

Summary
Pituitary neuroendocrine tumor (PitNET) is one of the most common intracranial tumors with variable recurrence rate. Currently, the recurrence prediction is unsatisfying and can be improved by understanding the cellular origins and differentiation status. Here, to comprehensively reveal the origin of PitNET, we perform comparative analysis of single-cell RNA sequencing data from 3 anterior pituitary glands and 21 PitNETs. We identify distinct genes representing major subtypes of well and poorly differentiated PitNETs in each lineage. To further verify the predictive value of differentiation biomarkers, we include an independent cohort of 800 patients with an average follow-up of 7.2 years. In both PIT1 and TPIT lineages, poorly differentiated groups show significantly higher recurrence rates while well-differentiated groups show higher recurrence rates in SF1 lineage. Our findings reveal the possible origin and differentiation status of PitNET based on which new differentiation classification is proposed and verified to predict tumor recurrence.
Keywords: anterior pituitary gland, pituitary neuroendocrine tumor, single-cell RNA sequencing, differentiation-related molecular classification, recurrence prediction
Graphical abstract
Highlights
-
•
Trajectory analysis in the APG reveals a progenitor PIT1 lineage cluster
-
•
The comparative analysis evaluates the differentiation status of PitNET
-
•
The differentiation-related markers could help predict the long-term recurrence
Zhang et al. provide a comparative analysis between anterior pituitary glands (APGs) and pituitary neuroendocrine tumors (PitNETs) at single-cell resolution. The differentiation status of PitNETs is evaluated, and the recurrence prediction values of differentiation-related markers are validated in an independent cohort of 800 patients.
Introduction
Pituitary neuroendocrine tumor (PitNET) is one of the most common intracranial tumors, with a prevalence of 14%–22%.1 PitNET originates from the anterior pituitary gland (APG),2,3 which secretes six hormones: prolactin (PRL), growth hormone (GH), thyroid-stimulating hormone (TSH), adrenocorticotropic hormone (ACTH), follicle-stimulating hormone (FSH), and luteinizing hormone (LH) (Figure S1A). PitNET exhibits a series of clinical manifestations due to excessive hormone secretion and local invasion of surrounding structures.4,5 The current pathological classification of PitNET, according to the latest 2022 WHO criteria, is based on histological hormone staining and the expression of three lineage-specific transcription factors (TFs), including PIT1 (also called POU1F1), TPIT (also called TBX19), and SF1 (also called NR5A1).6 The most widely used classification in clinical practice is the clinicopathological classification,7,8 which combines the immunohistochemistry (IHC) of three TFs and the levels of serum hormone. The clinicopathological classification categorizes PitNETs into PIT1 lineage (lactotroph, somatotroph, thyrotroph, and silent PIT1 tumors), TPIT lineage (corticotroph and silent TPIT tumors), SF1 lineage (gonadotroph and silent SF1 tumors), null cell, and plurihormonal tumors. Among these types, non-hormone-secreting PitNETs are classified as silent PIT1, silent TPIT, silent SF1, or null cell tumors based on the expression of three TFs. The present clinicopathological classification of PitNET is listed in Figure S1B.
The primary treatment of PitNET includes surgery, medication, and radiotherapy. Although great advances have been achieved in recent years, the recurrence, especially the long-term recurrence rate (varying from 10% to 30%), remains a big challenge in the clinic.9,10,11,12 The complete resection rate of the recurrent tumors was significantly lower and usually leads to visual defects and hypopituitarism, which needs lifelong hormone replacement therapy.13 For those patients with higher risks of recurrence, close follow-up and timely interventions should be taken. Therefore, precise prediction of recurrence is necessary for the individualized management of PitNET. However, the current clinicopathological classification cannot fulfill the above-mentioned needs, which only contains several risk factors for recurrence, including high Ki-67 index, lactotroph adenoma in men, silent corticotroph adenoma, and Crooke cell adenoma.6 The specific molecular markers that could precisely predict tumor recurrence remain lacking.
Understanding the cellular origin and differentiation status is fundamental for expanding and refining the current tumor classification, which may enhance its ability to predict tumor recurrence. The recent rapid progress in single-cell RNA sequencing (scRNA-seq) technologies provides an opportunity to comprehensively understand the cellular components and regulatory networks for heterogeneous tumors.14 For example, through the comparative analysis of scRNA-seq data between neural-crest-derived neuroblastoma (NB) and fetal adrenal glands, the tumor origin of NB was mapped to the noradrenergic chromaffin cells, of which proliferation/differentiation status have a critical prognostic effect on NB.15 Considering the diversity in both lineages and hormone-producing cells, APG and PitNET are ideal objects for scRNA-seq technologies to explore tumor origins and differentiated status.
Several publications have reported scRNA-seq for mouse pituitary,16,17,18,19,20,21,22,23,24 rat pituitary,25,26 human fetal pituitary,27 and human pituitary cells.28 The later study was performed on archived frozen pituitary tissues and thus at single-cell resolution, while the data of fresh adult human APG are limited. For tumors, one study showed the single-cell tagged reverse transcription sequencing data of 2,311 cells from 3 lineages of PitNETs, revealing a few novel tumor-related genes and intra-tumor heterogeneity.29 Another two scRNA-seq studies conducted by Zhang et al. focused on the secretion and invasion mechanism in ACTH and silent TPIT PitNETs, respectively.30,31 One recent publication elaborated in detail on the apoptosis-evading mechanisms in ACTH PitNETs.32 Despite these studies, an in-depth exploration combining APG and PitNET data to identify tumor origin and differentiation status at single-cell resolution is highly required.
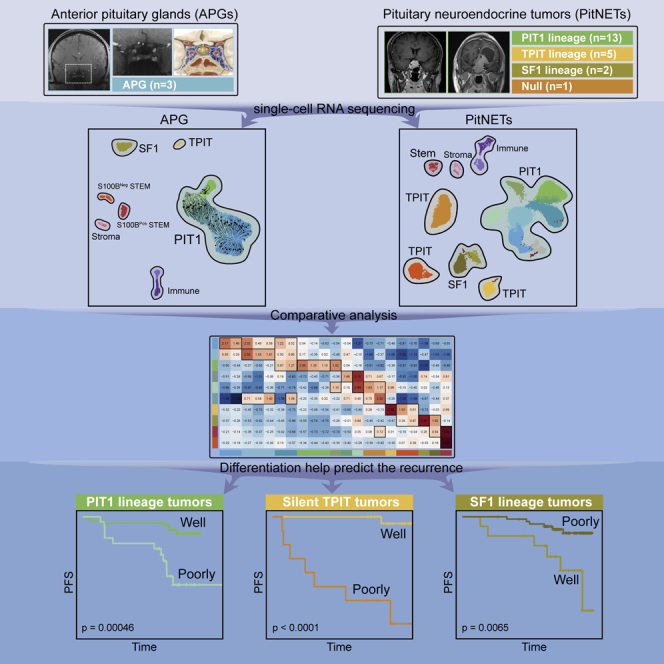
Therefore, we first performed scRNA-seq on three adult human APGs and established the transcriptional landscape at single-cell resolution. Then we used this unique resource to identify the transcriptional characteristics and possible origin of 21 PitNETs and use the data to classify their differentiation status. The novel molecular markers identified in each differentiated status PitNET were then utilized to predict tumor recurrence in an independent cohort of 800 tumors with an average follow-up duration of 7.2 years.
Results
Single-cell transcriptome profiling of adult human APGs
We obtained APGs from three adult donors (Figure 1A; Table S1), and these tissues were subjected to scRNA-seq. After stringent quality control, we obtained 6,589 cells in total, with the average number of detected genes per cell being 2,682 and 11,643 for the average number of detected unique molecular identifiers (UMIs) per cell (Figure S1C; STAR Methods). Based on canonical markers, six main cell populations were separated, including PIT1 lineage, TPIT lineage, SF1 lineage, stroma cells, immune cells, and stem cells (Figures 1B and S1D). These cell populations were further divided into 14 clusters by unsupervised clustering (LACTOHigh, LACTOMedium, PIT1_I, PIT1_II, SOMATO, THYRO, GONADO, CORTICO, S100BPos STEM, S100BNeg STEM, myeloid cells, lymphocytes, fibroblasts, and endothelial cells) (Figures 1B, S1E, and S2A; STAR Methods; Table S2). Notably, each cluster contained cells from different samples, indicating that cell types and gene expression patterns are broadly consistent across APG samples, and there are no obvious donor-specific subpopulations or batch effects (Figure S2B). The hormone-secreting cells were identified within each lineage based on the enriched gene expression levels of well-known markers as reported previously (Figures 1C and S2C). Some novel markers specifically expressed in each cell type are summarized in Figure 1D.
Figure 1.

Single-cell transcriptome profiling of three adult human APGs
(A) Overview of the study design and workflow.
(B) Integrated UMAP plot of all 6,589 cells from three APGs with clusters outlined by lineages and cells colored according to the corresponding cell types. LACTO, lactotrope; SOMATO, somatotrope; THYRO, thyrotrope; GONADO, gonadotrope.
(C) Integrated UMAP plot of APGs with cells colored according to the expression of classical marker genes of pituitary (PRL, GH1, TSHB, FSHB, POMC, and SOX2).
(D) Dot plot of novel marker genes in endocrine cell types of the APG. The color represents the scaled relative expression level of the marker genes in each cell type, and the size indicates the proportion of cells expressing the marker genes.
(E) mIHC staining of S100B, SOX2, CLDN4, SOX9, and VIM in sections from an adult APG. Tissues were counterstained with DAPI. The arrowheads indicate S100BPos STEM (SOX2+ S100B+). The arrows indicate S100BNeg STEM (SOX2+ S100B–). Scale bars, 50 μm. See also Figures S1 and S2.
We detected two population of cells with significantly high SOX2 expression and further identified these cells into S100B-positive cells (S100BPos STEM) and S100B-negative cells (S100BNeg STEM).26,28,33 Of note, several makers that had previously been identified in the stem cells of fetal pituitaries,27 such as CLDN4, also had enriched expression in these cells (Figure 1D). By multiplex IHC (mIHC) staining, we further verified the colocalization of adult pituitary stem cell markers (SOX2, SOX9, S100B, CLDN4, and VIM) in another APG sample, which showed different distribution patterns of two hypothetical stem cell clusters: (1) S100BPos STEM and S100BNeg STEM were sparsely scattered in the parenchymal of APGs; (2) S100BNeg STEM were continuously distributed at the edge of Rathke’s cleft (Figure 1E).34
Cellular diversity within PIT1 lineage cells of APGs
We noticed that two clusters of PIT1 lineage cells (PIT1_I and PIT1_II) showed low expression of GH1, PRL, and TSHB (Figures 1B and 1C). Particularly, PIT1_I shared several markers with S100BPos STEM, including PITX2 and LMO4 (Table S2). In addition, it presents the enriched expression of GATA3 (Figures 1D and S2D), a TF indicative of the poorly differentiated PIT1 lineage PitNETs in the 2022 WHO classification.6 Therefore, we speculated that this subpopulation comprises poorly differentiated PIT1 cells and could be the progenitor of hormone-secreting cells in the PIT1 lineage (Pro.PIT1).
To support our hypothesis, we performed trajectory analysis using scVelo and Monocle3.35,36 As we expected, these results showed the same differentiation pattern from Pro.PIT1 in three directions: GH-, PRL-, and TSH-secreting cells (Figures 2A, 2B, and S3A–S3C; Tables S2). We identified several genes, such as IGFBP7, GATA3, and LRRC4C, that were downregulated along differentiation (Figures 1D, 2C, and S2D). We also identified specific markers for well-differentiated GH- and PRL-secreting cells, including ENPP1 and NTS (Figures 1D and 2C). Moreover, we found PIT1_II to be a transient state along the trajectory from Pro.PIT1 cells to PRL-secreting cells. PIT1_II shared several markers with both Pro.PIT1 cells (STAT3, CCNL1) and PRL-secreting cells (NR4A1, ARC) (Figure S3D), suggesting the earlier differentiation status of PIT1_II (Pre. Lacto).
Figure 2.

Several differentiation statuses were identified based on the cellular diversity within APGs
(A) Partition-based graph abstraction connectivity among clusters of PIT1 lineage cells in APGs based on velocity-inferred directionality.
(B) Integrated UMAP plot of PIT1 lineage cells from APGs with cells colored by Monocle3-inferred pseudotime.
(C) Gene expression trends in particular genes along the differentiation trajectory from Pro.PIT1 toward SOMATO and LACTOHigh.
