
(A) U2OS cells transfected with indicated constructs were subjected to immunoprecipitation. Copurified HNRNPA2SUMO and HNRNPB1SUMO were examined by immunoblot using anti-HNRNPA2B1 antibody. (B) HEK293T cells were co-transfected with FLAG-HNRNPA2, HA-UBC9 and His-tagged SUMO1/2/3 as indicated. SUMO conjugated proteins were pulled down by Ni2+-NTA resin. (C) PLA assays were performed in U2OS cells using indicated antibodies. (D) GFP-HNRNPA2 and His-SUMO1, HA-UBC9 were transfected with or without FLAG-SENP1 into HEK293T cells. SUMO1 conjugated proteins were pulled down by Ni2+-NTA resin. (E) Knockdown of PIAS family SUMO E3 ligases by shRNAs and its effect on the expression of HNRNPA2B1SUMO were examined in U2OS cells. (F) Recombinant HNRNPA2 was incubated with SAE1, UBC9, SUMO1, ATP and an increasing concentration of PIAS2 for in vitro SUMOylation assay. (G) Wild-type (WT) and mutants of HNRNPA2 with His-SUMO1, HA-UBC9 were transfected into HEK293T cells as indicated. SUMO1 conjugated HNRNPA2 proteins enriched by Ni2+-NTA resin were immunoblotted. (H) Schematic representation of the human HNRNPA2 showing the RNA recognition motif (RRM) and prion-like domain (PrLD). The position of SUMOylation site K108 and the SUMOylation consensus sequence are indicated. (I) Bacterially expressed GST-HNRNPA2 WT or K108R were subjected to in vitro SUMOylation reactions in the presence or absence of SUMO1, SAE1, UBC9 and ATP as indicated. (J) PLA of the association of SUMO1 with FLAG-tagged WT and K108R mutant of HNRNPA2 in U2OS cells. Scale bars, 10 μm. See also Figure S1.
To further validate that HNRNPA2B1 is modified by SUMOylation, we transfected FLAG-HNRNPA2 together with His-tagged SUMO1 or SUMO2/3 and the SUMO-conjugating enzyme E2 UBC9 into HEK293T cells. As shown in Figure 1B, His-SUMO1 conjugated HNRNPA2 was pulled down by Ni2+-NTA resin under denaturing conditions with a size of ~47 kDa, which suggests HNRNPA2 covalently conjugated with one molecule of SUMO1 (HNRNPA2SUMO). However, we did not detect HNRNPA2 SUMOylation by His-tagged SUMO2/3. To examine that HNRNPA2B1 is also endogenously modified by SUMO1, co-immunoprecipitation (Co-IP) assays were performed with anti-SUMO1 antibody followed by immunoblotted with anti-HNRNPA2B1 antibody. The results showed that HNRNPA2B1 was modified by endogenous SUMO1 with a size of ~47 kDa (Figure S1B). N-Ethylmaleimide (NEM) can be used to stabilize SUMO conjugates, the absence of NEM results in almost complete loss of high molecular weight HNRNPA2SUMO (Figure S1C). Cells treated with ML-792, a potent and selective SUMO E1 enzyme inhibitor, significantly decreased the HNRNPA2SUMO level (Figure S1D).
To extend the above findings, we used a proximity-ligation assay (PLA) to analyze the association between HNRNPA2B1 and SUMO1 in cells. Fluorescent PLA dots indicative of close HNRNPA2B1/SUMO1 association were detected mainly in the nucleus (Figure 1C), where both HNRNPA2B1 and the SUMO machinery are enriched (Figure S1E). Altogether, these results demonstrated that HNRNPA2B1 is a SUMO1 substrate. Given that the shorter isoform HNRNPA2 is the main isoform accounting for ~90% of this protein in most tissues3, we focused on HNRNPA2 in the following studies.
UBC9 is the only well-characterized E2 conjugation enzyme for SUMO modification32, leading us to hypothesize that HNRNPA2 may physically interact with UBC9. As expected, Co-IP results revealed that HNRNPA2 indeed interacted with UBC9 (Figure S1F). We also checked HNRNPA2 SUMOylation in the presence of the deSUMOylating enzyme SENP1. As shown in Figure 1D, His-SUMO1 conjugated HNRNPA2 was pulled down and then immunoblotted to detect HNRNPA2SUMO. HNRNPA2SUMO was significantly enhanced by UBC9 overexpression, but this increase was efficiently reversed by co-expression with SENP1. To identify the putative HNRNPA2B1-specific SUMO E3 ligase, each of the E3 ligases PIAS1-4 was depleted using specific shRNAs. Among them, we observed PIAS2 to be a major E3 ligase involved in HNRNPA2B1 SUMOylation (Figure 1E). Knocking down PIAS2 led to a considerable reduction of HNRNPA2SUMO and HNRNPB1SUMO. To further confirm this finding, we performed in vitro SUMOylation assay. Incubation of recombinant GST-tagged HNRNPA2 in the presence of SAE1 (SUMO E1), UBC9 (SUMO E2), SUMO1 and an increasing concentration of PIAS2 efficiently drove a concentration-dependent SUMOylation of HNRNPA2 (Figures 1F and S1G), establishing PIAS2 as a SUMO E3 ligase for HNRNPA2.
To determine the potential SUMOylation site(s) of HNRNPA2, we utilized two independent computational programs: SUMOplot™ Analysis Program (https://www.abcepta.com/sumoplot) and GPS-SUMO33, to identify putative SUMOylation sites of HNRNPA2. Three lysine residues K108, K125 and K305 were observed with high prediction scores (Figures S1H and S1I). Furthermore, we found that mutation of lysine 108 to arginine (K108R), but not those of other two sites (K125R/K305R), inhibited HNRNPA2 SUMOylation (Figure 1G). SUMOylation generally occurs on the lysine residue within the consensus sequence [Ψ]-[K]-[X]-[E/D]22, where Ψ is a large hydrophobic residue and X represents any amino acid. K108 (corresponding to K120 of HNRNPB1) is embedded within a consensus SUMO sequence (Figure 1H). To further confirm that K108 is the SUMOylation site of HNRNPA2, we performed in vitro SUMOylation assays using recombinant wild-type (HNRNPA2WT) and K108R mutant (HNRNPA2K108R) proteins. As shown in Figure 1I, in contrast to HNRNPA2WT, the HNRNPA2SUMO band was abolished in our HNRNPA2K108R sample. Additionally, we obtained discrete PLA foci of HNRNPA2WT and SUMO1 (Figure 1J), but no such signals were visualized in HNRNPA2K108R expressing cells indicative of failed association. In summary, our results suggest that HNRNPA2 is SUMOylated at K108 by PIAS2. Moreover, our results are corroborated by previous mass spectrometry studies showing the presence of SUMOylation at HNRNPA2 (K108)34,35.
RPA1 associates with HNRNPA2B1SUMO through its SIM motif
HNRNPA2B1 is a well-known RNA-binding protein involved in multiple aspects of RNA metabolism. Interestingly, HNRNPA2B1 was listed as an RPA-ssDNA-binding candidate in a previous proteomic screening36, indicating HNRNPA2B1 might contribute to the cellular functions of RPA. This is in contrast to the reported role of HNRNPA2B1 in competing with RPA in telomere DNA binding21. We also found that RPA1 and RPA2, two subunits of RPA complex, were able to bind HNRNPA2B1 in cell extracts (Figure 2A). We demonstrated that these interactions were resistant to DNase I or RNase A treatment (Figure S2A), suggesting the DNA or RNA-independent associations between HNRNPA2B1 and RPA. Furthermore, we observed robust PLA signals between endogenous HNRNPA2B1 and RPA1/2, implying close proximity between HNRNPA2B1 and RPA in live cells (Figure 2B). It was previously reported that HNRNPA2B1 expression changes throughout the cell cycle37. Indeed, HNRNPA2B1 as well as HNRNPA2B1SUMO displayed a fluctuating expression pattern: with upregulation at S phase followed by a decline at G2/M, and even less expressing at G1 (Figures S2B and S2C). Accordingly, the interactions between HNRNPA2B1 and RPA1/2 were more efficiently detected at the S phase (Figure S2D).
Figure 2. RPA1 associates with HNRNPA2B1SUMO through its SIM motif.
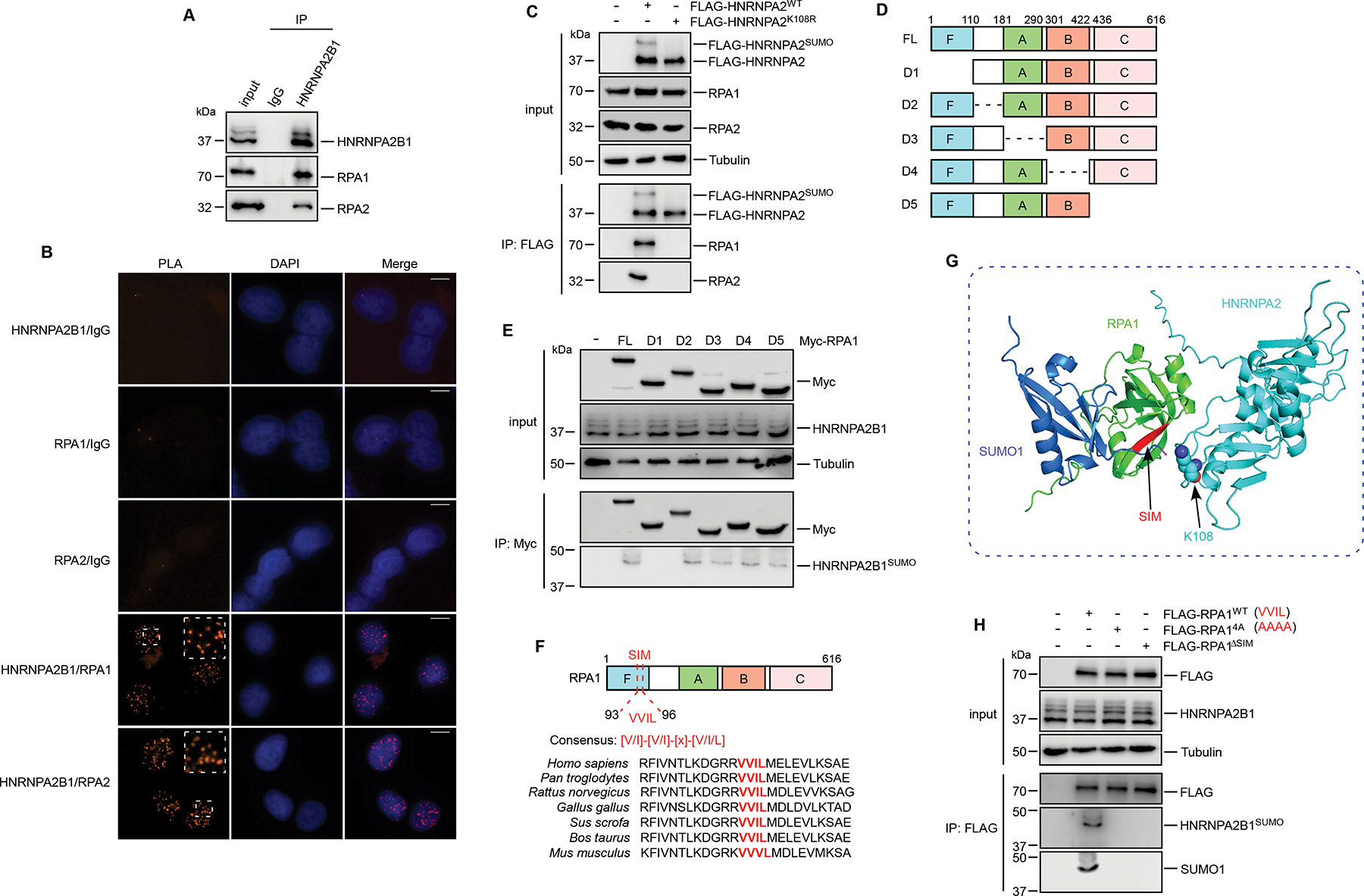
(A) U2OS cells were subjected to immunoprecipitation followed by immunoblots. (B) U2OS cells were analyzed by PLA with indicated antibodies. Scale bars, 10 um. (C) HEK293T cells transfected with HNRNPA2WT or HNRNPA2K108R were harvested for immunoprecipitation followed by immunoblots. (D-E) Schematic representation of the full-length (FL) RPA1 and the mutants used in this study (D). HEK293T cells transfected with the indicated constructs were prepared for the immunoprecipitation with anti-Myc antibody (E). (F) Schematic representation of the human RPA1 domains and sequence conservation of the SIM motif. (G) HNRNPA2K108 recognizes SIM (red) motif of RPA1 (green). The structural model of HNRNPA2SUMO-RPA1 complex was generated using AlphaFold2 online server by modeling a ternary complex of HNRNPA2-SUMO1-RPA1. (H) HEK293T cells transfected with the indicated constructs were collected for the immunoprecipitation. HNRNPA2B1SUMO was examined with anti-HNRNPA2B1 and anti-SUMO1 antibodies. See also Figure S2.
Given that there have been many studies reporting the involvement of SUMOylation in cellular activities, we set out to determine whether the interaction with RPA is SUMO-dependent. While HNRNPA2WT interacted with RPA, HNRNPA2K108R failed to associate with both RPA1 and RPA2 (Figure 2C), indicating the interactions between HNRNPA2 and RPA were SUMOylation-dependent. Next, a series of truncated or internally deleted mutants of RPA1 were utilized to further characterize the HNRNPA2B1SUMO-RPA1 interaction. RPA1 contains four oligonucleotide-binding (OB) folds designated F, A, B, and C domains, with A, B, and C domains being directly involved in ssDNA-binding17. The N-terminal F domain functions as an element of protein–protein interactions and participates in checkpoint activation17. As shown in Figures 2D and 2E, while full-length (FL) and other mutants of RPA1 could associate with HNRNPA2B1SUMO, the D1 mutant lacking the F domain failed to bind HNRNPA2B1SUMO, suggesting that RPA1 associates with HNRNPA2B1SUMO via its F domain. Since the F domain of RPA1 was previously demonstrated to be a platform for assembling the ATR-ATRIP kinase and its activators18,38, we hypothesized that the interaction of RPA1 with HNRNPA2B1SUMO may impair the recruitment of ATR-ATRIP complex, thus inhibiting local ATR activation during unperturbed replication.
As SUMO can create an interface for the recruitment of protein cofactors harboring short peptide sequences known as SUMO-interacting motifs (SIMs)27,28, we asked if RPA contains any amino acid motifs indicative of an interaction with the SUMO conjugation pathway. As expected, analysis of the RPA1 protein sequence using JASSA39 identified a putative SIM motif (amino acids 93–96) within its N-terminal F domain that could regulate interactions with SUMO-modified proteins (Figure S2E). Most SIMs feature a loose consensus sequence consisting of 3~4 aliphatic residues commonly flanked by acidic or phosphorylatable residues27, sequence alignment of RPA1 revealed a highly conserved VVIL consensus SIM motif across different species (Figure 2F). We next generated a structural model of HNRNPA2SUMO-RPA1 complex through modeling a ternary complex of HNRNPA2-SUMO1-RPA1. By mapping the position of the SIM on the structure of the N-terminal F domain of RPA1, we found that the SIM of RPA1 is near the K108 residue of HNRNPA2SUMO (Figure 2G), making it a possible module to connect HNRNPA2SUMO. To prove that RPA1 requires this SIM motif to interact HNRNPA2SUMO, we generated a SIM-deleted mutant (ΔSIM, RPA1ΔSIM) as well as a SIM-inactivated mutant (VVIL-AAAA, RPA14A) of RPA1. Both the SIM-deleted mutant and the SIM-inactivated mutant showed an almost complete loss of association with HNRNPA2B1SUMO as determined by Co-IP assays (Figure 2H). Consistently, compared with the wild-type RPA1, the SIM mutants significantly abolished the HNRNPA2B1/RPA1 PLA signals in cells (Figure S2F). These results indicate that RPA1 physically interacts with HNRNPA2B1SUMO through its SIM motif. Nevertheless, the assembly of the RPA complex and the associations between RPA with other HR repair factors including MRE11 and RAD51, were not affected by SIM mutants (Figures S2G and S2H), indicating the high substrate specificity for RPA1SIM.
