

Summary
Signal transduction induced by chimeric antigen receptors (CARs) is generally believed to rely on the activity of the SRC family kinase (SFK) LCK, as is the case with T cell receptor (TCR) signaling. Here, we show that CAR signaling occurs in the absence of LCK. This LCK-independent signaling requires the related SFK FYN and a CD28 intracellular domain within the CAR. LCK-deficient CAR-T cells are strongly signaled through CAR and have better in vivo efficacy with reduced exhaustion phenotype and enhanced induction of memory and proliferation. These distinctions can be attributed to the fact that FYN signaling tends to promote proliferation and survival, whereas LCK signaling promotes strong signaling that tends to lead to exhaustion. This non-canonical signaling of CAR-T cells provides insight into the initiation of both TCR and CAR signaling and has important clinical implications for improvement of CAR function.
Keywords: T cell receptor, signal transduction, co-stimulation, chimeric antigen receptor T cell, CAR, CAR-T, LCK independent signaling, CD28, FYN, allogeneic CAR-T, specificity
Graphical abstract
Highlights
-
•
Chimeric antigen receptors (CARs) can initiate signaling in T cells lacking LCK
-
•
CD28 domain and SRC family kinase FYN play a role in LCK-independent CAR signaling
-
•
Disrupting LCK in CAR-T cell (disLCK-CAR-T) improves the function and immunophenotype
-
•
DisLCK-CAR-T shows elevated anti-tumor efficacy and the potential for allogeneic use
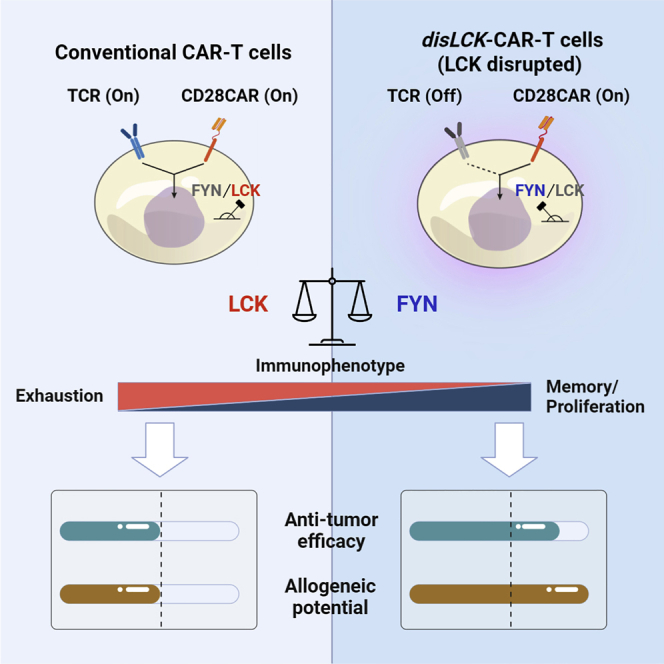
Wu et al. discover that LCK, an essential kinase for TCR signaling, is dispensable for CAR signaling. CD28 domain and the kinase FYN mediate this LCK-independent signaling. LCK-disrupted CAR-T shows increased in vivo anti-tumor efficacy—attributed to the improved memory/persistence and reduced exhaustion—and the potential for allogeneic use.
Introduction
Adoptive T cell therapy using chimeric antigen receptor T cells (CAR-T cells) shows great clinical success.1,2 The cytoplasmic domains of CD28 or CD137 (4-1BB) are widely used in CAR design.3 Nevertheless, the molecular mechanism of CAR signal transduction and how the co-stimulatory domain interplays with CD3ζ immunoreceptor tyrosine-based activation motif (ITAM) signals are poorly understood. In part, this is due to the assumption that CAR and T cell receptor (TCR) signaling are very similar because of the sharing of signaling domains. Phosphoproteome analysis of signaling by TCR, CD28-CD3ζ, and CD137-CD3ζ CARs did not find divergent signaling pathways.4 Rather, the differences were in signaling strength and kinetics.4,5,6,7
However, CAR signal transduction is potentially significantly different from what we understand of TCR signaling. Second- and later-generation CARs contain integral co-stimulatory elements, ensuring that co-stimulation occurs at the same time and places as antigen recognition, which is not the case for normal T cell activation. The formation of the CAR-T immunological synapse (IS) is different and less structured than that of normal T cells.3,5 The CD28 domain increases the phosphorylation rate of CD3ζ,8 whereas CD137 was found to recruit the THEMIS-SHP1 complex to the signalosome, resulting in decreased CAR signaling.9 It is therefore critical to understand how CAR transduces signaling such that therapeutic performance can be optimized.
In this work, we show that CARs with a CD28 domain, unlike TCRs, effectively transduce signaling in the absence of LCK, without which TCR signaling is totally abrogated and thymocyte development is profoundly blocked in Lck knockout mice.10,11 Here, we find that another SRC family kinase (SFK), FYN, can phosphorylate the CAR ITAMs. We demonstrate that LCK-disrupted CAR-T cells (disLCK-CAR-T) have special characteristics in vitro and in vivo. They only transduce signals through the CAR, not through endogenous TCRs. Therefore, disLCK-CAR-T cells could provide an alternative approach to generate allogeneic CAR-T cells without knocking out TCRs. We show that disLCK-CAR-T cells become more memory-like in vivo, with a less exhausted phenotype than conventional CAR-T cells, resulting in increased persistence and significantly improved efficacy. Signal network analysis indicates that the altered characteristics of disLCK-CAR-T cells could be caused by the increase of FYN-associated proliferation and survival pathways and by the decrease of strongly activated immune responses via LCK. In summary, this non-canonical LCK-independent CAR signaling sheds light on how CAR signaling is transduced and shows that we can use special aspects of CAR-T signal transduction to improve CAR-T technology.
Results
CAR-T cells can transduce T cell signaling without LCK
In our previous study, we compared signaling from a TCR with that from a second-generation CD28-CAR where both the target LMP2A peptide (L2)-major histocompatibility complex (MHC) complex (Figure S1A).12,13 We used CHO cells expressing single-chain peptide MHC (pMHC) as antigen-presenting cells (APCs) (Figures S1B and S1C).14 We found that anti-pMHC CARs have similar biophysical properties to TCR15 but that the CD8 co-receptor is dispensable for CAR signaling.14 CD8 is typically considered to be important for bringing LCK into the IS.16,17 Since CD8 was not able to enhance CAR-T signaling, we hypothesized that LCK might not be critical for CAR signaling, at least with regard to CD8-bound LCK. Surprisingly, unlike TCR, CAR was able to activate signaling in the absence of LCK, where CAR-Jcam cells produced interleukin-2 (IL-2) normally and showed phosphorylation of signaling molecules similar to TCR-Jurkat cells where LCK is present (Figures 1A, 1B, and S1D). We used CRISPR-Cas9 to perform homologous direct repair (HDR) to insert an anti-CD19 CD28-CAR construct, with either an Myc or His tag, into the LCK locus to disrupt its expression in primary CD8+ T cells (Figure 1C). Distinct populations of CAR+ CD8+ T cells were detected after HDR: CAR-Myc+, CAR-His+, and a double-positive population. Genotyping the insertion site of the total CAR+ population confirmed that the CAR construct was inserted into the LCK locus (Figure S1E). After sorting of each single and double CAR+ population, LCK expression was dramatically reduced in LCK-CAR-His and completely depleted in LCK-CAR-DP cells (Figures S1F and 1E). The LCK-CAR-DP cells seemed to have no significant difference in cytotoxicity compared with LCK-CAR-His and LCK-CAR-Myc cells. However, there was stronger cytokine secretion by LCK-CAR-DP cells (Figures 1F and 1G). These data imply that CAR signaling has an altered signal-triggering mechanism and that LCK, essential for TCR signaling, is dispensable for CAR signaling.
Figure 1.

CAR-T cells can transduce T cell signaling without LCK
(A) Top: western blot detection of LCK or FYN expression in CAR or TCR-Jcam1.6 cell. Bottom: CAR or TCR responsiveness in LCK-deficient Jcam1.6 cells. CHO-L2 is the artificial APC presenting the LMP2A426-434 (CLGGLLTMV) (L2) target peptide on HLA-A2. CHO-GAG presents the irrelevant GAG (SLYNTVATL) peptide.
(B) Phosphorylation of TCR signaling pathway molecules PLCγ1, ERK, and CD3ζ at different time points by LCK-deficient CD28CAR-Jcam cells. LCK-sufficient TCR-Jurkat cells were used as positive control. Phosphorylation of PLCγ1, ERK1/2, or CD3ζ was calculated relative to intensity of total PLCγ1, ERK, or ERK, respectively.
(C) Schematic of LCK locus-targeted homologous directed repair (HDR) by CRISPR-Cas9 editing. The anti-CD19-CD28CAR construct has either a Myc or His tag.
(D) CAR-His and CAR-Myc expression after LCK locus-targeted HDR.
(E) Western blot detection of LCK expression after sorting of CAR-His+, CAR-Myc+, and CAR-DP (Myc+/His+) CD8+-T cells. Control represents mock-edited CD8+-T cells.
(F) Cytotoxicity of CAR-His+, CAR-Myc+, and CAR-DP CD8+-T cells. Control group was the target cells with mock-edited CD8+-T cells.
(G) Cytokine secretion of CAR-His+, CAR-Myc+, and CAR-DP CD8+-T cells upon activation. CAR-T cells were incubated with CD19-expressing Daudi cells at a 1:1 ratio for 18 h, then stained for intracellular TNF or IFNγ. The left panel is the representative fluorescence-activated cell sorting (FACS) data, and the right panel is the statistical summary and analysis.
All data are representative of 3 independent experiments, plotted as mean ± SD of technical triplicates. ∗p < 0.05; ∗∗p < 0.01; ∗∗∗p < 0.001; ∗∗∗∗p < 0.0001; not significant (NS) p > 0.05. (A)–(G) were analyzed by Student’s t test.
See Figure S1 for additional data.
