

Graphical abstract
Abbreviations: 1-AG, 1-arachidonoyl glycerol; 1-LG, 1-linoleoyl glycerol; 2-AG, 2-arachidonoyl glycerol; 2-LG, 2- linoleoyl glycerol; ACN, acetonitrile; AEA, arachidonoyl ethanolamide; BHT, 2,6-di-tert-butyl-4-methylphenol; CAR, carnitine; EC, endocannabinoid; FC, fold change; FT, freezing temperature/storage in ice water; HETE, hydroxyeicosatetraenoate; HRMS, high-resolution mass spectrometry; IRB, Institutional Review Board; IS, internal standard; K3EDTA, tripotassium ethylenediaminetetraacetic acid; LC, liquid chromatography; LEA, linoleoyl ethanolamide; LLE, liquid–liquid extraction; LLOQ, lowest limit of quantification; LPA, lysophosphatidic acid; LPC, lysophosphatidylcholine; LPC O, lysophosphatidylcholine-ether; LPE, lysophosphatidylethanolamine; LPG, lysophosphatidylglycerol; LPI, lysophosphatic inositol; MeOH, methanol; MS/MS, tandem mass spectrometry; MTBE, methyl tertiary-butyl ether; OEA, oleoyl ethanolamide; PBS, phosphate-buffered saline; PC, phohsphatidylcholine; PE, phosphotidylethanolamine; PI, phosphatidylinositol; PEA, palmitoyl ethanolamide; QC, quality control; REC, Research Ethics Committee; Ref, reference sample; RT, room temperature; SEA, stearoyl ethanolamide; SPE, solid-phase extraction; STD, calibration standard; VEA, vaccenic acid ethanolamid; WB, whole blood
Keywords: Lipidomics, Metabolomics, K3EDTA plasma sampling, Sampling protocol, Pre-analytics
Highlights
-
•
Analysis of ex vivo distortion in K3ETDA whole blood and plasma.
-
•
Establishment of pre-analytical sample-handling protocols for clinical use.
-
•
Recommendation of analyte-specific sample handling protocols.
Abstract
The emerging disciplines of lipidomics and metabolomics show great potential for the discovery of diagnostic biomarkers, but appropriate pre-analytical sample-handling procedures are critical because several analytes are prone to ex vivo distortions during sample collection. To test how the intermediate storage temperature and storage period of plasma samples from K3EDTA whole-blood collection tubes affect analyte concentrations, we assessed samples from non-fasting healthy volunteers (n = 9) for a broad spectrum of metabolites, including lipids and lipid mediators, using a well-established LC-MS-based platform. We used a fold change-based approach as a relative measure of analyte stability to evaluate 489 analytes, employing a combination of targeted LC-MS/MS and LC-HRMS screening. The concentrations of many analytes were found to be reliable, often justifying less strict sample handling; however, certain analytes were unstable, supporting the need for meticulous processing. We make four data-driven recommendations for sample-handling protocols with varying degrees of stringency, based on the maximum number of analytes and the feasibility of routine clinical implementation. These protocols also enable the simple evaluation of biomarker candidates based on their analyte-specific vulnerability to ex vivo distortions. In summary, pre-analytical sample handling has a major effect on the suitability of certain metabolites as biomarkers, including several lipids and lipid mediators. Our sample-handling recommendations will increase the reliability and quality of samples when such metabolites are necessary for routine clinical diagnosis.
Introduction
LC-MS-based analysis of metabolites, including lipids and lipid mediators, is now a key component of biomarker research [1], [2], [3]. The relevance of lipidomics and metabolomics in such studies is supported by the complex biochemistry of endogenous compounds, reflecting highly individual conditions in health and disease [4]. For example, lipid biomarkers offer a better understanding of the complex metabolism in dyslipidemia [5], cancer [6], and immune-mediated inflammatory diseases [7]. The robust analysis of such metabolites, including lipids and lipid mediators, is therefore an important basis for the wider application of lipidomics and metabolomics in LC-MS-based clinical research, requiring standardized methods including pre-analytical sample handling [8], [9], [10], [11]. In such research projects, samples can be acquired prospectively in an ongoing clinical study or retrospectively by using existing samples, such as those from a biobank. Prospective sampling offers the great advantage that a suitable pre-analytical sample-handling protocol can be created in close cooperation with the clinicians, taking the instability of certain analytes into account while ensuring practical feasibility in a clinical setting. However, the planning and execution of such a project can be time consuming and expensive. In contrast, retrospective samples are advantageous because a sufficient set of samples already exists, which is important in studies focusing on rare diseases. Nevertheless, if samples have not been collected with due consideration for the pre-analytical instability of metabolites and lipids, they may not be reliable. In any case, comprehensive knowledge about factors that influence analyte concentrations during the pre-analytical phase is highly important.
Because several analytes are sensitive to ex vivo distortions, pre-analytics is a key challenge for the meaningful application of lipidomics and metabolomics [12]. If pre-analytical influences are unclear, the quality of samples and consequently the analytical data will be negatively affected [13]. This hinders the reproducibility of lipidomics and metabolomics in research [10], [14], which is necessary for the translation of clinical research into routine practice. In addition to factors related to the state of the subject, such as nutritional status [15], [16], exercise [17], comorbidities [18] and the time of day the blood is drawn [19], external factors during sample handling and intermediate storage can also affect sample quality [10]. Time-to-centrifugation, overall processing delay, and intermediate storage temperature are among the major factors responsible for analyte variability, but others include the choice of blood collection tubes [20], [21], centrifugation settings (e.g., speed, duration, and temperature [10]), prolonged venous compression [22], the final storage temperature, and the number of overall freeze–thaw cycles [21], [23], [24]. The most common clinical research specimens include plasma samples in K3EDTA tubes to prevent coagulation. However, the quality of plasma samples depends on many of the factors listed above [25]. In general, plasma should be separated from blood cells as soon as possible after drawing, and short-term storage in ice water is currently recommended before permanent storage in a deep freezer [10], [26], [27], [28], [29].
In clinical research projects, blood sample collection and pre-analytical sample handling are generally part of the routine clinical treatment of patients, so the sample quality can suffer from delayed processing because strict pre-analytical protocols are difficult to apply in such settings [30]. Therefore, sampling protocols need some flexibility to reflect clinical constraints while ensuring the pre-analytical stability of analytes. For retrospective studies, the sampling protocol was probably not selected based on the preservation of metabolites and lipids, limiting the usability of such samples. However, whereas some analytes are vulnerable to pre-analytical concentration changes in whole blood or plasma, ceramides, sphingomyelins and triglycerides are highly stable compared to other lipids such as endocannabinoids and lysophosphatidic acids (LPAs) [31]. Thus, samples taken using less stringent pre-analytical protocols may still be suitable for the analysis of many compounds.
The objective of this study was to investigate the influence of of pre-analytical factors, such as storage conditions and processing delays, on analyte variability in the context of a well-established metabolomics and lipidomics platform. This platform combines semi-quantitative lipid and metabolite LC-HRMS screening and quantitative LC-MS/MS for different metabolites, including lipids and lipid mediators. We recommend various sampling protocols to preserve as many unstable analytes as possible while ensuring the methods remain practicable in clinical settings. This is applicable to both prospective sampling and the reliability of samples acquired retrospectively, such as from a biobank.
Materials and methods
Chemicals
Water (LC-MS grade), acetonitrile (LC-MS grade, ≥99.95 %), hexane (UV/IR grade, ≥99.5 %), methanol (LC-MS grade ≥ 99.95 %), isopropanol (LC-MS grade, ≥99.95 %), acetone (LC-MS grade, ≥99.9 %), butanol (for synthesis, ≥99.5 %), trichloromethane/chloroform (UV/IR grade, ≥99.8 %) and citric acid (≥99.5 % p.a.) were purchased from Carl Roth (Karlsruhe, Germany), ethyl acetate (Reag. Ph. Eur. ≥ 99.9 %) was purchased from Biosolve (Valkenswaard, Netherlands), formic acid (98 %) and acetic acid (100 %) from Fisher Scientific (Schwerte, Germany), hydrochloric acid (Reag. Ph. Eur., 37 %) and disodium hydrogen phosphate (NormaPur) from VWR Chemicals (Darmstadt, Germany), and 2,6-di-tert-butyl-4-methylphenol (BHT, 99 %) from Sigma-Aldrich (Munich, Germany). Reference compounds and isotopically labeled internal standards (IS) were purchased from Cayman Chemicals (Ann Arbor, MI, USA) for endocannabinoids and oxylipins, or Avanti Polar Lipids (Alabaster, AL, USA) for sphingolipids, LPAs, and lipids analyzed by LC-HRMS. For polar compounds, tryptophan and related metabolites, isotopically labeled IS and reference compounds were purchased from Sigma-Aldrich or Cambridge Isotope Laboratories (Tewksbury, MA, USA). Detailed descriptions can be found in the supplementary material.
