

Summary
Predicting the clinical response to chemotherapeutic or targeted treatment in patients with locally advanced or metastatic lung cancer requires an accurate and affordable tool. Tumor organoids are a potential approach in precision medicine for predicting the clinical response to treatment. However, their clinical application in lung cancer has rarely been reported because of the difficulty in generating pure tumor organoids. In this study, we have generated 214 cancer organoids from 107 patients, of which 212 are lung cancer organoids (LCOs), primarily derived from malignant serous effusions. LCO-based drug sensitivity tests (LCO-DSTs) for chemotherapy and targeted therapy have been performed in a real-world study to predict the clinical response to the respective treatment. LCO-DSTs accurately predict the clinical response to treatment in this cohort of patients with advanced lung cancer. In conclusion, LCO-DST is a promising precision medicine tool in treating of advanced lung cancer.
Keywords: lung cancer, patient-derived organoid, drug sensitivity test, personalized medicine, real-world study, targeted therapy, chemotherapy
Graphical abstract
Highlights
-
•
Success rate of lung cancer organoids (LCOs) from malignant effusions exceeds 80%
-
•
LCOs faithfully reflect the sample’s pathologic and genomic features
-
•
LCO-based drug sensitivity tests accurately predict clinical response to treatment
-
•
LCO-based drug sensitivity tests represent temporal and spatial heterogeneity
Wang et al. generated lung cancer organoids (LCOs) derived from human malignant serous effusions and tissues. The LCOs accurately represented original tumor features. Drug sensitivity tests were performed to predict clinical response. The results illustrated enormous value of LCO as an in vitro platform for personalized medicine of lung cancer.
Introduction
Lung cancer is the second-most diagnosed cancer and the leading cause of cancer mortality, with over 600,000 deaths annually in China.1,2 Patients with advanced lung cancer who develop a malignant serous effusion (MSE) have a significantly worse prognosis, with an overall survival (OS) of 5.49 months, compared with 12.65 months for those without MSE.3 Over past decades, precision medicine, an approach that involves the formulation of regimens for treatment based on personalized factors and specific genetic targets, has been developed to improve therapeutic efficacy.4,5 Molecular-targeted therapy prolongs the OS of patients with advanced lung cancer and improves quality of life.6,7,8 Currently, the determination of personalized treatment regimens is strongly dependent on the results of molecular analytical approaches, particularly next-generation sequencing (NGS). NGS can detect tumor driver mutations that then allow the clinician to choose specific molecularly targeted drugs for cancer treatment.9 Molecular sequencing has historically relied on surgical and needle biopsy specimens. Studies have shown that the sensitivity and specificity of detecting epidermal growth factor receptor (EGFR) mutations in malignant pleural effusion (MPE) samples are equivalent to the measurements performed on tumor tissue samples (>80%).10 Further, MPE is a reliable sample for detecting kirsten rat sarcoma viral oncogene homolog (KRAS) mutations and anaplastic lymphoma kinase (ALK) rearrangements.11,12 It is also possible to detect any newly acquired genetic alterations using MSE.11,12
Lung cancer is a complex disease that exhibits phenotypic and genotypic diversity in different patients,13,14 presenting considerable challenges to the use of precision medicine. Non-small cell lung cancer (NSCLC) is the most extensively studied lung cancer subtype, although only 30% of patients harbor actionable mutations,15 and not all these patients can benefit from targeted therapy. Alternatively, some targeted anti-cancer agents have off-target effects as well.16 Patients without actionable mutations may benefit from targeted therapeutic drugs such as EGFR tyrosine kinase inhibitors (TKIs)17 or poly (ADP-ribose) polymerase (PARP) inhibitors.18 Therefore, it is imperative to establish a preclinical model that simulates the morphologic and genomic features of the original tissue to predict the clinical outcomes of targeted therapy.
There has been a considerable focus on establishing reliable preclinical models to evaluate a tumor’s response to chemotherapeutic or targeted agents. These models include cell lines,19 genetically engineered mouse models,20 organotypic tissue slice cultures,21 and patient-derived xenografts (PDXs).22,23 Although traditional tumor cell lines can be used to establish preclinical models, they are insufficient to represent complex tumors because of the lack of cell-to-cell interactions.24 Conversely, PDX can retain the structure and genetic characteristics of the original tumor and simulate a similar tumor microenvironment.25 However, PDXs, as well as genetically engineered models,26 are costly and time-consuming and require the sacrifice of a large number of experimental animals. Therefore, a preclinical model in which both maintenance and expansion are easily established is essential for translational medicine. Patient-derived organoids (PDOs) use three-dimensional (3D) structures that can accurately simulate the heterogeneity and diversity of tumors, show a high degree of genotypic and phenotypic consistency with the original clinical specimens, exhibit a response to antitumor drugs that mimics the patient’s expected response, and are available within a short period of time.27,28,29
In previous studies, organoids have been successfully cultured in a series of primary tumors, including colonic,30,31 uterine,32 ovarian,33 pancreatic,34,35 breast,36 and other solid tumors. Recently, organoids derived from primary lung tumors have been described.37,38,39 In the past, surgical specimens were used to establish lung cancer organoids (LCOs), but these were not easily available for advanced lung cancer. In addition to the establishment and validation of LCOs, studies focused on developing automated approaches, such as microarrays and microfluidic chips, to perform organoid-based drug tests in a short period of time.40,41 A recent study showed that the response outcomes of targeted therapy using lung adenocarcinoma (ADC) organoids were highly correlated with the real clinical response,42 revealing the potential of LCOs as a tool in personalized medicine.
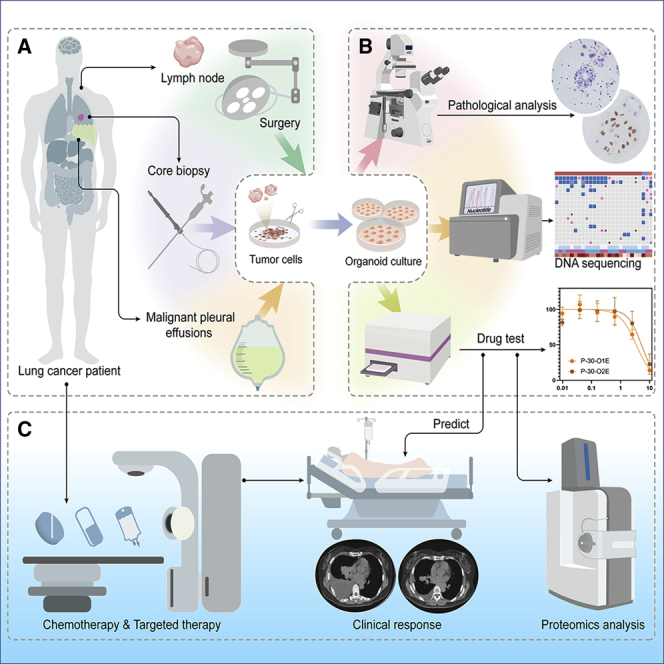
Success rates of LCO culture ranged from 7% to 87%, and MSE could be an ideal source.43 However, the accuracy of LCO-based drug sensitivity tests (LCO-DST) to predict clinical response remains unclear. In this study, we successfully generated 160 LCOs derived from tumor tissue specimens and MSE samples. We aimed to generate viable LCO models using tumor tissue or MSE samples, validate the reliability of the models with respect to their pathological and molecular features, and formulate personalized treatment strategies for advanced lung cancer using the results of the drug sensitivity tests (DSTs).
Results
Summary of the generation and pathological analysis of patient-derived lung cancer organoids
In our study, 214 samples for the generation of organoids were collected from 107 patients with advanced cancer between October 1, 2019, and September 30, 2021. LCOs were cultured using an existing method with slight modifications.44 We successfully established 162 PDOs (Figure 1A, Table S1) with a success rate of 75.7% (Figure S1). Following the exclusion of samples from two patients with breast cancer, 160 samples were finally included in the in-depth analysis. LCOs of different types were generated, including 127 ADCs, 10 squamous cell carcinomas (SCCs), 10 small cell lung carcinomas (SCLCs), 12 adenosquamous carcinomas (ASCs), and one pulmonary sarcomatous carcinoma. The LCOs in this cohort were mainly derived from MSE (132 of 162), including pleural fluid samples (103 of 132), ascitic fluid samples (15 of 132), and pericardial effusion samples (14 of 132). Further, 18 specimens were isolated from surgically resected biopsies of primary or metastatic leisions, which were mainly from lymph nodes. Ten specimens were obtained by core needle biopsy.
Figure 1.

Study flow chart and preservation of lung histopathology in LCOs
(A) Study workflow. See also Figure S1 and Tables S1–S3.
(B) Bright-field images of LCOs (left column), H&E staining of advanced lung cancer-derived LCOs (middle column), and the primary tumor malignant effusions (right column), including the pericardial effusion, pleural effusion (PE), and ascitic effusion (AE). Scale bars, 100 μm.
(C) IHC staining of the adenocarcinoma-derived LCOs as well as the original tissue with classic subtype markers TTF-1 (left column), CK7 (middle column), and Napsin A (right column). Scale bar, 50 μm.
(D) IHC staining of CK5/6, P40, and P63 in squamous cell carcinoma. Scale bar for the tissue, 50 μm. Scale bar for the LCO, 20 μm.
(E) IHC staining of TTF-1, Syn, CgA, and CD56 markers of small cell lung cancer. Scale bar, 50 μm.
We attempted to analyze the potenital causes of culture failure in 52 LCOs. Insufficient cells was one of the most common factors. Furthermore, univariate and multivariate analysis showed pathology and sampling type were independent influencing factors (Table S2). Lung adenocarcinoma and MSE are favorable for successful culture (Table S3).
Hematoxylin-eosin (H&E) staining and immunohistochemistry (IHC) were performed to validate the organoids and compare their morphology and pathology with the original tissue. IHC markers were selected from those that are routinely used for diagnosis of lung cancer subtypes. H&E staining (Figure 1B) and IHC (Figures 1C–1E) suggested that the LCOs retained the histopathologic characteristics of the original tumor tissue or MSE. For instance, ADC-derived LCOs were observed in clusters, with subtle cytologic features such as prominent nucleoli and cuboidal nuclear morphology, forming acinar structures and retaining the characteristics of primary ADC tissue (Figure 1B). Further, ADC-derived LCOs expressed classic ADC markers, including cytokeratin 7 (CK7), thyroid transcription factor 1 (TTF-1), and napsin A (Figure 1C).45 LCOs derived from SCC were obtained from surgically resected lung lesions (P-106) and showed strong expression of IHC indicators P40, P63, and CK5/6 (Figure 1D). The LCOs derived from SCLC showed small cell morphology and less periplasm, and expressed neuroendocrine markers such as CD56, synaptophysin (Syn), CgA, and TTF-1 (Figure 1E). These data indicate that LCOs can maintain the morphological and pathological features of the original tumor and mirror its individual characteristics.