LCK-independent CAR signaling relies on CD28 co-stimulatory domain
CAR differs from TCR in many ways, particularly in its chimeric design. To identify which domains in CAR enable LCK-independent signaling, we replaced domains in our CD28-CAR. For production simplicity, we collected total CAR+, including both single and double knockin populations, as disLCK-CAR-T cells. They showed a significant reduction of LCK expression (Figure S2A). LCK-independent CAR signaling was not determined by antigen specificity since anti-HER2, like anti-CD19 CAR, initiated strong signaling in disLCK-CAR-T cells (Figure 2A). The CD3ζ domain and the co-stimulatory domain were required to initiate CAR signaling in the absence of LCK (Figures S2B and S2C). The CD28 co-stimulatory domain was crucial to support LCK-independent CAR signaling, but the CD137 co-stimulatory domain did not support it (Figures 2B and S2D). In a third-generation CAR containing both CD28 and CD137 domains, disLCK-CAR-T cell activation in response to antigen was observed but was much weaker compared with CD28-CAR. The PI3K-binding motif and proline-rich region are critical in CD28 signal transduction.18 We tested CD28 domain deletion, mutation of PI3K-binding motif YMNM to FMNM, and alanine substitution of the prolines in the proline-rich region PYAP (where SFK binds) to AYAA.19 LCK-independent CAR signaling was strongly reduced without the CD28 intracellular domain and by AYAA mutation (Figures 2C and S2E). To further substantiate the importance of CD28 to LCK-independent signaling, we expressed CD80 and CD86 in CHO-APCs to enable endogenous CD28 signaling activation in trans (Figure S2F). When endogenous CD28 was co-activated by CD80/86, tumor necrosis factor (TNF) and interferon γ (IFNγ) secretion of disLCK-CD137-CAR-T cells was restored (Figure 2D). LCK-deficient CAR1-Jcam and TCR-Jcam cells also showed restored IL-2 production and downstream signaling when CD28 co-stimulation was provided (Figures S2G–S2I). Thus, LCK-independent CAR signaling required a CD28 co-stimulatory signal, mediated at least partly through the PYAP motif, either from CD28 intracellular domain in CAR or from endogenous CD28 molecules.
Figure 2.

LCK-independent CAR signaling relies on CD28 co-stimulatory domain
(A) The effects of recognition domain in disLCK-CAR-T cell signaling. Anti-CD19-CD28CAR construct has been replaced with anti-HER2 scFv. CHO-CD19- and HER2-expressing SKBR3 cells were used as target cells and mixed with its respective disLCK-CAR-T cells at a 1:1 ratio for 18 h. The top panel is the representative FACS data, and the bottom panel is the schematic of the constructs with different recognition domains and an analytical summary.
(B) The effects of intracellular domain in disLCK-CAR-T cell signaling. The top panel is the representative FACS data, and the bottom panel is the schematic of the constructs with different intracellular domains and an analytical summary.
(C) Cytokine secretion of disLCK-CAR-T cells with different CD28-CAR mutations. The top panel is the representative FACS data, and the bottom panel is the schematic of CD28-CAR mutations and an analytical summary.
(D) Cytokine secretion of disLCK-CD137-CAR-T with or without CD80/CD86 co-stimulation on endogenous CD28. The top panel is the representative FACS data, and the bottom panel is the analytical summary.
Data are representative of 3 independent experiments, plotted as mean ± SD of technical triplicates. p values denoted as in the Figure 1 legend by Student’s t test.
See Figure S2 for additional data.
SFK FYN mediates LCK-independent CAR signaling
To determine if any other SFK was responsible for initiation of LCK-independent CAR signaling, we used SFK pan-inhibitor PP2, resulting in inhibition of both CAR and TCR signaling in primary T cells as well as Jurkat cells (Figures 3A, S3A, and S3B). LCK and FYN are the main SFKs in T cells,20 so we used the selective inhibitors A770041 and SU6656 to target LCK and FYN, respectively.21,22 We initially used Jurkat T cells to study proximal TCR signaling,23,24 finding that LCK-independent CAR signaling in primary T cells was also found as in Jurkat T cells, as shown above (Figures 1, 2, S1, and S2). TCR-Jurkat was ∼7-fold more sensitive to the LCK inhibitor, whereas CAR-Jurkat was ∼1.5-fold more sensitive to the FYN inhibitor (Figures S3C and S3D). These data suggested that FYN could be the kinase activating downstream signaling from CAR. Western blot analysis in LCK-deficient Jcam cells showed that the activation site Y420 of FYN, corresponding to SFK phosphorylation site Y416, was phosphorylated after CAR-Jcam, but not TCR-Jcam, engagement with specific APCs, with a nearly 2-fold increase in FYN pY420 after 30 min (Figure 3B). To better validate the role of LCK and FYN, we used CRISPR-Cas9 to knock out (KO) LCK or FYN in Jurkat. After gRNA screening (Table S1) and single-cell sorting (Figure S3E), LCK-KO clone 20 and FYN-KO clone 8 were selected for further experiments (Figure 3C). The expression of CAR or TCR was equivalent in FYN-KO Jurkat, and lack of FYN was confirmed in CAR- or TCR-Jurkat FYN-KO (Figures S3F and S3G). In LCK-KO Jurkat, CAR, but not TCR, was still functional, consistent with the results above. Conversely, IL-2 production by CAR-Jurkat FYN-KO was dramatically reduced compared with the wild type (Figure 3D). Immunoprecipitation experiments showed that CD28-CAR associated with FYN upon activation (Figure 3E). This association weakened after mutation of the CD28 domain, and the strength of FYN association corresponded with the activation level, suggested by IL-2 production, supported by mutated CD28-CAR (Figure 3F). These data showed that CAR signaling with CD28 domain was more sensitive to, and could rely on, FYN instead of LCK.
Figure 3.

FYN is suggested to mediate CD28-CAR-T cell activation in the absence of LCK
(A) The cytotoxicity of primary CAR-T and TCR-T cells with or without PP2 inhibitor (10 μM). CAR-T targets CD19, and TCR-T targets HLA/A2-L2. Daudi cells, which express CD19 and were transduced with HLA/A2-L2, were used as target cells. The CAR-T cells were mixed with target cells at an E:T ratio of 4:1.
(B) Phosphorylation (pY420) of FYN in LCK-deficient CAR- or TCR-Jcam cells at different time points. APC stands for artificial antigen-presenting cell CHO-L2, to which the CAR and TCR responded. The number shown below indicates band intensity of pY420 relative to total FYN (FYN pY420 was stained by anti-pSRC family pY416).
(C) LCK and FYN expression in LCK or FYN-KO Jurkat clone after CRISPR-Cas9 editing. C-CBL was used as loading control.
(D) IL-2 production of TCR and CAR in LCK or FYN-KO Jurkat upon activation by CHO-L2.
(E) Association of FYN with CD28CAR variants. Jcam cells with different CD28CAR variants were activated by CHO-L2 for 5 min, and anti-Myc antibody was used to immunoprecipitate CAR. The association strength (intensity ratio) was calculated by the relative intensity of FYN to that of immunoprecipitated CAR.
(F) Correlation of FYN association strength with IL-2 production by Jcam cells with different CD28-CAR variants. The intensity ratio in abscissa is from (E), and the statistical significance is calculated by compare IL-2 production by CD28CAR variants with that of wild type (WT). The CAR and TCR mentioned above (B–E) both specifically target HLA/A2-L2, and CD28CAR was used.
Data are representative of 3 independent experiments (except 2 for E), plotted as mean ± SD of technical triplicates. p values denoted as in the Figure 1 legend by Student’s t test.
See Figure S3 for additional data.
disLCK-CAR-T cells selectively activate CAR signaling
Given that CD28-CAR and TCR signaling are mediated by different SFKs, we hypothesized that disLCK-CAR-T cells would not be activated via TCR. To test this hypothesis, our L2 peptide-specific CAR and a TCR with specificity against E183-91 (FLLTRILTI) epitope from hepatitis B virus (HBV), hence referred to as E183-TCR,25 were mono-transduced in Jurkat T cells designated as L2-CAR-T (1) and E183-TCR-T (2) or co-transduced into Jurkat (E183-TCR + L2-CAR-T [3]) or LCK-KO Jurkat (E183-TCR + L2-CAR-LCK KO-T [4]) (Figures 4A and S4). The E183-TCR + L2-CAR-T (iii) cells were able to produce IL-2 upon stimulation by either CHO-E183 or CHO-L2, showing that activation of CAR and TCR was unimpaired in a dual CAR + TCR system. However, CHO-L2, but not CHO-E183, induced IL-2 production in the E183-TCR + L2-CAR-LCK KO-T (iv) cells co-expressing CAR and TCR, showing that LCK deficiency allows signaling pathways for CAR, but not TCR, triggering (Figure 4A). In primary disLCK-CAR-T cells, anti-CD3 antibody (Ab) was used to activate endogenous TCR (Figure 4B). In contrast to CAR’s ability to activate disLCK-CAR-T cells, TCR signaling was strongly reduced, whereas TCR signaling was functional in conventional CAR-T cells (Figure 4C). Interestingly, CAR expression in conventional CAR-T cells was strongly downregulated upon endogenous TCR activation by anti-CD3 Ab but not in disLCK-CAR-T cells (Figure 4D). These data suggest that TCR and CAR signal triggering are differentiated in disLCK-CAR-T cells.
Figure 4.

disLCK-CAR-T cells selectively activate CAR signaling
(A) TCR and CAR responsiveness selectivity in LCK-sufficient or -deficient Jurkat T cells. The top panel shows the schematics of E183 peptide-specific TCR (E183-TCR) or LMP2 peptide-specific CAR (L2-CAR) expressed in LCK-sufficient (1–3) or -deficient Jurkat T cells (4). The bottom panel shows the responsiveness of each group against CHO-E183 or CHO-L2, respectively. CHO-E183 is a mono-peptide CHO APC presenting only the E183 peptide. Similarly, CHO-L2 presents only the LMP2A peptide.
(B) Schematics of activation of endogenous TCR in disLCK-CAR-T or conventional CAR-T cells. Anti-CD3 Ab was used to activate endogenous TCR.
(C) Cytokine secretion of CAR-T cells upon anti-CD3 activation. The left panel is the representative data, and the right panel is the statistical summary and analysis.
(D) CAR expression after anti-CD3 antibody activation. The left panel is the representative data, and the right panel is the statistical summary and analysis.
Data are representative of 3 independent experiments, plotted as mean ± SD of technical triplicates. p values denoted as in the Figure 1 legend by Student’s t test.