(D) Integrated UMAP plot and Slingshot developmental trajectories of integrated adult and fetal APGs with cells colored according to the corresponding cell type.
(E) mIHC staining of GH, NTS, PIT1, PRL, ENPP1, and TSHB in sections from adult APGs. Tissues were counterstained with DAPI. Scale bars, 50 μm
(F) mIHC staining of GH, IGFBP7, PRL, LRRC4C, PIT1, and TSHB in sections from adult APGs. Tissues were counterstained with DAPI. Scale bars, 50 μm. See also Figure S3.
We further reconstructed the developmental trajectory from the fetal to the adult APG by integrating our adult APG dataset with previously published fetal APG scRNA-seq data27 (Figures 2D and S3E). For both datasets, we identified five cell populations of hormone-secreting cells from three lineages and the stem cells (Figure S3F). We constructed the developmental trajectories for five hormone-secreting cells and found that the Adult.Pro.PIT1 and Adult.Pre.Lacto clusters are located at the earlier differentiation status of adult PIT1 lineage cells and adult PRL-secreting cells along the trajectory, respectively (Figures 2D, S3G, and S3H). Mapping Adult.Pro.PIT1 cells located at the entrance of the adult PIT1 lineage further validate the root cells’ position (Figure S3I).
The presence of poorly differentiated (Pro.PIT1) and well-differentiated (TSH-, GH-, or PRL-secreting) PIT1 cells was validated in adult human APGs by mIHC staining. NTS was colocalized with PRL-secreting cells, while ENPP1 was present in GH- and PRL-secreting cells (Figure 2E). Among Pro.PIT1 cells, IGFBP7 and LRRC4C were coexpressed in the cells with negative GH, PRL, or TSHB staining but positive PIT1 staining (Figure 2F).
Single-cell transcriptome profiling of PitNET
We generated a scRNA-seq landscape for three APGs and PitNETs from 21 patients, including 3 lactotroph, 5 somatotroph, 2 thyrotroph, 1 silent PIT1, 2 corticotroph, 3 silent TPIT, 2 silent SF1, 1 null cell, and 2 plurihormonal tumors (Figures 3A and S4A). All tumor tissues were also fixed for histopathological examination of canonical PitNET markers, including APG hormones and TFs. Among the tumors, 12 tumors were invasive and classified as Knosp grade37,38 3 or 4, while the Ki-67 index of 6 tumors was greater than 3%. Only one GH-secreting tumor (P04) had GNAS mutation and one corticotroph tumor (P20) carried USP8 mutation (Figure S4A). Additional sample details and patient demographics are summarized in Table S1.
Figure 3.

Comparison between PitNETs and APGs revealed the possible origin of each tumor cluster
(A) Integrated UMAP plot of all 64,937 cells from 21 PitNETs and 3 APGs with clusters outlined by cell types.
(B) Heatmap showing each PitNET cluster (columns) scored by each endocrine cluster in the APGs (rows). Scores were z-scaled. The black rectangles indicate the highest scores in each PitNET cluster. The cells from APGs were excluded from PitNET clusters.
(C) Integrated UMAP plot of cells from PitNETs with cells colored according to the corresponding classifications (left). These classifications were combined into four categories (right).
(D) Heatmap shows specific genes of the 12 main subtypes. See also Figures S4–S8.
After stringent quality control, 58,348 cells were obtained, with an average of 3,438 genes and 17,559 UMIs detected per cell (Figure S4B). Consistent with pathological IHC, we found similar expression patterns of PitNET classification markers (such as GH1, PRL, and TSHB) in scRNA-seq data (Figure S4A). We integrated the data of PitNET and APG cells and divided them into 29 clusters by unsupervised clustering, which could be generally grouped into 8 cellular populations based on canonical markers (Figures 3A and S4C; STAR Methods; Table S2). Surprisingly, cells from APGs were embedded within tumor cells, especially those from LACTOHigh, LACTOMedium, GONADO, and CORTICO (Figures S4D–S4F and S5A). In addition, tumor cells from the null cell tumor (P14) shared the same cluster with tumor cells from silent SF1 tumor (P15), implying the similarity between these two cell populations (Figures S4D, S4F, and S5A). Of note, a previous study suggested that the definition of SF1 lineage PitNETs should be extended to a partial null cell tumor.39 We observed variations of the proportions of immune and stroma cells among APG and tumor samples (Figure S5B). The endocrine cells of each tumor showed the monoclonal characteristics in both lineage composition and copy number variation state (Figures S5C and S6A). We next distinguished and excluded the non-tumor neuroendocrine cells in PitNET samples (Figures S5C, S6A, and S6B; STAR Methods).29,40,41
Heterogeneity of tumor stem-like cells in PitNET
The detection of cluster 21, which consisted of SOX2-expressing cells from APGs and PitNETs, implicated the presence of tumor stem-like cells (TSCs) in PitNETs (Figures 3A, S4C–S4F, and S5A).42,43 We conducted mIHC staining in another two PitNET tumor samples and showed the colocalization of multiple stem cell markers identified in cluster 21 (VIM, SOX2, KRT19, TROP2, and CLDN4) (Figures S6C and S6D). We also found the exclusive expression of SOX2 and MKI67 by both mIHC staining and scRNA-seq, suggesting that the TSCs in PitNETs tend to be in non-proliferative state (Figure S4C).
In the APG, the SOX2-expressing cells can be divided into S100BPos STEM and S100BNegSTEM. We wondered whether there were similar patterns in the PitNETs. To address this question, we performed a second round of clustering with only the 750 cells from cluster 21 and divided them into 10 clusters (Figures S7A–S7E; Tables S2 and S3). Regrouped clusters 0, 1, 2, 3, and 9 showed the expression of S100B and S100A1, while clusters 4, 5, 6, 7, and 8 were negative for the two markers of S100BPos STEM (Figure S7F). We next performed a comparative analysis to evaluate the similarity between TSCs and SOX2-expressing cells in APGs (Figure S7G). TSCs in clusters 0, 2, and 3 showed a high score of S100BPos STEM characteristics and were named tumor S100BPos STEM-like cells. TSCs in clusters 4, 6, 7, and 8 were then named tumor S100BNeg STEM-like cells. TSCs in clusters 1, 5, and 9 were considered intermediate states for showing comparable similarity with both S100BPos STEM and S100BNeg STEM. The epithelial-mesenchymal transition (EMT) process has been identified in stem cells of multiple cancers44 and the fetal APG.27,45 Therefore, we estimate the EMT scores for each cell as described previously (Figures S7G and S7H). Surprisingly, the epithelial and mesenchymal status of the TSCs were mutually distributed. Tumor S100BNeg STEM-like cells exhibit a high level of epithelial score and express epithelial markers such as CLDN4 and KRT19. Meanwhile, most tumor S100BPos STEM-like cells exhibit a higher level of mesenchymal score and express mesenchymal markers such as CXCR4 and VIM (Figures S7F and S7G). Moreover, both the epithelial and mesenchymal scores were decreased in the non-TSC PitNET tumor cells, suggesting the specific role of EMT in TSCs (Figures S7I and S7J).
Comparison between the PitNET and APG clusters uncovered novel characteristics of PitNET
To explore the tumor origin and differentiation status of PitNETs, we evaluated the similarity and distinguished the intra-lineage differences between the 21 clusters from the PitNETs with the 10 clusters from the APGs (Figure 3B). Cell scoring was applied to PitNETs clusters against the top 20 DEGs of each endocrine and stem cluster of APGs. The top DEGs were ranked according to the specificity and expression levels in each cluster (STAR Methods). The scores were z-scaled by row (each APG cluster) and the top 1–3 scored APG clusters within each column were considered the most similar and potential origin for each PitNETs cluster. Scoring PitNETs clusters using the top 15–30 DEGs was also calculated and produced similar results.
Based on scoring, the 21 tumor cell clusters could be merged into 13 subtypes (Figures 3B and 3C). We named each subtype according to its similarities to APG clusters, TF expression, differentiation status, and predominant clinicopathological subtypes: seven well-differentiated subtypes (PIT1_PRLHigh, PIT1_PRLMedium, PIT1_GH + PRL, PIT1_GH, PIT1_CGA, TPIT_Well-diff, and SF1_Well-diff); three poorly differentiated subtypes (PIT1_Poorly-diff, TPIT_Poorly-diff, and SF1_Poorly-diff); one stem cell cluster (SOX2_stem); and two extra subtypes (PIT1_GH + CGA and TPIT_PAX7) (Figure 3C).
To further confirm whether the molecular characteristics of each tumor cell cluster in the same subtype were similar, we performed Spearman’s correlation and hierarchical clustering of all PitNETs clusters, considering the transcriptomic similarities within the 21 PitNET clusters. We found that the clusters in the same subtype were highly consistent (Figure S8A).
Characteristics of well-differentiated subtypes
The seven well-differentiated subtypes showed similar expression patterns to those of their hypothesized origin APG clusters (Figure 3D; Table S2). PIT1_PRLHigh exhibited high expression of PRL, NTS, and VGF, which were also detected in the LACTOHigh cluster in APGs. Both LACTOMedium in APGs and PIT1_PRLMedium express SOX4 and ANGPT1. Compared with LACTOHigh and PIT1_PRLHigh, the PRL expression in LACTOMedium and PIT1_PRLMedium was lower, respectively. Several markers of SOMATO in APG, including GH1, GH2, ENPP1, and GNAS, were also highly expressed in PIT1_GH. PIT1_GH + PRL exhibited high expression of GHRHR, and the common markers of LACTOHigh (TENT5A and PCK1) and SOMATO (HES6 and RCN1) were also detected in this subtype (Figure S8B). The high expression of CGA (the α-subunit of TSH) was detected in PIT1_CGA, including tumor cells from clinical TSH-secreting PitNETs, which was consistent with the elevated circulating α-subunit in these patients. Well differentiated TPIT and SF1 lineages also expressed the corresponding hormone-related genes. Different endocrine cells in APG secrete hormones in distinct patterns, which may still exist in the derived PitNETs. The regulons of each cell were assessed using single-cell regulatory network inference and clustering (Figure 4A; Table S2).46 In well-differentiated subtypes, three lineage-specific regulons (PIT1, TPIT, and SF1) showed high activity, suggesting that they may be involved in the biological process of hormone secretion (Figure 4A). We tend to identify the molecules with great contribution to these hormone secretion pathways; we found that ENPP1, a member of “cellular response to peptide hormone stimulus,” was highly expressed in all GH-secreting cells from both APGs and PitNETs (Figures 4B and S9A). On the other hand, NTS was significantly highly expressed in lactotroph tumors and was also involved in “hormone activity” and “neuropeptide hormone activity” pathways (Figures 4B and S9A).
Figure 4.

The molecular features of well and poorly differentiated PitNETs
(A) Heatmap showing the activity of the specific single-cell regulatory network inference and clustering-inferred master regulons of the 12 subtypes. Poorly differentiated subtypes are highlighted in bold red font.
(B) Heatmap showing the representative pathways of the 12 subtypes. Pathways were retrieved from the HALLMARK, gene ontology, Kyoto Encyclopedia of Genes and Genomes, and REACTOME datasets. Lipid metabolic-related pathways are marked with red dotted rectangles. Poorly differentiated subtypes are highlighted in bold red font.
(C) RNA velocities of TPIT_Poorly diff cells (left) and gene expression trends in particular genes along scVelo latent time (right). The two clusters are the tumor cells of three silent TPIT tumors (P17, P18, and P19) shown in clusters 0 and 14 (Figure 3A).
(D) Diagram summarizes metabolic genes involved in glycolysis, the pentose phosphate pathway, the tricarboxylic acid cycle, cholesterol biosynthesis, and fatty acid metabolism. Alterations are defined by significant upregulation or downregulation of mRNA expression between PIT1_Poorly diff, TPIT_Poorly diff, and Pro.PIT1, Pre.Lacto. The alteration scores for each gene are presented as log ratios (fold-change, expressed as log2[ratio of the average mRNA expression value in each subtype versus the originate group in APGs]). Red, upregulated genes; blue, downregulated genes. Each element box, the top symbol was metabolic gene, the left is the alteration score of PIT1_Poorly-diff verse Pro.PIT1 and Pre.Lacto, the right is the alteration score of TPIT_Poorly-diff verse Pro.PIT1 and Pre.Lacto.
