

Summary
Myelin oligodendrocyte glycoprotein (MOG)-antibody (Ab)-associated disease (MOGAD) is an inflammatory demyelinating disease of the CNS. Although MOG is encephalitogenic in different mammalian species, the mechanisms by which human MOG-specific Abs contribute to MOGAD are poorly understood. Here, we use a systems-level approach combined with high-dimensional characterization of Ab-associated immune features to deeply profile humoral immune responses in 123 patients with MOGAD. We show that age is a major determinant for MOG-antibody-related immune signatures. Unsupervised clustering additionally identifies two dominant immunological endophenotypes of MOGAD. The pro-inflammatory endophenotype characterized by increased binding affinities for activating Fcγ receptors (FcγRs), capacity to activate innate immune cells, and decreased frequencies of galactosylated and sialylated immunoglobulin G (IgG) glycovariants is associated with clinically active disease. Our data support the concept that FcγR-mediated effector functions control the pathogenicity of MOG-specific IgG and suggest that FcγR-targeting therapies should be explored for their therapeutic potential in MOGAD.
Keywords: humoral signatures, myelin oligodendrocyte glycoprotein, MOGAD, antibody, Fc gamma receptors, demyelination
Graphical abstract
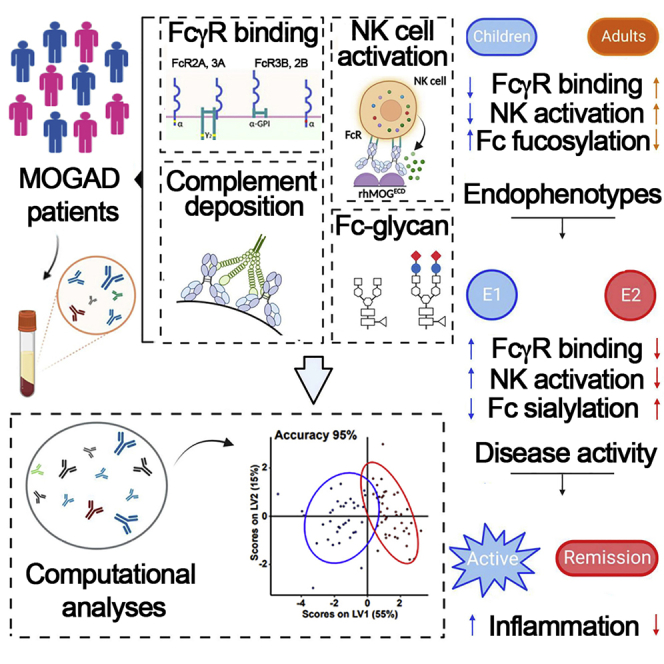
Highlights
-
•
Age is a major determinant of MOG-Ab immune signatures in MOG-Ab-associated disease
-
•
Increased FcγR-mediated functions of MOG-Abs are associated with disease activity
-
•
An experimental framework applicable to other autoantibody-mediated diseases
By profiling the biochemical and functional landscape of myelin oligodendrocyte glycoprotein (MOG) antibodies (Abs) in patients with MOG-Ab-associated disorder, Spatola et al. identify serological correlates of demographical and clinical disease features. A pro-inflammatory Ab signature characterized by increased binding affinities for activating FcγRs is associated with clinically active disease.
Introduction
Myelin oligodendrocyte glycoprotein (MOG)-targeting immunoglobulin G (IgG) antibodies (Abs) are consistently identified in children and adults with acquired CNS demyelinating syndromes, collectively termed MOG-Ab-associated disease (MOGAD).1,2 Although there are clinical phenotypic overlaps between MOGAD, multiple sclerosis (MS), and aquaporin-4 (AQP4)-IgG-positive neuromyelitis optica spectrum disorder (AQP4-IgG+ NMOSD), cumulative biological, clinical, and pathological evidence discriminates between these conditions. Presence of MOG-IgG in the serum assessed by cell-based assays (CBAs) confirms the diagnosis in patients with compatible clinical syndromes, as these Abs are rarely found in people with MS, AQP4-IgG+ NMOSD, or other neurological diseases or in healthy controls.3
The clinical phenotype associated with the presence of MOG-Abs changes with age from multifocal CNS demyelination often in the form of acute disseminated encephalomyelitis (ADEM) and optic neuritis (ON) in children to isolated ON with or without involvement of the spinal cord and brainstem in adults.4,5 While MOG is long known for its encephalitogenic potential in many different species, the mechanisms by which human MOG-specific Abs mediate pathology in patients with MOGAD are incompletely understood.2
Ab effector functions are largely mediated by the constant fragment crystallizable (Fc) domain of IgG molecules through ligation of cellular Fcγ receptors or binding of the complement component C1q.6 Apart from Ab isotype and subclass, the sugar moiety attached to the IgG constant heavy 2 (CH2) domain is critical for maintaining both the pro-inflammatory and anti-inflammatory effector functions of IgG molecules.7,8 Human MOG-Abs are almost exclusively of the complement fixing IgG1 isotype.5,9 While in vitro studies reported that human MOG-Abs can activate complement and cellular-dependent cytotoxicity,10,11,12,13,14 human MOG-Abs poorly recognize rodent MOG, and adoptive transfer of human MOG-Abs alone into rodents proved to be insufficient to induce CNS demyelination.12,15,16,17 Investigations on pathogenic mechanisms of human MOG-Abs in vivo have, therefore, been only partially successful.3 The goal of the present study was to systematically profile the biochemical and functional landscape of human MOG-Abs in pediatric and adult patients diagnosed with MOGAD using an unbiased, high-throughput systems serology platform and to identify humoral correlates for human MOG-Ab-associated clinical disease features.
Results
We included 123 individuals with MOGAD, among them 83 children and 40 adults. As expected, clinical phenotypes differed between both cohorts (Figure 1A; Table 1), with ADEM being the most frequent phenotype in children and ON (both unilateral and bilateral) being the most frequent in adults. A female predominance was observed in adults with MOGAD, whereas both genders were equally represented in children. Since serum MOG-IgG is rarely present in other neurological diseases or in healthy controls, we focused on profiling humoral signatures in patients with MOGAD stratified by age and demographical and clinical features. Using a systems serology approach,18,19,20 we next comprehensively determined IgG effector functions (Ab-dependent complement deposition [ADCD] and Ab-dependent natural killer cell activation [ADNKA]), Fc receptor (FcγR) binding affinities (FcγR 2A, 2B, 3A, 3B, and C1q components), and IgG-Fc glycosylation profiles to determine whether pediatric and adult patients with MOGAD differ in functional and Fc-biophysical profiles of MOG-specific Abs. In children, MOG-specific Abs had lower capacity to mediate ADNKA compared with adults. This decreased functionality was related to lower FcγR binding capacity, in particular FcγR3A highly expressed on NK cells (Figures 1B–1D).
Figure 1.

Distinct MOG antibody features discriminate between children and adult patients with MOGAD
(A) Clinical syndromes in pediatric and adult patients with MOGAD. ADEM, acute disseminated encephalomyelitis; ADEM/ON, simultaneous ADEM and ON; ADEM/ON/TM, Simultaneous ADEM, ON, and TM; BON, bilateral optic neuritis; BON/TM, simultaneous BON and TM; LETM, longitudinal extensive TM; MDEM, multiphasic disseminated EM; MOG-E, myelin oligodendrocyte glycoprotein encephalitis; ON/TM, simultaneous ON and TM; TM, transversal myelitis; UON, unilateral ON.
