

Abstract

A comprehensive understanding of the ligand field and its influence on the degeneracy and population of d-orbitals in a specific coordination environment are crucial for the rational design and enhancement of magnetic anisotropy of single-ion magnets (SIMs). Herein, we report the synthesis and comprehensive magnetic characterization of a highly anisotropic CoII SIM, [L2Co](TBA)2 (L is an N,N′-chelating oxanilido ligand), that is stable under ambient conditions. Dynamic magnetization measurements show that this SIM exhibits a large energy barrier to spin reversal Ueff > 300 K and magnetic blocking up to 3.5 K, and the property is retained in a frozen solution. Low-temperature single-crystal synchrotron X-ray diffraction used to determine the experimental electron density gave access to Co d-orbital populations and a derived Ueff, 261 cm–1, when the coupling between the dx2 – y2 and dxy orbitals is taken into account, in very good agreement with ab initio calculations and superconducting quantum interference device results. Powder and single-crystal polarized neutron diffraction (PNPD, PND) have been used to quantify the magnetic anisotropy via the atomic susceptibility tensor, revealing that the easy axis of magnetization is pointing along the N–Co–N′ bisectors of the N,N′-chelating ligands (3.4° offset), close to the molecular axis, in good agreement with complete active space self-consistent field/N-electron valence perturbation theory to second order ab initio calculations. This study provides benchmarking for two methods, PNPD and single-crystal PND, on the same 3d SIM, and key benchmarking for current theoretical methods to determine local magnetic anisotropy parameters.
Keywords: cobalt single-ion magnet, magnetic anisotropy, single-crystal polarized neutron diffraction, experimental electron density, polarized neutron powder diffraction, magnetic blocking, single-crystal synchrotron X-ray diffraction
Introduction
The rapid growth of the digital ecosystem in recent decades has made the need for powerful processors and high-density data storage systems more apparent, and single-molecule magnets (SMMs) are considered as having great potential in this regard.1−6 The last three decades have seen tremendous progress in this field, both on the synthetic side as well as in the development of computational methods and new advanced spectroscopic techniques that provide a direct correlation between experimental findings and theoretical models.7−15 In recent years, some excellent high-performance 3d- and 4f-ion-based SMMs have been reported that possess a very high effective energy barrier for magnetization reversal (Ueff) and blocking temperature (TB).16−25 In the case of SMMs based on 3d metal ions, magnetic bistability originates from an energy barrier (Ueff) that in turn results from the zero-field splitting (ZFS) of the ground spin multiplet. Current synthetic strategies thus target systems that minimize quenching of the angular momentum L, usually via ligand field design that preserves degeneracy of the (dx2 – y2, dxy) and/or (dxz, dyz) orbital pairs. Even in the case of largely quenched orbital angular momentum, however, sizable ZFS may result from spin–orbit coupling (SOC). Here, the axial ZFS parameter (D) and the total ground spin-state (S) define the energy barrier for a 3d SMM with Ueff = |D|S2 or Ueff = |D|(S2 – 0.25) for integer and non-integer spin systems, respectively.
Among the 3d transition metals, CoII ions are particularly exploited for the development of single-ion magnets (SIMs).26−28 The main emphasis is to increase the effect of SOC to enhance ZFS by designing systems with a defined geometry and coordination environment to achieve quasi-degenerate sets of orbitals.26 Prominent examples are two-coordinate nearly linear d7 complexes such as [FeI(C(SiMe3)3)2]− and [(sIPr)CoIINDmp] which feature a large orbital angular momentum contribution or the linear dialkyl complex CoII(C(SiMe2ONaph)3)2 exhibiting a limiting magnetic anisotropy for a CoII ion with a non-Aufbau ground state L = 3.16−18 However, the highly reactive nature and low stability of these complexes render them impractical for, e.g., anchoring on surfaces and for any device fabrication process. On the other hand, some four, six, and eight coordinate CoII complexes have been shown to serve as rugged and air-stable SIMs with moderate and high energy barriers;19,29−41 however, only a few of these CoII SIMs have been reported to exhibit slow relaxation dynamics in the absence of an applied magnetic field (see Table S5), the first reported one being the highly air-sensitive complex (Ph4P)2[Co(SPh)4].42
While beneficial design guidelines for conceiving SIMs with higher operating temperatures have emerged, further progress is still hampered by the difficulty of experimentally accessing the molecular magnetic susceptibility tensor, and by a limited understanding of the effects of local molecular and lattice vibrations and hyperfine coupling effects, etc., on the dynamics of the relaxation mechanism.43,44 Common theoretical methods still have their limitations as they mostly exclude various intermolecular interactions and packing effects in the crystalline state to avoid overparameterization, whereas experimental parameters obtained from superconducting quantum interference device (SQUID) magnetometry include the entirety of effects operative in the solid state. To obtain more precise local magnetic anisotropy parameters, sophisticated techniques such as low-temperature high-field high-frequency electron paramagnetic resonance (EPR), X-band EPR, variable field far-infrared (IR) spectroscopy, nuclear magnetic resonance (NMR) paramagnetic shift, and torque magnetometry have been used.9,11,13 These techniques not only enable direct estimation of the magnitude and sign of D but simultaneously allow for cross-verification of the theoretical models used.
In addition to the above-mentioned spectroscopic techniques, low-temperature single-crystal synchrotron X-ray diffraction has been employed to model an atom-centered experimental electron density (EED).7 EED studies have been recently applied in the field of molecular magnetism to obtain the experimental d-orbital population in SMMs.7,45 The d-orbital populations not only give an idea about the ligand field splitting but also a direct estimation of the ZFS. The strengths of the EED approach have also recently been exploited to quantify the oblateness of the 4f-valence EED in a DyIII SIM, and thus the magnetic easy axis as well as an estimate of the MJ-composition of the electronic ground state.8 In addition to EED studies, polarized neutron diffraction (PND) provides local atomic magnetic susceptibility tensors and the direction of the easy-axis or easy-plane with respect to the local coordination environment. This has been utilized in a few cases to study the correlation of magnetic anisotropy with the local coordination structure.12,46−50 The major drawback of this method is the requirement of +1 mm3 single crystals suitable for diffraction. However, it has recently been shown by some of us that the atomic susceptibility tensor can also be obtained reliably from powder PND measurements,46,51,52 substantially advancing this method as it is now pertinent to nearly all materials. These experimentally observable parameters also give confidence and reliability to the results obtained from theoretical calculations that are crucial for making significant progress toward the realization of high-performance SMMs. In particular, the magnetic easy axes derived from interpreting low-temperaturehigh-resolution synchrotron X-ray diffraction in two DyIII SIMs were found to deviate by only 13.1 and 8.7° compared to those obtained by ab initio calculations.8
In this report, we introduce an air-stable dianionic CoII complex derived from an N,N′-chelating bis(4-chlorophenyl)oxanilido ligand (H2L),53 [L2Co](TBA)2 (1), which exhibits a very high energy barrier for magnetization reversal under zero magnetic field and displays magnetic blocking up to 3.5 K. This new complex has been magnetically characterized both in solid state and frozen solution by direct current (dc) and alternating current (ac) magnetometry. A bouquet of advanced experimental and computational techniques including low-temperature single-crystal synchrotron X-ray diffraction with multipole refinement of the electron density (ED), powder PND, single-crystal PND, and ab initio calculations have then been applied to interrogate the signatures and the origin of the large magnetic anisotropy of 1. This study demonstrates that polarized neutron powder diffraction can be very useful to benchmark the local magnetic anisotropy of a CoII SIM, opening a way to bypass the requirement of large single crystals.
Results and Discussion
Synthesis, Characterization, and Molecular Structure
The CoII complex [L2Co](TBA)2 (1) and its ZnII analogue [L2Zn](TBA)2 (2) were synthesized by the reaction of anhydrous CoCl2 or ZnCl2, respectively, with the dipotassium salt of the ligand (H2L) under inert dinitrogen atmosphere and subsequent cation exchange with tetrabutylammonium bromide (TBABr; Scheme S1). Large block-shaped crystals were grown by slow diffusion of diethyl ether into MeCN solutions of the products. Complexes 1 and 2 were found to be stable under ambient aerobic conditions in the solid state for months without any significant decomposition.
After initial structure elucidation by standard single-crystal X-ray diffraction (see the SI for details), low-temperature high-resolution single-crystal X-ray synchrotron diffraction data of complex 1 were recorded at 25 K. 1 crystallizes in the triclinic space group P1̅ with the CoII ion in a distorted tetrahedral {N4} coordination environment with two dianionic ligands close to an orthogonal arrangement (Figure 1). The charge of the [L2Co]2– dianion is balanced by two TBA cations. The Co–N bond lengths lie in the narrow range 1.9766(4)–1.9969(5) Å. The N(1)–Co–N(2) and N(3)–Co–N(4) bite angles of the chelating oxanilido ligands [83.88(2) and 83.63(2)°] are considerably smaller than the other four N–Co–N angles [120.23(2)–129.50(2)°], thus giving rise to an approximate D2d symmetry or elongated tetrahedron (τ4 = 0.756, Table S9).54 The dihedral angle between the chelating N–Co–N planes is 87.88°, close to an orthogonal situation (Figure 1). The nearest metal ions in the lattice are separated by 11.82 Å. The isomorphous ZnII complex (2) displays similar structural features as complex 1 (see the SI for details). Selected bond lengths and angles of complexes 1 and 2 are listed in Table S10.