Sample collection and investigations of pre-analytical sample handling
Blood samples were collected from nine non-fasted healthy volunteers after informed consent was given. Sample collection was approved by the Research Ethics Committee/Institutional Review Board (REC/IRB) of the university hospital of Goethe-University Frankfurt without a formal IRB because no ethical concerns were raised over the use of human blood samples as anonymized material in this study. The ethnicity of study participants was deemed irrelevant for the determination of ex vivo influences on analyte concentrations and was not recorded. The volunteers comprised five male and four female participants (mean age = 36 ± 13). Venous blood samples were drawn using 2.7-mL K3EDTA S-Monovettes (Sarstedt, Nümbrecht, Germany). In addition to a baseline sample, 16 sampling tubes were drawn from each study participant, of which eight were immediately used to isolate plasma and eight were stored as whole blood (Fig. 1). Different storage times were tested for whole blood (20, 60, 120 and 240 min) and plasma samples (0, 20, 60, 120 and 240 min) at RT (20.4 ± 0.2 °C) and in ice-water (FT, 0.5 ± 0.2 °C). The immediately processed sample, which on average took 12 ± 2 min (0 min) to prepare from blood drawing until snap-freezing (including centrifugation), was regarded as the baseline sample for the corresponding measurement series (0 min, reference sample = Ref.). Samples were centrifuged at 2000 × g for 10 min at 4 °C. After the appropriate storage time (22 ± 2 min for 20 min, 63 ± 4 min for 60 min, 123 ± 3 min for 120 min and 244 ± 2 min for 240 min – all times excluding the duration of centrifugation), samples were centrifuged and separated from the cells (whole blood samples). In the latter case, whole blood samples were centrifuged immediately after drawing the blood, and the plasma was separated from the cells and stored for different periods of time at different temperatures as described above. Afterwards, plasma was aliquoted for the LC-MS methods and snap-frozen on dry ice. The schedule and procedure for sample generation is depicted in Fig. 1. All samples were stored at a temperature below –70 °C until analysis, resulting in a single freeze–thaw cycle. Thawing times were kept constant for all sample sets regardless of the analytical methods (Fig. 1).
Fig. 1.

Collected samples were stored as whole blood or plasma, depending on the time point of centrifugation. Differences in storage conditions were introduced by varying the temperature (RT or ice water) and delay in storage time after blood draw (20, 60, 120, or 240 min). A baseline sample for each study subject was processed immediately and was defined as the optimally treated reference sample.
Targeted LC-MS/MS analysis
Five different quantitative analysis methods were applied to determine the amounts of endocannabinoids, oxylipins, sphingolipids, LPAs and tryptophan-like metabolites, respectively. For each method, a calibration curve was generated. Calibration standards (STDs) and quality control samples (QCs) were prepared and extracted in method-dependent surrogate matrices. Pre-aliquoted plasma samples (endocannabinoids – 200 µL, sphingolipids – 10 µL, tryptophan-like metabolites – 100 µL, oxylipins – 200 µL, LPAs – 100 µL) were thawed in a refrigerator (4 °C) immediately before extraction.
Endocannabinoids: Standard preparation and sample extraction for endocannabinoids and endocannabinoid-like substances (described hereafter as endocannabinoids) was performed as previously described with minor modifications [32]. Plasma samples were spiked with 20 µL IS working solution whereas STDs and QCs in phosphate-buffered saline (PBS) as a surrogate matrix were spiked with 20 µL IS working solution and 20 µL of the respective standard working solution. Samples were vortexed (1 min) and centrifuged (20,000 × g, 1 min, 4 °C). Liquid–liquid extraction (LLE) was performed with 400 µL ethyl acetate:hexane (9:1 v/v). After vortexing (1 min) and centrifugation (20,000 × g, 3 min, 4 °C) the upper layer was transferred to a new polypropylene tube and evaporated (45 °C) under a gentle stream of nitrogen. Extracted and evaporated samples were reconstituted in 50 µL acetonitrile, vortexed (1 min), transferred into an LC-MS vial, and analyzed by LC-ESI-MS/MS using a triple quadrupole mass spectrometer QTRAP 6500 + with a Turbo Ion Spray source (both from Sciex, Darmstadt, Germany) in positive ESI mode. The LC system consisted of an Agilent 1290 Infinity II LC instrument with a binary HPLC pump, column oven, and autosampler (Agilent, Waldbronn, Germany). The 10-µL injected samples were separated using an Acquity UPLC BEH C18 2.1 × 100 mm column with an Acquity UPLC BEH C18 1.7 µm VanGuard pre-column 2.1 × 5 mm (both from Waters, Eschborn, Germany) and eluted in a gradient of solvent A (0.0025 % formic acid in water) and solvent B (0.0025 % formic acid in acetonitrile) with a total run time of 8 min. Twelve analytes were tested using this method. Detailed MS parameters and concentrations of STDs and QCs can be found in the supplementary material.
Oxylipins: Plasma samples were spiked with 20 µL IS working solution, whereas STDs and QCs (with 200 µL PBS as a surrogate matrix) were spiked with 20 µL IS working solution and 20 µL of the respective standard working solution. Samples were vortexed (1 min) and centrifuged (20,000 × g, 1 min, 4 °C), followed by protein precipitation and solid-phase extraction (SPE). For protein precipitation, 200 µL of acetonitrile:methanol (8:2 v/v) was added to each plasma sample. Samples were vortexed (1 min) and centrifuged (20,000 × g, 10 min, 4 °C). The supernatant was transferred to a 96-well plate for SPE with ABN cartridges (1 mL, 30 cc) using an Extrahera (Biotage, Uppsala, Sweden). The SPE protocol consisted of five steps (supplementary material). For conditioning (step 1), 1 mL of methanol was loaded onto the cartridges, followed by (step 2) 1 mL of 0.1 % formic acid for equilibration. For sample pretreatment, 600 µL of 1 % formic acid was added to each sample and mixed. After sample pretreatment, sample loading (step 3) involved the transfer of 1-mL aqueous samples to the cartridges, followed by a washing step (step 4) with methanol:water (40:60 v/v). For elution (step 5), 1 mL of acetonitrile was used. Samples were transferred to new polypropylene tubes, evaporated (45 °C) under a nitrogen stream, and reconditioned in 80 µL methanol:water (70:30 v/v) plus 0.0001 % BHT. Samples were vortexed (1 min), transferred to an LC-MS vial, and analyzed using the same LC-MS/MS system and apparatus described above, with a 17-min gradient elution to achieve sufficient analyte separation. Sixty-eight analytes were then measured in negative ESI mode. Detailed MS parameters and concentrations of STDs and QCs can be found in the supplementary material.
Sphingolipids: Standard preparation and extraction for sphingoid bases and ceramides (described hereafter as sphingolipids) was performed as previously described with minor modifications [33]. Plasma samples were spiked with 20 µL IS working solution whereas STDs and QCs in Z-buffer (200 µL) as a surrogate matrix were spiked with 20 µL IS working solution and 20 µL of the respective standard working solution. Z-buffer is 30 mM citric acid and 40 mM disodiumhydrogenphosphate in water. Plasma samples were vortexed (1 min) and centrifuged (20,000 × g, 1 min, RT). LLE was performed with 600 µL chloroform:methanol:240 mM hydrochloric acid (80:15:5 v/v/v), after diluting plasma samples with 200 µL Z-buffer. After vortexing (1 min) and centrifugation (20,000 × g, 5 min, RT), the lower layer was divided into two equal aliquots, transferred to two new polypropylene tubes, and evaporated (45 °C) under a gentle stream of nitrogen. Extracted and evaporated samples were resuspended in 50 µL methanol:formic acid (95:5 v/v) for sphingoid base measurements and in 50 µL THF:formic acid (99.8:0.2 v/v) + 10 mM ammonium formiate (9:1 v/v) for ceramide measurements. After reconstitution, samples were vortexed (1 min) and transferred to vials with inserts. For the analysis of sphingolipids, we used the same LC-MS/MS system described above in positive ESI mode, with an injection volume of 5 µL. However, sphingoid bases and ceramides were analyzed using different chromatographic columns and LC-MS/MS methods. For sphingoid bases, we used an UHPLC Zorbax Eclipse Plus C8 2.1 × 30 mm 1.8 µm column with an UHPLC guard C8 pre-column 2.1 × 3 mm 1.8 µm (Agilent). The fractions were eluted in a gradient of 0.5 % formic acid in water (solvent A) and 1 % formic acid in acetonitrile:isopropanol:acetone (50:30:20 v/v/v) (solvent B) for 4.5 min. To prevent carryover, two rinse injections were performed between each sample. For ceramides, we used an UHPLC Zorbax Eclipse Plus C18 2.1 × 50 mm 1.8 µm column with an UHPLC guard C18 pre-column 2.1 × 3 mm 1.8 µm (Agilent). The fractions were eluted in 10 mM ammonium formate + 0.2 % formic acid in water (solvent A) and 0.2 % formic acid in acetonitrile:isopropanol:acetone (50:30:20 v/v/v) for 7.5 min. Nine sphingoid bases and 16 ceramides were measured using this method. Detailed MS parameters and concentrations of STDs and QCs can be found in the supplementary material.
