

Abstract
External mechanical stress alters the nature of chemical bonds and triggers novel reactions, providing interesting synthetic protocols to supplement traditional solvent- or thermo-based chemical approaches. The mechanisms of mechanochemistry have been well studied in organic materials made of a carbon-centered polymeric framework and covalence force field. They convert stress into anisotropic strain which will engineer the length and strength of targeted chemical bonds. Here, we show that by compressing silver iodide in a diamond anvil cell, the external mechanical stress weakens the Ag–I ionic bonds and activate the global diffusion of super-ions. In contrast to conventional mechanochemistry, mechanical stress imposes unbiased influence on the ionicity of chemical bonds in this archetypal inorganic salt. Our combined synchrotron X-ray diffraction experiment and first-principles calculation demonstrate that upon the critical point of ionicity, the strong ionic Ag–I bonds break down, leading to the recovery of elemental solids from a decomposition reaction. Instead of densification, our results reveal the mechanism of an unexpected decomposition reaction through hydrostatic compression and suggest the sophisticated chemistry of simple inorganic compounds under extreme conditions.
Keywords: chemical reactions, ionic bond, mechanochemistry, silver iodide, decomposition
1. Introduction
Using solvents to activate or assist chemical reactions has long been accepted as an effective protocol for the industrial production of chemicals. Solvents dissolve and mix chemicals, allowing the exchange of energies between reagents to promote chemical reactions, which sometimes is necessary for the formation of specific crystals.1,2 However, solvent-free approaches or mechanochemistry, which employs mechanical forces to induce chemical reactions, hold key advantages in energy saving and environmental friendliness and may even provide access to novel structures.3 For example, the traditional ball-milling method has been established to synthesize a variety of materials including polymer,4,5 metal–organic framework,6 and pharmaceutical solids.7
Beyond hand grinding or mechanical milling, Yan et al. designed a molecular anvil to drive redox reactions in metal–organic chalcogenides, which enables mechanochemistry by hydrostatic pressure up to gigapascal pressure ranges.8 The invention of molecular anvil extends the scope of mechanochemistry, but is in line with its formulized definition in which the chemical reaction directly converts mechanical energy to chemistry potentials.9 However, mechanochemistry in inorganic materials has drawn far less attention than organic materials, despite its origin in 1820, when Faraday induced the mechanical reduction of AgCl with metals.10 In the atomistic level, inorganic materials are constructed through a variety of chemical bonds, including covalent, ionic, and metallic bonds.11 The complexity and variety in the combination of chemical bonds make the studies of inorganic mechanochemistry much more challenging.
In this work, we study the mechanochemistry of silver iodide (AgI) in an extended pressure range of 1 bar to 41.5 GPa. AgI is widely used as an additive to catalyze C–H functionalization reactions12,13 and a promising candidate for solid electrolytes.14−17 Pressure has previously induced polymorphism and superionic transitions in AgI. Under ambient conditions, AgI coexists in both wurtzite-type (AgI-II) and zinc-blende-type (AgI-II′) phases. AgI-II transforms to a rocksalt structure (namely, AgI-III, space group Fm-3m) at 76 K and 0.3 GPa.18 Meanwhile, powder neutron diffraction experiments observed that AgI-II′ transforms to AgI-III via an intermediate structure (named AgI-IV with a P4/nmm structure) which is only stable in a narrow pressure interval of 0.28–0.38 GPa.19 Using angular-dispersive X-ray diffraction (XRD), Hull and Keen found that the AgI-III undergoes a reconstructive transition to the KOH-type structure (named as AgI-V with P21/m space group) at 11.3 GPa,20 and is later predicted to proceed to orthorhombic TlI-type (space group Cmcm) and CsCl-type phases at higher pressures.21 AgI-V was measured to have an extraordinary diffusion coefficient in the range of 12–17 GPa and room temperature and was reported as the second superionic phase of AgI.22 While external stress generally shortens chemical bonds and induces polymorphic transition, it plays a key role in modulating the diffusion behavior of Ag+ ions, as is suggested in the literature.23 Approaching the stability limit of bond, the increasing enthalpy through compressing may eventually alter its chemical reactivity and even induce redox reactions.
Here, we employ a combination of synchrotron-based angular dispersive XRD and first-principles simulations to study AgI under pressure. The increase of pressure weakens the charge transfer between ions, and eventually leads to the decomposition of AgI into Ag and I elements.