(E) The top heatmap shows MuSiC-based deconvolution of an independent cohort of 134 PitNETs from Neou et al.39 The bottom heatmap shows corresponding clinical characteristics and signature gene expression levels. Poorly differentiated subtypes are highlighted in bold red font. The enrichment of lipid metabolism genes was marked using red dotted rectangles. The original classes (t1–t6), as a column from Neou et al.,39 are also shown in the “mRNA group.” See also Figures S9 and S10.
Characteristics of poorly differentiated subtypes
We also identified three subtypes with the different molecular characteristics of poorly differentiated PitNETs: PIT1_Poorly-diff, SF1_Poorly-diff, and TPIT_Poorly-diff. They were named based on two characteristics, including the close similarity to less differentiated clusters in APGs (Pro.PIT1- or SOX2-expressing cells include S100BPos STEM and S100BNeg STEM) and negative hormonal IHC staining. Each subtype had its features. PIT1_Poorly-diff was the PitNET subtype most similar to the Pro.PIT1 cluster in APGs, suggesting the low differentiation status of this subtype. These two clusters shared several markers, including IGFBP7, GATA3, and LRRC4C (Figure 3D). SF1_Poorly-diff showed similarity to both GONADO and S100BPos STEM clusters of APGs (Figure 3B). Compared with SF1_Well-diff, SF1_Poorly-diff showed decreased expression of FSHB (the β-subunits of FSH) (Figure S4C), suggesting lower hormone secretion in this subtype. Most TPIT_Poorly-diff cells were found in silent TPIT tumors (P17, P18, and P19), which is one of the PitNET subtypes with the worst prognosis.47,48,49,50 To further examine the heterogeneity and differentiation status within the TPIT_Poorly-diff subtype, we performed another scVelo analysis including only cells of this subtype (Figure 4C). Surprisingly, cells from each sample were distributed all along the trajectories, with only slightly different trends. We identified several downregulated markers during the latent time developmental progression, including ID4, NEAT1, BCL6, and CDH1, which were considered markers of the poorly differentiated TPIT_Poorly-diff subtype (Figures 4C and S9B; Table S2). The expression of TPIT_Poorly-diff-specific TFs, including NEUROD1 and ONECUT2, was also elevated at the beginning of the trajectory. On the other hand, the expression levels of several differentiated markers, such as TBX19 and CITED1, were elevated along the trajectory.
These three poorly differentiated subtypes also share common features. In pathway enrichment analysis, we identified dysregulation of several lipid metabolic pathways among the poorly differentiated subtypes (Figure 4B). To comprehensively investigate the metabolic reprogramming landscape, we applied scMetabolism51 for both the APG and PitNETs (Figure S9C). While all SF1 lineage tumors showed elevated metabolism regardless of differentiation status, PIT1_Poorly-diff and TPIT_Poorly-diff were characterized by upregulated metabolism, especially lipid metabolism. The relatively highly differentiated PitNET subtypes, such as PIT1_PRLHigh and PIT1_PRLMedium, were characterized by decreased metabolism, consistent with their hypothesized origin cells (LACTOHigh and LACTOMedium) in the APG (Figure S9C). Next, we focused on the difference in lipid metabolism pathways and the mRNA levels of the corresponding metabolic enzymes between two poorly differentiated PitNETs, PIT1_Poorly-diff and TPIT_Poorly-diff, and their hypothesized origin cells, Pro.PIT1 and Pre.Lacto in APGs (Figure 3B). TPIT_Poorly-diff was characterized by activated regulon of sterol regulatory element-binding TF 1 (SREBF1) (Figures 4A and 4D), which was proven to upregulate lipid metabolism-related pathways, including the “biosynthesis of unsaturated fatty acids” and “fatty acid elongation” pathways (Figure S9C).52 Representative metabolic enzymes such as fatty acid desaturase 1 (FADS1), fatty acid desaturase 2 (FADS2), and stearoyl-CoA desaturase (SCD) were upregulated in TPIT_Poorly-diff. PIT1_Poorly-diff was characterized by enriched steroid biosynthesis pathways, whose upregulated representative enzymes included acetyl-CoA acetyltransferase 1 (ACAT1), HMG-CoA reductase (HMGCR), and apolipoprotein E (APOE) (Figure 4D).
We validated these results in an independent cohort reported by Neou et al.39 Using multisubject single-cell deconvolution (MuSiC),53 we determined the proportions of the above-mentioned subtypes (Figures 3B and 3C) in the 134 samples and divided them into 10 groups (Figure 4E). The samples with predominantly poorly differentiated PitNET tumor cells (TPIT_Poorly-diff and PIT1_Poorly-diff) and SF1 lineage (SF1_Well-diff and SF1_Poorly-diff) tumor cells exhibited marked mRNA expression of lipid acid metabolism genes, including FADS1, FADS2, SCD, and SREBF1, which is consistent with our results (Figure 4D).
We also identified another subtype (TPIT_PAX7) marked by TBX19, PAX7, SOX2, and PCSK2 (Figures 4A, S10A, and S10B). All cells in this subtype were from a 51-year-old male patient (P21) suffering from ACTH-dependent Cushing’s syndrome. This patient underwent two surgeries. After the second surgery, the tumor relapsed quickly and increased to its preoperative size in only 3 months. He then received radiotherapy and temozolomide chemotherapy. Unfortunately, this combination treatment had no effects on controlling the tumor, and the patient expired 1 month later (Figures S10C and S10D). We noticed that the tumor cells in this subtype could be divided into two clusters based on UMAP (Figure 3A). One cluster showed similarity to CORTICO, while the other was more like GONADO (Figure 3B). Using mIHC, we confirmed the existence of two cell types in this TPIT lineage tumor: ACTH-positive SOX2-negative cells and ACTH-negative SOX2-positive cells (Figure S10A). Thus, we considered this tumor a rare PitNET with high intra-tumoral heterogeneity.
The differentiation status was related to long-term recurrence in a large retrospective cohort of 800 PitNETs
To investigate the clinical relevance of the newly discovered biomarkers in cell differentiation, we recruited an independent cohort of 800 PitNETs, including 298 (37.3%) PIT1 lineage tumors (76 lactotroph, 178 somatotroph, 13 thyrotroph, 26 silent PIT1, and 5 plurihormonal tumors), 125 (15.6%) TPIT lineage tumors (13 corticotroph and 112 silent TPIT tumors), 354 (44.3%) SF1 lineage tumors (44 gonadotroph and 310 silent SF1 tumors), and 23 (2.9%) null cell tumors. This cohort included similar numbers of female and male (368 versus 432) patients with a median age of 50 years (range: 12–83) and a median tumor size of 2.7 cm (range: 0.8–8.0). The follow-up was available in 624 (78.0%) patients. The median follow-up duration was 7.2 years, with a tumor recurrence rate of 7.1% (44/624) (Figure 5A; Table S4). The pituitary hormones and representative differentiation markers were detected in the cohort of 800 PitNETs by IHC, and the staining intensities of each antibody for each tumor were quantified as H score when blinded to patient clinical outcomes (Figures 5A and S11A; STAR Methods; Table S4).
Figure 5.

The differentiation status was related to long-term recurrence in an independent cohort of 800 patients
(A) Heatmap showing IHC staining intensities in an independent cohort of 800 PitNETs.
(B–D) Scatterplot showing the NTS H score (x axis) versus the PRL H score (y axis) (B), serum PRL concentration (ng/mL) (y axis) (C), and DRD2 H score(y axis) (D) in 115 pathologically PRL-positive tumors, respectively. The p value was calculated by Spearman’s correlation analysis.
(E and F) Kaplan-Meier PFS curves for 220 patients with PIT1 lineage tumors (E) and 61 patients with silent TPIT tumors (F) stratified by differentiation marker expression. The p value was calculated by the log rank test.
(G) The bar plot shows the gender proportion between well and poorly differentiated silent TPIT tumors. The p value was calculated by Fisher exact test.
(H) Kaplan-Meier PFS curves for 83 patients with silent TPIT tumors stratified by gender. The p value was calculated by the log rank test.
(I) Kaplan-Meier PFS curves for 281 patients with SF1 lineage tumors stratified by differentiation marker expression. The p value was calculated by the log rank test. See also Figure S11.
In the PIT1 lineage, the H score for NTS was positively correlated with PRL staining, preoperative serum PRL level, and DRD2 staining in PRL-positive tumors (Spearman’s correlation analysis; R = 0.75, p < 2.2e−16, R = 0.61, p = 2.2e−12, and R = 0.55, p = 3.7e−10, n = 115) (Figures 5B–5D). We defined PIT1 PitNETs with a high H score for IGFBP7, LRRC4C, or GATA3 as poorly differentiated PIT1 tumors and the remaining PIT1 PitNETs with a high H score for either ENPP1 or NTS as well-differentiated PIT1 tumors. Patients with poorly differentiated PIT1 tumors had a significantly worse progression-free survival (PFS) than those with well-differentiated PIT1 tumors (log rank test, p = 0.00046) (Figure 5E). Independent high H score for IGFBP7, LRRC4C, and GATA3 was also significantly related to worse PFS (log rank test, p = 0.037, p = 0.0027, and p = 0.043) (Figures S11B–S11D). Moreover, the H score for FADS1 was elevated in poorly differentiated PIT1 tumors (Wilcoxon rank-sum test, p = 2.3e−11, Figure S11E), suggesting upregulated unsaturated fatty acid metabolism.
We explored the pseudotime trajectory of TPIT_Poorly-diff and identified ID4 and CITED1 as markers for poorly and well-differentiated silent TPIT cells, respectively (Figure 4C). Here, in the validation cohort, we revealed the value of these two markers as a pair of recurrence prediction markers for silent TPIT tumors (log rank test, p < 0.0001 and p = 0.0054; Figures S11F and S11G). Since ID4 expression was elevated in poorly differentiated TPIT cells in scRNA-seq data, we defined silent TPIT PitNETs with high H scores for ID4 as poorly differentiated silent TPIT tumors, and the remaining silent TPIT PitNETs with high H scores for CITED1 as well-differentiated silent TPIT tumors. Patients with well- and poorly differentiated silent TPIT tumors showed a significant survival difference (log rank test, p < 0.0001, Figure 5F). Notably, while most patients with silent TPIT PitNETs were female (74/84, 88.1%), all silent TPIT tumors in male patients were classified into the poorly differentiated silent TPIT group (chi-square test, p < 0.001, Figure 5G), and exhibited strong predictive value (log rank test, p < 0.0001, Figure 5H), suggesting that male gender was a high risk factor for poorly differentiation status in silent TPIT PitNET. Poorly differentiated silent TPIT tumors showed higher H scores for FADS1 and HMGCR than well-differentiated silent TPIT tumors (Wilcoxon rank-sum test, p = 0.025 and p = 0.0066, Figures S11H and S11I), suggesting more synthesis of polyunsaturated fatty acid and cholesterol. However, well-differentiated silent TPIT tumors showed a higher H score for SCD (Wilcoxon rank-sum test, p = 0.00018, Figure S11J), indicating that even well-differentiated silent TPIT PitNETs preserve significant dysregulation of lipid metabolism.
In 354 SF1 lineage tumors, 73 (20.6%) SF1 tumors with high FSHB H scores were defined as well-differentiated SF1 tumors, while others were defined as poorly differentiated SF1 tumors. To our surprise, well-differentiated SF1 tumors, but not poorly differentiated tumors, exhibited a higher risk of recurrence (log rank test, p = 0.0065, Figure 5I).
Multiple factors used as predictors for the recurrence of PitNET were also evaluated in this cohort, including clinicopathological classification, Ki-67 index, tumor invasion (surgery or radiology), completeness of resection, tumor diameter, gender, and age (Table S4).12 To evaluate the predictive value of differentiation status, we compared it with previously reported factors using Cox regression survival analyses. We found that the Ki-67 index showed significant recurrence prediction capability in PIT1 lineage tumors in both univariable and multivariable analyses (p < 0.001) (Figure 6A). Furthermore, we performed survival analyses and consistently found that the Ki-67 index could predict the tumor recurrence only in PIT1 lineage rather than in silent TPIT and SF1 lineages (log rank test, p = 0.0023, p = 0.23, and p = 0.54, respectively; Figures 6B–6D). In PIT1 lineage, the Ki-67 index demonstrated the highest predictive value in well-differentiated tumors, while the predictive value is limited in poorly differentiated tumors (log rank test, p = 0.02 and p = 0.05, respectively; Figure 6E). However, in both silent TPIT and SF1 lineages, the Ki-67 index had no predictive value neither in well-differentiated nor poorly differentiated groups (p = 0.84 in well-differentiated silent TPIT, p = 0.95 in poorly differentiated silent TPIT, p = 0.34 in well-differentiated SF1, p = 0.19 in poorly differentiated SF1; Figures 6F and 6G). The predictive values of differentiation status and Ki-67 index are summarized in Figure 6H. However, neither tumor invasiveness nor tumor diameter were independent predictors for tumor recurrence in our cohort (Figure 6A).