Figure 1.

Molecular structure of the anion of 1 at 25 K (90% probability ellipsoids); hydrogen atoms, disordered parts, and cations have been omitted for clarity. The inset shows the nearly orthogonal arrangement of the Co-NCCN metallacycles.
The Vis–NIR spectrum of solid 1 reveals apparent absorption bands at 529 and 566 nm as well as a broad absorption extending from 800 to 1600 nm, centered around 1120 nm (Figure S8). In an environment of ideal Td symmetry, CoII ions with a4A ground state feature three spin-allowed transitions to the 4T2, 4T1(4F), and 4T1(4P) excited states. However, as evident from the crystal structure (Figure 1 inset and Table S9), the {N4} coordination sphere of the CoII ion in 1 is tetragonally elongated away from Td toward D2d symmetry, giving rise to multiple transitions that lead to broad absorption bands.32 The Vis–NIR spectrum of 1 in MeCN (Figure S9) shows similar bands at 530 and 568 as well as the broad feature centered around 1130 nm, indicating that the molecular structure is retained in solution. Rather low extinction coefficients (ε < 800 M–1 cm–1) corroborate that the absorption bands in the visible and NIR region originate from d–d transitions.
Magnetic Properties of 1
The static magnetic properties of complex 1 have been studied using a polycrystalline powdered sample covered with low-viscosity inert oil to prevent any torquing in the magnetic field. At 200 K, the χMT value of 3.16 cm3 K mol–1 is higher than the spin-only value of one isolated CoII ion (S = 3/2, g = 2.0; χMT = 1.875 cm3 K mol–1), evidencing significant orbital contributions to the magnetic moment (Figure 2). The χMT value decreases slowly upon lowering the temperature before decreasing more sharply below 6 K to finally reach a value of 2.32 cm3 K mol–1 at 2 K; the latter decrease indicates the presence of substantial magnetic anisotropy. The magnetization of 1 at 2.0 K rises steeply with magnetic fields up to 1.5 T before showing a more gradual further increase until 7.0 T, finally reaching 2.31 μB without complete saturation (Figure S15). This value is well below the expected Msat value of 3.0 μB for an isolated isotropic S = 3/2 system.
Figure 2.

Temperature dependence of χMT for 1 measured under an applied dc field of 0.5 T. The black solid line is the best fit to the spin Hamiltonian in eq 1. Inset: Zero field-cooled (ZFC) and field-cooled (FC) magnetic susceptibility measurements showing clear blocking at ca. 3.5 K for 1. The solid lines are only a guide to the eye.
To gain further insight, the magnetic susceptibility and variable-temperature variable-field magnetization data (Figure S14) were simultaneously simulated with the program julX_2s55 using the spin Hamiltonian in eq 1
| 1 |
The best fit was obtained for D = −113.0 cm–1, gx = gy = 2.25, and gz = 3.01. The large negative D indicates the presence of an easy axis of magnetic anisotropy and suggests an energy separation of ca. 226 cm–1 (325 K, |2D|) between the ground state mS = ±3/2 Kramers doublet (KD) and the excited state mS = ±1/2 KD. An ab initio theoretical calculation performed with the complete active space self-consistent field (CASSCF)/N-electron valence perturbation theory to second order (NEVPT2) method and def2-TZVP basis-sets using ORCA software56,57 and crystallographic atom coordinates give values of D = −118.1 cm–1 (E/D = 0.002) and gx = 1.94, gy = 1.95, gz = 3.3, in excellent agreement with experiment (only the anion [L2Co]2– and the major fraction of its disordered part was included in the calculations; details are provided in the SI). To explore the magnetization relaxation dynamics, zero field-cooled (ZFC) and field-cooled (FC) magnetic susceptibility measurements were carried out at a sweep rate of 2 K/min, revealing a blocking temperature (TB) of 3.5 K (Figure 2 inset). Temperature-dependent hysteresis measurements carried out at a sweep rate of 30 Oe/s show a butterfly-shaped open hysteresis loop up to 3.5 K, albeit with low coercivity (Figure S16). The sudden collapse of the hysteresis loop near zero-field can be attributed to quantum tunneling of magnetization (QTM), which is a common phenomenon in mononuclear CoII SMMs.16,19
Ac susceptibility measurements were performed in the frequency range 0.1–1000 Hz to quantify the dynamics of the slow relaxation of magnetization. Maxima in the frequency-dependent out-of-phase (χM″) component were observed from 4.5 to 20.0 K in the absence of any external dc field (Figure 3a). Cole–Cole plots of the ac susceptibilities show two merging semi-circular curves, indicating the existence of two distinct relaxation processes with one dominating at a lower temperature (Figure 3b). The χM″ versus χM′ curves were fitted with a generalized Debye function, and the relaxation rates (τ–1) for the fast and slow relaxation processes (FRP and SRP) were extracted (Table S1). Insight into the mechanism of magnetic relaxation was obtained by fitting the relaxation rates (τ–1) using the following function,
| 2 |
where the first term represents magnetic relaxation through QTM, the second and third terms correspond to relaxation via direct and Raman processes, and the last term accounts for relaxation through the Orbach mechanism. The fast relaxation process exhibits temperature-independent relaxation rates at lower temperatures indicating the presence of QTM (Figure S18). Fitting the relaxation rates of the FRP over the entire temperature range with a combination of Raman and QTM processes yields n = 4.73 ± 0.18 and C = 1.36 ± 0.07 × 10–2 s–1 K–n and τQTM = 2.04 ± 0.24 × 10–3 s (Figure S18d). Multiple attempts were made to fit the relaxation rates of the SRP using a combination of different relaxation processes, but a complete fit could not be obtained at lower temperatures. It is interesting to note that the relaxation rates obtained under zero dc field for a frozen dimethylformamide (DMF) solution of complex 1 could be satisfactorily fitted with a combination of Orbach and Raman relaxation pathways (vide infra, Figure 3d). A reasonable fit was obtained by treating the relaxation rates of the SRP with a combination of Orbach and Raman processes [Ueff = 325 K (fixed to |2D|)] with τ0 = 1.46 ± 0.13 × 10–10 s, n = 4.54 ± 0.04, and C = 4.92 ± 0.59 × 10–3 s–1 K–n. The inclusion of a QTM process did not influence the quality of the fit. The best-fit parameters obtained are comparable with those reported in the literature for CoII SIMs.16,18,19,35,58,59 Complex 1 displays a bite angle and dihedral angle close to those of highly anisotropic four-coordinate CoII SIMs reported in the literature (see Table S5). The application of an optimum dc field of 2000 Oe quenches the FRP, and only one relaxation process is evident from the Cole–Cole plots (Figure S19). The relaxation rates obtained under a dc field of 2000 Oe are comparable to the relaxation rates obtained (SRP) under a zero-dc field (Figure S30 and Table S2). As in the previous case, the relaxation rates could be fitted assuming a combination of Orbach and Raman mechanisms, yielding Ueff = 325 K (fixed to |2D|) with τ0 = 3.62 ± 0.08 × 10–11 s, n = 4.98 ± 0.05, and C = 1.47 ± 0.21 × 10–3 s–1 K–n (Figure S19d).
Figure 3.

(a) Out-of-phase (χM″) component of the frequency-dependent ac susceptibility measured in an oscillating ac field of 3.0 Oe and zero applied dc field for 1. (b) Cole–Cole plots for 1 under zero dc field revealing two relaxation processes. (c) Cole–Cole plots for the frozen DMF solution of 1 under zero dc field revealing a single relaxation process. (d) Plot of the relaxation rate τ–1 (logarithmic scale) versus T for 1 for the SRP (green circles) and for the frozen DMF solution of 1 under zero applied dc field. The solid blue line corresponds to the best fit including Orbach and Raman mechanisms.
To establish the intrinsic relaxation dynamics of 1 in the absence of intermolecular dipolar interactions, we prepared magnetically diluted samples containing ca. 5% (1′) or 2% (1″) of CoII complex 1 in the isomorphous diamagnetic matrix of ZnII complex 2. In contrast to the zero-field ac susceptibility measurements for the crystalline sample of pure 1, interestingly only a single relaxation process was observed for magnetically diluted 1′ throughout the whole temperature range (Figure S20, Table S3), similar to the ac susceptibility signature found for 1 upon application of a 2000 Oe dc field. Best-fit parameters are Ueff = 325 K (fixed to |2D|), τ0 = 1.37 ± 0.07 × 10–10 s, n = 4.75 ± 0.04, and C = 2.61 ± 0.29 × 10–3 s–1 K–n (Figure S20d). Overall, the relaxation rates (SRP) extracted for pure 1 and magnetically diluted 1′ are comparable over the entire temperature range (Figure S30 and Table S3). Temperature-dependent hysteresis measurements for 1″ (Figures S24 and S25) show slightly larger hysteresis loops with a minimal increase in coercivity compared to pure 1. In addition, ZFC and FC magnetic susceptibility measurements reveal no major change in the blocking temperature (TB) of 3.5 K (Figure S26), confirming the molecular origin of the SIM behavior.