The concordance for genomic profiling between MSE and MSE-derived LCOs
NGS was performed for 25 patients to test the concordance of genetic profiles between LCOs and the original samples. The clinical characteristics of the cohort are summarized in Table S4. Somatic mutational profiling of the effusion and LCO samples was performed. Alterations in the effusion and matched LCO samples are shown in Figure 2A. Collectively, 157 somatic alterations spanning 77 genes were identified in the effusion samples, including 100 single nucleotide variants (SNVs), 11 indels, 28 copy number variations (CNVs), four large genomic rearrangements (LGRs), and 14 fusions. Tumor protein 53 (TP53), EGFR, and retinoblastoma1 (RB1) were the most frequently mutated genes, occurring in 69% (n = 18), 69% (n = 18), and 19% (n = 5) of effusion samples (Figure S2A, Table S5). Moreover, 143 somatic mutations spanning 73 genes were identified in the LCO samples, including 93 SNVs, 11 indels, 26 CNVs, four LGRs, and nine fusions. EGFR, TP53, and RB1 were the most frequently mutated genes, occurring in 65% (n = 17), 58% (n = 15), and 15% (n = 4) of the LCO samples, respectively (Figure S2B). Among those alterations, 64 mutations (30.9%) were effusion-specific, 50 alterations (24.2%) were LCO-specific, and 93 mutations (44.9%) were shared between the two media. The alteration detection rates were comparable between the effusion and LCO samples (96.2% versus 80.7%, p = 0.19). No significant difference in the median maximum allele frequency (AF) between the effusion and LCO samples was observed (31.1% versus 34.0%, p = 0.98, Figure 2B). Next, the concordance of somatic alterations between MSE and LCOs was analyzed. Among the 20 effusion and matched LCO samples harbouring somatic alterations, 93 somatic alterations were detected in both effusion and LCO samples, 39 alterations were effusion-specific, and 41 alterations were LCO-specific. These data resulted in a by-variant sensitivity of 70.5% (93 of 132) with a positive predictive value of 69.4% (93 of 134) in LCO samples when the genomic profile of matched effusion samples was used as a reference (Figure 2C). Eleven matched LCOs and effusion samples had tumor mutation burden (TMB). A comparable TMB was observed between the two media (1.99 mutations/Mb for both, p = 0.89, Figure 2D). Further, LCO-based TMB was positively correlated with effusion-based TMB (Pearson’s r = 0.68, p = 0.02, Figure 2E). Cumulatively, these findings revealed acceptable concordance of genomic profiling between the effusion and LCO samples.
Figure 2.

Genomic profiling of LCO samples
(A) The overlap of somatic alteration in effusion and LCO samples. Blue indicates alterations detected from both sources, pink indicates alterations that were present only in the effusion samples, and scarlet indicates alterations were present only in the LCO samples. See also Figures S2A and S2B, and Tables S1 and S2.
(B) The difference of maxAF between effusion and LCO samples.
(C) The concordance for somatic alterations between effusion and LCO samples.
(D) The difference of TMB between effusion and LCO samples. p values are determined using the two-tailed t-test or Mann–Whitney U test for continuous variables.
(E) Correlation between effusion-based and LCO-based TMB (Pearson correlation coefficient, two-tailed). ADC, adenocarcinoma; ASC, adenosquamous carcinoma; CN, copy number; F, female; Indel, small insertion and deletion; LCO, lung cancer organoid; LGR, large genomic rearrangements; M, male; maxAF, maximum allele frequency; MSE, malignant serous effusion; SCC, squamous cell carcinoma; TMB, tumor mutational burden.
LCOs predict a personalized response to targeted therapy
In most patients, therapeutic regimens were personalized based on factors such as genetic features and previous treatments. Moreover, in vitro drug screening strategies were also tailored to individual patients. Fifty-four LCOs derived from 36 patients whose clinical regimens were identical to LCO-DST results were divided into four categories: osimertinib, chemotherapy, dual-targeted therapy, and other targeted therapy groups. The capability of LCO-DST to discriminate between clinically sensitive or resistant patients was determined using receiver operating characteristic (ROC) analysis. The predictor was LCO sensitivity to osimertinib, chemotherapy, dual targeted therapy, and other targeted therapy treatment. The concordances of response were 86.7% (13 of 15), 83.3% (10 of 12), 100% (10 of 10), and 70.6% (12 of 17), respectively (Table S6), with overall 84.0% (95% CI, 63.08%–94.75%) sensitivity, 82.8% (95% CI, 63.51%–93.47%) specificity, and 83.3% accuracy for LCO-DST (Figures 3A, 3B, S3A, and S3B). Drug response heatmap of six LCOs from four patients suggested personalized differences in patient responses to anti-cancer agents and a relatively high consistency in responses between different LCOs derived from the same patient. Generally, the half maximal inhibitory concentration (IC50) values of chemotherapy agents were higher than those of targeted therapeutic agents (Figure S3C).
Figure 3.

Comparison of LCO-based drug screening and clinical response
See also Figure S3 and Table S6.
(A) Flowchart of LCO-DST and clinical follow-ups in this study.
(B) The overall correlation between LCO-DST sensitivity and clinical response.
(C) Swimming graph of the progression-free survival (PFS) of patients who received osimertinib treatment.
(D) Dose-effect curves of LCO based on in vitro sensitivity of osimertinib.
(E) Violin plot of the IC50 values of osimertinib for clinical PR and PD groups.
(F) Receiver operating characteristic (ROC) analysis of osimertinib LCO drug tests showed an area under the curve (AUC) of 0.94 with a p value of 0.0047. p values are determined from the normal distribution (two-tail) for the comparison to a chance-level ROC curve (AUC = 0.5).
(G) Computed tomography (CT) scan of P-41 at the baseline, PR, and confirmed PR stages.
(H) CT scan of P-59 at the baseline, SD, and PD stages.
(I) Dose-effect curve of loratinib and SAF-189s for LCOs derived from P-63.
(J) CT scan of the brain and thoracic cavity of P-63 at baseline and PD stages.
(K) Dose-effect curves of the EP regimen (etoposide and paclitaxel) for LCOs derived from P-3, P-50, P-65, P-83.
∗: p <0.05.
In the osimertinib cohort, osimertinib was selected as the next-line treatment for 12 patients (Figure 3C), and the sensitivity of osimertinib was tested on 15 LCOs generated from these patients (Figure 3D). Generally, the IC50 of the progression disease (PD) and partial response (PR) groups could be distinguished significantly, with an area under the curve (AUC) value of 0.94 in ROC analysis (Figures 3E and 3F). The clinical response of most patients was accurately predicted using the LCO-DST. For example, P-41 was diagnosed with stage IVA ADC harboring EGFR 19del. First-line treatment was icotinib. After PD, the LCO-DST suggested the potential beneficial effect of osimertinib (IC50 = 0.10 μM). In the real world, osimertinib was used in the following treatment of this patient, and achieved sustained PR (Figure 3G). Further, the LCO-DST could also predict resistance to targeted therapy agents. For instance, P-59, with stage IVB ADC harboring EGFR L858R, progressed after treatment with erlotinib, icotinib, and gefitinib. EGFR T790M was found and osimertinib was chosen as the next-line treatment according to clinical guidelines. However, LCO-DST indicated resistance to osimertinib (IC50 = 4.37 μM) in vitro. Unfortunately, the disease progressed rapidly in only 4.5 months (Figure 3H). Furthermore, these results showed that the reduction in tumor volume was correlated with IC50 values (Figure S3D), suggesting that this approach may also predict the outcome after anti-cancer treatment.
Four patients with ALK fusion were enrolled in LCO-DST analysis, including one untreated patient and three treated patients. All LCO-DST results were consistent with the clinical response (100%, 5 of 5). For example, P-63 was diagnosed with stage IVA ADC harboring echinoderm microtubule-associated protein-like 4 (EML4)-ALK fusion. Second-generation ALK-TKIs, ceritinib and SAF-189s, were sequentially indicated and achieved PFS of 33.7 and 22.8 months. LCO-DST performed using MPE samples after the progress of second-line treatment predicted resistance to alectinib. SAF-189s was also ineffective in vitro (Figures 3I and 3J). Nevertheless, the LCO-DST were not considered when deciding the next-line treatment; alectinib was still decided as the third-line treatment. Intracranial metastases increased significantly, which led to disease progression. Another patient with ALK fusion (P-27) was previously resistant to alectinib and showed insensitivity to alectinib in the LCO-DST (Figure S3E).
Cases with other mutations, such as ROS1 (P-25), HER2 (P-75), MET exon 14 skipping (P-86 and P-105), and BRAF (P-95), were also enrolled in LCO-DST analysis (Table S6) and provided accurate predictions of the clinical response.
These results illustrate that LCOs may predict the clinical response to targeted therapy, both in untreated and treated patients. Although a limited number of regimens for each drug test may not accurately indicate which treatments are recommended, it is still critical for these patients to avoid ineffective therapies because of unnecessary side effects, time consumption, and resource expenditure.
LCOs predict a personalized response to chemotherapy
A correlation between the LCO drug screening test results with the real clinical response to chemotherapy has been previously reported.40 Here, we reported the data of nine patients in whom chemotherapy was recommended as the next-line course of action.
Of these, four patients had ADC and underwent treatment with TDM1(P-85), nab-paclitaxel (P-81), a PC regimen (pemetrexed and carboplatin) (P-25), and an EP regimen (etoposide and cisplatin) (P-3). Three out of four patients with SCLC underwent EP treatment (P-50, P-65, and P-83), and P-73 was treated with nab-paclitaxel. Among patients who underwent EP treatment, P-3 was diagnosed as ADC while others were diagnosed as SCLC. They were treated with the same regimen and had different outcomes. P-3 and P-65 both achieved PR while PD was observed in P-50 and P-83. The actual clinical response was predicted by the LCO-DSTs, where EP treatment inhibited both the LCOs derived from P-3 and P-65 but failed to inhibit the LCO derived from P-50 and P-83 (Figure 3K). These results suggested that in vitro testing of LCOs with chemotherapeutic agents can reflect the clinical response to chemotherapy.
LCOs derived from multiple samples reveal stability and heterogeneity
Apart from the success rate and the degree of purity of the models, another challenge in the use of tumor organoids for drug screening is experimental stability. Inter- and intra-tumor heterogeneity has been widely considered. In this study, we collected different samples from the same patient to explore the stability and heterogeneity of the LCO drug response. First, MSE samples were extracted from the patient at different times, the LCOs were established, and DSTs were performed. The cell count generally decreased with sampling time, but the establishment of the LCO models was not affected (Figure S4A). The genomic profiles of LCO samples with different culture durations were analyzed (Figure S4B). The time intervals between different sampling times ranged from 1 to 9 days. P-51 had three LCO samples collected on days 1, 2, and 3. P-78 and P-100 each had two LCO samples collected on days 1 and 2. P-101 had two LCO samples collected on days 4 and 9. For P-51, the consistency of somatic alterations among the three LCOs was 81.25% (13 of 16), with MYC amplification and WT1 amplification being identified only in the LCO collected on day 1, while IL7R amplification was not identified in the LCO collected on day 3. Moreover, the same alterations were observed in the LCO samples obtained from the remaining three patients (P-78, P-100, P-101). Collectively, these findings indicated a high concordance for genomic profiling among LCO samples obtained at different sampling times, suggesting that the LCO may be an optimized stable in vitro model of NSCLC (Figure 4A).