2. Materials and Methods
Pressure is applied by compressing samples in a diamond anvil cell (DAC). High purity AgI is commercially available at Alfa Aesar (Product # 7783-96-2, 99.999%). AgI powder is precompressed and cut into a thin petite size of 40 × 40 μm2. The sample chamber is a drilled hole with a diameter of 100 μm in a Re gasket, which is squeezed in between two pieces of 300 μm diamond culets. After placing the sample in the middle of sample chamber, the rest of the space is filled with paraffin oil as pressure medium. Pressurization is controlled by a membrane system in a LeToullec type DAC.24 Using a membrane-controlled system, the sample was slowly compressed to the target pressure, which was calibrated by the fluorescence of ruby. We then took in situ high-pressure XRD patterns at the sector ID15-B of European Synchrotron Radiation Facility. Diffraction data sets were collected on a large area EIGER2 X 9M CdTe flat panel detector and reduced by the Dioptas software (ver.0.5.0).25
For first-principles simulation, we performed global structural searches using the crystal structure analysis by particle swarm optimization code with the particle swarm optimization algorithm,26−28 which has successfully predicted structures of various systems ranging from elements to binary and ternary compounds.23,29−31 Geometrical optimization, total energy calculation, and electronic structure calculation were performed in the framework of density functional theory within the generalized gradient approximation Perdew–Burke–Ernzerhof32,33 in the Vienna Ab Initio Simulation Package (VASP) code.34 The projector-augmented-wave method35 was employed with the silver and iodine potentials which have 4d105s1 and 5s25p5 as valence states, respectively, adopted from the VASP potential library. A cut off energy of 700 eV for the plane-wave basis set and a Monkhorst–Pack36k-point grid with a spacing of 0.02 Å–1 were used to ensure the convergence for total energies less than 1 meV/atom. In order to study the high-pressure structural stability, we also conducted first-principles molecular dynamics (FPMD) simulations at high-pressure–temperature using the canonical NVT ensemble with the Nosé–Hoover thermostat37,38 and a time step of 1 fs. For FPMD simulation, only the gamma k-point was used. To analyze the interatomic interaction, the crystal orbital Hamilton populations (COHP)39 was calculated using the LOBSTER40 program.
3. Results
3.1. Structural and Phase Identification through XRD
We first compressed a piece of AgI powder-sample up to 41.5 GPa and tracked its structural changes using XRD (Figure 1a). The evolution of XRD patterns and two refined patterns of interests are plotted in Figure 1. Below 10.9 GPa (Figure 1b), the powder XRD data of AgI are readily indexed to the rocksalt-type structure (AgI-III) with the space group Fm-3m (lattice parameter a = 5.7816(2) Å). Once pressure increases to 11.9 GPa, as shown in Figure 1c, new sets of diffraction peaks appear. The pattern is still dominated by the face-centered cubic (FCC) AgI-III but the rest of peaks are not fully indexed to the previously reported AgI-V,20 instead, they can be associated with the FCC-Ag and the Immm-type I. Upon further compression, all diffraction peaks shift to lower d-spacings accordingly (Figure 1a). We shall note that although the space group P21/m of KOH-type AgI-V is a subgroup of the host FCC AgI-III, the transformation requires long-distance atomic displacement with multiple structure intermediates.41 Therefore, the structural transition is of first-order. With increasing pressure, the diffraction signals of AgI are gradually weakened, and the peaks of Ag are merged by I. The relatively broad peaks not only indicate that the polymorphic phase transition of I is rather sluggish, but the sample may be partially amorphized during the process of Ag–I decomposition.42,43 Our high-pressure experiments show that AgI becomes unstable at above 11.9 GPa and the entire process of mechanochemical reaction may cover a wide range of pressures up to 41.5 GPa.
Figure 1.

XRD patterns of compressed AgI. (a) Evolution of XRD patterns from 6.1 to 41.5 GPa. (b) FCC AgI-III at 10.9 GPa. Refinement achieved R1 = 5.60% and wR2 = 7.49%. Lattice parameter is a = 5.7816(2). (c) Mixture of AgI, Ag, and I at 11.9 GPa. AgI sample partially decomposes and the peaks of FCC-Ag and Immm-type I appear. Refinement converged at R1 = 7.57% and wR2 = 12.54%. Lattice parameters for each phase are AgI-III: a = 5.717(2), Ag: a = 4.018(1), I: a = 2.981(4), b = 3.698(1), c = 7.645(8). Red stars may partially come from the KOH-type AgI-V. A detailed analysis of AgI-V is supplied in Figure S1. Inset figures in (b) and (c) are caked two-dimensional diffraction patterns, whose y-axes are the azimuth angle. Wavelength of incident X-ray beam at the time of experiment is 0.4101 Å.
3.2. Structural Searching and Free Energy Analysis
The mechanochemical redox reaction originates from the free energy difference between AgI and the elementary solids. To investigate the mechanism of this mechanochemistry, we conducted exhaustive structure searching of stoichiometric AgxIy (x = 1–3; y = 1–3), whose simulation cell sizes are respect to 1–4 formula units (f.u.) in a pressure range of 0–100 GPa. The enthalpies of various structures of AgI relative to Ag and I are plotted as a function of pressure in Figure 2a,b. The calculated sequence of phase transformation is consistent with our experiment and previous individual studies,44 but our parallel predictions of I and Ag end members suggest that AgI will decompose to Ag and I above 41 GPa using static calculation. We further constrained the mechanical stability limit of AgI-III to 30 GPa, and the monoclinic AgI-V is at a stable phase above ∼12 GPa. We also calculated the formation enthalpy of various Ag–I stoichiometry under high pressure since pressure may create novel compound stoichiometry.30 However, as shown in Figure 2c, none of those unconventional Ag–I compounds are stable at the pressures we are interested in, except for AgI2 (space group P-4m2) at 0 GPa that was originally reported by Persson.45 To consider temperature effects, we calculated the vibration energy within the quasiharmonic approximation for both AgI-III and AgI-V. By calculating the Gibbs free energy of AgI-V, FCC-Ag, and Immm-I, we found that the decomposition pressure changes to ∼11 GPa at 334 K (Figure 3), reaching an agreement with the experiment. Increasing temperature will facilitate decompositions. While static enthalpy calculation works well in predicting the polymorphic phase transition from AgI-II through AgV, increasing the temperature can significantly lower the decomposition pressure. The phonon spectra of AgI-V (and also AgI-III, Figure S2) show a distinct softening of phonon modes at the A point of the Brillouin zone (Figure S2a–c), indicating lattice instability.