(B) Heatmap shows z scored values of MOG antibody functions (ADCD, ADNKA), binding to Fc gamma receptors (FcγRs) (FcγR2AR, 2AH, 2B, 3AF, 3AV, 3B), and complement component (C1q) in patients with MOGAD ordered by age. Each column corresponds to a single patient.
(C) MOG-antibody-specific functions (NK cell activation [ADNKA], complement deposition [ADCD]) and binding capacity to FcγRs and C1q; antibody Fc glycosylation (S, sialylation; B, bisecting N-glycan; G, galactosylation; F, fucosylation) in pediatric (n = 83) and adult (n = 40) patients. Each flower plot condenses all data for each patient group, and length of the petal represent the mean of the Z score value for each indicated feature.
(D) Univariate comparison of MOG-antibody features, including NK cell activation (ADNKA), binding to Fc gamma receptors (FcγR2A, 2B, 3A), and bulk IgG-Fc bisected and fucosylated N-glycans, between children and adults. Each dot represents an individual patient; data are presented as mean ± SD. Mann-Whitney test was used for statistical comparisons between age groups. p values indicate statistical significance (∗∗p ≤ 0.01, ∗∗∗p ≤ 0.001, ∗∗∗∗p ≤ 0.0001).
(E) Volcano plot shows the correlation of each MOG-antibody feature with age. Spearman correlation coefficients are indicated in the x axis (positive correlation on the right, red; negative correlation on the left, blue), and the statistical significance is indicated in the y axis (−log10[p values]). Values above black dashed line indicate statistically significant correlations (adjusted p value < 0.01, Benjamini-Hochberg correction for multiple comparisons).
(F) Multivariate analysis of MOG-antibody signatures in children and adults. Partial least-square discriminant analysis (PLSDA) on LASSO-selected features was used to resolve antibody profiles in children versus adults. Dots represent individual samples (children, blue; adults, red). Model accuracy 65% by cross-validation, model validation p < 0.001 by permutated test. Bar graph shows LASSO-selected features enriched in children or adults ranked by their variable importance in projection (VIP).
Each sample was run in duplicate (technical replicates), and results were obtained by averaging duplicates.
Table 1.
Demographical and clinical characteristics of patients with MOGAD
| All | Children | Adults | p | |
|---|---|---|---|---|
| N | 123 | 83 | 40 | |
| Female (n, %) | 74 (60) | 44 (53) | 30 (75) | 0.02 |
| Median age (y) (range; SD) | 13 (1–72; 18) | 8 (1–17; 5) | 42 (17.3–72; 14) | (<0.01) |
| Clinical phenotypes | ||||
| - Optic neuritis | 48 (39) | 21 (25) | 27 (67) | |
| - ADEM | 28 (23) | 28 (34) | 0 | <0.0001 |
| - Othersa | 47 (38) | 34 (41) | 13 (33) | |
| Disease status at time of sample (n, %)b | ||||
| - Exacerbation | 18/57 (31) | 13/43 (30) | 5/14 (36) | |
| - Remission | 39/57 (68) | 30/43 (70) | 9/14 (64) | 0.74 |
| Immunotherapy (%)c | ||||
| - Naive | 24/41 (59) | 24/41 (59) | ||
| - Steroids | 9/41 (22) | 9/41 (22) | – | – |
| - IVIG/SCIG | 7/41 (17) | 7/41 (17) | ||
| - IVIG + steroids | 1/41 (2) | 1/41 (2) | ||
ADEM, acute disseminated encephalomyelitis; IVIG, intravenous immunoglobulin; SCIG, subcutaneous immunoglobulin. p values refer to the comparison between children and adult patients. Fisher’s exact test or Wilcoxon test were used as appropriate.
Other clinical phenotypes included: simultaneous ADEM and optic neuritis; simultaneous ADEM, optic neuritis, and transverse myelitis; longitudinal extensive transverse myelitis; multiphasic disseminated encephalomyelitis; encephalitis; and simultaneous optic neuritis and transverse myelitis.
Total numbers here are different from N because we only had information about the relapse/remission disease status from a smaller portion of the cohort (57 individuals in total).
Information about immunotherapy was only available for 41 of 123 children with MOGAD.
Ab functionality and capacity to engage FcγRs depend on Ab class, subclass, and the type of N-linked glycans attached to the Fc part of Abs.8,21 Given that MOG-Abs are almost exclusively of IgG1 subclass,5,22 which is, along with IgG3, the most functional Ab subclass in humans,23 we next aimed to explore whether the observed age-related difference in functionality was related to Fc glycan profiles rather than IgG subclasses. Here, we observed a significant correlation between Fc glycan signatures and age (Figure 1E); in particular, fucosylation was negatively correlated with age, indicating higher levels in children than adults (Figure 1D). This is in line with previous observations of Fc fucosylation decrease during the lifetime24,25,26 and might explain the lower capacity of MOG-specific Abs to mediate cellular cytotoxicity by NK cells (Ab-dependent cell cytotoxicity [ADCC]). Indeed, Fc fucose is believed to prevent interaction between IgG and FcγR3A, thus reducing FcγR binding and capacity to engage NK activation.27 On the other hand, the presence of Fc bisecting GlcNAc, which has been described to improve ADCC,28 positively correlated with age (Figures 1D and 1E), supporting the observation that adults with MOGAD show improved Ab-mediated NK activation compared with children.
To define the minimal number of features that distinguished Abs in children versus adults, we next applied multivariate models (LASSO/partial least-square differential analysis [PLSDA]) to our MOG-specific Ab data. We observed separation in Ab profiles between our age groups (Figure 1F; model validation p < 0.0001 by permutated test, accuracy 65% by cross-validation). In agreement with the univariate analysis, MOG-specific ADNKA and FcγR binding (particularly, FcγR3A) were enriched in adults. Univariate and multivariate analyses of patients solely stratified by clinical phenotypes enriched within children or adults (ADEM versus non-ADEM and ON versus non-ON) or by gender (female versus male) independent from age groups, i.e., from all patients included, did not show significant differences in functional and Fc-biophysical profiles. These data indicate that MOG-specific Abs employ distinct innate effector pathways in children versus adults, in line with previous studies demonstrating that both biophysical25 and functional29 characteristics of IgG Abs change during aging. MOG-IgG-associated features were, therefore, analyzed separately for children and adults.
We next addressed whether the spectrum of MOG-Ab-associated features allows the identification of distinct immunophenotypic subgroups within our cohort. Using unsupervised cluster analysis, two dominant endophenotypes were identified in each age group (Figure 2A). Endophenotype 1 was characterized by increased MOG-specific Ab titers (in children) with high capacity to bind FcγRs and mediate ADNKA (in both children and adults), whereas endophenotype 2 was characterized by an attenuated MOG-specific profile with low titers and lower FcγR binding capacity. Also, in both children and adults, endophenotype 1 was enriched in Fc agalactosylation, whereas endophenotype 2 was enriched in Fc sialylation (Figure 2A). Supervised multivariate analyses using LASSO/PLSDA showed clear separation between endophenotypes (validation p < 0.01 for both children and adult models, accuracy 95% and 93%, respectively) and confirmed FcγR binding capacity as the most important feature separating endophenotypes 1 and 2 (Figure 2B).