Magnetic properties in solution have been reported only for a few SMMs,17,20,21,60−64 though it is a prerequisite for many potential SMM applications that they preserve their slow relaxation signatures as isolated molecules outside a crystal lattice. The lack of lattice solvent molecules facilitates a comparison of the magnetic properties of 1 in the solid state/powder and in solution. The high solubility of complex 1 prompted us to investigate a frozen 100 mM DMF solution of 1 (Figure S29), which indeed exhibits relaxation dynamics and hysteretic behavior very similar to the pristine solid sample of 1, though only a single relaxation is observed (Figure 3c). This corroborates that the relaxation dynamics observed for the solid material reflect an intrinsic property of the complex. The relaxation rates are comparable to those observed in the solid state (Figure S30 and Table S4) and could be fitted by a combination of Orbach and Raman relaxation mechanisms over the entire temperature range (Figure 3d); the best-fit parameters are Ueff = 325 K (fixed to |2D|) with τ0 = 7.38 ± 0.27 × 10–10 s, n = 4.13 ± 0.03, and C = 1.93 ± 0.20 × 10–2 s–1 K–n.
Experimental Electron Density
We have recently shown how the ED, and more specifically the d-orbital populations derived from it, can be exploited to extract the energy barrier for magnetic relaxation7 and facilitate a comparison with the values obtained using magnetization measurements. Therefore, the low-temperature high-resolution single-crystal X-ray synchrotron diffraction data65 of complex 1 in the ground electronic state have been used to refine a multipole model (MM) of the EED. A detailed account of the data collection and model refinement is given in the SI. We note that one of the cations is strongly disordered, even at this low temperature (25 K). However, the largest residual density features around the cations (∼1.3 eÅ–3) are much higher than those found near the [L2Co]2– dianion (∼0.6 eÅ–3), which gives confidence in the extracted d-orbital populations for the central Co ion.
It is well-known that for a metal ion in an elongated
tetrahedral coordination sphere with the unique axis along the elongation
axis, the d-orbital energies are ordered as follows: dz2 < dx2 – y2 < dxy < dxz, dyz.7 From the MM, d-orbital
populations for 1 were extracted as shown in Table 1. This table highlights
two conspicuous points. First, when the total population in the d-orbitals is taken into account, all orbitals show significant
deviation from integer populations. For dx2 – y2 and dxy, this clearly indicates
strong mixing of the ground (dxy1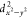 ) and excited state (dxy
) and excited state (dxy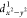 ) via SOC, while the deviations for the
other three orbitals are the results of ligand-to-metal charge transfer.
The second point is the asymmetry in the dxz and dyz orbital populations, which are
expected to be nearly degenerate. Table 1 also provides the d-orbital
populations resulting from a theoretical MM (theoretical electron
density, TED; for details, see the SI).
The overall distribution is similar but does not replicate the variations
in dxz and dyz, seen in the experimental populations. Note that the absolute values
for the d-orbital populations are slightly higher
in the EED than the TED due to the higher total d-electron count in the former. This discrepancy is likely caused
by experimental uncertainties; omission of the highly disordered countercations
in the theoretical calculation and thereby in the TED may also contribute
to the discrepancies between the two models.
) via SOC, while the deviations for the
other three orbitals are the results of ligand-to-metal charge transfer.
The second point is the asymmetry in the dxz and dyz orbital populations, which are
expected to be nearly degenerate. Table 1 also provides the d-orbital
populations resulting from a theoretical MM (theoretical electron
density, TED; for details, see the SI).
The overall distribution is similar but does not replicate the variations
in dxz and dyz, seen in the experimental populations. Note that the absolute values
for the d-orbital populations are slightly higher
in the EED than the TED due to the higher total d-electron count in the former. This discrepancy is likely caused
by experimental uncertainties; omission of the highly disordered countercations
in the theoretical calculation and thereby in the TED may also contribute
to the discrepancies between the two models.
Table 1. d-Orbital Populations on CoII in 1 from Various Methodsa.
| d-orbital | idealized | MM-experimental | MM-theoretical | ab initio (KD1) | ab initio (KD2) |
|---|---|---|---|---|---|
| dxz | 1 | 1.05 (13.6%) | 1.14 (15.4%) | 1.000 | 1.000 |
| dyz | 1 | 1.38 (17.9%) | 1.14 (15.4%) | 1.000 | 1.000 |
| dxy | 1 | 1.47 (19.1%) | 1.33 (17.9%) | 1.282 | 1.089 |
| dx2 – y2 | 2 | 1.97 (25.5%) | 1.75 (23.6%) | 1.715 | 1.903 |
| dz2 | 2 | 1.84 (23.8%) | 2.06 (27.7%) | 2.000 | 2.000 |
| total | 7 | 7.71 | 7.43 | 6.997 | 6.992 |
For ab initio, the d-orbital populations have been obtained by directly taking the weights of (1) and (2) as the face value of dxy and dx2 – y2. This has been done both for KD1 and KD2. For MM, this is the conversion from refined multipole parameters on CoII. See text for more details.
The procedure that allows us to calculate the energy difference between the two lowest KDs from the (experimental) d-orbital populations in an elongated tetrahedral coordination geometry has been described by Damgaard-Møller et al.7Scheme 1 shows the electronic structure of CoII obtained from the theoretical calculation. We exploit the nearly single Slater-determinant nature of the two lowest-lying and energetically isolated states, (1) and (2), prior to SOC. This paves the way to directly quantify the effect of SOC from the contribution, or weight, of (2) in the ground state, KD1. In addition, clearly given the discernible d-orbital distribution of (1) and (2), this weight is easily retrieved from the experimental d-orbital populations. We note that the experimental temperature of 25 K ensures that we have probed the ED of KD1 exclusively, with no contamination from KD2. The final step, converting the d-orbital distribution to an energy barrier involves an existing linear correlation between the energy barrier and weight of (2), see Scheme 1.
Scheme 1. Scheme Showing the Two Lowest-Lying CASSCF States and the Kramers Doublets Appearing Due to Spin–Orbit Couplinga.

Dashed lines show which CASSCF states contribute to the Kramers doublets (KDs). The dashed line between (2) and KD1 is colored as a guide for the eye due to the linear relation between this weight and the energy barrier, ΔK.
While the MM gives access to the d-orbital populations through the refined multipole parameters, both from the theoretical (TED) and the EED, the theoretical calculations lead directly to the weight of (2) in the ground state KD1, which is 0.282 (see the SI for more details). The d-orbital population from the ab initio calculation, the TED, and EED are summarized in Table 1.
We note that the weight of 0.282 of (2) in KD1 found for the theoretical calculation leads to an energy barrier of ΔK = 236.3 cm–1, in perfect accordance with the calculated D value of −118.1 cm–1 (vide supra). This confirms the accuracy of the linear relationship between ΔK and the weight of (2). The significant coupling between dx2 – y2 and dxy orbitals is clearly reflected in the populations, both from the ab initio calculation (1.715 and 1.282, respectively) and experimentally (1.967 and 1.472, respectively). The dz2 orbital is significantly depopulated in the EED relative to the expected 2.0 electrons, while the dyz, on the other hand, deviates from the expected population of 1e– with a higher value. All these deviations are signs of the mixing of states.
We use two different approaches for calculating the value of the energy barrier, ΔK, from the experimental d-orbital population. In the first approach (A), the population in the dx2 – y2 orbital (1.967) is used to represent the weight (0.967) of the ground state, (1), followed by a remaining contribution (0.033) which is the weight of the first excited state. This leads to the following wave function for the ground state: GS(A) : 0.967|(1), ± 3/2⟩ + 0.033|(2), ± 3/2⟩. With this composition, ΔK is calculated to be only 30.8 cm–1. In the second approach (B), we posit that the populations of dx2 – y2 and dxy represent the weights of the ground and first excited states, respectively. This leads to the wave function 0.967|(1), ± 3/2⟩ + 0.472|(2), ± 3/2⟩, which after normalization gives GS(B) : 0.672|(1), ± 3/2⟩ + 0.328|(2), ± 3/2⟩. This composition gives a ΔK value of 261 cm–1. In the first approach (A), where the wave function is based solely on the population of the dx2 – y2 orbital, the contribution to the first excited state is very low, which underestimates the energy barrier, ΔK. The reason is the large total d-electron count, making this approach less reliable. In the second approach (B), where populations of both the dx2 – y2 and dxy orbitals are taken into account, the effect of the total d-electron count is removed and leads to a more realistic value compared to magnetic measurements and theoretical calculations. This shows that the coupling of the two orbitals is important to consider when the energy barrier is estimated from the EED.