Figure 2.

Enthalpy analysis of AgI polymorphs. (a) Enthalpies of the formation of AgI under high pressure relative to Ag and I. (b) Highlight of enthalpy of formation in the pressure range of 0–10 GPa. (c) Enthalpies of formation of Ag–I compounds under high pressure. Solid lines show the convex hulls. Dotted lines through the neighboring points residing above the convex hull are guides for the eyes. All the calculations are performed at static conditions.
Figure 3.

Free energy calculation. (a) Formation Gibbs free energy (ΔG) of AgI-V relative to FCC Ag and Immm-type I. (b) Stability field of AgI-V and its decomposing products. (c) ΔG of AgI-III relative to FCC Ag and Immm-type I.
The pressure dependences of ΔH, ΔU, and PΔV for both AgI-III and V are plotted in Figure S3a,b. A case in point is the surge of the PV term with increasing pressure. In particular, gains of enthalpy from the PV of the AgI compound are greater than that of the sum of Ag and I combined when the pressure is above 2 GPa. Therefore, the PV term is the principal contributor to the increment of enthalpy in the AgI and governs its thermodynamic stability. We also calculated ΔH, ΔU, and PΔV versus pressure for the possible local minima phases of AgI, as shown in Figure S4a in the Supplementary Materials, which exhibit the same tendency.
3.3. MD Simulation
Since the AgI-V was reported to show superionic behavior with a soaring diffusion coefficient, we continued to perform FPMD simulations at relevant P–T conditions. Judging from the mean squared displacement, AgI is a well-defined ionic solid at 300 K, even after prolonged 40 ps simulation time (Figure S5). At 12 GPa and 700 K, AgI-V enters the superionic state with a soaring mean-squared displacement of Ag+, which is consistent with the literature.22 However, with increasing pressure, e.g., at 50 GPa and 500 K, we observed enhanced atomic diffusion of Ag as well as I atoms (Figure S6c,f) and also for AgI-III, Figure S7). In contrast to the conventional superionic phase in which the cation ion (Ag+) freely diffuses in the host lattice, AgI exhibited interdiffusion and consequently, we observed local domains with metallic Ag or I bonds. Within the finite simulation box, we clearly located such domains for evidence of elemental dissociation (inset of Figure 4).
Figure 4.
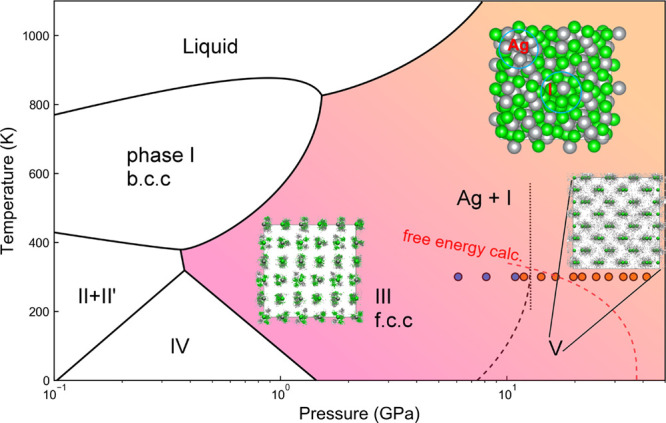
Phase diagram of the Ag–I compound. Solid lines are taken from ref (22). Blue dotted line connects the transition pressure predicted by static enthalpy calculation in Figure 2 and the room-temperature experiment of ref (22). Black dotted line is the transition boundary of AgI-III and V proposed by the electrical conductivity experiment (ref (22)). Blue (AgI solid) and orange (decomposition) solid circles are from XRD data of this work. Red dashed line is from free energy calculation considering vibrational parts. Inset figures are trajectories of the supercell structure taken from MD simulation, with ordering solid in AgI-III, superionic solid in AgI-V and decomposed AgI at the end of MD simulation. Green balls (atomic radius) or dots are for iodine atoms, silver balls (atomic radius) or dots are for silver atoms.
On the basis of our experiment and calculation, we constructed a P–T phase diagram of AgI up to 41.5 GPa (Figure 4). In different pressure regimes, AgI exhibits distinct chemical behaviors. The proceedings of polymorphic transition from AgI-II to AgI-V comply with increasing chemical instability. This intriguing mechanochemistry motivate us to investigate the evolution of Ag–I ionic bonds, which is critical to reveal their reaction mechanism.
