

Abstract
A specter is haunting the world, the specter of obesity. During the last decade, this pandemia has skyrocketed threatening children, adolescents and lower income families worldwide. Although driven by an increase in the consumption of ultraprocessed edibles of poor nutritional value, the obesogenic changes in contemporary human lifestyle affect people differently, revealing that some individuals are more prone to develop increased adiposity. During the last years, we performed a variety of genetic, evolutionary, biochemical and behavioral experiments that allowed us to understand how a group of neurons present in the arcuate nucleus of the hypothalamus regulate the expression of the proopiomelanocortin (Pomc) gene and induce satiety. We disentangled the neuronal transcriptional code of Pomc by identifying the cis-acting regulatory elements and primary transcription factors controlling hypothalamic Pomc expression and determined their functional importance in the regulation of food intake and adiposity. Altogether, our studies reviewed here shed light on the power and limitations of the mammalian central satiety pathways and may contribute to the development of individual and collective strategies to reduce the debilitating effects of the self-induced obesity pandemia.
Keywords: enhancer, epigenetics, exaptation, gene expression, melanocortins, mutant mice, transcription, transgenic mouse
Introduction to the central regulation of food intake
Body weight and energy balance are controlled in vertebrate animals by brain circuits that promote foraging, food intake or satiety after integrating multiple metabolic and environmental signals with current and future metabolic demands. Peripheral organs, such as the adipose tissue, pancreas, liver and gastrointestinal tract, release hormones in response to nutrient flux. This circulating information about the energy status of the organism is then sensed and interpreted by specialized arrays of neurons located in the arcuate nucleus of the ventromedial hypothalamus and in the dorsal medulla. Molecular cloning of the leptin gene [1], mutations of which are responsible for an autosomal recessive form of early-onset extreme obesity, propelled the discovery of hypothalamic circuits and genes directly involved in the central regulation of food intake [2]. Leptin is a circulating hormone mainly secreted by adipocytes and its plasma levels correlate with the amount of triglycerides present in the adipose tissue. Thus, circulating leptin provides a direct readout of the available energy stored as fat in the organism.
Two distinct populations of neurons present in the arcuate nucleus of the hypothalamus express leptin receptors and also respond to other humoral metabolic signals such as ghrelin, glucose and insulin. In simple terms, neurons expressing the agouti-related protein (AGRP) promote food intake and those expressing the proopiomelanocortin (POMC) gene induce satiety to limit food intake. Thus, these two interrelated and functionally opposite sets of arcuate neurons are considered the yin and yang of food intake regulation [3]. POMC neurons release melanocortin peptides that induce anorexia upon stimulation of Gs-coupled melanocortin 4 receptors (MC4R), whereas, AGRP released from AGRP neurons promotes feeding by acting as a competitive MC4R antagonist. Stereotaxic application of MC4R antagonists into the cerebral ventricles [4] as well as optogenetic activation of AGRP neurons [5] promote food intake even in well fed mice, whereas pharmacological stimulation of MC4R in the hypothalamus [4] and optogenetic activation of POMC neurons promote satiety [5]. This general view of POMC and AGRP neurons playing contrasting anorexigenic and orexigenic responses has been challenged recently by novel provocative results based on photometric detection of hypothalamic neuronal activity in freely behaving mice [6]. Just the presentation of food to fasted mice, or even food cues without any consumption, produced an immediate activation of POMC neurons and a concomitant inactivation of AGRP neurons [6]. These opposing activity patterns were maintained during food consumption and then switched back when feeding was interrupted by abrupt removal of the food [6], suggesting that the endogenous release of AGRP and melanocortins anticipate consummatory and satiety behavior, respectively.
The increasing knowledge gained during the last 25 years concerning the genetics and physiology involved in food intake regulation has been ironically mirrored by a dramatic increase in the prevalence of adult and childhood overweight and obesity. This worldwide burden prompted the World Health Organization and Public Health systems of many countries to declare obesity as a pandemia [7]. Pandemic obesity has been driven primarily by pervasive changes in the global food system that instigate people from all countries, ages and socioeconomic status to consume high calorie, ultraprocessed, affordable and ubiquitously marketed edibles with poor nutritional value. However, the obesogenic changes in contemporary human lifestyle do not affect all individuals equally, revealing that some individuals have an inherently greater predisposition to develop overweight and increased adiposity. The genetic contribution to body weight determined in family studies, adopted children and twins reared apart [8,9] has consistently been reported at 40–70% [10], placing weight as a highly heritable trait only slightly below that of height [11,12]. As expected, homozygous null mutations in the human leptin [13] and leptin receptor [14] genes lead to early-onset extreme obesity syndromes remarkably similar to the phenotypes found in mice with mutations in the orthologous genes. The rare existing cases of leptin deficiency have responded therapeutically to recombinant leptin replacement treatment [15]. However, leptin treatment is largely ineffective at promoting weight loss in patients with common obesity, probably because they already have high serum leptin levels and develop an as yet incompletely defined resistance to further leptin action. More recent studies suggest that leptin sensitizing agents may be potentially employed in leptin pharmacotherapy [16]. Similarly, rare, null allele mutations in the human POMC gene lead to severe hyperphagia, early-onset obesity and adrenal insufficiency [17], whereas mutations in MC4R gene are the most common monogenic disorders causing obesity [18]. A recently developed MC4R agonist named setmelanotide has induced significant weight loss in two POMC-deficient patients [19]. Although this drug has potential in broader populations of obese patients, caution is warranted as it may act at other melanocortin receptors involved in autonomic functions and skin pigmentation.
In summary, human monogenic obesity syndromes are rare and most familial cases of obesity appear to be driven by several coexisting low-frequency polymorphic alleles, each of them contributing a small percentage to the overall phenotype. A large number of genome-wide association studies has been performed worldwide during the last several years and identified nearly 100 different loci associated with high body mass index, type II diabetes, increased adiposity or high leptin levels [20–22]. However, the individual contribution of most of these variants is practically irrelevant except for a few like the one reported in the FTO locus [23,24]. Another exceptional single locus is POMC. Despite the fact that humans deficient in POMC are rare, the POMC locus has been strongly associated to obesity. A number of genome-wide studies have found highly significant linkage scores between obesity-related traits and a genomic segment in chromosome 2 near POMC [25–27]. Because polymorphisms in POMC coding sequences do not appear to account for these associations [28], it is likely that mutations in noncoding regulatory elements may alter POMC transcript levels and modify the relative amount of central melanocortins.
Regulation of POMC gene expression
POMC is expressed at high levels in endocrine cells of the pituitary gland and neurons present in the arcuate nucleus of the hypothalamus. POMC encodes a prohormone that after cell-specific post-translational processing generates several biologically active peptides that orchestrate the mammalian stress response [29]. In the anterior pituitary, corticotrophs release adenocorticotropic hormone (ACTH), whereas in hypothalamic neurons the POMC prohormone is further processed to generate the melanocortins α-, β- and γ- melanocyte stimulating hormones (MSH) and the analgesic endogenous opioid β-endorphin [29,30] (Fig. 1).
Fig. 1.