To further assess the ability of RPA1SIM to recognize SUMO, we performed in vitro pull-down assays. Bacterially expressed recombinant RPA1WT or RPA14A were incubated with SUMO1 protein in the presence of increasing NaCl concentration and the bound SUMO1 was eluted for subsequent analysis. We observed that RPA1WT and SUMO1 showed consistently strong binding even under high NaCl concentration conditions (Figure S2I). However, the RPA14A mutant failed to bind SUMO1 in vitro (Figure S2I). Interestingly, the RPA1 SIM mutant was defective for its interaction with ATRIP (Figure S2J), a SUMOylated partner induced by ultraviolet radiation40, indicating the requirement of SIM for the binding of RPA1 with its SUMOylated substrate. Moreover, HNRNPA2 could be recognized by recombinant RPA1 as revealed by His pull-down assays (Figure S2K). Although the ability of HNRNPA2K108R to recognize the recombinant RPA1 was severely compromised compared to the HNRNPA2 counterpart, a signal of HNRNPA2 was detected after long exposure (Figure S2K), implying that other sequence of HNRNPA2 besides SUMOylation site also contributes to the association between HNRNPA2 and RPA1. In sum, these results prompt us to conclude that RPA1 recognizes SUMOylated HNRNPA2B1 through its SIM motif and SUMOylation of HNRNPA2B1 plays a significant part in its association with RPA1. However, an uncharacterized module of HNRNPA2B1 might also be required to impart specificity for SUMOylated HNRNPA2B1.
HNRNPA2B1SUMO prevents RPA accumulation from replication forks during unperturbed DNA replication
Since RPA plays essential roles during normal DNA replication, we wondered whether HNRNPA2B1SUMO could affect RPA loading onto ssDNA during unperturbed replication. To this end, we performed isolation of proteins on nascent DNA (iPOND) assays to analyze the impact of HNRNPA2B1 loss by CRISPR/Cas9-based targeted knockout (KO) on the RPA bound to the nascent replication forks (Figure S3A). The result revealed that compared to WT counterparts, depletion of HNRNPA2B1 in U2OS cells markedly increased RPA accumulation at replication forks (Figure 3A). Notably, previous iPOND-MS studies identified the enrichment of HNRNPA2B1 on nascent DNA41,42. Indeed, we observed that HNRNPA2B1SUMO, but not the unmodified HNRNPA2B1, could be recruited to nascent DNA (Figure 3A), possibly via its interaction with RPA. We next performed DNA fiber assays to monitor the progression of individual replication forks in HNRNPA2B1 KO U2OS cells. Newly synthesized DNA was labeled with iododeoxyuridine (ldU) followed by a second labelling with chlorodeoxyuridine (CIdU) and the length of individual tracts was quantified. WT and HNRNPA2B1 KO cells revealed no significant difference in fork progression speed (Figure 3B) or in the percentage of replication origins firing (Figure S3B), suggesting that loss of HNRNPA2B1 did not affect unperturbed DNA replication.
Figure 3. HNRNPA2B1SUMO prevents RPA accumulation from replication forks during unperturbed DNA replication.
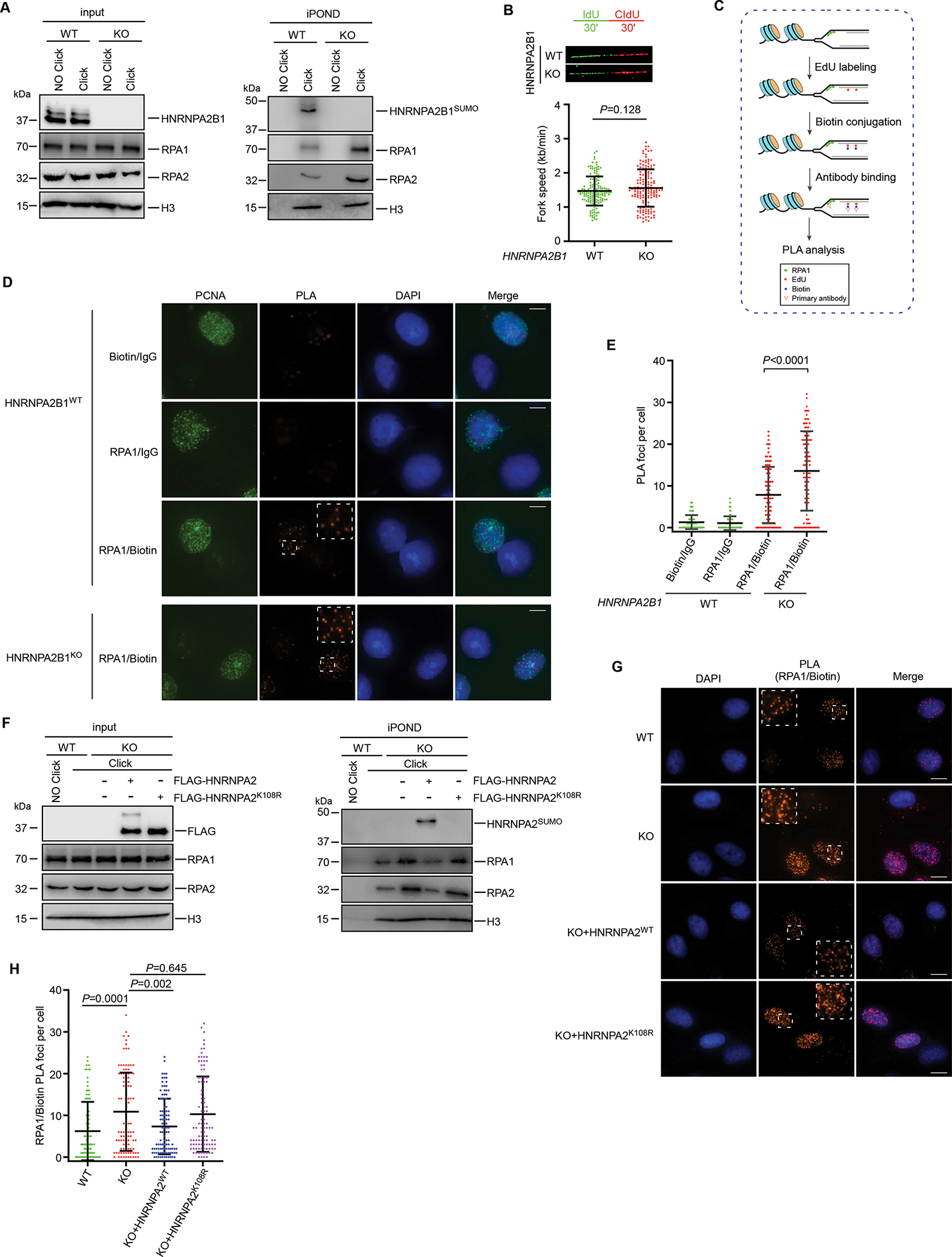
(A) Replication fork bound proteins were isolated by the iPOND assay with HNRNPA2B1 WT and KO U2OS cells. (B) Schematic for measuring replication fork progression by DNA fiber analysis. Representative images were shown and 150 events were counted for each condition. (C) Schematic of the PLA utilized to detect the association of proteins with nascent DNA as described in the main text. (D-E) Representative images of PLA foci obtained in the WT or HNRNPA2B1 KO U2OS cells according to the protocol depicted in (C). Quantification of the number of PLA foci per cell was shown in (E). (F) HNRNPA2B1 KO U2OS cells were reconstituted with FLAG-tagged HNRNPA2 or HNRNPA2K108R. iPOND assay was performed as described in (A). (G-H) HNRNPA2B1 KO U2OS cells were reconstituted with FLAG-tagged HNRNPA2 or HNRNPA2K108R. PLA assay was performed as described in (D). Quantification of the number of PLA foci per cell was shown in (H). Values represent mean and s.e.m. of three independent experiments. Scale bars, 10 μm. Statistical analysis was performed using two-tailed unpaired t-tests. See also Figure S3.
To further demonstrate the inhibitory effect of HNRNPA2B1 on accumulation of RPA at forks, we applied a PLA-based approach that measures the association of proteins at nascent ssDNA located at replication forks. In this assay, U2OS cells were first labeled with EdU for 15 min. Biotin was then conjugated to EdU-labeled DNA by using click chemistry, and PLA analysis was performed to detect proteins that are bound to biotin-labeled nascent DNA (Figure 3C). Using this approach, we observed robust HNRNPA2B1/Biotin PLA foci (Figure S3C) and these PLA foci were clearly compromised by PIAS2 knockdown (Figures S3C and S3D). This suggests that HNRNPA2B1 associates with nascent DNA in a SUMOylation-dependent manner and fits with our observations from the iPOND data (Figure 3A). As expected, knockout of HNRNPA2B1 led to a remarkable increase in the number of RPA/Biotin PLA foci (Figures 3D and 3E), indicating that HNRNPA2B1 prevents RPA association with nascent DNA during unperturbed replication. Next, we estimated the percentage of RPA bound by HNRNPA2B1, our data revealed that as much as ~43% of RPA1 in human cells were bound by HNRNPA2B1 (Figure S3E), supporting HNRNPA2B1 as an essential regulator of RPA1 during DNA replication.
To further assess the requirement of HNRNPA2B1 SUMOylation for the association between HNRNPA2B1 and RPA at replication forks, we stably re-expressed HNRNPA2WT or HNRNPA2K108R in HNRNPA2B1 KO cells. iPOND analysis showed that HNRNPA2WT but not the HNRNPA2K108R mutant protein could be recruited to the nascent replication forks (Figure 3F). Importantly, reconstitution of HNRNPA2B1 KO cells with HNRNPA2WT but not HNRNPA2K108R restored the inhibitory effect of HNRNPA2B1 on RPA accumulation at forks as revealed by iPOND assay (Figure 3F) and the number of RPA/Biotin PLA foci (Figures 3G and 3H), without causing aberrations in the cell cycle (Figure S3F). Collectively, these data indicate that HNRNPA2B1SUMO functions as an endogenous inhibitor of RPA to prevent RPA accumulation at forks during normal replication.
To determine whether the HNRNPA2B1-mediated RPA is connected to the ATR-based control of RPA at replication forks, we knocked down ATR in U2OS cells or treated cells with ATR inhibitors (ATRi) and analyzed the SUMOylation of HNRNPA2B1. As shown in Figure S3G, the SUMOylation of HNRNPA2B1 was not affected by ATR depletion or inhibition of ATR activity, suggesting that ATR is not required for HNRNPA2B1 SUMOylation. The associations between HNRNPA2B1 and RPA were not significantly altered by ATRi treatment (Figure S3H). Furthermore, depletion of HNRNPA2B1 consistently elicited a strong increase in RPA accumulation at forks in the presence of ATRi as illustrated by iPOND assay (Figure S3I) and RPA/Biotin PLA foci (Figures S3J and S3K), indicating that the HNRNPA2B1-mediated regulation of RPA at forks is not dependent on ATR pathway.
DNA damage dissociates RPA from HNRNPA2B1SUMO by reducing HNRNPA2B1SUMO
The intimate association of HNRNPA2B1 with RPA prompted us to hypothesize that HNRNPA2B1 might also participate in the DNA damage response (DDR). To test this, we analyzed the widely used genomic instability signature weighted genomic integrity index (wGII) in patients of multiple cancer types from the cancer genome atlas (TCGA) database. The results revealed that patients with breast cancer (Figure S4A), bladder cancer (Figure S4B), lung cancer (Figure S4C) or ovarian cancer (Figure S4D) with high HNRNPA2B1 expression exhibit higher wGII indicative of increased genomic instability. To further explore the performance of HNRNPA2B1 in the DDR, we employed laser micro-irradiation assay to examine the HNRNPA2B1 cellular localization following DNA damage. As shown in Figures 4A and S4E, HNRNPA2B1 was excluded from sites of DNA breaks, highlighting a potential functional role for HNRNPA2B1 in the DDR. Since HNRNPA2B1SUMO was found to be recruited to nascent strand chromatin (Figures 3A and S3C), this raised the possibility that HNRNPA2B1SUMO is repressed by DNA damage. In line with this hypothesis, cells treated with camptothecin (CPT), hydroxyurea (HU) or ionizing radiation (IR) showed a markedly decreased HNRNPA2B1SUMO (Figures 4B and S4F). Similarly, HU treatment severely decreased His-SUMO1 conjugated to HNRNPA2 as demonstrated by Ni2-NTA pull down assays (Figure S4G). Extending this observation, treatment with CPT, HU and IR considerably reduced proximity labelling of endogenous HNRNPA2B1 and SUMO1 in cells (Figures 4C and 4D), which was consistent with our biochemical analysis.
Figure 4. DNA damage disrupts HNRNPA2B1SUMO/RPA association by repressing HNRNPA2B1SUMO.
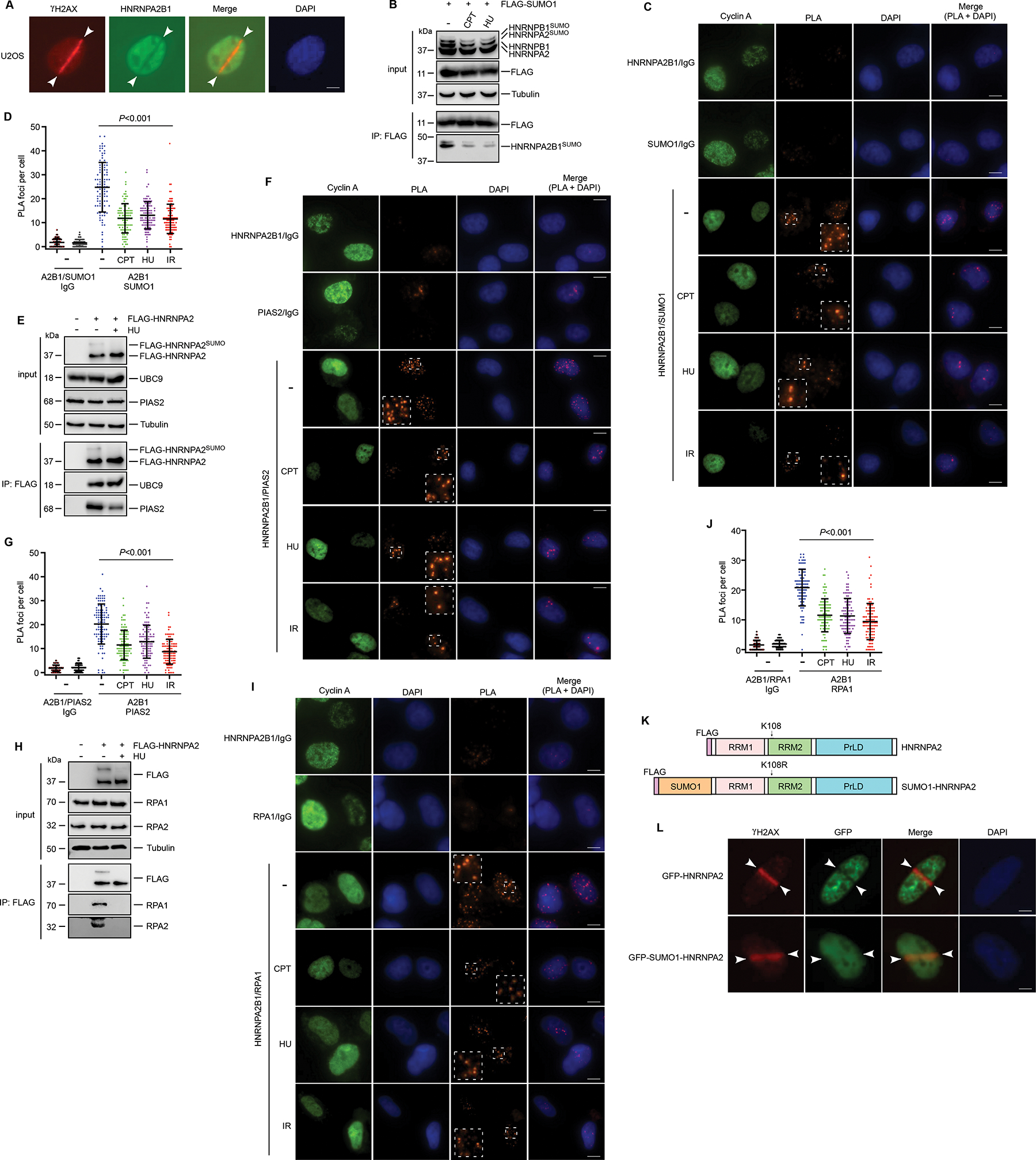
(A) U2OS cells were subjected to laser microirradiation followed by immunofluorescence. (B) HEK293T cells transfected with FLAG-SUMO1 were treated with CPT (1 uM, 1h) or HU (10 mM, 2h) followed by immunoprecipitation. (C-D) U2OS cells either untreated or exposed to CPT (1 uM, 1h), HU (10 mM, 2h) or irradiation (10 Gy, 1h) were analyzed by PLA (C). 100 cells quantified in each condition were shown in (D). (E) HEK293T cells transfected with FLAG-HNRNPA2 were treated with HU as indicated. Immunoprecipitation was performed followed by immunoblots. (F-G) U2OS cells either untreated or exposed to CPT, HU or irradiation and then analyzed by PLA (F). 100 cells quantified in each condition were shown in (G). (H) HEK293T cells transfected with FLAG-HNRNPA2 were treated with HU as indicated. Immunoprecipitation was performed followed by immunoblots. (I-J) U2OS cells either untreated or exposed to CPT, HU or irradiation were analyzed by PLA (I). 100 cells quantified in each condition were shown in (J). (K) Schematic representation of FLAG-HNRNPA2WT and FLAG-SUMO1-HNRNPA2K108R proteins. (L) U2OS cells transiently transfected with GFP-HNRNPA2 or GFP-SUMO1-HNRNPA2K108R were subjected to laser microirradiation followed by immunofluorescence. Scale bar, 10 μm (C, F and I); 5 um (A and L). Values represent mean and s.e.m. of three independent experiments. Statistical analysis was performed using two-tailed unpaired t-tests. See also Figure S4.