Figure 6.

The recurrence prediction capability of Ki-67 index for PitNET was stratified by the new differentiation classification
(A) Cox regression survival analyses reveal the recurrence predictive values of multiple factors in three lineages. HR, hazard ratios; N, number of patients; ∗p < 0.05, ∗∗p < 0.01, ∗∗∗p < 0.001.
(B–D) Kaplan-Meier PFS curves for PIT1 lineage tumors (B), silent TPIT tumors (C), and SF1 lineage tumors (D) stratified by Ki-67 index. The p value was calculated by the log rank test.
(E–G) Kaplan-Meier PFS curves for patients with PIT1 lineage tumors (E), silent TPIT tumors (F), and SF1 lineage tumors (G) stratified by differentiation status and Ki-67 index. The p value was calculated by the log rank test.
(H) Recurrence prediction value of differentiation status and Ki-67 index in each lineage. n.s., not significant.
Discussion
PitNET is the most common neuroendocrine tumor, characterized by high tumor heterogeneity and diverse clinical manifestations. At present, the prediction of tumor recurrence is a tremendous challenge, as the current classification system has limited prediction value. In this study, we conducted a comparative analysis of normal adult APG and PitNET at single-cell resolution and evaluated the differentiation status of PitNET (Figure 7). Then, single-cell transcriptomic signatures were unfolded in the three major lineages of the current clinicopathological classification. ENPP1, NTS, GATA3, IGFBP7, and LRRC4C are specific markers for PIT1 lineage; ID4 and CITED1 are specific markers for TPIT lineage; FSHB is a specific marker for SF1 lineage. Expanding the current classification with new transcriptome signatures and markers is the principal scientific novelty of this study, which may enhance its clinical translation and application.
Figure 7.

Diagram of the tumor origin and differentiation status of PitNET
The top yellow box, middle green box, and bottom brown box represent TPIT, PIT1, and SF1 lineage, respectively. The left panel represents the self-renewal trajectory of adult APG. The middle and right panels represent PitNETs in well and poorly differentiation status, respectively. The solid line represents the development process of endocrine cells in adult APG. The dashed line represents the potential tumorigenesis process of PitNET. The markers were annotated below each APG or PitNET subtype. Characteristics of each tumor subtype are listed below the diagram, with higher recurrence subtypes emphasized in bold red font. The speculated Pre.TPIT and Pre.SF1 are labeled with gray dashed line circles since they were not detected in our study.
Furthermore, based on the differentiation status of PitNETs tumor cells, a new differentiation classification was proposed: well and poorly differentiated groups. In a separate large cohort of 800 patients with a median follow-up duration of 7.2 years, we validated these markers identified from two differentiated groups. Poorly differentiated groups showed significantly higher recurrence rates in both PIT1 and TPIT lineage, while well-differentiated groups showed significantly higher recurrence rates in SF1 lineage. Together, these demonstrate the potential application of the new differentiation classification in recurrence prediction.
In addition, the new differentiation classification could also stratify the predictive ability of the Ki-67 index, a proliferative marker widely used as a predictor of tumor recurrence.54,55,56 We found that, in well-differentiated PIT1 PitNETs, Ki-67 holds excellent predictive value for tumor recurrence, while in poorly differentiated PIT1, TPIT, or SF1 PitNETs, the predictive value for tumor recurrence was limited. These may explain the conflicting results of Ki-67 prediction value in gonadotroph tumors or nonfunctioning pituitary adenomas.57,58,59,60 Collectively, establishing a new differentiation classification that could precisely predict tumor recurrence with specific molecular markers was the clinical innovation of this study, which may help to create individualized follow-up strategies.
We revealed the developmental trajectory in the APG and identified a cluster named Pro.PIT1. After merging our adult APG data with the fetal APG data, we found that large numbers of fetal PIT1 cells were in a poor differentiation status and similar to the Pro.PIT1 cells in the adult APG. In particular, most lactotropes in the fetal cells were immature, suggesting the postnatal development of this cell type.27 For TPIT and SF1 lineages, cells in adult APG were similar to the corresponding fetal cells, while the progenitor cells of these two lineages were not identified in this study. We speculate that the progenitor cells of these two lineages may be located at the marginal zone or the remnant of Rathke’s cleft. Another scRNA-seq study including all pituitary tissues (anterior lobe, Rathke’s pouch, and posterior lobe) should be processed to understand adult pituitary heterogeneity further.
We focused on 750 SOX2-expressing cells (296 from APGs and 454 from PitNETs) and found that activation of EMT-related pathways was shared across individuals and decreased along with degree of differentiation. We also identified and validated several cell surface markers shared by most SOX2-expressing cells (e.g., CLDN4), which could be used for flow sorting. Interestingly, we found that most TSCs in PitNETs were not proliferating. On the other hand, an in vitro study suggests that isolated PitNET TSCs could grow as spheroids.61 These results suggested the TSCs were in a non-proliferative state in vivo and had the potency to proliferate and differentiate.
By comparing PitNETs with APGs, we estimated the possible cellular origin and differentiation status of each tumor cluster. Well-differentiated PitNETs showed activation of hormone biosynthesis- and secretion-related pathways. On the other hand, most poorly differentiated PitNETs were silent tumors and showed upregulation of metabolism-related pathways, especially lipid metabolism. This pattern may be due to the high proliferation of these subtypes and the necessity of lipid metabolites for multiple mitosis-related processes, such as cytomembrane synthesis and intracellular signal transduction.62 The molecular features of well- and poorly differentiated PitNETs were also verified in a cohort of 134 PitNETs published by Neou et al.39 The characteristic alterations may provide ideas for future therapeutic target exploration and drug development for these highly recurring PitNET subtypes.
Notably, we sequenced a rare tumor coexpressing SOX2 and PAX7, indicating its poorly differentiation status.16,63 This patient relapsed quickly and responded poorly to treatments (Figure S10). These features supported the importance of evaluating tumor differentiation status, especially for those clinically invasive tumors. Further study accumulating more cases with poorly differentiated tumors should be performed to focus on the clinical characteristics, molecular features, and high proliferation mechanism, which may identify the prognostic value of this new subtype and provide new treatment targets.
In conclusion, we reveal the single-cell transcriptomic landscape of three adult APG and 21 PitNET tumors and estimate the differentiation status of each PitNET. We further found that the markers of each differentiation status could help predict the recurrence of PitNETs.
Limitations of the study
There are still some limitations of this study. First, death may have potential impact on the transcriptome program of APG cells, leading to the unknown bias of data. Second, the number of PitNETs is still limited for each subtype. Additional studies that include a higher number of samples and their data integration are needed to get deeper insights. Third, although our study inferred the possible origin of the PitNET by comparative analysis, lineage-tracing confirmation studies of the PitNET models are required to confirm the origin of the PitNETs studied.
STAR★Methods
Key resources table
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| Mouse monoclonal anti-SOX2 (clone 20G5) | Abcam | Cat#ab171380; RRID:AB_2732072 |
| Mouse monoclonal anti-VIM (clonal E−5) | Santa Cruz Biotechnology | Cat#sc-373717; RRID:AB_10917747 |
| Rabbit monoclonal anti-S100B (clone EP1576Y) | Abcam | Cat#ab52642; RRID:AB_882426 |
| Rabbit monoclonal anti-SOX9 (clone EPR14335-78) | Abcam | Cat#ab185966; RRID:AB_2728660 |
| Rabbit monoclonal anti-CLDN4 (clone EPRR17575) | Abcam | Cat#ab210796; RRID:AB_2732879 |
| Rabbit monoclonal anti-PCSK2 (clone D1E1S) | Cell Signaling Technology | Cat#14013; RRID:AB_2631285 |
| Mouse monoclonal anti-GH (clonal E−7) | Santa Cruz Biotechnology | Cat#sc-374266; RRID:AB_10989917 |
| Rabbit polyclonal anti-NTS | Abclonal | Cat#A12326; RRID:AB_2759176 |
| Mouse monoclonal anti-PIT1 (clonal G-2) | Santa Cruz Biotechnology | Cat#sc-25258; RRID:AB_628135 |
| Rabbit monoclonal anti-SF1 (clonal EPR19744) | Abcam | Cat#ab217317; RRID:AB_2920891 |
| Mouse monoclonal anti-TPIT (clonal 2G1) | OriGene | Cat#TA800683; RRID:AB_2625610 |
| Mouse monoclonal anti-PRL (clonal A-7) | Santa Cruz Biotechnology | Cat#sc-46698; RRID:AB_628174 |
| Rabbit monoclonal anti-ENPP1 (clone EPR22262-22) | Abcam | Cat#ab223268; RRID:AB_2928116 |
| Rabbit monoclonal anti-TSHB (clone EPR8198) | Abcam | Cat#ab155958; RRID:AB_2928117 |
| Rabbit monoclonal anti-IGFBP7 (clone EPR11912(B)) | Abcam | Cat#ab171085; RRID:AB_2928118 |
| Rabbit polyclonal anti-LRRC4C | Proteintech | Cat#13845-1-AP; RRID:AB_2137623 |
| Rabbit monoclonal anti-TROP2 (clone EPR20043) | Abcam | Cat#ab214488; RRID:AB_2811182 |
| Rabbit monoclonal anti-KRT19 (clone EP1580Y) | Abcam | Cat#ab52625; RRID:AB_2281020 |
| Mouse monoclonal anti-Ki-67 (clonal 8D5) | Cell Signaling Technology | Cat#9449; RRID:AB_2797703 |
| Rabbit monoclonal anti-FSHB (clone EPR24439-141) | Abcam | Cat#ab281562; RRID:AB_2928119 |
| Rabbit monoclonal anti-LHB (clone EPR8196) | Abcam | Cat#ab150416; RRID:AB_2928120 |
| Mouse monoclonal anti-ACTH (clonal B427) | Santa Cruz Biotechnology | Cat#sc-57021; RRID:AB_785253 |
| Mouse monoclonal anti-GATA3 (clone L50-823) | Abcam | Cat#ab282110; RRID:AB_2928121 |
| Mouse monoclonal anti-CITED1 (clonal 5H6) | Abcam | Cat#ab87978; RRID:AB_2040929 |
| Rabbit polyclonal anti-ID4 | LSBio | Cat#LS-C368822; RRID:AB_2928122 |
| Rabbit monoclonal anti-FADS1 (clonal EPR6898) | Abcam | Cat#ab126706; RRID:AB_11130088 |
| Rabbit monoclonal anti-SCD (clonal EPR21963) | Abcam | Cat#ab236868; RRID:AB_2928123 |
| Rabbit monoclonal anti-FASN (clonal C20G5) | Cell Signaling Technology | Cat#3180; RRID:AB_2100796 |
| Mouse monoclonal anti-HMGCR (clonal CL0260) | Abcam | Cat#ab242315; RRID:AB_2928124 |
| Biological samples | ||
| Patient-derived PitNET tissues | Huashan Hospital of Fudan University | Table S1 |
| The human anterior pituitary gland | Donors | Table S1 |
| Critical commercial assays | ||
| Human Tumor Dissociation Kit | Miltenyi Biotec | 130-095-929 |
| Chromium Single Cell 3′ Reagent Kits v3 | 10x Genomics | N/A |
| multiplex mIHC kit | PerkinElmer | NEL861001KT |
| Deposited data | ||
| Single-cell RNA-seq data | This paper | NGDC: PRJCA009690 |
| Fetal pituitary single-cell RNA-seq data | Zhang et al.27 | GEO: GSE142653 |
| PitNETs bulk RNA-seq data | Neou et al.39 | EMBL-EBI: E-MTAB-7768 (https://www.ebi.ac.uk/) |
| Software and algorithms | ||
| Cell Ranger v3.1.0 | 10x Genomics | https://support.10xgenomics.com/single-cell-gene-expression/software/pipelines/3.1/installation |
| Cell Bender | Fleming et al.64 | https://github.com/broadinstitute/CellBender |
| R version v4.0.4 | The R Project for Statistical Computing | https://www.r-project.org/ |
| Seurat v4.1.0 | Satija Lab65 | https://satijalab.org/seurat/ |
| scDblFinder | Germain et al.66 | https://github.com/plger/scDblFinder |
| InferCNV v1.10.0 | inferCNV of the Trinity CTAT Project. | https://github.com/broadinstitute/inferCNV |
| Scanpy v1.7.1 | Wolf et al.67 | https://scanpy.readthedocs.io/en/stable/ |
| Scanorama v1.7.1 | Hie et al.68 | https://github.com/brianhie/scanorama |
| pySCENIC v0.11.2 | Van de Sande et al.46 | https://aertslab.org/#scenic |
| velocyto v0.17.17 | La Manno et al.69 | https://velocyto.org/ |
| scVelo v0.2.4 | Bergen et al.35 | https://scvelo.readthedocs.io/ |
| Survival v3.2_13 | N/A | https://CRAN.R-project.org/package=survival |
| clusterProfiler v3.18.1 | Yu et al.70 | https://guangchuangyu.github.io/software/clusterProfiler/ |
| MSigDB v7.4 | Subramanian et al.71 | http://www.gsea-msigdb.org/gsea/msigdb/index.jsp |
| Monocle3 v1.0.0 | Cao et al.36 | https://github.com/cole-trapnell-lab/monocle3 |
| slingshot v2.2.0 | Street et al.72 | https://github.com/kstreet13/slingshot |
| PAGA | Wolf et al.73 | https://github.com/theislab/paga |
| UMAP | McInnes et al.74 | N/A |
| scMetabolism | Wu et al.51 | https://github.com/wu-yc/scMetabolism |
| MuSiC v0.2.0 | Wang et al.53 | https://xuranw.github.io/MuSiC/index.html |
| DESeq2 v1.34.0 | Love et al.75 | https://bioconductor.org/packages/release/bioc/html/DESeq2.html |
| ggpubr v0.4.0 | N/A | https://rpkgs.datanovia.com/ggpubr/index.html |
| tableone v0.13.0 | N/A | https://CRAN.R-project.org/package=tableone |
| QuPath v0.3.0 | Bankhead et al.76 | https://qupath.github.io/ |
Resource availability
Lead contact
Further information and requests for resources should be directed to and will be fulfilled by the lead contact, Yao Zhao (zhaoyaohs@vip.sina.com).