Figure 2.

MOG-antibody-specific and Fc gamma receptor binding affinity signatures define two endophenotypes in MOGAD
(A) 2D projection of k-means separating MOG-specific features in 2 clusters (or endophenotypes) in children and adults. Heatmaps indicating the mean value (z scored) for each antibody feature in endophenotype 1 (E1) and E2. Comparisons between endophenotypes for each age group were carried out using Mann-Whitney test, ∗p ≤ 0.05, ∗∗p ≤ 0.01, ∗∗∗p ≤ 0.001, ∗∗∗∗p ≤ 0.0001.
(B) Multivariate analysis comparing MOG-antibody signatures between E1 and E2 in children and adults. PLSDA on LASSO-selected features was used to resolve antibody profiles in E1 versus E2. Dots represent individual samples (E1, blue; E2, red). Model accuracy 95% (children) and 93% (adults) by cross-validation, model validation p < 0.001 by permutated test. Bar graph shows LASSO-selected features enriched in E1 or E2 in children or adults ranked by their VIP.
Each sample was run in duplicate (technical replicates), and results were obtained by averaging duplicates.
We next aimed to understand whether these Ab-related endophenotypes tracked with disease activity or distinct clinical features in each age group. Endophenotype 1 was enriched in children experiencing active disease compared with patients in clinical remission and absent in adults in remission (Figure 3A). Neither endophenotypes were enriched in female versus male patients or were attributable to clinical syndromes at disease onset (ADEM, ON, others). We additionally investigated the possibility that immunotherapy might account for the differential Ab signatures observed across endophenotypes. In children, endophenotype 1 was enriched for patients receiving immunotherapy, whereas endophenotype 2 was enriched for immunotherapy-naive patients. Although immunotherapy in our study cohort was not recorded on a regular basis and is, therefore, incomplete, these data do not support the hypothesis that immunotherapy is a main driver of the attenuated inflammatory signature observed in endophenotype 2 (Figure S1). Notably, correlation among Ab features, in particular between titers, FcγR binding, functions, and Fc glycosylation profiles, was higher during active disease than during remission (Figure 3B) regardless of the age group, indicating a more coordinated humoral response during active disease. Thus, disease activity in MOGAD tracks with unique MOG-specific Ab signatures, characterized by highly coordinated humoral response with higher FcγR binding affinities, Ab-mediated NK cell activation, increased titers, and an inflammatory glycan signature characterized by Fc agalactosylation and asialylation.
Figure 3.

E1 is characterized by active disease in MOGAD patients
(A) Frequency of active disease versus remission in E1 and E2 in children and adults. Fisher’s exact test was used for proportion comparisons in each age group (children p = 0.0047 and adults p = 0.2308). Numbers of individuals with active disease versus remission are indicated in each sector.
(B) Correlation matrix across MOG-specific ADNKA, titers, C1q, and activating FcgR (FcgR 2A, 3A, 3B) binding capacity, and Fc glycosylation profiles (G, galactosylation; S, sialylation) during active disease (left) or remission (right). Correlation strength is proportional to color intensity (r from −1 = negative correlation, blue, to +1 = positive correlation, purple). Features that were not significantly correlated in either group (age, B = bisecting GlcNAC, ADCD, inhibitory FcgR2B) are not reported (z scored, Spearman r correlation, Benjamini-Hochberg correction for multiple comparisons; ∗p ≤ 0.05; ∗∗p ≤ 0.01; ∗∗∗p ≤ 0.001; ∗∗∗∗p ≤ 0.0001).
Discussion
Our study identified serological signatures of human MOG-Ab-associated clinical disease features. Increased binding affinity for activating FcγRs strongly contributed to the clinical disease activity-associated endophenotype. These data indicate that the capability of MOG-IgG to recruit and instruct innate immune cells expressing FcγR is instrumental in driving the pathology of MOGAD. Indeed, neuropathological hallmarks of MOGAD include perivenous and confluent MOG-dominant myelin loss with predominant CD4+ T cell and granulocytic inflammation aside from complement deposition.17,30,31,32 Furthermore, adoptive transfer of human MOG-IgG cross-reacting with rodent MOG can induce CNS demyelination in vivo provided that myelin-specific CD4+ T cells are co-transferred.33 Studies in experimental autoimmune encephalomyelitis (EAE) models provide a mechanistic explanation for these observations. In an EAE model induced by co-transfer of MOG-specific CD4+ T cells and B cells, myelin-reactive auto-Abs accumulated in CNS-resident phagocytes, resulting in increased capacity of these cells to present MOG and to reactivate CNS-invading MOG-specific CD4+ T cells.34 In an independent MOG T cell receptor transgenic model, transfer of MOG-specific IgG triggered activation and expansion of MOG-specific CD4+ T cells in an Fc-dependent manner, thereby facilitating spontaneous EAE development.35 Also, adoptive transfer of affinity-purified MOG-IgG derived from patients with MOGAD together with MOG-specific CD4+ T cells into rodents augmented T cell activation and CNS infiltration.33 The aforementioned studies indicate that MOG-Ab-mediated opsonization followed by IgG Fc-dependent internalization augments antigen presentation and boosts activation of autoreactive immune cells. Using an unbiased computational approach in patients with MOGAD, our data strongly support the concept—established in MOG-induced experimental models of CNS demyelination—that MOG-IgG Fc-mediated cellular reactivation contributes to the development and disease activity of human MOGAD.
In patients with active disease, increased binding affinity for FcγRs tracked higher frequencies of agalactosylated and asialylated IgG glycovariants and increased titers of MOG-specific Abs. The latter is in line with the observation that MOG-IgG titers may drop to undetectable levels during remission from MOGAD attacks36,37 and that persistent MOG-IgG seropositivity is a predictor for recurrent disease activity.38 Decrease in the level of IgG galactosylation, and hence an increase in the abundance of agalactosylated G0 glycoforms, is one of the most prominent and established changes in IgG glycosylation at the level of total-serum and antigen-specific IgGs in a broad spectrum of chronic inflammatory and autoimmune diseases, such as rheumatoid arthritis (RA), systemic lupus erythematosus (SLE), autoimmune vasculitis, and Crohn’s disease.39 Total serum levels of agalactosylated G0 glycoforms have also been reported to correlate with clinical disease activity in RA.39 Although the Fc-linked sugar domain only contains a minor fraction of sialylated sugar structures, asialylated glycovariants are reported to be increased in patients with RA, SLE, and autoimmune vasculitis and to precede disease relapses.39 Our finding that higher levels of agalactosylated and asialylated IgG is associated with clinical disease activity in MOGAD is, therefore, supported by observations made in other autoimmune diseases. Whether lack of IgG-Fc terminal galactose or sialic acid residues translate into higher functional pro-inflammatory activity of Abs, or merely reflects chronic inflammatory processes without actively contributing to autoimmune disease development and severity, has not been conclusively clarified. Data obtained in mice, however, suggest that lack of terminal galactosylation or sialylation changes IgG structure and increases the affinity for activating FcγRs.40,41,42,43,44,45,46
Randomized clinical trials that guide therapy for patients with MOGAD are lacking, and early studies of B cell-depleting strategies show potentially limited efficacy in preventing relapses.47 Based on retrospective data, intravenous Igs (IVIGs) and oral corticosteroids are considered the most effective and tolerable therapies for MOGAD.48,49 Data form several experimental models of autoimmune diseases, and human studies provide strong evidence that modulation of FcγR expression and function, lowering the activation of FcγR-expressing immune cells, is a key mechanism for the clinical efficacy of IVIGs.40,50,51,52,53,54 Therapeutic platforms targeting the activation or signaling of FcγRs such as Bruton’s tyrosine kinase (BTK) inhibitors are currently being evaluated for their efficacy and safety in MS and other autoimmune diseases.55,56,57,58 Our data suggest that biologics targeting FcγR function might potentially restrain MOG-Ab-induced pathology and improve clinical outcomes for patients with MOGAD.