Given PIAS2 was the SUMO E3 ligase responsible for the HNRNPA2B1 SUMOylation, we next tested whether HNRNPA2B1 interacts with PIAS2 after DNA damage. As shown in Figure 4E, lysates from HEK293T cells transfected with FLAG-HNRNPA2 were used for Co-IP with anti-FLAG antibody. Subsequent immunoblotting analysis showed that HNRNPA2 indeed interacted with PIAS2. Of note, HU treatment dramatically reduced the association between HNRNPA2 and PIAS2, however, the HNRNPA2-UBC9 interaction was not affected by HU treatment (Figure 4E). We also applied PLA using primary antibodies against HNRNPA2B1 and PIAS2. In untreated cells, we obtained discrete proximity labelling of HNRNPA2B1 and PIAS2 (Figure 4F), corroborating the notion that these two proteins are in close proximity. Similarly, the HNRNPA2B1/PIAS2 PLA foci were markedly impaired by treatment with CPT, HU or IR in cells (Figures 4F and 4G). This suggests that DNA-damaging agents disrupt the interaction between HNRNPA2B1 and PIAS2, thus decreasing PIAS2-mediated SUMOylation of HNRNPA2B1.
Our finding that SUMOylation regulates a HNRNPA2B1-RPA interaction prompted us to interrogate this interaction under DNA damage. Interestingly, while HNRNPA2SUMO is able to bind both RPA1 and RPA2, the interaction between HNRNPA2SUMO and RPA in cells was clearly abolished by HU (Figure 4H), consistent with the decreased HNRNPA2/B1 SUMOylation. In addition to HU, CPT and IR also diminished HNRNPA2B1/RPA association as revealed by PLA (Figures 4I and 4J). In accordance with the cell cycle-dependent expression of HNRNPA2B1, the PLA foci were more efficiently detected in Cyclin A positive cells (Figures 4C, 4F and 4I). Altogether, these results indicate that DNA damage induced a decline of HNRNPA2B1SUMO that drives the exclusion of HNRNPA2B1SUMO from chromatin as well as dissociation of RPA from HNRNPA2B1SUMO.
To establish a direct link between HNRNPA2B1SUMO and DNA damage, we generated an N-terminal SUMO1-fusion of HNRNPA2K108R (termed SUMO1-HNRNPA2) (Figure 4K). Interestingly, laser microirradiation showed that compared to the HNRNPA2, the SUMO1-HNRNPA2 fusion could not be excluded from the chromatin (Figure 4L). We then examined the potential of the SUMO1-HNRNPA2 to rescue the genetically ablated HNRNPA2B1. HNRNPA2B1 KO cells were stably complemented with FLAG-tagged HNRNPA2 (termed HNRNPA2) or SUMO1-HNRNPA2 and analyzed for cell viability (Figures S4H–S4J). Clonogenic analysis showed that HNRNPA2B1-deficient U2OS and MDA-MB-231 cells were more resistant to HU treatment, whereas re-expression of HNRNPA2 would reverse this phenomenon (Figures S4K and S4L). Intriguingly, cells restored of SUMO1-HNRNPA2 displayed significantly less survival following exposure to HU (Figures S4K and S4L). These data indicate that the turnover of HNRNPA2SUMO is essential for proper cell response to DNA damage.
Forced expression of SUMO1-HNRNPA2 limits RPA availability during DNA replication stress
During DNA replication stress, RPA is the first responder in cells coordinating cellular response and checkpoint activation43. Given that RPA is responsible for the majority of ssDNA-binding activity16, we next explored whether HNRNPA2B1 regulates RPA1 function during replication stress. To examine this, HNRNPA2B1 KO cells complemented with HNRNPA2 or SUMO1-HNRNPA2 were used for Co-IP assays. As shown in Figure 5A, HU treatment abolished the HNRNPA2-RPA interaction in HNRNPA2 rescued cells. Conversely, expression of SUMO1-HNRNPA2 in HNRNPA2B1 KO cells elicited a persistent association of HNRNPA2/RPA with or without HU treatment (Figure 5A). Furthermore, the chromatin accumulation of RPA was significantly reduced in SUMO1-HNRNPA2 reconstituted cells compared to HNRNPA2 expressing cells (Figure 5B). RPA-ssDNA functions as an essential upstream module in orchestrating ATR activation17, accordingly we observed almost abrogated chromatin accumulation of ATR and ATRIP in HU-treated SUMO1-HNRNPA2 expressing cells (Figure 5B). Intriguingly, we noted an increase in unmodified HNRNPA2 within the nuclear soluble pool of HNRNPA2 expressing cells after HU treatment (Figure 5B), reflecting the dynamics of HNRNPA2 in response to genotoxic stress.
Figure 5. Forced expression of SUMO1-HNRNPA2 impairs ATR activation and the replication stress response by limiting RPA availability.
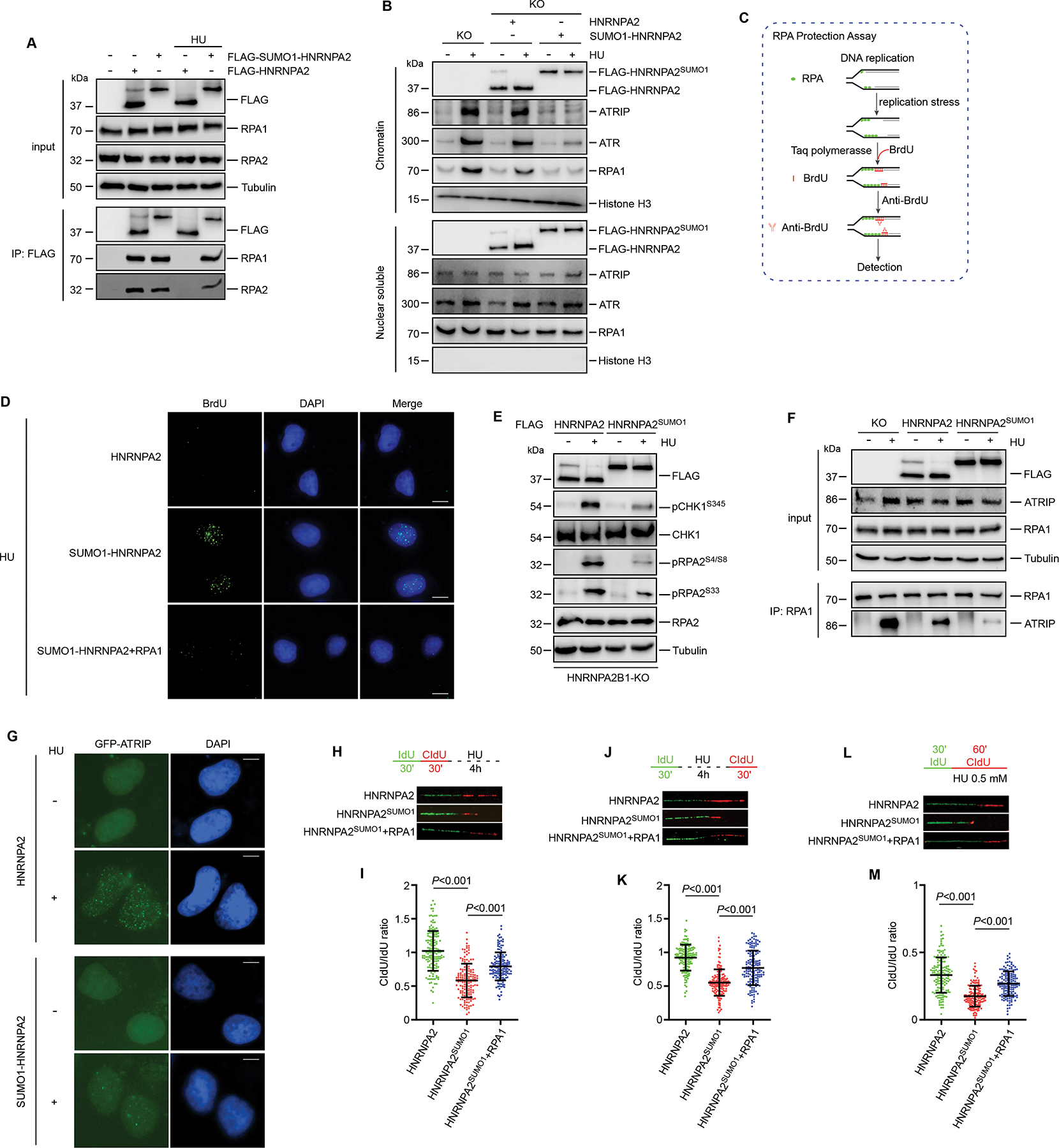
(A) HNRNPA2B1 KO U2OS cells were complemented with HNRNPA2 or SUMO1-HNRNPA2 and then treated with HU followed by immunoprecipitation. (B) HNRNPA2B1 KO U2OS cells were complemented with HNRNPA2 or SUMO1-HNRNPA2. Chromatin and nuclear soluble fractions were separated after HU treatment for immunoblot analysis. (C) Diagram showing the principle of RPA protection assay as described in the main text. (D) HNRNPA2B1 KO U2OS cells complemented with HNRNPA2, SUMO1-HNRNPA2 together with or without RPA1 were subjected to RPA protection assay. Representative images were shown. (E) HNRNPA2B1 KO U2OS cells were stably complemented with HNRNPA2 or SUMO1-HNRNPA2. Cells were treated with or without HU and harvested for immunoblots. (F) HNRNPA2B1 KO U2OS cells were stably restored with HNRNPA2 or SUMO1-HNRNPA2. Cells were treated with or without HU for immunoprecipitation. (G) HNRNPA2B1 KO U2OS cells were stably complemented with HNRNPA2 or SUMO1-HNRNPA2. GFP-ATRIP transiently transfected cells were treated with or without HU before immunofluorescence analysis. (H-I) Schematic for measuring nascent DNA degradation at stalled replication forks by DNA fiber analysis and representative images of DNA fibers (H). 150 events quantified in each condition were shown in (I). (J-K) Schematic for measuring replication fork restart by DNA fiber analysis and representative images of DNA fibers (J). 150 events quantified in each condition were shown in (K). (L-M) Schematic for measuring fork progression in cells treated with low dose HU by DNA fiber analysis and representative images of DNA fibers (L). 150 events quantified in each condition were shown in (M). Values represent mean and s.e.m. of three independent experiments. Scale bars, 10 μm. Statistical analysis was performed using two-tailed unpaired t-tests. See also Figure S5.
The above findings prompted us to hypothesize that SUMO1-HNRNPA2 would limit the ability of nuclear soluble RPA to localize to chromatin upon DNA replication stress. To this end, we performed RPA protection assays in HNRNPA2B1 KO cells complemented with HNRNPA2 or SUMO1-HNRNPA2 as described previously44,45. In this assay, replication stress-generated ssDNA fragments, which are not protected by RPA due to the limiting free RPA, would serve as a template for polymerization of 5-bromo-2’-deoxyuridine (BrdU)-labelled DNA by exogenous Taq polymerase (Figure 5C). Upon treatment with HU, anti-BrdU antibody detected few punctate foci in HNRNPA2 expressing cells, but significantly more in SUMO1-HNRNPA2 expressing cells indicating more unprotected tracks of ssDNA (Figure 5D). Notably, co-expression of RPA1 with SUMO1-HNRNPA2 could markedly reduce the ssDNA-derived BrdU foci (Figures 5D, S5A and S5B). Collectively, these results strongly support the idea that forced expression of SUMO1-HNRNPA2 limits RPA availability during DNA replication stress through restricting the access of nuclear soluble RPA to chromatin, thus generating high levels of RPA-unprotected ssDNA, implying that HNRNPA2B1SUMO functions as a “monitor” of RPA that regulates RPA dynamics during normal replication and stress response.
Forced expression of SUMO1-HNRNPA2 impairs ATR activation and the replication stress response
If HNRNPA2B1SUMO functions as a “sponge” of RPA, it should be vital for RPA-ssDNA-directed ATR activation. Immunoblotting analysis revealed that HNRNPA2B1 KO markedly enhanced the phosphorylation of CHK1S345 and RPA2S33 (Figure S5C), which are well-known substrates of ATR46,47. Similar results were observed using a phosphorylation-specific antibody against RPA2S4/S8 for fluorescent immunostaining in HU-treated cells. Depletion of HNRNPA2B1 exacerbated phospho-RPA2S4/S8 foci formation (Figures S5D and S5E)48,49. Furthermore, HNRNPA2 or SUMO1-HNRNPA2 was stably re-expressed in HNRNPA2B1 KO cells. We observed that restoring with SUMO1-HNRNPA2 resulted in reduced phosphorylation of CHK1S345, RPA2S33 and RPA2S4/S8 after HU treatment (Figure 5E). Consistently, immunofluorescence staining revealed that phospho-RPA2S4/S8 foci were efficiently suppressed by SUMO1-HNRNPA2 re-expression when compared with HNRNPA2 (Figures S5F and S5G).
An early event of ATR activation is the recruitment of the ATR-ATRIP complex to damaged sites which depends on RPA1-ATRIP interactions18. The N-terminus of RPA1 is specifically important for the activation of ATR17. Our finding that HNRNPA2B1SUMO associates with the N-terminus of RPA1 led us to ask if lower HNRNPA2B1SUMO is required for ATR-ATRIP recruitment to sites of DNA damage. Indeed, depletion of HNRNPA2B1 boosted HU-induced focus formation of GFP-ATRIP (Figures S5H and S5I). Co-IP assays were performed with anti-RPA1 antibody followed by immunoblotting using HNRNPA2B1 KO cells restored with HNRNPA2 or SUMO1-HNRNPA2. As shown in Figure 5F, in SUMO1-HNRNPA2 expressing cells the RPA1-ATRIP interaction was significantly disrupted compared to HNRNPA2 expressing cells. In addition, GFP-ATRIP foci were clearly induced by HU in HNRNPA2 expressing cells, but largely suppressed by SUMO1-HNRNPA2 expression (Figures 5G and S5J). Together, these data suggest that during replication stress, HNRNPA2B1 dissociates from RPA1 thus allowing for the assembly of the ATR-ATRIP complex at damage sites. Forced SUMO1-HNRNPA2 expression would restrict the access of ATRIP to RPA-ssDNA and undermine ATR activation.