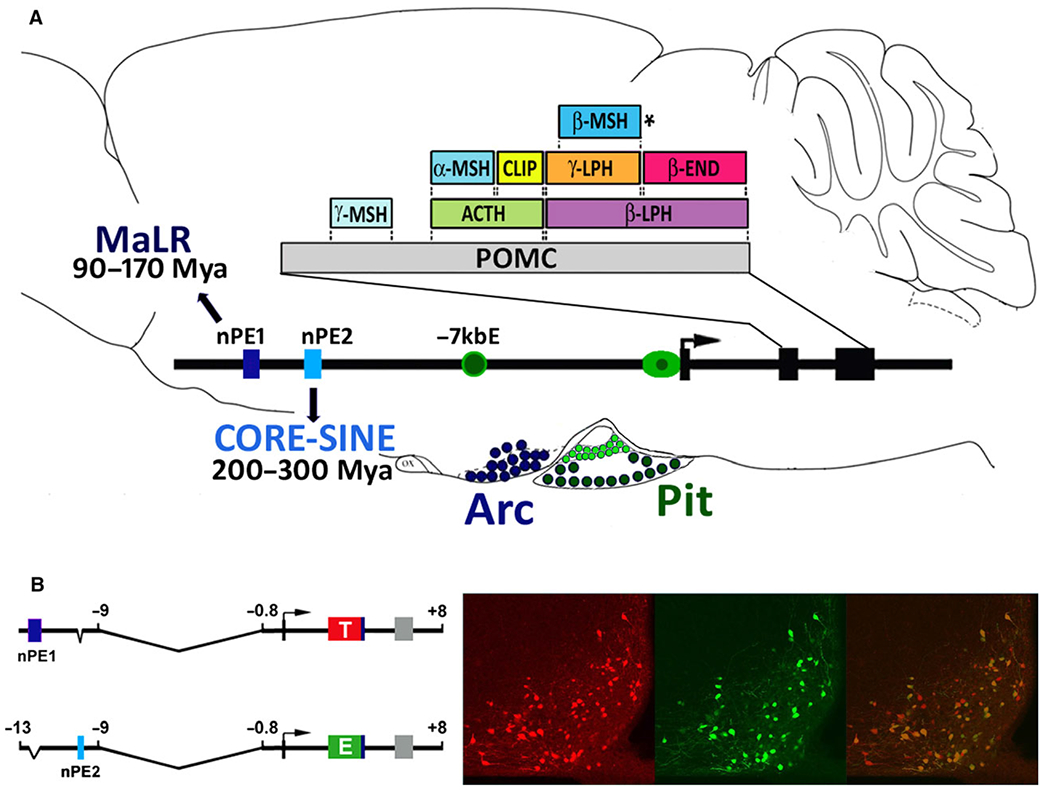
(A) Schematic of Pomc gene expression and peptide profiles in the rodent brain and pituitary. Pituitary expression (Pit, green color) is driven by the proximal promoter (mainly to intermediate lobe melanotrophs) and the −7 kb enhancer (mainly to anterior lobe corticotrophs). Pomc expression in the arcuate nucleus of the hypothalamus (Arc, blue color) is controlled by the distal neuronal enhancers nPE1 and nPE2 that were exapted from a Mammalian-apparent LTR and a CORE-SINE retroposon, respectively, at different time points along the lineage leading to mammals. Pomc exons are in black boxes. Coding sequences for the POMC prohormone and the peptides obtained after endoproteolytic cleavage are indicated in colored boxes. The asterisk next to β-MSH denotes that this peptide is not released in mice. (B) Schematic of the transgenes nPE1Pomc-tomato and nPE2Pomc-EGFP described in Ref. [64]. Expression of tomato (left), EGFP (center), and a merged photograph (right) in a coronal hemisection of the arcuate nucleus of an adult compound transgenic mouse showing extensive colocalization of both reporter fluorescent proteins.
The transcriptional regulation of POMC also follows distinct cell-specific cis/trans codes. By combining in vitro transfection assays in immortalized pituitary cell lines [31] and reporter gene expression in transgenic mice, it was possible to initiate and advance studies on regulation of POMC expression in pituitary cell types [32,33]. These early studies demonstrated that pituitary-specific expression of rat or mouse Pomc is controlled by several cis-acting elements localized within 500 bp proximal to the transcriptional start site [32,33]. A number of transcription factors (TFs) including Nurr77, NeuroD1, Ascl1, Pitx1, SP1 and Tpit have been shown to participate in the control of pituitary Pomc expression and/or the determination of corticotropic and melanotropic cell lineages [33–38]. In addition to the proximal promoter, a conserved pituitary-specific Pomc enhancer was identified 7 kb upstream of the mouse Pomc gene transcriptional start site [39]. This distal pituitary enhancer recruits the same subset of TFs as the proximal promoter, except Pitx1, and is highly dependent on Tpit for activity [39]. When tested in transgenic mice, the −7 kb enhancer was shown to be more active in corticotrophs than in melanotrophs, contrary to what has been observed for the proximal Pomc promoter [39] (Fig. 1A). More recently, the TF Pax7 has been identified as an essential pioneer factor to establish the melanotroph lineage [40]. Early expression of Pax7 remodels the chromatin along specific enhancers allowing access of factors like Tpit to otherwise hidden DNA binding sites. This Pax7-induced epigenetic remodeling drives expression of melanotroph-specific genes, including those encoding the prohormone convertase PC2 and the dopamine D2 receptor [40]. For a recent comprehensive review about the transcriptional regulation of Pomc in pituitary cells and related human diseases, see Ref. [41].
Identification of the enhancers and TFs that control hypothalamic Pomc expression experienced a relative delay mainly due to the lack of an authentic neuronal cell line able to express Pomc with transcriptional fidelity and with which to perform reliable expression studies. In addition, early transgenic mouse expression studies showed that the proximal Pomc promoter drives cell-specific expression of reporter genes only to pituitary POMC cells but not to the hypothalamus [32,42,43]. Therefore, our initial attempts to search for regulatory elements controlling Pomc expression in the arcuate nucleus of the hypothalamus in the pregenomic era were performed blindly by chromosome walking upstream and downstream of the Pomc gene and testing the isolated flanking fragments in transgenic mouse expression assays. Using this approach, we identified a 4 kb fragment located between −13 and −9 kb upstream of the mouse Pomc promoter that when included in transgenic constructs allowed authentic neural-specific and developmentally regulated reporter expression in hypothalamic POMC neurons [44] (Fig. 1). The possibility to specifically target POMC neurons prompted us to drive enhanced green fluorescent protein (EGFP) expression to the arcuate nucleus and perform the first electrophysiological recordings in readily identified bright fluorescent POMC neurons in hypothalamic slices [45]. Double immunofluorescence performed in brain slices of Pomc-EGFP transgenic mice confirmed coexpression of EGFP and POMC peptides in more than 99% of these neurons [45]. Electrophysiological studies in Pomc-EGFP mouse arcuate slices led to the discovery that leptin increases the frequency of action potentials in POMC neurons by two concurrent mechanisms: depolarization through a leptin-activated nonspecific cation channel and reduced inhibition by local orexigenic Agrp/GABA neurons [45]. We also found that melanocortins play an autoinhibitory effect on this circuit. The discovery that leptin activates arcuate POMC neurons revealed a fundamental signaling entry point from the periphery to the brain through the blood–brain barrier. Stimulation of leptin receptors expressed on arcuate POMC neurons increases their activity leading to the release of POMC-encoded peptides. Central melanocortins, in turn, stimulate melanocortin receptors to induce satiety and increase metabolic rate [2,46]. Altogether, the increase in leptin levels due to accumulation of triglyceride stores triggers negative feedback mechanisms that reduce further food consumption. Pomc-EGFP transgenic mice have been extensively used during the last 15 years and are the gold standard system for interrogation of the physiological and pharmacological properties of various neurotransmitters and peptides, such as 5-HT and PYY3-36, that modulate the activity of POMC neurons [47,48]. However, not all hypothalamic POMC neurons express leptin receptors [49]; neither are all POMC neurons involved in the control of food intake. In fact, anatomical, histological, pharmacological, electrophysiological and molecular studies have accumulated extensive evidence supporting the idea that hypothalamic POMC neurons are highly heterogeneous and that several phenotypically and functionally distinctive subpopulations seem to coexist. From the arcuate nucleus, POMC neurons project toward a wide array of rostral nuclei including the septum, striatum, thalamus and hypothalamus and, caudally, to the brainstem and medulla [50]. In addition, subpopulations of POMC neurons are distributed along the anteroposterior and medial–lateral axes of the arcuate nucleus and corelease either the fast neurotransmitters GABA or glutamate [51], while partially overlapping populations express the serotonin 5HT2c receptor [47], leptin or insulin receptors [52] and melanocortin autoreceptors MC3R or MC4R. A recent single-cell RNA sequencing study performed on FACS sorted green fluorescent hypothalamic neurons taken from Pomc-EGFP mice revealed the great level of heterogeneity exhibited by this population of around 3000 neurons expressing Pomc [53].