See Figure S4 for additional data.
disLCK-CAR-T cells enhance in vivo therapeutic efficacy
To investigate functional consequences of LCK-independent CAR signaling and whether disLCK-CAR-T cells could have practical applications, we compared disLCK-CAR-T with conventional CAR-T cells in vitro and vivo. disLCK-CAR-T cells with comparable CAR expression to conventional CAR-T showed significantly higher activation for cytokine secretion (Figures 5A and S5A) but no significant difference in cytotoxicity (Figures 5B, 5C, and S5B–S5D). However, in vivo efficacy of CAR-T cells was enhanced after disruption of LCK expression, as evaluated in both liquid and solid tumor mouse models (Figure 5D). In a standard mouse leukemia model, where 5 × 105 CD19-expressing Nalm-6 cells were injected, disLCK-CAR-T significantly improved in vivo efficacy compared with conventional CAR-T cells (Figure 5E). To further validate the impact from LCK disruption, we implemented a short hairpin RNA (shRNA) method to knock down LCK expression in CAR-T cells, with a non-targeting shRNA as control. The shLCK-CAR-T cells had comparable CAR expression to non-targeting contro (shNTC)-CAR-T cells, but the LCK expression in shLCK-CAR-T cells was greatly reduced (Figures S5E and S5F). At the dose that CAR-T cells showed significant therapeutic efficacy versus control, the shLCK-CAR-T cells significantly improved the in vivo efficacy compared with shNTC-CAR-T cells in terms of mouse survival and Nalm-6 cell numbers in vivo, as detected by luciferase signal (Figures 5E, S5G, and S5H). In a solid tumor mouse model using a breast cancer cell line, MCF7 cells expressing HER2 (Figure 5D), shLCK-CAR-T cells showed significantly enhanced in vivo performance. Tumor growth in the shLCK-CAR-T cell-treated group was reduced more dramatically than the shNTC-CAR-T cell-treatment group (Figure 5F).
Figure 5.

LCK-disrupted CAR-T cells enhance in vivo therapeutic efficacy
(A) Cytokine secretion of disLCK-CAR-T and conventional CAR-T (generated by lentivirus transduction) upon activation by target cells. The left panel is the representative FACS data, and the right panel is the analytical summary.
(B) Comparison of cytotoxicity of disLCK-CAR-T and conventional CAR-T to CD19 expressing Daudi cells at a serial E:T ratio.
(C) Real-time killing curve of disLCK-CAR and conventional CAR-T to CHO cells overexpressing CD19. Cell index represents the cell growth. Arrow points show when the CAR-T cells were added. The respective E:T ratio is indicated in parentheses.
(D) Schematic of in vivo mouse model of liquid cancer or solid tumor.
(E) Kaplan-Meier analysis of survival in standard leukemia cancer model (Nalm-6 cells and CAR-T cells were administered at 0.5 × 106 and 3 × 106 cells, respectively, log rank Mantel-Cox test). The CAR-T cells in left graph were generated as described in (A) (n = 3–4 mice per group) and were generated by lentivirus transduction. The right graph shows treatment with cells containing both shRNA (either non-targeting control [shNTC] or LCK targeting [shLCK]) and CAR expression cassette (n = 7 mice per CAR-T group, n = 4 for control group treated by PBS).
(F) In vivo efficacy of anti-solid tumor mouse model. 2 × 106 breast cancer cell line MCF7-HER2 cells were administered subcutaneously (s.c.) at day 0, and 8 × 106 anti-HER2CAR-T cells were administered on day 5. Tumor size was measured and monitored over time (n = 5 mice per group).
Data in (A)–(C) are representative of 3 independent experiments, plotted as mean ± SD of technical triplicates. p values denoted as in the Figure 1 legend using Student’s t test for (A) and two-way ANOVA for (B), (C), and (F).
See Figure S5 for additional data.
disLCK-CAR-T cell activation by FYN leads to a more proliferative, memory-retaining, and exhaustion-reduced state
FYN and LCK were reported to negatively regulate each other’s activation.20 This was observed when FYN and LCK were knocked out individually (Figure 3D). It is therefore possible that the activation profile of TCR signaling might be altered by more FYN activity when LCK is disrupted, leading to a distinct immunophenotype. This is predicted by analyzing which molecules in TCR signaling distinctly interact with FYN compared with LCK. We cross referenced and filtrated among different gene databases and curated a “TCR signaling molecules pool,” which contains 1,440 molecules involved in TCR signaling. Among them, 330 molecules were identified to be linked with FYN or LCK. Their respective link scores to FYN and LCK were acquired via the String webtool. These molecules were further divided into FYN uniquely linked (58), FYN more-strongly linked (11), FYN and LCK shared linked (149), LCK more-strongly linked (10), and LCK uniquely linked (102) (Figure 6A). The more-strongly linked molecule is defined by difference between link score to FYN and to LCK >0.3, and the uniquely linked are the ones showing connections only either to FYN or to LCK. FYN uniquely and more-strongly linked molecules (69) or LCK uniquely and more-strongly linked molecules (112) were grouped and analyzed in the KEGG signal pathway database to show their distinct impacts on TCR signaling pathways. FYN-group molecules were more involved in proliferation and survival signaling pathways, including PI3K-AKT signaling, Rap1 signaling, MAPK signaling, and Ras signaling. LCK-group molecules were more involved in immune action pathways, such as Th1, Th2, and Th17 cell differentiation. These signal pathway involvements were further substantiated by clustering the molecular links within the FYN or LCK groups (Figures S6A and S6B). This result implies that disLCK-CAR-T cells could be more prone to proliferation and survival by enhanced FYN activity and would prevent exhaustion from overactivated immune responses.
Figure 6.

disLCK-CAR-T cell activation by FYN leads to a more proliferative, memory-retaining, and exhaustion-reduced state
(A) Signal pathway analysis of FYN and LCK group molecules. The 330 molecules linked to either FYN or LCK can be divided into FYN unique (blue, 58 molecules); FYN more-strongly linked (light blue, 11 molecules); shared linked (dark gray, 149 molecules); LCK more-strongly linked (orange, 10 molecules); and LCK unique (red, 102 molecules). FYN or LCK unique and strongly linked molecules are grouped in the FYN or LCK group, respectively, and analyzed in the KEGG signal pathway database. The pathways are ranked by the number of genes in the respective pathways.
(B) CAR-T cell proliferation upon target cell activation. The proliferation event was detected by FACS after 5-day incubation with Nalm-6 cells. The left panel shows the representative graph, and the right panel is the statistical summary.
(C) CAR, exhaustion, and memory marker expression in resting T cells (5 days after sorting and restimulation by feeder cells).
(D) Radar chart summary of exhaustion, and memory marker expression in T cells after encountering target cells at different E:T ratios. Axes show the percentage of expression of each marker.
(E) Memory subtypes of CAR-T cells after stimulation at different E:T ratios. T cells were stained with anti-CD45RA and anti-CD62L after 18-h incubation with Daudi cells at the corresponding E:T ratio.
Data are representative of 3 (B) or 2 (C–E) independent experiments, plotted as mean ± SD of technical triplicates. p values denoted as in the Figure 1 legend calculated by Student’s t test.
See Figures S6 and S7 for additional data.
In vitro evaluation of disLCK-CAR-T supported this implication. As shown in Figure 6B, the disLCK-CAR-T proliferated strongly upon antigen stimulation. Differences in the quiescent state were evident: conventional CAR-T cells had a lower expression of the central and stem-cell memory marker CD62L (Figures 6C and S7A). After stimulation, we observed that conventional CAR-T cells had higher expression of exhaustion molecules and lower CD62L expression (Figure 6D). These changes were more dramatic as the effector-to-target (E:T) ratio decreased. disLCK-CAR-T cells had more of a memory phenotype than conventional CAR-T cells, with a larger proportion of the less-differentiated stem cell memory (TSCM: CD45RA+CD62L+) and central memory (Tcm: CD45RA–CD62L+) T cells (Figure 6E). The induction of enhanced memory and reduced exhaustion due to targeting the LCK gene were verified by comparison with targeting another gene locus: β2-microglobulin (B2M). Similar to conventional CAR-T, the disB2M-CAR-T cells showed a more differentiated memory state and more exhaustion marker upregulation upon stimulation than disLCK-CAR-T cells (Figures S7B–S7E).
disLCK-CAR-T cells have an enhanced therapeutic profile in vivo
To reflect the distinct immune-state in vitro, disLCK-CAR-T and conventional CAR-T cells were immunophenotyped ex vivo after administration (Figure 7A). disLCK-CAR-T cells showed a significantly higher expression of CD45RO+ memory cells at both days 11 and 18 compared with conventional CAR-T cells (Figure 7B). In addition, conventional CAR-T cells upregulated exhaustion markers PD-1, TIM-3, and LAG3 more than did disLCK-CAR-T cells. The percentage of exhaustion marker-negative disLCK-CAR-T cells was notably higher than that of conventional CAR-T cells (Figure 7C). Besides this different immunophenotype, the number of disLCK-CAR-T cells was significantly higher at days 10 and 16 in vivo compared with conventional CAR-T cells (Figures 7D and S7F). We consolidated the in vivo performance data and performed principal-component analysis (PCA). disLCK-CAR-T and conventional cells can be distinguished as different groups (Figure 7E). disLCK-CAR-T cells were enriched in the more persistent, memory-prone, and exhaustion marker-reduced cluster, contrasting significantly with conventional CAR-T cells (Figure 7F). In summary, these data suggest that tilting CD28-CAR-T activation toward FYN via disruption of LCK expression, results in a more proliferative, memory-retaining, and less exhausted phenotype, explaining the enhanced therapeutic performance of disLCK-CAR-T cells in vivo.
Figure 7.

disLCK-CAR-T cells have an enhanced therapeutic profile in vivo
(A) Schematic of the ex vivo analysis of CAR-T cells after administration in vivo.
(B) Bone marrow CAR-T cells were analyzed at day 11 or 18 after cancer cells injected, and percentage of memory T cells (CD45RO+) was detected by FACS. The left panel shows the representative FACS data, and the right panel shows the statistical summary. n = 4 mice per group, each dot = one mouse, CAR-T = blue square, disLCK-CAR-T = red round dot.
(C) Exhaustion marker expression on the CAR-T cells from bone marrow analyzed on day 10. The left panel shows the representative FACS data, and the right panel shows the statistical summary. n = 4 mice per group, each dot = one mouse, CAR-T = blue square, disLCK-CAR-T = red round dot. Cells with 3, 2, or 1 exhaustion marker expression (TIM-3, LAG-3, and PD-1) are designated as exhaustion (+), and cells without exhaustion marker expression are designated as exhaustion (−).
(D) Cell number of the CAR-T cells from bone marrow and spleen at days 10 and 16 after cancer cells were injected. 1.8 × 106 CAR-T cells were administered in each mouse at day 4 after 1.5 × 106 Nalm-6 cells were injected at day 0. n = 4 mice per group.
(E) Principal-component analysis (PCA) of conventional CAR-T and disLCK-CAR-T cell in vivo performance. Each dot = one mouse, disLCK-CAR-T = red round dots, conventional CAR-T = blue square dots.
(F) Heatmap of the in vivo performances of CAR-T cells after PCA. The columns are biological repeats. Each row represents PCA values of ex vivo data of each factor, memory is the CD45RO+ group, and exhaustion (−) and (+) are defined the same as in (C).
Data in (B)–(D) are means ± SD. Boxes in (B) (right panels) represent 95% confidence intervals. p values denoted as in the Figure 1 legend using Student’s t test.
See Figure S7F for additional data.
