

Abstract
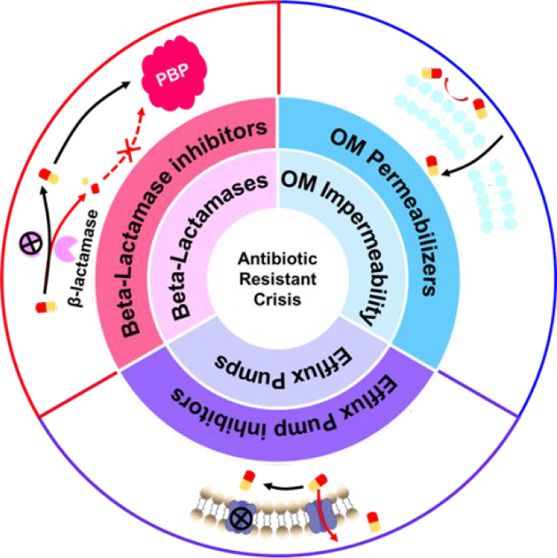
The antimicrobial resistance crisis is a global health issue requiring discovery and development of novel therapeutics. However, conventional screening of natural products or synthetic chemical libraries is uncertain. Combination therapy using approved antibiotics with inhibitors targeting innate resistance mechanisms provides an alternative strategy to develop potent therapeutics. This review discusses the chemical structures of effective β-lactamase inhibitors, outer membrane permeabilizers, and efflux pump inhibitors that act as adjuvant molecules of classical antibiotics. Rational design of the chemical structures of adjuvants will provide methods to impart or restore efficacy to classical antibiotics for inherently antibiotic-resistant bacteria. As many bacteria have multiple resistance pathways, adjuvant molecules simultaneously targeting multiple pathways are promising approaches to combat multidrug-resistant bacterial infections.
Keywords: antimicrobial resistance, combination therapy, β-lactamase inhibitors, outer membrane permeabilizers, efflux pump inhibitors
Introduction
The discovery of new antibiotic classes in the past three decades has been slow. The reasons include limited investment in drug discovery endeavors by major pharmaceutical companies, depletion of low-hanging fruits from the previous decade, and exhaustion of new drug candidates because of the use of the same drug compound libraries.1 The use of combination therapies to treat bacterial infections is still an emerging field of research that may provide a promising complementary strategy to fight antimicrobial resistance in pathogenic bacteria. However, rational design of such adjuvants requires an understanding of the correlation between the chemistry of adjuvant molecules and the biology of the resistance mechanism. In this review, we seek to summarize the chemical basis of combination therapies to combat antimicrobial resistance.
The four general classes of antibiotic resistance mechanisms include (a) enzymatic degradation, (b) outer membrane impermeability, (c) the presence of efflux pumps, and (d) target mutation.2,3 In the case of target mutation, no adjuvant drugs have been discovered to counteract this mechanism. The most clinically important class of evolved resistance is enzymatic degradation of β-lactam antibiotics by β-lactamases.4 These enzymes are encoded by genes on mobilized plasmids, which can be easily transferred between bacterial species, resulting in the spread of drug resistance.2 In addition to the enzymatic degradation of drugs, bacteria also evolve resistance to antibiotics via enzymatic modification of the antibiotics by introducing various chemical groups, such as acyl, phosphate, nucleotidyl, and ribitoyl groups,5 resulting in preventing the antibiotics from binding to their targets due to steric hindrance.2 Aminoglycosides are particularly vulnerable to enzymatic modification since they possess many modifiable amine and hydroxyl groups. N-Acetyltransferases, O-nucleotidyltransferases, and O-phosphotransferases are the three main aminoglycoside-modifying enzymes conferring resistance to aminoglycosides.6 Strategies to overcome aminoglycoside modifying enzymes have been thoroughly reviewed by Garneau-Tsodikova et al.7 and Zhou et al.8 Furthermore, the potency of most antibiotics is highly restricted by intracellular accumulation of the drugs inside bacteria, which is controlled by the passage of drugs to their targets and their accumulation at the target site. Gram-negative bacteria have intrinsic resistance to many classes of antibiotics due to the impermeability of their outer membrane,9 which limits the passage of antibiotics to their targets. Few antibiotics are sufficiently potent to treat Gram-negative bacterial infections compared with those that can treat Gram-positive bacterial infections. Moreover, accumulation