Regulation of hypothalamic Pomc expression
The completion of the first vertebrate genome projects in the early 2000s allowed us to perform phylogenetic footprinting analyses along the 4 kb distal mouse Pomc module necessary for reporter gene expression in hypothalamic POMC neurons of transgenic mice [54]. This type of bioinformatic algorithm globally compares multiple short overlapping local alignments to detect high identity cross species sequences. Thus, this strategy can be used as a proxy to identify transcriptional enhancers embedded in intergenic or intronic regions based on the concept that the rate of molecular evolution in functional sequences is more constrained due to selective pressure and mutations accumulate much slower than in nonfunctional residues the evolve at neutral rates. Following this strategy, we detected two highly conserved sequences within the 4 kb distal module which are located at − 12 and −10 kb of the mouse Pomc 5′-flanking region [54]. We named these putative neuronal Pomc enhancers (nPE) nPE1 and nPE2. To determine whether nPE1 and nPE2 had neuron-specific transcriptional enhancer function, we performed a comprehensive deletional transgenic mouse analysis of the distal module cloned immediately upstream of either the mouse Pomc promoter or a heterologous minimal promoter. In addition, we tested a human chromosomal region orthologous to the distal 4 kb mouse Pomc module. This study led to the following conclusions: (a) a genomic region located between −13 and −9 kb of mouse Pomc is necessary and sufficient to direct authentic reporter gene expression to hypothalamic POMC neurons; (b) this genomic neuron-specific enhancer region contains two highly conserved short sequences that we named nPE1 and nPE2 which are present in mammalian genomes but not in those of birds, amphibians or fishes; (c) either nPE1 or nPE2 are able to drive cell-specific reporter expression to POMC arcuate neurons, whereas the simultaneous deletion of both enhancers abolishes reporter expression in POMC neurons; (d) a human genomic fragment carrying nPE1 and nPE2 also drives reporter gene expression to pomc hypothalamic neurons of transgenic mice proving that mouse and human orthologs are functionally conserved; (e) brain and pituitary Pomc expression are independently controlled by distinct sets of enhancers [54] (Fig. 1A).
The comparative genomic analyses at the Pomc locus used in this study also revealed that the teleost fishes Tetraodon, Fugu and zebrafish possess two pomc genes, which we called pomca and pomcb, unlike the human, mouse and chicken genomes that have only one POMC gene [55]. We found that these two fish pomc paralogs originated in the whole-genome duplication specific to the teleost lineage over 300 million years ago (Mya). Since pomca has been found to be expressed in the equivalent teleost arcuate nucleus of the hypothalamus and pomcb in the preoptic area of the brain, we concluded that the pomc paralogs underwent a process of subfunctionalization of their expression domains during teleost evolutionary history [55]. In addition, we found evidence for subfunctionalization of the peptide domains encoded by the two pomc paralogs, although it is unknown if these distinct processes were concurrent [55].
Molecular evolution of neuronal Pomc enhancers
Although Pomc is expressed in the ventromedial hypothalamus of all jawed vertebrates, we failed to find nPE1 and nPE2 paralog sequences in non-mammalian vertebrate Classes. To investigate this discrepancy, we searched for the evolutionary history of nPE1 and nPE2 sequences. We first found that the enhancer nPE2 is more ancient than nPE1. While nPE2 is highly conserved across Prototheria (monotremes), Metatheria (marsupials) and Eutheria (placental mammals), nPE1 is a placental novelty. To reconstruct the origin of functional novelties, we developed an in silico paleogenomics strategy based on systematic and progressive searches for nPE1 and nPE2 paralogs in all available genome databases that could reveal evolutionary relics. We did not find any sequence similar to nPE2 in any of the available vertebrate genomes except the opossum, where we identified three short sequences similar to opossum nPE2 [56]. When used as queries in further BLAST searches, those four sequences showed to be similar to members of the marsupial MAR1 family of CORE-SINE retroposons [56]. Mobile elements have been shown to be involved in gene and genome evolution by providing a large reservoir of raw sequences that, upon acquiring functional mutations, may become adaptive and fixed in their host genomes, a process called exaptation [57,58]. Thus, our results suggest that an ancient CORE-SINE retroposon was inserted into the POMC locus and exapted as a neuronal Pomc enhancer in the mammalian lineage, before the marsupial/placental split that occured around 170 Mya (Fig. 1A). It is impossible to confirm whether or not the CORE-SINE inserted upstream POMC functioned immediately as an enhancer, as is assumed for Alu elements carrying potential binding sites for nuclear receptors [59–61], or that the inserted mobile element accumulated mutations until evolving into a novel neuronal POMC enhancer that became fixed during mammalian evolution [62].
To discover the evolutionary origin of nPE1, we again performed in silico paleogenomics using human nPE1 as a query and detected 15 high identity hits in the human genome, annotated as sequences derived from the family of mammalian-apparent LTR retro-transposons (MaLR) [63]. MaLRs originated before the radiation of eutherians, between 80 to 100 Mya [62], a period that matches well with the hypothesis that nPE1 constitutes a placental novelty. Altogether, the distal hypothalamic enhancer module located upstream of the Pomc gene contains two highly conserved, but evolutionarily unrelated, sequences originated after the sequential exaptation of two different types of retroposons [64] (Fig. 1A).
Based on the independent and distinct evolutionary history of nPE1 and nPE2, we wanted to determine whether both enhancers drove overlapping or distinct spatiotemporal expression patterns in the ventromedial hypothalamus. To this end, we designed two structurally similar transgenes in which nPE1 and nPE2 drove the expression of the red fluorescent proteins tomato or EGFP, respectively (Fig. 1B). Analysis of nPE1-tomato.nPE2-EGFP compound mice showed more than 85% of coexpression of both fluorescent markers throughout the entire anteroposterior and medial–lateral axes of the arcuate nucleus [64] (Fig. 1B). The onset of both transgenes was coincidental within the same array of neurons located at the base of the developing hypothalamus of compound nPE1-tomato.nPE2-EGFP e10.5 embryos, matching the initial spatiotemporal expression of mouse Pomc [65]. These results led us to conclude that nPE1 and nPE2 control Pomc expression in the entire population of hypothalamic neurons that normally express this gene [64]. The independent evolutionary origin and identical enhancer function of nPE1 and nPE2 indicate that these two regulatory elements are functional analogs that underwent a process of convergent evolution that started during basal mammalian evolution, more than 166 Mya [62], and finished after the marsupial/placental split around 147 Mya [62]. To our knowledge, this is the first and probably still the only functionally documented example of authentic convergent molecular evolution of cell-specific enhancers [64] (Fig. 1). It has been reported that around half of the early population of immature POMC neurons that originate in the developing hypothalamus stop expressing Pomc at midgestation and adopt different neuropeptide phenotypes including the expression of Agrp/Npy [66] or kisspeptin [67]. The mechanisms by which Pomc expression is silenced and whether the enhancers nPE1 and nPE2 play any role during this phenotypic transition are still unknown.
The presence of two apparently redundant enhancers controlling neuronal Pomc expression suggested to us that one of them is likely to be under lower selective pressure [68,69]. To challenge this hypothesis, we calculated the rate of molecular evolution of nPE1 and nPE2 by performing, first, a long-scale interspecies’ mammalian orthologs comparison to assess the sequence variation during the last 100 million years, and, second, a short-scale intraspecies’ human genetics population study. The former calculation showed that nPE1 is evolving 2.64 times faster than nPE2 and also faster than sequences coding for bioactive POMC peptides present in exon 3 [64]. At the human population level, we found three polymorphic sites at nPE1 and the absence of polymorphisms in nPE2. These results showed that the regulatory elements nPE1 and nPE2 are evolving slower than coding sequences, although almost all exon 3 SNPs do not introduce missense mutations in the amino acid sequences of any bioactive POMC peptides processed from the prohormone [64]. Altogether, these interspecies’ and intraspecies’ comparisons show that the more ancient nPE2 is under stronger selective pressure than the younger nPE1, precluding the hypothesis that the exaptation of nPE1 relaxed the selective pressure on nPE2 [64].
The existence of two overlapping enhancers controlling gene expression in the same cell types has been observed in several developmental genes [69,70]. This level of redundancy may be critical to buffer environmental perturbations and control the spatiotemporal expression pattern of developmental genes in a precise and stable manner during embryogenesis. A recent study indicates that overlapping enhancers are a predominant feature in Drosophila developmental genes [71]. So far, nPE1 and nPE2 appear to be the only reported case of overlapping enhancers controlling expression of a gene that plays important physiological roles throughout the postnatal life. Carrying two apparently redundant enhancers may constitute an adaptation to allow for higher transcriptional rates and/or lower fluctuations. These two possible alternatives may be illustrated by a double-scull rowboat metaphor in which the two rowers act together to attain a faster transcriptional speed, and also maintain a lower cruise control level with the participation of only one rower. It is interesting to note that nPE1 and nPE2 are both present in all placental mammalian Orders, indicating that both enhancers coexist upstream of Pomc under purifying selection since the radiation of mammals around 90 Mya [72]. This finding strongly suggests that both enhancers play important roles in Pomc transcription and mammalian physiology.