Discussion
Although huge successes have been seen with CAR-T therapy in the clinic, CAR signaling itself has not yet been fully defined.3 To this end, we intended to identify molecules specifically relevant to CAR, but not TCR, signaling on the basis that this could reveal potentially useful properties specific to CAR signaling. Surprisingly, CAR signaling was triggered normally even in the absence of LCK. In addition, the cytokine release by disLCK-CAR-T cells was significantly higher than that of conventional CAR-T cells. This could be related to the signal regulatory function of LCK.26 On the other hand reduced LCK expression significantly decreases TCR signaling strength,27 explaining why endogenous TCR signaling was dramatically inhibited in disLCK-CAR-T cells. This trait of splitting CAR and TCR signal activation in the absence of LCK could be exploited to generate allogeneic CAR-T. Unlike knocking out TCR in CAR-T cells, which results in lower persistence in vivo,28 TCR-retaining disLCK-CAR-T cells could potentially sustain tonic signaling, thus prolonging survival of allogeneic CAR-T cells.29 This point could be inferred from our observation that ERK in LCK-deficient TCR-T cells was phosphorylated upon stimulation by pMHC, perhaps due to a PLCγ1-independent signaling pathway.30,31
This non-canonical CAR signaling pathway has not previously been reported in CAR-T studies. Systematic scrutinization of CAR domains revealed that the CD28 co-stimulatory domain plays a critical role. FYN, the other SFK active in T cell signaling, has been shown to bind to TCR in proximal signaling.32,33 It could also be important in non-canonical CAR signaling. Deletion of FYN or LCK in Jurkat cells using CRISPR-Cas9 showed that CAR signaling relies more on FYN than on LCK. Despite the limitations of Jurkat cells, proximal TCR signaling mechanisms in Jurkat resemble those in primary T cells.23,24 Hence, these results suggested that FYN could be the major kinase orchestrating CAR signal transduction under physiological conditions. We showed that LCK-independent CAR signaling is mediated by the interplay between FYN and the CD28 domain. The role of CD28 in CAR is not primarily related to a spacer effect—by which CD3ζ is distanced from the cell membrane—because this spacer effect was not sufficient to trigger LCK-independent signaling for CD137-CAR, and the reduction of LCK-independent CAR signaling was observed with point mutations in CD28. Nonetheless, the spacer effect may make CD3ζ more accessible to FYN compared with LCK. FYN is localized more freely than LCK, including association with the endoplasmic reticulum (ER) or cytoskeleton.34,35,36,37 This could favor CD3ζ phosphorylation by FYN given that CD3ζ is the preferred substrate for FYN, whereas LCK preferentially phosphorylates CD3ε.24 We attribute LCK-independent CAR signaling primarily to signal functions of the CD28 module, such as the FYN recruitment by CD28 shown in this study. This could explain why the third-generation CAR showed LCK-independent signaling, but to a limited degree, where CD28 signaling was disrupted when the CD137 domain was between CD28 and CD3ζ domains. The importance of the transmembrane has not been explored in this study, but it may play a role in mediating the LCK-independent CAR signaling to a certain degree given that it has been reported to influence downstream CAR signaling.38 Interestingly, first-generation CAR or TCR, as well as CD137-CAR, was able to resume signal activation in the absence of LCK when endogenous CD28 was co-stimulated in trans. Based on our findings, we hypothesize that CD28 may bring FYN to the IS once co-stimulated, partially to fulfill LCK’s function. The kinase switch in CD28-containing CAR compared with CD137-containing CAR potentially explains their distinct performances as seen in previous studies,20,27,37 FYN and LCK are distinct in terms of signaling kinetics, pathways, and strength. FYN-mediated CD28-CAR signaling would, therefore, show different responses than CD137-CAR signaling, where their initiations are controlled by different kinases. Although CD28 co-stimulatory signaling has been studied intensively, it has seldom been explored under LCK-deficient conditions.18 Since LCK and FYN have both been shown to interact with CD28,39 CD28 signaling might be different when only FYN can interact with it. However, to what degree the CD28 co-stimulatory signaling pathways, such as the PI3K, PKCθ, and AKT pathways, have been affected after disruption of LCK is unknown.
The balance of FYN and LCK in T cells could naturally favor FYN signaling after LCK disruption, resulting in a favorable therapeutic performance that increases the overall in vivo efficacy. FYN and LCK both have uniquely linked and more-strongly linked molecules (FYN or LCK group) in TCR signaling. Signal pathway analysis reveals that the interactions within the FYN-specific group would provide T cells with the tendency to proliferate and survive. This analysis is consistent with previous studies on FYN where FYN controls cell growth.40,41 On the other hand, the LCK-specific signaling partners are more relevant in immune activity. It is likely that CAR-T cells with LCK present are more easily overactivated and thus exhausted. For example, overactivated and chronic nuclear factor κB (NF-κB) signaling—which is more involved in the LCK-unique system in T cells—would lead to cellular senescence and apoptosis.42,43 This could be important for CD28-CAR-T cells, which may be chronically activated, leading to early exhaustion.44 Treating CD28-CAR-T cells with the LCK inhibitor dasatinib during culture or changing to a CD137 co-stimulatory domain, which would recruit THEMIS-SHP1 to inhibit LCK activity, could avoid early exhaustion.9,45 Dasatinib, however, has broad inhibition of the SFK and other kinases, including both LCK and FYN. This could obscure the fact identified in this study that FYN could be more important to CD28-CAR. Nevertheless, it will not affect its application to control CAR signaling and as a pharmacological switch since LCK and FYN are both inhibited by using dasatinib.46 Overall, disLCK-CAR-T cells became more proliferative, showed resistance to exhaustion, and tended to retain memory phenotype, leading to their increased in vivo efficacy and suggesting significant benefits for translation in clinical settings.
In conclusion, we report that CAR-T signaling can be triggered without the presence of LCK. We show that this non-canonical signaling pathway bypassing LCK is related to the CD28 co-stimulatory domain. Moreover, FYN is revealed to be more crucial than LCK in CD28-CAR signal transduction. This kinase switch provides distinct properties for disLCK-CAR-T cells, including tolerance to inhibitory signals and a propensity to proliferate and differentiate to memory cells. The disLCK-CAR-T cells could provide a more durable and effective CAR-T therapy for clinical use and suggest that editing signaling molecules may be a fruitful avenue to construct the next generation of CAR-T technologies.
Limitations of the study
The study primarily focuses on CD8+ T cells. CD4+ T cells were not part of the study. Although LCK-independent CAR signaling should be replicable, the impact to CD4+-CAR-T cells could be different compared with CD8+-CAR-T cells. Future studies should include both CD4+ and CD8+ T cells. In addition, omics studies such as transcriptome and phosphoproteomics were not performed. With the disruption of LCK in CAR-T cells, cell fate and signaling associated with CD28 co-stimulatory signaling after activation could be distinct. A better understanding of the disLCK-CAR-T cells can be acquired after such omics studies.
STAR★Methods
Key resources table
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| Myc-Tag (9B11) Mouse mAb (PE Conjugate) | Cell Signaling Technology | Cat# 3739S; RRID:AB_10889248 |
| Myc-Tag (71D10) Rabbit mAb (Alexa Fluor® 700 Conjugate) | Cell Signaling Technology | Cat# 42136S; RRID:AB_2799214 |
| Myc-Tag (9B11) Mouse mAb (Alexa Fluor® 647 Conjugate) | Cell Signaling Technology | Cat# 2233S; RRID:AB_823474 |
| Phospho-Src Family (Tyr416) (D49G4) Rabbit mAb | Cell Signaling Technology | Cat# 6943S; RRID:AB_10013641 |
| Phospho-PLCγ1 (Tyr783) Antibody | Cell Signaling Technology | Cat# 2821S; RRID:AB_330855 |
| Phospho p44/42 MAPK (Erk1) (Tyr204)/(Erk2) (Tyr187) (D1H6G) Mouse mAb | Cell Signaling Technology | Cat# 5726S; RRID:AB_2797617 |
| Phospho-CD3ζ (Tyr142) Antibody | Cell Signaling Technology | Cat# 67748S |
| c-Cbl Antibody | Cell Signaling Technology | Cat# 2747S; RRID:AB_2275284 |
| PLC-γ1 Antibody | Cell Signaling Technology | Cat# 2822S; RRID:AB_2163702 |
| Purified Mouse Anti-ERK1 Clone MK12 (RUO) | BD Biosciences | Cat# 610031; RRID:AB_397448 |
| Anti-Human CD8 BUV395 | BD Biosciences | Cat# 563795; RRID:AB_2722501 |
| BV421 Mouse Anti-Human IFN-γ | BD Biosciences | Cat# 562988; RRID:AB_2737934 |
| Purified anti-Fyn [FYN-59] | Biolegend | Cat# 626502; RRID:AB_2278824 |
| Anti-Human CD28 Alexa 488 | Biolegend | Cat# 302916; RRID:AB_493100 |
| PE anti-human CD80 [2D10] | Biolegend | Cat# 305207; RRID:AB_314503 |
| FITC anti-human CD19 [4G7] | Biolegend | Cat# 392507; RRID:AB_2750098 |