Polarized Neutron Diffraction
While the EED approach can estimate the energy barrier with good precision, it assumes the existence of a magnetic anisotropy axis as the d-orbitals are defined according to a coordinate system in which the z axis is along the elongation direction and the x,y axes point in-between ligands. Indeed, information on these axes is not directly accessible via the EED. In contrast, the local magnetic anisotropy in paramagnetic compounds can be quantified using PND via the site susceptibility model.66 The method has been successfully used to determine the atomic susceptibility tensors in several SMMs.12,46,48,51,67−69 Recently, the applicability of the method was extended to powder samples (polarized neutron powder diffraction, PNPD),50 alleviating the demanding need for large single-crystals and time-consuming measurements at multiple crystal orientations. However, this remarkable progress has so far only been exploited on a single occasion to study the atomic susceptibility tensor,51 without independent verification from the single-crystal method. Importantly, the present work thus includes benchmarking the two methods, PNPD and single crystal PND, on the same compound. PNPD data of 1 were collected at the D20 diffractometer of ILL200,70 using neutrons of a 2.41 Å wavelength at 2 K and an applied field of 1 T. Single-crystal PND data of 1 were collected at the HB-3A/DEMAND71 diffractometer of Oak Ridge National Laboratory (ORNL), high flux isotope reactor (HFIR) using neutrons of a 1.5424 Å wavelength at 4 K and an applied field of 0.48 T. Susceptibility tensors were refined using the Python-package Cryspy (0.5.9)/Cryspy_Editor (1.6.0).50 The synchrotron X-ray structure discussed earlier was used to simulate the nuclear structure factors. Describing the tensor required the refinement of six independent susceptibility parameters due to the site symmetry of the CoII ion (for details see the SI). In Figure 4, the refined susceptibility tensor for the Co site in 1 is visualized as an ellipsoid overlaid on the molecular structure, in a manner similar to depicting atomic displacement parameters.
Figure 4.

Magnetic susceptibility tensor for 1 derived from (a) PNPD and (b) joint PND/PNPD refinement. Negative eigenvalues are shown as positive. Ellipsoid is scaled arbitrarily, with the relative magnitudes corresponding to the relative eigenvalues in μBT–1. Counterions and hydrogen atoms are omitted for clarity. Thermal ellipsoids are drawn with a 50% probability.
For the PNPD model, the 2θ range 7.6–32.1° was used. Background, shape parameters, asymmetry parameters, scale factor, and beam offset were refined using the sum and difference patterns. These parameters were then fixed, and the susceptibility parameters were modeled using only the difference pattern. The obtained susceptibility tensor leads to a prolate surface and thus a clearly discernible easy axis anisotropy of 1. The eigenvector corresponding to the most magnetically susceptible direction nearly bisects the bridging C–C bond of the amides of the oxanilido ligand, thus lying close to the molecular axis (3.8° offset to the N1–N2 midpoint). The eigenvalues of the susceptibility tensor [3.35(17), −0.12(15), 0.56(18) μBT–1] indicate a strong axial magnetic anisotropy of 1. These results agree with the magnetization data and ab initio calculations, which also suggest easy-axis anisotropy. We find a close agreement (7.3° deviation) between the easy-axis directions from CASSCF calculations and PNPD, Figure 5. As the PNPD measurements were performed in the linear region of the magnetization curve, the powder-averaged magnetization can be estimated using the expression 1/3(χx + χy + χz)H, resulting in a value of 1.3(1) μB. This value is somewhat lower than the expected value of ∼1.65 μB at 2 K and 1 T obtained from the bulk magnetization curve shown in Figure S15. It should be noted that the crystal structure coordinates used in the refining of the PND data were based on an X-ray synchrotron diffraction experiment, which may explain the discrepancy of the estimated powder averaged magnetization using local anisotropy parameters with the expected value. Indeed, it is well known that neutrons are more sensitive to the hydrogen position than X-rays. Thus, since the polarized signal is proportional to the product of nuclear and magnetic structure factors, uncertainty in the estimation of the former can cause a systematic error in the calculation of the atomic susceptibility tensor.
Figure 5.

Visualization of the magnetic easy axis of 1 from CASSCF calculations (blue), PNPD (red), and joint PND/PNPD refinement (green). Counterions and hydrogen atoms are omitted for clarity.
A refinement model using only single-crystal PND data was not possible, as we could only extract flipping ratios from one magnetic field direction for the single-crystal PND experiment. This was because the crystal broke during the multiple heating and cooling cycles. However, it has been possible to refine a joint PND/PNPD model using the 38 extracted flipping ratios together with the PNPD data. From this, a similar susceptibility ellipsoid was found, Figure 4 (bottom), as well as a similar gz direction, Figure 5 (10.1° offset to PNPD easy axis). The found discrepancies between easy axis directions from ab initio calculations and PND/PNPD measurements are of similar magnitudes to those found by other studies.12,51
Conclusions
In summary, we have employed a set of sophisticated techniques, including static and dynamic magnetometry, EED based on high-quality, low-temperature synchrotron diffraction data, powder and single-crystal PND, and ab initio electronic structure analysis to decipher the origin and quantify the large magnetic anisotropy of a new CoII based SIM. This CoII SIM with two chelating diamido ligands and strongly distorted local tetrahedral geometry around CoII features a large energy separation of over 300 K between the ground and excited-state KDs and shows magnetic hysteresis up to 3.5 K. The slow relaxation dynamics are preserved upon magnetic dilution and in frozen solution, indicating that it is an intrinsic molecular property of 1. From the EED, we were able to extract d-orbital populations for the CoII ion, which show a clear coupling between the dx2 – y2 and dxy orbitals. Calculations of the energy barrier between the two lowest Kramers’ doublets were performed based on results from the EED. When this coupling is taken into account, the resulting energy barrier is 261 cm–1, which correlates well with both magnetic measurements and theoretical calculations. Results from PNPD and PND allow experimental determination of the magnetic susceptibility tensor, showing clear easy axis anisotropy for 1; the direction of the easy axis being in good agreement with the ab initio results. The present fundamental study demonstrates the unique strengths of combining a complete magnetic characterization with advanced diffraction methods, in particular single-crystal synchrotron X-ray diffraction and PNPD, in the assessment of SMMs.
Experimental and Computational Methods
Materials
All solvents employed in the syntheses were dried and distilled before use according to standard procedures. Syntheses of the complexes were carried out under a dry and inert nitrogen atmosphere. Unless otherwise stated, all chemicals were purchased from commercial sources and used as received. The proligand H2L, N,N′-bis(4-chlorophenyl)oxanilide, was synthesized following a previously reported procedure.53 Complexes 1 and 2 were found to be stable under ambient atmospheric conditions.
Instruments for Spectroscopic and Analytical Characterization
1H NMR and 13C NMR spectra were recorded on Bruker Avance 400 spectrometers at room temperature. Chemical shifts are reported in parts per million (ppm) relative to residual proton and carbon signals of the solvent. IR spectra were recorded on a Cary 630 FTIR spectrometer equipped with Dial Path Technology and analyzed by FTIR MicroLab software. Vis–NIR (visible–near-IR) spectra of solutions and solid material were recorded with a Varian Cary 5000 spectrophotometer. ESI mass spectra were collected using a Bruker HCT ultra spectrometer. Elemental analyses were performed by the analytical laboratory of the Institute of Inorganic Chemistry at the University of Göttingen using an Elementar Vario EL III instrument.
Synthesis Protocols
[L2Co](TBA)2 (1)
H2L (309 mg, 1.0 mmol) was dissolved in dry THF (20.0 mL) and KH (80.0 mg, 2.0 mmol) was added, leading to the evolution of dihydrogen. The solution was stirred overnight and solid anhydrous CoCl2 (65 mg, 0.5 mmol) was then added. The reaction mixture was stirred at room temperature for 24 h. Tetrabutylammonium bromide (354 mg, 1.1 mmol) was added and stirring continued for a further 24 h. The red solution was allowed to settle and then filtered. The volume of the filtrate was reduced under vacuum to around 4.0 mL. Diffusion of diethyl ether into the solution led to the formation of reddish pink blocked-shaped crystals (Figure S1) within a few days. Yield: (475 mg, 82% based on Co). Elemental analysis calculated for C60H88Cl4CoN6O4 C 62.23; H 7.66; N 7.26. Found C 61.99; H 7.72; N 7.28. FTIR (cm–1) 2961 (w), 2934 (w), 2874 (w), 1646 (w), 1604 (s), 1582 (s), 1561 (m), 1480 (s), 1458 (m), 1403 (w), 1373 (m), 1302 (s), 1291 (s), 1265 (s), 1211 (w), 1164 (w),1101 (w), 1087 (m), 1031 (w), 1008 (w), 974 (w), 940(w), 919 (m), 881 (w), 857 (w), 831 (s), 801 (m), 738 (w), 706 (w), 677 (w), 589 (w), 522 (w), 502 (m), 471 (m). ESI-MS (negative ion mode, CH3CN): m/z 915.1 [M-TBA]−, 336.3 [M-2TBA]2–. Vis–NIR (in CH3CN, λmax in nm [ε in M–1 cm–1]) 530 [318], 568 [797], 1130 [101]. Vis–NIR (solid state, λmax in nm) 529, 566, 1120.