Taking together, our study provides a framework for correlating biosignatures of auto-Abs with clinical disease features and support the concept that FcγR-mediated effector functions of human MOG-specific IgG critically contribute to the development and clinical activity of MOGAD.
Limitations of the study
Our study has limitations, including the relatively low number of samples, incomplete information on immunotherapy, lack of longitudinal data from patients to allow intraindividual Ab signature comparison, and the use of total serum instead of antigen-specific IgG for Fc-glycan analyses, mainly due to limited availability of samples. Longitudinal measurements with integrated analysis of clinical parameters are needed to better evaluate the predictive value of MOG-Ab-associated signatures and features for relapses, disability outcomes, and in guiding treatment decisions.
STAR★Methods
Key resources table
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| Anti-guinea pig complement C3 goat IgG fraction, fluorescein-conjugated | MP Biomedicals | Cat #0855385 |
| PE-Cy5 Mouse anti-human CD107a (clone H4A3) | BD Biosciences | Cat #555802 |
| APC-Cy7 Mouse anti-human CD16 (clone 3G8) | BD Biosciences | Cat #557758 |
| PE-Cy7 Mouse anti-human CD56 (clone B159) | BD Biosciences | Cat #557747 |
| AF700 Mouse anti-human CD3 (clone UCHT-1) | BD Biosciences | Cat #557943 |
| Biological samples | ||
| Human serum | Recruited from tertiary hospitals in Australia, New Zealand, Austria and Germany | N/A |
| NK cells from human buffy coats | Blood donors from Massachusetts General Hospital Research | N/A |
| Chemicals, peptides, and recombinant proteins | ||
| Recombinant human FcγR, FcγR2AR, FcγR2AH, FcγR2B, FcγR3AF, FcγR3AV, FcγR3B, and C1q | Duke Protein Production facility | N/A |
| Recombinant human myelin oligodendrocyte glycoprotein extracellular domain (rhMOGECD) | Provided by Prof. Edgar Meinl, Lüdwig Maximilians-Universität München | N/A |
| Acetonitrile | Fisher | Cat#A9984 |
| Protein A/G beads (magnetic) | Thermo Fisher Scientific | Cat#26162 |
| IdeZ enzyme | New England Biolabs | Cat#P0770S |
| Critical commercial assays | ||
| EZ-link Sulfo-NHS-LC-LC-Biotin | Thermo Fisher Scientific | Cat #21338 |
| Zebra Spin Desalting Columns | Thermo Fisher Scientific | Cat #PI-89883 |
| FluoSpheres NeutrAvidin-labeled Microspheres, 1 μm, red fluorescent | Thermo Fisher Scientific | Cat #F8775 |
| Low-Tox complement guinea pig | Cederlane | Cat #CL4051 |
| NK cell isolation kit, Human | Miltenyi | Cat #130-092657 |
| GolgiStop protein transport inhibitor | BD Biosciences | Cat #554724 |
| Brefeldin A solution, 1000X | Biolegend | Cat #420601 |
| Fix & Perm cell fixation kit & Permeabilization kit | Thermo Fisher Scientific | Cat #00-5523-00 |
| Glycan AssureHyPerformance APTS kit | Applied Biosystem | Cat#A33953 |
| Software and algorithms | ||
| R version 4.1.0 | Freely available online | N/A |
| Prism GraphPad Version 9.4.1 (458) | GraphPad | N/A |
| original data and code | Mendeley database | https://doi.org/10.17632/dj43gnzjyr.1 |
| original data and code | Github | https://github.com/LoosC/systemsseRology |
Resource availability
Lead contact
Further information and requests for resources and reagents should be directed to and will be fulfilled by the lead contact, Jan Lünemann (jan.luenemann@ukmuenster.de).
Materials availability
Reagents generated and used in this study will be made available on request, but we may require a payment and/or a completed Materials Transfer Agreement (MTA). There are restrictions to the availability of FcγRs due to MTA.
Experimental model and subject details
Biological samples and human subjects
Patients of all age diagnosed with MOG-antibody associated disorders (MOGAD) were recruited from tertiary hospitals in Australia, New Zealand, Austria and Germany. Diagnosis of MOGAD was established by clinical criteria and identification of MOG antibodies in serum samples, collected at disease onset, remission, or relapse, by cell-based assays (as explained below) in Sydney, Australia and Innsbruck, Austria. Clinical information included demographics (age, gender), disease course (relapsing versus monophasic), phenotype (including acute demyelinating encephalomyelitis, ADEM, neuromyelitis optica spectrum disorder, optic neuritis, ON, longitudinally extended transverse myelitis, LETM, and encephalitis, MOG-E), and immunotherapy. Children were defined as ≤ 18 years of age. Informed consent was obtained from patients or carers in the case of pediatric patients. Hospital Ethics Committee approval was obtained at each center (NEAF 12/SCHN/395, University of Sydney, Australia, and AM3041A and AM4059, Medical University of Innsbruck, Austria and University Witten/Herdecke, Germany, respectively), according to the Declaration of Helsinki in its current version. In patients with active disease, blood samples were taken within the first 2 weeks after the onset of the clinical episode. In patients receiving immunotherapy (steroids, intravenous immunoglobulin, or subcutaneously applied immunoglobulin) blood was drawn before therapy initiation. Upon venipuncture, samples were kept 30 min at room temperature for clot retraction, then centrifuged at 4°C and resulting sera was frozen at −80°C. Samples were thawed on ice prior to experiments. Each sample was run in duplicate, and results were obtained by averaging duplicates. Presence of MOG antibodies in patient sera was identified by cell-based assays (CBA).5,59
Method details
Antibody-dependent complement deposition (ADCD)
Patient's sera were heat inactivated for 30 min at 56°C. Purified rhMOGECD60 was biotinylated using EZ-link Sulfo-NHS-LC-LC-Biotin (Cat. N° 21,338, Thermo Fisher Scientific) following the manufacturer's instructions and remaining biotin excess was removed using Zebra Spin Desalting Columns (Cat. N° PI-89883, Thermo Fisher Scientific). Biotinylated rhMOGECD was coupled onto 1 μm fluorescent neutravidin-labeled beads (Cat. N° F8775, Thermo Fisher Scientific) in a proportion of 10 μg protein to 10 μL of beads. Incubation of 10 μL PBS-diluted patient serum (1:10) with an equal volume of rhMOGECD -coated beads for 2 h at 37°C in V-bottom 96-well plates. Unbound antibodies were washed by centrifugation. Lyophilized guinea pig complement (Cat. N° CL4051, Cedarlane) was reconstituted according the manufacturers recommendations, then diluted 1:50 in RPMI supplemented with 10% FCS (R10) and 200 μL were added to each well and incubated at 37°C for 15 min. Beads were washed with 15 mM EDTA and stained using anti-guinea pig complement C3 (Cat. N° 0,855,385, MP Biomedicals). All samples were assayed in duplicates. Analysis was carried out using a CytoFLEX (Beckman Coulter). Complement deposition score were calculated as %C3+ beads multiplied C3+ Median fluorescence intensity (MedFI) divided by 104. Quality checks were performed to ensure that known positive samples were at least 2 times higher than negative controls and that replicate values correlated. After the quality checks, values from replicates were averaged.61