| Brilliant Violet 421™ anti-human CD86 Antibody | Biolegend | Cat# 374211; RRID:AB_2728393 |
| APC anti-human CD3 [OKT3] | Biolegend | Cat# 317318; RRID:AB_1937212 |
| PE anti-human CD279 (PD-1) Antibody | Biolegend | Cat# 329905; RRID:AB_940481 |
| Brilliant Violet 711™ anti-human CD279 (PD-1) Antibody | Biolegend | Cat# 367427; RRID:AB_2721554 |
| Fluor® 488 anti-human CD279 (PD-1) Antibody | Biolegend | Cat# 329935; RRID:AB_2563593 |
| PE Mouse Anti-Mouse CD366 (TIM-3) | Biolegend | Cat# 345006; RRID:AB_2116576 |
| Brilliant Violet 421™ anti-human CD223 (LAG-3) [11C3C65] | Biolegend | Cat# 369314; RRID:AB_2629797 |
| Brilliant Violet 421™ anti-human CD62L [DREG-56] | Biolegend | Cat# 304828; RRID:AB_2562914 |
| PE/Cy7 anti-human CD45RO [UCHL1] | Biolegend | Cat# 304229; RRID:AB_11203903 |
| anti-human CD8A APC (REAfinity) | Miltenyi biotech | Cat# 130-110-679; RRID:AB_2659237 |
| Anti-Human TNF alpha PE | eBioscience | Cat# 12-7349-82; RRID:AB_466208 |
| Anti-Human CD3 Functional Grade Purified | eBioscience | Cat# 16-0037-85; RRID:AB_468855 |
| Anti-Human LCK (3A5) | Santa Cruz Biotechnology | Cat# sc-433; RRID:AB_627880 |
| goat anti-mouse IgG (H + L) secondary antibody Alexa Fluor 647 | Thermo Fisher Scientific | Cat# A-21235; RRID:AB_2535804 |
| IRDye 800CW Goat anti-Mouse IgG2b | LI-COR | Cat# 926–32352; RRID:AB_2782999 |
| IRDye 680LT Goat anti-Rabbit | LI-COR | Cat# 926-68021; RRID:AB_10706309 |
| HLA-A2-L2 TCR-like antibody | Paul A. MacAry lab13 | N/A |
| Bacterial and virus strains | ||
| NEB Stable Competent E.coli (High Efficiency) | New England Biolabs (NEB) | Cat# C3040H |
| Biological samples | ||
| human peripheral blood mononuclear cells (PBMCs) | Healthy donors | N/A |
| Chemicals, peptides, and recombinant proteins | ||
| SFK inhibitor PP2 | Sigma-Aldrich | Cat# P0042 |
| Lectin from Phaseolus vulgaris (red kidney bean) | Sigma-Aldrich | Cat# L9017 |
| polyethylenimine (PEI) | Sigma-Aldrich | Cat# 408727 |
| NP-40 Alternative | Sigma-Aldrich | Cat# 492016 |
| LCK inhibitor A770041 | MedChemExpress | Cat# HY-11011 |
| FYN inhibitor SU6656 | SelleckChem | Cat# S7774 |
| Indo-1, AM | Thermo Fisher Scientific | Cat# I1223 |
| CellTrace™ Violet | Thermo Fisher Scientific | Cat# C34557 |
| Dynabeads™ human T-activator CD3/CD28 | Thermo Fisher Scientific | Cat# 11132D |
| GAG (SLYNTVATL) peptide | GenScript | N/A |
| LMP2A426-434 (CLGGLLTMV) peptide | GenScript | N/A |
| D-Luciferin | PerkinElmer | Cat# 122799 |
| Biotarget medium | Biological Industry | Cat# 05-080-1A |
| Human platelet lysate (Ultra-GRO™-Advanced) | AventaCell | Cat# HPCFDCRL50 |
| Human IL-2 | R&D System | Cat# 202-IL-050 |
| Human IL-7 | R&D System | Cat# 207-IL-025 |
| Human IL-15 | R&D System | Cat# 247-ILB-025 |
| LIVE/DEAD® Fixable Near-IR Dead Cell Stain Kit | Life Technologies | Cat# L34976 |
| In-Fusion HD cloning kit | Clontech | Cat# 639648 |
| Q5 mutagenesis Kit | New England Biolabs (NEB) | Cat# E0554S |
| Cas9-NLS protein | UC Berkeley-Qb3 lab | N/A |
| Protein Transport Inhibitor (Containing Brefeldin A) | BD | Cat# 555029 |
| Protein G Dynabeads | Invitrogen | Cat# 10003D |
| Critical commercial assays | ||
| IL-2 ELISA assay | Invitrogen | Cat# 88-7025-88 |
| CytoTox 96® Non-Radioactive Cytotoxicity Assay | Promega | Cat# G1780 |
| 96-well RTCA E-plates | Agilent | Cat# 300601010 |
| Experimental models: Cell lines | ||
| Jurkat 76 | Mirjam Heemskerk47 | N/A |
| Raji | ATCC | Cat# CCL-86 |
| Nalm-6 | ATCC | Cat# CRL-3273 |
| Daudi | ATCC | Cat# CCL-213 |
| MCF-7 | ATCC | Cat# HTB-22 |
| SKBR3 | ATCC | Cat# HTB-30 |
| Jurkat cam1.6 | ATCC | Cat# CRL-2063 |
| Lenti-X | Takara Bio | Cat# 632180 |
| Nalm-6-Luc | This manuscript | N/A |
| MCF-7-HER2 | This manuscript | N/A |
| Experimental models: Organisms/strains | ||
| NOD.Cg-PrkdcscidIL2Rgtm1wjl/SzJlnv (NSG) | InVivos | Strain code: 005557 |
| Oligonucleotides | ||
| gRNA sequence for Cas9-KO | See Table S1 | N/A |
| shLCK sequence: CTGCAAGACAACCTGGTTATC |
Methi et al.48 | PMID: 16301647 |
| shNTC sequence: CCTAAGGTTAAGTCGCCCTCG |
Sarbassov et al.49 | PMID: 15718470 |
| Recombinant DNA | ||
| Anti-CD19 scFv (FMC63) | Laurence Cooper lab50 | N/A |
| Anti-HER2 scFv (4D5-5) | Richard Morgan lab51 | N/A |
| LMP2A specific TCR-like scFv | Paul A. MacAry lab13 | N/A |
| CD28CAR | This manuscript | N/A |
| CD137CAR | This manuscript | N/A |
| pLV-shRNA-CAR | This manuscript | N/A |
| pLV | Vectorbuilder | N/A |
| pRP[Exp]-CMV > gag:pol:RRE | Vectorbuilder | VB160226-10009 |
| pRP[Exp]-RSV > Rev | Vectorbuilder | VB160226-10011 |
| pRP[Exp]-CMV > VSVG | Vectorbuilder | VB160226-10010 |
| pcDNA3-Clover | Addgene | Cat# 40259 |
| Software and algorithms | ||
| FlowJo 10.0 | FlowJo, LLC | https://www.flowjo.com/ |
| GraphPad Prism version 7.0 | GraphPad software | https://www.graphpad.com |
| RTCA 2.0 | Agilent | https://www.agilent.com |
| ClustVis | University of Tartu | https://biit.cs.ut.ee/clustvis/ |
| GSEA | Broad Institute | https://www.gsea-msigdb.org/gsea/index.jsp |
| GO | GO Consortium | http://geneontology.org/ |
| The Human Protein Atlas | The Human Protein Atlas | https://www.proteinatlas.org/ |
| DICE | La Jolla Institute for Immunology | https://dice-database.org/ |
| String webtool | String Consortium | https://string-db.org/ |
| DAVID Bioinformatics Resource | Laboratory of Human Retrovirology and Immunoinformatics | https://david.ncifcrf.gov/summary.jsp |
Resource availability
Lead contact
Further information and requests for resources and reagents should be directed to and will be fulfilled by the lead contact, Nicholas R.J. Gascoigne (micnrjg@nus.edu.sg).
Materials availability
Reagents generated in this study will be made available on request, but we may require a payment and/or a completed Materials Transfer Agreement based on NUS’s legal requirement if there is potential for commercial application.
Experimental model and subject details
Cell lines and cell culture
Endogenous TCR and co-receptor-deficient Jurkat76 was a kind gift from Prof Mirjam Heemskerk47 (Leiden Univ, NL). Other cell lines (Raji, Nalm-6, Daudi, MCF-7, SKBR3 including LCK-deficient Jurkat cam1.6) were from the American Type Culture Collection (ATCC). No commonly misidentified cell lines were used (ICLAC database). Jurkat lines were maintained in RPMI-1640 media (Hyclone) supplemented with 10% fetal bovine serum (Hyclone), 2 mM L-glutamine (Gibco) and MEM non-essential amino acid (Gibco) (cRPMI) in humidified 5% CO2 incubator at 37°C. Human embryonic kidney epithelial cells (HEK293) were cultured in DMEM (Hyclone) supplemented with 10% fetal bovine serum (Hyclone), 2 mM L-glutamine (Gibco) and MEM non-essential amino acid (Gibco). Tetracycline-Regulated Expression (T-REx) CHO cell line was purchased from Invitrogen and used for the generation of artificial APC. The CHO cells were cultured in Ham’s F-12 (Gibco) medium with 10% fetal bovine serum (Hyclone), 2 mM L-glutamine (Gibco) and MEM non-essential amino acid (Gibco). Transfections of Single-chain trimer MHC constructs into CHO were performed using the polyethylenimine (PEI) method. After transfection, cells were drug-selected (1.0 mg/mL G418, Hyclone), then single cell sorted. The pMHC complex expression was checked regularly by flow cytometry, and cell lines used were checked routinely to detect mycoplasma contamination.
Primary CD8+ T cell activation and culture
Naive CD8+ T-cells were isolated isolated from blood using RosetteSep human CD8+ T cell enrichment cocktail (Stemcell) and Ficoll (GE Healthcare Life Sciences) gradient centrifugation. Naive CD8+ T-cells were stimulated by anti-CD3+CD28 Dynabeads (ThermoFisher) in Biotarget medium (Biological Industry) supplemented with 4% human platelet lysate (Ultra-GRO-Advanced, AventaCell) with 100U/mL IL-2 (R&D System) for 48hrs. Activated CD8+ T-cells were maintained in medium containing 100 U/mL IL-2, 10 ng/mL IL15 and 10 ng/mL IL7 (R&D System). T-cells were restimulated every 14-20 days by feeder cells, which were freshly isolated peripheral blood mononuclear cells (PBMC) and irradiated at 30 Gy. PBMC and T-cells were resuspended at a ratio of 2:1 in the medium with concentrations of 100U/mL IL-2, 10 ng/mL IL7, 10 ng/mL IL15 and 1.5 μg/mL of Lectin from Phaseolus vulgaris (Sigma-Aldrich). Blood collection protocol was approved by NUS IRB. Informed consent was obtained from all donors.
In vivo animal model
NOD.Cg-PrkdcscidIL2Rgtm1wjl/SzJlnv (NSG) female mice, 6-8 weeks old, (InVivos) were used for in a standard leukemia model using Nalm-6 cells, which has been transduced with luciferase, and a solid tumor model using MCF7-HER2 cells. CAR-T-cells were sorted by FACSFusion sorter and restimulated by feeder cells at day −3. Cancer cells were administered intravenously (i.v.) via tail (Nalm-6) or subcutaneously (MCF7- HER2). Both cell lines produced even tumor burdens in our experiments, and no mice were excluded before treatment. Expanded CAR-T-cells were administered i.v. at day 4 after cancer cell injection or at day 5 after solid tumor initial growth. The mice were euthanized when paralysis was observed or tumor size exceeded 2000 mm3. For ex vivo CAR-T-cell phenotyping, bone marrow and spleen cells were extracted and analyzed by FACS. Animal Protocols were approved by NUS IACUC.