Lysophosphatidic acids: Plasma samples were spiked with 20 µL of IS working solution whereas STDs and QCs in 200 µL Z-buffer as a surrogate matrix were spiked with 20 µL of IS working solution and 20 µL of the respective standard working solution. Samples were vortexed (1 min) and centrifuged (20,000 × g, 1 min, 4 °C). LLE was performed with 400 µL of butanol. After vortexing (1 min) and centrifugation (20,000 × g, 3 min, 4 °C), the upper layer was transferred to a new polypropylene tube and evaporated (45 °C) under a gentle stream of nitrogen. Samples were reconstituted in 50 µL of methanol, vortexed (1 min) and transferred to vials with inserts. An LC-MS/MS system was used for the analysis of LPAs in negative ESI mode with an injection volume of 5 µL. Samples were fractionated on an HPLC EVO C18 2.1 × 50 mm 1.7 µm column with a UPLC C18 pre-column 2.1 × 5 mm (Waters) and were eluted in a gradient of 50 mM ammonium acetate + 0.2 % formic acid in water (solvent A) and 0.2 % formic acid in acetonitrile:isopropanol (50:50 v/v) (solvent B) for 5 min. Seven analytes were measured using this method. Detailed MS parameters and concentrations of STDs and QCs can be found in the supplementary material.
Tryptophan-like metabolites: Plasma samples were spiked with 20 µL of IS working solution, whereas STDs and QCs in 100 µL PBS as a surrogate matrix were spiked with 20 µL of the IS working solution and 20 µL of the respective standard working solution. Samples were vortexed (1 min) and then centrifuged (20,000 × g, 1 min, 4 °C) prior to protein precipitation in 300 µL of cold methanol. After vortexing (30 s), samples were stored below –20 °C for 30 min to facilitate protein precipitation, and then centrifuged (20,000 × g, 5 min, 4 °C). The supernatants were transferred to brown vials, evaporated (45 °C) under a stream of nitrogen, and reconstituted in 50 µL of a water:acetic acid (99.9:0.1 v/v) solution. After reconstitution, samples were vortexed (1 min) and transferred into vials with inserts. The same LC-MS/MS system as previously described was used for the analysis of tryptophan-like metabolites in positive ESI mode with an injection volume of 2.5 µL. Samples were fractionated on an UHPLC Hypersil Gold aQ 2.1 × 100 mm 1.9 µm column with a UHPLC pre-column 2.1 mm filter (Thermo Fisher Scientific, Dreieich, Germany) using a gradient of 0.1 % formic acid in water (solvent A) and 0.1 % formic acid in methanol (solvent B) for 9 min. Seven analytes were measured using this method; details on MS parameters and concentrations of standards and QCs can be found in the supplementary material.
Data acquisition and evaluation: Data were acquired and processed for all quantitative methods using Analyst™ v1.7.1 and MultiQuant™ v3.0.3 (both Sciex, Darmstadt, Germany). Each analytical run consisted of a standard calibration curve and a set of QC samples. A QC set consisted of two QC samples per concentration level – lowest limit (LLQC), low (LQC), medium (MQC) and high (HQC) concentrations) that surrounded the unknown samples. LLQC samples are not mandatory in routine LC-MS/MS analytics and were, therefore, only included for methods covering a wider concentration range, such as endocannabinoids (see Supplementary Materials). Additionally, two pre-aliquoted control samples were used to ensure comparability between analytical runs and were evaluated using a method-specific control chart. All unknown samples were measured in a randomized order within and between study participants, so that subject individual samples were not separated during analysis. Acceptance criteria per analytical run were applied as follows: (i) the back-calculated concentrations of the calibration standards should be within ± 15 % of the nominal value, except for the LLOQ, which should be within ± 20 %; (ii) at least 75 % of the calibration standards, with a minimum of six, must fulfill the accuracy criterion; (iii) the accuracy values of the QC samples should be within ± 15 % of the nominal values; and (iv) for every set of QC samples, at least 67 % QC samples and at least 50 % at each concentration level must comply with this criterion. Not all analytes fulfilled all acceptance criteria, so the results were divided into quantitative (analytes fulfilling all acceptance criteria) and semi-quantitative data (analytes not fulfilling all acceptance criteria). Detailed information about the fulfillment of acceptance criteria and applied classifications for every analyte can be found in the Supplementary Materials.
Screening of lipids and polar metabolites
Lipids and polar metabolites were extracted from 10 µL aliquots of the same plasma samples discussed above. Samples were processed together with blank samples and pooled QC samples. Samples were prepared by LLE following the addition of 75 µL of IS in methanol (see supplementary material) followed by 250 µL of methyl tertiary-butyl ether (MTBE) and 50 µL of 50 mM ammonium formate. The samples were then vortexed (1 min) and centrifuged (20,000 × g, 5 min, RT). The upper phase was transferred and the lower phase re-extracted in 100 µL MTBE:methanol:water (10:3:2.5 v/v/v), followed by centrifugation as above. For the measurement of lipids, the combined upper phases were evaporated under a gentle stream of nitrogen at 45 °C, stored at < –70 °C, and reconstituted in 100 µL methanol before analysis. For the measurement of polar metabolites, 200 µL acetonitrile were added to the lower phases and mixed by gentle shaking. The mixture was then dried under a nitrogen stream at 45 °C and reconstituted in 100 µL acetonitrile:water (50:50 v/v) before analysis. QC samples were created by pooling equal volumes of all unknown samples, vortexing and aliquoting. Lipids and polar metabolites were fractionated on a Vanquish Horizon UHPLC system and detected with an Orbitrap Exploris 480 (both Thermo Fisher Scientific) using positive and negative ionization modes.
Screening for lipids: Lipids were separated on a Zorbax RRHD Eclipse Plus C8 2.1 × 50 mm, 1.8 µm column, with a guard column of the same type (Agilent), using a gradient of 10 mM ammonium formate + 0.1 % formic acid in water (solvent A) and 0.1 % formic acid in acetonitrile:isopropanol (2:3 v/v) (solvent B). Further details can be found in the Supplementary Material.
Screening for polar metabolites: Polar metabolites were separated on a SeQuant ZIC-HILIC, 3.5 µm, 100 mm × 2.1 mm I.D. column (Merck, Darmstadt, Germany) coupled to a guard column with the same chemistry and an inline filter in a gradient of 0.1 % formic acid in water (solvent A) and 0.1 % formic acid in acetonitrile (solvent B). Further details can be found in the Supplementary Material.
Data acquisition and evaluation: Data were acquired using XCalibur v4.4 (Thermo Fisher Scientific). The measurement sequences started with solvent blank, extracted blank, zero, two QC and a control chart sample, continuing with one QC sample after every-eight samples and two additional QCs and a control chart sample at the end of the run. The samples were measured in a randomized order as described above. For HILIC separation the system was equilibrated with 10 injections of extracted plasma samples. Each sample was injected twice, switching ionization polarity between samples. Raw data were evaluated using TraceFinder v5.1 (Thermo Fisher Scientific) with target lists of 665 previously identified lipids derived from different matrices (not solely human K3EDTA plasma) and 276 polar metabolites. Peak integration was performed with a mass tolerance of ± 5 ppm and either the Genesis or ICIS integration algorithms were applied. Analytes whose identity could not be confirmed using exact mass, isotope ratio, and MS/MS fragmentation matching LipidBlast [34] or mzCloud offline library (for mzVault 2.3_Omics_2020A) were excluded. Furthermore, compounds for which (i) < 80 % of values exceeded the signal in the extracted blank sample by at least a factor of 2, or (ii) if peak integration could not be performed reproducibly, defined by a coefficient of variance in QC samples > 20 % (lipids) or > 30 % (polar metabolites), were excluded from further evaluation. Sphingolipids, which were already quantified using LC-MS/MS, were also omitted. Applying these criteria, 296 lipids and 122 polar metabolites were considered for further evaluation. Results for polar metabolites were normalized using median-based probabilistic quotient normalization (PQN) [35] with the 24 QC samples after imputing missing values with half of the minimum of the respective analyte. Lipids were normalized via one internal standard per lipid class. More detailed information, including the target lists, can be found in the supplementary material.