4. Discussion
4.1. Theoretical Chemistry on the Ionic Ag–I Bonds
We evaluate the charge transfer between Ag and I atoms through Bader analysis, as shown in Figure 5a. The results for all energetically favored structures at given pressure are shown in Figure S8a of the Supplementary Materials. The charge transfer decreases dramatically under compression, indicating that the strength of the Ag–I ionic bond is gradually weakened. Under high pressure, the shortened bonding lead to orbital overlaps between atoms and the emergence of covalent action.46 Through COHP analysis, which quantifies the overlap strength and crystal orbital overlap population, we calculate the negative integrated COHP (−ICOHP), negative COHP (−COHP), the projected density of states (PDOS), and the bond type descriptor Q defined by Rahm and Hoffmann47,48 to explore the bonding properties for both AgI-III and V in Figure 5b–e.
| 1 |
where n is the total number of electrons, χ̅ is the average binding energy of a collection of electrons, E is the total energy, and χ̅ can be obtained from the DOS:
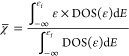 |
2 |
Figure 5.

Bond chemistry of AgI. (a) Net charge of Ag in KOH-type AgI-V and FCC-AgI-III. Charge transfer decreases with increasing pressures, which means that the strength of the Ag–I ionic bond gradually decreases at high pressures. (b)–ICOHP for Ag and I atoms in KOH-type AgI-V and FCC-AgI-III. Value of −ICOHP increases with increasing pressure, which means the orbital overlap between Ag and I increase at high pressure. (c) Calculated PDOS and −COHP for FCC-AgI at 5, and 50 GPa, respectively. States are aligned at the Fermi level (vertical dashed lines). (d) Band gaps and Q value calculated as a function of pressure for the KOH-type AgI-V and FCC-AgI-III. (e) Calculated PDOS for FCC-AgI around Fermi level at 50 GPa.
where εf is the fermi energy in eq 2. The corresponding calculations for ground state structure at given pressure have been plotted in Figure S8b–d and exhibited the same tendency as the AgI-III. The calculated PDOS and −COHP show a large hybridization and an obvious overlap between 4d of Ag and 5p of I orbitals, which indicate a weak covalent interaction between Ag and I atoms (Figure 5d,e). For comparison, the −COHP value of a typical C–C covalent bond in fullerene C60 is 5.9.49 In the meantime, the values of Q, which signifies the ionicity of the Ag–I bond, sharply decrease by a factor of 3 (Figure 5d). The value of −ICOHP continues to increase with pressure, showing the strengthening of covalent interaction between Ag and I atoms. We find that hindered charge transfer weakens the ionicity of the Ag–I bond, but facilitate orbital hybridization, all of which point to the instability of ionic bonding. Energetic-wise, the softened ionicity may play a more substantial role than orbital overlapping during the mechanochemical redox reaction of AgI.
4.2. Mechanism of Mechanochemistry in Compressed AgI
Applying hydrostatic pressure stabilizes novel stoichiometric compounds and sometimes, high pressure is necessary to trigger certain chemical reactions.23,50,51 For example, noble gases are chemically inert under ambient conditions, but readily to form stable compounds under high pressure with various elements include alkali metal,52 alkaline earth metal,53 transition metal,54−56 and nonmetal.46,57−59 In those reports, pressure acts as a global thermodynamic variable that imposes unbiased effects on the chemical bonds. This is in stark contrast with organic mechanochemistry that stretch specific bonds to initiate chemical reactions. In our case of AgI, applying pressure breaks the chemical bond by weakening the ionicity of all Ag–I bonds. As a result, both Ag and I atoms enter large-scale interdiffusion at temperatures far below the theoretical melting temperature of ionic solids (Figure 5). This mechanism of mechanochemistry is readily applicable to other ionic compounds like In(OH)3 and CaLi2, which have been experimentally observed to decompose into elemental solids.60 Our results suggest that the interaction between atoms is important to stabilize ionic compounds and their high-P–T physical phenomena. In the special case of superionic solids, ionic bonding is partially destroyed but still interact to in sustain a solid framework. By losing its ionicity, the solid framework is no longer stable and those previously engaged diffusive ions start to concentrate, form metallic bonds and eventually, trigger the mechanochemical redox reaction and lead to the recovery of elemental solids. We further calculated the formation enthalpies of AgBr and AgCl under high pressures. The progressive weakening of ionic interactions and mechanochemical decomposition are readily applied to these two silver halides, both of which will decompose into elemental solids. Our calculations predict the decomposition pressure at static conditions is 116 GPa for AgBr and 323 GPa for AgCl. Compared to these more genuine ionic solids, the critical pressure in breaking the ionicity of AgI is much lower, and thus more technically achievable through experiment. In addition to ionic compounds, it is also possible that compressed methane and alloys (e.g., lanthanum,61 and Ca–Si62) have a similar decomposition mechanism under external pressure stimuli.
5. Conclusions
Our results suggest that the pressure effects on chemical bonds in materials are rather complex. In principle, the ionicity strongly depends on the relative change of the orbital energies under pressure, and it is not definitive that the ionicity should always reduce, although it seems so for many compounds, especially those consisting of metal atoms that possess low-energy unfilled d levels. Under compression, large charges transfer from anions to these d orbitals, reducing the ionicity.62 In some other cases like alkali metal compounds, the ionicity can increase because of the d-orbital effect.63−66
In this work, both Ag and I have fully filled d orbitals (Figure S8). We show that AgI is a typical example of progressive weakening of ionic interactions between ions under pressurization. At a pressure below 11.9 GPa, the polymorphic phase transition of AgI is driven by the enthalpy change of different phases. Above the critical pressure, the temperature effect sets in, which substantially lowers the decomposition pressure predicted by enthalpy calculation. The decrease of ionicity bypasses further polymorphic phase transition from structural searching (e.g., the Cmcm-type phase) and decompose to end member elements. We conclude that our combined experiment and computation have illustrated the evolution of ionic bonds in AgI under compression at gigapascals conditions. Such a mechanism of pressure in controlling the structural and interatomic interactions may readily introduce to a variety of silver halides and potentially other ionic compounds.