To investigate the importance of having two enhancers and the individual contributions of nPE1 and nPE2 to hypothalamic Pomc expression, we generated mutant mice lacking nPE1, nPE2 or both enhancers simultaneously [72] (Fig. 2A). Quantitative analysis of Pomc mRNA levels during various developmental stages and adulthood in wild-type and the different homozygous enhancer mutants showed that: (a) only the concurrent presence of nPE1 and nPE2 in both Pomc alleles assures normal hypothalamic Pomc mRNA levels; (b) the simultaneous deletion of nPE1 and nPE2 induces a profound deficit in Pomc mRNA levels that leads to hyperphagia and obesity; (c) at the early start of Pomc expression, both enhancers act synergistically to attain typical Pomc mRNA levels in the presumptive hypothalamus. The synergism is evident when measuring the much higher Pomc mRNA levels present in wild-type e10.5 embryos in comparison with the sum of the levels detected in the individual enhancer mutants; (d) during adulthood, however, the individual participation of each enhancer seems to be additive with nPE1 and nPE2 contributing with ~ 80% and ~ 20% of the total Pomc mRNA levels, respectively; (e) nPE1 ablation revealed its main contribution to Pomc mRNA adult levels since its absence impairs satiety and, consequently, induces obesity (Fig. 2B); and (f) deletion of nPE2 does not seem to alter the control of food intake in the presence of nPE1, at least in laboratory conditions. However, ablation of nPE2 in the absence of nPE1 triggers hyperphagia and extreme obesity [72](Fig. 2B,C).
Fig. 2.
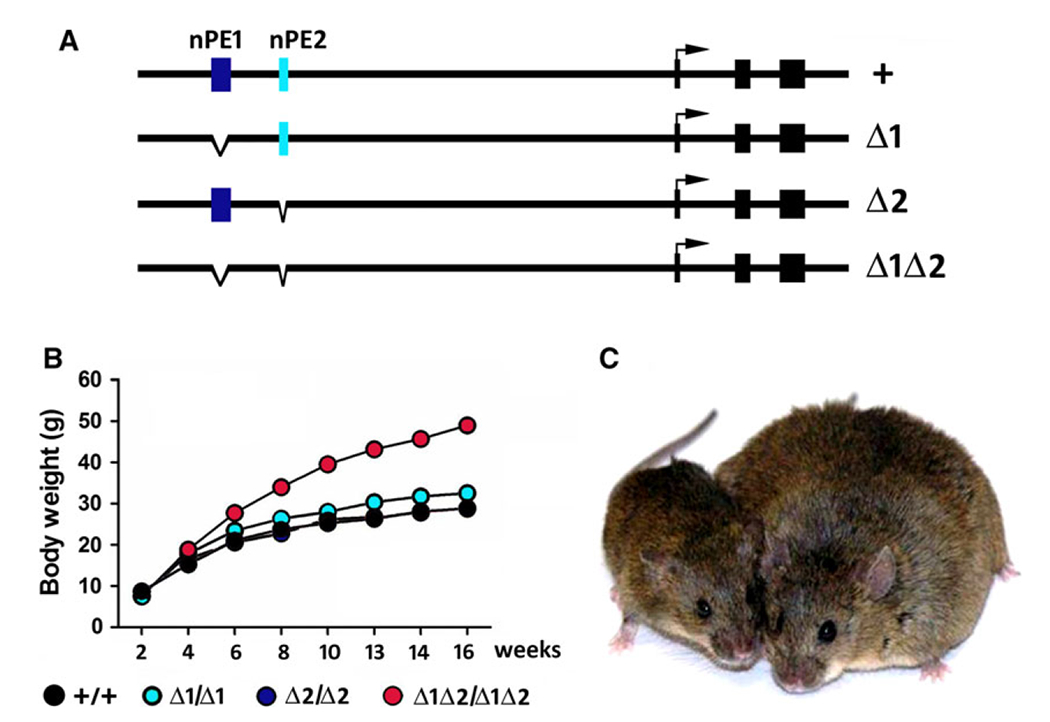
(A) Schematic of the mouse Pomc and nPE mutant Pomc alleles as shown in Ref. [72]. (B) Body weight of wild-type, and homozygous mutants for nPE1, nPE2 and for both enhancers along the first 16 weeks. nPE1 mutants are mildly overweight and nPE1/nPE2 mutants are obese [72]. (C) Comparison of a wild-type and a homozygous adult mutant sibling male lacking both nPE1 and nPE2 [72].
In general, we have observed that once Pomc mRNA levels drop below ~ 35% of its normal values mice exhibit satiety control deficits and excessive fat accumulation. Although lacking nPE2 does not alter food intake regulation, a 20% reduction in Pomc mRNA levels may increase the risk of an insufficient control of energy balance mechanisms [72]. Hyperphagia and excessive body weight are highly maladaptive in the wild because a greater body mass and unnecessary foraging increases the likelihood of encounters with predators while limiting escape efficiency. Therefore, the adaptive value of maintaining Pomc expression above a critical threshold seems to be essential for reproductive success. This comprehensive study highlights the importance of evaluating the participation of individual enhancers to gene expression in their native genomic context to then study the effects of each mutant allele in general physiology and fitness [72] (Fig. 2).
The cis-trans code controlling hypothalamic Pomc expression
An emerging conclusion drawn by the discovery that nPE1 and nPE2 are two evolutionarily convergent enhancers that regulate Pomc expression in all POMC hypothalamic neurons [64] is that these two functional analogs are likely to share DNA motifs that recruit similar TFs. In fact, we detected a 21-bp imperfect palindromic motif in nPE1 which is highly similar to a sequence present in nPE2 [72]. These sequences carry two TAAT inverted motifs, known to be recognized by homeodomain-TFs (HDTFs). Another sequence present in nPE2 also showed two inverted homeodomain (HD) binding sites. These similar pairs of inverted TAAT motifs concur with a canonical TCAAG/T motif probably recognized by a HD-TF of the NKX family [72]. All these HD-binding sites showed high identity levels between humans, mice and most other mammals. We investigated the putative functional relevance of these shared conserved sequences in expression studies performed in transgenic mice with transition mutations in either nPE1 or nPE2. All transgenic mouse founders carrying mutated nPE1 or nPE2 failed to drive EGFP expression to the arcuate nucleus of the hypothalamus in contrast to the control transgenes carrying wild-type nPE1 or nPE2 that drove clear EGFP expression to this brain region [72]. These results suggested to us that a common array of cis-acting motifs, probably recognized by the same TFs, are responsible for the functional analogy between nPE enhancers and prompted us to search for TFs involved in specification of the POMC hypothalamic neuronal lineage.
To detect HDTFs that may recognize these DNA elements, we looked for the preferred DNA binding motifs of each mouse HD obtained in a massive in vitro binding study [73]. Using the nPE sequences as queries, we generated an initial list of candidate HDTFs that was later reduced by discarding those showing expression patterns incompatible with that of Pomc based on gene expression data obtained from the Allen Brain Atlas at different mouse ages [74]. The final TF candidates were tested in gel mobility shift assays to determine their ability to bind in vitro to particular motifs present in nPE1 and/or nPE2. We selected the LIM-HD-TF Islet1 (ISL1) because its expression pattern in the ventromedial hypothalamus is similar to that of Pomc since its expression onset and throughout the adult mouse life. After combining molecular, genetic, physiological and evolutionary approaches, we demonstrated that ISL1 directly plays a fundamental regulatory role in hypothalamic Pomc expression [75]. In this work we concluded that: (a) Pomc and Isl1 coexpress during embryonic development, postnatal life and adulthood; (b) gel-shift and ChIP assays demonstrated that ISL1 binds to precise sequences present in nPE1 and nPE2 which are essential for their enhancer function; (c) ISL1 plays a critical role for Pomc expression during the early stages of hypothalamic development; (d) after birth, the lack of Isl1 expression induces a remarkable deficit in hypothalamic Pomc expression levels; (e) the absence of Isl1 expression in POMC neurons of adult mice reduces Pomc expression and causes an energy balance phenotype including obesity; and (f) hypothalamic Pomc depends on ISL1 in mice and zebrafish demonstrating that this regulation is conserved throughout vertebrate evolution [75]. Altogether, our results indicate that ISL1 acts as a terminal differentiation gene establishing the specific identity of POMC neurons after their birth and accumulation in the mantle zone and before the arcuate nucleus is formed. In addition, ISL1 controls hypothalamic Pomc expression in adulthood and therefore plays a fundamental role in food intake and body weight regulation [75]. More recently, it has been found that ISL1 not only plays an essential role in the differentiation of POMC neurons but also in other neuronal subtypes present in the arcuate nucleus such as those expressing Agrp, Ghrh and somatostatin [76].