ATR signaling plays a pivotal role in regulating cellular replication stress response50. Thus, we carried out DNA fiber assay to monitor replication fork dynamics in HNRNPA2B1 KO cells complemented with HNRNPA2 or SUMO1-HNRNPA2. The ratio of CIdU to IdU track lengths was measured to reflect DNA synthesis in the presence of stress. Compared to HNRNPA2 expressing cells, stalled forks in SUMO1-HNRNPA2 expressing cells were prone to be shorter with HU (4 mM) treatment (Figures 5H and 5I) and failed to recover after release from replication stress (Figures 5J and 5K). Additionally, in the presence of low HU doses (0.5 mM) forks progressed particularly slowly in SUMO1-HNRNPA2 expressing cells compared to HNRNPA2 expressing cells (Figures 5L and 5M). Importantly, we observed that SUMO1-HNRNPA2 expressing-induced fork defects could be largely compensated by co-expression of RPA1 (Figures 5H–5M). Collectively, we concluded that forced expression of SUMO1-HNRNPA2 affects not only the availability of nuclear soluble RPA to chromatin, but also ATR activation by restricting ATR-ATRIP from the damage sites, suggesting that reduced HNRNPA2B1SUMO and HNRNPA2B1SUMO/RPA dissociation are required for efficient replication stress response.
HNRNPA2B1 impedes HR repair via limiting RPA availability
While RPA is a crucial regulator of DNA replication and the replication stress response, RPA is also implicated in error-free repair of DNA double-strand breaks (DSBs) by HR19. Our finding that HNRNPA2B1 limits RPA availability raises an interesting possibility that HNRNPA2B1 might function in HR repair. To test this possibility, we analyzed HR deficiency (HRD) score and three independent measures of genomic instability acting as the biomarker of tumor HRD: large-scale state transitions (LST), loss of heterozygosity (LOH) and telomeric allelic imbalance (TAI), in breast cancer samples. we observed that high expression of HNRNPA2B1 is associated with an increased HRD score (Figure 6A), LST (Figure 6B), LOH (Figure 6C) and TAI (Figure 6D), indicating that HNRNPA2B1 inhibits HR repair. To further confirm this finding, we employed a well-established DR-GFP HR reporter assay in which expression of I-SceI endonuclease generates a DSB that when repaired by HR restores GFP expression (Figure 6E)51. In both HEK293T and U2OS cells, loss of HNRNPA2B1 enhanced HR efficiency (Figures 6F and S6A–S6D), directly suggesting that HNRNPA2B1 impedes HR repair. Notably, we did not find significant changes in cell cycle distributions in asynchronous cell populations (Figure S6E), suggesting that the observed effect was not a result of changes in cell cycle regulation.
Figure 6. HNRNPA2B1 impedes HR repair via limiting RPA availability.
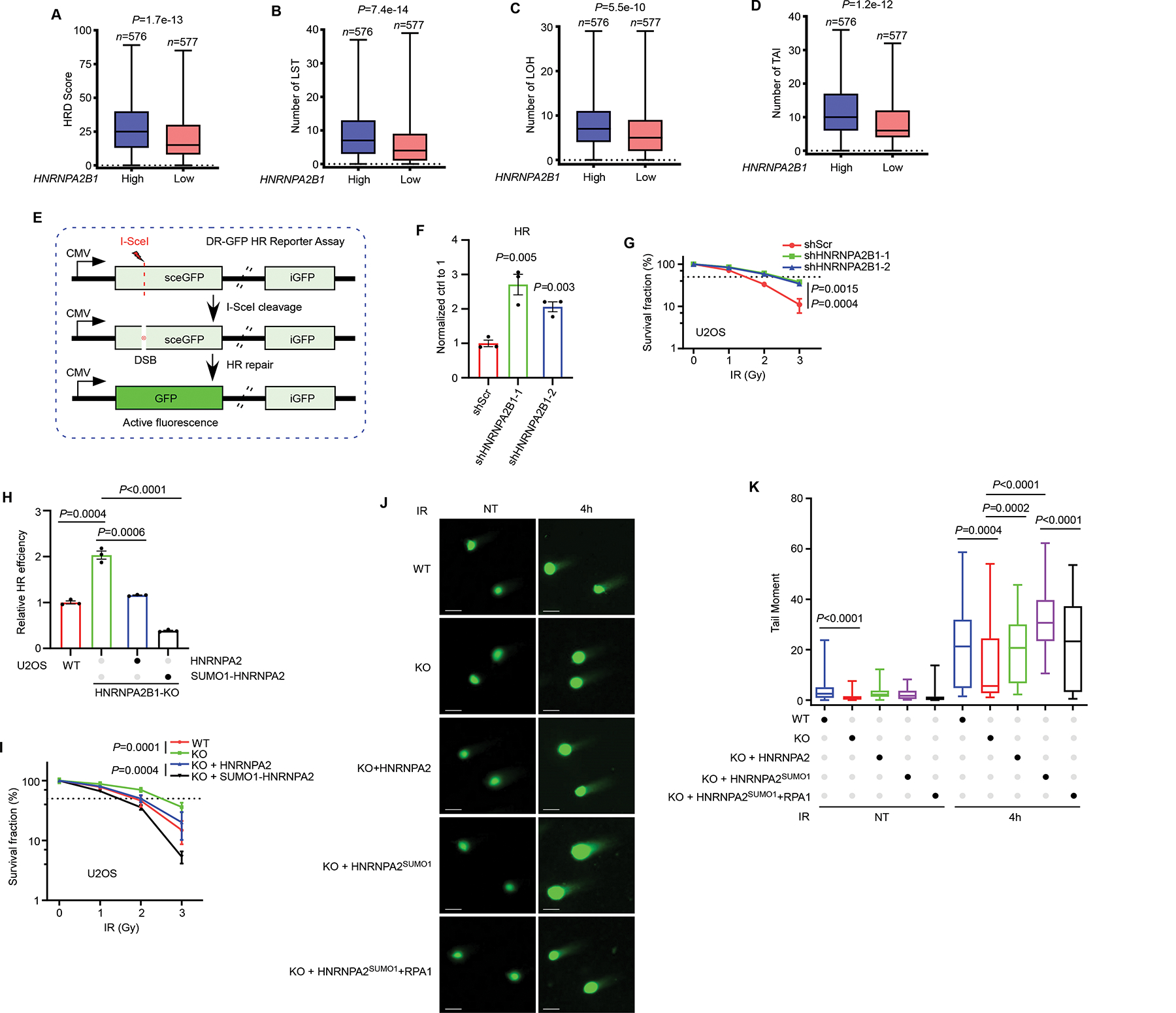
(A-D) Analysis of HRD score (A), LST (B), LOH (C) and TAI (D) in breast cancer samples (See Methods section). (E) Schematic for the DR-GFP HR reporter system. (F) HR reporter assay was performed to assess HR activity in HNRNPA2B1 knockdown U2OS cells. (G) The sensitivity of HNRNPA2B1 knockdown U2OS cells to IR was assessed by clonogenic assay. (H) HNRNPA2B1 KO cells stably complemented with HNRNPA2 or SUMO1-HNRNPA2 were subjected to HR reporter assay. HR activity was normalized to WT cells. (I) HNRNPA2B1 KO cells were stably re-expressed with HNRNPA2 or SUMO1-HNRNPA2 to assess the sensitivity to IR by clonogenic assay. (J-K) HNRNPA2B1 KO U2OS cells were stably restored with HNRNPA2 or SUMO1-HNRNPA2 together with or without RPA1. Cells were exposed to IR (5 Gy) followed by neutral comet assay (J). 100 cells quantified in each condition were shown in (K). Values represent mean and s.e.m. of three independent experiments. Statistical analysis was performed using two-tailed unpaired t-tests (Figures 6F, 6G and 6H, 6I). See also Figure S6.
To further dissect the role of HNRNPA2B1 in the DDR, we examined both U2OS and MDA-MB-231 cells depleted of HNRNPA2B1 for sensitivity to IR and cisplatin (Figures S6C and S6F), which can induce DSBs directly or indirectly. Loss of HNRNPA2B1 caused cells resistance to both IR and cisplatin (Figures 6G and S6G–S6I), suggesting that HNRNPA2B1 is functional for cells responding to DSB-inducing agents. We next assessed the kinetics of IR-induced γH2AX foci, a marker of DNA damage, in HNRNPA2B1 depleted or overexpressed cells (Figure S6J). As shown in Figures S6K and S6L, HNRNPA2B1 knockdown cells displayed decreased accumulation of γH2AX foci at late time points (8 h) when compared with control cells. Conversely, ectopically expressed HNRNPA2 remarkably delayed the resolution of γH2AX foci at 16 h post IR (Figures S6M and S6N). Taken together, these results provide strong evidence showing that HNRNPA2B1 hinders DNA repair.
Next, we wondered if the function of HNRNPA2B1 in DNA repair is dependent on its SUMOylation. To this end, we restored HNRNPA2 and SUMO1-HNRNPA2 expression in HNRNPA2B1 KO cells, respectively, and performed HR reporter assays. We observed that re-expression of HNRNPA2 restored the HR capability to that of parental U2OS cells (Figure 6H). However, expression of SUMO1-HNRNPA2 severely compromised HR repair (Figure 6H). Furthermore, clonogenic analysis showed that introduction of HNRNPA2 restored sensitivity to IR in HNRNPA2B1 KO cells (Figure 6I), while expression of SUMO1-HNRNPA2 markedly reduced survival of HNRNPA2B1 KO cells following exposure to various doses of IR (Figure 6I). To provide direct evidence that HNRNPA2B1SUMO plays a role in DSB repair in individual cells, we performed neutral comet assays in HNRNPA2B1 KO U2OS cells complemented with HNRNPA2 or SUMO1-HNRNPA2 that were exposed to IR. Depletion of HNRNPA2B1 suppressed IR-induced DSBs as measured by tail moments, indicating that HNRNPA2B1 prevents DSB repair (Figures 6J and 6K). Notably, restoration of SUMO1-HNRNPA2 significantly delayed the resolution of DSBs, as evidenced by larger tail moments than HNRNPA2 expressing cells. However, co-expression of RPA1 reduced the tail moment, indicating improved DSB repair (Figures 6J and 6K). Altogether, these results indicate that the HR inhibitory effect of HNRNPA2B1 is dependent on its SUMOylation in limiting RPA availability.
High HNRNPA2B1 expression predicts better patient survival in breast cancer
We next analyzed the cancer genome atlas (TCGA) database to study whether HNRNPA2B1 expression has an impact in the clinical setting. We observed that HNRNPA2B1 expression was upregulated across multiple tumor types (13 of 15), including breast cancer (Figures 7A and S7A). Moreover, based on Kaplan-Meier plot data, high expression of HNRNPA2B1 was associated with better overall survival in breast cancer (Figure 7B), bladder cancer (Figure S7B), lung cancer (Figure S7C) and ovarian cancer (Figure S7D). To further investigate the pathological relevance of HNRNPA2B1, we performed immunohistochemistry (IHC) staining using a human breast cancer tissue microarray containing tumor samples at different clinical stages (Figure S7E). Analysis of 89 breast tumor tissues and 61 adjacent non-tumor tissue samples revealed a pronounced upregulation of HNRNPA2B1 in breast tumors (Figures 7C and 7D). Upregulation of HNRNPA2B1 was observed in 62.9% (56 of 89) of breast tumors, whereas only 8.20% (5 of 61) of adjacent non-tumor tissues exhibited high expression of HNRNPA2B1 (Figure 7C). In addition, we assessed whether the expression of HNRNPA2B1 is associated with clinical outcomes based on our microarray analysis. We found that breast cancer patients with high HNRNPA2B1 expression had better overall survival (Figure 7E) as well as improved disease-free survival (Figure 7F). Additionally, Kaplan-Meier survival analysis showed that patients with lower HNRNPA2B1 expression were at increased risk of breast cancer-related death compared to those with higher HNRNPA2B1 expression (Figure 7G). Taken together, these results strongly suggest that HNRNPA2B1 expression may function as an independent predictor of good prognosis for breast cancer patients.
Figure 7. High HNRNPA2B1 expression predicts better survival in breast cancer patients.
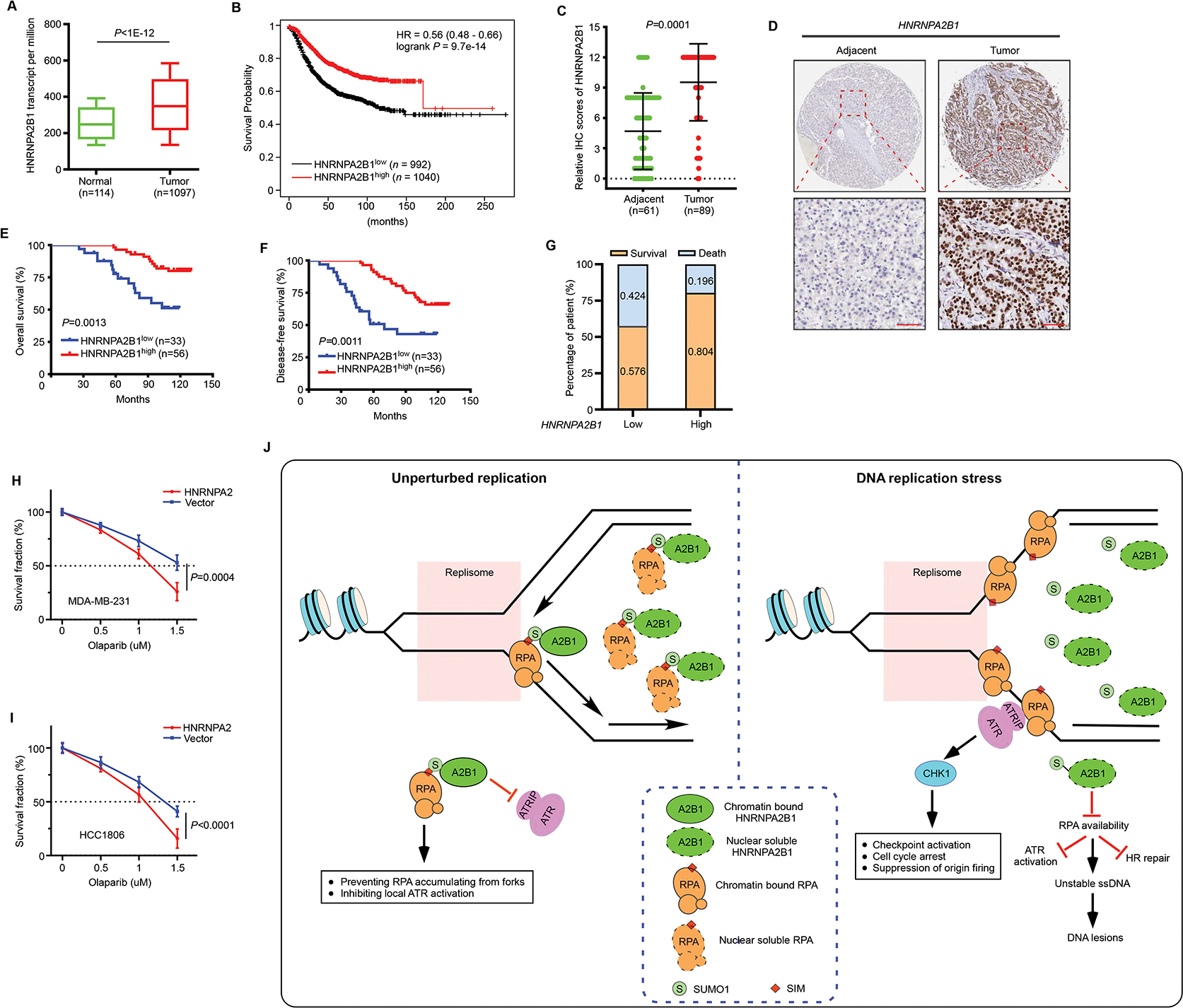
(A) HNRNPA2B1 expression in breast cancer and matched normal tissues was shown based on TCGA data. P values were from Wilcoxon tests. (B) Kaplan Meier plotter reveals the prognostic value of HNRNPA2B1 expression in patients with breast cancer. (C-D) IHC staining of HNRNPA2B1 was evaluated in adjacent tissues and breast tumor tissues. The calculated results based on the staining intensity were shown in (C) and representative images of HNRNPA2B1 IHC staining were shown in (D). Scale bar, 100 uM. (E-F) Kaplan–Meier estimates the overall survival (E) and disease-free survival (F) of breast cancer patients with low or high expression of HNRNPA2B1. Scores of <= 9 were defined as low expression, scores of >= 12 were defined as high expression. The log-rank test was used to compare the survival curves between groups. (G) The survival and death rates for breast cancer patients with low and high HNRNPA2B1 expression were shown. (H-I) Sensitivity of control and HNRNPA2 overexpressed MDA-MB-231 (H) and HCC1806 (I) cells to Olaparib was assessed by clonogenic assay. (J) Working model depicting the role of HNRNPA2B1SUMO during unperturbed DNA replication and stress response (See discussion). Values represent mean and s.e.m. of three independent experiments and statistical analysis was performed using two-tailed unpaired t-tests (Figures 7H and 7I). See also Figure S7.