Acknowledgments
This work was supported by the National Natural Science Foundation of China under Grants No. 11974154, the Natural Science Foundation of Shandong Province No. ZR2022MA004, ZR2022MF342, and ZR2022MA023. Q.H. is supported by the CAEP Research Project (CX20210048) and a Tencent Xplorer Prize (XPLORER-2020-1013). M.M. was supported by the National Science Foundation under Grant Nos. DMR-1848141 and OAC-2117956.
Supporting Information Available
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/jacsau.2c00550.
Computational details, unrefined diffraction pattern, phonon dispersion curves, ΔH, ΔU, and PΔV versus pressure, dynamic behavior of Ag and I atoms within AgI at different pressures and temperatures, electronic properties of AgI, and energy versus pressure of AgBr and AgCl (PDF)
Author Contributions
J.L., Q.H., and X.W. designed research; J.L., Y.G., Z.X., P.Z., G.G., M.M., Q.H., and X.W. performed research; J.L., Q.H., M.M., and X.W. analyzed data; and J.L., Q.H., and X.W. wrote the paper.
The authors declare no competing financial interest.
Supplementary Material
References
- Krusenbaum A.; Grätz S.; Tigineh G. T.; Borchardt L.; Kim J. G. The Mechanochemical Synthesis of Polymers. Chem. Soc. Rev. 2022, 51, 2873–2905. 10.1039/D1CS01093J. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shan N.; Toda F.; Jones W. Mechanochemistry and Co-Crystal Formation: Effect of Solvent on Reaction Kinetics. Chem. Commun. 2002, 20, 2372–2373. 10.1039/b207369m. [DOI] [PubMed] [Google Scholar]
- Muñoz-Batista M. J.; Rodriguez-Padron D.; Puente-Santiago A. R.; Luque R. Mechanochemistry: Toward Sustainable Design of Advanced Nanomaterials for Electrochemical Energy Storage and Catalytic Applications. ACS Sustainable Chem. Eng. 2018, 6, 9530–9544. 10.1021/acssuschemeng.8b01716. [DOI] [Google Scholar]
- Kargin V. A.; Plate N. A. Chemical Grafting on Crystal Surfaces. Vysok. Soed 1959, 1, 330. [Google Scholar]
- Ravnsbæk J. B.; Swager T. M. Mechanochemical Synthesis of Poly(Phenylene Vinylenes). ACS Macro Lett. 2014, 3, 305–309. 10.1021/mz500098r. [DOI] [PubMed] [Google Scholar]
- Do J.-L.; Friščić T. Mechanochemistry: A Force of Synthesis. ACS Cent. Sci. 2017, 3, 13–19. 10.1021/acscentsci.6b00277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tan D.; Loots L.; Friščić T. Towards Medicinal Mechanochemistry: Evolution of Milling from Pharmaceutical Solid Form Screening to the Synthesis of Active Pharmaceutical Ingredients (APIs). Chem. Commun. 2016, 52, 7760–7781. 10.1039/C6CC02015A. [DOI] [PubMed] [Google Scholar]
- Yan H.; Yang F.; Pan D.; Lin Y.; Hohman J. N.; Solis-Ibarra D.; Li F. H.; Dahl J. E. P.; Carlson R. M. K.; Tkachenko B. A.; Fokin A. A.; Schreiner P. R.; Galli G.; Mao W. L.; Shen Z.-X.; Melosh N. A. Sterically Controlled Mechanochemistry under Hydrostatic Pressure. Nature 2018, 554, 505–510. 10.1038/nature25765. [DOI] [PubMed] [Google Scholar]
- Horie K.; Barón M.; Fox R. B.; He J.; Hess M.; Kahovec J.; Kitayama T.; Kubisa P.; Maréchal E.; Mormann W.; Stepto R. F. T.; Tabak D.; Vohlídal J.; Wilks E. S.; Work W. J. Definitions of Terms Relating to Reactions of Polymers and to Functional Polymeric Materials (IUPAC Recommendations 2003). Pure Appl. Chem. 2004, 76, 889–906. 10.1351/pac200476040889. [DOI] [Google Scholar]
- Faraday M. Literature and the Arts. Q. J. Sci. 1820, 8, 374. [Google Scholar]
- Scano A.; Ennas G. 3 Rediscovering Mechanochemistry for Inorganic Materials. In Mechanochemistry; De Gruyter, 2020; pp 23–40. [Google Scholar]