Antibody-dependent NK cell activation (ADNKA)
Assay was carried out in ELISA plates coated with 100 μL of rhMOGECD at a final concentration of 4 μg/mL and then blocked with 5% PBS-BSA. Patient’s sera were diluted 1:60 and 100 μL were added to each well and incubated for 2 h at 37°C. NK cells isolation was carried out from buffy coat of two healthy individuals using NK isolation Kit (Cat. N° 130–092657, Miltenyi) and cells were incubated in RPMI media containing 1 ng/mL IL-15 at 37°C. NK cells were incubated with anti-CD107a (Cat. N° 555,802, BD Biosciences), GolgiStop (Cat. N° 554,724, BD Biosciences) and brefeldin A (Cat. N° 420,601, Biolegend) for 5 h at 37°C. Then, NK cells were stained with anti-CD16 (Cat. N° 557,758, BD Biosciences), anti-CD56 (Cat. N° 557,747, BD Biosciences) and anti-CD3 (Cat. N° 557,943, BD Biosciences). Cells were fixed and permeabilized using Fix/Perm kit (Cat. N° 00-5523-00, Thermo Fisher Scientific). NK cells were analyzed on a CytoFLEX cytometer. NK cells were identified as CD3−CD16+CD56+. Each serum sample was assayed in parallel for both healthy NK cell donors. Values from each replicate were averaged. Quality checks were performed to ensure that known positive samples were at least 2 times higher than negative controls and that replicate values correlated. After the quality checks, values from replicates were averaged.61
Fc receptors and complement binding analyses
FcR-binding and complement-binding capacity were measured using a customized multiplexed Luminex assay, as reported.62 Briefly, MOG was coupled to fluorescent carboxyl-modified Luminex microspheres. Antigen-coupled microspheres were incubated with serum (dilution 1:20-1:40) for 16 h at 4°C. Immunocomplexes were then washed. Recombinant human FcαR, FcγR2AR, FcγR2AH, FcγR2B, FcγR3AF, FcγR3AV, FcγR3B, and C1q (Duke Protein Production facility) were biotinylated, conjugated to streptavidin-PE, and incubated with the immunocomplexes. Binding was detected using a Bio-Plex System (IntelliCyt, iQue Screener Plus), and Median Fluorescence Intensity (MFI) was reported and compared to PBS levels.
IgG-Fc glycan analysis
Samples were heat inactivated by incubation at 56°C for 60 min. Samples were spun at 20,000 g for 10 min and the supernatant was used for glycan analysis assay. Heat-inactivated samples were incubated for 30 min with uncoupled streptavidin magnetic beads to remove non-specific bead binding. Sample was then removed from the magnetic beads and added to protein A/G beads for 1 h at 37°C to isolate the IgG fraction. The IgG fraction was then washed and incubated with IDEZ (NEB) to cleave off the Fc from IgG antibodies overnight at room temperature. The antibodies-Fc were then deglycosylated using PNGase and labeled with APTS per manufacturer’s protocol (Glycan Assure APTS kit). Glycans were analyzed using a 3500xL genetic analyzer (Applied Biosystems). N-glycan fucosyl, afucosyl and afucosyl-fucosyl mixed libraries were also run with the samples to enable identification of different glycan families. The relative frequency of glycan families was determined using GlycanAssure software.
Quantification and statistical analysis
Statistical and computational analyses
Univariate comparative analyses of antibody features between groups was performed using non-parametric tests (Mann-Whitney, Kruskal-Wallis, Friedman test) and Fisher exact test for frequencies comparison between active versus remission disease in patients, analyses were carried out using in GraphPad Prism with corrections for multiple comparisons. Spearman correlation coefficients were calculated, and p values were obtained for two-tailed tests.
Multivariate analyses were performed with R (version 4.1.0). Luminex data were log10-transformed. All data were z-scored. Missing values were imputed using R package ‘DMwR’. A partial least square discriminant analysis (PLSDA) with Least Absolute Shrinkage Selection Operator (LASSO) was used to select antibody features that contributed most to discriminate children versus adults, and between endophenotypes.63 Antibody features were ranked according to their Variable Importance in Projection (VIP) score, for which higher values indicate higher contribution to the model, and first latent variable loadings (LV1) were plotted. 5-fold cross validation was performed. The model was validated by comparing it to a null model, for which the modeling approach was repeated 100x with permutated labels. P-values were obtained from the tail probability of the accuracy distribution of the control model. This procedure was repeated for 10 replicates of cross-validation, and median p values were reported.
K-means clustering was performed for children and adult data separately. All antibody features, including glycan data, FcγR binding data, and gender, were used for the clustering analysis. Number of clusters, 2, was determined heuristically. Implementation and visualization of k-means clustering was done with “ClusterR” package and “factoextra” package, respectively, in R (version 4.1.0).
Acknowledgments
M.S. receives research support in the form of Neuroscience Research Scholarship by the American Academy of Neurology (USA), by the Swiss National Science Foundation (Switzerland), and by the La Caixa Foundation (Spain). The authors acknowledge support by the Collaborative Research Center TR-128 “Initiating/Effector versus Regulatory Mechanisms in Multiple Sclerosis – Progress toward Tackling the Disease” (to E.M., H.W., and J.D.L.). The graphical abstract was created with BioRender.com.
Author contributions
Conceptualization, J.D.L.; methodology, M.S., O.C., and W.J.; software, M.S. and W.J.; formal analysis, M.S. and O.C.; investigation, O.C. and C.W.K.; resources, M.R., K.R., and F.B.; writing – original draft, M.S., O.C., and J.D.L.; writing – review & editing, M.S., O.C., J.A.L., E.-M.W., S.R., C.W.K., T.H., E.M., M.R., R.C.D., H.W., D.A.L., K.R., F.B., G.A., and J.D.L.
Declaration of interests
The authors declare no competing interests.
Inclusion and diversity
We support inclusive, diverse, and equitable conduct of research.
Published: January 19, 2023
Footnotes
Supplemental information can be found online at https://doi.org/10.1016/j.xcrm.2022.100913.
Contributor Information
Marianna Spatola, Email: marianna.spatola@gmail.com.
Jan D. Lünemann, Email: jan.luenemann@ukmuenster.de.
Supplemental information
Data and code availability
Data reported in this paper will be shared by the lead contact upon request.
All original data and code are available at: Mendeley database: https://doi.org/10.17632/dj43gnzjyr.1
https://github.com/LoosC/systemsseRology.
Any additional information required to reanalyze the data reported in this paper is available from the lead contact upon request.