of small hydrophilic antibiotics that cross the bacterial cell wall via porins may be prevented by efflux pumps in the bacterial membrane,10,11 leading to efflux-mediated antibiotic resistance to multiple antibiotics.12 Worryingly, the three resistance mechanisms, i.e., β-lactamases, outer membrane impermeability, and efflux pumps, can also work in concert,13,14 leading to a high level of antibiotic resistance, as observed in some deadly ESKAPE pathogens (Enterococcus faecium, Staphylococcus aureus, Klebsiella pneumoniae, Acinetobacter baumannii, Pseudomonas aeruginosa, and Enterobacter species), which are responsible for most nosocomial infection cases.15
A relatively unexploited strategy to treat bacterial infections involves combining antibiotics with adjuvant molecules.16,17 Many possibilities exist for combination therapies, but only a few limited cases of pairing antibiotics with adjuvants have been translated to clinical practice. The most successful combination therapy is Augmentin, a combination of the β-lactamase inhibitor clavulanic acid and the β-lactam antibiotic amoxicillin.18 Herein, we review the chemical structure of three broad classes of antibiotic adjuvant molecules, specifically (a) β-lactamase inhibitors, (b) outer membrane permeabilizers, and (c) efflux pump inhibitors; discuss their mechanism of action; and highlight the most promising adjuvants under development. Insights and future prospects are also proposed in the hope that they will contribute to future progress in the development of antibiotic/adjuvant combinations to fight the antibiotic resistance crisis.
β-Lactamase Inhibitors
β-Lactam antibiotics target essential penicillin-binding proteins involved in peptidoglycan synthesis. They have been a therapeutic mainstay for over 70 years.19 However, the production of β-lactamases by many bacteria has caused many β-lactam antibiotic treatments to become futile.20 β-Lactamases have evolved with the development of newer generations of β-lactam antibiotics, culminating in the carbapenemase New Delhi metallo-β-lactamase-1 (NDM-1), which causes treatment failure of some last-resort carbapenem antibiotics against Gram-negative bacteria.21 According to the World Health Organization, carbapenem-resistant Gram-negative bacteria are the pathogens for which new antibiotics are the most critically needed.22
β-Lactamases generally hydrolyze antibiotics through the formation of tetrahedral intermediates (Figure 1). Four classes of β-lactamases are produced by bacteria. They are distinguished by specific sequence motifs and are classified as class A, B, C, and D β-lactamases.23 Class A, C, and D β-lactamases utilize serine to hydrolyze β-lactam antibiotics via covalent acylation resulting from the addition of the nucleophilic amino acid serine (Figure 1A).24 Class B β-lactamases are metalloenzymes that require divalent Zn2+ ions to hydrolyze β-lactams via a metal-catalyzed water nucleophilic reaction (Figure 1B).24
Figure 1.

Mechanism of β-lactam antibiotic hydrolysis by (A) serine β-lactamases (Ser-BLs) and (B) metallo-β-lactamases (MBLs).
β-Lactamase inhibitors are chemically classified according to their chemical structures: (i) β-lactam-ring-containing inhibitors, which are the earliest generation of inhibitors and are effective against class A β-lactamases; (ii) diazabicyclooctane-containing inhibitors, which broaden the efficacy against class A, C, and D β-lactamases; (iii) metallo-β-lactamase (MBL) inhibitors, which include a variety of chemical structures to combat class B β-lactamases; and (iv) other polymeric inhibitors (Table 1 and Scheme 1). The common characteristic of these β-lactamase inhibitors is rapid formation of stable intermediates with β-lactamases via either nucleophilic addition or competitive displacement of Zn2+ ions by the functional moieties (Figure 2), such as the β-lactam ring, diazabicyclooctane, chelating groups, and boronic acid.