Materials availability
This study did not generate new unique reagents.
Experimental model and subject details
Patient recruitments
Two cohorts of patients were recruited: one for scRNA-seq analysis (21 PitNETs) and another for IHC validation (800 PitNETs). All patients received surgery at the Department of Neurosurgery at Huashan Hospital, an affiliate of Shanghai Medical College, Fudan University. The 21 PitNETs cohort received surgery between 2018 and 2021, while the 800 PitNETs cohort were collected between 2010 and 2014. Both cohorts only included subjects without previous malignancies. Patients’ clinical data for all patients were collected from medical records retrospectively (Tables S1 and S4). Four adult anterior pituitary gland tissues were obtained from cadaveric organ donors without evidence of any endocrine disease (Table S1). All patients gave written informed consent, and the ethics committee at Huashan Hospital approved the study.
Method details
Preparation of single-cell suspensions
Each specimen was equally cut into at least two fragments. One fragment of the provided tissues was processed for scRNA-seq, and the remainder was processed for histopathological assessment. In brief, fresh tumor tissues were mechanically chopped with scalpels on a plate and enzymatically digested with Human Tumor Dissociation Kit (Miltenyi Biotec, Bergisch Gladbach, Germany) per manufacturer guidelines. Cell viability was confirmed to be >70% in all tissues using trypan blue exclusion. Cell suspensions were filtered using a 70 μm strainer (ThermoFisher Scientific), and dissociated cells were pelleted and re-suspended in PBS with 1% BSA.
Multiplex immunohistochemistry
Formalin-fixed and paraffin-embedded tissue sections (3μm) were de-paraffinized and rehydrated. Next, heat-induced epitope retrieval (HIER) was performed, followed by blocking with 3% hydrogen peroxide in TBST for 10 min and staining with the multiplex mIHC kit (PerkinElmer, NEL861001KT, Shanghai Kelin Institute). Briefly, after the first primary antibody staining, slides were incubated using the HRP-polymer detection system for 10 min, then visualization using Opal TSA working solution (1:100) for another 10 min. Afterward, antigen retrieval was conducted again to prepare the slides for the next antibody. Using this Opal staining method, primary antibodies were applied sequentially. Lastly, slides were counterstained with DAPI (Sigma, 1:1000) for nuclei visualization and subsequently coverslipped using the Hardset mounting media (VectaShield, H-1400).
All tissue sections that underwent multiplex fluorescent staining for each fluorophore were imaged using the Vectra Polaris imaging system (PerkinElmer, Shanghai Kelin Institute) under the appropriate fluorescent filters to produce the spectral library required for multispectral analysis. A whole slide scan of the multiplex tissue sections produced multispectral fluorescent images visualized in Phenochart (PerkinElmer) and imaging at 200× magnification.
Immunohistochemistry staining of tissue sections
PitNET tissues were fixed in 4% paraformaldehyde (PFA) for 24 h and processed for paraffin embedding tissue sections of 3μm. Following deparaffinization and rehydration, heat-induced epitope retrieval (HIER) was performed by submerging the slides in antigen unmasking solution (Solarbio).
After blocking endogenous peroxidase and nonspecific binding sites (0.3% H2O2 and 5% normal goat serum, sequentially), primary antibodies were applied at 4°C overnight. Slides were incubated with Dako REAL EnVision HRP rabbit/mouse (belong to K5007, DAKO, Glostrup, Denmark) at RT for 20min, followed by treatment with Dako REAL DAB + CHROMOGEN and Dako REAL substrate buffer (belong to K5007, DAKO, Glostrup, Denmark) to visualize staining signals under light microscopy, finally counterstained using hematoxylin solution. Stained slides were scanned using Ocus (Grundium, Tampere, Finland) and analyzed with Qupath software (see below).
IHC image analysis
Stained slides were scanned using Ocus (Grundium, Tampere, Finland) and analyzed with Qupath software v0.3.076. Images were preprocessed by the build-in stain vector estimator. Cells with shape and stain parameters in each area were identified by build-in cell detection via nucleus stain (hematoxylin). The mean DAB optical density (OD) thresholds for positivity were decided according to the staining pattern and intensities on all images for each antibody. The H-score was calculated as the percentage of tumor cells with positive staining multiplied by the average intensity of positive staining. Scripts of the whole-slide images analysis protocol above were created, batch performed on each set of images, and further checked by two expert pathologists. All quantifications were evaluated blinded to patient clinical information and outcomes.
DNA extraction and targeted exome sequencing
Total DNA (0.3 μg) was extracted from the 10 mg FFPE tissues using QIAamp DNA FFPE Tissue Kit (Qiagen). Sequencing libraries were generated using the probe independently designed by Medical Laboratory of Nantong ZhongKe Co, Ltd. Then the DNA libraries were sequenced on Illumina Novaseq 6000 platform. Fastp was used to control the data quality of the obtained raw data (FASTQ). Valid sequencing data was mapped to the reference human genome (UCSC hg19) by Burrows-Wheeler Aligner (BWA) software77 to get the original mapping results stored in BAM format. Then, SAMtools78 and Sambamba were used to recalibrate the BAM file quality, and calculated the sequence coverage and depth. Single-nucleotide polymorphisms (SNPs) and small insertions and deletions (InDels) of each exome were detected using the HaplotypeCaller tool of Genome Analysis Toolkit (GATK). The somatic SNVs and InDels were detected by muTect2.79 Filter criteria for high quality variants were defined as the allelic fraction more than 15%.
Droplet-based single-cell RNA-seq
The scRNA-seq was performed by Jiayin Biotechnology Ltd. (Shanghai, China), according to the manufacturer’s instructions of the Chromium Single Cell 3′ Reagent Kits v3. Single-cell suspensions were loaded on the Chromium Controller (10x Genomics, Pleasanton) to generate Gel Bead-In-Emulsions (GEMs). Next, barcoded sequencing libraries were conducted following the instruction manual of the Chromium Single Cell 3′ Reagent Kits v3 (10x Genomics). Following the library preparation, the sequencing was performed with paired-end sequencing of 150nt each end on one lane of NovaSeq 6000 per tissue.
Processing scRNA-seq data
Raw sequencing data were processed using the CellRanger (10X Genomics, v3.1.0) with default parameters. Reads were aligned to the human genome reference sequence (GRCh38). Uniquely mapped reads were used for UMI counting, and gene expression levels were further quantified for each barcode detected.
These raw feature-barcode matrices were processed by CellBender64 with default parameters to remove ambient RNA molecules for each sample. The clean corrected expression matrices were then processed and analyzed using Seurat package (v4.1.0).65 To filter out low-quality cells, cells that failed to meet the following criteria were discarded: 1) the number of detected genes more than 500, 2) the number of detected UMIs more than 3,000, and 3) a fraction of mitochondrial UMIs counts less than 30%. Doublets were identified by scDblFinder66 with the parameter dbr.SD = 1, and carefully removed based on the expression patterns of cell type/lineage-specific markers.
Data integration, clustering, and normalization
To remove potential batch effects, consensus variable features among samples, including APGs, APGs and PitNETs, human adult APGs and fetal APGs, were first identified. For individual samples, highly variable features were selected. These selected features were merged, and consensus features were generated based on hits across samples. Scanorama (v1.7.1)68 was used for dimension reduction with these consensus features and to integrate these datasets with default parameters. The graph-based clustering was then performed using functions FindNeighbors and FindClusters. In this study, raw counts were scaled to 10,000 and log2-transformed. Gene expression levels were defined as log2(normalized counts +1).
Identifying differentially expressed genes (DEGs) and cell classification
DEGs were identified with the FindAllMarkers function in the Seurat package (one-tailed Wilcoxon rank-sum test, p values adjusted using the Bonferroni correction). These DEGs with adjusted p value less than 0.01 were considered significant DEGs.
Cells in APGs were first classified based on the expression of classical marker genes.17,20,26,32 The expression percentage of each marker gene in the given cell type is annotated in parentheses. Endothelial cells were identified by specific expression of PLVAP (92.3%), ESAM (86.5%), PECAM1 (80.8%), CLEC14A (84.6%), EMCN (90.4%), FLT1 (84.6%), ADGRL4 (84.6%), CAVIN2 (94.2%) or KDR (84.6%); fibroblasts were identified by COL1A1 (95.2%), COL1A2 (100%), COL3A1 (95.2%), COL5A1 (100%), DCN (95.2%), LUM (95.2%), ACTA2 (90.5%), FN1 (95.2%) or FBLN1 (95.2%); lymphocytes were identified by PTPRC (86.0%), CD3E (82.2%), CD3D (70.1%), CD52 (81.3%) or CD48 (85.0%); Myeloid cells were identified by FCER1G (97.8%), CD68 (89.6%), CD14 (80.8%), CD74 (99.5%), RGS1 (87.4%), CSF1R (81.9%), C1QA (84.1%), C1QB (80.8%) or C1QC (81.9%). Pituitary endocrine cells were identified by SCG2 (88.8%), SCG3 (78.1%), CHGB (97.7%), SEZ6L2 (82.9%), SYP (63.6%), SNAP25 (79.8%), or UCHL1 (91.5%), including the following cell type-specific markers: CORTICO were identified by POMC (100.0%), TBX19 (94.0%), GAL (98.8%), RAB3B (90.4%) or ASCL1 (38.6%), GONADO were identified by NR5A1 (46.4%), CGA (65.4%), LHB (33.5%), FSHB (54.4%), TGFBR3L (90.6%) or GNRHR (21.2%); SOMATO were identified by POU1F1 (59.0%), GH1 (100.0%), GH2 (27.1%) or GHRHR (77.7%); LACTO (LACTOHigh and LACTOMedium) were identified by POU1F1 (80.1%), PRL (100.0%), DRD2 (52.9%), or VGF (50.4%); THYRO were identified by POU1F1 (83.0%), TSHB (52.3%), CGA (100.0%). Pituitary stem cells were identified by SOX2 (86.9%), AGR3 (78.5%), SLPI (97.7%), KRT19 (75.2%), TACSTD2 (45.6%), MGST1 (74.2%), including the following cell type-specific markers: S100BPos STEM were identified by S100B (33.2%), S100A1 (63.8%), SOX9 (46.2%), FABP7 (69.8%), TTYH1 (72.4%), LYPD1 (84.4%), or RFX4 (63.8%).