- Anand M.; Sunoj R. B.; Schaefer H. F. Non-Innocent Additives in a Palladium(II)-Catalyzed C-H Bond Activation Reaction: Insights into Multimetallic Active Catalysts. J. Am. Chem. Soc. 2014, 136, 5535–5538. 10.1021/ja412770h. [DOI] [PubMed] [Google Scholar]
- Bay K. L.; Yang Y.-F.; Houk K. N. Multiple Roles of Silver Salts in Palladium-Catalyzed C–H Activations. J. Organomet. Chem. 2018, 864, 19–25. 10.1016/j.jorganchem.2017.12.026. [DOI] [Google Scholar]
- Farrington G. C.; Briant J. L. Fast Ionic Transport in Solids. Science 1979, 204, 1371–1379. 10.1126/science.204.4400.1371. [DOI] [PubMed] [Google Scholar]
- Guo Y. G.; Lee J. S.; Maier J. AgI Nanoplates with Mesoscopic Superionic Conductivity at Room Temperature. Adv. Mater. 2005, 17, 2815–2819. 10.1002/adma.200501215. [DOI] [Google Scholar]
- Makiura R.; Yonemura T.; Yamada T.; Yamauchi M.; Ikeda R.; Kitagawa H.; Kato K.; Takata M. Size-Controlled Stabilization of the Superionic Phase to Room Temperature in Polymer-Coated AgI Nanoparticles. Nat. Mater. 2009, 8, 476–480. 10.1038/nmat2449. [DOI] [PubMed] [Google Scholar]
- Zhang H.; Tsuchiya T.; Liang C.; Terabe K. Size-Controlled AgI/Ag Heteronanowires in Highly Ordered Alumina Membranes: Superionic Phase Stabilization and Conductivity. Nano Lett. 2015, 15, 5161–5167. 10.1021/acs.nanolett.5b01388. [DOI] [PubMed] [Google Scholar]
- Hanson R. C.; Fjeldly T. A.; Hochheimer H. D. Raman Scattering from Five Phases of Silver Iodide. Phys. Status Solidi 1975, 70, 567–576. 10.1002/pssb.2220700216. [DOI] [Google Scholar]
- Keen D. A.; Hull S. A Powder Neutron Diffraction Study of the Pressure-Induced Phase Transitions within Silver Iodide. J. Phys.: Condens. Matter 1993, 5, 23–32. 10.1088/0953-8984/5/1/005. [DOI] [Google Scholar]
- Hull S.; Keen D. Pressure-Induced Phase Transitions in AgCl, AgBr, and AgI. Phys. Rev. B: Condens. Matter Mater. Phys. 1999, 59, 750–761. 10.1103/PhysRevB.59.750. [DOI] [Google Scholar]
- Catti M. First-Principles Landau Potential for the Rocksalt to KOH to TlI-Type Phase Transitions of AgI. Phys. Rev. B: Condens. Matter Mater. Phys. 2006, 74, 174105 10.1103/PhysRevB.74.174105. [DOI] [Google Scholar]
- Han Y. H.; Wang H. B.; Troyan I. A.; Gao C. X.; Eremets M. I. Pressure Induced Ionic-Superionic Transition in Silver Iodide at Ambient Temperature. J. Chem. Phys. 2014, 140, 044708 10.1063/1.4862824. [DOI] [PubMed] [Google Scholar]
- Miao M.; Sun Y.; Zurek E.; Lin H. Chemistry under High Pressure. Nat. Rev. Chem. 2020, 4, 508–527. 10.1038/s41570-020-0213-0. [DOI] [Google Scholar]
- Letoullec R.; Pinceaux J. P.; Loubeyre P. The Membrane Diamond Anvil Cell: A New Device for Generating Continuous Pressure and Temperature Variations. High Press. Res. 1988, 1, 77–90. 10.1080/08957958808202482. [DOI] [Google Scholar]
- Prescher C.; Prakapenka V. B. DIOPTAS: A Program for Reduction of Two-Dimensional X-Ray Diffraction Data and Data Exploration. High Press. Res. 2015, 35, 223–230. 10.1080/08957959.2015.1059835. [DOI] [Google Scholar]
- Wang Y.; Lv J.; Zhu L.; Ma Y. Crystal Structure Prediction via Particle-Swarm Optimization. Phys. Rev. B: Condens. Matter Mater. Phys. 2010, 82, 094116 10.1103/PhysRevB.82.094116. [DOI] [Google Scholar]
- Wang Y.; Lv J.; Zhu L.; Ma Y. CALYPSO: A Method for Crystal Structure Prediction. Comput. Phys. Commun. 2012, 183, 2063–2070. 10.1016/j.cpc.2012.05.008. [DOI] [Google Scholar]
- Gao B.; Gao P.; Lu S.; Lv J.; Wang Y.; Ma Y. Interface Structure Prediction via CALYPSO Method. Sci. Bull. 2019, 64, 301–309. 10.1016/j.scib.2019.02.009. [DOI] [PubMed] [Google Scholar]