Figure 4.

Stability and heterogeneity of LCOs
(A) Concordance for somatic alterations detected in MSE and LCO samples at different sampling times in four patients. See also Figure S4. ADC, adenocarcinoma; CN, copy number; D, day; F, female; Indel, small insertion and deletion; LCO, lung cancer organoid; LGR, large genomic rearrangements; M, male; MSE, malignant serous effusion; P, patient; SCC, squamous cell carcinoma.
(B) The concordance of SNVs between MSE and LCO of P-51.
(C) The concordance of SNVs between MSE and LCO of P-87.
(D) The concordance of SNVs between MSE and LCO of P-96.
(E) The concordance of SNVs between MSE and LCO of P-60.
(F) The concordance of SNVs between MSE and LCO of P-100.
(G) CT scan of P-51 presents the location of the pleural effusion (PE) and bright-field images of the PE-derived LCOs on days 1, 2, and 3.
(H) Dose-response curve of Nab-PTX plus carboplatin and GDC-0941 for LCOs of P-51.
(I) CT scans of P-51 at the baseline and after Nab-PTX + carboplatin treatment.
(J) CT scan of P-33 shows the location of the PE and lymph node (LN) and bright-field images of the PE- and LN-derived LCOs.
(K) Dose-response curve of crizotinib, alectinib, and loratinib for P-33 LCOs.
(L) CT scan of P-33 at the baseline and after alectinib treatment.
(M) CT scan of P-30 shows the location of the ascitic effusion (AE) and PE, as well as the bright-field images of the AE- and PE-derived LCOs.
(N) Dose-response curve of osimertinib and osimertinib + cabozantinib for P-30 LCOs.
Next, we investigated the concordance for genomic profiling of tissue samples, effusion samples, and LCOs obtained from five patients. In P-51, 13 SNVs were detected in the tumor tissue samples, and 12 SNVs were detected in both MSE and LCOs; however, only two out of 12 CNVs were detected in both MSE and LCOs (Figure 4B). High concordance for SNVs between tissue and effusion/LCO samples was also observed in P-87 and P-96 (Figures 4C and 4D). Low concordance for CNVs between tissue and effusion/LCO samples was observed in P-60 and P-100 (Figures 4E and 4F). These findings suggest that SNVs, rather than CNVs, may be accurately detected in effusion/LCO samples.
In terms of the in vitro LCO drug response, stability was maintained across the continuous samples. P-51, with advanced SCC, had a large volume of MPE that was collected over 3 consecutive days. Cell density from the MPE decreased over time, and the number of organoid formations was also affected, although the cell density in the LCO cultures remained the same (Figure 4G). The LCO-DST yielded stable results for different samples. Three LCOs were minimally inhibited by the combination of nab-paclitaxel, which had also been proven to be clinically ineffective, while a phosphoinositide 3-kinase (PI3K) inhibitor, GDC-0941, showed a stronger inhibitory effect (Figures 4H and 4I). The LCOs derived from MSE samples seemed to exhibit large differences from tissue-derived LCOs, such as the LCOs derived from the MPE of P-33, which were loose and irregular. The LCOs derived from the lymph nodes of the same patient showed tighter and more regular shapes (Figure 4J). Lymph node-derived LCOs were also more sensitive to DST (Figure 4K), and the patient who underwent treatment with alectinib achieved a PR on clinical evaluation, consistent with the drug sensitivity test result (Figure 4L). In the case of different MSE samples, the LCO-DST often showed the same trend with respect to the dose-effect relationship. For instance, two samples were collected from MPE and ascitic effusion, and the morphological features of LCOs were slightly different: the MPE-derived LCOs showed vacuolated and solid appearances, while the ascitic effusion-derived LCOs were solid (Figure 4M). The DST of ascitic effusion-derived LCOs were slightly more sensitive than those of the MPE-derived LCOs, although the difference between the sensitivities was not significant (Figure 4N).
LCO drug screening represents the clinical response of dual-targeted therapy
Resistance to a single anti-cancer agent is common in patients with advanced lung cancer. It would be helpful if LCO-DST could predict the effect of combinational therapy in vitro. Several cases in our study presented with progressive lung disease. LCOs were obtained, and combination therapy regimens were tested. P-60 was diagnosed as Stage IVB lung ADC (Figures 5A and S5A) with EGFR L858R mutation and MET copy number gain (5.0) detected in cerebrospinal fluid at baseline. This patient experienced worsened dizziness and vomiting with enlarged brain metastases after taking osimertinib; it seemed that osimertinib may not the optimal choice for him. One LCO (P-60-O1T) derived from lymph node and three (P-60-O2E, P-60-O3E, and P-60-O4E) derived from consecutive pericardial effusion (day 1, 4, 5) were successfully cultured and DST were performed (Figure 5B). IC50 of osimertinib combined with savolitinib were 1.32, 0.71, 0.25, and 0.24 in these four LCOs, respectively. Compared with IC50 of osimertinib (1.82, 1.15, 0.25, and 1.30, respectively) and savolitinib (only performed in P-60-O3E and P-60-O4E, which were 11.59 and 17.61), the result revealed that osimertinib plus savolitinib may be superior to osimertinib or savolitinib monotherapy for this patient. Interestingly, the patient was given with osimertinib plus savolitinib since MET amplification were further confirmed in MSE and LCO via fluorescence in situ hybridization (FISH), immunofluorescence staining, and IHC (Figures 5C, 5D, and S5B), and achieved confirmed PR (Figure 5E). P-61 was diagnosed as stage IVA lung ADC with EGFR 19del at baseline (Figure 5F). The patient underwent first-line treatment with icotinib and second-line treatment with osimertinib because of an acquired T790M mutation. After the development of drug resistance, the patient underwent dual-targeted therapy, including osimertinib and cabozantinib. Tumor evaluation revealed PR after 45 days of dual-drug therapy (Figure 5G). NGS confirmed the presence of rearranged during transfection (RET)-CCDC6 fusion (Figure 5H) besides EGFR 19del and T790M. DST of LCOs derived from pleural effusion (P-61-O1E, P-61-O2E, P-61-O3E) and ascitic effusion (P-61-O4E, P-61-O5E) during the same period showed that IC50 of osimertinib combined with cabozantinib were 0.15, 0.22, 0.16, 0.33, and 0.44, respectively, while IC50 of osimertinib combined with BLU667 were 0.03, 0.02, 0.01, 0.03, and 0.24, which indicated that osimertinib combined with cabozantinib/BLU-667 significantly inhibited tumor growth (Figure 5I). These cases suggested that the LCO-DST has the potential to predict effective combination treatment regimens.
Figure 5.

LCOs predict dual-targeted therapy for drug-resistant cases
See also Figure S5.
(A) Disease progression of P-60.
(B) Dose-effect curve of LCOs derived from the lymph node (LN) and pericardial effusion of P-60.
(C) FISH of MET amplification of the LN, pericardial effusion, and LCOs derived from pericardial effusion and the LN.
(D) Immunofluorescence staining of MET and DAPI, and immunohistochemistry staining of EGFR L858R for LCOs derived from pericardial effusion and lymph nodes of P-60. Scale bar, 20 μm.
(E) CT scans indicating the progression of the disease of P-60.
(F) Disease progression of P-61.
(G) CT scans of P-61.
(H) FISH of RET rearrangement in pleural effusion (PE) and ascitic effusion (AE)-derived organoids of P-61.
(I) Dose-effect curve of LCOs derived from the PE and AE of P-61.
Proteomics landscape of LCOs reveals the molecular mechanisms of dual-targeted therapy
Although the effect of combination treatment with EGFR and RET inhibitors has been demonstrated in LCO drug screening assays, the underlying molecular mechanisms remain unclear. Therefore, 4D label-free high-throughput proteomic analysis was performed. We identified 39,312 unique peptides in the 287,153 spectra. A total of 4,865 proteins were identified and, of these, 4,833 were quantified. The quantified proteins were divided into four groups (4818, 4787, 4785, and 4833), in which organoids were treated with osimertinib, BLU-667 combination treatment with osimertinib, BLU-667 (combo group), and 0.1% dimethyl sulfoxide, respectively. The results of the principal-component analysis (PCA) indicated that the expression levels of the different treatment groups were different, suggesting diverse effects (Figure 6A). In terms of the differentially expressed proteins (DEPs), 119 proteins were significantly altered in the combo regimen compared with the control group (fold change >1.2, p < 0.05), including 26 upregulated proteins and 93 downregulated proteins (Figure 6B). However, when treated with a single agent, osimertinib or BLU-667, the number of DEPs was 154 (Figure 6C) and 285 (Figure 6D). In terms of the cellular location of the DEPs, all three treatment groups showed the same trend of protein expression, of which the number of nuclear proteins was the highest in the DEPs, followed by that of cytoplasmic and cytomembrane proteins. These results suggest that the alteration of protein expression with all three treatments aimed to regulate nuclear functions, including gene expression.
Figure 6.

Proteomic profiles of pleural fluid-derived organoids indicated drug resistance mechanisms
(A) PCA plot of the protein expression levels of different groups. The control group is shown in blue. The combination group, in which the LCOs were treated with both osimertinib and BLU-667, is shown in green. The groups treated with osimertinib (Osi) and BLU-667 alone are shown in purple and yellow, respectively.
(B) Volcano plots of differentially expressed proteins (DEPs) are shown for the osimertinib versus control group.
(C) Volcano plot of DEPs is shown for the BLU-667 versus control group.
(D) Volcano plot of DEPs is shown for the combo versus control group.
(E) Expression levels of the caspase family in osimertinib versus control (orange), BLU-667 versus control (blue), and combo versus control (light purple).
(F) An overview of the alteration of the signaling cascade of EGFR and RET; arrows indicate the trend of the expression of related proteins.
(G) Changes in the expression levels of key proteins among the osimertinib versus control, BLU-667 versus control, and combo versus control groups. See also Figure S6.