Statistical data evaluation
Statistical analysis was performed with GraphPad Prism v9.2.0 (3 3 2) 64-bit (GraphPad Software, San Diego, CA, USA) and RStudio v1.4.1717 64-bit (R Foundation for Statistical Computing, Vienna, Austria) with R v4.1.2 (2021–11-02) (RStudio, Boston, MA, USA) using the packages “readxl” [36], “dplyr” [37], “tidyverse” [38] and “reshape2″ [39] for data wrangling and ”ggplot2″ [40], “ggplot2″ [40], “gplots” [41], “ComplexHeatmaps” [42], “irr” [43] and “circlize” [44] for data visualization. For all measurements, we omitted from further analysis those analytes with a missing value proportion > 20 % and those with a proportion of measurements > 20 % under the limit of quantification. This resulted in the omission of 48 analytes (eight sphingolipids, one endocannabinoid, three LPAs and 36 oxylipins). Two analytes of the endocannabinoid class, namely 2- linoleoyl glycerol (2-LG) and oleoyl ethanolamide (OEA), showed > 20 % of values exceeding the upper limit of their calibration curves, but were still included in the evaluation using the extrapolated values above the ULOQ, because linearity of the method was expected to be sufficient to calculate these concentrations. For OEA, this can be explained by insufficient chromatographic separation from the isomer vaccenic acid ethanolamide (VEA). Therefore, OEA was provided as the sum parameter of OEA and VEA.
Results displayed for sample concentrations and relative changes generally represent the mean of nine samples, except where individual measurements were missing. For previously filtered analytes with > 20 % of missing values, a chi-squared test was performed (α = 0.05, with FDR correction) to determine if missing values significantly deviated from the expected distribution across all study participants. Significant results indicated that given analytes could mostly be measured in specific study participants, and it was assumed that the missing values could be attributed to inter-individual variability. Despite the lack of data for some analytes, calculation of fold changes was still applicable to the remaining values. This was true for 13 analytes of the oxylipin class, which are highlighted in the Supplementary Material.
Commonalities and differences between the present study and published data [26] regarding pre-analytical sample stability were identified by highlighting the different analyte coverages, experimental designs, and the agreement between the studies. While reanalyzing 195 analytes from the previous study, an additional 294 analytes, mainly consisting of oxylipins, certain sphingolipids, tryptophan and its metabolites, as well as polar metabolites in general, were measured and evaluated in the present study. A total of 61 analytes included in the previous study (mainly diglycerides and triglycerides as well as certain fatty acids) were not evaluated in the current study, because they failed the acceptance criteria set out above. For the assessment of concordance between this study and the earlier publication [26], we calculated Cohen’s unweighted kappa as a measure for interrater reliability [45]. Detailed results of this comparison are provided in the supplementary information.
Analysis of stability data and provision of blood sampling protocol recommendations
Fold change was the basis for stability evaluation in the following analyses. Fold changes of analytes within tested conditions (matrix, temperature and period of intermediate storage) were obtained by calculating the quotient of the condition-wise mean concentrations and the mean concentration of the baseline samples (). Analytes were deemed unstable for a specific storage condition at a specific time if their fold change compared to the baseline exceeded ± 20 % in the following analysis, which matches the common acceptance threshold coefficient of variance in lipidomics [46]. Furthermore, the entire process of pre-analytical processing (represented by concentration fold changes before and after centrifugation) was assessed for each analyte, by evaluating whether either one of the two fold changes exceeded ± 20 %, or the sum of both exceeded ± 30 %. The critical threshold of ± 20 % for the evaluation of individual fold changes was extended to ± 30 % for the sum of two fold changes because the impact of combined pre-analytical influences was not tested in the present study due to restricted sample size, and thus two individual measurements had to be combined.
The aim of this analysis was to assign analyte-specific pre-analytical processing recommendations that would maximize clinical flexibility. We assigned each analyte to the last protocol where the stability thresholds defined were not exceeded, going from the most strict to the least strict pre-processing recommendation (Fig. 2). We evaluated individual fold changes, as well as their sum, to account for effects that are unique to either of the matrices (whole blood or plasma) as well as matrix-independent influences, such as temperature during processing.
Fig. 2.

Process for matching individual analytes with protocol recommendations based on their experimental stability data: First, the experimentally-determined fold changes representing the protocols were selected. Then, each protocol was evaluated for the stability criteria, starting with the most to the least strict protocol. Finally, an analyte was assigned to the last protocol that met the stability criteria.
Results and discussion
The susceptibility of metabolites, including lipids and lipid mediators, to ex vivo distortions has been comprehensively investigated [26], [28], [47], but is rarely a standardized part of LC-MS-based clinical research studies focusing on lipidomics and metabolomics. This is partly due to difficulties in establishing the strict pre-analytical protocols that are necessary for endocannabinoids, lysophosphatidycholines [48], eicosanoids [47] or LPAs [31] in clinical research projects. However, a concept is needed to ensure that lipidomics and metabolomics remain feasible analytical approaches in clinical research even when clinical working practices do not allow the fastest pre-analytical sample processing protocols. Because not all analytes are prone to ex vivo distortions, even if samples are handled at RT with delayed processing, they can be analyzed after following a less strict protocol, still covering a broad range of metabolites, including several lipids and lipid mediators. To generate a comprehensive data-based definition of these protocols, we conducted experiments focusing on the two main factors influencing the pre-analytical handling of whole blood and plasma samples, namely the temperature and delay during sample processing.
Analyte variability caused by pre-analytical sample handling
Our experiments to determine pre-analytical stability focused on temperature (samples stored in ice water or at RT) and the period of intermediate storage (20, 60, 120, or 240 min as whole blood or plasma) during the preparation of plasma samples from K3EDTA whole-blood collection tubes. We measured 1070 analytes using targeted LC-MS/MS methods for 25 sphingolipids, 68 oxylipins, 12 endocannabinoids, seven LPAs, as well as tryptophan and six related metabolites, in combination with an LC-HRMS screening approach applying target lists for 665 lipids and 276 polar metabolites. Having evaluated acceptance criteria for each analyte, the datasets comprised 489 analytes in total. During this process, 468 analytes of the 1070 measured analytes were excluded from further evaluation due to values below the LLOQ (targeted methods) or threshold for signal intensity (screening methods), and 119 analytes were excluded due to missed acceptance criteria in the screening methods. For targeted LC-MS/MS analytics, 15 analytes missed the acceptance criteria and were evaluated as semi-quantitative data, but were not excluded from further evaluation. Detailed information on the analytes and corresponding results, including the data for assessing the acceptance criteria, can be found in the supplementary material.
The obtained data confirmed the considerable effects of different pre-analytical factors on K3EDTA plasma concentrations of various metabolites, including lipids and lipid mediators (Fig. 3). In some cases, the influences of ex vivo distortion affected whole lipid classes or groups simultaneously, as shown for ethanolamides (Fig. 4). In other cases, only single molecules were affected – for example, 12-HETE levels increased significantly compared to other hydroxyeicosatetraenoates (HETEs) included in the study when whole blood or plasma were stored at RT (Fig. 4). These results emphasize the need for careful sample handling in LC-MS-based clinical studies focusing on metabolites, including lipids and lipid mediators, as detailed and analyte-specific knowledge is necessary to ensure robust measurements and selection of an appropriate sampling protocol.
Fig. 3.

Fold changes in the abundance of 489 analytes during sample processing were analyzed. Each analyte is represented by a single column, depicting 16 analyte-specific fold changes from the inside to the outside of the plot. For simplification purposes, fold changes were capped at 50 % and 200 %, respectively. The circular heat map consists of four tracks, each representing a different pre-analytical condition. From the innermost to the outermost tracks, blood specimens were stored as plasma in ice water, plasma at RT, whole blood in ice water, and whole blood at RT. For each tested condition, four fold changes corresponding to four different time points (20, 60, 120, and 240 min) are depicted from the center to the periphery of each track. The heat map is encircled by a legend indicating the metabolite class or lipid species. Clockwise, the lysophospholipids include LPA (4), LPC O- (5), LPE (12), LPG (1), LPI (4) and LPC (19). The phospholipids encompass PE (27), PI (14) and PC (74). Finally, the sphingolipids are composed of sphingoid bases (3), ceramides (19) and sphingomyelins (40).
Fig. 4.

Comparison of analyte concentrations across multiple pre-analytical conditions was conducted, specifically focusing on the ethanolamides and structurally isomeric HETEs. The results indicated that the ethanolamides were similarly influenced by pre-analytical conditions across the whole group; however, the HETE analytes displayed much more diverse changes.
Notably, not all compounds were similarly or at all influenced by the same conditions; a large proportion of the investigated analytes remained stable even when stored at RT for the longest intermediate storage period of 240 min. Fig. 5 shows the number of unstable analytes when samples were handled according to the different protocols, grouped in accordance with the methods used and in total. An analyte was counted as unstable if the fold change of concentration compared to the baseline exceeded ± 20 % for the different experiments using whole blood or plasma. The 20 % threshold is a common acceptance measure for the relative deviation of QC samples in lipidomics [46] and was therefore used in this study.