- Wang X.; Wang Y.; Miao M.; Zhong X.; Lv J.; Cui T.; Li J.; Chen L.; Pickard C. J.; Ma Y. Cagelike Diamondoid Nitrogen at High Pressures. Phys. Rev. Lett. 2012, 109, 175502 10.1103/PhysRevLett.109.175502. [DOI] [PubMed] [Google Scholar]
- Zhang L.; Wang Y.; Lv J.; Ma Y. Materials Discovery at High Pressures. Nat. Rev. Mater. 2017, 2, 17005. 10.1038/natrevmats.2017.5. [DOI] [Google Scholar]
- Lin J.; Wang F.; Rui Q.; Li J.; Wang Q.; Wang X. A Novel Square Planar N42– Ring with Aromaticity in BeN4. Matter Radiat. Extrem. 2022, 7, 038401 10.1063/5.0084802. [DOI] [Google Scholar]
- Perdew J. P.; Burke K.; Ernzerhof M. Generalized Gradient Approximation Made Simple. Phys. Rev. Lett. 1996, 77, 3865–3868. 10.1103/PhysRevLett.77.3865. [DOI] [PubMed] [Google Scholar]
- Patton D. C.; Pederson M. R. Application of the Generalized-Gradient Approximation to Rare-Gas Dimers. Phys. Rev. A: At., Mol., Opt. Phys. 1997, 56, R2495–R2498. 10.1103/PhysRevA.56.R2495. [DOI] [Google Scholar]
- Kresse G.; Furthmüller J. Efficient Iterative Schemes for Ab Initio Total-Energy Calculations Using a Plane-Wave Basis Set. Phys. Rev. B: Condens. Matter Mater. Phys. 1996, 54, 11169–11186. 10.1103/PhysRevB.54.11169. [DOI] [PubMed] [Google Scholar]
- Blöchl P. E. Projector Augmented-Wave Method. Phys. Rev. B: Condens. Matter Mater. Phys. 1994, 50, 17953–17979. 10.1103/PhysRevB.50.17953. [DOI] [PubMed] [Google Scholar]
- Monkhorst H. J.; Pack J. D. Special Points for Brillouin-Zone Integrations. Phys. Rev. B: Solid State 1976, 13, 5188–5192. 10.1103/PhysRevB.13.5188. [DOI] [Google Scholar]
- Nosé S. A Unified Formulation of the Constant Temperature Molecular Dynamics Methods. J. Chem. Phys. 1984, 81, 511–519. 10.1063/1.447334. [DOI] [Google Scholar]
- Hoover W. G. Canonical Dynamics: Equilibrium Phase-Space Distributions. Phys. Rev. A: At., Mol., Opt. Phys. 1985, 31, 1695–1697. 10.1103/PhysRevA.31.1695. [DOI] [PubMed] [Google Scholar]
- Dronskowski R.; Bloechl P. E. Crystal Orbital Hamilton Populations (COHP): Energy-Resolved Visualization of Chemical Bonding in Solids Based on Density-Functional Calculations. J. Phys. Chem. 1993, 97, 8617–8624. 10.1021/j100135a014. [DOI] [Google Scholar]
- Maintz S.; Deringer V. L.; Tchougréeff A. L.; Dronskowski R. LOBSTER: A Tool to Extract Chemical Bonding from Plane-Wave Based DFT. J. Comput. Chem. 2016, 37, 1030–1035. 10.1002/jcc.24300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ivantchev S.; Kroumova E.; Madariaga G.; Pérez-Mato J. M.; Aroyo M. I. SUBGROUPGRAPH: A Computer Program for Analysis of Group–Subgroup Relations between Space Groups. J. Appl. Crystallogr. 2000, 33, 1190–1191. 10.1107/S0021889800007135. [DOI] [Google Scholar]
- Zhu S.-C.; Chen G.-W.; Zhang D.; Xu L.; Liu Z.-P.; Mao H.; Hu Q. Topological Ordering of Memory Glass on Extended Length Scales. J. Am. Chem. Soc. 2022, 144, 7414–7421. 10.1021/jacs.2c01717. [DOI] [PubMed] [Google Scholar]
- Bu K.; Hu Q.; Qi X.; Wang D.; Guo S.; Luo H.; Lin T.; Guo X.; Zeng Q.; Ding Y.; Huang F.; Yang W.; Mao H.-K.; Lü X. Nested Order-Disorder Framework Containing a Crystalline Matrix with Self-Filled Amorphous-like Innards. Nat. Commun. 2022, 13, 4650. 10.1038/s41467-022-32419-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li Y.; Zhang L. J.; Cui T.; Li Y. W.; Wang Y.; Ma Y. M.; Zou G. T. First-Principles Studies of Phonon Instabilities in AgI under High Pressure. J. Phys.: Condens. Matter 2008, 20, 195218 10.1088/0953-8984/20/19/195218. [DOI] [Google Scholar]
- Persson K.Materials Data on AgI2 (SG:115) by Materials Project; 2014.