In terms of the function of DEPs, alterations in several protein families are closely related to the survival of tumor cells. The combination of osimertinib and BLU-667 resulted in a dramatic increase in the expression of caspase 3, the critical factor triggering cell apoptosis, while the expression of other caspase family members, such as caspase 8 and caspase 10, also increased after treatment with the combination regimen (Figure 6E). RET rearrangement is a critical factor in EGFR-TKI resistance because of the common downstream cascade. RET TKIs were also combined with the third-generation EGFR-TKI osimertinib as a treatment regimen for patients with EGFR-RET duo mutation. However, the underlying molecular mechanisms remain unclear. Proteomic analysis by 4D liquid chromatography-tandem mass spectrometry (LC-MS/MS) showed that the expression of the downstream proteins of EGFR and RET were downregulated, such as RAC, PI3K, and MEK (Figure 6F), suggesting their role in tumor cell survival. Another key pathway altered in the treatment was the Hippo pathway, which is critical for tumor growth and survival. Both MOB1 and MST1/2 levels were significantly decreased by either separate or combined treatment with BLU-667 and osimertinib. The level of YAP, a key transcription factor for survival-related genes, decreased after treatment with the combination regimen of BLU-667 and osimertinib (Figure 6G). These results suggested that the combination treatment significantly triggered cell death compared with treatment with single reagents (Figures S6A–S6F).
Discussion
There are limitations in formulating treatment strategies based on the results of genetic profile and other biomarkers.46,47 In many cases, particularly with primary drug-resistant patients, the molecular mechanisms of the response to therapy, or lack thereof, are unclear. Therefore, a precise approach to accurately predict the clinical response is imperative in the use of precision medicine for the treatment of lung cancer. Previous studies have indicated that MSE-derived organoids and other in vitro models are potential materials for predicting the clinical efficacy of treatment.48,49 Our results suggest that the LCO models provide a personalized platform for the application of both targeted therapy and chemotherapy for the treatment of patients with advanced lung cancer. A total of 162 organoids, most of which were derived from MSE samples, were established in our cohort. The success rate of tissue-derived and MSE-derived organoids was 57.8% (30 of 52) and 81.4% (132 of 162), implying that MSE could provide an opportunity to bridge this gap in the success rate. Tumor cell count is often insufficient in organoid cultures obtained from core needle biopsies and lymph node resections. Furthermore, one limitation of models obtained from surgical and biopsy samples is the overgrowth of normal airway epithelial cells.50 However, an MSE consists primarily of tumor cells rather than non-malignant epithelial or mesothelial cells and can be obtained by a relatively minimally invasive manner.51 Therefore, LCOs derived from MSE tend to be purer tumor organoids, allowing these models to be excellent candidates for DST. LCOs derived from either tissue or MSE samples faithfully reflected the pathological and molecular characteristics of the original tumor, and these results provided a reliable basis for the subsequent DST. The LCO-DST results are highly consistent with the clinical response to therapy, and the method is promising as a significant prediction tool for use in personalized medicine to build individualized treatment regimens for lung cancer.
Whether LCOs derived from MSEs can represent the entire tumor profile is a critical challenge in the application of this method. Previous studies have shown that the accordance of genetics profile between MSE liquid biopsy and tissue biopsy was higher than 80%.52 Thus, in some cases, MSEs may replace tissue biopsy in diagnostic occasions. However, in this study, the LCO-DST results of P-17 suggesting sensitive to osimertinib failed to predict the over clinical response (PR in the lung, while brain metastases PD), and the mixed response to osimertinib may be a confounding factor. The results of several patients revealed that metastatic lesion biopsy derived LCOs can predict the clinical response of local lesions to therapeutic agents, but sometimes cannot give an overall prediction because of heterogeneity.
In the context of modern precision medicine, NGS presents good analytical validity to detect clonally dominant alterations.53 It has recently been applied in clinics with the aim of identifying targetable alterations. However, the emergence of drug resistance is the greatest challenge in targeted therapy.54 In this context, it is important to identify a preclinical model suitable for new drug research in lung cancer. For instance, patients P-59 and P-70, who both exhibited EGFR-TKI-resistant disease, did not show any resistant mutations. Thus, the DST results are critical for the selection of a more effective treatment regimen.
Continuous sampling was performed for some patients in this cohort. Genetic features and drug responses in MSE and LCOs exhibited high concordance. These results indicated that the genetic profile and LCO-DST provided a stable readout, which was highly correlated with clinical efficacy. However, fewer living cells could be collected from the samples obtained during the later dates, which may have affected the success of LCO cultures. Therefore, collecting effusions during the first few days is recommended. Moreover, because of heterogeneity, the drug test assays may show different results from LCOs derived from samples collected at different dates from the same patient. Another finding of this study is that previous treatments cannot be accurately detected by LCOs, although this may not affect the real-world application of LCO-DST, which is to detect the sensitivity of unused drugs, as in this study.
As a functional model that can include the multi-omics characteristics of the original tumor and simulate drug sensitivity in patients, tumor organoids can also be applied in the development of novel drugs as an in vitro tumor replacement.55 In this study, proteomics analysis was applied to analyze the molecular landscape of combinational therapies and present the molecular details that explain the clinical efficacy of combination treatment. Tumor organoids have several unique advantages: they are representative of the morphological and molecular characteristics of the original tumor and can partially simulate the physiological and drug sensitivity characteristics of the original tumor.4,38,56,57,58,59,60 Further, they exhibit rapid proliferation, a short culture cycle, and a high success rate of culture when drug sensitivity tests are performed. As mentioned above, the results of LCO-DST can guide clinical treatment regimens.
In the clinical use of anti-cancer agents, appropriate drugs should be selected according to the specific characteristics of each patient, such as the genetic mutations involved and other factors. The formulation of the drug sensitivity scheme followed the principles of clinical drug use to maximize the results of LCO drug sensitivity to guide the subsequent clinical diagnosis and treatment of patients. Our studies showed that the concordance for overall consistency of drug sensitivity with the clinical response reached 83.33%. A certain consistency exists between DST and clinical efficacy, which was validated in our cohort. For instance, the results of LCO-DST of a patient reflected the fact that the dual-targeted drugs showed high tumor control rates, which was later confirmed during the clinical treatment. In conclusion, our study gave an overview of the applications of LCO-DST to predict therapeutic response in advanced lung cancer and found the LCO-DST a potential in precision medicine, further research can focus on the standardization of the approaches, such as large-scale clinical trials, to define the precise cutoff values or other perimeters in different types of therapeutic agents.
Limitations of the study
The major limitation of this study was the enrolled cohort. As a real-world study, it did not have a designed enrollment plan, and the genomic background and treatment regimen of the patients in our cohort varied. Small number of samples included for drug sensitivity analyses and inconsistent drug regimens in three groups except for osimertinib group also account for our study’s limitations in many other regimens, the cutoff values of IC50 of the effective and resistant groups were not yet defined. Moreover, the drug screening panel differed among the patients. Furthermore, many similar studies have focused on the long-term culture of LCOs and the establishment of living biobanks. In this study, to evaluate LCO-DST as a translational medicine approach, most organoids in our cohort were cultured and tested within a short period of time; therefore, not all organoids underwent genomic and pathological analyses. For similar reasons, not all organoids could be used in intensive investigation such as proteomics analysis and further biochemical analysis. In terms of proteomics, a further limitation is that all LCOs were used for LC-MS/MS tests, and there were insufficient samples for validation of the DEPs such as those in the Hippo pathway. As EGFR mutation combined with RET amplification is a rare genotype, no patient could be enrolled for validation. Owing to these limitations, this study cannot be classified as a “clinical trial.” Nonetheless, this study reflects the real application of organoid technology in precision therapy for various subtypes of lung cancer and lays a foundation for further clinical trials.
STAR★Methods
Key resources table
| REAGENT or SOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| Rabbit anti TTF-1 | Abcam | Cat# ab133638;RRID: AB_2734144 |
| Rabbit anti Napsin A | Abcam | Cat# ab129189; RRID:AB_11143020 |
| Rabbit anti CK-7 | Abcam | Cat# ab181598;RRID: AB_2783822 |
| Rabbit anti P63 | Abcam | Cat# ab124762; RRID: AB_10971840 |
| Rabbit anti P40 | Abcam | Cat# ab273135; RRID: AB_2927663 |
| Rattbit anti CK5/6 | Abcam | Cat# ab52635;RRID: AB_869890 |
| Rattbit anti CD56 | Abcam | Cat# ab220360; RRID: AB_2927664 |
| Rattbit anti Synaptophysin | Abcam | Cat# ab32127; RRID: AB_2286949 |
| Rabbit anti Chromogranin | Abcam | Cat# ab254322; RRID: AB_2910555 |
| Rabbit anti EGF Receptor (L858R Mutant Specific) (43B2) | ewEast | Cat# 26082; RRID: AB_2629297 |
| Goat Anti-Rabbit IgG H&L (HRP) | Abcam | Cat# ab205718;RRID: AB_2819160 |
| Goat Anti-Mouse IgG H&L (HRP) | Abcam | Cat# ab205719;RRID: AB_2755049 |
| Goat Anti-Rabbit IgG H&L (Alexa Fluor® 488) | Abcam | Cat# ab150077;RRID: AB_2630356 |
| Biological samples | ||
| Human tumor tissue samples | This study | N/A |
| Human effusion samples | This study | N/A |
| Chemicals, peptides, and recombinant proteins | ||
| B27 supplement (-Vit A) | GIBCO | Cat# 17504044 |
| N2 supplement | GIBCO | Cat# 17502048 |
| GlutaMax | GIBCO (ThermoFisher) | Cat# 35050061 |
| Wnt3A | Novoprotein | Cat# C06D |
| R-Spondin1 | Novoprotein | Cat# CX83 |
| Noggin | Novoprotein | Cat# CB89 |
| EGF | Peprotech | Cat# AF-100-15 |
| FGF-basic | Peprotech | Cat# 100-18C |
| Y27632 | MCE | Cat# HY-10071 |
| A83-01 | Sigma-Aldrich | Cat# SML0788 |
| DMEM/F12 | GIBCO | Cat# 12634028 |
| Green Chromogen | Leica | Cat# DC9913 |
| BOND IHC polymer detection kit | Leica | Cat# DS9800 |
| FBS | GIBCO | Cat# 10099141 |
| PBS | Sangon | Cat# E607008-0500 |
| HEPES | Sigma-Aldrich | Cat# H6147 |
| HBSS | GIBCO | Cat# 14175095 |
| Penicillin-Streptomycin | Sigma-Aldrich | Cat# V900929 |
| Amphotericin B | GIBCO | Cat# 15290026 |
| Cell recovery solution | Corning | Cat# 354253 |
| Matrigel | Corning | Cat# 356231 |
| DNase | Sigma-Aldrich | Cat# 11284932001 |
| Collagenase/dispase | Roche | Cat# 10269638001 |
| BOND IHC polymer detection kit | Leica | Cat# DS9800 |
| Afatinib | MCE | Cat# HY-10261 |
| Osimertinib | MCE | Cat# HY-15772 |
| Erlotinib | MCE | Cat# HY-50896 |
| Gefitinib | MCE | Cat# HY-50895 |
| Dacomitinib | MCE | Cat# HY-13272 |
| Almonertinib | MCE | Cat# HY-112823 |
| Icotinib | MCE | Cat# HY-15164A |
| SAF-189s | Fochon Pharmaceuticals | N/A |
| Crizotinib | MCE | Cat# HY-50878 |
| Alectinib | MCE | Cat# HY-13011 |
| Ceritinib | MCE | Cat# HY-15656 |
| Brigatinib | MCE | Cat# HY-12857 |
| Lorlatinib | MCE | Cat# HY-12215 |
| Entrectinib | MCE | Cat# HY-12678 |
| Dabrafenib | MCE | Cat# HY-14660 |
| Pyrotinib | MCE | Cat# HY-104065 |
| Larotrectinib | MCE | Cat# HY-12866 |
| Savolitinib | MCE | Cat# HY-15959 |
| Bozitinib | MCE | Cat# HY-125017 |
| BLU667 | MCE | Cat# HY-112301 |
| Docetaxel | MCE | Cat# HY-B0011 |
| Paclitaxel | MCE | Cat# HY-B0015 |
| Gemcitabine | MCE | Cat# HY-17026 |
| Etoposide | MCE | Cat# HY-13629 |
| Pemetrexed | MCE | Cat# HY-10820 |
| Cisplatin | MCE | Cat# HY-17394 |
| Carboplatin | MCE | Cat# HY-17393 |
| Irinotecan | MCE | Cat# HY-16562 |
| Experimental models:Organisms/Strains | ||
| Human lung cancer organoid samples | This study | N/A |
| Software and algorithms | ||
| R | R Development Core Team, 2008 | https://www.r-project.org/ |
| Critical commercial assays | ||
| CellTiter-Glo® 3D Cell Viability Assay | Promega | Cat# G9681 |
| Deposited data | ||
| DNA sequencing | This paper | National Genomics Data Center (HRA003231) |
| LC-MS/MS | This paper | ProteomeXchange Consortium (PXD037076) |
| Others | ||
| Cytation 5 Cell Imaging Multimode Reader | Agilent | Cat# BTCYT5M |
| Fluorescence microscope | Olympus | Cat# 29560 |
| BOND-III automated stainer | Leica | Cat# 22.2201 |
Resource availability
Lead contact
Further information and requests for resources and reagents should be directed to and will be fulfilled by the lead contact, Jin-Ji Yang (yangjinji@gdph.org.cn).