It is conceivable that other TFs and motifs participate in establishing the neuronal-specific expression of Pomc so further work is necessary to reveal the identity of the full molecular complex. Other motifs present in the hypothalamic enhancers probably bind TFs related to the hormonal regulation of Pomc. For example, we identified a conserved element in nPE2 that can bind TFs of the nuclear receptor family with a zinc-finger DNA binding domain, and a ligand-binding domain that recruits several types of steroid hormones [77]. We found that the estrogen receptor alpha (ESR1) is a candidate nuclear receptor factor to regulate neuronal Pomc expression since it binds to this nPE2 motif in vitro and is expressed in POMC neurons during development and adulthood [77]. Thus, it is possible that estrogen exerts its anorectic effect by controlling POMC expression. We have also identified a conserved canonical STAT3 binding site in nPE1 which is likely to participate in leptin-induced activation of Pomc expression, although the importance of this site remains to be explored.
Hypothalamic Pomc expression in teleosts
Our phylogenetic footprinting analysis showed that nPE1 and nPE2 are highly conserved sequences in mammals but are absent from the genomes of all other Classes of vertebrates despite the fact that fishes, amphibians, reptiles and birds express Pomc in functionally homologous neurons of the ventromedial hypothalamus. This apparent discrepancy raises two intriguing questions: (a) what are the regulatory elements that control the expression of Pomc in the hypothalamus of non-mammalian vertebrates? and (b) are nPE1 and/or nPE2 able to function as transcriptional enhancers in Pomc neurons of non-mammalian vertebrates? We have answered these two questions by studying teleost fishes, the most distant evolutionary Class of vertebrates from mammals that express Pomc in the hypothalamus. We found that the proximal promoter sequences of zebrafish pomca (the teleost paralog expressed in the ventromedial hypothalamus) drove reporter gene expression only to the pituitary gland of transgenic zebrafish, similar to what we had previously found for mammalian Pomc [78]. However, insertion of the distal mouse Pomc module containing nPE1 and nPE2 upstream of the zebrafish promoter construct allowed reporter gene expression in POMC neurons of the zebrafish ventromedial hypothalamus [78]. These data indicate that although the mammalian POMC hypothalamic enhancers are mammalian specific, they nonetheless contain a transcriptional code that has remained conserved for more than 450 million years of vertebrate evolution. Furthermore, when nPE2 and nPE1 originated de novo during early mammalian evolution, the newly created cis/trans interactions were similar to the ancestral ones. This result matched our previous finding that hypothalamic ISL1 is essential for Pomc expression in the ventromedial hypothalamus of mice and zebrafish [75]. Deletional analysis showed that, as in transgenic mice, the sole presence of nPE1 or nPE2 was able to drive EGFP expression in zebrafish hypothalamic POMC neurons, but the concurrent removal of both enhancers completely inactivated the 4 kb mouse distal Pomc module. By performing chromosome walking upstream of zebrafish pomca, we identified a 1.2 kb fragment, nonhomologous to the mammalian Pomc enhancers, similarly capable of targeting EGFP expression to pomca hypothalamic neurons [78]. Within this fragment, we found two short motifs containing TAAT sequences that showed 85% identity with a 50 bp critical fragment of nPE2. Strikingly, transgenic zebrafish embryos bearing substitution mutations in the TAAT motifs failed to express EGFP in the brain, indicating that they play a critical role in pomca neuronal expression controlled by the intact 1.2 kb region [78]. The TAAT motifs present in zfnPE are within an annotated hAT Charlie DNA transposon, a family of DNA transposons that is highly abundant in the zebrafish genome. Because the pomca loci of medaka, stickleback, Tetraodon and Fugu are devoid of sequences derived from a hAT Charlie transposon we hypothesized that insertion of this mobile DNA element upstream of zebrafish pomca was a relatively recent event. The completion of additional genome projects will allow to determine whether exaptation of zfnPE from a hAT Charlie DNA transposon occurred only in zebrafish, in Cypriniformes or in the lineage leading to Ostariophysi. Altogether, this type of comparative genomics sheds light on the molecular evolution of gene expression regulation by showing how enhancers undergo extensive turnover while maintaining the ancestral transcriptional features.
Physiological importance of hypothalamic POMC function in the control of food intake and obesity
Although pituitary POMC is a pan-vertebrate feature, hypothalamic Pomc expression is an evolutionary novelty originated in the lineage leading to jawed vertebrates. The functional independence of two different territories and cell types expressing the same Pomc gene is supported by the modular architecture of the transcriptional cis-acting domains. While pituitary expression uses the proximal Pomc promoter and a −7 kb enhancer, the hypothalamic neurons rely on the distal enhancer module carrying nPE1 and nPE2 [39,54]. To further investigate the functional relevance of this distal module, we generated a strain of mutant mice lacking the ability to express Pomc selectively in the arcuate nucleus [79]. The targeted insertion of a heterologous cassette into the neural enhancer domain of the Pomc locus prevented hypothalamic Pomc expression but had no effect on expression levels in pituitary cells [79]. In mice lacking arcuate Pomc knockout (ArcPomcKO), hypothalamic POMC neurons develop normally and project to the typical target areas, indicating that POMC-derived peptides are not critical for normal development and maintenance of the neuronal circuitry per se. ArcPomcKO mice were hyperphagic and developed early-onset extreme obesity when consuming a standard low-fat chow, in contrast to high-fat or high-sucrose diet-induced obesity models that produce changes in dopaminergic reward centers [79]. ArcPomcKO mutants also exhibited hyperinsulinemia, hyperleptinemia, hepatic steatosis and decreased locomotor activity. In contrast, heterozygous arcPomc+/− mice expressing only 50% of hypothalamic Pomc mRNA levels showed normal body weight and daily food intake, and also all other measured parameters, including leptin and insulin levels, were normal [79].
The genetic cassette inserted in the Pomc locus that led to the generation of arcPomcKO mice was flanked by two loxP sites. The use of this type of genetic switch allows the reactivation of hypothalamic Pomc expression using a temporally controlled cre recombinase and allowed us to answer a fundamental question: is established obesity reversible? This question is particularly timely given the alarming prevalence of obesity in young and adult people, most of which fail to achieve a normal weight after dieting, probably due to compensatory reductions in metabolic rate [80–82]. Diet-induced obesity in rodents also showed to permanently tune-up the body weight set point once animals return to normal food consumption [83–85]. Using a tamoxifen activable cre transgene, we rescued hypothalamic Pomc expression in mice carrying different levels of overweight and found a remarkable improvement in food intake, body weight and fat deposits, including cases of extreme obesity. However, we found that the ability of mice to regain a normal body weight progressively decreased as the overweight at the time of genetic rescue was higher. To evaluate whether obesity itself was directly involved in the resistance to regain normal body weight after restoration of Pomc expression in older mice, we food restricted arcPomcKO mice from weaning until age P60. After hypothalamic Pomc expression rescue, the mice were given free access to chow. Interestingly, we found that food-restricted nor-moweight arcPomcKO mice completely maintained their normal body weight once Pomc expression was rescued even when eating ad libitum [79]. In contrast, non-tamoxifen rescued arcPomcKO mice acquired hyperphagic state that promoted an accelerated weight gain similar to that of naive arcPomcKO mice. These results indicated that the historical body weight at the time of arcuate Pomc rescue, and not the age of the mice, determines the body weight set point and, therefore, the food intake level necessary to achieve those values [79]. This finding is in agreement with clinical [80,82] and rodent data [83–85] obtained during high-fat diet–induced obesity in which chronic overweight has led to secondary metabolic adaptations that act to maintain obesity despite caloric intake reductions. Because in these experiments, we controlled for genetic and environmental factors including standard chow of low hedonic value, our results indicate that the onset of obesity may cause a permanent change in the body weight set point by promoting a maladaptive allostatic state that will defend a greater than necessary body weight.