References
- 1.Fadda G., Armangue T., Hacohen Y., Chitnis T., Banwell B. Paediatric multiple sclerosis and antibody-associated demyelination: clinical, imaging, and biological considerations for diagnosis and care. Lancet Neurol. 2021;20:136–149. doi: 10.1016/S1474-4422(20)30432-4. [DOI] [PubMed] [Google Scholar]
- 2.Marignier R., Hacohen Y., Cobo-Calvo A., Pröbstel A.K., Aktas O., Alexopoulos H., Amato M.P., Asgari N., Banwell B., Bennett J., et al. Myelin-oligodendrocyte glycoprotein antibody-associated disease. Lancet Neurol. 2021;20:762–772. doi: 10.1016/S1474-4422(21)00218-0. [DOI] [PubMed] [Google Scholar]
- 3.Reindl M., Waters P. Myelin oligodendrocyte glycoprotein antibodies in neurological disease. Nat. Rev. Neurol. 2019;15:89–102. doi: 10.1038/s41582-018-0112-x. [DOI] [PubMed] [Google Scholar]
- 4.Jurynczyk M., Messina S., Woodhall M.R., Raza N., Everett R., Roca-Fernandez A., Tackley G., Hamid S., Sheard A., Reynolds G., et al. Clinical presentation and prognosis in MOG-antibody disease: a UK study. Brain. 2017;140:3128–3138. doi: 10.1093/brain/awx276. [DOI] [PubMed] [Google Scholar]
- 5.Tea F., Lopez J.A., Ramanathan S., Merheb V., Lee F.X.Z., Zou A., Pilli D., Patrick E., van der Walt A., Monif M., et al. Characterization of the human myelin oligodendrocyte glycoprotein antibody response in demyelination. Acta Neuropathol. Commun. 2019;7:145. doi: 10.1186/s40478-019-0786-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Bournazos S., Wang T.T., Dahan R., Maamary J., Ravetch J.V. Signaling by antibodies: recent progress. Annu. Rev. Immunol. 2017;35:285–311. doi: 10.1146/annurev-immunol-051116-052433. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Arnold J.N., Wormald M.R., Sim R.B., Rudd P.M., Dwek R.A. The impact of glycosylation on the biological function and structure of human immunoglobulins. Annu. Rev. Immunol. 2007;25:21–50. doi: 10.1146/annurev.immunol.25.022106.141702. [DOI] [PubMed] [Google Scholar]
- 8.Wang T.T., Ravetch J.V. Functional diversification of IgGs through Fc glycosylation. J. Clin. Invest. 2019;129:3492–3498. doi: 10.1172/JCI130029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Waters P., Woodhall M., O'Connor K.C., Reindl M., Lang B., Sato D.K., Juryńczyk M., Tackley G., Rocha J., Takahashi T., et al. MOG cell-based assay detects non-MS patients with inflammatory neurologic disease. Neurol. Neuroimmunol. Neuroinflamm. 2015;2:e89. doi: 10.1212/NXI.0000000000000089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Brilot F., Dale R.C., Selter R.C., Grummel V., Kalluri S.R., Aslam M., Busch V., Zhou D., Cepok S., Hemmer B. Antibodies to native myelin oligodendrocyte glycoprotein in children with inflammatory demyelinating central nervous system disease. Ann. Neurol. 2009;66:833–842. doi: 10.1002/ana.21916. [DOI] [PubMed] [Google Scholar]
- 11.Keller C.W., Lopez J.A., Wendel E.M., Ramanathan S., Gross C.C., Klotz L., Reindl M., Dale R.C., Wiendl H., Rostásy K., et al. Complement activation is a prominent feature of MOGAD. Ann. Neurol. 2021;90:976–982. doi: 10.1002/ana.26226. [DOI] [PubMed] [Google Scholar]
- 12.Peschl P., Schanda K., Zeka B., Given K., Böhm D., Ruprecht K., Saiz A., Lutterotti A., Rostásy K., Höftberger R., et al. Human antibodies against the myelin oligodendrocyte glycoprotein can cause complement-dependent demyelination. J. Neuroinflammation. 2017;14:208. doi: 10.1186/s12974-017-0984-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Zhou D., Srivastava R., Nessler S., Grummel V., Sommer N., Brück W., Hartung H.P., Stadelmann C., Hemmer B. Identification of a pathogenic antibody response to native myelin oligodendrocyte glycoprotein in multiple sclerosis. Proc. Natl. Acad. Sci. USA. 2006;103:19057–19062. doi: 10.1073/pnas.0607242103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Mader S., Gredler V., Schanda K., Rostasy K., Dujmovic I., Pfaller K., Lutterotti A., Jarius S., Di Pauli F., Kuenz B., et al. Complement activating antibodies to myelin oligodendrocyte glycoprotein in neuromyelitis optica and related disorders. J. Neuroinflammation. 2011;8:184. doi: 10.1186/1742-2094-8-184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Mayer M.C., Breithaupt C., Reindl M., Schanda K., Rostásy K., Berger T., Dale R.C., Brilot F., Olsson T., Jenne D., et al. Distinction and temporal stability of conformational epitopes on myelin oligodendrocyte glycoprotein recognized by patients with different inflammatory central nervous system diseases. J. Immunol. 2013;191:3594–3604. doi: 10.4049/jimmunol.1301296. [DOI] [PubMed] [Google Scholar]
- 16.Saadoun S., Waters P., Owens G.P., Bennett J.L., Vincent A., Papadopoulos M.C. Neuromyelitis optica MOG-IgG causes reversible lesions in mouse brain. Acta Neuropathol. Commun. 2014;2:35. doi: 10.1186/2051-5960-2-35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Spadaro M., Gerdes L.A., Mayer M.C., Ertl-Wagner B., Laurent S., Krumbholz M., Breithaupt C., Högen T., Straube A., Giese A., et al. Histopathology and clinical course of MOG-antibody-associated encephalomyelitis. Ann. Clin. Transl. Neurol. 2015;2:295–301. doi: 10.1002/acn3.164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Bartsch Y.C., Wang C., Zohar T., Fischinger S., Atyeo C., Burke J.S., Kang J., Edlow A.G., Fasano A., Baden L.R., et al. Humoral signatures of protective and pathological SARS-CoV-2 infection in children. Nat. Med. 2021;27:454–462. doi: 10.1038/s41591-021-01263-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Atyeo C., Pullen K.M., Bordt E.A., Fischinger S., Burke J., Michell A., Slein M.D., Loos C., Shook L.L., Boatin A.A., et al. Compromised SARS-CoV-2-specific placental antibody transfer. Cell. 2021;184:628–642.e10. doi: 10.1016/j.cell.2020.12.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Spatola M., Loos C., Cizmeci D., Webb N., Gorman M.J., Rossignol E., Shin S., Yuan D., Fontana L., Mukerji S.S., et al. Functional compartmentalization of antibodies in the central nervous system during chronic HIV infection. J. Infect. Dis. 2022;226:738–750. doi: 10.1093/infdis/jiac138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Hayes J.M., Cosgrave E.F.J., Struwe W.B., Wormald M., Davey G.P., Jefferis R., Rudd P.M. Glycosylation and Fc receptors. Curr. Top. Microbiol. Immunol. 2014;382:165–199. doi: 10.1007/978-3-319-07911-0_8. [DOI] [PubMed] [Google Scholar]