Inferring CNVs from single-cell RNA-seq data
The single-cell CNVs were inferred by a moving averaged expression profiles across chromosomal intervals using InferCNV as previously reported.80,81,82,83,84 In particular, the endocrine and stem cells identified in APGs were considered as the reference cells,15 and average CNVs value for these cells was subtracted from all other cells. When excluding suspicious normal APG cells in P10, we scored “CNV correlation”, which refers to the correlation between the CNV profile of each cell and the average CNA profile of all neuroendocrine cells from the corresponding tumor. Cells were then classified as suspicious normal APG cells if they had CNV correlation less than 0.340,41.
Exclude suspicious normal APG cells in PitNET samples
Three approaches were combined to distinguish the non-tumor neuroendocrine cells in PitNET samples. First, through the lineage annotation of tumor cells in each sample, we detected cells with more than one lineage in one tumor sample (P10) (Figure S5C). Since APGs are composed of heterogeneous endocrine cells while PitNETs were considered monoclonal origin,2,85 the identification of cells from multiple lineages suggests the possible presence of normal APGs cells in this tumor sample. Next, to distinguish suspicious normal APG cells in P10, we inferred CNVs and excluded tumor cells with CNV correlation less than 0.3. Finally, to double-check any normal APG cells left, we merged the residual cells from PitNETs through unsupervised clustering (Figure S6B). Clusters from multiple tumor samples were annotated by canonical markers, and none of them were neuroendocrine cells, while all neuroendocrine cells were mainly constituted of one tumor, suggesting all suspicious normal APG cells were excluded before downstream tumor origin analysis.
Calculating gene expression score
The gene scores were calculated using the AddModuleScore function in the Seurat R package based on a certain predefined expression program.
The epithelial (EPI) and mesenchymal (MESEN) signatures used for evaluating the epithelial-mesenchymal transition (EMT) status of SOX2 expressing cells were retrieved from Tan et al.86
In Figure 3B, PitNET clusters were scored by the top-20 DEGs identified in the endocrine and stem cluster of APGs. The top DEGs were ranked according to the formula pct.1∗(1-pct.2)∗avg_log2FC, to consider both the expression and specificity of DEGs. The pct.1, pct.2, and avg_log2FC in the formula were precalculated by FindAllMarkers function in Seurat within all endocrine and stem cells in APGs. Scoring PitNETs clusters using top 15 to 30 DEGs was also calculated and produced similar results.
Pathway enrichment analysis
Pathway enrichment analyses were performed by GSEA, enricher, and compareCluster functions implemented in clusterProfiler (v3.18.1)70 with pathway database MSigDB (v7.4)71. The gene sets were retrieved from the following categories: hallmark, KEGG, Reactome, GO biological processes, GO cellular components, and GO molecular functions.
Inferring activated transcription factors by SCENIC analysis
The Single-Cell rEgulatory Network Inference and Clustering (SCENIC) analysis was performed using the python pipeline pySCENIC (v0.11.2)46 with default parameters. The co-expression modules were identified by GRNBoost. The input matrix was the normalized expression matrix for cells of interest. And the output AUCell matrix of regulon activities was further binarized. A human TF gene list was retrieved from the resources of pySCENIC (https://github.com/aertslab/pySCENIC/tree/master/resources). The motifs database for Homo sapiens was downloaded from the cisTargetDBs (https://resources.aertslab.org/cistarget/).
Inferring cell state transition by RNA velocity
RNA velocities were calculated using velocyto (v0.17.17)69 and scVelo (v0.2.4)35. Briefly, spliced/unspliced/ambiguous reads of each sample were annotated by velocyto.py with CellRanger generating BAM files. RNA velocities were inferred by scVelo and projected as a field onto the pre-computed UMAP embedding for visualization.
Pseudotime analysis and lineage trajectories construction
The R package Monocle3 (v1.0.0)36 was applied to construct the trajectory of the integrated adult and fetal APG cells, and PIT1 lineage cells from APGs. The counts matrix and UMAP space were transferred into Monocle3. The trajectories were learned by cluster_cells and learn_graph functions. The root cells of adult and fetal APG cells were placed at the region of stem cell cluster (Figure S3H). The root cells of PIT1 lineage cells were located between the THRYO and SOMATO in the Pro.PIT1 cluster (Figure S3C).
The lineage trajectories of integrated human adult and fetal APGs were constructed by Slingshot (v2.2.0)72. The start cluster was set as Adult.STEM.
The trajectory of cell populations identified in the integrated human adult and fetal APG cells was constructed using the Partition-based graph abstraction (PAGA) method73 based on the UMAP embedding generated by Scanorama. The trajectory for PIT1 lineage cells in APGs was inferred by RNA velocity directionality.
scMetabolism analysis
To quantify metabolism activity at the single-cell resolution, scMetabolism51 was applied to the endocrine and SOX2 expressing cells of APGs and PitNETs. VISION was selected as the quantitative method, and GSEA was chosen to provide metabolism pathways.
Processing bulk RNA-seq data for PitNET samples
The mRNA-seq dataset previously published by Neou et al.39 was downloaded. DESeq2 (v1.34.0)75 was applied for the mRNA-seq read counts to get a normalized matrix.
Deconvolution analysis
The deconvolution analysis of bulk PitNETs RNA-seq data against each subtype was performed using MuSiC (v0.2.0).53 The signature gene input was generated by differential gene analysis using Seurat’s one-tailed Wilcoxon rank-sum test. To eliminate the variants of differentiated expression gene numbers of each subtype, the top 20 signature genes were selected as described above.
Survival analysis
The tissue microarray (TMA) dataset (n = 800) was used to evaluate the recurrence predictive performance of well or poorly differentiated genes. Tumors with Ki-67 index ≥2.0% were defined as high Ki-67 group, and the remaining were low Ki-67 group. Kaplan–Meier survival curves were created and compared among subgroups using log rank tests, and Cox proportional hazards models were performed to evaluate the recurrence prediction value of multiple factors, and both methods were implemented in the R package Survival (v3.2_13). The scan function was used to yield a cutoff where the difference in survival was most significant. These cutoff levels were then used to generate Kaplan–Meier graphs.
Quantification and statistical analysis
Statistical analysis was performed using R package ggpubr (v0.4.0) and tableone (v0.13.0). The Wilcoxon rank-sum test, Spearman’s correlation test, Fisher exact test, and log rank test were used in this study. Detailed descriptions of statistical tests were specified in the results section and the figure legends.
Acknowledgments
This work is supported by the National Key R&D Program of China (2022YFA1303200) (to Y. Zhao); the China Pituitary Adenoma Specialist Council (CPASC) (to Y. Zhao); the National High Technology Research and Development Program of China (863 program, 2014AA020611) (to Y. Zhao); the Chang Jiang Scholars Program (to Y. Zhao); the National Program for Support of Top-Notch Young Professionals (to Y. Zhao); the National Science Fund for Distinguished Young Scholars (81725011) (to Y. Zhao); CAMS Innovation Fund for Medical Sciences (2021-I2M-C&T-A-025) (to Y. Zhao); the Clinical Research Plan of SHDC (SHDC2020CR2004A) (to Y. Zhao); the National Natural Science Foundation of China (U21A20389 to Y. Zhao, 82170841 and 81970716 to Z.Z., 81802495 to Q.Z., 81800720 to M.H., 82073640 to N.Q., 82201776 to Z.C.); the National Key Research and Development Program of China (2018YFA0107701 to F.G.); Shanghai Sailing program (18YF1403400) (to Q.Z.); Shanghai Chenguang Scholar (19CG08) (to Q.Z.). We thank doctoral student Hongzhe Zhao of Sun Yat-sen University Cancer Center, State Key Laboratory of Oncology in South China and Collaborative Innovation Center for Cancer Medicine for helpful advice.
Author contributions
Conceptualization, Q.Z., Z.Z., F.G., and Y. Zhao; supervision, Q.Z., Z.Z., F.G., and Y. Zhao; resources (clinical data and follow-up information collection), Z.C., M.H., Yue Wu, N.Q., Z.M., Zhao Ye, Y. Zhang, S.Y., H.Y., Z.Z., Yongfei Wang, Y.L., and Y. Zhao; resources (samples collection), N.Q., Z.M., Zhao Ye, Ye Wang, Yongfei Wang, and Y. Zhao; resources (radiology review), Yue Wu, N.Q., Z.M., and Zhao Ye; investigation (samples handling), Z.C. and M.H.; investigation (single-cell RNA-seq data generation), B.Y., X.L., F.G., and J.C.; investigation (IHC staining and analyses), B.Y., H. Cheng, and H. Chen; formal analysis (bioinformatics and statistical analyses), Q.Z., B.Y., X.L., and F.G.; writing – original draft preparation, Q.Z., B.Y., X.L., Z.C., M.H., and Yue Wu; writing – review & editing, all authors.
Declaration of interests
The authors declare no competing interests.
Published: February 7, 2023
Footnotes
Supplemental information can be found online at https://doi.org/10.1016/j.xcrm.2023.100934.
Contributor Information
Zhaoyun Zhang, Email: zhaoyunzhang@fudan.edu.cn.
Fan Guo, Email: guofan@ioz.ac.cn.
Yao Zhao, Email: zhaoyaohs@vip.sina.com.
Supplemental information
Data and code availability
-
•
All raw scRNA-seq data have been deposited at the National Genomics Data Center (NGDC, https://bigd.big.ac.cn/) under the accession number PRJCA009690.
-
•
This study did not generate original codes. All software and algorithms used in this study are publicly available and listed in the key resources table.
-
•
Any additional information required to reanalyze the data reported in this paper is available from the lead contact upon request.