Materials availability
This study did not generate new unique reagents.
Experimental model and subject details
Primary or metastatic tumor tissues were obtained from patients with advanced lung cancer by performing fine-needle aspiration or surgically resected biopsies at Guangdong Provincial People’s Hospital. Malignant effusion samples were also collected. The collection of patient data and biospecimens for lung cancer organoid (LCO) culture, drug tests, and further studies has been performed according to the guidelines of Research Ethics Committee of Guangdong Provincial People’s Hospital, Guangdong Academy of Medical Sciences (Ethics project number: No.GDREC2019323H), following both national and local laws. Written informed consent was also obtained from all patients. The main inclusion criteria were patients with clinically locally advanced or metastatic lung cancer, aged 18 years or older, with fresh tissues available through either biopsy or surgical resection of the primary or metastatic lesions, and with malignant effusion samples collected using a sterile drainage bag. Clinical information is available in Table S1. Drug sensitivity tests were performed after the generation of sufficient LCOs. However, only patients with concurrent or subsequent clinical drug response evaluations, which were consistent with the LCO-based drug test results, were enrolled in the observational clinical research.
Method details
Patient sample collection and processing
The primary inclusion criteria were patients with clinically locally advanced or metastatic lung cancer, aged 18 years or older, with fresh tissues available from either biopsy or surgical resection of the primary or metastatic lesions, and with malignant effusion samples collected using a sterile drainage bag. Drug sensitivity tests were performed after generating sufficient LCOs. However, only patients with concurrent or subsequent clinical drug response evaluations, consistent with the LCO-based drug test results, were enrolled in the observational clinical research.
Primary or metastatic tumor tissues were obtained from patients with advanced lung cancer by performing fine-needle aspiration or surgically resected biopsies at Guangdong Provincial People’s Hospital. Malignant effusion samples were also collected. Written informed consent was obtained from all patients. The research protocol was approved by the Research Ethics Committee of the Guangdong Provincial People’s Hospital, Guangdong Academy of Medical Sciences (Guangzhou, China). The samples were diagnosed based on pathological assessment.
LCO cultures
Patient-derived LCO cultures were obtained as previously described.37 MSE samples (200–1000 mL) were obtained by thoracentesis and collected aseptically in heparinized (10 U/mL) sterile bottles or bags. These were subsequently transferred to the laboratory on ice for further processing within 4 h. Samples were centrifuged at 300 ×g for 5 min at 4°C, and the cell pellets were resuspended in LCO medium (LCOM, DMEM/F12, supplied with 20 ng/mL bFGF, 50 ng/mL human EGF, N2, B27, GlutaMAX, 10 μM Y-27632, and 1% penicillin-streptomycin, Gibco, New York, NY, USA). For the tumor tissue samples, tumor fragments were generated using sterilized ophthalmic scissors, sized approximately 1 mm3. These were suspended in cold Hank’s Balanced Salt Solution (HBSS) with antibiotics and transported to the laboratory on ice within 1 h of resection. After washing thrice with cold HBSS containing antibiotics and sectioning with sterile blades, the samples were incubated in DMEM/F12 supplemented with 0.001% DNase, 1 mg/mL collagenase/dispase, penicillin-streptomycin, and 0.5 mg/mL amphotericin B at 37°C for 2 h with gentle agitation and intermittent resuspension. The digested tissue suspension was then titrated via pipetting and passed through a 70 μm filter.
The cell suspension generated from either MSE or tissue samples was centrifuged at 112 rpm for 3 min. The pellet was then resuspended in LCOM. Thereafter, 200 μL Matrigel (Corning, New York, NY, USA) was added to 100 μL of the cell suspension to establish organoids, and the resulting suspension was allowed to solidify upside-down in pre-warmed 6-well culture plates (Corning) at 37°C for 30 min. After gelation, 3 mL LCOM was added to each well. The medium was changed every two to three days.
Histology and immunohistochemistry
Organoids and their corresponding parental tumors were fixed in 4% paraformaldehyde, followed by paraffin embedding, sectioning, deparaffinization, dehydration, and hematoxylin and eosin staining. Immunohistochemistry (IHC) was performed using antibodies targeting the thyroid transcription factor (TTF-1), Cytokeratin 7 (CK7), and Napsin A for ADC; Cytokeratin 5/6 (CK5/6), P40, and P63 for SCC; TTF-1, synaptophysin (Syn), chromogranin A (CgA), and CD56 for SCLC. Green Chromogen and the BOND IHC polymer detection kit were used for color development. The IHC staining was performed using the BOND-III automated stainer (Leica, Wetzlar, Germany). Images were captured using the IX73 microscope (Olympus, Tokyo, Japan).
Fluorescence in situ hybridization
Dual-color fluorescence in situ hybridization (FISH) was performed using the MET probe, labeled with spectrum red, and the chromosome 17 specific centromere (D17Z1) probe, labeled with spectrum green, on sections cut from the same block. Deparaffinization, in situ hybridization, and staining were performed using the PathVysion kit (Abbott-Vysis Lab, Abbott Park, IL, USA). Fluorescent signals in at least 60 non-overlapping interphase nuclei with intact morphology were scored using a Zeiss Axioplan 2 microscope with a × 100 planar objective using a triple band-pass filter permissive to blue, green, and red colors.
FISH for RET was performed using the ZytoLight SPEC RET Dual-Color Break Apart Probe Kit (CliniSciences, Montrouge, France). At least 100 tumor nuclei were analyzed, and a case was considered positive when the number of neoplastic nuclei with a split signal or with a single 3′ signal was above a minimum of 20% of the observed neoplastic nuclei.
DNA extraction
DNA was extracted from tumor tissues and effusion-derived NSCLC organoids (also known as patient-derived LCOs) using the QIAamp DNA FFPE tissue kit (Qiagen, Valencia, CA, USA) and the QIAamp DNA kit (Qiagen, Valencia, CA, USA) according to the manufacturer’s instructions. Cell-free DNA was extracted from effusion samples using the QIAamp Circulating Nucleic Acid Kit (Qiagen, Valencia, CA, USA), according to the manufacturer’s instructions. The concentration of DNA was measured with the Qubit 2.0 Fluorometer with the Qubit double-stranded DNA assay kit (Life Technologies, Carlsbad, CA, USA).
Next-generation sequencing library preparation and capture-based targeted sequencing
DNA extracted from the tumor, effusion, and LCO samples was fragmented using the Covaris M220 focused ultrasonicator (Covaris, Inc., Woburn, MA, USA) or Bioruptor ultrasonicator (diagenode sa, Inc., Seraing, Belgium), followed by end repair, phosphorylation, dA addition, and adaptor ligation for library construction. The samples were subsequently subjected to capture-based targeted sequencing using a panel of 520, 425, 168, or 139 cancer-related genes (panels of 520 or 168 belongs to Burning Rock Biotech, Guangzhou, China, while panels of 425 or 139 belongs to Nanjing Geneseeq Technology Inc.). DNA was hybridized with capture probe baits, selected with magnetic beads, and amplified using PCR. A bioanalyzer high-sensitivity DNA assay was performed to assess the quality and size of the fragments. Indexed samples were sequenced on the Nextseq500 sequencer (Illumina, Inc., San Diego, CA, USA) or HiSeq 4000 sequencer (Illumina, Inc., San Diego, CA, USA) with paired-end reads.
Sequence data analysis
Raw sequencing data were preprocessed using Trimmomatic 0.36 for trimming adaptors and low-quality reads. Preprocessed sequencing data were then mapped to the human genome (hg19) using Burrows–Wheeler Aligner 0.7.10. Variant calling and annotation were performed using the GATK 3.2, MuTect, and VarScan. According to the ExAC, 1,000 Genomes, dbSNP, and ESP6500SI-V2 databases, variants with a population frequency over 0.1% were grouped as single nucleotide polymorphisms and excluded from further analysis. The remaining variants were annotated using ANNOVAR and SnpEff v3.6. Copy number variations (CNVs) and large genomic rearrangements (LGRs) were detected using in-house analysis scripts based on the depth of coverage data of capture intervals, as previously described.62,63 DNA translocation analysis was performed using both Tophat2 and Factera 1.4.3. The maximum allele frequency (maxAF) for each mutation was calculated.
Tumor mutation burden (TMB) was defined as somatic single nucleotide variants (SNVs) and small insertions and deletions (indels) located in the coding region and its 20 bp upstream/downstream regions. TMB was calculated using the following equation:
Differences between the two groups were assessed using the two-tailed t-test or Mann–Whitney U test for continuous variables. Statistical significance was set at p < 0.05. The Pearson correlation coefficient was used to assess the correlation between tumor and LCO-based TMB. Bivariant sensitivity and positive predictive value in LCO samples were calculated when the genomic profile of effusion samples was used as a reference. All statistical analyses were performed using R 3.3.3.