What condition prevents obese arcPomcKO mice to recover a normal body weight even when eating normal chow? Is it possible to reprogram their body weight set point? To address these questions, we exposed obese adult arcPomcKO mice weighing almost 60 g to a calorie restricted diet for 8–12 weeks, a period sufficient for all the mice to reach normal body weight (~ 28 g). At that point, we activated the Cre-inducible genetic switch that rescues hypothalamic Pomc expression and found that the mice had reprogrammed their body weight set point to normal values and were able to sustain a normal body weight for a prolonged time eating regular chow ad libitum [86]. The readjustment of body weight set point was prevented by long-lasting PASylated leptin given days before and during the hypothalamic rescue by tamoxifen treatment, indicating that leptin resistance is a key factor preventing normalization of the body weight set point [86]. These results led us to the conclusion that restoration of hypothalamic leptin sensitivity is a necessary condition, together with normal Pomc expression, for obese mice to achieve and sustain normal metabolic homeostasis, whereas deficits in either parameter set a maladaptive allostatic balance that defends increased adiposity and body weight [86].
Different to autonomic and endocrine homeostatic mechanisms that work to maintain a physiological variable close to a fixed set point, body weight and energy storage follow an allostatic regulation that includes voluntary actions like foraging and food intake which may be activated in anticipation of future caloric needs. The allostatic control of body growth starts during early fetal development and continues postnatally until sexual maturation is achieved, and only beyond this point body weight is maintained as a relatively fixed parameter [87]. Exceptional periods occur during particular physiological conditions such as pregnancy, lactation, or hibernation in which allostatic mechanisms regain the scene to induce short-term adaptive hyperphagia that promotes fat accumulation and body weight gain. However, these parameters revert to normal once environmental conditions become less demanding. Unlike allostatic hyperphagia triggered by pregnancy or hibernation to induce ‘viability through change’ [88], when chronic overfeeding followed by obesity is detached from physiological or environmental needs, it generates a state that is maladaptive to the health of an organism.
In conclusion, during the last 25 years, we have generated extensive genetic, evolutionary and functional evidence that has contributed to a better understanding of the role of hypothalamic Pomc in the control of food intake and body weight.
Acknowledgements
The authors are grateful to the numerous students, post-doctoral fellows and colleagues in our laboratories who contributed to the original studies referenced in this review. This work was supported by Consejo Nacional de Investigaciones Científicas y Técnicas (MR), Agencia Nacional de Promotión Cientifíca y Tecnólogica, Argentina (MR) and Universidad de Buenos Aires, Argentina (MR), and the National Institutes of Health, USA with grants DK068400 (MJL and MR), and DK066604 (MJL).
Abbreviations
- ACTH
adenocorticotropic hormone
- Agrp
agouti-related protein
- arcPomcKO
arcuate Pomc knockout
- EGFP
enhanced green fluorescent protein
- ESR1
estrogen receptor alpha
- HD
homeodomain
- ISL1
Islet1
- MaLR
mammalian-apparent LTR retrotransposon
- MC4R
melanocortin 4 receptor
- MSH
melanocyte stimulating hormone
- nPE
neuronal Pomc enhancer
- POMC
proopiomelanocortin
- TF
transcription factor
References
- 1.Zhang Y, Proenca R, Maffei M, Barone M, Leopold L and Friedman JM (1994) Positional cloning of the mouse obese gene and its human homologue. Nature 372, 425–432. [DOI] [PubMed] [Google Scholar]
- 2.Schwartz MW, Woods SC, Porte D, Seeley RJ and Baskin DG (2000) Central nervous system control of food intake. Nature 404, 661–671. [DOI] [PubMed] [Google Scholar]
- 3.Seeley RJ and Berridge KC (2015) The hunger games. Cell 160, 805–806. [DOI] [PubMed] [Google Scholar]
- 4.Fan W, Boston BA, Kesterson RA, Hruby VJ and Cone RD (1997) Role of melanocortinergic neurons in feeding and the agouti obesity syndrome. Nature 385, 165–168. [DOI] [PubMed] [Google Scholar]
- 5.Aponte Y, Atasoy D and Sternson SM (2011) AGRP neurons are sufficient to orchestrate feeding behavior rapidly and without training. Nat Neurosci 14, 351–355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Chen Y, Lin Y-C, Kuo T-W and Knight Z (2015) Sensory detection of food rapidly modulates arcuate feeding circuits. Cell 160, 829–841. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.James WPT (2008) WHO recognition of the global obesity epidemic. Int J Obes 32, S120–S126. [DOI] [PubMed] [Google Scholar]
- 8.Maes HH, Neale MC and Eaves LJ (1997) Genetic and environmental factors in relative body weight and human adiposity. Behav Genet 27, 325–351. [DOI] [PubMed] [Google Scholar]
- 9.Stunkard AJ, Harris JR, Pedersen NL and McClearn GE (1990) The body-mass index of twins who have been reared apart. N Engl J Med 322, 1483–1487. [DOI] [PubMed] [Google Scholar]
- 10.Barsh GS, Farooqi IS and O’Rahilly S (2000) Genetics of body-weight regulation. Nature 404, 644–651. [DOI] [PubMed] [Google Scholar]
- 11.Silventoinen K, Kaprio J and Lahelma E (2000) Genetic and environmental contributions to the association between body height and educational attainment: a study of adult Finnish twins. Behav Genet 30, 477–485. [DOI] [PubMed] [Google Scholar]
- 12.Carmichael CM and McGue M (1995) A cross-sectional examination of height, weight, and body mass index in adult twins. J Gerontol A Biol Sci Med Sci 50, B237–B244. [DOI] [PubMed] [Google Scholar]
- 13.O’Rahilly S, Montague CT, Farooqi IS, Whitehead JP, Soos MA, Rau H, Wareham NJ, Sewter CP, Digby JE, Mohammed SN et al. (1997) Congenital leptin deficiency is associated with severe early-onset obesity in humans. Nature 387, 903–908. [DOI] [PubMed] [Google Scholar]
- 14.Froguel P, Clément K, Vaisse C, Lahlou N, Cabrol S, Pelloux V, Cassuto D, Gourmelen M, Dina C, Chambaz J et al. (1998) A mutation in the human leptin receptor gene causes obesity and pituitary dysfunction. Nature 392, 398–401. [DOI] [PubMed] [Google Scholar]
- 15.Farooqi IS, Matarese G, Lord GM, Keogh JM, Lawrence E, Agwu C, Sanna V, Jebb SA, Perna F, Fontana S et al. (2002) Beneficial effects of leptin on obesity, T cell hyporesponsiveness, and neuroendocrine/metabolic dysfunction of human congenital leptin deficiency. J Clin Invest 110, 1093–1103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Quarta C, Sánchez-Garrido MA, Tschöp MH and Clemmensen C (2016) Renaissance of leptin for obesity therapy. Diabetologia 59, 920–927. [DOI] [PubMed] [Google Scholar]
- 17.Krude H, Biebermann H, Luck W, Horn R, Brabant G and Grüters A (1998) Severe early-onset obesity, adrenal insufficiency and red hair pigmentation caused by POMC mutations in humans. Nat Genet 19, 155–157. [DOI] [PubMed] [Google Scholar]
- 18.Coll AP, Farooqi IS and O’Rahilly S (2007) The hormonal control of food intake. Cell 129, 251–262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Kühnen P, Clément K, Wiegand S, Blankenstein O, Gottesdiener K, Martini LL, Mai K, Blume-Peytavi U, Grüters A and Krude H (2016) Proopiomelanocortin deficiency treated with a melanocortin-4 receptor agonist. N Engl J Med 375, 240–246. [DOI] [PubMed] [Google Scholar]
- 20.Wheeler E, Huang N, Bochukova EG, Keogh JM, Lindsay S, Garg S, Henning E, Blackburn H, Loos RJF, Wareham NJ et al. (2013) Genome-wide SNP and CNV analysis identifies common and low-frequency variants associated with severe early-onset obesity. Nat Genet 45, 513–517. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Thorleifsson G, Walters GB, Gudbjartsson DF, Steinthorsdottir V, Sulem P, Helgadottir A, Styrkarsdottir U, Gretarsdottir S, Thorlacius S, Jonsdottir I et al. (2009) Genome-wide association yields new sequence variants at seven loci that associate with measures of obesity. Nat Genet 41, 18–24 [DOI] [PubMed] [Google Scholar]