Fig. 5.

Comparison of the total number of analytes with fold changes exceeding given acceptance criteria for different storage conditions was undertaken. The proportion of fold changes above ± 20 % for individual conditions and ± 30 % for combined conditions is indicated by the colors of the tiles. Numbers within the tiles represent the number of analytes with out-of-range concentration changes. The first four columns represent the tested conditions (combination of storage matrix, temperature, and duration). The last two columns display a combined evaluation of fold changes, depending on the combination of pre-analytical sample conditions (whole blood and plasma stored in ice water or at RT, respectively, for different periods of time).
We found that fewer changes in analyte concentrations occurred in blood samples that were centrifuged immediately after sampling and stored intermediately as plasma in ice water (Fig. 5). This supports the recommendation from an earlier publication [1], suggesting that, if the intermediate storage of blood samples between collection and deep freezing is unavoidable, immediate centrifugation and cooling of the samples is generally advisable. Under these conditions, only 21 analytes had concentrations that deviated by > 20 % in 240 min of storage in ice water. In contrast, when the plasma samples were stored for 240 min at RT, 67 analytes had concentrations that deviated by > 20 %. Notable increases were found among the endocannabinoids, LPAs and LPCs, aclycarnitines, oxylipins and some amino acids, including leucine and isoleucine. Furthermore, the concentration of some individual analytes (e.g., 1-lactoglobulin) nearly doubled when the plasma was stored at RT, indicating that major changes can occur due to changes in storage temperature.
Whereas only 21 analytes were unstable when samples were stored as plasma in ice water, this increased to 38 when the samples were stored as whole blood. Therefore, plasma provides greater stability than whole blood, however the effect of temperature is greater, with 67 analytes being unstable in RT plasma samples. It is therefore very important to cool blood samples immediately after drawing. Nonetheless, major fold changes of up to 500 % were observed for some analytes even in the cooled whole-blood samples, especially ethanolamines, acylcarnitines and metabolites such as nicotinamide and lactic acid. The most drastic fold changes in analyte concentrations (up to 700 %) were observed in whole blood samples stored at RT. After 240 min, 87 analytes were found to deviate by > 20 %, the most severely affected being sphingoid bases, endocannabinoids and LPA, which agrees with previously published studies [32], [48], [49].
To verify our findings, we compared our data with a lipid-focused study on the ex vivo distortion of analytes [26]. In order to assess the alignment in stability between the two studies, fold changes were calculated for the 195 intersecting analytes under conditions that were investigated in both studies. For each storage condition, analytes deviating by more than ± 20 % from the baseline were identified, and Cohen’s unweighted kappa index was calculated in order to estimate concordance between the study results [45]. Overall, our work supported the findings from the previous study regarding the instability of specific analytes, including the extreme vulnerability of endocannabinoids under all pre-analytical conditions, the increase in LPA concentrations in whole blood samples and plasma at RT, and the sensitivity of sphingosine phosphate concentrations in whole blood stored at RT. Especially in scenarios that exposed analyte instability (whole blood stored at RT), the concordance of the studies was substantial based on Cohen’s unweighted kappa ( = 0.701). For plasma samples stored at RT and whole blood samples stored on ice water, the interrater agreement was still very good ( = 0.556 and = 0.534). However, the measurements showed only slight agreement ( = 0.154) for samples stored as plasma on ice water. This reflects the fact that under these conditions, the overall stability of analytes is expected to be high, and any observed increases beyond fold change limits are much more likely to result from statistical fluctuations in measurement, leading in random differences between studies. Therefore, the overall findings concerning analytes and analyte groups that are prone to instability (e.g., endocannabinoids, LPA, sphingolipids) are in good accordance. However, when using Cohen’s unweighted kappa to compare situations where analytes are generally stable, incidental technical variance can obscure measurement concordance. The comparative data are summarized in the supplementary material.
Definition of viable pre-analytical sample handling protocols
The main objective of this study was to provide sample-handling protocols applicable in clinical settings that maximize the number of stable analytes. Fig. 5 summarizes the number of analytes that cannot be measured reliably when applying particular pre-analytical conditions as well as the combination of these conditions, combining data for intermediate storage of whole blood and plasma before the final storage of samples. In addition to the evaluation of storage as whole blood or plasma samples, an acceptable fold change of ± 30 % was used for the sum of two fold changes. The higher criterion was used to evaluate the combination of storing whole blood and plasma, representing the corresponding protocol, and can be justified by the need to combine multiple measurement results. Comparing the results for the strictest protocol ([I] 20 + 20 min cooled storage of whole blood/plasma – 28 unstable compounds/5.9 % of evaluated compounds) with the least strict protocol ([VIII] 240 + 240 min RT storage of whole blood/plasma – 113 unstable compounds/23.1 % of evaluated compounds), it becomes clear that 85 analytes cannot be measured due to the different pre-analytical sampling protocols, accounting for 17.3 % of the total number of evaluated analytes.
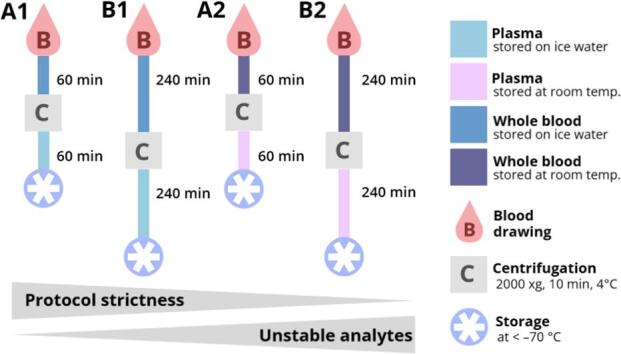
Considering that clinical application is as relevant as the number of compounds that can be measured, we compared two pre-analytical sampling protocols for clinical research projects, [I] (20 + 20 min cooled storage of whole blood/plasma) and [II] (60 + 60 min of cooled storage of whole blood/plasma). The less strict protocol (longer storage time) resulted in an additional 18 compounds becoming unstable (total = 46) but it was more feasible in a clinical setting. Therefore, protocol [II] was chosen as the strictest protocol (A1) for the final protocol recommendations shown in Fig. 6. Sample cooling may not be possible in all settings, so protocol [VI] (60 + 60 min non-cooled storage of whole blood/plasma) was defined as the next strictest protocol (A2). To reduce the number of protocols and keep the recommendations clear, the number of unstable analytes for the three remaining periods of intermediate storage was compared, regardless of the storage temperature. This revealed little difference between the storage of samples for 120 + 120 or 240 + 240 min, so protocols B1 and B2 were defined as those involving storage for 240 + 240 min in ice water or at RT, respectively (protocols [IV] and [VIII] in Fig. 5). Our recommendations therefore comprise a series of four protocols that differ in the level of strictness and feasibility for routine clinical practice. Because temperature is more important than storage duration in terms of analyte variability, strictness is ranked as follows: A1 > B1 > A2 > B2 (Fig. 6).
Fig. 6.

After filtering, 489 metabolites and lipids were evaluated for their stability. Pre-analytical protocols were compiled by comparing the fold changes of the individually tested conditions to baseline samples. The stability was assessed by evaluating if one of the two analyte-specific and condition-specific fold changes exceeded ± 20 % or if the sum of both exceeded ± 30 %. If an above-threshold fold change was observed for any analyte when using any given protocol, it was considered problematic for less strict protocols (e.g., longer preprocessing time, warmer storage temperature), and was counted as such. The figure illustrates the proportion of analytes deemed unstable for each of the protocols.
The high number of stable analytes, even after a total storage time of up to 8 h at RT, in combination with previous studies [26] indicating the stability of several lipids for up to 24 h, gives rise to the conclusion that an even less strict protocol, with prolonged delay until final storage, might be feasible for lipidomics and metabolomics analysis. However, highly relevant analytes, such as sphingosine phosphates, several oxylipins, acylcarnitines and LPA certainly require stricter protocols when of interest, due to their inherent pre-analytical instability. Analyte stability during longer time intervals was not investigated in this study and is an obvious limitation caused by the maximum number of blood collection tubes per participant and the focus on the rather fast sample processing with shorter delays. In order to avoid prolonged venous compression, as well as increasing time delays between the acquisition of multiple samples, the total number of tubes was limited to 17 and the investigation of very long storage delays was omitted. The testing of the suggested protocols, instead of evaluating the usability of the protocols by combining the results for whole blood and plasma storage, was not performed for the same reason and will be covered in a future study. Future studies will also include a larger number of volunteers covering a broader spectrum of cofactors such as age, body mass index and/or eating behaviors.