Drug treatments and sensitivity tests
Treatments with anti-cancer drugs and sensitivity tests were performed as described previously,64,65 with some modifications. Briefly, organoids cultured for 1–2 weeks were harvested and dissociated using 1× TrypLe (Gibco). The dissociated organoids were mixed in LCOM with Matrigel (Corning) (1:1 ratio) and transferred onto 384-well white plates. Approximately, 2000 cells were seeded in each well. After gelation, 30 μL LCOM was added to each well. The organoids were cultured for 48 h. Thereafter, a dilution series of each compound (10, 2.5, 0.625, 0.156, 0.039, and 0.0097 μM) was dispensed using liquid-handling robots. The cell viability was assayed using 3D CellTiter-Glo (Promega, Madison, WI, USA) after 96 h of drug incubation. The plates were agitated for 30 min at RT prior to luminescence measurement. The IC50 values were determined using GraphPad Prism 7.0 (GraphPad Software, La Jolla, CA, USA).
Proteomics
Protein extraction and digestion for LC-MS/MS
LCOs were transferred into a 6-well plate (Corning), incubated for 7 days, and then treated using osimertinib (0.6 μM), BLU-667 (1 μM), and the combination of osimertinib and BLU-667 (0.6 μM and 1 μM, respectively). The organoids were harvested using the Cell Recovery Solution (Corning) and stored in liquid nitrogen until further use for protein extraction.
SDT (4% SDS, 100 mM Tris-HCl, 1 mM DTT, pH 7.6) buffer was used for sample lysis and protein extraction. The amount of protein was quantified using the BCA Protein Assay Kit (Bio-Rad, Hercules, CA, USA). Protein digestion using trypsin was performed according to the filter-aided sample preparation (FASP) procedure described by Matthias Mann.66 The digest peptides of each sample were desalted on C18 Cartridges [Empore™ SPE Cartridges C18 (standard density), bed I.D., 7 mm; volume, 3 mL; Sigma], concentrated using vacuum centrifugation, and reconstituted in 40 μL of 0.1% (v/v) formic acid.
FASP digestion procedure
A total of 200 μg of proteins for each sample were incorporated into 30 μL SDT buffer (4% SDS, 100 mM DTT, 150 mM Tris-HCl, pH 8.0). The detergent, DTT, and other low-molecular-weight components were removed using the UA buffer (8 M urea, 150 mM Tris-HCl, pH 8.0) by repeated ultrafiltration (Microcon units, 10 kD). Subsequently, 100 μL iodoacetamide (100 mM IAA in UA buffer) was added to block reduced cysteine residues, and the samples were incubated for 30 min in the dark. The filters were washed with 100 μL UA buffer three times and then using 100 μL of 25 mM NH4HCO3 buffer twice. Finally, the protein suspensions were digested using 4 μg of trypsin (Promega) in 40 μL of 25 mM NH4HCO3 buffer overnight at 37°C. The resulting peptides were collected as a filtrate. The peptides of each sample were desalted on C18 Cartridges [Empore™ SPE Cartridges C18 (standard density), bed I.D., 7 mm; volume, 3 mL; Sigma], concentrated by vacuum centrifugation, and reconstituted in 40 μL of 0.1% (v/v) formic acid. The peptide content was estimated by UV light spectral density at 280 nm using an extinction coefficient of 1.1 of 0.1% (g/L) solution that was calculated based on the frequency of tryptophan and tyrosine in vertebrate proteins.
LC-MS/MS analysis
LC-MS/MS analysis was performed using the timsTOF Pro mass spectrometer (Bruker, USA) coupled to Nanoelute (Bruker Daltonics) for 60 min. The peptides were loaded onto a reverse-phase trap column (Thermo Scientific Acclaim PepMap100, 100 μm∗2 cm, nanoViper C18) connected to the C18-reversed-phase analytical column (Thermo Scientific Easy Column, 10 cm long, 75 μm inner diameter, 3 μm resin) in buffer A (0.1% formic acid) and separated at a linear gradient of buffer B (84% acetonitrile and 0.1% formic acid) at a flow rate of 300 nL/min controlled by IntelliFlow technology. The mass spectrometer was operated in positive ion mode. The mass spectrometer recorded ion mobility MS spectra over a mass range of m/z 100–1700 and 1/k0 of 0.6–1.6. Subsequently, 10 cycles of PASEF MS/MS were performed with a target intensity of 1.5k and a threshold of 2500. The active exclusion was enabled with a release time of 0.4 min.
Protein identification and quantification
The raw MS data for each sample were combined and searched using MaxQuant 1.5.3.17 software for identification and quantitation analysis.
Bioinformatic analysis of proteomics data
Cluster analysis of phosphorylated peptides
Cluster 3.0 (http://bonsai.hgc.jp/∼ mdehoon/software/cluster/software.htm) and Java Treeview software (URL: http://jtreeview. sourceforge.net) were used to perform hierarchical clustering analysis. The Euclidean distance algorithm for similarity measurement and average linkage clustering algorithm (clustering uses the centroids of the observations) for clustering were selected when performing hierarchical clustering. A heatmap is often presented as a visual aid, in addition to the dendrogram.
Gene ontology (GO) annotation
The protein sequences of the selected DEPs were locally searched using the NCBI BLAST + client software (ncbi-blast-2.2.28+-win32.exe) and InterProScan to find homologous sequences, GO terms were mapped, and sequences were annotated using the Blast2GO software (BioBam Bioinformatics, Valencia, Spain). The GO annotation results were plotted using R scripts.
Kyoto encyclopedia of genes and genomes (KEGG) annotation
Following the annotation steps, the studied proteins were BLASTed against the online KEGG database (http://geneontology.org/) to retrieve their KEGG orthology identifications and were subsequently mapped to KEGG pathways.
Enrichment analysis
Enrichment analysis was performed based on the Fisher exact test, considering all quantified proteins as the background dataset. The Benjamini–Hochberg correction for multiple testing was applied to adjust the derived p values. Only functional categories and pathways with p values under a threshold of 0.05 were considered significant.
Quantification and statistical analysis
The capability of LCO-DST to discriminate between clinically effective or resistant patients was determined using receiver operating characteristic analysis. The predictor was organoid sensitivity to osimertinib, chemotherapy, dual targeted therapy, and other targeted therapy treatment, which is the ratio of the ATP luminance value of treated organoids to that of untreated organoids on day 4 after treatment. The diagnostic performance of the LCOs in predicting the patients’ clinical response to different treatments were evaluated based on area under the curve, sensitivity, specificity, and diagnostic accuracy.
Acknowledgments
We thank all patients who participated in this study as well as their families. We thank Prof. Chuxia Deng from University of Macau for the helpful discussions. We also thank Burning Rock Biotech, Nanjing Geneseeq Technology Inc., and Tongshu Biotechnology Co., LTD. for their help with data analysis. The authors also thank Accurate International Biotechnology Co. for their assistance with the organoid techniques. We also thank Shanghai Applied Protein Technology Co. for the help with proteomics analysis. This study was supported by the High-level Hospital Construction Project (grant no. DFJH201809 to J.J.Y.); the National Natural Science Foundation of China (grant no. 81972164 to J.J.Y.); the Natural Science Foundation of Guangdong Province (grant no. 2019A1515010931 to J.J.Y.); and the Key Lab System Project of Guangdong Science and Technology Department – Guangdong Provincial Key Laboratory of Translational Medicine in Lung Cancer (grant no. 2017B030314120 to Y.L.W.).
Author contributions
Conceptualization, J.J.Y.,Y.L.W., and H.J.C.; methodology, Z.X.C., Y.F.L., and C.X.; investigation, W.F.L., Q.Y.G., S.L.Z., J.W.S., and Y.Q.C.; writing - original draft, H.M.W., C.Y.Z., K.C.P., J.W.S., and Z.X.C.; writing - review & editing, X.C.Z. and J.J.Y.; visualization, H.M.W., C.Y.Z., and K.C.P.; funding acquisition, J.J.Y.; resources, Q.Y.G., S.L.Z., J.W.S.; supervision, Q.Z., C.R.X., Z.W., and J.S.; validation, Q.Z., C.R.X., Z.W., and J.S.; formal analysis, H.H.Y.
Declaration of interests
The authors declare no competing interests.
Published: January 18, 2023
Footnotes
Supplemental information can be found online at https://doi.org/10.1016/j.xcrm.2022.100911.
Contributor Information
Hua-Jun Chen, Email: chenhuajun@gdph.org.cn.
Yi-Long Wu, Email: syylwu@live.cn.
Jin-Ji Yang, Email: yangjinji@gdph.org.cn.
Supplemental information
Data and code availability
-
•
The next-generation DNA sequencing dataset generated during this study is available at the National Genomics Data Center: HRA003231 (URL: https://ngdc.cncb.ac.cn). The mass spectrometry proteomics data reported in this paper have been deposited to the ProteomeXchange Consortium: PXD037076(http://proteomecentral.proteomexchange.org) via iProx partner repository61.
-
•
This paper does not report the original code.
-
•
Any additional information required to reanalyze the data reported in this work paper is available from the lead contact upon request.