- Peng F.; Wang Y.; Wang H.; Zhang Y.; Ma Y. Stable Xenon Nitride at High Pressures. Phys. Rev. B: Condens. Matter Mater. Phys. 2015, 92, 094104 10.1103/PhysRevB.92.094104. [DOI] [Google Scholar]
- Rahm M.; Hoffmann R. Toward an Experimental Quantum Chemistry: Exploring a New Energy Partitioning. J. Am. Chem. Soc. 2015, 137, 10282–10291. 10.1021/jacs.5b05600. [DOI] [PubMed] [Google Scholar]
- Rahm M.; Hoffmann R. Distinguishing Bonds. J. Am. Chem. Soc. 2016, 138, 3731–3744. 10.1021/jacs.5b12434. [DOI] [PubMed] [Google Scholar]
- Maintz S.; Deringer V. L.; Tchougréeff A. L.; Dronskowski R. Analytic Projection from Plane-Wave and PAW Wavefunctions and Application to Chemical-Bonding Analysis in Solids. J. Comput. Chem. 2013, 34, 2557–2567. 10.1002/jcc.23424. [DOI] [PubMed] [Google Scholar]
- Yoo C.-S. Chemistry under Extreme Conditions: Pressure Evolution of Chemical Bonding and Structure in Dense Solids. Matter Radiat. Extrem. 2020, 5, 018202 10.1063/1.5127897. [DOI] [Google Scholar]
- Tse J. S. A Chemical Perspective on High Pressure Crystal Structures and Properties. Natl. Sci. Rev. 2020, 7, 149–169. 10.1093/nsr/nwz144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dong X.; Oganov A. R.; Goncharov A. F.; Stavrou E.; Lobanov S.; Saleh G.; Qian G.-R.; Zhu Q.; Gatti C.; Deringer V. L.; Dronskowski R.; Zhou X.-F.; Prakapenka V. B.; Konôpková Z.; Popov I. A.; Boldyrev A. I.; Wang H.-T. A Stable Compound of Helium and Sodium at High Pressure. Nat. Chem. 2017, 9, 440–445. 10.1038/nchem.2716. [DOI] [PubMed] [Google Scholar]
- Miao M.; Wang X.; Brgoch J.; Spera F.; Jackson M. G.; Kresse G.; Lin H. Anionic Chemistry of Noble Gases: Formation of Mg–NG (NG = Xe, Kr, Ar) Compounds under Pressure. J. Am. Chem. Soc. 2015, 137, 14122–14128. 10.1021/jacs.5b08162. [DOI] [PubMed] [Google Scholar]
- Zhu L.; Liu H.; Pickard C. J.; Zou G.; Ma Y. Reactions of Xenon with Iron and Nickel Are Predicted in the Earth’s Inner Core. Nat. Chem. 2014, 6, 644–648. 10.1038/nchem.1925. [DOI] [PubMed] [Google Scholar]
- Liu Z.; Botana J.; Hermann A.; Valdez S.; Zurek E.; Yan D.; Lin H. Q.; Miao M. S. Reactivity of He with Ionic Compounds under High Pressure. Nat. Commun. 2018, 9, 951. 10.1038/s41467-018-03284-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dewaele A.; Pépin C. M.; Geneste G.; Garbarino G. Reaction between Nickel or Iron and Xenon under High Pressure. High Press. Res. 2017, 37, 137–146. 10.1080/08957959.2016.1267165. [DOI] [Google Scholar]
- Kim M.; Debessai M.; Yoo C.-S. Two- and Three-Dimensional Extended Solids and Metallization of Compressed XeF2. Nat. Chem. 2010, 2, 784–788. 10.1038/nchem.724. [DOI] [PubMed] [Google Scholar]
- Braïda B.; Hiberty P. C. The Essential Role of Charge-Shift Bonding in Hypervalent Prototype XeF2. Nat. Chem. 2013, 5, 417–422. 10.1038/nchem.1619. [DOI] [PubMed] [Google Scholar]
- Dewaele A.; Worth N.; Pickard C. J.; Needs R. J.; Pascarelli S.; Mathon O.; Mezouar M.; Irifune T. Synthesis and Stability of Xenon Oxides Xe2O5 and Xe3O2 under Pressure. Nat. Chem. 2016, 8, 784–790. 10.1038/nchem.2528. [DOI] [PubMed] [Google Scholar]
- Gurlo A.; Dzivenko D.; Andrade M.; Riedel R.; Lauterbach S.; Kleebe H.-J. J. Pressure-Induced Decomposition of Indium Hydroxide. J. Am. Chem. Soc. 2010, 132, 12674–12678. 10.1021/ja104278p. [DOI] [PubMed] [Google Scholar]
- Yang X.; Li H.; Liu H.; Wang H.; Yao Y.; Xie Y. Pressure-Induced Decomposition of Binary Lanthanum Intermetallic Compounds. Phys. Rev. B 2020, 101, 184113 10.1103/PhysRevB.101.184113. [DOI] [Google Scholar]
- Gao G.; Ashcroft N. W.; Miao M.; Hoffmann R. Novel Si Networks in the Ca/Si Phase Diagram under Pressure. J. Phys. Chem. C 2014, 118, 25167–25175. 10.1021/jp507813y. [DOI] [Google Scholar]
- Li X.; Hermann A.; Peng F.; Lv J.; Wang Y.; Wang H.; Ma Y. Stable Lithium Argon Compounds under High Pressure. Sci. Rep. 2015, 5, 16675. 10.1038/srep16675. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rogachev A. Y.; Miao M.; Merino G.; Hoffmann R. Molecular CsF5 and CsF2+. Angew. Chem., Int. Ed. 2015, 54, 8275–8278. 10.1002/anie.201500402. [DOI] [PubMed] [Google Scholar]
- Botana J.; Brgoch J.; Hou C.; Miao M. Iodine Anions beyond −1: Formation of LinI(N=2–5) and Its Interaction with Quasiatoms. Inorg. Chem. 2016, 55, 9377–9382. 10.1021/acs.inorgchem.6b01561. [DOI] [PubMed] [Google Scholar]
- Liu Z.; Botana J.; Miao M.; Yan D. Unexpected Xe Anions in XeLin Intermetallic Compounds. EPL 2017, 117, 26002. 10.1209/0295-5075/117/26002. [DOI] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.