Extensive studies have reported that DDR pathway alterations affect cancer chemotherapeutic response52. Defective HR has been demonstrated as an important therapeutic target as exemplified by the advent of poly(ADP-ribose) polymerase (PARP) inhibitors displaying synthetic lethality when utilized in HR-deficient cells53,54. To explore whether HNRNPA2B1 impacts breast cancer response to chemotherapy, we overexpressed HNRNPA2 in two breast cancer cell lines MDA-MB-231 and HCC1806, which showed a relatively low expression of HNRNPA2B1 (Figures S7F–S7H). Clonogenic analysis revealed that overexpression of HNRNPA2 markedly sensitized cells to the PARP inhibitor Olaparib (Figures 7H and 7I), suggesting that high expression of the HNRNPA2B1 renders breast cancer cells vulnerable to PARP inhibitor. Therefore, we infer that HNRNPA2B1 may serve as a prospective biomarker for response prediction to PARP inhibitor in breast cancer.
Discussion
In this study, we uncovered that HNRNPA2B1SUMO functions as an endogenous inhibitor of RPA during unperturbed replication (Figure 7J). We found that RPA1 contains a SIM motif located in its N-terminal F domain (Figure 2F). Interestingly, this SIM motif recognizes HNRNPA2B1SUMO, which is SUMOylated at K108 by PIAS2 (Figure 1). During unperturbed DNA replication, HNRNPA2B1SUMO and RPA1 form a stable nuclear soluble complex in the SUMO-dependent manner, preventing RPA accumulation at forks (Figures 2 and 3). Previous studies documented that the N-terminus of RPA1 binds to multiple checkpoint proteins, including ATRIP17. Our results support the view that HNRNPA2B1SUMO/RPA interaction at chromatin would restrict the access of ATR-ATRIP complex to the RPA-ssDNA platform, such that the HNRNPA2B1SUMO/RPA association impedes local ATR activation at active replication forks (Figure 7J). Following DNA damage, RPA dissociates from HNRNPA2B1SUMO through reducing the levels of HNRNPA2B1SUMO, thus enabling the release of RPA to bind ssDNA (Figures 4, 5 and 7J). Loss of HNRNPA2B1 or uncoupling of HNRNPA2B1SUMO could license the recruitment of ATR activating complex to the RPA-ssDNA and allow for the ensuing ATR activation (Figure 7J). Consistent with this idea, enforced expression of a SUMO1-HNRNPA2 fusion could hinder ATR activation as well as the replication stress response (Figure 5). These data place HNRNPA2B1SUMO at the core of ATR activation.
While cells reserve ~8 fold higher of RPA than the amount needed to support normal replication, unscheduled firing of dormant origins can exhaust the RPA pool and result in replication catastrophe20, suggesting an RPA threshold exists. Motivated by the phenomena that enforced expression of SUMO1-HNRNPA2 fusion leads to exposed ssDNA and compromises the replication stress response (Figure 5), we propose that cells sustain RPA availability during replication stress via reducing HNRNPA2B1SUMO. A recent paper describes that the basal activity level of ATR monitors and regulates the amounts of RPA at replication forks during unperturbed replication55. Here, our findings highlight HNRNPA2B1SUMO as an endogenous RPA inhibitor via recognizing the highly conserved SIM motif of RPA1 (Figures 2 and 3). Thus, the presence of the SIM within RPA1 may provide a different modality to deal with genotoxic stress by coupling to SUMO modified components.
Genomic instability is a well-documented hallmark of most cancers56. In this study, we observed that HNRNPA2B1 was upregulated in multiple tumor types (Figure S7A), and high expression of HNRNPA2B1 was linked to increased genomic instability (Figures S4A–S4D), suggesting a potential functional role of HNRNPA2B1 in tumor development. Based on the findings described here, we reasoned that the HR repair inhibition characteristic of HNRNPA2B1 elicits inefficient DNA damage repair, which contributes to genomic instability and predisposes to the progression of tumors. Genomic instability has previously been linked to unfavorable clinical outcomes57, however, our analysis revealed that higher expression of HNRNPA2B1 was associated with better cancer patient survival (Figures 7B, 7E, 7F and S7B–S7D). This prompts us to hypothesize that tumors with a higher level of HNRNPA2B1 might be more sensitive to DNA-damaging agents considering its inhibitory role in the HR and replication stress. Indeed, forced expression of HNRNPA2B1 renders breast cancer cells sensitive to PARP inhibition (Figures 7H and 7I). More importantly, tumors highly expressing HNRNPA2B1 show HR repair deficiency and display potential vulnerability to the synthetic lethality therapy. Therefore, our work identifies HNRNPA2B1 as a prospective response biomarker for PARP inhibitor treatment, which may lead to more effective targeting therapeutic approaches for specific patient groups.
Limitations of the study
Although our data demonstrated that HNRNPA2B1 exists as a SUMOylated protein, our study lacks a specific HNRNPA2B1SUMO antibody to unambiguously monitor the behaviors of HNRNPA2B1SUMO in both the chromatin and nuclear soluble pools. This issue also limits us from further investigating the physiological role of HNRNPA2B1SUMO in vivo. Another limitation of our study is the lack of direct evidence regarding how the association between HNRNPA2B1SUMO and RPA counteracts the local activation of the ATR pathway during unperturbed replication. The use of SUMO fusion protein to mimic protein SUMOylation might not completely represent physiological SUMOylation. Finally, while we generated a model of HNRNPA2SUMO-RPA1 complex, a more detailed structural study of the complex is still needed to understand its working mode during unperturbed replication and genotoxic stress response.
STAR★Methods
Resource Availability
Lead contact
Further information and requests for resources and reagents should be directed to Zhenkun Lou (Lou.Zhenkun@mayo.edu).
Materials availability
All unique reagents generated in this study are available from the lead contact with a completed Materials Transfer Agreement.
Data and code availability
Original western blot images and microscopy data reported in this paper have been deposited at Mendeley. The DOI is listed in the Key Resources Table.
Any additional information required to reanalyze the data reported in this paper is available from the lead contact upon reasonable request.
This paper does not report original code.
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| Mouse Anti-HNRNPA2B1 | Santa Cruz | Cat# sc-374053; RRID: AB_10947257 |
| Rabbit Anti-HNRNPA2B1 | GeneTex | Cat# GTX127928; RRID: AB_2616069 |
| Mouse Anti-phospho-Histone H2A.X (Ser139) | Millipore | Cat# 05-636; RRID: AB_309864 |
| Mouse Monoclonal Anti-FLAG® M2 | Sigma-Aldrich | Cat# F1804; RRID: AB_262044 |
| Rabbit Anti-RPA1 | Bethyl | Cat# A300-241A; RRID: AB_2180681 |
| Mouse Anti-RPA2 | Santa Cruz | Cat# sc-56770; RRID: AB_785534 |
| Rabbit Anti-ATR | Cell Signaling Technology | Cat# 2790; RRID: AB_2227860 |
| Rabbit Anti-ATRIP | Cell Signaling Technology | Cat# 2737; RRID: AB_823659 |
| Mouse Anti-Cyclin A | Santa Cruz | Cat# sc-239; RRID: AB_627334 |
| Mouse Anti-HA | Sigma-Aldrich | Cat# H9658; RRID: AB_260092 |
| Rabbit Anti-His-Tag (D3I1O) | Cell Signaling Technology | Cat# 12698; RRID: AB_2744546 |
| Rabbit Anti-PIAS1 | Cell Signaling Technology | Cat# 3550; RRID: AB_1904090 |
| Rabbit Anti-PIAS2 | GeneTex | Cat# GTX115180; RRID: AB_11162657 |
| Rabbit Anti-PIAS3 | Cell Signaling Technology | Cat# 9042; RRID: AB_2797692 |
| Rabbit Anti-PIAS4 | Cell Signaling Technology | Cat# 4392; RRID: AB_10547884 |
| Rabbit Anti-UBC9 | Cell Signaling Technology | Cat# 4786; RRID: AB_10559206 |
| Mouse Anti-CHK1 | Cell Signaling Technology | Cat# 2360; RRID: AB_2080320 |
| Rabbit Anti-Phospho-CHK1 (Ser345) | Cell Signaling Technology | Cat# 2341; RRID: AB_330023 |
| Rabbit Anti-Phospho-RPA32 (Ser4, Ser8) | Bethyl | Cat# A300-245A; RRID: AB_210547 |
| Rabbit Anti-Phospho-RPA32 (S33) | Bethyl | Cat# A300-246A; RRID: AB_2180847 |
| Mouse Anti-BrdU | BD Biosciences | Cat# 347580; RRID: AB_10015219 |
| Rat Anti-BrdU | Abcam | Cat# ab6326; RRID: AB_305426 |
| Rabbit Anti-RAD51 | GeneTex | Cat# GTX100469; RRID: AB_1951602 |
| Mouse Anti-SUMO1 | Thermo Fisher Scientific | Cat# 33-2400; RRID: AB_2533109 |
| Mouse Anti-Biotin | Jackson ImmunoResearch Labs | Cat# 200-002-211; >RRID: AB_2339006 |
| Rabbit Anti-Phospho-Histone H3 (Ser10) | Cell Signaling Technology | Cat# 9701; RRID: AB_331535 |
| Mouse Anti- PCNA (Alexa Fluor 488 Conjugate) | Cell Signaling Technology | Cat# 8580; RRID: AB_11178664 |
| Alexa Fluor 488-AffiniPure Goat Anti-Mouse IgG (H+L) | Jackson ImmunoResearch Labs | Cat# 115-545-062; RRID: AB_2338845 |
| Alexa Fluor 488-AffiniPure Goat Anti-Rabbit IgG (H+L) | Jackson ImmunoResearch Labs | Cat# 111-545-045; RRID: AB_2338049 |
| Rhodamine Red-X-AffiniPure Goat Anti-Mouse IgG (H+L) | Jackson ImmunoResearch Labs | Cat# 115-295-146; RRID: AB_2338766 |
| Rhodamine Red-X-AffiniPure Donkey Anti-Rat IgG (H+L) | Jackson ImmunoResearch Labs | Cat# 712-295-153; RRID: AB_2340676 |
| Bacterial and virus strains | ||
| BL21 (DE3) Escherichia coli | Transgen Biotech | Cat# CD601-02 |
| Chemicals, peptides, and recombinant proteins | ||
| Cisplatin | Sigma-Aldrich | Cat# P4394 |
| Olaparib (AZD-2281) | LC laboratories | Cat# O-9201 |
| ML-792 | MedChemExpress | Cat# HY-108702 |
| Isopropyl b-D-1-thiogalactopyranoside (IPTG) | Thermo Fisher Scientific | Cat# 15529019 |
| 5-Iodo-2′-deoxyuridine | Sigma-Aldrich | Cat# I7125 |
| 5-Chloro-2′-deoxyuridine | Sigma-Aldrich | Cat# C6891 |
| N-Ethylmaleimide | Sigma-Aldrich | Cat# E3876 |
| BrdU | Biolegend | Cat# 423401 |
| 5-ethynyl-2’-deoxyuridine | Cayman Chemical | Cat# 20518 |
| TransIT-X2® Dynamic Delivery System | Mirus | Cat# MIR6004 |
| DAPI | Thermo Fisher Scientific | Cat# D1306 |
| Dynabeads™ Protein G | Thermo Fisher Scientific | Cat# 10003D |
| Anti-Flag M2 Affinity Gel | Sigma-Aldrich | Cat# A2220 |
| PrimeSTAR HS DNA Polymerase | TaKaRa | Cat# R040A |
| Hydroxyurea | Sigma-Aldrich | Cat# H8627 |
| Biotin-azide | Cayman Chemical | Cat# 13040 |
| Berzosertib (VX970) | Selleck | Cat# S7102 |
| (+)-Sodium L-ascorbate | Sigma-Aldrich | Cat# A7631 |
| SUMO1 | Boston Biochem | Cat# UL-712 |
| PIAS2 | NOVUS Biologicals | Cat# H00009063-P01 |
| Pierce™ Glutathione Agarose | Thermo Fisher Scientific | Cat# 16102BID |
| Ni-NTA Agarose | QIAGEN | Cat# 30210 |
| SAE1 | Boston Biochem | Cat# E-315 |
| UBC9 | Boston Biochem | Cat# E2-645 |
| Critical commercial assays | ||
| Duo-link in situ PLA Kit | Sigma-Aldrich | Cat# DUO92101 |
| QuickChange Site-Directed Mutagenesis Kit | Stratagene | Cat# 200519 |
| CometAssay kit | Trevigen | Cat# 4250-050-K |
| Deposited data | ||
| Raw data files | This paper | Mendeley Data: https://doi.org/10.17632/vzgpmfmt9t.1 |
| Experimental models: Cell lines | ||
| Human: HEK293T | ATCC | Cat# CRL-11268 |
| Human: U2OS | ATCC | Cat# HTB-96 |
| Human: MDA-MB-231 | ATCC | Cat# HTB-26 |
| Human: HCC1806 | ATCC | Cat# CRL-2335™ |
| Oligonucleotides | ||
| shHNRNPA2B1-1 | Sigma-Aldrich | TRCN0000001058 |
| shHNRNPA2B1-2 | Sigma-Aldrich | TRCN0000001059 |
| shATR | Sigma-Aldrich | TRCN0000196538 |
| shPIAS1-1 | Sigma-Aldrich | TRCN0000231898 |
| shPIAS1-2 | Sigma-Aldrich | TRCN0000231900 |
| shPIAS2 | Sigma-Aldrich | TRCN0000230128 |
| shPIAS3 | Sigma-Aldrich | TRCN0000274337 |
| HNRNPA2B1 KO | GenScript | TCTCTTGCTACAGCACGTTT |
| HNRNPA2-forward | This paper | ATGGAGAGAGAAAAGGAACAG |
| HNRNPA2-reverse | This paper | GTATCGGCTCCTCCCACC |
| RPA1-forward | This paper | ATGGTCGGCCAACTGAGC |
| RPA1-reverse | This paper | CATCAATGCACTTCTCC |
| SUMO1-forward | This paper | ATGTCTGACCAGGAGGC |
| SUMO1-reverse | This paper | AACTGTTGAATGACC |
| shPIAS4-1 | Sigma-Aldrich | TRCN0000231892 |
| shPIAS4-2 | Sigma-Aldrich | TRCN0000004115 |
| Recombinant DNA | ||
| PLVX3-FLAG-HNRNPA2 | This paper | N/A |
| PLVX6-EGFP-HNRNPA2 | This paper | N/A |
| pGEX-4T2-HNRNPA2 | This paper | N/A |
| PLVX3-FLAG-RPA1 | This paper | N/A |
| PLVX3-FLAG-RPA1 (ΔSIM) | This paper | N/A |
| Myc-RPA1 | This paper | N/A |
| pTEV-His-RPA1 (4A) | This paper | N/A |
| PLVX3-FLAG-RPA1 (4A) | This paper | N/A |
| PLVX3-FLAG-SUMO1 | This paper | N/A |
| PLVX3-FLAG-SUMO1-HNRNPA2 | This paper | N/A |
| PLVX3-FLAG-HNRNPA2 (K108R) | This paper | N/A |
| pFLAG-CMV-SENP1 | Addgene | Cat# 17357 |
| pCMV-HA-hUBC9 | Addgene | Cat# 14438 |
| Software and algorithms | ||
| GraphPad Prism | GraphPad | https://www.graphpad.com |
| ImageJ | ImageJ | https://imagej.nih.gov/ij/ |
| FlowJo (10.1) | FlowJo LLC | http://www.flowjo.com/ |
Experimental Model and Subject Details
Cell culture conditions
MDA-MB-231, HCC1806, U2OS and HEK293T cell lines were purchased from ATCC. MDA-MB-231 and HEK293T were cultured in Dulbecco’s Modified Eagle Medium (DMEM) with 10% fetal bovine serum (FBS). U2OS and HCC1806 cells were cultured in McCoy’s 5 A and RPMI1640 with 10% FBS, respectively. The ER-mCherry-LacI-FokI-DD U2OS cell line was a gift from R. A. Greenberg (University of Pennsylvania). All cell culture reagents were obtained from Life technologies. All cell lines have been identified by the medical genome facility center of Mayo Clinic (Rochester, MN) and were routinely tested for mycoplasma contamination. All cells were incubated in a humidified Thermo Fisher incubator with 5% CO2.