References
- 1.Sung H., Ferlay J., Siegel R.L., Laversanne M., Soerjomataram I., Jemal A., Bray F. Global cancer statistics 2020: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA. Cancer J. Clin. 2021;71:209–249. doi: 10.3322/caac.21660. [DOI] [PubMed] [Google Scholar]
- 2.Wang N., Mengersen K., Tong S., Kimlin M., Zhou M., Wang L., Hu W. Lung cancer mortality in China: spatial and temporal trends among subpopulations. Chest. 2019;156:972–983. doi: 10.1016/j.chest.2019.07.023. [DOI] [PubMed] [Google Scholar]
- 3.Porcel J.M., Gasol A., Bielsa S., Civit C., Light R.W., Salud A. Clinical features and survival of lung cancer patients with pleural effusions. Respirology. 2015;20:654–659. doi: 10.1111/resp.12496. [DOI] [PubMed] [Google Scholar]
- 4.Pauli C., Hopkins B.D., Prandi D., Shaw R., Fedrizzi T., Sboner A., Sailer V., Augello M., Puca L., Rosati R., et al. Personalized in vitro and in vivo cancer models to guide precision medicine. Cancer Discov. 2017;7:462–477. doi: 10.1158/2159-8290.CD-16-1154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Wu L., Wang Y., Xu X., Liu Y., Lin B., Zhang M., Zhang J., Wan S., Yang C., Tan W. Aptamer-based detection of circulating targets for precision medicine. Chem. Rev. 2021;121:12035–12105. doi: 10.1021/acs.chemrev.0c01140. [DOI] [PubMed] [Google Scholar]
- 6.Hahn W.C., Bader J.S., Braun T.P., Califano A., Clemons P.A., Druker B.J., Ewald A.J., Fu H., Jagu S., Kemp C.J., et al. An expanded universe of cancer targets. Cell. 2021;184:1142–1155. doi: 10.1016/j.cell.2021.02.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Wu Y.L., Cheng Y., Zhou X., Lee K.H., Nakagawa K., Niho S., Tsuji F., Linke R., Rosell R., Corral J., et al. Dacomitinib versus gefitinib as first-line treatment for patients with EGFR-mutation-positive non-small-cell lung cancer (ARCHER 1050): a randomised, open-label, phase 3 trial. Lancet Oncol. 2017;18:1454–1466. doi: 10.1016/S1470-2045(17)30608-3. [DOI] [PubMed] [Google Scholar]
- 8.Arbour K.C., Riely G.J. Systemic therapy for locally advanced and metastatic non-small cell lung cancer: a review. JAMA. 2019;322:764–774. doi: 10.1001/jama.2019.11058. [DOI] [PubMed] [Google Scholar]
- 9.Cobain E.F., Wu Y.M., Vats P., Chugh R., Worden F., Smith D.C., Schuetze S.M., Zalupski M.M., Sahai V., Alva A., et al. Assessment of clinical benefit of integrative genomic profiling in advanced solid tumors. JAMA Oncol. 2021;7:525–533. doi: 10.1001/jamaoncol.2020.7987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Liu X., Lu Y., Zhu G., Lei Y., Zheng L., Qin H., Tang C., Ellison G., McCormack R., Ji Q. The diagnostic accuracy of pleural effusion and plasma samples versus tumour tissue for detection of EGFR mutation in patients with advanced non-small cell lung cancer: comparison of methodologies. J. Clin. Pathol. 2013;66:1065–1069. doi: 10.1136/jclinpath-2013-201728. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Cai G., Wong R., Chhieng D., Levy G.H., Gettinger S.N., Herbst R.S., Puchalski J.T., Homer R.J., Hui P. Identification of EGFR mutation, KRAS mutation, and ALK gene rearrangement in cytological specimens of primary and metastatic lung adenocarcinoma. Cancer Cytopathol. 2013;121:500–507. doi: 10.1002/cncy.21288. [DOI] [PubMed] [Google Scholar]
- 12.Wang W., Tang Y., Li J., Jiang L., Jiang Y., Su X. Detection of ALK rearrangements in malignant pleural effusion cell blocks from patients with advanced non-small cell lung cancer: a comparison of Ventana immunohistochemistry and fluorescence in situ hybridization. Cancer Cytopathol. 2015;123:117–122. doi: 10.1002/cncy.21510. [DOI] [PubMed] [Google Scholar]
- 13.Sacher A.G., Dahlberg S.E., Heng J., Mach S., Jänne P.A., Oxnard G.R. Association between younger age and targetable genomic alterations and prognosis in non-small-cell lung cancer. JAMA Oncol. 2016;2:313–320. doi: 10.1001/jamaoncol.2015.4482. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Chen Z., Fillmore C.M., Hammerman P.S., Kim C.F., Wong K.K. Non-small-cell lung cancers: a heterogeneous set of diseases. Nat. Rev. Cancer. 2014;14:535–546. doi: 10.1038/nrc3775. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Byeon S., Lee B., Park W.Y., Choi Y.L., Jung H.A., Sun J.M., Ahn J.S., Ahn M.J., Park K., Lee S.H. Benefit of targeted DNA sequencing in advanced non-small-cell lung cancer patients without EGFR and ALK alterations on conventional tests. Clin. Lung Cancer. 2020;21:e182–e190. doi: 10.1016/j.cllc.2019.11.006. [DOI] [PubMed] [Google Scholar]
- 16.Lin A., Giuliano C.J., Palladino A., John K.M., Abramowicz C., Yuan M.L., Sausville E.L., Lukow D.A., Liu L., Chait A.R., et al. Off-target toxicity is a common mechanism of action of cancer drugs undergoing clinical trials. Sci. Transl. Med. 2019;11:eaaw8412. doi: 10.1126/scitranslmed.aaw8412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Stinchcombe T.E. The use of EGFR tyrosine kinase inhibitors in EGFR wild-type non-small-cell lung cancer. Curr. Treat. Options Oncol. 2016;17:18. doi: 10.1007/s11864-016-0394-4. [DOI] [PubMed] [Google Scholar]
- 18.Cartwright D., Roxburgh P., Stanley B., Brown J., Mclaren A., Coulter S., Forte C., Glasspool R. 510 Niraparib outcomes in brca wild-type platinum sensitive recurrent ovarian cancer: a comparison of real-world data to the nova trial. Int. J. Gynecol. Cancer. 2020;30:A85–A86. doi: 10.1136/ijgc-2020-ESGO.146. [DOI] [Google Scholar]
- 19.Gazdar A.F., Girard L., Lockwood W.W., Lam W.L., Minna J.D. Lung cancer cell lines as tools for biomedical discovery and research. J. Natl. Cancer Inst. 2010;102:1310–1321. doi: 10.1093/jnci/djq279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Pan Y., Han H., Labbe K.E., Zhang H., Wong K.K. Recent advances in preclinical models for lung squamous cell carcinoma. Oncogene. 2021;40:2817–2829. doi: 10.1038/s41388-021-01723-7. [DOI] [PubMed] [Google Scholar]
- 21.de Witte C.J., Espejo Valle-Inclan J., Hami N., Lõhmussaar K., Kopper O., Vreuls C.P.H., Jonges G.N., van Diest P., Nguyen L., Clevers H., et al. Patient-derived ovarian cancer organoids mimic clinical response and exhibit heterogeneous inter- and intrapatient drug responses. Cell Rep. 2020;31:107762. doi: 10.1016/j.celrep.2020.107762. [DOI] [PubMed] [Google Scholar]
- 22.Wang D., Pham N.A., Tong J., Sakashita S., Allo G., Kim L., Yanagawa N., Raghavan V., Wei Y., To C., et al. Molecular heterogeneity of non-small cell lung carcinoma patient-derived xenografts closely reflect their primary tumors. Int. J. Cancer. 2017;140:662–673. doi: 10.1002/ijc.30472. [DOI] [PubMed] [Google Scholar]
- 23.Meehan T.F., Conte N., Goldstein T., Inghirami G., Murakami M.A., Brabetz S., Gu Z., Wiser J.A., Dunn P., Begley D.A., et al. PDX-MI: minimal information for patient-derived tumor xenograft models. Cancer Res. 2017;77:e62–e66. doi: 10.1158/0008-5472.CAN-17-0582. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Nagle P.W., Plukker J.T.M., Muijs C.T., van Luijk P., Coppes R.P. Patient-derived tumor organoids for prediction of cancer treatment response. Semin. Cancer Biol. 2018;53:258–264. doi: 10.1016/j.semcancer.2018.06.005. [DOI] [PubMed] [Google Scholar]
- 25.Jung J., Seol H.S., Chang S. The generation and application of patient-derived xenograft model for cancer research. Cancer Res. Treat. 2018;50:1–10. doi: 10.4143/crt.2017.307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Becher O.J., Holland E.C. Genetically engineered models have advantages over xenografts for preclinical studies. Cancer Res. 2006;66:3355–3358. doi: 10.1158/0008-5472.CAN-05-3827. [DOI] [PubMed] [Google Scholar]
- 27.Chen H., Gotimer K., De Souza C., Tepper C.G., Karnezis A.N., Leiserowitz G.S., Chien J., Smith L.H. Short-term organoid culture for drug sensitivity testing of high-grade serous carcinoma. Gynecol. Oncol. 2020;157:783–792. doi: 10.1016/j.ygyno.2020.03.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Hill S.J., Decker B., Roberts E.A., Horowitz N.S., Muto M.G., Worley M.J., Jr., Feltmate C.M., Nucci M.R., Swisher E.M., Nguyen H., et al. Prediction of DNA repair inhibitor response in short-term patient-derived ovarian cancer organoids. Cancer Discov. 2018;8:1404–1421. doi: 10.1158/2159-8290.CD-18-0474. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Fatehullah A., Tan S.H., Barker N. Organoids as an in vitro model of human development and disease. Nat. Cell Biol. 2016;18:246–254. doi: 10.1038/ncb3312. [DOI] [PubMed] [Google Scholar]
- 30.van de Wetering M., Francies H.E., Francis J.M., Bounova G., Iorio F., Pronk A., van Houdt W., van Gorp J., Taylor-Weiner A., Kester L., et al. Prospective derivation of a living organoid biobank of colorectal cancer patients. Cell. 2015;161:933–945. doi: 10.1016/j.cell.2015.03.053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Vlachogiannis G., Hedayat S., Vatsiou A., Jamin Y., Fernández-Mateos J., Khan K., Lampis A., Eason K., Huntingford I., Burke R., et al. Patient-derived organoids model treatment response of metastatic gastrointestinal cancers. Science. 2018;359:920–926. doi: 10.1126/science.aao2774. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Tamura H., Higa A., Hoshi H., Hiyama G., Takahashi N., Ryufuku M., Morisawa G., Yanagisawa Y., Ito E., Imai J.I., et al. Evaluation of anticancer agents using patient-derived tumor organoids characteristically similar to source tissues. Oncol. Rep. 2018;40:635–646. doi: 10.3892/or.2018.6501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Zhao J.P., Hu Y., Du R.H., Chen Z.S., Jin Y., Zhou M., Zhang J., Qu J.M., Cao B. [Expert consensus on the use of corticosteroid in patients with 2019-nCoV pneumonia] Zhonghua Jiehe He Huxi Zazhi. 2020;43:E007. doi: 10.3760/cma.j.issn.1001-0939.2020.0007. [DOI] [PubMed] [Google Scholar]
- 34.Holokai L., Chakrabarti J., Lundy J., Croagh D., Adhikary P., Richards S.S., Woodson C., Steele N., Kuester R., Scott A., et al. Murine- and human-derived autologous organoid/immune cell Co-cultures as pre-clinical models of pancreatic ductal adenocarcinoma. Cancers. 2020;12:3816. doi: 10.3390/cancers12123816. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Boj S.F., Hwang C.I., Baker L.A., Chio I.I.C., Engle D.D., Corbo V., Jager M., Ponz-Sarvise M., Tiriac H., Spector M.S., et al. Organoid models of human and mouse ductal pancreatic cancer. Cell. 2015;160:324–338. doi: 10.1016/j.cell.2014.12.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Sachs N., de Ligt J., Kopper O., Gogola E., Bounova G., Weeber F., Balgobind A.V., Wind K., Gracanin A., Begthel H., et al. A living biobank of breast cancer organoids captures disease heterogeneity. Cell. 2018;172:373–386.e10. doi: 10.1016/j.cell.2017.11.010. [DOI] [PubMed] [Google Scholar]