[L2Zn](TBA)2 (1)
H2L (309 mg, 1.0 mmol) was dissolved in dry THF (20.0 mL), and KH (80.0 mg, 2.0 mmol) was added, leading to the evolution of dihydrogen. The solution was stirred overnight and solid anhydrous ZnCl2 (68 mg, 0.5 mmol) was then added. The reaction mixture was stirred at room temperature for 24 h. Tetrabutylammonium bromide (354 mg, 1.1 mmol) was added and stirring continued for 24 h. The colorless solution was allowed to settle and then filtered. The volume of the reaction mixture was reduced under vacuum to around 4.0 mL. Diffusion of diethyl ether into the solution led to the formation of colorless blocked-shaped crystals within a few days. Yield: (490 mg, 84% based on Zn). Elemental analysis calculated for C60H88Cl4N6O4Zn C 61.88; H 7.62; N 7.22. Found C 62.21; H 7.71; N 7.16. 1H NMR (400 MHz, CD3CN) δ 7.75–7.67 (m, 1H), 7.09–7.01 (m, 1H), 3.13–3.04 (m, 2H), 1.58 (p, J = 7.8 Hz, 2H), 1.32 (h, J = 7.4 Hz, 2H), 0.94 (t, J = 7.3 Hz, 3H) ppm. 13C NMR (100 MHz, CD3CN) δ 167.6, 149.0, 128.8, 125.4, 125.2, 59.3, 24.3, 20.3, 13.8 ppm. FTIR (cm–1) 2962 (w), 2935 (w), 2874 (w), 1650 (w), 1603 (s), 1582 (s), 1560 (m), 1481 (s), 1460 (m), 1404 (w), 1374 (m), 1307 (s), 1293 (s), 1265 (s), 1212 (w), 1168 (w),1102 (w), 1087 (m), 1031 (w), 1008 (w), 972 (w), 939 (w), 921 (m), 881 (w), 858 (w), 831 (s), 802 (m), 737 (w), 706 (w), 676 (w), 627 (w), 590 (w), 520 (w), 502 (m), 481 (w), 468 (m). ESI-MS (negative ion mode, in CH3CN): m/z 922.0 [M-TBA]−, 338.8 [M-2TBA]2–.
Preparation of the Magnetically Diluted Samples [L2Co0.05Zn0.95](TBA)2 (1′) and [L2Co0.02Zn0.98](TBA)2 (1″)
Appropriately weighed amounts of complexes 1 and 2 (by molar ratio) were dissolved in a minimum amount of MeCN, and the solution was stirred for 1 h. Diethyl ether was allowed to diffuse into the solution, leading to the near quantitative isolation of bright light pink crystals within a week.
Magnetic Measurements
Magnetic measurements were carried out with a Quantum-Design MPMS3 SQUID magnetometer equipped with a 7 T magnet. dc magnetic susceptibility measurements were performed under an applied dc field of 0.5 T with powdered polycrystalline samples in the range from 200.0 to 2.0 K. The powdered samples were packed in a polycarbonate or gelatine capsule and covered with low-viscosity perfluoropolyether-based inert oil Fomblin Y45 in a non-magnetic sample holder. Each raw data for the measured magnetic moment was corrected for the diamagnetic contribution of the capsules (including the inert oil) according to Mdia(capsule) = χg·m·H, with an experimentally obtained gram susceptibility of the capsules including the inert oil. The diamagnetic contribution of the compounds was corrected using Pascal’s constants. Magnetic measurements of frozen solutions were carried out by dissolving the sample in DMF in an NMR tube and sealing the tube under a vacuum to exclude dioxygen. Experimental data were modeled using a fitting procedure to the spin Hamiltonian:
Full-matrix diagonalization of the spin Hamiltonian for ZFS and Zeeman splitting was performed with the julX_2s program.55 ac susceptibility measurements were carried out in an oscillating ac field of 3.0 Oe and frequencies ranging from 0.1 to 1000 Hz.
Single-Crystal Structure Determination
In-house X-ray data were collected on an STOE IPDS II diffractometer (graphite monochromated Mo-Kα radiation, λ = 0.71073 Å) by use of ω scans at −140 °C. To obtain high-quality data necessary for modeling the ED of 1, single crystal X-ray diffraction was measured at BL02B1 at the SPring-8 synchrotron. ω scans were performed from 0 to 180° in steps of 0.1°, with fixed χ assuming values of 0, 20, and 40°, respectively, and 2θ at 0 and −20°, resulting in 6 scans in total with 1800 frames in each. The scan time was 0.44 s per frame and the diffracted data was collected with a PILATUS3 X 1M CdTe detector. The crystal was cooled to 25 K and a wavelength of 0.24830 Å was employed. Further experimental details as well as details of the structure solutions are provided in the Supporting Information. CCDC 2212249 (1, STOE IPDS), 2212250 (2, STOE IPDS), and 2212086 (1, SPring-8 synchrotron) contain the supplementary crystallographic data for this paper. These data can be obtained free of charge via www.ccdc.cam.ac.uk/data_request/cif, by emailing data_request@ccdc.cam.ac.uk, or by contacting The Cambridge Crystallographic Data Centre, 12 Union Road, Cambridge CB2 1EZ, UK; fax: +44 1223 336033.
Electron Density Modeling
The ED was modeled based on the synchrotron data with the program XD201672 using the Hansen–Coppens MM formalism. The z axis for Co was chosen to point in-between N(1) and N(2), and the x-axis in-between N(1) and N(3). Hydrogen atoms have been positioned using tabulated bond distances and described with isotropic ADPs. Hexadecapolar angular functions were used to describe the ED for all non-H atoms, while selected quadropoles were used for hydrogen atoms. Core expansion/contraction parameters were included based on the results of multipole modeling from theoretical structure factors.
Polarized Neutron Diffraction
PNPD data were collected at the high-flux powder diffractometer D20 of the Institut Laue-Langevin (ILL)70 on a powdered sample of 1. Data were measured at 2 K and an applied magnetic field of 1 T, with neutrons of a 2.41 Å wavelength, using the angular range 2θ = 7.6–32.1°. The flipping difference pattern was used to model the atomic site susceptibility tensor for Co using the Python-package Cryspy (v. 0.5.9) through Cryspy_Editor (v. 1.6.0).50 Single-crystal PND data were measured at the ORNL HFIR four-circle single-crystal neutron diffractometer HB-3A/DEMAND.71 Data were measured with a 1.5424 Å wavelength at 4 K and a field of 0.48 T. Only one crystal orientation provided data of sufficient quality, and a joint PND/PNPD model was refined.
Theoretical Calculations
Ab initio calculations were performed in ORCA software,56,57,73 using the atom positions obtained from the EED. Prior to the calculation, the two counterions were removed from the structure, so the calculation is only performed on the [L2Co]2– dianion. Furthermore, the disorder within the anion was removed, that is, one of the peripheral 4-Cl-phenyl groups (see Figure S33a) was removed and the occupancy of the other group was set to 1. The CASSCF method was used in the calculation with the seven d-electrons in the CoII ion in five active orbitals, together with the NEVPT2 method. In the calculations, all 10 S = 3/2 states and 40 S = 1/2 states were included, and the def2-TZVP basis set was used. The alignment of the molecule was performed according to the pseudo-D2d symmetry with the z axis for Co between the N(1) and N(2) atoms, and the x axis for Co between the N(1) and N(3) atoms.
Acknowledgments
S.K.G. is associated with the Research Training Group BENCh (RTG 2455) funded by the Deutsche Forschungsgemeinschaft (DFG, German Research Foundation, 389479699). S.K.G. thanks the Alexander von Humboldt Foundation and the International Centre of the University of Göttingen for postdoctoral research fellowships. The purchase of the SQUID magnetometer was supported by the DFG (project number INST 186/1329-1 FUGG) and the Niedersächsische Ministerium für Wissenschaft und Kultur (MWK). J.O. acknowledges funding from Danscatt, the Danish Ministry for Higher Education and Science (SMART and Q-MAT Lighthouses), Villum Foundation, and the Novo Nordisk Foundation. E.F. and H.B.C. are supported by the US Department of Energy (DOE), Office of Science, Office of Basic Energy Sciences, Early Career Research Program Award KC0402020, under Contract No. DE-AC05-00OR22725. This research used resources at the High Flux Isotope Reactor and the Spallation Neutron Source, the DOE Office of Science User Facility operated by ORNL. We thank Zachery Morgan for assistance with single crystal PND data reduction. The numerical results presented in this work were obtained at the Centre for Scientific Computing, Aarhus (http://phys.au. dk/forskning/cscaa). Support by the Open Access Publication Funds of the University of Göttingen is gratefully acknowledged.
Supporting Information Available
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/jacsau.2c00575.
Author Contributions
S.K.G. carried out the synthesis, spectroscopic characterization and magnetic measurements and their analyses. H.H.N. carried out the synchrotron X-ray data collection, theoretical calculation and ED modeling. A.M.T. collected and analyzed PNPD and PND data, with contributions from E.A.K. E.F. and H.B.C. helped collect PND data. T.C.H., E.L.B., and A.G. helped collect PNPD data. I.K. assisted with the PNPD analysis. S.D. and S.D. helped with the in-house single-crystal X-ray data and magnetic data collection and analysis. J.O. and F.M. conceived the study and supervised the experiments and analyses. The manuscript was written through the contributions of all authors. All authors have approved the final version of the manuscript. S.K.G., H.H.N., and A.M.T. contributed equally. CRediT: Sandeep K. Gupta data curation, formal analysis, investigation, visualization, writing-original draft; Hannah Hedegaard Nielsen data curation, formal analysis, investigation, visualization, writing-original draft; Andreas Munch Thiel data curation, formal analysis, investigation, visualization, writing-original daft; Emil Andreasen Klahn data curation, formal analysis; Erxi Feng data curation, investigation; Huibo B. Cao data curation, investigation; Thomas Christian Hansen data curation, investigation; Eddy Lelièvre-Berna data curation, investigation; Arsen Gukasov data curation, investigation; Iurii Kibalin data curation, formal analysis, investigation, validation, writing-review & editing; Sebastian Dechert data curation, investigation; Serhiy Demeshko data curation, formal analysis, investigation; Jacob Overgaard conceptualization, funding acquisition, supervision, writing-original draft; Franc Meyer conceptualization, funding acquisition, resources, supervision, writing-review & editing.