- 22.Mariotto S., Ferrari S., Monaco S., Benedetti M.D., Schanda K., Alberti D., Farinazzo A., Capra R., Mancinelli C., De Rossi N., et al. Clinical spectrum and IgG subclass analysis of anti-myelin oligodendrocyte glycoprotein antibody-associated syndromes: a multicenter study. J. Neurol. 2017;264:2420–2430. doi: 10.1007/s00415-017-8635-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Vidarsson G., Dekkers G., Rispens T. IgG subclasses and allotypes: from structure to effector functions. Front. Immunol. 2014;5:520. doi: 10.3389/fimmu.2014.00520. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.de Haan N., Reiding K.R., Driessen G., van der Burg M., Wuhrer M. Changes in healthy human IgG Fc-glycosylation after birth and during early childhood. J. Proteome Res. 2016;15:1853–1861. doi: 10.1021/acs.jproteome.6b00038. [DOI] [PubMed] [Google Scholar]
- 25.Gudelj I., Lauc G., Pezer M. Immunoglobulin G glycosylation in aging and diseases. Cell. Immunol. 2018;333:65–79. doi: 10.1016/j.cellimm.2018.07.009. [DOI] [PubMed] [Google Scholar]
- 26.Pucic M., Muzinic A., Novokmet M., Skledar M., Pivac N., Lauc G., Gornik O. Changes in plasma and IgG N-glycome during childhood and adolescence. Glycobiology. 2012;22:975–982. doi: 10.1093/glycob/cws062. [DOI] [PubMed] [Google Scholar]
- 27.Shields R.L., Lai J., Keck R., O'Connell L.Y., Hong K., Meng Y.G., Weikert S.H.A., Presta L.G. Lack of fucose on human IgG1 N-linked oligosaccharide improves binding to human Fcgamma RIII and antibody-dependent cellular toxicity. J. Biol. Chem. 2002;277:26733–26740. doi: 10.1074/jbc.M202069200. [DOI] [PubMed] [Google Scholar]
- 28.Hodoniczky J., Zheng Y.Z., James D.C. Control of recombinant monoclonal antibody effector functions by Fc N-glycan remodeling in vitro. Biotechnol. Prog. 2005;21:1644–1652. doi: 10.1021/bp050228w. [DOI] [PubMed] [Google Scholar]
- 29.Collier D.A., Ferreira I.A.T.M., Kotagiri P., Datir R.P., Lim E.Y., Touizer E., Meng B., Abdullahi A., CITIID-NIHR BioResource COVID-19 Collaboration. Elmer A., et al. Age-related immune response heterogeneity to SARS-CoV-2 vaccine BNT162b2. Nature. 2021;596:417–422. doi: 10.1038/s41586-021-03739-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Höftberger R., Guo Y., Flanagan E.P., Lopez-Chiriboga A.S., Endmayr V., Hochmeister S., Joldic D., Pittock S.J., Tillema J.M., Gorman M., et al. The pathology of central nervous system inflammatory demyelinating disease accompanying myelin oligodendrocyte glycoprotein autoantibody. Acta Neuropathol. 2020;139:875–892. doi: 10.1007/s00401-020-02132-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Takai Y., Misu T., Kaneko K., Chihara N., Narikawa K., Tsuchida S., Nishida H., Komori T., Seki M., Komatsu T., et al. Myelin oligodendrocyte glycoprotein antibody-associated disease: an immunopathological study. Brain. 2020;143:1431–1446. doi: 10.1093/brain/awaa102. [DOI] [PubMed] [Google Scholar]
- 32.Kothur K., Wienholt L., Tantsis E.M., Earl J., Bandodkar S., Prelog K., Tea F., Ramanathan S., Brilot F., Dale R.C. B cell, Th17, and neutrophil related cerebrospinal fluid cytokine/chemokines are elevated in MOG antibody associated demyelination. PLoS One. 2016;11:e0149411. doi: 10.1371/journal.pone.0149411. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Spadaro M., Winklmeier S., Beltrán E., Macrini C., Höftberger R., Schuh E., Thaler F.S., Gerdes L.A., Laurent S., Gerhards R., et al. Pathogenicity of human antibodies against myelin oligodendrocyte glycoprotein. Ann. Neurol. 2018;84:315–328. doi: 10.1002/ana.25291. [DOI] [PubMed] [Google Scholar]
- 34.Flach A.C., Litke T., Strauss J., Haberl M., Gómez C.C., Reindl M., Saiz A., Fehling H.J., Wienands J., Odoardi F., et al. Autoantibody-boosted T-cell reactivation in the target organ triggers manifestation of autoimmune CNS disease. Proc. Natl. Acad. Sci. USA. 2016;113:3323–3328. doi: 10.1073/pnas.1519608113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Kinzel S., Lehmann-Horn K., Torke S., Häusler D., Winkler A., Stadelmann C., Payne N., Feldmann L., Saiz A., Reindl M., et al. Myelin-reactive antibodies initiate T cell-mediated CNS autoimmune disease by opsonization of endogenous antigen. Acta Neuropathol. 2016;132:43–58. doi: 10.1007/s00401-016-1559-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.López-Chiriboga A.S., Majed M., Fryer J., Dubey D., McKeon A., Flanagan E.P., Jitprapaikulsan J., Kothapalli N., Tillema J.M., Chen J., et al. Association of MOG-IgG serostatus with relapse after acute disseminated encephalomyelitis and proposed diagnostic criteria for MOG-IgG-associated disorders. JAMA Neurol. 2018;75:1355–1363. doi: 10.1001/jamaneurol.2018.1814. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Waters P., Fadda G., Woodhall M., O'Mahony J., Brown R.A., Castro D.A., Longoni G., Irani S.R., Sun B., Yeh E.A., et al. Serial anti-myelin oligodendrocyte glycoprotein antibody analyses and outcomes in children with demyelinating syndromes. JAMA Neurol. 2020;77:82–93. doi: 10.1001/jamaneurol.2019.2940. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Oliveira L.M., Apóstolos-Pereira S.L., Pitombeira M.S., Bruel Torretta P.H., Callegaro D., Sato D.K. Persistent MOG-IgG positivity is a predictor of recurrence in MOG-IgG-associated optic neuritis, encephalitis and myelitis. Mult. Scler. 2019;25:1907–1914. doi: 10.1177/1352458518811597. [DOI] [PubMed] [Google Scholar]
- 39.Seeling M., Brückner C., Nimmerjahn F. Differential antibody glycosylation in autoimmunity: sweet biomarker or modulator of disease activity? Nat. Rev. Rheumatol. 2017;13:621–630. doi: 10.1038/nrrheum.2017.146. [DOI] [PubMed] [Google Scholar]
- 40.Kaneko Y., Nimmerjahn F., Ravetch J.V. Anti-inflammatory activity of immunoglobulin G resulting from Fc sialylation. Science. 2006;313:670–673. doi: 10.1126/science.1129594. [DOI] [PubMed] [Google Scholar]
- 41.Malhotra R., Wormald M.R., Rudd P.M., Fischer P.B., Dwek R.A., Sim R.B. Glycosylation changes of IgG associated with rheumatoid arthritis can activate complement via the mannose-binding protein. Nat. Med. 1995;1:237–243. doi: 10.1038/nm0395-237. [DOI] [PubMed] [Google Scholar]