- 22.Gaulton KJ, Ferreira T, Lee Y, Raimondo A, Mägi R, Reschen ME, Mahajan A, Locke A, Rayner NW, Robertson N et al. (2015) Genetic fine mapping and genomic annotation defines causal mechanisms at type 2 diabetes susceptibility loci. Nat Genet 47, 1415–1425. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Scuteri A, Sanna S, Chen W-M, Uda M, Albai G, Strait J, Najjar S, Nagaraja R, Orrú M, Usala G et al. (2007) Genome-wide association scan shows genetic variants in the FTO gene are associated with obesity-related traits. PLoS Genet 3, e115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Loos RJF and Bouchard C (2008) FTO: the first gene contributing to common forms of human obesity. Obes Rev 9, 246–250. [DOI] [PubMed] [Google Scholar]
- 25.Comuzzie AG, Hixson JE, Almasy L, Mitchell BD, Mahaney MC, Dyer TD, Stern MP, MacCluer JW and Blangero J (1997) A major quantitative trait locus determining serum leptin levels and fat mass is located on human chromosome 2. Nat Genet 15, 273–276. [DOI] [PubMed] [Google Scholar]
- 26.Rotimi CN, Comuzzie AG, Lowe WL, Luke A, Blangero J and Cooper RS (1999) The quantitative trait locus on chromosome 2 for serum leptin levels is confirmed in African-Americans. Diabetes 48, 643–644. [DOI] [PubMed] [Google Scholar]
- 27.Delplanque J, Barat-Houari M, Dina C, Gallina P, Clément K, Guy-Grand B, Vasseur F, Boutin P and Froguel P (2000) Linkage and association studies between the proopiomelanocortin (POMC) gene and obesity in Caucasian families. Diabetologia 43, 1554–1557. [DOI] [PubMed] [Google Scholar]
- 28.Hixson JE, Almasy L, Cole S, Birnbaum S, Mitchell BD, Mahaney MC, Stern MP, MacCluer JW, Blangero J and Comuzzie AG (1999) Normal variation in leptin levels is associated with polymorphisms in the proopiomelanocortin gene, POMC. J Clin Endocrinol Metab 84, 3187–3191. [DOI] [PubMed] [Google Scholar]
- 29.Raffin-Sanson ML, de Keyzer Y and Bertagna X (2003) Proopiomelanocortin, a polypeptide precursor with multiple functions: from physiology to pathological conditions. Eur J Endocrinol 149, 79–90. [DOI] [PubMed] [Google Scholar]
- 30.Rubinstein M, Mogil JS, Japón M, Chan EC, Allen RG and Low MJ (1996) Absence of opioid stress-induced analgesia in mice lacking beta-endorphin by site-directed mutagenesis. Proc Natl Acad Sci USA 93, 3995–4000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Newell-Price J (2003) Proopiomelanocortin gene expression and DNA methylation: implications for Cushing’s syndrome and beyond. J Endocrinol 177, 365–372. [DOI] [PubMed] [Google Scholar]
- 32.Liu B, Hammer GD, Rubinstein M, Mortrud M and Low MJ (1992) Identification of DNA elements cooperatively activating proopiomelanocortin gene expression in the pituitary glands of transgenic mice. Mol Cell Biol 12, 3978–3990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Liu B, Mortrud M and Low MJ (1995) DNA elements with AT-rich core sequences direct pituitary cell-specific expression of the pro-opiomelanocortin gene in transgenic mice. Biochem J 312 (Pt 3), 827–832. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Lamolet B, Pulichino AM, Lamonerie T, Gauthier Y, Brue T, Enjalbert A and Drouin J (2001) A pituitary cell-restricted T box factor, Tpit, activates POMC transcription in cooperation with Pitx homeoproteins. Cell 104, 849–859. [DOI] [PubMed] [Google Scholar]
- 35.Philips A, Maira M, Mullick A, Chamberland M, Lesage S, Hugo P and Drouin J (1997) Antagonism between Nur77 and glucocorticoid receptor for control of transcription. Mol Cell Biol 17, 5952–5959. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Poulin G, Turgeon B and Drouin J (1997) NeuroD1/beta2 contributes to cell-specific transcription of the proopiomelanocortin gene. Mol Cell Biol 17, 6673–6682. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Pulichino A-M, Vallette-Kasic S, Tsai JP-Y, Couture C, Gauthier Y and Drouin J (2003) Tpit determines alternate fates during pituitary cell differentiation. Genes Dev 17, 738–747. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Tremblay JJ, Lanctôt C and Drouin J (1998) The pan-pituitary activator of transcription, Ptx1 (pituitary homeobox 1), acts in synergy with SF-1 and Pit1 and is an upstream regulator of the Lim-homeodomain gene Lim3/Lhx3. Mol Endocrinol 12, 428–441. [DOI] [PubMed] [Google Scholar]
- 39.Langlais D, Couture C, Sylvain-Drolet G and Drouin J (2011) A pituitary-specific enhancer of the POMC gene with preferential activity in corticotrope cells. Mol Endocrinol 25, 348–359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Budry L, Balsalobre A, Gauthier Y, Khetchoumian K, L’Honore A, Vallette S, Brue T, Figarella-Branger D, Meij B and Drouin J (2012) The selector gene Pax7 dictates alternate pituitary cell fates through its pioneer action on chromatin remodeling. Genes Dev 26, 2299–2310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Drouin J (2016) 60 years of POMC: transcriptional and epigenetic regulation of POMC gene expression. J Mol Endocrinol 56, T99–T112. [DOI] [PubMed] [Google Scholar]
- 42.Hammer GD, Fairchild-Huntress V and Low MJ (1990) Pituitary-specific and hormonally regulated gene expression directed by the rat proopiomelanocortin promoter in transgenic mice. Mol Endocrinol 4, 1689–1697. [DOI] [PubMed] [Google Scholar]
- 43.Rubinstein M, Mortrud M, Liu B and Low MJ (1993) Rat and mouse proopiomelanocortin gene sequences target tissue-specific expression to the pituitary gland but not to the hypothalamus of transgenic mice. Neuroendocrinology 58, 373–380. [DOI] [PubMed] [Google Scholar]
- 44.Young JI, Otero V, Cerdán MG, Falzone TL, Chan EC, Low MJ and Rubinstein M (1998) Authentic cell-specific and developmentally regulated expression of pro-opiomelanocortin genomic fragments in hypothalamic and hindbrain neurons of transgenic mice. J Neurosci 18, 6631–6640. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Cowley MA, Smart JL, Rubinstein M, Cerdán MG, Diano S, Horvath TL, Cone RD and Low MJ (2001) Leptin activates anorexigenic POMC neurons through a neural network in the arcuate nucleus. Nature 411, 480–484. [DOI] [PubMed] [Google Scholar]
- 46.Bjørbaek C and Kahn BB (2004) Leptin signaling in the central nervous system and the periphery. Recent Prog Horm Res 59, 305–331. [DOI] [PubMed] [Google Scholar]
- 47.Heisler LK, Cowley MA, Tecott LH, Fan W, Low MJ, Smart JL, Rubinstein M, Tatro JB, Marcus JN, Holstege H et al. (2002) Activation of central melanocortin pathways by fenfluramine. Science 297, 609–611. [DOI] [PubMed] [Google Scholar]
- 48.Batterham RL, Cowley MA, Small CJ, Herzog H, Cohen MA, Dakin CL, Wren AM, Brynes AE, Low MJ, Ghatei MA et al. (2004) Physiology: does gut hormone PYY3–36 decrease food intake in rodents? (reply). Nature 430, 650–654. [DOI] [PubMed] [Google Scholar]
- 49.Lam DD, Attard CA, Mercer AJ, Myers MG, Rubinstein M and Low MJ (2015) Conditional expression of Pomc in the Lepr-positive subpopulation of POMC neurons is sufficient for normal energy homeostasis and metabolism. Endocrinology 156, 1292–1302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Cone RD (2005) Anatomy and regulation of the central melanocortin system. Nat Neurosci 8, 571–578. [DOI] [PubMed] [Google Scholar]
- 51.Jarvie BC and Hentges ST (2012) Expression of GABAergic and glutamatergic phenotypic markers in hypothalamic proopiomelanocortin neurons. J Comp Neurol 520, 3863–3876. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Williams KW, Margatho LO, Lee CE, Choi M, Lee S, Scott MM, Elias CF and Elmquist JK (2010) Segregation of acute leptin and insulin effects in distinct populations of arcuate proopiomelanocortin neurons. J Neurosci 30, 2472–2479. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Lam BYH, Cimino I, Polex-Wolf J, Nicole Kohnke S, Rimmington D, Iyemere V, Heeley N, Cossetti C, Schulte R, Saraiva LR et al. (2017) Heterogeneity of hypothalamic pro-opiomelanocortin-expressing neurons revealed by single-cell RNA sequencing. Mol Metab 6, 383–392. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.de Souza FSJ, Santangelo AM, Bumaschny V, Avale ME, Smart JL, Low MJ and Rubinstein M (2005) Identification of neuronal enhancers of the proopiomelanocortin gene by transgenic mouse analysis and phylogenetic footprinting. Mol Cell Biol 25, 3076–3086. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.de Souza FSJ, Bumaschny VF, Low MJ and Rubinstein M (2005) Subfunctionalization of expression and peptide domains following the ancient duplication of the proopiomelanocortin gene in teleost fishes. Mol Biol Evol 22, 2417–2427. [DOI] [PubMed] [Google Scholar]