The authors declare no competing financial interest.
Supplementary Material
References
- Gatteschi D.; Sessoli R.; Villain J.. Molecular Nanomagnets; Oxford University Press: New York, 2011. [Google Scholar]
- Cervetti C.; Rettori A.; Pini M. G.; Cornia A.; Repollés A.; Luis F.; Dressel M.; Rauschenbach S.; Kern K.; Burghard M.; Bogani L. The classical and quantum dynamics of molecular spins on graphene. Nat. Mater. 2016, 15, 164–168. 10.1038/nmat4490. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shiddiq M.; Komijani D.; Duan Y.; Gaita-Ariño A.; Coronado E.; Hill S. Enhancing coherence in molecular spin qubits via atomic clock transitions. Nature 2016, 531, 348–351. 10.1038/nature16984. [DOI] [PubMed] [Google Scholar]
- Bogani L.; Wernsdorfer W. Molecular spintronics using single-molecule magnets. Nat. Mater. 2008, 7, 179–186. 10.1038/nmat2133. [DOI] [PubMed] [Google Scholar]
- Urdampilleta M.; Klyatskaya S.; Cleuziou J. P.; Ruben M.; Wernsdorfer W. Supramolecular spin valves. Nat. Mater. 2011, 10, 502–506. 10.1038/nmat3050. [DOI] [PubMed] [Google Scholar]
- Aravena D.; Ruiz E. Spin dynamics in single-molecule magnets and molecular qubits. Dalton Trans. 2020, 49, 9916–9928. 10.1039/D0DT01414A. [DOI] [PubMed] [Google Scholar]
- Damgaard-Møller E.; Krause L.; Tolborg K.; Macetti G.; Genoni A.; Overgaard J. Quantification of the Magnetic Anisotropy of a Single-Molecule Magnet from the Experimental Electron Density. Angew. Chem., Int. Ed. 2020, 59, 21203–21209. 10.1002/anie.202007856. [DOI] [PubMed] [Google Scholar]
- Gao C.; Genoni A.; Gao S.; Jiang S.; Soncini A.; Overgaard J. Observation of the asphericity of 4f-electron density and its relation to the magnetic anisotropy axis in single-molecule magnets. Nat. Chem. 2020, 12, 213–219. 10.1038/s41557-019-0387-6. [DOI] [PubMed] [Google Scholar]
- Bone A. N.; Widener C. N.; Moseley D. H.; Liu Z.; Lu Z.; Cheng Y.; Daemen L. L.; Ozerov M.; Telser J.; Thirunavukkuarasu K.; Smirnov D.; Greer S. M.; Hill S.; Krzystek J.; Holldack K.; Aliabadi A.; Schnegg A.; Dunbar K. R.; Xue Z.-L. Applying Unconventional Spectroscopies to the Single-Molecule Magnets, Co(PPh3)2X2 (X=Cl, Br, I): Unveiling Magnetic Transitions and Spin-Phonon Coupling. Chem. – Eur. J. 2021, 27, 11110–11125. 10.1002/chem.202100705. [DOI] [PubMed] [Google Scholar]
- Craven M.; Nygaard M. H.; Zadrozny J. M.; Long J. R.; Overgaard J. Determination of d-Orbital Populations in a Cobalt(II) Single-Molecule Magnet Using Single-Crystal X-ray Diffraction. Inorg. Chem. 2018, 57, 6913–6920. 10.1021/acs.inorgchem.8b00513. [DOI] [PubMed] [Google Scholar]
- Perfetti M.; Cucinotta G.; Boulon M.-E.; El Hallak F.; Gao S.; Sessoli R. Angular-Resolved Magnetometry Beyond Triclinic Crystals Part II: Torque Magnetometry of Cp*ErCOT Single-Molecule Magnets. Chem. – Eur. J. 2014, 20, 14051–14056. 10.1002/chem.201404218. [DOI] [PubMed] [Google Scholar]
- Klahn E. A.; Gao C.; Gillon B.; Gukasov A.; Fabrèges X.; Piltz R. O.; Jiang S.-D.; Overgaard J. Mapping the Magnetic Anisotropy at the Atomic Scale in Dysprosium Single-Molecule Magnets. Chem. – Eur. J. 2018, 24, 16576–16581. 10.1002/chem.201803300. [DOI] [PubMed] [Google Scholar]
- Zhang Y.; Krylov D.; Rosenkranz M.; Schiemenz S.; Popov A. A. Magnetic anisotropy of endohedral lanthanide ions: paramagnetic NMR study of MSc2N@C80-Ih with M running through the whole 4f row. Chem. Sci. 2015, 6, 2328–2341. 10.1039/C5SC00154D. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Atanasov M.; Aravena D.; Suturina E.; Bill E.; Maganas D.; Neese F. First principles approach to the electronic structure, magnetic anisotropy and spin relaxation in mononuclear 3d-transition metal single molecule magnets. Coord. Chem. Rev. 2015, 289-290, 177–214. 10.1016/j.ccr.2014.10.015. [DOI] [Google Scholar]
- Luo Q.-C.; Zheng Y.-Z. Methods and Models of Theoretical Calculation for Single-Molecule Magnets. Magnetochemistry 2021, 7, 107. 10.3390/magnetochemistry7080107. [DOI] [Google Scholar]
- Bunting P. C.; Atanasov M.; Damgaard-Møller E.; Perfetti M.; Crassee I.; Orlita M.; Overgaard J.; Slageren J. V.; Neese F.; Long J. R. A linear cobalt(II) complex with maximal orbital angular momentum from a non-Aufbau ground state. Science 2018, 362, eaat7319 10.1126/science.aat7319. [DOI] [PubMed] [Google Scholar]
- Zadrozny J. M.; Xiao D. J.; Atanasov M.; Long G. J.; Grandjean F.; Neese F.; Long J. R. Magnetic blocking in a linear iron(I) complex. Nat. Chem. 2013, 5, 577–581. 10.1038/nchem.1630. [DOI] [PubMed] [Google Scholar]
- Yao X.-N.; Du J.-Z.; Zhang Y.-Q.; Leng X.-B.; Yang M.-W.; Jiang S.-D.; Wang Z.-X.; Ouyang Z.-W.; Deng L.; Wang B.-W.; Gao S. Two-Coordinate Co(II) Imido Complexes as Outstanding Single-Molecule Magnets. J. Am. Chem. Soc. 2017, 139, 373–380. 10.1021/jacs.6b11043. [DOI] [PubMed] [Google Scholar]
- Rechkemmer Y.; Breitgoff F. D.; van der Meer M.; Atanasov M.; Hakl M.; Orlita M.; Neugebauer P.; Neese F.; Sarkar B.; van Slageren J. A four-coordinate cobalt(II) single-ion magnet with coercivity and a very high energy barrier. Nat. Commun. 2016, 7, 10467. 10.1038/ncomms10467. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goodwin C. A. P.; Ortu F.; Reta D.; Chilton N. F.; Mills D. P. Molecular magnetic hysteresis at 60 kelvin in dysprosocenium. Nature 2017, 548, 439–442. 10.1038/nature23447. [DOI] [PubMed] [Google Scholar]
- Yu K.-X.; Kragskow J. G. C.; Ding Y.-S.; Zhai Y.-Q.; Reta D.; Chilton N. F.; Zheng Y.-Z. Enhancing Magnetic Hysteresis in Single-Molecule Magnets by Ligand Functionalization. Chem 2020, 6, 1777–1793. 10.1016/j.chempr.2020.04.024. [DOI] [Google Scholar]
- Guo F.-S.; Day Benjamin M.; Chen Y.-C.; Tong M.-L.; Mansikkamäki A.; Layfield Richard A. Magnetic hysteresis up to 80 kelvin in a dysprosium metallocene single-molecule magnet. Science 2018, 362, 1400–1403. 10.1126/science.aav0652. [DOI] [PubMed] [Google Scholar]
- Rinehart J. D.; Fang M.; Evans W. J.; Long J. R. A N23– Radical-Bridged Terbium Complex Exhibiting Magnetic Hysteresis at 14 K. J. Am. Chem. Soc. 2011, 133, 14236–14239. 10.1021/ja206286h. [DOI] [PubMed] [Google Scholar]
- Gould C. A.; McClain K. R.; Reta D.; Kragskow J. G. C.; Marchiori D. A.; Lachman E.; Choi E.-S.; Analytis J. G.; Britt R. D.; Chilton N. F.; Harvey B. G.; Long J. R. Ultrahard magnetism from mixed-valence dilanthanide complexes with metal-metal bonding. Science 2022, 375, 198–202. 10.1126/science.abl5470. [DOI] [PubMed] [Google Scholar]
- Guo F.-S.; Day B. M.; Chen Y.-C.; Tong M.-L.; Mansikkamäki A.; Layfield R. A. A Dysprosium Metallocene Single-Molecule Magnet Functioning at the Axial Limit. Angew. Chem., Int. Ed. 2017, 56, 11445–11449. 10.1002/anie.201705426. [DOI] [PubMed] [Google Scholar]