Table 1. Summary of the Potentiating Effects of β-Lactamase Inhibitors.
| no. | β-lactamase Inhibitors | molecular structure | combined antibiotic drug | class | microbial species | MICa of drug alone | MICa in combination | fold decrease | FIC | ref |
|---|---|---|---|---|---|---|---|---|---|---|
| a | tazobactam | Scheme 1a | piperacillin | A | K. pneumoniae | >200 | 3.13 | >64 | N.A. | (25) |
| b | sulbactam | Scheme 1b | piperacillin | A | K. pneumoniae | >200 | 50 | >4 | N.A. | (25) |
| c | clavulanic acid | Scheme 1c | piperacillin | A | K. pneumoniae | >200 | 1.56 | >128 | N.A. | (25) |
| d | AAI101 | Scheme 1d | cefepime | A | Enterobacteriaceae | >64 | 0.13 | >492 | N.A. | (29) |
| e | LN-1-255 | Scheme 1e | imipenem, | A, D | K. pneumoniae | 64 | 4 | 16 | N.A. | (31) |
| f | avibactam | Scheme 1f | ceftazidime | A, C, D | Enterobacteriaceae | 32 | 0.5 | 64 | N.A. | (32) |
| g | nacubactam | Scheme 1g | meropenem | A, C, D | P. aeruginosa | 8 | 2 | 4 | <0.375 | (37) |
| h | ETX2514 | Scheme 1h | meropenem | A, C, D | A. baumannii | 64 | 0.5 | 128 | <0.5 | (38) |
| i | bisthiazolidines | Scheme 1i | imipenem | B | A. baumannii | 128 | 16 | 8 | N.A. | (39) |
| j | MMTZs | Scheme 1 j | imipenem | B | Enterobacteriaceae | 1-64 | 0.25–16 | ≥4 | N.A. | (41) |
| k | thanatin | Scheme 1k | meropenem | B | E. coli | 144* | 18* | 8 | 0.625 | (42) |
| l | ebselen | Scheme 1l | meropenem | B | E. coli | 333* | 0.65* | 512 | <0.1 | (43) |
| m | auranofin | Scheme 1m | meropenem | B | E. coli | 16 | 0.5 | 32 | 0.126 | (44) |
| n | PRX7009 | Scheme 1n | meropenem | B | Enterobacteriaceae | >32 | 1 | >32 | N.A. | (45) |
| o | QPX7728 | Scheme 1o | meropenem | A, B, C, D | Enterobacteriaceae | >32 | 0.125 | >256 | N.A. | (46) |
| p | cobaltocenium-containing polymers | Scheme 1p | cefazolin | N.A. | MRSA | N.A.b | N.A.b | N.A.b | N.A.b | (47) |
| q | CSM5-K5 | Scheme 1q | oxacillin | N.A. | E. faecalis | 32 | 2 | 16 | 0.25–0.31 | (48) |
The MIC is given as μg/mL, except when specified by “*”, which indicates that the MIC is given as μM.
The synergy was determined by a disc diffusion assay. N.A., not available.
Scheme 1. Chemical Structure of β-Lactamase Inhibitors: (a)–(e) β-Lactam-Ring-Containing Inhibitors; (f)–(h) Diazabicyclooctane-Containing inhibitors; (i)–(o) Metallo-β-Lactamase Inhibitors; (p) and (q) Other Polymeric Inhibitors.

Figure 2.

General mechanism of β-lactamase inhibitors. (A) Acylation of serine-β-lactamases (Ser-BLs) by tazobactam to form an ester linkage. (B) Reversible acylation of Ser-BLs by avibactam to form a carbamoyl linkage. (C) Bisthiazolidines bind to the dizinc centers of metallo-β-lactamases (MBLs) via a free thiol group. (D) Boronic acid inhibitors form tetrahedral intermediates with β-lactamases.
Tazobactam, sulbactam, and clavulanic acid (Table 1a–c, Scheme 1a–c) are three important clinically approved β-lactamase inhibitors.25 They are β-lactam-ring-containing β-lactamase inhibitors that can irreversibly acylate β-lactamases by attacking the serine group of the enzyme26 (Figure 2A), which prevents hydrolysis of the coadministered β-lactam antibiotics by serine-β-lactamases (Ser-BLs). However, they are active only against class A β-lactamases.27 Moreover, their efficacy has been compromised by the evolution of newer β-lactamases, such as those found in K. pneumoniae carbapenemases (KPCs).28 AAI101 (Table 1d, Scheme 1d) is prepared by quaternization of the triazole ring of tazobactam and has shown improved inhibition against class A KPCs.29. It is under clinical Phase III development in combination with cefepime30. The optimization of this type of β-lactamase inhibitor through 6-alkylidene and 2′-substitution of tazobactam led to LN-1-255 (Table 1e, Scheme 1e), which inhibits not only class A β-lactamases but also class D β-lactamases,31 including oxacillinase (OXA)-24 and OXA-48.