Method details
Plasmid constructions
Full length HNRNPA2 was cloned from HEK293T cDNA and inserted into the pLVX3-3XFLAG, pLVX6-EGFP and pGEX-4T-2 vectors and confirmed by sequencing. Myc-tagged RPA1 truncation mutants were gifts from Jingsong Yuan (Columbia University). GFP-ATRIP plasmid was a gift from Shan Zha (Columbia University). His-RPA1 plasmid was a gift from Donna D. Zhang (University of Arizona). SUMO1 and SUMO2/3 plasmids were stored in our lab. HA-UBC9 (14438) and FLAG-SENP1 (17357) were got from Addgene. The RPA14A, RPA1 ΔSIM mutants and HNRNPA2K108R were generated by site-directed mutagenesis and subcloned into pLVX3-3XFLAG or pGEX-4T2 vector as indicated. SUMO1 was subcloned into pLVX3-3XFLAG-HNRNPA2K108R to get the SUMO1-HNRNPA2 fusion construct.
RNA interference
HNRNPA2B1, PIAS1, PIAS2, PIAS3, PIAS4 and ATR-specific shRNA constructs were commercially got from Sigma. Lentivirus for shHNRNPA2B1 as well as the control (shScr) were packaged with pMD2.G and pSPAX2. Briefly, 3 μg pMD2.G, 9 μg pSPAX2 and 12 μg lentivirus plasmids were co-transfected into HEK293T cells in 10 cm cell culture dish using ProFection Mammalian Transfection System (Promega). The lentivirus particles were harvested at 48h after transfection and concentrated. For infection, the concentrated lentivirus particles were added into the medium with 8 ug/ml polybrene and cultured at 37°C for 24–48 hours before they were selected with puromycin (8 μg/mL). The knockdown cells were confirmed by Western blot and used for further experiments.
HNRNPA2B1 knockout (KO) cells were generated using Lenti-CRISPR V2 (GenScript) that contains a guide (g)RNA targeting human HNRNPA2B1 (TCTCTTGCTACAGCACGTTT) by CRISPR/Cas9 approach. The lentiviruses were harvested at 48h post transfection and were utilized to infect indicated cells. The infected cells were then selected with puromycin (8 μg/mL). The KO cells were confirmed by Western blot and used for further experiments. For the HNRNPA2, HNRNPA2K108R and SUMO1-HNRNPA2 reconstituted cells, following 48h post lentivirus transfection, cells were then screened and confirmed by Western blot. The indicated HNRNPA2B1 KO cells were maintained under the same conditions as their wild-type control cells.
Immunofluorescence
Briefly, cells were first fixed with 4% paraformaldehyde (PFA) then permeabilized with 0.25% Triton X-100. Then cells were blocked by 2% BSA followed by incubation with primary antibody. Fluorescent secondary antibody and DAPI were then incubated with cells to stain the targeted proteins and nucleus, respectively. The cover slips were mounted onto glass slides with Prolong Diamond Antifade Mountant (P36961, Thermo). Cells were visualized using ImageXpress Micro Confocal (Molecular Devices) and at least 100 cells were counted in each condition. To stain RPA2, cells were fixed with cold methanol /acetone (1/1) at −20 °C for 15 min, then washed twice using PBS and fixed again with 4% PFA at room temperature for 15 min before incubation with primary antibody.
In situ proximity ligation assay (PLA)
U2OS cells were fixed with 4% PFA for 30 min at room temperature followed by permeabilization with 0.25% Triton X-100 for 20 min. Then PLA experiments were performed using Duo-link in situ PLA kit (DUO92008, Sigma) according to the manufacturer’s protocol. Briefly, samples were first blocked with blocking solution at 37 °C for 1h and then incubated with the mixture of primary antibodies at 4 °C overnight. Then, the probes were incubated at 37 °C for 1h, followed by hybridization, ligation and amplification. Cellular nuclei were stained with DAPI. The cover slips were mounted onto glass slides with Prolong Diamond Antifade Mountant and visualized using ImageXpress Micro Confocal (Molecular Devices) and at least 100 cells were counted in each condition.
For EdU incorporation, cells were first labeled with EdU (10 uM, 15 min) followed by fixation and permeabilization. Then, click reactions (combine the following reagents in the listed order: 0.1 X PBS, 2 mM copper (II) sulfate, 5 uM biotin azide, 10 mM sodium ascorbate) were performed at room temperature for 15 min. Cells were then washed with PBS before PLA assay. Anti-Biotin antibody was used to detect EdU signal.
Laser microirradiation
U2OS or MDA-MA-231 cells were cultured on glass-bottom dishes (Wuxi NEST Biotechnology). Laser microirradiation was performed using a Micropoint Laser Illumination and Ablation system (Photonic Instruments). A high-energy ultraviolet laser (170 mJ at 10 Hz) generated DNA breaks in nuclei. Cells were then fixed with 4% PFA for primary antibody staining. Images were taken using the IX71 microscope with cellSens software (Olympus) or the Nikon Eclipse 80i fluorescence microscope.
wGII signature analysis
Segmented copy number data and HNRNPA2B1 RNA expression data as log-transformed transcripts per million (TPM) in TCGA breast cancer cohort (n = 1092 samples) were downloaded from the UCSC xenabrowser58. The chromatin instability signature wGII for all samples was recalculated as described in Andrea et al59. The samples were subgrouped based on the median expression level of HNRNPA2B1. Statistical significance was tested using two-sided Mann–Whitney–Wilcoxon tests.
Western blot and Co-immunoprecipitation (Co-IP)
Cells were harvested and lysed with RIPA lysis buffer (CST). After centrifugation at 12000 rpm for 10 min (4°C), the supernatants were collected for immunoblot with indicated antibodies.
For Co-IP assays, HEK293T or U2OS cells transfected with indicated constructs were collected, washed once in PBS and lysed in Cell Lysis Buffer (CST) or RIPA lysis buffer (CST) with freshly added protease inhibitor cocktail (Roche) for 10 min at 4°C. The lysates were briefly sonicated and clarified by centrifugation at 12000 rpm for 10 min (4°C). The supernatants were incubated with Dynabeads protein G beads (Life technologies) and indicated antibody at 4°C for 4 h with rotation. Beads were washed 3 times in lysis buffer and bound proteins were eluted by heating at 95°C for 10 min in 1× LDS sample buffer (Life technologies). The eluates were analyzed by immunoblotting.
RPA-protection assay
RPA-protection assays were performed as described previously44,45. Briefly, the indicated U2OS cells cultured on glass coverslips were treated with HU to induce replication stress. The cells were first permeabilized with 0.25% Triton X-100 in PBS for 20 min on ice then fixed in 4% PFA at room temperature for 15 min. Then, the cells were inverted onto Parafilm placed in a sealed chamber and incubated with Taq DNA polymerase (M0273S, NEB), dNTP (0.2 mM) and BrdU (0.4 mM) at 72°C for 5 min. After that, the slides were incubated with a specific antibody against BrdU (347580, BD) at 4°C overnight, followed by incubation with a secondary antibody for 1h at room temperature before immunofluorescence microscopy.
DNA fiber assay
DNA fiber assays were performed as described by Quinet et al60 with some changes. In brief, the indicated U2OS cells were pulse labeled with 25 μM IdU for 30 min, washed with PBS (37 °C), refed with media (37 °C) containing 250 μM CldU and cultured for 30 min. Cells were then washed in PBS and diluted with cold PBS. Cells (~100,000) were lysed in 15 μl fiber lysis buffer (200 mM Tris-HCl, pH 7.4, 50 mM EDTA, and 0.5% SDS) on a microscope slide, and the slide was tilted to a 15° angle to allow fibers to spread along the length of the slide. Fibers were fixed in 3:1 methanol/acetic acid for 10 min and then air-dried. The slides were washed twice with PBS and denatured in 2.5 M hydrochloric acid overnight at 4 °C. Slides were washed twice with PBS, blocked in 2% BSA in PBS for 30 min, and stained with primary antibodies overnight at 4 °C: Mouse anti-BrdU (1/25, BD Biosciences, 347580), Rat anti-BrdU (1/1000, Abcam, ab6326) diluted in 5% BSA in PBS). The next morning, slides were washed twice with PBS, stained with secondary antibodies Alexafluor 594 anti-mouse (1/500, Invitrogen, A11032) and Alexafluor 488 anti-rat (1/500, Invitrogen, A110066) for 1h at room temperature and washed three times with PBS. SlowFade Gold 30 (μL) (ThermoFisher, S36936) was added, and coverslips were applied. Images of fibers were acquired at 63× magnification on a Nikon eclipse 80i Fluorescence microscope. For the DNA replication elongation rates, the lengths of the DNA fibers (only CldU tracks connected with IdU tracks) were measured and DNA replication elongation rates were calculated as fiber length divided by pulse labeling time. Fiber lengths were measured using ImageJ, and micrometer values were expressed in kilobases using the following conversion factor: 1 μm = 2.59 kb.
iPOND (isolation of proteins on nascent DNA)
iPOND assays were performed according to protocol with some changes. Briefly, about 1× 108 cells per sample were labeled with 10 μM EdU for 15 min and then washed with PBS for two times. Cells were then fixed with 1% formaldehyde in PBS for 20 min at room temperature followed by quench crosslinking reaction by adding 1.25 M glycine. Cells were then washed and permeabilized with 0.25% Triton X-100 for 1h at 4 °C before incubating with the click reaction buffer (10 uM biotin azide, 2 mM CuSO4 and 10 mM sodium L-ascorbate in PBS) for 2 h at room temperature. After click reaction, cell pellets were resuspended in lysis buffer (1% SDS in 50 mM Tris-HCl, pH 8.0) and subjected to sonication. Cell lysates were then incubated with streptavidin beads and rotated the biotin captures overnight at 4 °C before being washed twice with cold lysis buffer, once with 1 ml of 1 M NaCl and repeated the cold lysis buffer washes two more times. Beads were incubated with 1× LDS loading buffer and heated at 95 °C for 10 min. The eluates were collected for SDS-PAGE analysis and immunoblotted with the indicated antibodies.
Expression and purification of recombinant proteins
HNRNPA2WT and HNRNPA2K108R were subcloned into pGEX-4T2 vector and transformed into BL21(DE3) Escherichia coli cells. Protein expression was induced with isopropyl-β-D-thiogalactoside (IPTG, 0.4 mM) at 16 °C for 16h. The bacteria were sonicated in the lysis buffer (20 mM Tris-HCl pH 8.0, 500 mM NaCl, 1 mM EDTA, 0.5 mM PMSF and 1% Triton X-100). Lysates were cleared by centrifugation at 4 °C for 30 min and applied to batch purification columns containing 1 ml bed volume of glutathione agarose (GE) and incubated for 4 h at 4 °C. The flow through was discarded and the beads were washed 5 times with lysis buffer. Proteins were eluted using elution buffer containing 100 mM Tris-HCl pH 8.0, 150 mM NaCl and 30 mM reduced glutathione (GSH). Proteins were dialyzed against buffer containing 50 mM Tris-HCl pH 7.5, 100 mM NaCl and 10% glycerol for subsequent experiments.
For the His-tagged RPA1 protein purification, the bacteria were sonicated in lysis buffer (20 mM Tris-HCl pH 8.0, 600 mM NaCl, 1 mM EDTA, 10 mM imidazole, 0.5 mM PMSF and 1% Triton X-100). Lysates were cleared by centrifugation (14000 rpm) at 4 °C for 45 min and applied to DNase I (5 ug/ml) treatment on ice for 15 min. Then, lysates were incubated with 1 ml Ni-NTA His-affinity resin and rotated for 2h at 4 °C. The flow through was discarded and the beads were washed 5 times with lysis buffer. Proteins were eluted using elution buffer containing 100 mM Tris-HCl pH 8.0, 150 mM NaCl and 250 mM imidazole. Proteins were dialyzed against buffer containing 50 mM Tris-HCl pH 7.5, 100 mM NaCl and 10% glycerol for subsequent experiments. For the His-RPA1 pull-down assay, purified His-RPA1 was incubated with SUMO1 (Boston Biochem) in NETN buffer for 2h at 4 °C, washed and eluted before subsequent analysis.
SUMOylation assay under denature conditions
SUMOylation analysis by Ni2+-NTA pull-downs was performed as previously described with some changes61. Briefly, the indicated cells were harvested and pellets were washed once in PBS. Then cells were lysed in lysis buffer (8 M Urea, 0.1 M NaH2PO4, 300 mM NaCl and 10 mM Tris pH 8.0). Lysates were briefly sonicated to shear DNA and incubated with Ni2+-NTA agarose beads (QIAGEN) for 1h at room temperature. Beads were washed 5 times with lysis buffer. Input and beads were boiled for 10 min in 1 X LDS loading buffer and subjected to immunoblotting.
The SUMOylation analysis by Co-immunoprecipitation (Co-IP) under denaturing conditions was conducted to detect both exogenous and endogenous SUMOylation of HNRNPA2B1. Briefly, cells were lysed with lysis buffer S (20 mM sodium phosphate pH 7.4, 150 mM NaCl, 1% SDS, 1% Triton, 0.5% sodium deoxycholate, 5 mM EDTA, 5 mM EGTA, 20 mM N-ethylmaleimide (NEM), 5 mM DTT and Protease inhibitor cocktail), boiled for 15 min and then sonicated until the lysate became fluid. The lysates were clarified by centrifugation at 13000 rpm for 10 min at 4°C, diluted 1:10 with lysis buffer S without SDS, and then incubated with the indicated antibodies and Protein G beads 4h at 4°C. Beads were washed 5 times with lysis buffer, boiled for 10 min in 1 X LDS loading buffer and subsequently analyzed by Western blotting.
In vitro SUMOylation assay
In vitro SUMOylation assays were performed as previously described with some changes61. In brief, SUMO modification reactions were typically performed in a total volume of 100 μl with 200 ng of SUMO-activating enzyme (SAE1) (Boston Biochem), 100 ng of UBC9 (Boston Biochem), 1 μg of SUMO1 (Boston Biochem), 100 ng of PIAS2 (NOVUS), and recombinant GST-HNRNPA2 bound to GST-sepharose (20 μg). The 10X reaction buffer (Boston Biochem) was added with 5 mM ATP-Mg. Reactions were incubated at 37°C for 1h and stopped by adding 10 mM EDTA. The beads were washed with NETN buffer (20 mM Tris-HCl, pH 8.0, 100 mM NaCl, 1 mM EDTA, 0.5% Nonidet P-40 with protease inhibitors) for 3 times and boiled in 1X LDS loading buffer. Finally, the samples were analyzed by immunoblotting.
DR-GFP HR reporter assay
To examine the repair of I-SceI-generated DSBs by HR in cells, the indicated HEK293T or U2OS cells were transfected with DR-GFP, pCBA-I-SecI and pmCherry. The HR efficiency was determined at 48h post-transfection. The double positive (GFP and mCherry-positive) cells were quantified by Attune NxT Flow Cytometry (Thermo Fisher). Results were normalized to control group. The gating strategies were shown in Supplementary Figure. S5B.