- 56.Santangelo AM, De Souza FSJ, Franchini LF, Bumaschny VF, Low MJ and Rubinstein M (2007) Ancient exaptation of a CORE-SINE retroposon into a highly conserved mammalian neuronal enhancer of the proopiomelanocortin gene. PLoS Genet 3, 1813–1826. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Brosius J (2003) The contribution of RNAs and retroposition to evolutionary novelties. Genetica 118, 99–116. [PubMed] [Google Scholar]
- 58.Kazazian HH (2004) Mobile elements: drivers of genome evolution. Science 303, 1626–1632. [DOI] [PubMed] [Google Scholar]
- 59.Norris J, Fan D, Aleman C, Marks JR, Futreal PA, Wiseman RW, Iglehart JD, Deininger PL and McDonnell DP (1995) Identification of a new subclass of Alu DNA repeats which can function as estrogen receptor-dependent transcriptional enhancers. J Biol Chem 270, 22777–22782. [DOI] [PubMed] [Google Scholar]
- 60.Laperriere D, Wang T-T, White JH and Mader S (2007) Widespread Alu repeat-driven expansion of consensus DR2 retinoic acid response elements during primate evolution. BMC Genom 8, 23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Babich V, Aksenov N, Alexeenko V, Oei SL, Buchlow G and Tomilin N (1999) Association of some potential hormone response elements in human genes with the Alu family repeats. Gene 239, 341–349. [DOI] [PubMed] [Google Scholar]
- 62.Bininda-Emonds ORP, Cardillo M, Jones KE, MacPhee RDE, Beck RMD, Grenyer R, Price SA, Vos RA, Gittleman JL and Purvis A (2007) The delayed rise of present-day mammals. Nature 446, 507–512. [DOI] [PubMed] [Google Scholar]
- 63.Smit AF (1993) Identification of a new, abundant superfamily of mammalian LTR-transposons. Nucleic Acids Res 21, 1863–1872. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Franchini LF, Lopez-Leal R, Nasif S, Beati P, Gelman DM, Low MJ, de Souza FJS and Rubinstein M (2011) Convergent evolution of two mammalian neuronal enhancers by sequential exaptation of unrelated retroposons. Proc Natl Acad Sci 108, 15270–15275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Japón MA, Rubinstein M and Low MJ (1994) In situ hybridization analysis of anterior pituitary hormone gene expression during fetal mouse development. J Histochem Cytochem 42, 1117–1125. [DOI] [PubMed] [Google Scholar]
- 66.Padilla SL, Carmody JS and Zeltser LM (2010) Pomc-expressing progenitors give rise to antagonistic neuronal populations in hypothalamic feeding circuits. Nat Med 16, 403–405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Sanz E, Quintana A, Deem JD, Steiner RA, Palmiter RD and McKnight GS (2015) Fertility-regulating Kiss1 neurons arise from hypothalamic POMC-expressing progenitors. J Neurosci 35, 5549–5556. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Hong J-W, Hendrix DA and Levine MS (2008) Shadow enhancers as a source of evolutionary novelty. Science 321, 1314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Perry MW, Boettiger AN, Bothma JP and Levine M (2010) Shadow enhancers foster robustness of Drosophila gastrulation. Curr Biol 20, 1562–1567. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Frankel N, Davis GK, Vargas D, Wang S, Payre F and Stern DL (2010) Phenotypic robustness conferred by apparently redundant transcriptional enhancers. Nature 466, 490–493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Cannavò E, Khoueiry P, Garfield DA, Geeleher P, Zichner T, Gustafson EH, Ciglar L, Korbel JO and Furlong EEM (2016) Shadow enhancers are pervasive features of developmental regulatory networks. Curr Biol 26, 38–51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Lam DD, de Souza FSJ, Nasif S, Yamashita M, López Leal R, Otero-Corchon V, Meece K, Sampath H, Mercer AJ, Wardlaw SL et al. (2014) Partially redundant enhancers cooperatively maintain mammalian pomc expression above a critical functional threshold. PLoS Genet 11, e1005133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Berger MF, Badis G, Gehrke AR, Talukder S, Philippakis AA, Peña-Castillo L, Alleyne TM, Mnaimneh S, Botvinnik OB, Chan ET et al. (2008) Variation in homeodomain DNA binding revealed by high-resolution analysis of sequence preferences. Cell 133, 1266–1276. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Thompson CL, Ng L, Menon V, Martinez S, Lee C-K, Glattfelder K, Sunkin SM, Henry A, Lau C, Dang C et al. (2014) A high-resolution spatiotemporal atlas of gene expression of the developing mouse brain. Neuron 83, 309–323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Nasif S, de Souza FSJ, González LE, Yamashita M, Orquera DP, Low MJ and Rubinstein M (2015) Islet 1 specifies the identity of hypothalamic melanocortin neurons and is critical for normal food intake and adiposity in adulthood. Proc Natl Acad Sci 112, E1861–E1870. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Lee B, Lee S, Lee S-K and Lee JW (2016) The LIM-homeobox transcription factor Isl1 plays crucial roles in the development of multiple arcuate nucleus neurons. Development 143, 3763–3773. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.de Souza FSJ, Nasif S, López-Leal R, Levi DH, Low MJ and Rubinstein M (2011) The estrogen receptor α colocalizes with proopiomelanocortin in hypothalamic neurons and binds to a conserved motif present in the neuron-specific enhancer nPE2. Eur J Pharmacol 660, 181–187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Domené S, Bumaschny VF, de Souza FSJ, Franchini LF, Nasif S, Low MJ and Rubinstein M (2013) Enhancer turnover and conserved regulatory function in vertebrate evolution. Philos Trans R Soc Lond B Biol Sci 368, 20130027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Bumaschny VF, Yamashita M, Casas-Cordero R, Otero-Corchón V, de Souza FSJ, Rubinstein M and Low MJ (2012) Obesity-programmed mice are rescued by early genetic intervention. J Clin Invest 122, 4203–4212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Rosenbaum M, Goldsmith R, Bloomfield D, Magnano A, Weimer L, Heymsfield S, Gallagher D, Mayer L, Murphy E and Leibel RL (2005) Low-dose leptin reverses skeletal muscle, autonomic, and neuroendocrine adaptations to maintenance of reduced weight. J Clin Invest 115, 3579–3586. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Bray GA and Greenway FL (2007) Pharmacological treatment of the overweight patient. Pharmacol Rev 59, 151–184. [DOI] [PubMed] [Google Scholar]
- 82.Tremblay A and Chaput J-P (2009) Adaptive reduction in thermogenesis and resistance to lose fat in obese men. Br J Nutr 102, 488–492. [DOI] [PubMed] [Google Scholar]
- 83.Rolls BJ, Rowe EA and Turner RC (1980) Persistent obesity in rats following a period of consumption of a mixed, high energy diet. J Physiol 298, 415–427. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Levin BE and Dunn-Meynell AA (2000) Defense of body weight against chronic caloric restriction in obesity-prone and -resistant rats. Am J Physiol Regul Integr Comp Physiol 278, R231–R237. [DOI] [PubMed] [Google Scholar]
- 85.Guo J, Jou W, Gavrilova O and Hall KD (2009) Persistent diet-induced obesity in male C57BL/6 mice resulting from temporary obesigenic diets. PLoS ONE 4, e5370. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Chhabra KH, Adams JM, Jones GL, Yamashita M, Schlapschy M, Skerra A, Rubinstein M and Low MJ (2016) Reprogramming the body weight set point by a reciprocal interaction of hypothalamic leptin sensitivity and Pomc gene expression reverts extreme obesity. Mol Metab 5, 869–881. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Leibel RL (2008) Molecular physiology of weight regulation in mice and humans. Int J Obes 32, S98–S108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Schulkin J (2003) Allostasis: a neural behavioral perspective. Horm Behav 43, 21–27; discussion 28–30. [DOI] [PubMed] [Google Scholar]