References
- 1.Ezzat S., Asa S.L., Couldwell W.T., Barr C.E., Dodge W.E., Vance M.L., McCutcheon I.E. The prevalence of pituitary adenomas: a systematic review. Cancer. 2004;101:613–619. doi: 10.1002/cncr.20412. [DOI] [PubMed] [Google Scholar]
- 2.Herman V., Fagin J., Gonsky R., Kovacs K., Melmed S. Clonal origin of pituitary adenomas. J. Clin. Endocrinol. Metab. 1990;71:1427–1433. doi: 10.1210/jcem-71-6-1427. [DOI] [PubMed] [Google Scholar]
- 3.Melmed S., Kaiser U.B., Lopes M.B., Bertherat J., Syro L.V., Raverot G., Reincke M., Johannsson G., Beckers A., Fleseriu M., et al. Clinical biology of the pituitary adenoma. Endocr. Rev. 2022;43:1003–1037. doi: 10.1210/endrev/bnac010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Molitch M.E. Diagnosis and treatment of pituitary adenomas: a review. JAMA. 2017;317:516–524. doi: 10.1001/jama.2016.19699. [DOI] [PubMed] [Google Scholar]
- 5.Melmed S. Pituitary-tumor endocrinopathies. N. Engl. J. Med. 2020;382:937–950. doi: 10.1056/NEJMra1810772. [DOI] [PubMed] [Google Scholar]
- 6.Asa S.L., Mete O., Perry A., Osamura R.Y. Overview of the 2022 WHO classification of pituitary tumors. Endocr. Pathol. 2022;33:6–26. doi: 10.1007/s12022-022-09703-7. [DOI] [PubMed] [Google Scholar]
- 7.Drummond J., Roncaroli F., Grossman A.B., Korbonits M. Clinical and pathological aspects of silent pituitary adenomas. J. Clin. Endocrinol. Metab. 2019;104:2473–2489. doi: 10.1210/jc.2018-00688. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Trouillas J., Jaffrain-Rea M.L., Vasiljevic A., Raverot G., Roncaroli F., Villa C. How to classify the pituitary neuroendocrine tumors (PitNET)s in 2020. Cancers. 2020;12 doi: 10.3390/cancers12020514. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Tampourlou M., Ntali G., Ahmed S., Arlt W., Ayuk J., Byrne J.V., Chavda S., Cudlip S., Gittoes N., Grossman A., et al. Outcome of nonfunctioning pituitary adenomas that regrow after primary treatment: a study from two large UK centers. J. Clin. Endocrinol. Metab. 2017;102:1889–1897. doi: 10.1210/jc.2016-4061. [DOI] [PubMed] [Google Scholar]
- 10.Patil C.G., Prevedello D.M., Lad S.P., Vance M.L., Thorner M.O., Katznelson L., Laws E.R., Jr. Late recurrences of Cushing's disease after initial successful transsphenoidal surgery. J. Clin. Endocrinol. Metab. 2008;93:358–362. doi: 10.1210/jc.2007-2013. [DOI] [PubMed] [Google Scholar]
- 11.Subramanian V., Lee R.S.M., Howell S., Gregson S., Lahart I.M., Kaushal K., Pappachan J.M. Non-functioning pituitary macroadenomas: factors affecting postoperative recurrence, and pre- and post-surgical endocrine and visual function. Endocrine. 2021;73:407–415. doi: 10.1007/s12020-021-02713-1. [DOI] [PubMed] [Google Scholar]
- 12.Guaraldi F., Zoli M., Righi A., Gibertoni D., Marino Picciola V., Faustini-Fustini M., Morandi L., Bacci A., Pasquini E., Mazzatenta D., Asioli S. A practical algorithm to predict postsurgical recurrence and progression of pituitary neuroendocrine tumours (PitNET)s. Clin. Endocrinol. 2020;93:36–43. doi: 10.1111/cen.14197. [DOI] [PubMed] [Google Scholar]
- 13.Esquenazi Y., Essayed W.I., Singh H., Mauer E., Ahmed M., Christos P.J., Schwartz T.H. Endoscopic endonasal versus microscopic transsphenoidal surgery for recurrent and/or residual pituitary adenomas. World Neurosurg. 2017;101:186–195. doi: 10.1016/j.wneu.2017.01.110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Svensson V., Vento-Tormo R., Teichmann S.A. Exponential scaling of single-cell RNA-seq in the past decade. Nat. Protoc. 2018;13:599–604. doi: 10.1038/nprot.2017.149. [DOI] [PubMed] [Google Scholar]
- 15.Dong R., Yang R., Zhan Y., Lai H.D., Ye C.J., Yao X.Y., Luo W.Q., Cheng X.M., Miao J.J., Wang J.F., et al. Single-cell characterization of malignant phenotypes and developmental trajectories of adrenal neuroblastoma. Cancer Cell. 2020;38:716–733.e6. doi: 10.1016/j.ccell.2020.08.014. [DOI] [PubMed] [Google Scholar]
- 16.Mayran A., Sochodolsky K., Khetchoumian K., Harris J., Gauthier Y., Bemmo A., Balsalobre A., Drouin J. Pioneer and nonpioneer factor cooperation drives lineage specific chromatin opening. Nat. Commun. 2019;10:3807. doi: 10.1038/s41467-019-11791-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Ho Y., Hu P., Peel M.T., Chen S., Camara P.G., Epstein D.J., Wu H., Liebhaber S.A. Single-cell transcriptomic analysis of adult mouse pituitary reveals sexual dimorphism and physiologic demand-induced cellular plasticity. Protein Cell. 2020;11:565–583. doi: 10.1007/s13238-020-00705-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Ruf-Zamojski F., Zhang Z., Zamojski M., Smith G.R., Mendelev N., Liu H., Nudelman G., Moriwaki M., Pincas H., Castanon R.G., et al. Single nucleus multi-omics regulatory landscape of the murine pituitary. Nat. Commun. 2021;12:2677. doi: 10.1038/s41467-021-22859-w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Chen Q., Leshkowitz D., Blechman J., Levkowitz G. Single-cell molecular and cellular architecture of the mouse neurohypophysis. eNeuro. 2020;7 doi: 10.1523/ENEURO.0345-19.2019. ENEURO.0345, 19.2019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Cheung L.Y.M., George A.S., McGee S.R., Daly A.Z., Brinkmeier M.L., Ellsworth B.S., Camper S.A. Single-cell RNA sequencing reveals novel markers of male pituitary stem cells and hormone-producing cell types. Endocrinology. 2018;159:3910–3924. doi: 10.1210/en.2018-00750. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Vennekens A., Laporte E., Hermans F., Cox B., Modave E., Janiszewski A., Nys C., Kobayashi H., Malengier-Devlies B., Chappell J., et al. Interleukin-6 is an activator of pituitary stem cells upon local damage, a competence quenched in the aging gland. Proc. Natl. Acad. Sci. USA. 2021;118 doi: 10.1073/pnas.2100052118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Laporte E., Hermans F., De Vriendt S., Vennekens A., Lambrechts D., Nys C., Cox B., Vankelecom H. Decoding the activated stem cell phenotype of the neonatally maturing pituitary. Elife. 2022;11 doi: 10.7554/eLife.75742. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Kučka M., Gonzalez-Iglesias A.E., Tomić M., Prévide R.M., Smiljanic K., Sokanovic S.J., Fletcher P.A., Sherman A., Balla T., Stojilkovic S.S. Calcium-prolactin secretion coupling in rat pituitary lactotrophs is controlled by PI4-kinase alpha. Front. Endocrinol. 2021;12 doi: 10.3389/fendo.2021.790441. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Lopez J.P., Brivio E., Santambrogio A., De Donno C., Kos A., Peters M., Rost N., Czamara D., Brückl T.M., Roeh S., et al. Single-cell molecular profiling of all three components of the HPA axis reveals adrenal ABCB1 as a regulator of stress adaptation. Sci. Adv. 2021;7 doi: 10.1126/sciadv.abe4497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Fletcher P.A., Smiljanic K., Prévide R.M., Constantin S., Sherman A.S., Coon S.L., Stojilkovic S.S. The astroglial and stem cell functions of adult rat folliculostellate cells. Glia. 2023;71:205–228. doi: 10.1002/glia.24267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Fletcher P.A., Smiljanic K., Maso Prévide R., Iben J.R., Li T., Rokic M.B., Sherman A., Coon S.L., Stojilkovic S.S. Cell type- and sex-dependent transcriptome profiles of rat anterior pituitary cells. Front. Endocrinol. 2019;10:623. doi: 10.3389/fendo.2019.00623. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Zhang S., Cui Y., Ma X., Yong J., Yan L., Yang M., Ren J., Tang F., Wen L., Qiao J. Single-cell transcriptomics identifies divergent developmental lineage trajectories during human pituitary development. Nat. Commun. 2020;11:5275. doi: 10.1038/s41467-020-19012-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Zhang Z., Zamojski M., Smith G.R., Willis T.L., Yianni V., Mendelev N., Pincas H., Seenarine N., Amper M.A.S., Vasoya M., et al. Single nucleus transcriptome and chromatin accessibility of postmortem human pituitaries reveal diverse stem cell regulatory mechanisms. Cell Rep. 2022;38 doi: 10.1016/j.celrep.2022.110467. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Cui Y., Li C., Jiang Z., Zhang S., Li Q., Liu X., Zhou Y., Li R., Wei L., Li L., et al. Single-cell transcriptome and genome analyses of pituitary neuroendocrine tumors. Neuro Oncol. 2021;23:1859–1871. doi: 10.1093/neuonc/noab102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Zhang D., Hugo W., Bergsneider M., Wang M.B., Kim W., Vinters H.V., Heaney A.P. Single-cell RNA sequencing in silent corticotroph tumors confirms impaired POMC processing and provides new insights into their invasive behavior. Eur. J. Endocrinol. 2022;187:49–64. doi: 10.1530/EJE-21-1183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Zhang D., Hugo W., Redublo P., Miao H., Bergsneider M., Wang M.B., Kim W., Yong W.H., Heaney A.P. A human ACTH-secreting corticotroph tumoroid model: novel Human ACTH-Secreting Tumor Cell in vitro Model. EBioMedicine. 2021;66 doi: 10.1016/j.ebiom.2021.103294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Asuzu D.T., Alvarez R., Fletcher P.A., Mandal D., Johnson K., Wu W., Elkahloun A., Clavijo P., Allen C., Maric D., et al. Pituitary adenomas evade apoptosis via noxa deregulation in Cushing's disease. Cell Rep. 2022;40 doi: 10.1016/j.celrep.2022.111223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Andoniadou C.L., Matsushima D., Mousavy Gharavy S.N., Signore M., Mackintosh A.I., Schaeffer M., Gaston-Massuet C., Mollard P., Jacques T.S., Le Tissier P., et al. Sox2(+) stem/progenitor cells in the adult mouse pituitary support organ homeostasis and have tumor-inducing potential. Cell Stem Cell. 2013;13:433–445. doi: 10.1016/j.stem.2013.07.004. [DOI] [PubMed] [Google Scholar]
- 34.Kato Y., Yoshida S., Kato T. New insights into the role and origin of pituitary S100beta-positive cells. Cell Tissue Res. 2021;386:227–237. doi: 10.1007/s00441-021-03523-7. [DOI] [PubMed] [Google Scholar]
- 35.Bergen V., Lange M., Peidli S., Wolf F.A., Theis F.J. Generalizing RNA velocity to transient cell states through dynamical modeling. Nat. Biotechnol. 2020;38:1408–1414. doi: 10.1038/s41587-020-0591-3. [DOI] [PubMed] [Google Scholar]
- 36.Cao J., Spielmann M., Qiu X., Huang X., Ibrahim D.M., Hill A.J., Zhang F., Mundlos S., Christiansen L., Steemers F.J., et al. The single-cell transcriptional landscape of mammalian organogenesis. Nature. 2019;566:496–502. doi: 10.1038/s41586-019-0969-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Knosp E., Steiner E., Kitz K., Matula C. Pituitary adenomas with invasion of the cavernous sinus space: a magnetic resonance imaging classification compared with surgical findings. Neurosurgery. 1993;33:610–617. doi: 10.1227/00006123-199310000-00008. [DOI] [PubMed] [Google Scholar]
- 38.Micko A.S.G., Wöhrer A., Wolfsberger S., Knosp E. Invasion of the cavernous sinus space in pituitary adenomas: endoscopic verification and its correlation with an MRI-based classification. J. Neurosurg. 2015;122:803–811. doi: 10.3171/2014.12.JNS141083. [DOI] [PubMed] [Google Scholar]
- 39.Neou M., Villa C., Armignacco R., Jouinot A., Raffin-Sanson M.L., Septier A., Letourneur F., Diry S., Diedisheim M., Izac B., et al. Pangenomic classification of pituitary neuroendocrine tumors. Cancer Cell. 2020;37:123–134.e5. doi: 10.1016/j.ccell.2019.11.002. [DOI] [PubMed] [Google Scholar]
- 40.Neftel C., Laffy J., Filbin M.G., Hara T., Shore M.E., Rahme G.J., Richman A.R., Silverbush D., Shaw M.L., Hebert C.M., et al. An integrative model of cellular states, plasticity, and genetics for glioblastoma. Cell. 2019;178:835–849.e21. doi: 10.1016/j.cell.2019.06.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Ma L., Hernandez M.O., Zhao Y., Mehta M., Tran B., Kelly M., Rae Z., Hernandez J.M., Davis J.L., Martin S.P., et al. Tumor cell biodiversity drives microenvironmental reprogramming in liver cancer. Cancer Cell. 2019;36:418–430.e6. doi: 10.1016/j.ccell.2019.08.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Nys C., Lee Y.L., Roose H., Mertens F., De Pauw E., Kobayashi H., Sciot R., Bex M., Versyck G., De Vleeschouwer S., et al. Exploring stem cell biology in pituitary tumors and derived organoids. Endocr. Relat. Cancer. 2022;29:427–450. doi: 10.1530/ERC-21-0374. [DOI] [PubMed] [Google Scholar]