Method details
Constructs and sequences
CARs with CD28 and CD137 costimulatory sequences were synthesized and cloned into lentiviral vector pLV. This and associated packaging plasmids, bearing Gag/Pol, Rev, and VSV-G respectively, were supplied by Vectorbuilder. Human HER2 genes were cloned from a human cDNA library in-house. The scFv constructs for TCR-like antibodies13 were produced in P.A.M.’s lab, and FMC63 scFv and 4D5 scFv were used for anti-CD19CAR and anti-HER2CAR, respectively.50,51 TCR specific for HLA-A∗02:01 with peptide LMP2A426-434 (from EBV) was a generous gift from Hans Stauss (University College London). Single-chain trimer GAG-HLA-A2 was a gift from Keith Gould (Imperial College London). Peptide mutagenesis: GAG (SLYNTVATL) to LMP2A426-434 (CLGGLLTMV) (L2); CD28 Y170F, P187,190A, and other mutations were made with Q5 mutagenesis Kit (New England Biolabs). Molecular cloning work used In-Fusion HD cloning kit (Clontech), and single-chain trimer MHC constructs were cloned into pcDNA3-Clover (Addgene plasmid #40259) to generate artificial antigen presenting CHO cells.52,53,54 shRNA-CAR constructs were based on pLV-CAR vector described above, in which a cassette of the human U6 and shRNA sequence were added. shLCK sequence48:5′-CTGCAAGACAACCTGGTTATC-3′ shNTC sequence49: 5′-CCTAAGGTTAAGTCGCCCTCG-3′.
Lentivirus production and transduction
6.5 × 105 HEK293-Lenti-X cells/well were seeded onto 6 well plates one day before transfection and incubated at 37°C with 5% CO2. The cells were transfected with packaging plasmids and lentiviral vector using polyethylenimine (PEI), and the medium replaced after 12h. Viral supernatant was harvested twice in the following 2days. The collected viral supernatant was titred, filtered through 0.45μm membrane (Millipore) and concentrated by ultracentrifugation filter (100kDa, Amicon). For lentivirus transductions, polybrene and HEPES were added at 8-10 μg/mL and 10mM, respectively, with Jurkat or CD8+ T-cells after 48h initial activation by anti-CD3+CD28 Dynabeads (ThermoFisher) at 106/mL, followed by spinoculation at 2,500 rpm for 2 hrs at 32°C. For CHO cell transduction, viral solution was directly added without spinoculation for 24h. Cells and viral solutions were separated after transduction.
CRISPR-Cas9 knock-out and knock-in genetic editing
Cas9 lentivector was obtained from Addgene (#52961).55 The gRNA sequences for FYN and LCK were retrieved from http://chopchop.cbu.uib.no/and are shown in the Table S1. Cas9 sequence was linked with mTagBFP by P2A cleavable linker. The Cas9 lentivector was used to make lentivirus for transduction of Jurkat. Single cells were sorted by mTagBFP and western blotting was used to screen LCK or FYN KO Jurkat clones. In primary CD8+ T cells, LCK targeted homologous directed repair (HDR) was performed as previously described.56 The LCK gRNA2, GCCGGGAAAAGTGATTCGAG, was selected and chemically modified. Full sgRNA sequence was, 5′-G∗C∗C∗GGGAAAAGUGAUUCGAGGUUUUAGAGCUAGAAAUAGCAAGUUAAAA UAAGGCUAGUCCGUUAUCAACUUGAAAAAGUGGCACCGAGUCGGUGCU∗U∗U∗U-3’. Asterisk (∗) represents 2′-O-methyl 3′ phosphorothioate. The Cas9-NLS protein (New England Biology) was incubated with sgRNA at a molecular ratio of 1:2 at 37°C for 30min to form ribonucleoprotein (RNP) complexes. The dsDNA donor was designed so that 1kb of the homologous arm was flanking the CAR construct on each side. 120pmol of RNP and 2μg of dsDNA were electroporated into 106 activated primary CD8+ T-cells by Amaxa P3 Primary Cell Kit (V4XP-3024) and Amaxa 4D electroporation system (Lonza) via program EH115. The CAR + population was stained by anti-Myc tag conjugates (Cell Signaling Technology) and later sorted by FACSFusion sorter (BD) in flow cytometry core facility team of The Life Sciences Institute (LSI), NUS.
Calcium flux assay
The calcium flux assay was performed as previously described.57 In brief, cells were suspended at 107/mL in PBS (PBS) and loaded with 2 μM Indo-1 AM for 30 min at 37°C, followed by washing twice with cRPMI. Cells were pre-warmed to 37°C for 10min before analysis and kept at 37°C during analysis. For cell stimulation, an HLA-A2-L2 monomer was pre-refolded, biotinylated58 and crosslinked with streptavidin Alexa 647 (ThermoFisher) to form tetramer, that was used to stimulate cells. Mean fluorescence ratio of Indo-1high/Indo-1low was calculated using FlowJo kinetics program.
T cell stimulation and cytokine secretion assay
This was performed largely as described.53,54 Artificial antigen presenting CHO cells (APC-CHO) were seeded onto a 96 well flat plate at 2–3x104/well one day before use. Each CAR-T-cell or TCR-T-cell sample was counted and 2 × 105 cells/well was added into APC-CHO pre-seeded plate for 18h. After incubation, supernatant was collected for IL-2 ELISA assay (Invitrogen). T cell pellets were resuspended for immunophenotyping or for another round of stimulation. For primary CAR-T-cell cytokine secretion, target cells (at 1:1 ratio) or precoated anti-CD3 antibody (5 μg/mL) were used to activate CAR signaling or endogenous TCR signaling, respectively. CAR-T-cells were stimulated together with Brefeldin A in one well of a 96 well plate for 6h. After stimulation, cells were collected and stained with anti-CD8 for T cell gating and anti-Myc to measure CAR expression. T-cells were then further permeabilized and stained with anti-TNF and IFNγ before FACS analysis. For proliferation experiments, CAR-T-cells labeled with CellTrace Violet and Nalm-6 cells were mixed at a ratio of 1:1 at 5 × 104 cells/well in 96 well plates. Cells were collected after 5 days and analyzed by FACS.
Cytotoxicity assay
Target cells were seeded at 4 × 104/well in U-bottom 96 well plates, followed by adding 4 × 103 to 4 × 105 CAR-T-cells at effector to target (E:T) ratio 0.1:1 to 10:1. Cells were cocultured for 18 h at 37°C, 5% CO2. The supernatant was then collected, and the CytoTox 96 Non-Radioactive Cytotoxicity Assay (Promega) was used. Cytotoxicity (%) was calculated by formula (LDHrelease-negative)/(LDHmaxrelease-negative). The cell pellet was suspended and stained with antibody conjugates to do immunophenotyping. For real-time killing experiments, xCelligence (Agilent) was used. 2 × 104 CD19-transduced CHO cells (CHO-CD19) were pre-seeded in 96-well RTCA E-plates and incubated for 24h. CAR-T-cells were added at designated ratio. Cell growth was monitored for additional 36h.
Western blotting and immunoprecipitation (IP)
106 cells/sample were lysed in NP-40 lysis buffer. Cell debris was pelleted, supernatant collected and heated with reducing protein loading buffer (Nacalai-Tesque). To detect phosphorylation after stimulation, 105 APC-CHO cells were seeded per well in a 24-well plate one day before stimulation. 106 CAR-T or TCR-T-cells were added into each well and cocultured for the designated duration. Cells were collected and prepared as above. To detect FYN association with CAR, 107 CAR-Jcam cells and 2 × 106 CHO-L2 cells were mixed for 5min in 1mL cRPMI. Stimulation was stopped by adding 10mL pre-cooled PBS. Cells were collected and lysed in 200μL NP-40 lysis buffer. Lysate was incubated overnight with protein G Dynabeads (Invitrogen, 10003D) with anti-Myc. IP samples were washed in lysis buffer and heated with reducing protein loading buffer. All samples were loaded in a 4-12% Bis-Tris gradient gel (NuPAGE, Invitrogen) and transferred to a PVDF membrane (Immobilon-FL Transfer Membrane, Millipore). The membrane was blocked using blocking buffer (Odyssey, LI-COR). The membrane was probed with different primary antibodies, followed after washing by secondary antibodies: IRDye 800CW Goat anti-Mouse IgG2b (Cat# 926–32,352, LI-COR) and IRDye 680LT Goat anti-Rabbit (Cat#926-68021). Visualization and quantification of the blot was by the LI-COR Odyssey infrared imaging system.
Flow cytometry and cell sorting
Flow cytometry experiments were conducted on BD LSR Fortessa X-20 (Becton Dickinson). Cell sorting was conducted on either Mo-Flo XDP (Beckman Coulter, Inc.) or SY3200 (Sony Biotechnology Inc.) by Flow Cytometry Laboratory, Immunology Program, National University of Singapore. Data analysis was performed on FlowJo.
Signal pathway analysis
The “T-cell receptor pathway”, “T cell signaling”, “T cell activation”, “T cell stimulation”, “T cell costimulation”, and “T cell inhibitory signaling” were queried in GSEA (https://www.gsea-msigdb.org/gsea/index.jsp), GO (http://geneontology.org/), and NCBI to create a total TCR signaling-related molecules pool. The molecules in the pool were later screened by their expression in Naive or activated CD8+ T-cells via The Human Protein Atlas (https://www.proteinatlas.org/) and Database of Immune Cell Expression (DICE) (https://dice-database.org/), where the proteins with low or no expression in CD8 T cells were filtered out. These filtered signaling molecule pools were then the “TCR signaling molecules pool”. Signaling molecules within the “TCR signaling molecules pool” that interact to FYN or LCK and their respective link score were acquired via String webtool (https://string-db.org/). Signal pathway identification was done by analyzing the FYN or LCK group molecules (described in the main text) in KEGG pathway database via DAVID Bioinformatics Resource (https://david.ncifcrf.gov/summary.jsp).
Statistical information and data analysis
Two-tailed Student’s t test or two-way ANOVA analysis was performed for column data or curve data respectively by using GraphPad Prism 7. The data meet the assumptions of the tests. Variance is similar between the compared groups. For principal component analysis (PCA), in vivo performance factors were consolidated, and the data analyzed by ClustVis (https://biit.cs.ut.ee/clustvis/), obtaining converted PCA values for each factor acquired.
Acknowledgments
We thank the flow cytometry core facility team of The Life Sciences Institute (LSI), NUS, for sorting and Professors Hans Stauss, Antonio Bertoletti, and Keith Gould for providing TCR and scHLA constructs; Professors Dario Campana and Haiyan Liu for support and helpful suggestions on animal studies; and Dr. Gan Shu Uin for helpful suggestions on virus production. This work was funded by NUS ILO TAP grant TAP2002019-04-25, Singapore Ministry of Health’s National Medical Research Council MOH-000523, and Ministry of Education NUHSRO/2020/110/T1/SEED-MAR/06. L.W., Q.W., and J.L. were supported by research scholarships from Yong Loo Lin School of Medicine.