- 42.Nimmerjahn F., Anthony R.M., Ravetch J.V. Agalactosylated IgG antibodies depend on cellular Fc receptors for in vivo activity. Proc. Natl. Acad. Sci. USA. 2007;104:8433–8437. doi: 10.1073/pnas.0702936104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Quast I., Keller C.W., Maurer M.A., Giddens J.P., Tackenberg B., Wang L.X., Münz C., Nimmerjahn F., Dalakas M.C., Lünemann J.D. Sialylation of IgG Fc domain impairs complement-dependent cytotoxicity. J. Clin. Invest. 2015;125:4160–4170. doi: 10.1172/JCI82695. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Rademacher T.W., Williams P., Dwek R.A. Agalactosyl glycoforms of IgG autoantibodies are pathogenic. Proc. Natl. Acad. Sci. USA. 1994;91:6123–6127. doi: 10.1073/pnas.91.13.6123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Scallon B.J., Tam S.H., McCarthy S.G., Cai A.N., Raju T.S. Higher levels of sialylated Fc glycans in immunoglobulin G molecules can adversely impact functionality. Mol. Immunol. 2007;44:1524–1534. doi: 10.1016/j.molimm.2006.09.005. [DOI] [PubMed] [Google Scholar]
- 46.Sondermann P., Pincetic A., Maamary J., Lammens K., Ravetch J.V. General mechanism for modulating immunoglobulin effector function. Proc. Natl. Acad. Sci. USA. 2013;110:9868–9872. doi: 10.1073/pnas.1307864110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Whittam D.H., Cobo-Calvo A., Lopez-Chiriboga A.S., Pardo S., Gornall M., Cicconi S., Brandt A., Berek K., Berger T., Jelcic I., et al. Treatment of MOG-IgG-associated disorder with rituximab: an international study of 121 patients. Mult. Scler. Relat. Disord. 2020;44:102251. doi: 10.1016/j.msard.2020.102251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Wang X., Kong L., Zhao Z., Shi Z., Chen H., Lang Y., Lin X., Du Q., Zhou H. Effectiveness and tolerability of different therapies in preventive treatment of MOG-IgG-associated disorder: a network meta-analysis. Front. Immunol. 2022;13:953993. doi: 10.3389/fimmu.2022.953993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Chen J.J., Huda S., Hacohen Y., Levy M., Lotan I., Wilf-Yarkoni A., Stiebel-Kalish H., Hellmann M.A., Sotirchos E.S., Henderson A.D., et al. Association of maintenance intravenous immunoglobulin with prevention of relapse in adult myelin oligodendrocyte glycoprotein antibody-associated disease. JAMA Neurol. 2022;79:518–525. doi: 10.1001/jamaneurol.2022.0489. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Samuelsson A., Towers T.L., Ravetch J.V. Anti-inflammatory activity of IVIG mediated through the inhibitory Fc receptor. Science. 2001;291:484–486. doi: 10.1126/science.291.5503.484. [DOI] [PubMed] [Google Scholar]
- 51.Bruhns P., Samuelsson A., Pollard J.W., Ravetch J.V. Colony-stimulating factor-1-dependent macrophages are responsible for IVIG protection in antibody-induced autoimmune disease. Immunity. 2003;18:573–581. doi: 10.1016/s1074-7613(03)00080-3. [DOI] [PubMed] [Google Scholar]
- 52.Tanaka M., Krutzik S.R., Sieling P.A., Lee D.J., Rea T.H., Modlin R.L. Activation of Fc gamma RI on monocytes triggers differentiation into immature dendritic cells that induce autoreactive T cell responses. J. Immunol. 2009;183:2349–2355. doi: 10.4049/jimmunol.0801683. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Tackenberg B., Jelcic I., Baerenwaldt A., Oertel W.H., Sommer N., Nimmerjahn F., Lünemann J.D. Impaired inhibitory Fcgamma receptor IIB expression on B cells in chronic inflammatory demyelinating polyneuropathy. Proc. Natl. Acad. Sci. USA. 2009;106:4788–4792. doi: 10.1073/pnas.0807319106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Quast I., Cueni F., Nimmerjahn F., Tackenberg B., Lünemann J.D. Deregulated Fcgamma receptor expression in patients with CIDP. Neurol. Neuroimmunol. Neuroinflamm. 2015;2:e148. doi: 10.1212/NXI.0000000000000148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Lee D.S.W., Rojas O.L., Gommerman J.L. B cell depletion therapies in autoimmune disease: advances and mechanistic insights. Nat. Rev. Drug Discov. 2021;20:179–199. doi: 10.1038/s41573-020-00092-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Zarrin A.A., Bao K., Lupardus P., Vucic D. Kinase inhibition in autoimmunity and inflammation. Nat. Rev. Drug Discov. 2021;20:39–63. doi: 10.1038/s41573-020-0082-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Ruck T., Nimmerjahn F., Wiendl H., Lünemann J.D. Next-generation antibody-based therapies in neurology. Brain. 2022;145:1229–1241. doi: 10.1093/brain/awab465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Zuercher A.W., Spirig R., Baz Morelli A., Rowe T., Käsermann F. Next-generation Fc receptor-targeting biologics for autoimmune diseases. Autoimmun. Rev. 2019;18:102366. doi: 10.1016/j.autrev.2019.102366. [DOI] [PubMed] [Google Scholar]
- 59.Reindl M., Schanda K., Woodhall M., Tea F., Ramanathan S., Sagen J., Fryer J.P., Mills J., Teegen B., Mindorf S., et al. International multicenter examination of MOG antibody assays. Neurol. Neuroimmunol. Neuroinflamm. 2020;7:e674. doi: 10.1212/NXI.0000000000000674. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Macrini C., Gerhards R., Winklmeier S., Bergmann L., Mader S., Spadaro M., Vural A., Smolle M., Hohlfeld R., Kümpfel T., et al. Features of MOG required for recognition by patients with MOG antibody-associated disorders. Brain. 2021;144:2375–2389. doi: 10.1093/brain/awab105. [DOI] [PubMed] [Google Scholar]
- 61.Boudreau C.M., Yu W.H., Suscovich T.J., Talbot H.K., Edwards K.M., Alter G. Selective induction of antibody effector functional responses using MF59-adjuvanted vaccination. J. Clin. Invest. 2020;130:662–672. doi: 10.1172/JCI129520. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Brown E.P., Licht A.F., Dugast A.S., Choi I., Bailey-Kellogg C., Alter G., Ackerman M.E. High-throughput, multiplexed IgG subclassing of antigen-specific antibodies from clinical samples. J. Immunol. Methods. 2012;386:117–123. doi: 10.1016/j.jim.2012.09.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Westerhuis J.A., van Velzen E.J.J., Hoefsloot H.C.J., Smilde A.K. Multivariate paired data analysis: multilevel PLSDA versus OPLSDA. Metabolomics. 2010;6:119–128. doi: 10.1007/s11306-009-0185-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
Data reported in this paper will be shared by the lead contact upon request.
All original data and code are available at: Mendeley database: https://doi.org/10.17632/dj43gnzjyr.1
https://github.com/LoosC/systemsseRology.
Any additional information required to reanalyze the data reported in this paper is available from the lead contact upon request.