Avibactam (Table 1f, Scheme 1f),32 which is a β-lactamase inhibitor without the β-lactam ring, consists of a diazabicyclooctane scaffold. It shows potency against not only class A β-lactamases, including KPC-type carbapenemases, but also class C and a few class D β-lactamases.4 Avibactam has a bicyclic core connected by a urea bond, representing a structural difference from β-lactam. The advanced β-lactamase inhibitors should rapidly acylate β-lactamases with slow hydrolysis. Avibactam can rapidly acylate the serine group in Ser-BLs to form a carbamoyl linkage (Figure 2B(i)), which is much more resistant to hydrolysis than the ester linkage of β-lactam-ring-containing β-lactamase inhibitors, such as tazobactam (Figure 2A).33 The absence of the β-lactam ring scaffold in the molecular structure of avibactam may contribute to its evasion of the mechanism that leads to resistance to β-lactam-ring-containing inhibitors.34 Moreover, because the ring strain energy of the five-membered ring in diazabicyclooctane is low, avibactam can be regenerated through reverse deacylation of the carbamoyl linkage due to the intramolecular ring closure (Figure 2B(ii)), which can subsequently deactivate other β-lactamases. These properties promoted the approval, in 2015, of avibactam and ceftazidime combination therapy to treat serious infections caused by Gram-negative bacteria.35
Another diazabicyclooctane compound under development is nacubactam (Table 1g, Scheme 1g), which is prepared via substitution of the carboxamide side chain of avibactam with a 2-aminoethoxy group. Nacubactam showed interesting properties of not only inhibiting Ser-BLs but also targeting penicillin-binding protein 2 (PBP2) to directly inhibit peptidoglycan synthesis.36,37 Because of its dual inhibitory mechanisms, nacubactam displays strong synergistic activity with various β-lactam antibiotics, including piperacillin, cefepime, aztreonam, and meropenem. Furthermore, ETX2514 (Table 1h, Scheme 1h), which is a rationally designed molecule produced by modification of the piperidine ring of avibactam to 4-methyl-1,2,3,6-tetrahydropyridine to increase the chemical reactivity and binding efficacy to β-lactamases, also shows dual inhibition of both Ser-BLs and PBP2 with an intrinsic antimicrobial potency against E. coli and K. pneumoniae. Compared to other diazabicyclooctane β-lactamase inhibitors, ETX2514 exhibited faster and better inhibition efficacy against all Ser-BLs studied.38
Although success of the translation of β-lactam-ring-containing β-lactamase inhibitors and various diazabicyclooctane derivatives to clinical application provides more treatment options for serious infections caused by β-lactam antibiotic-resistant bacteria, neither is effective against class B metallo-β-lactamases (MBLs). These enzymes are extremely important because they can hydrolyze all clinically useful β-lactam antibiotics, including carbapenems (except for monobactams).26 Nevertheless, monobactams are degradable by Ser-BLs, which are frequently found in pathogens along with MBLs. The ineffectiveness of the β-lactam inhibitors discussed above against MBLs may arise from the different hydrolytic mechanisms of MBLs (Figure 1B). Structurally, all MBLs contain conserved motifs to coordinate Zn2+ ions, which are essential for catalytic hydrolysis.24 In fact, most MBL inhibitors act via binding or chelating Zn2+ ions. Bisthiazolidines (Table 1i, Scheme 1i),39 which are thiol-containing bicyclic molecules, bind to the dizinc centers of MBLs via free thiol groups40 to replace the hydroxide of dizinc clusters during the hydrolytic process (Figure 2C). In addition to thiolate Zn2+ ion clusters, 2-mercaptomethyl-thiazolidines (MMTZs) (Table 1j, Scheme 1j) can form sulfur–aromatic interactions between the thioether and the binding pocket of MBLs,41 which further stabilizes the thiolated Zn2+ ion clusters. Thus, MMTZs exhibit more potent inhibition of MBLs than do bisthiazolidines.41