Colony-formation assay
U2OS, MDA-MB-231 or HCC1806 cells (1000) stably expressing the indicated constructs were plated in triplicates in each well of six-well plates for 24h before treated with indicated DNA-damaging agents for 10–14 days at 37 °C to allow colony formation. Colonies were stained with Giemsa and counted. Results were normalized to plating efficiencies.
Cell cycle analysis
The indicated cells were prepared and fixed in 70% ice-cold ethanol. Then the fixed cells were washed with PBS and resuspended in PI/RNase solution (Thermo Fisher Scientific) at room temperature for 30 min. The samples were analyzed using Attune NxT Flow Cytometer (Thermo Fisher Scientific) and the results were analyzed by FlowJo v.10. For cell synchronization by double thymidine block and release experiments, U2OS cells or the indicated cells were treated with 2 mM thymidine for 18h and released for 9h, followed by the second 18h treatment before being released into thymidine-free medium for the indicated periods of time. Cells were collected at 0, 4, 9h for analysis of cell cycle by DNA staining using PI or analysis of protein by Western blot using indicated antibodies.
Neutral Comet Assay
Neutral comet assays were performed using a Single Cell Gel Electrophoresis Assay Kit (TREVIGEN, 4250-050-K) based on the manufacturer’s protocols. In brief, the indicated cells were prepared for irradiation (5 Gy) and recovered for 4h. Then, 1 × 105/ ml cells were combined with LMAgarose (at 37°C) 1% at a ratio of 1:10 (v/v) and immediately pipetted onto slides. Slides were immersed in the lysis solution at 4°C overnight followed by electrophoresis at 28 V for 1h and stained with SYBR Gold (Invitrogen, 1/10000) for 30 min at room temperature. Images were acquired on a Nikon Eclipse 80i Fluorescence microscope. Comet tail moment were analyzed using OpenComent plugin.
HRD score, LOH, LST and TAI analysis
TCGA breast cancer cohort HRD scores and the three subscores (TAI, LST, LOH) have been published previously and were downloaded from Genomic Data Commons (GDC)62,63. Expression data (n = 1153 samples) as log-transformed transcripts per million (TPM) were downloaded from the UCSC xenabrowser58. Samples were subgrouped by median expression level of HNRNPA2B1. For the results shown in Figure 6, the box outlines show the 25th and 75th percentiles, the solid lines show the median value and the whiskers extending to the most extreme data point that is no more than 1.5 times the interquartile range. Statistical significance was tested using two-sided Mann–Whitney–Wilcoxon test.
Structural modelling
The structural model of HNRNPA2SUMO-RPA1 complex was generated using AlphaFold2 online server (https://colab.research.google.com/github/sokrypton/ColabFold/blob/main/AlphaFold2.ipynb) by modeling a ternary complex of HNRNPA2-SUMO1-RPA1. A set of 5 models was generated. Interface between HNRNPA2 and RPA1 was converged in all 5 models, while SUMO1 adopted different conformations. The one exhibiting closest distance between Lys108 of HNRNPA2 and Gly83 of SUMO1 was used for further analysis. Figure was prepared using PyMol v.2.3.2.
Quantification and statistical analysis
Values in bar and line graphs were presented as mean ± S.E.M of three independent experiments. Statistical analysis was performed using two-tailed Student’s t-tests unless mentioned otherwise. All statistical analysis was performed using Prism v.8.0 (GraphPad) or Microsoft Excel 2019.
Supplementary Material
Highlights.
RPA1 associates with HNRNPA2B1SUMO through its SIM motif
HNRNPA2B1SUMO prevents RPA accumulation at unperturbed replication forks
HNRNPA2B1SUMO limits RPA availability – impacting the replication stress response and HR
Declined HNRNPA2B1SUMO is required for ATR activation during DNA replication stress
Acknowledgements
We thank Dr. R.A. Greenberg (University of Pennsylvania) for providing the ER-mCherry-LacI-FokI-DD U2OS cell line and Dr. Jingsong Yuan (Columbia University) for providing the RPA1 truncation plasmids. We also thank Dr. Shan Zha (Columbia University) for providing the GFP-ATRIP plasmid and Dr. Donna D. Zhang (University of Arizona) for providing the His-RPA1 plasmid. We thank members of our lab for helpful discussions. This work was supported by CA264600 and CA217183. J.A.K. was supported by T32GM65841.
Footnotes
Declaration of Interests
The authors declare no competing interests.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Krecic AM, and Swanson MS (1999). hnRNP complexes: composition, structure, and function. Curr. Opin. Cell Biol. 11, 363–371. 10.1016/s0955-0674(99)80051-9. [DOI] [PubMed] [Google Scholar]
- 2.Geuens T, Bouhy D, and Timmerman V (2016). The hnRNP family: insights into their role in health and disease. Human Genet. 135, 851–867. 10.1007/s00439-016-1683-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Kozu T, Henrich B, and Schäfer KP (1995). Structure and expression of the gene (HNRPA2B1) encoding the human hnRNP protein A2/B1. Genomics 25, 365–371. 10.1016/0888-7543(95)80035-k. [DOI] [PubMed] [Google Scholar]
- 4.Burd CG, Swanson MS, Görlach M, and Dreyfuss G (1989). Primary structures of the heterogeneous nuclear ribonucleoprotein A2, B1, and C2 proteins: a diversity of RNA binding proteins is generated by small peptide inserts. Proc. Natl. Acad. Sci. USA 86, 9788–9792. 10.1073/pnas.86.24.9788. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Keene JD (2007). RNA regulons: coordination of post-transcriptional events. Nat. Rev. Genet. 8, 533–543. 10.1038/nrg2111. [DOI] [PubMed] [Google Scholar]
- 6.Glisovic T, Bachorik JL, Yong J, and Dreyfuss G (2008). RNA-binding proteins and post-transcriptional gene regulation. FEBS Lett. 582, 1977–1986. 10.1016/j.febslet.2008.03.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Alarcón CR, Goodarzi H, Lee H, Liu X, Tavazoie S, and Tavazoie SF (2015). HNRNPA2B1 Is a Mediator of m(6)A-Dependent Nuclear RNA Processing Events. Cell 162, 1299–1308. 10.1016/j.cell.2015.08.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Villarroya-Beltri C, Gutiérrez-Vázquez C, Sánchez-Cabo F, Pérez-Hernández D, Vázquez J, Martin-Cofreces N, Martinez-Herrera DJ, Pascual-Montano A, Mittelbrunn M, and Sánchez-Madrid F (2013). Sumoylated hnRNPA2B1 controls the sorting of miRNAs into exosomes through binding to specific motifs. Nat. Commun. 4, 2980. 10.1038/ncomms3980. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Zhou J, Allred DC, Avis I, Martínez A, Vos MD, Smith L, Treston AM, and Mulshine JL (2001). Differential expression of the early lung cancer detection marker, heterogeneous nuclear ribonucleoprotein-A2/B1 (hnRNP-A2/B1) in normal breast and neoplastic breast cancer. Breast Cancer Res. Treat. 66, 217–224. 10.1023/a:1010631915831. [DOI] [PubMed] [Google Scholar]
- 10.Tauler J, Zudaire E, Liu H, Shih J, and Mulshine JL (2010). hnRNP A2/B1 modulates epithelial-mesenchymal transition in lung cancer cell lines. Cancer Res. 70, 7137–7147. 10.1158/0008-5472.can-10-0860. [DOI] [PubMed] [Google Scholar]
- 11.Stockley J, Villasevil ME, Nixon C, Ahmad I, Leung HY, and Rajan P (2014). The RNA-binding protein hnRNPA2 regulates β-catenin protein expression and is overexpressed in prostate cancer. RNA Biol. 11, 755–765. 10.4161/rna.28800. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Moran-Jones K, Grindlay J, Jones M, Smith R, and Norman JC (2009). hnRNP A2 regulates alternative mRNA splicing of TP53INP2 to control invasive cell migration. Cancer Res. 69, 9219–9227. 10.1158/0008-5472.can-09-1852. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Clower CV, Chatterjee D, Wang Z, Cantley LC, Vander Heiden MG, and Krainer AR (2010). The alternative splicing repressors hnRNP A1/A2 and PTB influence pyruvate kinase isoform expression and cell metabolism. Proc. Natl. Acad. Sci. USA 107, 1894–1899. 10.1073/pnas.0914845107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Wang L, Wen M, and Cao X (2019). Nuclear hnRNPA2B1 initiates and amplifies the innate immune response to DNA viruses. Science 365. 10.1126/science.aav0758. [DOI] [PubMed] [Google Scholar]
- 15.Shishkin SS, Kovalev LI, Pashintseva NV, Kovaleva MA, and Lisitskaya K (2019). Heterogeneous Nuclear Ribonucleoproteins Involved in the Functioning of Telomeres in Malignant Cells. Int. J. Mol. Sci. 20. 10.3390/ijms20030745. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Wold MS (1997). Replication protein A: a heterotrimeric, single-stranded DNA-binding protein required for eukaryotic DNA metabolism. Annu. Rev. Biochem. 66, 61–92. 10.1146/annurev.biochem.66.1.61. [DOI] [PubMed] [Google Scholar]
- 17.Maréchal A, and Zou L (2015). RPA-coated single-stranded DNA as a platform for post-translational modifications in the DNA damage response. Cell Res. 25, 9–23. 10.1038/cr.2014.147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Zou L, and Elledge SJ (2003). Sensing DNA damage through ATRIP recognition of RPA-ssDNA complexes. Science 300, 1542–1548. 10.1126/science.1083430. [DOI] [PubMed] [Google Scholar]
- 19.Ciccia A, and Elledge SJ (2010). The DNA damage response: making it safe to play with knives. Mol. Cell 40, 179–204. 10.1016/j.molcel.2010.09.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Toledo LI, Altmeyer M, Rask MB, Lukas C, Larsen DH, Povlsen LK, Bekker-Jensen S, Mailand N, Bartek J, and Lukas J (2013). ATR prohibits replication catastrophe by preventing global exhaustion of RPA. Cell 155, 1088–1103. 10.1016/j.cell.2013.10.043. [DOI] [PubMed] [Google Scholar]
- 21.Flynn RL, Centore RC, O’Sullivan RJ, Rai R, Tse A, Songyang Z, Chang S, Karlseder J, and Zou L (2011). TERRA and hnRNPA1 orchestrate an RPA-to-POT1 switch on telomeric single-stranded DNA. Nature 471, 532–536. 10.1038/nature09772. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Zhao X (2018). SUMO-Mediated Regulation of Nuclear Functions and Signaling Processes. Mol. Cell 71, 409–418. 10.1016/j.molcel.2018.07.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Johnson ES (2004). Protein modification by SUMO. Annu. Rev. Biochem. 73, 355–382. 10.1146/annurev.biochem.73.011303.074118. [DOI] [PubMed] [Google Scholar]
- 24.Kunz K, Piller T, and Müller S (2018). SUMO-specific proteases and isopeptidases of the SENP family at a glance. J. Cell Sci. 131. 10.1242/jcs.211904. [DOI] [PubMed] [Google Scholar]
- 25.Pichler A, Fatouros C, Lee H, and Eisenhardt N (2017). SUMO conjugation - a mechanistic view. Biomol. Concepts 8, 13–36. 10.1515/bmc-2016-0030. [DOI] [PubMed] [Google Scholar]
- 26.Saitoh H, and Hinchey J (2000). Functional heterogeneity of small ubiquitin-related protein modifiers SUMO-1 versus SUMO-2/3. J. Biol. Chem. 275, 6252–6258. 10.1074/jbc.275.9.6252. [DOI] [PubMed] [Google Scholar]
- 27.Kerscher O (2007). SUMO junction-what’s your function? New insights through SUMO-interacting motifs. EMBO Rep. 8, 550–555. 10.1038/sj.embor.7400980. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Yau TY, Sander W, Eidson C, and Courey AJ (2021). SUMO Interacting Motifs: Structure and Function. Cells 10. 10.3390/cells10112825. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Vassileva MT, and Matunis MJ (2004). SUMO modification of heterogeneous nuclear ribonucleoproteins. Mol. Cell. Biol. 24, 3623–3632. 10.1128/mcb.24.9.3623-3632.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Pelisch F, Pozzi B, Risso G, Muñoz MJ, and Srebrow A (2012). DNA damage-induced heterogeneous nuclear ribonucleoprotein K sumoylation regulates p53 transcriptional activation. J. Biol. Chem. 287, 30789–30799. 10.1074/jbc.M112.390120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Lee SW, Lee MH, Park JH, Kang SH, Yoo HM, Ka SH, Oh YM, Jeon YJ, and Chung CH (2012). SUMOylation of hnRNP-K is required for p53-mediated cell-cycle arrest in response to DNA damage. EMBO J. 31, 4441–4452. 10.1038/emboj.2012.293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Johnson ES, and Blobel G (1997). Ubc9p is the conjugating enzyme for the ubiquitin-like protein Smt3p. J. Biol. Chem. 272, 26799–26802. 10.1074/jbc.272.43.26799. [DOI] [PubMed] [Google Scholar]
- 33.Zhao Q, Xie Y, Zheng Y, Jiang S, Liu W, Mu W, Liu Z, Zhao Y, Xue Y, and Ren J (2014). GPS-SUMO: a tool for the prediction of sumoylation sites and SUMO-interaction motifs. Nucleic Acids Res. 42, W325–330. 10.1093/nar/gku383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Lumpkin RJ, Gu H, Zhu Y, Leonard M, Ahmad AS, Clauser KR, Meyer JG, Bennett EJ, and Komives EA (2017). Site-specific identification and quantitation of endogenous SUMO modifications under native conditions. Nat. Commun. 8, 1171. 10.1038/s41467-017-01271-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Hendriks IA, D’Souza RC, Yang B, Verlaan-de Vries M, Mann M, and Vertegaal AC (2014). Uncovering global SUMOylation signaling networks in a site-specific manner. Nat. Struct. Mol. Biol. 21, 927–936. 10.1038/nsmb.2890. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Maréchal A, Li JM, Ji XY, Wu CS, Yazinski SA, Nguyen HD, Liu S, Jiménez AE, Jin J, and Zou L (2014). PRP19 transforms into a sensor of RPA-ssDNA after DNA damage and drives ATR activation via a ubiquitin-mediated circuitry. Mol. Cell 53, 235–246. 10.1016/j.molcel.2013.11.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.He Y, Brown MA, Rothnagel JA, Saunders NA, and Smith R (2005). Roles of heterogeneous nuclear ribonucleoproteins A and B in cell proliferation. J. Cell Sci. 118, 3173–3183. 10.1242/jcs.02448. [DOI] [PubMed] [Google Scholar]