Analyte specific sampling protocol recommendations
Biomarker candidates should inherently reflect a clinical outcome in their concentration levels. In the search for clinical biomarkers relevant to disease, analytes of interest must be examined for their validity [50]. In this context, analytical validity relates to robustness of measurement, including repeatability and precision of the marker. Additionally, biomarkers should be highly specific to the observed outcome, while staying mostly unaffected by other influences such as temperature, storage conditions, and laboratory handling, among others [51]. The need for high reproducibility and low error rates pose strict restrictions on clinical biomarkers. In the context of hypothesis-driven investigations, the selection of pre-analytical protocols, or the evaluation of results from a completed study, might be necessary on an analyte-specific level. Therefore, every analyte was evaluated regarding pre-analytical vulnerability by assigning one of the four defined protocols as easy-to-use evaluation criteria. In order to assign individual analytes to one of the four proposed protocols (Fig. 6), we evaluated the two specific fold changes that are derived from concentration changes specific to the conditions represented by the respective protocols. If either of the two fold changes exceeded 20 % or the sum of both fold changes exceeded 30 % in either direction, the protocol was deemed unsuitable. Going from the strictest (A1), through B1 and A2 to the least strict protocol (B2), which would result in less demanding conditions for clinical personnel, each of the 489 evaluated compounds was assigned to the last protocol where the fold change did not exceed one of the defined thresholds. The least strict protocol (B2) is suitable for 377 of the 489 evaluated analytes. Increasing the requirements and accounting for potentially less stable analytes, protocol A2 accommodates an additional 51 analytes, including acylcarnitines (CAR 10:0 and CAR 16:0), which have been shown to indicate various metabolic deficiencies, as well as cardiopulmonary disease [52], and homocysteine, which has been associated with myocardial infarction, Alzheimer’s disease, and depression [48]. Protocol B1 introduces intermediate storage in ice water, which accommodates 24 additional analytes over A2, resulting in a total of 452 compounds. The analytes that require cooled intermediate storage include sphingoid bases, which have recently been linked to community-acquired pneumonia [53], LPAs, which are candidate biomarkers for gynecological cancers [54], [55] and rheumatic diseases such as arthritis [56] and chronic pain [57], as well lactate, which is a well-established biomarker [58]. The strictest conditions (protocol A1) are required for four additional analytes, including nicotinamide and hypoxanthine, a promising biomarker in sports medicine [59], [60]. However, some analytes (including endocannabinoids) cannot be measured within the defined thresholds even when applying the strictest protocol. Faster sample processing is generally unfeasible in clinical research, so the use of these analytes in K3EDTA plasma is unreliable and the data should be interpreted with caution, as previously discussed [61]. Detailed information for suggested pre-analytical protocols for every analyte evaluated in this study can be found in the supplementary material.
Given that clinical research is often based on existing samples, for example those stored in biobanks, it is important to evaluate the usability of such samples for lipidomics and metabolomics analysis. Therefore, the four different protocols provide orientation to evaluate the stability of analytes or analyte groups in biobank samples. Although not necessarily designed with metabolomics or lipidomics in mind, biobanks usually adhere to sample protocols, which can vary depending on the institution. Examples of biobank sampling requirements can be found in the literature [49], where biobanks A and B process their blood samples from collection through plasma sample processing at 4 °C until sample freezing within 2 h. Compared to protocol A1 (60 + 60 min processing time on ice water = 2 h) most metabolites and lipids in these samples can be assumed to remain stable, with the exception of endocannabinoids in general, specific oxylipins such as 5,6-EpETrE and 9-HOTrE, and specific acylcarnitines such as CAR 14:1 and CAR 18:0. Biobank C, described in the same publication [49], reportedly processed their samples after 4–9 h. Assessing this sample-handling procedure with protocol B1 (240 + 240 min processing time on ice water = 8 h), the stability of more analytes and analyte classes is affected unequivocally, including tryptophan-related metabolites such as 3-OH-Ana and nicotinamide, as well as certain fatty acids (e.g., FA 16:1, FA 20:5 and FA 24:1). When intermediate storage is assumed to take place at RT, many different analyte classes become unstable, including oxylipins (e.g., 12-HETE, 20-HETE and 22-HDHA), acylcarnitines (e.g., CAR 16:0, CAR 18:1 and CAR 20:4), essential and non-essential amino acids (e.g., Glu, Ile, Orn and 4-oxoproline), other polar metabolites (e.g., xanthine and orotidine), and almost all LPAs. These sample handling recommendations illustrate the usefulness of the data presented herein and offer protocols to evaluate the usability of samples from biobanks A, B and C to improve confidence in lipidomics and metabolomics data.
Conclusion
LC-MS-based clinical research focusing on metabolites, including lipids and lipid mediators, is a promising approach for the development of new biomarkers. However, non-standardized pre-analytical sample handling hinders the reproducibility and wider application of these analyses in clinical research, thereby preventing their translation from clinical research projects into routine clinical practice. The main objective of this study was to define pre-analytical sample handling protocols that ensure the stability of a maximum number of analytes and are feasible for implementation in the clinic. After evaluating different pre-analytical conditions for a total of 489 metabolites, including lipids and lipid mediators, four protocols were recommended with varying strictness levels in terms of storage temperature and sample processing duration. Our results confirm that pre-analytics has a major impact on several analytes or analyte groups and support the conclusions of previous studies, but also reveal that many compounds remain stable even when K3EDTA whole-blood samples are processed at RT and stored for up to 4 h before and after centrifugation. These findings can be used to plan prospective sampling in clinical research projects and to estimate analyte stability in retrospective samples. Furthermore, they enable the assessment of whether lipidomics or metabolomics data are reliable or potentially influenced by pre-analytical sample handling due to analyte-specific vulnerability.
Declaration of Competing Interest
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
Acknowledgements
This work was supported by the Deutsche Forschungsgemeinschaft Sonderforschungsbereich SFB 1039/Z01 “Krankheitsrelevante Signaltransduktion durch Fettsäurederivate und Sphingolipide”, by Fraunhofer Cluster of Excellence for Immune Mediated diseases CIMD and the HIPPOCRATES project. HIPPOCRATES has received funding from the Innovative Medicines Initiative 2 Joint Undertaking (JU) under grant agreement no. 101007757. The JU receives support from the European Union’s Horizon 2020 research and innovation program and EFPIA. The HRMS instrument was co-funded by the Deutsche Forschungsgemeinschaft (DFG, German Research Foundation) no. 445757098. The authors would like to thank Richard M. Twyman, Daniel Kratz and Olivia Engelbach for their careful proofreading of the manuscript as well as Sandra Trautmann, Carlo Angioni, Yannick Schreiber and Viktoria Wagner for technical support in performing the experiments.
Footnotes
Peer review under responsibility of “MSACL”.
Supplementary data to this article can be found online at https://doi.org/10.1016/j.jmsacl.2023.02.002.