Author contributions
L.W. designed, conducted experiments, and analyzed the data; J.B. and N.R.J.G. provided discussion and co-designed experiments; Q.W. provided discussion and support on LCK analysis; P.D.S.V., J.X.H.O., L.-z.W., Y.S., J.C.T., J.Y., V.J.Y.T., Y.L.C., and T.Y.Y.T. provided technical or practical help; C.K.T.K. prepared the TCR signal molecule pool for bioinformatic analysis. J.L. and P.A.M. provided Ab sequences and reagents; and L.W., J.B., and N.R.J.G. wrote the paper with input and final approval from all authors.
Declaration of interests
National University of Singapore has filed patents based on these findings. P.A.M. is a shareholder and advisory board member of Gen Y Biologics Pte., Ltd. (company registration number: 202005553Z). Patent title: Engineered Immune Cells (PCT/SG2020/050090). Published September 10, 2020: WO 2020/180243 (L.W., N.R.J.G., and J.B.).
Inclusion and diversity
We support inclusive, diverse, and equitable conduct of research.
Published: January 24, 2023
Footnotes
Supplemental information can be found online at https://doi.org/10.1016/j.xcrm.2023.100917.
Supplemental information
Data and code availability
All data needed to evaluate the conclusions in the paper are present in the paper or the supplemental information. This paper does not report original code. Any additional information required to re-analyze the data reported in this work paper is available from the lead contact upon request.
References
- 1.Kochenderfer J.N., Yu Z., Frasheri D., Restifo N.P., Rosenberg S.A. Adoptive transfer of syngeneic T cells transduced with a chimeric antigen receptor that recognizes murine CD19 can eradicate lymphoma and normal B cells. Blood. 2010;116:3875–3886. doi: 10.1182/blood-2010-01-265041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Kalos M., Levine B.L., Porter D.L., Katz S., Grupp S.A., Bagg A., June C.H. T cells with chimeric antigen receptors have potent antitumor effects and can establish memory in patients with advanced leukemia. Sci. Transl. Med. 2011;3:95ra73. doi: 10.1126/scitranslmed.3002842. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Wu L., Wei Q., Brzostek J., Gascoigne N.R.J. Signaling from T cell receptors (TCRs) and chimeric antigen receptors (CARs) on T cells. Cell. Mol. Immunol. 2020;17:600–612. doi: 10.1038/s41423-020-0470-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Salter A.I., Ivey R.G., Kennedy J.J., Voillet V., Rajan A., Alderman E.J., Voytovich U.J., Lin C., Sommermeyer D., Liu L., et al. Phosphoproteomic analysis of chimeric antigen receptor signaling reveals kinetic and quantitative differences that affect cell function. Sci. Signal. 2018;11:eaat6753. doi: 10.1126/scisignal.aat6753. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Davenport A.J., Cross R.S., Watson K.A., Liao Y., Shi W., Prince H.M., Beavis P.A., Trapani J.A., Kershaw M.H., Ritchie D.S., et al. Chimeric antigen receptor T cells form nonclassical and potent immune synapses driving rapid cytotoxicity. Proc. Natl. Acad. Sci. USA. 2018;115:E2068–E2076. doi: 10.1073/pnas.1716266115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Gulati P., Rühl J., Kannan A., Pircher M., Schuberth P., Nytko K.J., Pruschy M., Sulser S., Haefner M., Jensen S., et al. Aberrant lck signal via CD28 costimulation augments antigen-specific functionality and tumor control by redirected T cells with PD-1 blockade in humanized mice. Clin. Cancer Res. 2018;24:3981–3993. doi: 10.1158/1078-0432.CCR-17-1788. [DOI] [PubMed] [Google Scholar]
- 7.Xiong W., Chen Y., Kang X., Chen Z., Zheng P., Hsu Y.H., Jang J.H., Qin L., Liu H., Dotti G., Liu D. Immunological synapse predicts effectiveness of chimeric antigen receptor cells. Mol. Ther. 2018;26:963–975. doi: 10.1016/j.ymthe.2018.01.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Rohrs J.A., Zheng D., Graham N.A., Wang P., Finley S.D. Computational model of chimeric antigen receptors explains site-specific phosphorylation kinetics. Biophys. J. 2018;115:1116–1129. doi: 10.1016/j.bpj.2018.08.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Sun C., Shou P., Du H., Hirabayashi K., Chen Y., Herring L.E., Ahn S., Xu Y., Suzuki K., Li G., et al. THEMIS-SHP1 recruitment by 4-1BB tunes LCK-mediated priming of chimeric antigen receptor-redirected T cells. Cancer Cell. 2020;37:216–225.e6. doi: 10.1016/j.ccell.2019.12.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Molina T.J., Kishihara K., Siderovski D.P., van Ewijk W., Narendran A., Timms E., Wakeham A., Paige C.J., Hartmann K.U., Veillette A., et al. Profound block in thymocyte development in mice lacking p56lck. Nature. 1992;357:161–164. doi: 10.1038/357161a0. [DOI] [PubMed] [Google Scholar]
- 11.Straus D.B., Weiss A. Genetic evidence for the involvement of the lck tyrosine kinase in signal transduction through the T cell antigen receptor. Cell. 1992;70:585–593. doi: 10.1016/0092-8674(92)90428-f. [DOI] [PubMed] [Google Scholar]
- 12.Lai J., Choo J.A.L., Tan W.J., Too C.T., Oo M.Z., Suter M.A., Mustafa F.B., Srinivasan N., Chan C.E.Z., Lim A.G.X., et al. TCR-like antibodies mediate complement and antibody-dependent cellular cytotoxicity against Epstein-Barr virus-transformed B lymphoblastoid cells expressing different HLA-A∗02 microvariants. Sci. Rep. 2017;7:9923. doi: 10.1038/s41598-017-10265-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Sim A.C.N., Too C.T., Oo M.Z., Lai J., Eio M.Y., Song Z., Srinivasan N., Tan D.A.L., Pang S.W., Gan S.U., et al. Defining the expression hierarchy of latent T-cell epitopes in Epstein-Barr virus infection with TCR-like antibodies. Sci. Rep. 2013;3:3232. doi: 10.1038/srep03232. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Wu L., Brzostek J., Sankaran S., Wei Q., Yap J., Tan T.Y.Y., Lai J., MacAry P.A., Gascoigne N.R.J. Targeting CAR to the peptide-MHC complex reveals distinct signaling compared to that of TCR in a Jurkat T cell model. Cancers. 2021;13:867. doi: 10.3390/cancers13040867. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Banik D., Hamidinia M., Brzostek J., Wu L., Stephens H.M., MacAry P.A., Reinherz E.L., Gascoigne N.R.J., Lang M.J. Single molecule force spectroscopy reveals distinctions in key biophysical parameters of alphabeta T-cell receptors compared with chimeric antigen receptors directed at the same ligand. J. Phys. Chem. Lett. 2021;12:7566–7573. doi: 10.1021/acs.jpclett.1c02240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Gascoigne N.R.J., Casas J., Brzostek J., Rybakin V. Initiation of TCR phosphorylation and signal transduction. Front. Immunol. 2011;2:72. doi: 10.3389/fimmu.2011.00072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Artyomov M.N., Lis M., Devadas S., Davis M.M., Chakraborty A.K. CD4 and CD8 binding to MHC molecules primarily acts to enhance Lck delivery. Proc. Natl. Acad. Sci. USA. 2010;107:16916–16921. doi: 10.1073/pnas.1010568107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Esensten J.H., Helou Y.A., Chopra G., Weiss A., Bluestone J.A. CD28 costimulation: from mechanism to therapy. Immunity. 2016;44:973–988. doi: 10.1016/j.immuni.2016.04.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Moeller M., Haynes N.M., Trapani J.A., Teng M.W.L., Jackson J.T., Tanner J.E., Cerutti L., Jane S.M., Kershaw M.H., Smyth M.J., Darcy P.K. A functional role for CD28 costimulation in tumor recognition by single-chain receptor-modified T cells. Cancer Gene Ther. 2004;11:371–379. doi: 10.1038/sj.cgt.7700710. [DOI] [PubMed] [Google Scholar]
- 20.Palacios E.H., Weiss A. Function of the src-family kinases, lck and fyn, in T-cell development and activation. Oncogene. 2004;23:7990–8000. doi: 10.1038/sj.onc.1208074. [DOI] [PubMed] [Google Scholar]
- 21.Blake R.A., Broome M.A., Liu X., Wu J., Gishizky M., Sun L., Courtneidge S.A. SU6656, a selective src family kinase inhibitor, used to probe growth factor signaling. Mol. Cell Biol. 2000;20:9018–9027. doi: 10.1128/mcb.20.23.9018-9027.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Stachlewitz R.F., Hart M.A., Bettencourt B., Kebede T., Schwartz A., Ratnofsky S.E., Calderwood D.J., Waegell W.O., Hirst G.C. A-770041, a novel and selective small-molecule inhibitor of Lck, prevents heart allograft rejection. J. Pharmacol. Exp. Ther. 2005;315:36–41. doi: 10.1124/jpet.105.089169. [DOI] [PubMed] [Google Scholar]
- 23.Abraham R.T., Weiss A. Jurkat T cells and development of the T-cell receptor signalling paradigm. Nat. Rev. Immunol. 2004;4:301–308. doi: 10.1038/nri1330. [DOI] [PubMed] [Google Scholar]
- 24.Li L., Guo X., Shi X., Li C., Wu W., Yan C., Wang H., Li H., Xu C. Ionic CD3-Lck interaction regulates the initiation of T-cell receptor signaling. Proc. Natl. Acad. Sci. USA. 2017;114:E5891–E5899. doi: 10.1073/pnas.1701990114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Sastry K.S.R., Too C.T., Kaur K., Gehring A.J., Low L., Javiad A., Pollicino T., Li L., Kennedy P.T.F., Lopatin U., et al. Targeting hepatitis B virus-infected cells with a T-cell receptor-like antibody. J. Virol. 2011;85:1935–1942. doi: 10.1128/JVI.01990-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Salmond R.J., Filby A., Qureshi I., Caserta S., Zamoyska R. T-cell receptor proximal signaling via the Src-family kinases, Lck and Fyn, influences T-cell activation, differentiation, and tolerance. Immunol. Rev. 2009;228:9–22. doi: 10.1111/j.1600-065X.2008.00745.x. [DOI] [PubMed] [Google Scholar]
- 27.Denny M.F., Patai B., Straus D.B. Differential T-cell antigen receptor signaling mediated by the Src family kinases Lck and Fyn. Mol. Cell Biol. 2000;20:1426–1435. doi: 10.1128/mcb.20.4.1426-1435.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Stenger D., Stief T.A., Kaeuferle T., Willier S., Rataj F., Schober K., Vick B., Lotfi R., Wagner B., Grünewald T.G.P., et al. Endogenous TCR promotes in vivo persistence of CD19-CAR-T cells compared to a CRISPR/Cas9-mediated TCR knockout CAR. Blood. 2020;136:1407–1418. doi: 10.1182/blood.2020005185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Seddon B., Legname G., Tomlinson P., Zamoyska R. Long-term survival but impaired homeostatic proliferation of Naive T cells in the absence of p56lck. Science. 2000;290:127–131. doi: 10.1126/science.290.5489.127. [DOI] [PubMed] [Google Scholar]