- Sarkar A.; Dey S.; Rajaraman G. Role of Coordination Number and Geometry in Controlling the Magnetic Anisotropy in FeII, CoII, and NiII Single-Ion Magnets. Chem. – Eur. J. 2020, 26, 14036–14058. 10.1002/chem.202003211. [DOI] [PubMed] [Google Scholar]
- Murrie M. Cobalt(II) single-molecule magnets. Chem. Soc. Rev. 2010, 39, 1986–1995. 10.1039/b913279c. [DOI] [PubMed] [Google Scholar]
- Gómez-Coca S.; Aravena D.; Morales R.; Ruiz E. Large magnetic anisotropy in mononuclear metal complexes. Coord. Chem. Rev. 2015, 289-290, 379–392. 10.1016/j.ccr.2015.01.021. [DOI] [Google Scholar]
- Vaidya S.; Tewary S.; Singh S. K.; Langley S. K.; Murray K. S.; Lan Y.; Wernsdorfer W.; Rajaraman G.; Shanmugam M. What Controls the Sign and Magnitude of Magnetic Anisotropy in Tetrahedral Cobalt(II) Single-Ion Magnets?. Inorg. Chem. 2016, 55, 9564–9578. 10.1021/acs.inorgchem.6b01073. [DOI] [PubMed] [Google Scholar]
- Saber M. R.; Dunbar K. R. Ligands effects on the magnetic anisotropy of tetrahedral cobalt complexes. Chem. Commun. 2014, 50, 12266–12269. 10.1039/C4CC05724D. [DOI] [PubMed] [Google Scholar]
- Yao X.-N.; Yang M.-W.; Xiong J.; Liu J.-J.; Gao C.; Meng Y.-S.; Jiang S.-D.; Wang B.-W.; Gao S. Enhanced magnetic anisotropy in a tellurium-coordinated cobalt single-ion magnet. Inorg. Chem. Front. 2017, 4, 701–705. 10.1039/C6QI00543H. [DOI] [Google Scholar]
- Zadrozny J. M.; Telser J.; Long J. R. Slow magnetic relaxation in the tetrahedral cobalt(II) complexes [Co(EPh)4]2– (E = O, S, Se). Polyhedron 2013, 64, 209–217. 10.1016/j.poly.2013.04.008. [DOI] [Google Scholar]
- Carl E.; Demeshko S.; Meyer F.; Stalke D. Triimidosulfonates as Acute Bite-Angle Chelates: Slow Relaxation of the Magnetization in Zero Field and Hysteresis Loop of a CoII Complex. Chem. – Eur. J. 2015, 21, 10109–10115. 10.1002/chem.201406083. [DOI] [PubMed] [Google Scholar]
- Vallejo J.; Pardo E.; Viciano-Chumillas M.; Castro I.; Amorós P.; Déniz M.; Ruiz-Pérez C.; Yuste-Vivas C.; Krzystek J.; Julve M.; Lloret F.; Cano J. Reversible solvatomagnetic switching in a single-ion magnet from an entatic state. Chem. Sci. 2017, 8, 3694–3702. 10.1039/C6SC05188J. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cui H.-H.; Lu F.; Chen X.-T.; Zhang Y.-Q.; Tong W.; Xue Z.-L. Zero-Field Slow Magnetic Relaxation and Hysteresis Loop in Four-Coordinate CoII Single-Ion Magnets with Strong Easy-Axis Anisotropy. Inorg. Chem. 2019, 58, 12555–12564. 10.1021/acs.inorgchem.9b01175. [DOI] [PubMed] [Google Scholar]
- Boča R.; Miklovič J.; Titiš J. Simple Mononuclear Cobalt(II) Complex: A Single-Molecule Magnet Showing Two Slow Relaxation Processes. Inorg. Chem. 2014, 53, 2367–2369. 10.1021/ic5000638. [DOI] [PubMed] [Google Scholar]
- Novikov V. V.; Pavlov A. A.; Nelyubina Y. V.; Boulon M.-E.; Varzatskii O. A.; Voloshin Y. Z.; Winpenny R. E. P. A Trigonal Prismatic Mononuclear Cobalt(II) Complex Showing Single-Molecule Magnet Behavior. J. Am. Chem. Soc. 2015, 137, 9792–9795. 10.1021/jacs.5b05739. [DOI] [PubMed] [Google Scholar]
- Yao B.; Deng Y.-F.; Li T.; Xiong J.; Wang B.-W.; Zheng Z.; Zhang Y.-Z. Construction and Magnetic Study of a Trigonal-Prismatic Cobalt(II) Single-Ion Magnet. Inorg. Chem. 2018, 57, 14047–14051. 10.1021/acs.inorgchem.8b02692. [DOI] [PubMed] [Google Scholar]
- Vallejo J.; Castro I.; Ruiz-García R.; Cano J.; Julve M.; Lloret F.; De Munno G.; Wernsdorfer W.; Pardo E. Field-Induced Slow Magnetic Relaxation in a Six-Coordinate Mononuclear Cobalt(II) Complex with a Positive Anisotropy. J. Am. Chem. Soc. 2012, 134, 15704–15707. 10.1021/ja3075314. [DOI] [PubMed] [Google Scholar]
- Saber M. R.; Singh M. K.; Dunbar K. R. Geometrical control of the magnetic anisotropy in six coordinate cobalt complexes. Chem. Commun. 2020, 56, 8492–8495. 10.1039/D0CC03238G. [DOI] [PubMed] [Google Scholar]
- Chen L.; Wang J.; Wei J.-M.; Wernsdorfer W.; Chen X.-T.; Zhang Y.-Q.; Song Y.; Xue Z.-L. Slow Magnetic Relaxation in a Mononuclear Eight-Coordinate Cobalt(II) Complex. J. Am. Chem. Soc. 2014, 136, 12213–12216. 10.1021/ja5051605. [DOI] [PubMed] [Google Scholar]
- Zadrozny J. M.; Long J. R. Slow Magnetic Relaxation at Zero Field in the Tetrahedral Complex [Co(SPh)4]2–. J. Am. Chem. Soc. 2011, 133, 20732–20734. 10.1021/ja2100142. [DOI] [PubMed] [Google Scholar]
- Atanasov M.; Neese F. Computational Studies on Vibronic Coupling in Single Molecule Magnets: Impact on the Mechanism of Magnetic Relaxation. J. Phys.: Conf. Ser. 2018, 1148, 012006 10.1088/1742-6596/1148/1/012006. [DOI] [Google Scholar]
- Lunghi A.; Sanvito S. Multiple spin–phonon relaxation pathways in a Kramer single-ion magnet. J. Chem. Phys. 2020, 153, 174113. 10.1063/5.0017118. [DOI] [PubMed] [Google Scholar]
- Thomsen M. K.; Nyvang A.; Walsh J. P. S.; Bunting P. C.; Long J. R.; Neese F.; Atanasov M.; Genoni A.; Overgaard J. Insights into Single-Molecule-Magnet Behavior from the Experimental Electron Density of Linear Two-Coordinate Iron Complexes. Inorg. Chem. 2019, 58, 3211–3218. 10.1021/acs.inorgchem.8b03301. [DOI] [PubMed] [Google Scholar]
- Tripathi S.; Vaidya S.; Ahmed N.; Andreasen Klahn E.; Cao H.; Spillecke L.; Koo C.; Spachmann S.; Klingeler R.; Rajaraman G.; Overgaard J.; Shanmugam M. Structure-property correlation in stabilizing axial magnetic anisotropy in octahedral Co(II) complexes. Cell Rep. Phys. Sci. 2021, 2, 100404 10.1016/j.xcrp.2021.100404. [DOI] [Google Scholar]
- De S.; Flambard A.; Garnier D.; Herson P.; Köhler F. H.; Mondal A.; Costuas K.; Gillon B.; Lescouëzec R.; Le Guennic B.; Gendron F. Probing the Local Magnetic Structure of the [FeIII(Tp)(CN)3]− Building Block Via Solid-State NMR Spectroscopy, Polarized Neutron Diffraction, and First-Principle Calculations. Chem. – Eur. J. 2019, 25, 12120–12136. 10.1002/chem.201902239. [DOI] [PubMed] [Google Scholar]
- Ridier K.; Mondal A.; Boilleau C.; Cador O.; Gillon B.; Chaboussant G.; Le Guennic B.; Costuas K.; Lescouëzec R. Polarized Neutron Diffraction to Probe Local Magnetic Anisotropy of a Low-Spin Fe(III) Complex. Angew. Chem., Int. Ed. 2016, 55, 3963–3967. 10.1002/anie.201511354. [DOI] [PubMed] [Google Scholar]
- Ridier K.; Gillon B.; Gukasov A.; Chaboussant G.; Cousson A.; Luneau D.; Borta A.; Jacquot J.-F.; Checa R.; Chiba Y.; Sakiyama H.; Mikuriya M. Polarized Neutron Diffraction as a Tool for Mapping Molecular Magnetic Anisotropy: Local Susceptibility Tensors in CoII Complexes. Chem. – Eur. J. 2016, 22, 724–735. 10.1002/chem.201503400. [DOI] [PubMed] [Google Scholar]