Appendix A. Supplementary data
The following are the Supplementary data to this article:
References
- 1.Yang K., Han X. Lipidomics: Techniques, Applications, and Outcomes Related to Biomedical Sciences. Trends Biochem. Sci. 2016;41:954–969. doi: 10.1016/j.tibs.2016.08.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Gonzalez-Covarrubias V., Martínez-Martínez E., Del Bosque-Plata L. The Potential of Metabolomics in Biomedical Applications. Metabolites. 2022;12 doi: 10.3390/metabo12020194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Sethi S., Brietzke E. Recent advances in lipidomics: Analytical and clinical perspectives. Prostaglandins Other Lipid Mediat. 2017;128–129:8–16. doi: 10.1016/j.prostaglandins.2016.12.002. [DOI] [PubMed] [Google Scholar]
- 4.Ishikawa M., Maekawa K., Saito K., Senoo Y., Urata M., Murayama M., Tajima Y., Kumagai Y., Saito Y. Plasma and serum lipidomics of healthy white adults shows characteristic profiles by subjects' gender and age. PLoS One. 2014;9:e91806. doi: 10.1371/journal.pone.0091806. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Kontush A., Lhomme M., Chapman M.J. Unraveling the complexities of the HDL lipidome. J. Lipid Res. 2013;54:2950–2963. doi: 10.1194/jlr.R036095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Naz S., Moreira dos Santos D.C., García A., Barbas C. Analytical protocols based on LC-MS, GC-MS and CE-MS for nontargeted metabolomics of biological tissues. Bioanalysis. 2014;6:1657–1677. doi: 10.4155/bio.14.119. [DOI] [PubMed] [Google Scholar]
- 7.Dorochow E., Köhm M., Hahnefeld L., Gurke R. Metabolic Profiling in Rheumatoid Arthritis, Psoriatic Arthritis, and Psoriasis: Elucidating Pathogenesis, Improving Diagnosis, and Monitoring Disease Activity. J. Pers. Med. 2022;12 doi: 10.3390/jpm12060924. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Liebisch G., Fahy E., Aoki J., Dennis E.A., Durand T., Ejsing C.S., Fedorova M., Feussner I., Griffiths W.J., Köfeler H., Merrill A.H., Murphy R.C., O'Donnell V.B., Oskolkova O., Subramaniam S., Wakelam M.J.O., Spener F. Update on LIPID MAPS classification, nomenclature, and shorthand notation for MS-derived lipid structures. J. Lipid Res. 2020;61(12):1539–1555. doi: 10.1194/jlr.S120001025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Burla B., Arita M., Arita M., Bendt A.K., Cazenave-Gassiot A., Dennis E.A., Ekroos K., Han X., Ikeda K., Liebisch G., Lin M.K., Loh T.P., Meikle P.J., Orešič M., Quehenberger O., Shevchenko A., Torta F., Wakelam M.J.O., Wheelock C.E., Wenk M.R. MS-based lipidomics of human blood plasma: a community-initiated position paper to develop accepted guidelines. J. Lipid Res. 2018;59:2001–2017. doi: 10.1194/jlr.S087163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Lehmann R. From bedside to bench-practical considerations to avoid pre-analytical pitfalls and assess sample quality for high-resolution metabolomics and lipidomics analyses of body fluids. Anal. Bioanal. Chem. 2021 doi: 10.1007/s00216-021-03450-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Kirwan J.A., Brennan L., Broadhurst D., Fiehn O., Cascante M., Dunn W.B., Schmidt M.A., Velagapudi V. Preanalytical Processing and Biobanking Procedures of Biological Samples for Metabolomics Research: A White Paper, Community Perspective (for “Precision Medicine and Pharmacometabolomics Task Group”-The Metabolomics Society Initiative) Clin. Chem. 2018;64:1158–1182. doi: 10.1373/clinchem.2018.287045. [DOI] [PubMed] [Google Scholar]
- 12.Hyötyläinen T., Orešič M. Bioanalytical techniques in nontargeted clinical lipidomics. Bioanalysis. 2016;8:351–364. doi: 10.4155/bio.15.244. [DOI] [PubMed] [Google Scholar]
- 13.Liebisch G. Biochimica et biophysica acta BBA. 2017;62:636. doi: 10.1016/0006-3002(62)90265-2. [DOI] [Google Scholar]
- 14.Lukowski J.K., Pamreddy A., Velickovic D., Zhang G., Pasa-Tolic L., Alexandrov T., Sharma K., Anderton C.R. Storage Conditions of Human Kidney Tissue Sections Affect Spatial Lipidomics Analysis Reproducibility. J. Am. Soc. Mass Spectrom. 2020;31(12):2538–2546. doi: 10.1021/jasms.0c00256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Castro-Castro M.-J., Candás-Estébanez B., Esteban-Salán M., Calmarza P., Arrobas-Velilla T., Romero-Román C., Pocoví-Mieras M., Aguilar-Doreste J.-Á. Removing Lipemia in Serum/Plasma Samples: A Multicenter Study. Ann. Lab. Med. 2018;38(6):518–523. doi: 10.3343/alm.2018.38.6.518. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Simundic A.M. Standardization of collection requirements for fasting samples For the Working Group on Preanalytical Phase (WG-PA) of the European Federation of Clinical Chemistry and Laboratory Medicine (EFLM) Clin. Chim. Acta. 2013;50:160. doi: 10.1016/0009-8981(74)90091-6. [DOI] [PubMed] [Google Scholar]
- 17.Pedersen B.K. Exercise and cytokines. Immunol. Cell Biol. 2000;78(5):532–535. doi: 10.1111/j.1440-1711.2000.t01-11-.x. [DOI] [PubMed] [Google Scholar]
- 18.Pietzner M., Stewart I.D., Raffler J., Khaw K.-T., Michelotti G.A., Kastenmüller G., Wareham N.J., Langenberg C. Plasma metabolites to profile pathways in noncommunicable disease multimorbidity. Nat. Med. 2021;27:471–479. doi: 10.1038/s41591-021-01266-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Sprenger R.R., Hermansson M., Neess D., Becciolini L.S., Sørensen S.B., Fagerberg R., Ecker J., Liebisch G., Jensen O.N., Vance D.E., Færgeman N.J., Klemm R.W., Ejsing C.S. Lipid molecular timeline profiling reveals diurnal crosstalk between the liver and circulation. Cell Rep. 2021;34 doi: 10.1016/j.celrep.2021.108710. [DOI] [PubMed] [Google Scholar]
- 20.Kratz D., Sens A., Schäfer S.M.G., Hahnefeld L., Geisslinger G., Thomas D., Gurke R. Pre-analytical challenges for the quantification of endocannabinoids in human serum. J. Chromatogr. B Anal. Technol. Biomed. Life Sci. 2022;1190:123102. doi: 10.1016/j.jchromb.2022.123102. [DOI] [PubMed] [Google Scholar]
- 21.Jonasdottir H.S., Brouwers H., Toes R.E.M., Ioan-Facsinay A., Giera M. Effects of anticoagulants and storage conditions on clinical oxylipid levels in human plasma. Biochim. Biophys. Acta Mol. Cell Biol. Lipids. 1863;2018:1511–1522. doi: 10.1016/j.bbalip.2018.10.003. [DOI] [PubMed] [Google Scholar]
- 22.Brunkhorst R., Pfeilschifter W., Patyna S., Büttner S., Eckes T., Trautmann S., Thomas D., Pfeilschifter J., Koch A. Preanalytical Biases in the Measurement of Human Blood Sphingolipids. Int. J. Mol. Sci. 2018;19 doi: 10.3390/ijms19051390. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Fliniaux O., Gaillard G., Lion A., Cailleu D., Mesnard F., Betsou F. Influence of common preanalytical variations on the metabolic profile of serum samples in biobanks. J. Biomol. NMR. 2011;51:457–465. doi: 10.1007/s10858-011-9574-5. [DOI] [PubMed] [Google Scholar]
- 24.S. Cuhadar, M. Koseoglu, A. Atay, A. Dirican, The effect of storage time and freeze-thaw cycles on the stability of serum samples, Biochem. Med. (Zagreb) 23 (2013) 70–77. 10.11613/bm.2013.009. [DOI] [PMC free article] [PubMed]
- 25.Lippi G., Chance J.J., Church S., Dazzi P., Fontana R., Giavarina D., Grankvist K., Huisman W., Kouri T., Palicka V., Plebani M., Puro V., Salvagno G.L., Sandberg S., Sikaris K., Watson I., Stankovic A.K., Simundic A.-M. Preanalytical quality improvement: from dream to reality. Clin. Chem. Lab. Med. 2011;49:1113–1126. doi: 10.1515/CCLM.2011.600. [DOI] [PubMed] [Google Scholar]
- 26.Hahnefeld L., Gurke R., Thomas D., Schreiber Y., Schäfer S.M.G., Trautmann S., Snodgrass I.F., Kratz D., Geisslinger G., Ferreirós N. Implementation of lipidomics in clinical routine: Can fluoride/citrate blood sampling tubes improve preanalytical stability? Talanta. 2020;209 doi: 10.1016/j.talanta.2019.120593. [DOI] [PubMed] [Google Scholar]
- 27.Cao Z., Kamlage B., Wagner-Golbs A., Maisha M., Sun J., Schnackenberg L.K., Pence L., Schmitt T.C., Daniels J.R., Rogstad S., Beger R.D., Yu L.-R. An Integrated Analysis of Metabolites, Peptides, and Inflammation Biomarkers for Assessment of Preanalytical Variability of Human Plasma. J. Proteome Res. 2019;18:2411–2421. doi: 10.1021/acs.jproteome.8b00903. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Kamlage B., Maldonado S.G., Bethan B., Peter E., Schmitz O., Liebenberg V., Schatz P. Quality markers addressing preanalytical variations of blood and plasma processing identified by broad and targeted metabolite profiling. Clin. Chem. 2014;60:399–412. doi: 10.1373/clinchem.2013.211979. [DOI] [PubMed] [Google Scholar]
- 29.Bervoets L., Louis E., Reekmans G., Mesotten L., Thomeer M., Adriaensens P., Linsen L. Influence of preanalytical sampling conditions on the 1H NMR metabolic profile of human blood plasma and introduction of the Standard PREanalytical Code used in biobanking. Metabolomics. 2015;11:1197–1207. doi: 10.1007/s11306-015-0774-y. [DOI] [Google Scholar]
- 30.Fantz C.R., Greene D.N. Where Are the Preanalytical Stability Standards? J. Appl. Lab. Med. 2018;2:830–832. doi: 10.1373/jalm.2018.026062. [DOI] [PubMed] [Google Scholar]
- 31.Onorato J.M., Shipkova P., Minnich A., Aubry A.-F., Easter J., Tymiak A. Challenges in accurate quantitation of lysophosphatidic acids in human biofluids. J. Lipid Res. 2014;55:1784–1796. doi: 10.1194/jlr.D050070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Gurke R., Thomas D., Schreiber Y., Schäfer S.M.G., Fleck S.C., Geisslinger G., Ferreirós N. Determination of endocannabinoids and endocannabinoid-like substances in human K3EDTA plasma - LC-MS/MS method validation and pre-analytical characteristics. Talanta. 2019;204:386–394. doi: 10.1016/j.talanta.2019.06.004. [DOI] [PubMed] [Google Scholar]
- 33.Spohner A.K., Jakobi K., Trautmann S., Thomas D., Schumacher F., Kleuser B., Lütjohann D., El-Hindi K., Grösch S., Pfeilschifter J., Saba J.D., D. Meyer Zu Heringdorf, Mouse Liver Compensates Loss of Sgpl1 by Secretion of Sphingolipids into Blood and Bile. Int. J. Mol. Sci. 2021;22 doi: 10.3390/ijms221910617. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Kind T., Liu K.-H., Lee D.Y., DeFelice B., Meissen J.K., Fiehn O. LipidBlast in silico tandem mass spectrometry database for lipid identification. Nat. Methods. 2013;10:755–758. doi: 10.1038/nmeth.2551. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Dieterle F., Ross A., Schlotterbeck G., Senn H. Probabilistic Quotient Normalization as Robust Method to Account for Dilution of Complex Biological Mixtures. Application in 1 H NMR Metabonomics. Anal. Chem. 2006;78(13):4281–4290. doi: 10.1021/ac051632c. [DOI] [PubMed] [Google Scholar]
- 36.Wickham H, readxl: Read Excel Files: https://readxl.tidyverse.org, https://github.com/tidyverse/readxl. (2022).