- 30.Shan X., Balakir R., Criado G., Wood J.S., Seminario M.C., Madrenas J., Wange R.L. Zap-70-independent Ca(2+) mobilization and Erk activation in Jurkat T cells in response to T-cell antigen receptor ligation. Mol. Cell Biol. 2001;21:7137–7149. doi: 10.1128/MCB.21.21.7137-7149.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Kortum R.L., Rouquette-Jazdanian A.K., Miyaji M., Merrill R.K., Markegard E., Pinski J.M., Wesselink A., Nath N.N., Alexander C.P., Li W., et al. A phospholipase C-gamma1-independent, RasGRP1-ERK-dependent pathway drives lymphoproliferative disease in linker for activation of T cells-Y136F mutant mice. J. Immunol. 2013;190:147–158. doi: 10.4049/jimmunol.1201458. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Gauen L.K., Zhu Y., Letourneur F., Hu Q., Bolen J.B., Matis L.A., Klausner R.D., Shaw A.S. Interactions of p59fyn and ZAP-70 with T-cell receptor activation motifs: defining the nature of a signalling motif. Mol. Cell Biol. 1994;14:3729–3741. doi: 10.1128/mcb.14.6.3729-3741.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Samelson L.E., Phillips A.F., Luong E.T., Klausner R.D. Association of the fyn protein-tyrosine kinase with the T-cell antigen receptor. Proc. Natl. Acad. Sci. USA. 1990;87:4358–4362. doi: 10.1073/pnas.87.11.4358. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Ley S.C., Marsh M., Bebbington C.R., Proudfoot K., Jordan P. Distinct intracellular localization of Lck and Fyn protein tyrosine kinases in human T lymphocytes. J. Cell Biol. 1994;125:639–649. doi: 10.1083/jcb.125.3.639. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Frasson M., Vitadello M., Brunati A.M., La Rocca N., Tibaldi E., Pinna L.A., Gorza L., Donella-Deana A. Grp94 is Tyr-phosphorylated by Fyn in the lumen of the endoplasmic reticulum and translocates to Golgi in differentiating myoblasts. Biochim. Biophys. Acta. 2009;1793:239–252. doi: 10.1016/j.bbamcr.2008.10.001. [DOI] [PubMed] [Google Scholar]
- 36.Liang J., Lyu J., Zhao M., Li D., Zheng M., Fang Y., Zhao F., Lou J., Guo C., Wang L., et al. Tespa1 regulates T cell receptor-induced calcium signals by recruiting inositol 1,4,5-trisphosphate receptors. Nat. Commun. 2017;8:15732. doi: 10.1038/ncomms15732. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Lovatt M., Filby A., Parravicini V., Werlen G., Palmer E., Zamoyska R. Lck regulates the threshold of activation in primary T cells, while both Lck and Fyn contribute to the magnitude of the extracellular signal-related kinase response. Mol. Cell Biol. 2006;26:8655–8665. doi: 10.1128/MCB.00168-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Majzner R.G., Rietberg S.P., Sotillo E., Dong R., Vachharajani V.T., Labanieh L., Myklebust J.H., Kadapakkam M., Weber E.W., Tousley A.M., et al. Tuning the antigen density requirement for CAR T-cell activity. Cancer Discov. 2020;10:702–723. doi: 10.1158/2159-8290.CD-19-0945. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Raab M., Cai Y.C., Bunnell S.C., Heyeck S.D., Berg L.J., Rudd C.E. p56Lck and p59Fyn regulate CD28 binding to phosphatidylinositol 3-kinase, growth factor receptor-bound protein GRB-2, and T cell-specific protein-tyrosine kinase ITK: implications for T-cell costimulation. Proc. Natl. Acad. Sci. USA. 1995;92:8891–8895. doi: 10.1073/pnas.92.19.8891. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Posadas E.M., Al-Ahmadie H., Robinson V.L., Jagadeeswaran R., Otto K., Kasza K.E., Tretiakov M., Siddiqui J., Pienta K.J., Stadler W.M., et al. FYN is overexpressed in human prostate cancer. BJU Int. 2009;103:171–177. doi: 10.1111/j.1464-410X.2008.08009.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Saito Y.D., Jensen A.R., Salgia R., Posadas E.M. Fyn: a novel molecular target in cancer. Cancer. 2010;116:1629–1637. doi: 10.1002/cncr.24879. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Zhi H., Yang L., Kuo Y.L., Ho Y.K., Shih H.M., Giam C.Z. NF-kappaB hyper-activation by HTLV-1 tax induces cellular senescence, but can be alleviated by the viral anti-sense protein HBZ. PLoS Pathog. 2011;7:e1002025. doi: 10.1371/journal.ppat.1002025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Krishna S., Xie D., Gorentla B., Shin J., Gao J., Zhong X.P. Chronic activation of the kinase IKKbeta impairs T cell function and survival. J. Immunol. 2012;189:1209–1219. doi: 10.4049/jimmunol.1102429. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Long A.H., Haso W.M., Shern J.F., Wanhainen K.M., Murgai M., Ingaramo M., Smith J.P., Walker A.J., Kohler M.E., Venkateshwara V.R., et al. 4-1BB costimulation ameliorates T cell exhaustion induced by tonic signaling of chimeric antigen receptors. Nat. Med. 2015;21:581–590. doi: 10.1038/nm.3838. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Weber E.W., Parker K.R., Sotillo E., Lynn R.C., Anbunathan H., Lattin J., Good Z., Belk J.A., Daniel B., Klysz D., et al. Transient rest restores functionality in exhausted CAR-T cells through epigenetic remodeling. Science. 2021;372:eaba1786. doi: 10.1126/science.aba1786. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Mestermann K., Giavridis T., Weber J., Rydzek J., Frenz S., Nerreter T., Mades A., Sadelain M., Einsele H., Hudecek M. The tyrosine kinase inhibitor dasatinib acts as a pharmacologic on/off switch for CAR T cells. Sci. Transl. Med. 2019;11:eaau5907. doi: 10.1126/scitranslmed.aau5907. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Heemskerk M.H.M., Hoogeboom M., de Paus R.A., Kester M.G.D., van der Hoorn M.A.W.G., Goulmy E., Willemze R., Falkenburg J.H.F. Redirection of antileukemic reactivity of peripheral T lymphocytes using gene transfer of minor histocompatibility antigen HA-2-specific T-cell receptor complexes expressing a conserved alpha joining region. Blood. 2003;102:3530–3540. doi: 10.1182/blood-2003-05-1524. [DOI] [PubMed] [Google Scholar]
- 48.Methi T., Ngai J., Mahic M., Amarzguioui M., Vang T., Tasken K. Short-interfering RNA-mediated Lck knockdown results in augmented downstream T cell responses. J. Immunol. 2005;175:7398–7406. doi: 10.4049/jimmunol.175.11.7398. [DOI] [PubMed] [Google Scholar]
- 49.Sarbassov D.D., Guertin D.A., Ali S.M., Sabatini D.M. Phosphorylation and regulation of Akt/PKB by the rictor-mTOR complex. Science. 2005;307:1098–1101. doi: 10.1126/science.1106148. [DOI] [PubMed] [Google Scholar]
- 50.Jena B., Maiti S., Huls H., Singh H., Lee D.A., Champlin R.E., Cooper L.J.N. Chimeric antigen receptor (CAR)-specific monoclonal antibody to detect CD19-specific T cells in clinical trials. PLoS One. 2013;8:e57838. doi: 10.1371/journal.pone.0057838. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Zhao Y., Wang Q.J., Yang S., Kochenderfer J.N., Zheng Z., Zhong X., Sadelain M., Eshhar Z., Rosenberg S.A., Morgan R.A. A herceptin-based chimeric antigen receptor with modified signaling domains leads to enhanced survival of transduced T lymphocytes and antitumor activity. J. Immunol. 2009;183:5563–5574. doi: 10.4049/jimmunol.0900447. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Hoerter J.A.H., Brzostek J., Artyomov M.N., Abel S.M., Casas J., Rybakin V., Ampudia J., Lotz C., Connolly J.M., Chakraborty A.K., et al. Coreceptor affinity for MHC defines peptide specificity requirements for TCR interaction with coagonist peptide-MHC. J. Exp. Med. 2013;210:1807–1821. doi: 10.1084/jem.20122528. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Zhao X., Sankaran S., Yap J., Too C.T., Ho Z.Z., Dolton G., Legut M., Ren E.C., Sewell A.K., Bertoletti A., et al. Nonstimulatory peptide-MHC enhances human T-cell antigen-specific responses by amplifying proximal TCR signaling. Nat. Commun. 2018;9:2716. doi: 10.1038/s41467-018-05288-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Zhao X., Hamidinia M., Choo J.A.L., Too C.T., Ho Z.Z., Ren E.C., Bertoletti A., MacAry P.A., Gould K.G., Brzostek J., Gascoigne N.R.J. Use of single chain MHC technology to investigate Co-agonism in human CD8+ T cell activation. J. Vis. Exp. 2019 doi: 10.3791/59126. [DOI] [PubMed] [Google Scholar]
- 55.Sanjana N.E., Shalem O., Zhang F. Improved vectors and genome-wide libraries for CRISPR screening. Nat. Methods. 2014;11:783–784. doi: 10.1038/nmeth.3047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Roth T.L., Puig-Saus C., Yu R., Shifrut E., Carnevale J., Li P.J., Hiatt J., Saco J., Krystofinski P., Li H., et al. Reprogramming human T cell function and specificity with non-viral genome targeting. Nature. 2018;559:405–409. doi: 10.1038/s41586-018-0326-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Fu G., Gascoigne N.R.J. Multiplexed labeling of samples with cell tracking dyes facilitates rapid and accurate internally controlled calcium flux measurement by flow cytometry. J. Immunol. Methods. 2009;350:194–199. doi: 10.1016/j.jim.2009.07.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Choo J.A.L., Thong S.Y., Yap J., van Esch W.J.E., Raida M., Meijers R., Lescar J., Verhelst S.H.L., Grotenbreg G.M. Bioorthogonal cleavage and exchange of major histocompatibility complex ligands by employing azobenzene-containing peptides. Angew. Chem. Int. Ed. Engl. 2014;53:13390–13394. doi: 10.1002/anie.201406295. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
All data needed to evaluate the conclusions in the paper are present in the paper or the supplemental information. This paper does not report original code. Any additional information required to re-analyze the data reported in this work paper is available from the lead contact upon request.