- 43.Vankelecom H., Roose H. The stem cell connection of pituitary tumors. Front. Endocrinol. 2017;8:339. doi: 10.3389/fendo.2017.00339. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Lambert A.W., Weinberg R.A. Linking EMT programmes to normal and neoplastic epithelial stem cells. Nat. Rev. Cancer. 2021;21:325–338. doi: 10.1038/s41568-021-00332-6. [DOI] [PubMed] [Google Scholar]
- 45.Yoshida S., Kato T., Kato Y. EMT involved in migration of stem/progenitor cells for pituitary development and regeneration. J. Clin. Med. 2016;5 doi: 10.3390/jcm5040043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Van de Sande B., Flerin C., Davie K., De Waegeneer M., Hulselmans G., Aibar S., Seurinck R., Saelens W., Cannoodt R., Rouchon Q., et al. A scalable SCENIC workflow for single-cell gene regulatory network analysis. Nat. Protoc. 2020;15:2247–2276. doi: 10.1038/s41596-020-0336-2. [DOI] [PubMed] [Google Scholar]
- 47.Ben-Shlomo A., Cooper O. Silent corticotroph adenomas. Pituitary. 2018;21:183–193. doi: 10.1007/s11102-018-0864-8. [DOI] [PubMed] [Google Scholar]
- 48.Xu Z., Ellis S., Lee C.C., Starke R.M., Schlesinger D., Lee Vance M., Lopes M.B., Sheehan J. Silent corticotroph adenomas after stereotactic radiosurgery: a case-control study. Int. J. Radiat. Oncol. Biol. Phys. 2014;90:903–910. doi: 10.1016/j.ijrobp.2014.07.013. [DOI] [PubMed] [Google Scholar]
- 49.Jahangiri A., Wagner J.R., Pekmezci M., Hiniker A., Chang E.F., Kunwar S., Blevins L., Aghi M.K. A comprehensive long-term retrospective analysis of silent corticotrophic adenomas vs hormone-negative adenomas. Neurosurgery. 2013;73:8–17. doi: 10.1227/01.neu.0000429858.96652.1e. [DOI] [PubMed] [Google Scholar]
- 50.Lopes M.B.S. The 2017 World Health Organization classification of tumors of the pituitary gland: a summary. Acta Neuropathol. 2017;134:521–535. doi: 10.1007/s00401-017-1769-8. [DOI] [PubMed] [Google Scholar]
- 51.Wu Y., Yang S., Ma J., Chen Z., Song G., Rao D., Cheng Y., Huang S., Liu Y., Jiang S., et al. Spatiotemporal immune landscape of colorectal cancer liver metastasis at single-cell level. Cancer Discov. 2022;12:134–153. doi: 10.1158/2159-8290.CD-21-0316. [DOI] [PubMed] [Google Scholar]
- 52.Shimano H., Sato R. SREBP-regulated lipid metabolism: convergent physiology - divergent pathophysiology. Nat. Rev. Endocrinol. 2017;13:710–730. doi: 10.1038/nrendo.2017.91. [DOI] [PubMed] [Google Scholar]
- 53.Wang X., Park J., Susztak K., Zhang N.R., Li M. Bulk tissue cell type deconvolution with multi-subject single-cell expression reference. Nat. Commun. 2019;10:380. doi: 10.1038/s41467-018-08023-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Marchevsky A.M., Hendifar A., Walts A.E. The use of Ki-67 labeling index to grade pulmonary well-differentiated neuroendocrine neoplasms: current best evidence. Mod. Pathol. 2018;31:1523–1531. doi: 10.1038/s41379-018-0076-9. [DOI] [PubMed] [Google Scholar]
- 55.Zhang Y., Zhou Y., Mao F., Yao R., Sun Q. Ki-67 index, progesterone receptor expression, histologic grade and tumor size in predicting breast cancer recurrence risk: a consecutive cohort study. Cancer Commun. 2020;40:181–193. doi: 10.1002/cac2.12024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Asano D., Kudo A., Akahoshi K., Maekawa A., Murase Y., Ogawa K., Ono H., Ban D., Tanaka S., Tanabe M. Curative surgery and ki-67 value rather than tumor differentiation predict the survival of patients with high-grade neuroendocrine neoplasms. Ann. Surg. 2022;276:e108–e113. doi: 10.1097/SLA.0000000000004495. [DOI] [PubMed] [Google Scholar]
- 57.Salehi F., Agur A., Scheithauer B.W., Kovacs K., Lloyd R.V., Cusimano M. Ki-67 in pituitary neoplasms: a review--part I. Neurosurgery. 2009;65:429–437. doi: 10.1227/01.NEU.0000349930.66434.82. [DOI] [PubMed] [Google Scholar]
- 58.Guadagno E., D'Avella E., Cappabianca P., Colao A., Del Basso De Caro M. Ki67 in endocrine neoplasms: to count or not to count, this is the question! A systematic review from the English language literature. J. Endocrinol. Invest. 2020;43:1429–1445. doi: 10.1007/s40618-020-01275-9. [DOI] [PubMed] [Google Scholar]
- 59.Dubois S., Guyétant S., Menei P., Rodien P., Illouz F., Vielle B., Rohmer V. Relevance of Ki-67 and prognostic factors for recurrence/progression of gonadotropic adenomas after first surgery. Eur. J. Endocrinol. 2007;157:141–147. doi: 10.1530/EJE-07-0099. [DOI] [PubMed] [Google Scholar]
- 60.Iglesias P., Arcano K., Triviño V., García-Sancho P., Díez J.J., Cordido F., Villabona C. Non-functioning pituitary adenoma underwent surgery: a multicenter retrospective study over the last four decades (1977-2015) Eur. J. Intern. Med. 2017;41:62–67. doi: 10.1016/j.ejim.2017.03.023. [DOI] [PubMed] [Google Scholar]
- 61.Würth R., Barbieri F., Pattarozzi A., Gaudenzi G., Gatto F., Fiaschi P., Ravetti J.L., Zona G., Daga A., Persani L., et al. Phenotypical and pharmacological characterization of stem-like cells in human pituitary adenomas. Mol. Neurobiol. 2017;54:4879–4895. doi: 10.1007/s12035-016-0025-x. [DOI] [PubMed] [Google Scholar]
- 62.Röhrig F., Schulze A. The multifaceted roles of fatty acid synthesis in cancer. Nat. Rev. Cancer. 2016;16:732–749. doi: 10.1038/nrc.2016.89. [DOI] [PubMed] [Google Scholar]
- 63.Budry L., Balsalobre A., Gauthier Y., Khetchoumian K., L'honoré A., Vallette S., Brue T., Figarella-Branger D., Meij B., Drouin J. The selector gene Pax7 dictates alternate pituitary cell fates through its pioneer action on chromatin remodeling. Genes Dev. 2012;26:2299–2310. doi: 10.1101/gad.200436.112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Fleming S.J., Marioni J.C., Babadi M. CellBender remove-background: a deep generative model for unsupervised removal of background noise from scRNA-seq datasets. bioRxiv. 2019 doi: 10.1101/791699. Preprint at. [DOI] [Google Scholar]
- 65.Hao Y., Hao S., Andersen-Nissen E., Mauck W.M., 3rd, Zheng S., Butler A., Lee M.J., Wilk A.J., Darby C., Zager M., et al. Integrated analysis of multimodal single-cell data. Cell. 2021;184:3573–3587.e29. doi: 10.1016/j.cell.2021.04.048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Germain P., Lun A., Macnair W., Robinson M. Doublet identification in single-cell sequencing data using scDblFinder [version 1; peer review: 1 approved, 1 approved with reservations. F1000Research. 2021;10 doi: 10.12688/f1000research.73600.1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Wolf F.A., Angerer P., Theis F.J. SCANPY: large-scale single-cell gene expression data analysis. Genome Biol. 2018;19:15. doi: 10.1186/s13059-017-1382-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Hie B., Bryson B., Berger B. Efficient integration of heterogeneous single-cell transcriptomes using Scanorama. Nat. Biotechnol. 2019;37:685–691. doi: 10.1038/s41587-019-0113-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.La Manno G., Soldatov R., Zeisel A., Braun E., Hochgerner H., Petukhov V., Lidschreiber K., Kastriti M.E., Lönnerberg P., Furlan A., et al. RNA velocity of single cells. Nature. 2018;560:494–498. doi: 10.1038/s41586-018-0414-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Yu G., Wang L.G., Han Y., He Q.Y. clusterProfiler: an R package for comparing biological themes among gene clusters. OMICS. 2012;16:284–287. doi: 10.1089/omi.2011.0118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Subramanian A., Tamayo P., Mootha V.K., Mukherjee S., Ebert B.L., Gillette M.A., Paulovich A., Pomeroy S.L., Golub T.R., Lander E.S., Mesirov J.P. Gene set enrichment analysis: a knowledge-based approach for interpreting genome-wide expression profiles. Proc. Natl. Acad. Sci. USA. 2005;102:15545–15550. doi: 10.1073/pnas.0506580102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Street K., Risso D., Fletcher R.B., Das D., Ngai J., Yosef N., Purdom E., Dudoit S. Slingshot: cell lineage and pseudotime inference for single-cell transcriptomics. BMC Genom. 2018;19:477. doi: 10.1186/s12864-018-4772-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Wolf F.A., Hamey F.K., Plass M., Solana J., Dahlin J.S., Göttgens B., Rajewsky N., Simon L., Theis F.J. PAGA: graph abstraction reconciles clustering with trajectory inference through a topology preserving map of single cells. Genome Biol. 2019;20:59. doi: 10.1186/s13059-019-1663-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.McInnes L., Healy J., Melville J. Umap: uniform manifold approximation and projection for dimension reduction. arXiv. 2018 doi: 10.48550/arXiv.1802.03426. Preprint at. [DOI] [Google Scholar]
- 75.Love M.I., Huber W., Anders S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 2014;15:550. doi: 10.1186/s13059-014-0550-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Bankhead P., Loughrey M.B., Fernández J.A., Dombrowski Y., McArt D.G., Dunne P.D., McQuaid S., Gray R.T., Murray L.J., Coleman H.G., et al. QuPath: open source software for digital pathology image analysis. Sci. Rep. 2017;7 doi: 10.1038/s41598-017-17204-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Li H., Durbin R. Fast and accurate short read alignment with Burrows-Wheeler transform. Bioinformatics. 2009;25:1754–1760. doi: 10.1093/bioinformatics/btp324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Li H., Handsaker B., Wysoker A., Fennell T., Ruan J., Homer N., Marth G., Abecasis G., Durbin R., 1000 Genome Project Data Processing Subgroup The sequence alignment/map format and SAMtools. Bioinformatics. 2009;25:2078–2079. doi: 10.1093/bioinformatics/btp352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Cibulskis K., Lawrence M.S., Carter S.L., Sivachenko A., Jaffe D., Sougnez C., Gabriel S., Meyerson M., Lander E.S., Getz G. Sensitive detection of somatic point mutations in impure and heterogeneous cancer samples. Nat. Biotechnol. 2013;31:213–219. doi: 10.1038/nbt.2514. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Venteicher A.S., Tirosh I., Hebert C., Yizhak K., Neftel C., Filbin M.G., Hovestadt V., Escalante L.E., Shaw M.L., Rodman C., et al. Decoupling genetics, lineages, and microenvironment in IDH-mutant gliomas by single-cell RNA-seq. Science. 2017;355 doi: 10.1126/science.aai8478. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Puram S.V., Tirosh I., Parikh A.S., Patel A.P., Yizhak K., Gillespie S., Rodman C., Luo C.L., Mroz E.A., Emerick K.S., et al. Single-cell transcriptomic analysis of primary and metastatic tumor ecosystems in head and neck cancer. Cell. 2017;171:1611–1624.e24. doi: 10.1016/j.cell.2017.10.044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Tirosh I., Venteicher A.S., Hebert C., Escalante L.E., Patel A.P., Yizhak K., Fisher J.M., Rodman C., Mount C., Filbin M.G., et al. Single-cell RNA-seq supports a developmental hierarchy in human oligodendroglioma. Nature. 2016;539:309–313. doi: 10.1038/nature20123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Tirosh I., Izar B., Prakadan S.M., Wadsworth M.H., 2nd, Treacy D., Trombetta J.J., Rotem A., Rodman C., Lian C., Murphy G., et al. Dissecting the multicellular ecosystem of metastatic melanoma by single-cell RNA-seq. Science. 2016;352:189–196. doi: 10.1126/science.aad0501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Patel A.P., Tirosh I., Trombetta J.J., Shalek A.K., Gillespie S.M., Wakimoto H., Cahill D.P., Nahed B.V., Curry W.T., Martuza R.L., et al. Single-cell RNA-seq highlights intratumoral heterogeneity in primary glioblastoma. Science. 2014;344:1396–1401. doi: 10.1126/science.1254257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Jacoby L.B., Hedley-Whyte E.T., Pulaski K., Seizinger B.R., Martuza R.L. Clonal origin of pituitary adenomas. J. Neurosurg. 1990;73:731–735. doi: 10.3171/jns.1990.73.5.0731. [DOI] [PubMed] [Google Scholar]
- 86.Tan T.Z., Miow Q.H., Miki Y., Noda T., Mori S., Huang R.Y.J., Thiery J.P. Epithelial-mesenchymal transition spectrum quantification and its efficacy in deciphering survival and drug responses of cancer patients. EMBO Mol. Med. 2014;6:1279–1293. doi: 10.15252/emmm.201404208. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
-
•
All raw scRNA-seq data have been deposited at the National Genomics Data Center (NGDC, https://bigd.big.ac.cn/) under the accession number PRJCA009690.
-
•
This study did not generate original codes. All software and algorithms used in this study are publicly available and listed in the key resources table.
-
•
Any additional information required to reanalyze the data reported in this paper is available from the lead contact upon request.