The antimicrobial peptide thanatin (Table 1k, Scheme 1k) inhibits NDM-1 by extracting Zn2+ ions via the strong chelating power of its guanidine group.42 Another interesting MBL inhibitor is the selenium-containing molecule ebselen (Table 1l, Scheme 1l).43 Ebselen functions as a Zn2+ ion competitor that binds to a conserved motif in NDM-1.43 Similarly, Li et al. found that the antirheumatic drug auranofin (Table 1m, Scheme 1m) restored meropenem activity against NDM-1-positive Enterobacteriaceae due to its strong thiophilicity, forming Au-NDM-1 complexes.44 Boronic acid can mimic the tetrahedral intermediate formed during the hydrolysis process (Figure 1B) to inhibit both Ser-BLs and MBLs (Figure 2D). Vaborbactam (RPX7009) (Table 1n, Scheme 1n) is the leading boronic acid-containing β-lactamase inhibitor and is now undergoing clinical Phase III trials.45 This drug candidate is used in combination with meropenem against carbapenemase-producing Gram-negative bacteria. Recently, Hecker et al. discovered an ultrabroad-spectrum β-lactamase inhibitor cyclic boronic acid QPX7728 (Table 1o, Scheme 1o) that displayed remarkable potency against all classes of β-lactamases with potential for both intravenous and oral administration.46 QPX7728 boosted meropenem against extended-spectrum beta-lactamase producing Enterobacteriaceae even better than vaborbactam and has advanced to late-stage preclinical development.
Unlike small-molecule inhibitors, cationic cobaltocenium-containing polymers (Table 1p, Scheme 1p) potentiate the action of multiple β-lactam antibiotics against methicillin-resistant S. aureus (MRSA) by forming bioconjugates with anionic β-lactam antibiotics via electrostatic interactions;47 these bioconjugates are highly resistant to hydrolysis by β-lactamases. Kline et al. found that CSM5-K5 (Table 1q, Scheme 1q), a chitosan derivative synthesized by Chan-Park et al., restored oxacillin activity against MRSA USA300 by targeting the large surface protein Ebh, which is associated with resistance of S. aureus to oxacillin.48 The increased potency of cationic polymer inhibitors compared to that of small-molecule inhibitors may result from the initial high-affinity binding to the bacterial envelope through electrostatic interaction.
β-Lactams comprise an important antibiotic class in the current antibiotic arsenal, and combination therapy using β-lactam antibiotics with β-lactamase inhibitors represents a validated strategy to overcome β-lactamase resistance in bacteria, which has extended the life of β-lactam antibiotics. Although many promising β-lactamase inhibitors have been discovered, resistance to these new inhibitors and their combinations has already been detected in both the laboratory and clinical settings,20 which may be attributed to the specific protein targets of these inhibitors. Therefore, continuous exploration of new β-lactamase inhibitors is needed, which should be accompanied by strict controls on the use of these new inhibitors to avoid, as much as possible, the emergence of resistance to these drugs. Despite fruitful progress in the discovery of β-lactamase inhibitors, the most significant challenge lies in the development of new class B MBL inhibitors, which are not in clinical use at present.49 Although several MBL inhibitors such as RPX7009 have entered clinical trials, it is still too early to judge their clinical promise. Thus, additional efforts to discover novel MBL inhibitors are needed.