- 37.Chen J.H., Chu X.P., Zhang J.T., Nie Q., Tang W.F., Su J., Yan H.H., Zheng H.P., Chen Z.X., Chen X., et al. Genomic characteristics and drug screening among organoids derived from non-small cell lung cancer patients. Thorac. Cancer. 2020;11:2279–2290. doi: 10.1111/1759-7714.13542. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Shi R., Radulovich N., Ng C., Liu N., Notsuda H., Cabanero M., Martins-Filho S.N., Raghavan V., Li Q., Mer A.S., et al. Organoid cultures as preclinical models of non-small cell lung cancer. Clin. Cancer Res. 2020;26:1162–1174. doi: 10.1158/1078-0432.CCR-19-1376. [DOI] [PubMed] [Google Scholar]
- 39.Li Z., Qian Y., Li W., Liu L., Yu L., Liu X., Wu G., Wang Y., Luo W., Fang F., et al. Human lung adenocarcinoma-derived organoid models for drug screening. iScience. 2020;23:101411. doi: 10.1016/j.isci.2020.101411. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Hu Y., Sui X., Song F., Li Y., Li K., Chen Z., Yang F., Chen X., Zhang Y., Wang X., et al. Lung cancer organoids analyzed on microwell arrays predict drug responses of patients within a week. Nat. Commun. 2021;12:2581. doi: 10.1038/s41467-021-22676-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Ding S., Hsu C., Wang Z., Natesh N.R., Millen R., Negrete M., Giroux N., Rivera G.O., Dohlman A., Bose S., et al. Patient-derived micro-organospheres enable clinical precision oncology. Cell Stem Cell. 2022;29:905–917.e6. doi: 10.1016/j.stem.2022.04.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Kim S.Y., Kim S.M., Lim S., Lee J.Y., Choi S.J., Yang S.D., Yun M.R., Kim C.G., Gu S.R., Park C., et al. Modeling clinical responses to targeted therapies by patient-derived organoids of advanced lung adenocarcinoma. Clin. Cancer Res. 2021;27:4397–4409. doi: 10.1158/1078-0432.CCR-20-5026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Lee D., Kim Y., Chung C. Scientific validation and clinical application of lung cancer organoids. Cells. 2021;10 doi: 10.3390/cells10113012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Kim M., Mun H., Sung C.O., Cho E.J., Jeon H.-J., Chun S.-M., Jung D.J., Shin T.H., Jeong G.S., Kim D.K., et al. Patient-derived lung cancer organoids as in vitro cancer models for therapeutic screening. Nat. Commun. 2019;10:3991. doi: 10.1038/s41467-019-11867-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Terry J., Leung S., Laskin J., Leslie K.O., Gown A.M., Ionescu D.N. Optimal immunohistochemical markers for distinguishing lung adenocarcinomas from squamous cell carcinomas in small tumor samples. Am. J. Surg. Pathol. 2010;34:1805–1811. doi: 10.1097/PAS.0b013e3181f7dae3. [DOI] [PubMed] [Google Scholar]
- 46.Tannock I.F., Hickman J.A. Limits to personalized cancer medicine. N. Engl. J. Med. 2016;375:1289–1294. doi: 10.1056/NEJMsb1607705. [DOI] [PubMed] [Google Scholar]
- 47.Vargas A.J., Harris C.C. Biomarker development in the precision medicine era: lung cancer as a case study. Nat. Rev. Cancer. 2016;16:525–537. doi: 10.1038/nrc.2016.56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Wu C.G., Chiovaro F., Curioni-Fontecedro A., Casanova R., Soltermann A. In vitro cell culture of patient derived malignant pleural and peritoneal effusions for personalised drug screening. J. Transl. Med. 2020;18:163. doi: 10.1186/s12967-020-02331-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Li J., Xu H., Zhang L., Song L., Feng D., Peng X., Wu M., Zou Y., Wang B., Zhan L., et al. Malignant ascites-derived organoid (MADO) cultures for gastric cancer in vitro modelling and drug screening. J. Cancer Res. Clin. Oncol. 2019;145:2637–2647. doi: 10.1007/s00432-019-03004-z. [DOI] [PubMed] [Google Scholar]
- 50.Dijkstra K.K., Monkhorst K., Schipper L.J., Hartemink K.J., Smit E.F., Kaing S., de Groot R., Wolkers M.C., Clevers H., Cuppen E., Voest E.E. Challenges in establishing pure lung cancer organoids limit their utility for personalized medicine. Cell Rep. 2020;31:107588. doi: 10.1016/j.celrep.2020.107588. [DOI] [PubMed] [Google Scholar]
- 51.Carneiro F.P., Muniz-Junqueira M.I., Pittella-Silva F., Carneiro M.d.V., Takano G.H.S., Vianna L.M.d.S., De Andrade L.B., De Castro T.M.M.L., Peres I., Dos Santos Borges T.K., et al. A panel of markers for identification of malignant and non-malignant cells in culture from effusions. Oncol. Rep. 2017;38:3538–3544. doi: 10.3892/or.2017.6022. [DOI] [PubMed] [Google Scholar]
- 52.Jiao X.D., Ding L.R., Zhang C.T., Qin B.D., Liu K., Jiang L.P., Wang X., Lv L.T., Ding H., Li D.M., et al. Serum tumor markers for the prediction of concordance between genomic profiles from liquid and tissue biopsy in patients with advanced lung adenocarcinoma. Transl. Lung Cancer Res. 2021;10:3236–3250. doi: 10.21037/tlcr-21-543. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Frampton G.M., Fichtenholtz A., Otto G.A., Wang K., Downing S.R., He J., Schnall-Levin M., White J., Sanford E.M., An P., et al. Development and validation of a clinical cancer genomic profiling test based on massively parallel DNA sequencing. Nat. Biotechnol. 2013;31:1023–1031. doi: 10.1038/nbt.2696. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Lim Z.F., Ma P.C. Emerging insights of tumor heterogeneity and drug resistance mechanisms in lung cancer targeted therapy. J. Hematol. Oncol. 2019;12:134. doi: 10.1186/s13045-019-0818-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Sato T., Stange D.E., Ferrante M., Vries R.G.J., Van Es J.H., Van den Brink S., Van Houdt W.J., Pronk A., Van Gorp J., Siersema P.D., Clevers H. Long-term expansion of epithelial organoids from human colon, adenoma, adenocarcinoma, and Barrett's epithelium. Gastroenterology. 2011;141:1762–1772. doi: 10.1053/j.gastro.2011.07.050. [DOI] [PubMed] [Google Scholar]
- 56.Weeber F., van de Wetering M., Hoogstraat M., Dijkstra K.K., Krijgsman O., Kuilman T., Gadellaa-van Hooijdonk C.G.M., van der Velden D.L., Peeper D.S., Cuppen E.P.J.G., et al. Preserved genetic diversity in organoids cultured from biopsies of human colorectal cancer metastases. Proc. Natl. Acad. Sci. USA. 2015;112:13308–13311. doi: 10.1073/pnas.1516689112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Huang L., Holtzinger A., Jagan I., BeGora M., Lohse I., Ngai N., Nostro C., Wang R., Muthuswamy L.B., Crawford H.C., et al. Ductal pancreatic cancer modeling and drug screening using human pluripotent stem cell- and patient-derived tumor organoids. Nat. Med. 2015;21:1364–1371. doi: 10.1038/nm.3973. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Chua C.W., Shibata M., Lei M., Toivanen R., Barlow L.J., Bergren S.K., Badani K.K., McKiernan J.M., Benson M.C., Hibshoosh H., Shen M.M. Single luminal epithelial progenitors can generate prostate organoids in culture. Nat. Cell Biol. 2014;16:951–961. doi: 10.1038/ncb3047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Mullenders J., de Jongh E., Brousali A., Roosen M., Blom J.P.A., Begthel H., Korving J., Jonges T., Kranenburg O., Meijer R., Clevers H.C. Mouse and human urothelial cancer organoids: a tool for bladder cancer research. Proc. Natl. Acad. Sci. USA. 2019;116:4567–4574. doi: 10.1073/pnas.1803595116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Roelofs C., Hollande F., Redvers R., Anderson R.L., Merino D. Breast tumour organoids: promising models for the genomic and functional characterisation of breast cancer. Biochem. Soc. Trans. 2019;47:109–117. doi: 10.1042/BST20180375. [DOI] [PubMed] [Google Scholar]
- 61.Ma J., Chen T., Wu S., Yang C., Bai M., Shu K., Li K., Zhang G., Jin Z., He F., et al. iProX: an integrated proteome resource. Nucleic Acids Res. 2019;47:D1211–D1217. doi: 10.1093/nar/gky869. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Wu D., Xie Y., Jin C., Qiu J., Hou T., Du H., Chen S., Xiang J., Shi X., Liu J. The landscape of kinase domain duplication in Chinese lung cancer patients. Ann. Transl. Med. 2020;8:1642. doi: 10.21037/atm-20-7408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Li Y.S., Jiang B.Y., Yang J.J., Zhang X.C., Zhang Z., Ye J.Y., Zhong W.Z., Tu H.Y., Chen H.J., Wang Z., et al. Unique genetic profiles from cerebrospinal fluid cell-free DNA in leptomeningeal metastases of EGFR-mutant non-small-cell lung cancer: a new medium of liquid biopsy. Ann. Oncol. 2018;29:945–952. doi: 10.1093/annonc/mdy009. [DOI] [PubMed] [Google Scholar]
- 64.Jin Y., Xue Q., Shen X., Zheng Q., Chen H., Zhou X., Li Y. PD-L1 expression and comprehensive molecular profiling predict survival in nonsmall cell lung cancer: a real-world study of a large Chinese cohort. Clin. Lung Cancer. 2022;23:43–51. doi: 10.1016/j.cllc.2021.08.009. [DOI] [PubMed] [Google Scholar]
- 65.Ding R.B., Chen P., Rajendran B.K., Lyu X., Wang H., Bao J., Zeng J., Hao W., Sun H., Wong A.H.H., et al. Molecular landscape and subtype-specific therapeutic response of nasopharyngeal carcinoma revealed by integrative pharmacogenomics. Nat. Commun. 2021;12:3046. doi: 10.1038/s41467-021-23379-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Wiśniewski J.R., Mann M. Consecutive proteolytic digestion in an enzyme reactor increases depth of proteomic and phosphoproteomic analysis. Anal. Chem. 2012;84:2631–2637. doi: 10.1021/ac300006b. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
-
•
The next-generation DNA sequencing dataset generated during this study is available at the National Genomics Data Center: HRA003231 (URL: https://ngdc.cncb.ac.cn). The mass spectrometry proteomics data reported in this paper have been deposited to the ProteomeXchange Consortium: PXD037076(http://proteomecentral.proteomexchange.org) via iProx partner repository61.
-
•
This paper does not report the original code.
-
•
Any additional information required to reanalyze the data reported in this work paper is available from the lead contact upon request.