- Kibalin I. A.; Gukasov A. Local magnetic anisotropy by polarized neutron powder diffraction: Application of magnetically induced preferred crystallite orientation. Phys. Rev. Res. 2019, 1, 033100 10.1103/PhysRevResearch.1.033100. [DOI] [Google Scholar]
- Klahn E. A.; Damgaard-Moller E.; Krause L.; Kibalin I.; Gukasov A.; Tripathi S.; Swain A.; Shanmugam M.; Overgaard J. Quantifying magnetic anisotropy using X-ray and neutron diffraction. IUCrJ 2021, 8, 833–841. 10.1107/S2052252521008290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kibalin I.; Gukasov A. Quantifying Magnetic Anisotropy Using Polarized Neutron Powder Diffraction. Neutron News 2021, 32, 26–27. 10.1080/10448632.2021.1997311. [DOI] [Google Scholar]
- Santana M. D.; García G.; Vicente-Hernández C.; García L.; Pérez J.; Rojo T.; Lezama L. N,N′-bis(substituted-phenyl)oxamides and their dinuclear pentacoordinate nickel(II) complexes. J. Organomet. Chem. 2008, 693, 2009–2016. 10.1016/j.jorganchem.2008.03.001. [DOI] [Google Scholar]
- Yang L.; Powell D. R.; Houser R. P. Structural variation in copper(I) complexes with pyridylmethylamide ligands: structural analysis with a new four-coordinate geometry index, τ4. Dalton Trans. 2007, 9, 955–964. 10.1039/B617136B. [DOI] [PubMed] [Google Scholar]
- Bill E.julX_2s, Simulation of molecular magnetic data software; Max-Planck Institute for Bioinorganic Chemistry: Mülheim, Ruhr, 2014. [Google Scholar]
- Neese F. Software update: the ORCA program system, version 4.0. WIREs Comput. Mol. Sci. 2017, 8, e1327 10.1002/wcms.81. [DOI] [Google Scholar]
- Neese F. The ORCA program system. WIREs Comput. Mol. Sci. 2012, 2, 73–78. 10.1002/wcms.81. [DOI] [Google Scholar]
- Bamberger H.; Albold U.; Dubnická Midlíková J.; Su C.-Y.; Deibel N.; Hunger D.; Hallmen P. P.; Neugebauer P.; Beerhues J.; Demeshko S.; Meyer F.; Sarkar B.; van Slageren J. Iron(II), Cobalt(II), and Nickel(II) Complexes of Bis(sulfonamido)benzenes: Redox Properties, Large Zero-Field Splittings, and Single-Ion Magnets. Inorg. Chem. 2021, 60, 2953–2963. 10.1021/acs.inorgchem.0c02949. [DOI] [PubMed] [Google Scholar]
- Legendre C. M.; Damgaard-Møller E.; Overgaard J.; Stalke D. The Quest for Optimal 3d Orbital Splitting in Tetrahedral Cobalt Single-Molecule Magnets Featuring Colossal Anisotropy and Hysteresis. Eur. J. Inorg. Chem. 2021, 2021, 3108–3114. 10.1002/ejic.202100465. [DOI] [Google Scholar]
- da Cunha T. T.; Jung J.; Boulon M.-E.; Campo G.; Pointillart F.; Pereira C. L. M.; Le Guennic B.; Cador O.; Bernot K.; Pineider F.; Golhen S.; Ouahab L. Magnetic Poles Determinations and Robustness of Memory Effect upon Solubilization in a DyIII-Based Single Ion Magnet. J. Am. Chem. Soc. 2013, 135, 16332–16335. 10.1021/ja4089956. [DOI] [PubMed] [Google Scholar]
- Boskovic C.; Brechin E. K.; Streib W. E.; Folting K.; Bollinger J. C.; Hendrickson D. N.; Christou G. Single-Molecule Magnets: A New Family of Mn12 Clusters of Formula [Mn12O8X4(O2CPh)8L6]. J. Am. Chem. Soc. 2002, 124, 3725–3736. 10.1021/ja012403k. [DOI] [PubMed] [Google Scholar]
- Schlegel C.; Burzurí E.; Luis F.; Moro F.; Manoli M.; Brechin E. K.; Murrie M.; van Slageren J. Magnetic Properties of Two New Fe4 Single-Molecule Magnets in the Solid State and in Frozen Solution. Chem. – Eur. J. 2010, 16, 10178–10185. 10.1002/chem.200903505. [DOI] [PubMed] [Google Scholar]
- Zaleski C. M.; Tricard S.; Depperman E. C.; Wernsdorfer W.; Mallah T.; Kirk M. L.; Pecoraro V. L. Single Molecule Magnet Behavior of a Pentanuclear Mn-Based Metallacrown Complex: Solid State and Solution Magnetic Studies. Inorg. Chem. 2011, 50, 11348–11352. 10.1021/ic2008792. [DOI] [PubMed] [Google Scholar]
- El Hallak F.; van Slageren J.; Gómez-Segura J.; Ruiz-Molina D.; Dressel M. High-frequency ESR and frequency domain magnetic resonance spectroscopic studies of single molecule magnets in frozen solution. Phys. Rev. B 2007, 75, 104403 10.1103/PhysRevB.75.104403. [DOI] [Google Scholar]
- Krause L.; Tolborg K.; Grønbech T. B. E.; Sugimoto K.; Iversen B. B.; Overgaard J. Accurate high-resolution single-crystal diffraction data from a Pilatus3 X CdTe detector. J. Appl. Crystallogr. 2020, 53, 635–649. 10.1107/S1600576720003775. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gukasov A.; Brown P. J. Determination of atomic site susceptibility tensors from polarized neutron diffraction data. J. Phys.: Condens. Matter 2002, 14, 8831. 10.1088/0953-8984/14/38/307. [DOI] [PubMed] [Google Scholar]
- Klahn E. A.; Thiel A. M.; Degn R. B.; Kibalin I.; Gukassov A.; Wilson C.; Canaj A. B.; Murrie M.; Overgaard J. Magnetic anisotropies of Ho(III) and Dy(II) single-molecule magnets experimentally determined via polarized neutron diffraction. Dalton Trans. 2021, 50, 14207–14215. 10.1039/D1DT01959G. [DOI] [PubMed] [Google Scholar]
- Iasco O.; Chumakov Y.; Guégan F.; Gillon B.; Lenertz M.; Bataille A.; Jacquot J.-F.; Luneau D. Mapping the Magnetic Anisotropy inside a Ni4 Cubane Spin Cluster Using Polarized Neutron Diffraction. Magnetochemistry 2017, 3, 25. 10.3390/magnetochemistry3030025. [DOI] [Google Scholar]
- Borta A.; Gillon B.; Gukasov A.; Cousson A.; Luneau D.; Jeanneau E.; Ciumacov I.; Sakiyama H.; Tone K.; Mikuriya M. Local magnetic moments in a dinuclear Co2+ complex as seen by polarized neutron diffraction: Beyond the effective spin-1/2 model. Phys. Rev. B 2011, 83, 184429 10.1103/PhysRevB.83.184429. [DOI] [Google Scholar]
- Lelievre-Berna E.; Wills A. S.; Bourgeat-Lami E.; Dee A.; et al. Powder diffraction with spin polarized neutrons. Meas. Sci. Technol. 2010, 21, 055106. 10.1088/0957-0233/21/5/055106. [DOI] [Google Scholar]
- Hansen T. C.; Henry P. F.; Fischer H. E.; Torregrossa J.; Convert P. The D20 instrument at the ILL: a versatile high-intensity two-axis neutron diffractometer. Meas. Sci. Technol. 2008, 19, 034001 10.1088/0957-0233/19/3/034001. [DOI] [Google Scholar]
- Cao H.; Chakoumakos B. C.; Andrews K. M.; Wu Y.; Riedel R. A.; Hodges J.; Zhou W.; Gregory R.; Haberl B.; Molaison J.; Lynn G. W. DEMAND, a Dimensional Extreme Magnetic Neutron Diffractometer at the High Flux Isotope Reactor. Crystals 2019, 9, 5. 10.3390/cryst9010005. [DOI] [Google Scholar]
- Volkov A.; Macchi P.; Farrugia L. J.; Gatti C.; Mallinson P.; Richter T.; Koritsanszky T., XD2016 - A Computer Program Package for Multipole Refinement, Topological Analysis of Charge Densities and Evaluation of Intermolecular Energies from Experimental and Theoretical Structure Factors; University at Buffalo, State University of New York: New York, USA, 2016. [Google Scholar]
- Neese F.; Wennmohs F.; Becker U.; Riplinger C. The ORCA quantum chemistry program package. J. Chem. Phys. 2020, 152, 224108. 10.1063/5.0004608. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.