- 38.Choi JH, Lindsey-Boltz LA, Kemp M, Mason AC, Wold MS, and Sancar A (2010). Reconstitution of RPA-covered single-stranded DNA-activated ATR-Chk1 signaling. Proc. Natl. Acad. Sci. USA 107, 13660–13665. 10.1073/pnas.1007856107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Beauclair G, Bridier-Nahmias A, Zagury JF, Saïb A, and Zamborlini A (2015). JASSA: a comprehensive tool for prediction of SUMOylation sites and SIMs. Bioinformatics 31, 3483–3491. 10.1093/bioinformatics/btv403. [DOI] [PubMed] [Google Scholar]
- 40.Wu CS, Ouyang J, Mori E, Nguyen HD, Maréchal A, Hallet A, Chen DJ, and Zou L (2014). SUMOylation of ATRIP potentiates DNA damage signaling by boosting multiple protein interactions in the ATR pathway. Genes Dev. 28, 1472–1484. 10.1101/gad.238535.114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Ribeyre C, Zellweger R, Chauvin M, Bec N, Larroque C, Lopes M, and Constantinou A (2016). Nascent DNA Proteomics Reveals a Chromatin Remodeler Required for Topoisomerase I Loading at Replication Forks. Cell Rep. 15, 300–309. 10.1016/j.celrep.2016.03.027. [DOI] [PubMed] [Google Scholar]
- 42.Lopez-Contreras AJ, Ruppen I, Nieto-Soler M, Murga M, Rodriguez-Acebes S, Remeseiro S, Rodrigo-Perez S, Rojas AM, Mendez J, Muñoz J, and Fernandez-Capetillo O (2013). A proteomic characterization of factors enriched at nascent DNA molecules. Cell Rep. 3, 1105–1116. 10.1016/j.celrep.2013.03.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Chen R, and Wold MS (2014). Replication protein A: single-stranded DNA’s first responder: dynamic DNA-interactions allow replication protein A to direct single-strand DNA intermediates into different pathways for synthesis or repair. BioEssays 36, 1156–1161. 10.1002/bies.201400107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Guan J, Lu C, Jin Q, Lu H, Chen X, Tian L, Zhang Y, Ortega J, Zhang J, Siteni S, et al. (2021). MLH1 Deficiency-Triggered DNA Hyperexcision by Exonuclease 1 Activates the cGAS-STING Pathway. Cancer Cell 39, 109–121.e105. 10.1016/j.ccell.2020.11.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Ibler AEM, ElGhazaly M, Naylor KL, Bulgakova NA, F.E.-K S., and Humphreys D. (2019). Typhoid toxin exhausts the RPA response to DNA replication stress driving senescence and Salmonella infection. Nat. Commun. 10, 4040. 10.1038/s41467-019-12064-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Liu Q, Guntuku S, Cui XS, Matsuoka S, Cortez D, Tamai K, Luo G, Carattini-Rivera S, DeMayo F, Bradley A, et al. (2000). Chk1 is an essential kinase that is regulated by Atr and required for the G(2)/M DNA damage checkpoint. Genes Dev. 14, 1448–1459. [PMC free article] [PubMed] [Google Scholar]
- 47.Vassin VM, Anantha RW, Sokolova E, Kanner S, and Borowiec JA (2009). Human RPA phosphorylation by ATR stimulates DNA synthesis and prevents ssDNA accumulation during DNA-replication stress. J. Cell Sci. 122, 4070–4080. 10.1242/jcs.053702. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Anantha RW, Vassin VM, and Borowiec JA (2007). Sequential and synergistic modification of human RPA stimulates chromosomal DNA repair. J. Biol. Chem. 282, 35910–35923. 10.1074/jbc.M704645200. [DOI] [PubMed] [Google Scholar]
- 49.Shiotani B, Nguyen HD, Håkansson P, Maréchal A, Tse A, Tahara H, and Zou L (2013). Two distinct modes of ATR activation orchestrated by Rad17 and Nbs1. Cell Rep. 3, 1651–1662. 10.1016/j.celrep.2013.04.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Saldivar JC, Cortez D, and Cimprich KA (2017). The essential kinase ATR: ensuring faithful duplication of a challenging genome. Nat. Rev. Mol. Cell Biol. 18, 622–636. 10.1038/nrm.2017.67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Pierce AJ, Johnson RD, Thompson LH, and Jasin M (1999). XRCC3 promotes homology-directed repair of DNA damage in mammalian cells. Genes Dev. 13, 2633–2638. 10.1101/gad.13.20.2633. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Konstantinopoulos PA, Ceccaldi R, Shapiro GI, and D’Andrea AD (2015). Homologous Recombination Deficiency: Exploiting the Fundamental Vulnerability of Ovarian Cancer. Cancer Discov. 5, 1137–1154. 10.1158/2159-8290.cd-15-0714. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Litton JK, Rugo HS, Ettl J, Hurvitz SA, Gonçalves A, Lee KH, Fehrenbacher L, Yerushalmi R, Mina LA, Martin M, et al. (2018). Talazoparib in Patients with Advanced Breast Cancer and a Germline BRCA Mutation. New Engl. J. Med. 379, 753–763. 10.1056/NEJMoa1802905. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Robson M, Goessl C, and Domchek S (2017). Olaparib for Metastatic Germline BRCA-Mutated Breast Cancer. New Engl. J. Med. 377, 1792–1793. 10.1056/NEJMc1711644. [DOI] [PubMed] [Google Scholar]
- 55.Yin Y, Lee WTC, Gupta D, Xue H, Tonzi P, Borowiec JA, Huang TT, Modesti M, and Rothenberg E (2021). A basal-level activity of ATR links replication fork surveillance and stress response. Mol. Cell 81, 4243–4257.e4246. 10.1016/j.molcel.2021.08.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Hanahan D, and Weinberg RA (2011). Hallmarks of cancer: the next generation. Cell 144, 646–674. 10.1016/j.cell.2011.02.013. [DOI] [PubMed] [Google Scholar]
- 57.Andor N, Maley CC, and Ji HP (2017). Genomic Instability in Cancer: Teetering on the Limit of Tolerance. Cancer Res. 77, 2179–2185. 10.1158/0008-5472.can-16-1553. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Goldman MJ, Craft B, Hastie M, Repečka K, McDade F, Kamath A, Banerjee A, Luo Y, Rogers D, Brooks AN, et al. (2020). Visualizing and interpreting cancer genomics data via the Xena platform. Nat. Biotech. 38, 675–678. 10.1038/s41587-020-0546-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Marquard AM, Eklund AC, Joshi T, Krzystanek M, Favero F, Wang ZC, Richardson AL, Silver DP, Szallasi Z, and Birkbak NJ (2015). Pan-cancer analysis of genomic scar signatures associated with homologous recombination deficiency suggests novel indications for existing cancer drugs. Biomark. Res. 3, 9. 10.1186/s40364-015-0033-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Quinet A, Carvajal-Maldonado D, Lemacon D, and Vindigni A (2017). DNA Fiber Analysis: Mind the Gap! Methods Enzymol. 591, 55–82. 10.1016/bs.mie.2017.03.019. [DOI] [PubMed] [Google Scholar]
- 61.Luo K, Zhang H, Wang L, Yuan J, and Lou Z (2012). Sumoylation of MDC1 is important for proper DNA damage response. EMBO J. 31, 3008–3019. 10.1038/emboj.2012.158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Knijnenburg TA, Wang L, Zimmermann MT, Chambwe N, Gao GF, Cherniack AD, Fan H, Shen H, Way GP, Greene CS, et al. (2018). Genomic and Molecular Landscape of DNA Damage Repair Deficiency across The Cancer Genome Atlas. Cell Rep. 23, 239–254.e236. 10.1016/j.celrep.2018.03.076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Telli ML, Timms KM, Reid J, Hennessy B, Mills GB, Jensen KC, Szallasi Z, Barry WT, Winer EP, Tung NM, et al. (2016). Homologous Recombination Deficiency (HRD) Score Predicts Response to Platinum-Containing Neoadjuvant Chemotherapy in Patients with Triple-Negative Breast Cancer. Clin. Cancer Res. 22, 3764–3773. 10.1158/1078-0432.ccr-15-2477. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
Original western blot images and microscopy data reported in this paper have been deposited at Mendeley. The DOI is listed in the Key Resources Table.
Any additional information required to reanalyze the data reported in this paper is available from the lead contact upon reasonable request.
This paper does not report original code.
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| Mouse Anti-HNRNPA2B1 | Santa Cruz | Cat# sc-374053; RRID: AB_10947257 |
| Rabbit Anti-HNRNPA2B1 | GeneTex | Cat# GTX127928; RRID: AB_2616069 |
| Mouse Anti-phospho-Histone H2A.X (Ser139) | Millipore | Cat# 05-636; RRID: AB_309864 |
| Mouse Monoclonal Anti-FLAG® M2 | Sigma-Aldrich | Cat# F1804; RRID: AB_262044 |
| Rabbit Anti-RPA1 | Bethyl | Cat# A300-241A; RRID: AB_2180681 |
| Mouse Anti-RPA2 | Santa Cruz | Cat# sc-56770; RRID: AB_785534 |
| Rabbit Anti-ATR | Cell Signaling Technology | Cat# 2790; RRID: AB_2227860 |
| Rabbit Anti-ATRIP | Cell Signaling Technology | Cat# 2737; RRID: AB_823659 |
| Mouse Anti-Cyclin A | Santa Cruz | Cat# sc-239; RRID: AB_627334 |
| Mouse Anti-HA | Sigma-Aldrich | Cat# H9658; RRID: AB_260092 |
| Rabbit Anti-His-Tag (D3I1O) | Cell Signaling Technology | Cat# 12698; RRID: AB_2744546 |
| Rabbit Anti-PIAS1 | Cell Signaling Technology | Cat# 3550; RRID: AB_1904090 |
| Rabbit Anti-PIAS2 | GeneTex | Cat# GTX115180; RRID: AB_11162657 |
| Rabbit Anti-PIAS3 | Cell Signaling Technology | Cat# 9042; RRID: AB_2797692 |
| Rabbit Anti-PIAS4 | Cell Signaling Technology | Cat# 4392; RRID: AB_10547884 |
| Rabbit Anti-UBC9 | Cell Signaling Technology | Cat# 4786; RRID: AB_10559206 |
| Mouse Anti-CHK1 | Cell Signaling Technology | Cat# 2360; RRID: AB_2080320 |
| Rabbit Anti-Phospho-CHK1 (Ser345) | Cell Signaling Technology | Cat# 2341; RRID: AB_330023 |
| Rabbit Anti-Phospho-RPA32 (Ser4, Ser8) | Bethyl | Cat# A300-245A; RRID: AB_210547 |
| Rabbit Anti-Phospho-RPA32 (S33) | Bethyl | Cat# A300-246A; RRID: AB_2180847 |
| Mouse Anti-BrdU | BD Biosciences | Cat# 347580; RRID: AB_10015219 |
| Rat Anti-BrdU | Abcam | Cat# ab6326; RRID: AB_305426 |
| Rabbit Anti-RAD51 | GeneTex | Cat# GTX100469; RRID: AB_1951602 |
| Mouse Anti-SUMO1 | Thermo Fisher Scientific | Cat# 33-2400; RRID: AB_2533109 |
| Mouse Anti-Biotin | Jackson ImmunoResearch Labs | Cat# 200-002-211; >RRID: AB_2339006 |
| Rabbit Anti-Phospho-Histone H3 (Ser10) | Cell Signaling Technology | Cat# 9701; RRID: AB_331535 |
| Mouse Anti- PCNA (Alexa Fluor 488 Conjugate) | Cell Signaling Technology | Cat# 8580; RRID: AB_11178664 |
| Alexa Fluor 488-AffiniPure Goat Anti-Mouse IgG (H+L) | Jackson ImmunoResearch Labs | Cat# 115-545-062; RRID: AB_2338845 |
| Alexa Fluor 488-AffiniPure Goat Anti-Rabbit IgG (H+L) | Jackson ImmunoResearch Labs | Cat# 111-545-045; RRID: AB_2338049 |
| Rhodamine Red-X-AffiniPure Goat Anti-Mouse IgG (H+L) | Jackson ImmunoResearch Labs | Cat# 115-295-146; RRID: AB_2338766 |
| Rhodamine Red-X-AffiniPure Donkey Anti-Rat IgG (H+L) | Jackson ImmunoResearch Labs | Cat# 712-295-153; RRID: AB_2340676 |
| Bacterial and virus strains | ||
| BL21 (DE3) Escherichia coli | Transgen Biotech | Cat# CD601-02 |
| Chemicals, peptides, and recombinant proteins | ||
| Cisplatin | Sigma-Aldrich | Cat# P4394 |
| Olaparib (AZD-2281) | LC laboratories | Cat# O-9201 |
| ML-792 | MedChemExpress | Cat# HY-108702 |
| Isopropyl b-D-1-thiogalactopyranoside (IPTG) | Thermo Fisher Scientific | Cat# 15529019 |
| 5-Iodo-2′-deoxyuridine | Sigma-Aldrich | Cat# I7125 |
| 5-Chloro-2′-deoxyuridine | Sigma-Aldrich | Cat# C6891 |
| N-Ethylmaleimide | Sigma-Aldrich | Cat# E3876 |
| BrdU | Biolegend | Cat# 423401 |
| 5-ethynyl-2’-deoxyuridine | Cayman Chemical | Cat# 20518 |
| TransIT-X2® Dynamic Delivery System | Mirus | Cat# MIR6004 |
| DAPI | Thermo Fisher Scientific | Cat# D1306 |
| Dynabeads™ Protein G | Thermo Fisher Scientific | Cat# 10003D |
| Anti-Flag M2 Affinity Gel | Sigma-Aldrich | Cat# A2220 |
| PrimeSTAR HS DNA Polymerase | TaKaRa | Cat# R040A |
| Hydroxyurea | Sigma-Aldrich | Cat# H8627 |
| Biotin-azide | Cayman Chemical | Cat# 13040 |
| Berzosertib (VX970) | Selleck | Cat# S7102 |
| (+)-Sodium L-ascorbate | Sigma-Aldrich | Cat# A7631 |
| SUMO1 | Boston Biochem | Cat# UL-712 |
| PIAS2 | NOVUS Biologicals | Cat# H00009063-P01 |
| Pierce™ Glutathione Agarose | Thermo Fisher Scientific | Cat# 16102BID |
| Ni-NTA Agarose | QIAGEN | Cat# 30210 |
| SAE1 | Boston Biochem | Cat# E-315 |
| UBC9 | Boston Biochem | Cat# E2-645 |
| Critical commercial assays | ||
| Duo-link in situ PLA Kit | Sigma-Aldrich | Cat# DUO92101 |
| QuickChange Site-Directed Mutagenesis Kit | Stratagene | Cat# 200519 |
| CometAssay kit | Trevigen | Cat# 4250-050-K |
| Deposited data | ||
| Raw data files | This paper | Mendeley Data: https://doi.org/10.17632/vzgpmfmt9t.1 |
| Experimental models: Cell lines | ||
| Human: HEK293T | ATCC | Cat# CRL-11268 |
| Human: U2OS | ATCC | Cat# HTB-96 |
| Human: MDA-MB-231 | ATCC | Cat# HTB-26 |
| Human: HCC1806 | ATCC | Cat# CRL-2335™ |
| Oligonucleotides | ||
| shHNRNPA2B1-1 | Sigma-Aldrich | TRCN0000001058 |
| shHNRNPA2B1-2 | Sigma-Aldrich | TRCN0000001059 |
| shATR | Sigma-Aldrich | TRCN0000196538 |
| shPIAS1-1 | Sigma-Aldrich | TRCN0000231898 |
| shPIAS1-2 | Sigma-Aldrich | TRCN0000231900 |
| shPIAS2 | Sigma-Aldrich | TRCN0000230128 |
| shPIAS3 | Sigma-Aldrich | TRCN0000274337 |
| HNRNPA2B1 KO | GenScript | TCTCTTGCTACAGCACGTTT |
| HNRNPA2-forward | This paper | ATGGAGAGAGAAAAGGAACAG |
| HNRNPA2-reverse | This paper | GTATCGGCTCCTCCCACC |
| RPA1-forward | This paper | ATGGTCGGCCAACTGAGC |
| RPA1-reverse | This paper | CATCAATGCACTTCTCC |
| SUMO1-forward | This paper | ATGTCTGACCAGGAGGC |
| SUMO1-reverse | This paper | AACTGTTGAATGACC |
| shPIAS4-1 | Sigma-Aldrich | TRCN0000231892 |
| shPIAS4-2 | Sigma-Aldrich | TRCN0000004115 |
| Recombinant DNA | ||
| PLVX3-FLAG-HNRNPA2 | This paper | N/A |
| PLVX6-EGFP-HNRNPA2 | This paper | N/A |
| pGEX-4T2-HNRNPA2 | This paper | N/A |
| PLVX3-FLAG-RPA1 | This paper | N/A |
| PLVX3-FLAG-RPA1 (ΔSIM) | This paper | N/A |
| Myc-RPA1 | This paper | N/A |
| pTEV-His-RPA1 (4A) | This paper | N/A |
| PLVX3-FLAG-RPA1 (4A) | This paper | N/A |
| PLVX3-FLAG-SUMO1 | This paper | N/A |
| PLVX3-FLAG-SUMO1-HNRNPA2 | This paper | N/A |
| PLVX3-FLAG-HNRNPA2 (K108R) | This paper | N/A |
| pFLAG-CMV-SENP1 | Addgene | Cat# 17357 |
| pCMV-HA-hUBC9 | Addgene | Cat# 14438 |
| Software and algorithms | ||
| GraphPad Prism | GraphPad | https://www.graphpad.com |
| ImageJ | ImageJ | https://imagej.nih.gov/ij/ |
| FlowJo (10.1) | FlowJo LLC | http://www.flowjo.com/ |