- 37.Hadley Wickham, Romain François, Lionel Henry, Kirill Müller, dplyr: A Grammar of Data Manipulation.
- 38.H. Wickham, M. Averick, J. Bryan, W. Chang, L. McGowan, R. François, G. Grolemund, A. Hayes, L. Henry, J. Hester, M. Kuhn, T. Pedersen, E. Miller, S. Bache, K. Müller, J. Ooms, D. Robinson, D. Seidel, V. Spinu, K. Takahashi, D. Vaughan, C. Wilke, K. Woo, H. Yutani, Welcome to the Tidyverse, JOSS 4 (2019) 1686. 10.21105/joss.01686.
- 39.Wickham H. Reshaping Data with the reshape Package. J. Stat. Softw. 2007;21(12):1–20. http://www.jstatsoft.org/v21/i12/ [Google Scholar]
- 40.Wickham H. Springer-Verlag; New York: 2016. ggplot2: Elegant Graphics for Data Analysis. ISBN 978-3-319-24277-4, [Google Scholar]
- 41.Warnes G.R., Bolker B., Bonebakker L., Gentleman R., Huber W., Liaw A., Lumley T., Maechler M., Magnusson A., Moeller S., Schwartz M., Venables B. gplots: Various R Programming Tools for Plotting Data. R package version. 2022;3(1):3. https://CRAN.R-project.org/package=gplots [Google Scholar]
- 42.Z. Gu, R. Eils, M. Schlesner, Complex heatmaps reveal patterns and correlations in multidimensional genomic data, Bioinformatics 32 (2016) 2847–2849. 10.1093/bioinformatics/btw313. [DOI] [PubMed]
- 43.Matthias Gamer, <puspendra.pusp22@gmail.com> (2019). irr: Various Coefficients of Interrater Reliability and Agreement. R package version 0.84.1. https://CRAN.R-project.org/package=irr.
- 44.Gu Z., Gu L., Eils R., Schlesner M., Brors B. circlize Implements and enhances circular visualization in R. Bioinformatics. 2014;30:2811–2812. doi: 10.1093/bioinformatics/btu393. [DOI] [PubMed] [Google Scholar]
- 45.Gisev N., Bell J.S., Chen T.F. Interrater agreement and interrater reliability: key concepts, approaches, and applications. Res. Soc. Adm. Pharm. 2013;9:330–338. doi: 10.1016/j.sapharm.2012.04.004. [DOI] [PubMed] [Google Scholar]
- 46.Ghorasaini M., Mohammed Y., Adamski J., Bettcher L., Bowden J.A., Cabruja M., Contrepois K., Ellenberger M., Gajera B., Haid M., Hornburg D., Hunter C., Jones C.M., Klein T., Mayboroda O., Mirzaian M., Moaddel R., Ferrucci L., Lovett J., Nazir K., Pearson M., Ubhi B.K., Raftery D., Riols F., Sayers R., Sijbrands E.J.G., Snyder M.P., Su B., Velagapudi V., Williams K.J., de Rijke Y.B., Giera M. Cross-Laboratory Standardization of Preclinical Lipidomics Using Differential Mobility Spectrometry and Multiple Reaction Monitoring. Anal. Chem. 2021;93:16369–16378. doi: 10.1021/acs.analchem.1c02826. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Heiling S., Knutti N., Scherr F., Geiger J., Weikert J., Rose M., Jahns R., Ceglarek U., Scherag A., Kiehntopf M. Metabolite Ratios as Quality Indicators for Pre-Analytical Variation in Serum and EDTA Plasma. Metabolites. 2021;11 doi: 10.3390/metabo11090638. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Yin P., Lehmann R., Xu G. Effects of pre-analytical processes on blood samples used in metabolomics studies. Anal. Bioanal. Chem. 2015;407:4879–4892. doi: 10.1007/s00216-015-8565-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Liu X., Hoene M., Yin P., Fritsche L., Plomgaard P., Hansen J.S., Nakas C.T., Niess A.M., Hudemann J., Haap M., Mendy M., Weigert C., Wang X., Fritsche A., Peter A., Häring H.-U., Xu G., Lehmann R. Quality Control of Serum and Plasma by Quantification of (4E,14Z)-Sphingadienine-C18-1-Phosphate Uncovers Common Preanalytical Errors During Handling of Whole Blood. Clin. Chem. 2018;64:810–819. doi: 10.1373/clinchem.2017.277905. [DOI] [PubMed] [Google Scholar]
- 50.Hayes D.F. Biomarker validation and testing. Mol. Oncol. 2015;9:960–966. doi: 10.1016/j.molonc.2014.10.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.R.L. Holland, What makes a good biomarker?, Adv Precis Med 1 (2016) 66. 10.18063/APM.2016.01.007.
- 52.Dambrova M. Acylcarnitines: Nomenclature, Biomarkers, Therapeutic Potential, DrugTargets, andClinical Trials. Pharmacol. Rev. 2022;74 doi: 10.1124/pharmrev.121.000408. [DOI] [PubMed] [Google Scholar]
- 53.Hsu S.-C., Chang J.-H., Hsu Y.-P., Bai K.-J., Huang S.-K., Hsu C.-W. Circulating sphingosine-1-phosphate as a prognostic biomarker for community-acquired pneumonia. PLoS One. 2019;14:e0216963. doi: 10.1371/journal.pone.0216963. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Yagi T., Shoaib M., Kuschner C., Nishikimi M., Becker L.B., Lee A.T., Kim J. Challenges and Inconsistencies in Using Lysophosphatidic Acid as a Biomarker for Ovarian Cancer. Cancers (Basel) 2019;11 doi: 10.3390/cancers11040520. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Brian De La Franier and Michael Thompson, Detection of the Ovarian Cancer Biomarker Lysophosphatidic Acid in Serum. [DOI] [PMC free article] [PubMed]
- 56.Orosa B., García S., Conde C. The autotaxin-lysophosphatidic acid pathway in pathogenesis of rheumatoid arthritis. Eur. J. Pharmacol. 2015;765:228–233. doi: 10.1016/j.ejphar.2015.08.028. [DOI] [PubMed] [Google Scholar]
- 57.Ueda H. LPA receptor signaling as a therapeutic target for radical treatment of neuropathic pain and fibromyalgia. Pain Manag. 2020;10:43–53. doi: 10.2217/pmt-2019-0036. [DOI] [PubMed] [Google Scholar]
- 58.Adeva-Andany M. Comprehensive review on lactate metabolism in human health. Mitochondrion. 2014 doi: 10.1016/j.mito.2014.05.007. [DOI] [PubMed] [Google Scholar]
- 59.Farthing D.E., Farthing C.A., Xi L. Inosine and hypoxanthine as novel biomarkers for cardiac ischemia: from bench to point-of-care. Exp. Biol. Med. (Maywood) 2015;240:821–831. doi: 10.1177/1535370215584931. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Mahanty A., Xi L. Utility of cardiac biomarkers in sports medicine: Focusing on troponin, natriuretic peptides, and hypoxanthine. Sports Med. Health Sci. 2020;2:65–71. doi: 10.1016/j.smhs.2020.05.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Kratz D., Thomas D., Gurke R. Endocannabinoids as potential biomarkers: It's all about pre-analytics. J. Mass Spectrom. Adv. Clin. Lab. 2021;22:56–63. doi: 10.1016/j.jmsacl.2021.11.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.