Outer Membrane Permeabilizers
The presence of an outer membrane in Gram-negative bacteria is responsible for intrinsic resistance to many antibiotic classes.9 The outer leaflet of the asymmetric bilayer of the outer membrane contains lipopolysaccharide (LPS) molecules that have three structural regions (Figure 3A): (i) an O-antigen consisting of repeating oligosaccharide units on the outside region, (ii) the core oligosaccharide, which is attached directly to lipid A and commonly contains various sugars, and (iii) lipid A, a glycolipid that is the hydrophobic anchor of the LPS. Lipid A, which has two glucosamine residues linked by a β-1,6 linkage and one phosphate group on each sugar residue, is conserved among various Gram-negative bacteria. The 5–7 carbon acyl chains in lipid A allow it to be inserted into the hydrophobic core of the bilayer, and divalent ions (e.g., Ca2+ and Mg2+) mediate ionic cross-linking of phosphate groups on adjacent molecules.50−53 The typical E. coli lipid A includes six acyl chains with four directly linking to the sugar units and the other two links to the β-hydroxyl groups on the acyl chain (Figure 3A). In P. aeruginosa, the lipid A acyl tail forms an increased number of hydrogen bonds due to the existence of three free β-hydroxyl groups (Figure 3B). This additional interaction makes the outer membrane of P. aeruginosa 10–100 times less permeable than that of other bacteria.54 In addition to the hydrogen bonds, the hydrophobic interaction between the acyl lipid tails also substantially contributes to the impermeability of the outer membrane to antibiotics. The relatively long acyl lipid chain length in K. pneumoniae makes it highly impermeable to many large antibiotics (Figure 3C).55
Figure 3.

(A) Chemical structure of the E. coli lipopolysaccharide (LPS). The O-antigen, core oligosaccharide, and lipid A, which forms ionic cross-links with divalent ions, are displayed. Lipid A structures of P. aeruginosa (B) and K. pneumoniae (C) are shown.
Divalent cationic ions cross-link LPS molecules by forming ionic bridges with negatively charged phosphate groups in lipid A and are essential for the outer membrane integrity of Gram-negative bacteria (Figure 3A).50 Various molecules can compromise the physical integrity of the outer membrane of Gram-negative bacteria by removal of or competition with divalent ions, therefore breaking the cross-linked structure between divalent cationic ions and LPS molecules. Examples of these molecules include charge-containing small-molecular-weight drugs,56 cationic antimicrobial peptides,57,58 chelating agents, and cationic polymers.42
Screening of 1440 approved drugs revealed the antiprotozoal drug pentamidine (Table 2a, Scheme 2a) as the most potent candidate to increase outer membrane permeability through disruption of the cation bridge holding the LPS molecules in E. coli and A. baumannii. Pentamidine sensitized both bacteria to large antibiotics such as novobiocin and rifampicin.56 However, pentamidine was inactive against P. aeruginosa, probably due to the much lower permeability of the outer membrane of P. aeruginosa than those of E. coli and A. baumannii.54
Table 2. Summary of the Potentiating Effects of Outer Membrane Permeabilizers.
| no. | OM permeabilizers | molecular structure | combined drug | microbial species | proposed mechanisms | MICa of drug alone | MICa in combination | fold decrease | FIC | ref |
|---|---|---|---|---|---|---|---|---|---|---|
| a | pentamidine | Scheme 2a | novobiocin | A. baumannii | displace the ion cross-linkers in the OM | 12 | <0.2 | >60 | <0.258 | (56) |
| b | colistin | Scheme 2b | rifampicin | P. aeruginosa | displace the ion cross-linkers in the OM | 64 | 4 | 16 | 0.31 | (59) |
| c | PMBN | Scheme 2c | fusidic acid | E. coli | displace the ion cross-linkers in the OM | 100 | 1 | 100 | <0.03 | (67) |
| d | SPR741 | Scheme 2d | rifampicin | A. baumannii. | displace the ion cross-linkers in the OM | 4 | 0.5 | 8 | 0.14 | (71) |
| e | SLAP-S25 | Scheme 2e | colistin | Providencia alcalifaciens | displace the ion cross-linkers in the OM | 16 | 0.016 | 1000 | 0.002 | (73) |
| f | thanatin | Scheme 1k | meropenem | E. coli | sequester the ion cross-linkers in the OM | 144* | 18* | 8 | 0.625 | (42) |
| g | LABv2.1 | Scheme 2f | erythromycin | E. coli | target 1-phosphor-GlcNAc of lipid A | 200 | <0.2 | >1000 | <0.251 | (75) |
| h | LL-37 | N.D. | histone | E. coli | pore formation in the OM and IM | N.A.b | N.A.b | N.A.b | N.A.b | (79) |
| i | PAS8-b-PDM12 | Scheme 2g | rifampicin | E. coli | target lipid A in the OM | 12.5 | 0.25 | 50 | 0.25 | (82) |
| j | 2,6-DAC | Scheme 2h | novobiocin | A. baumannii | enhance antibiotic penetration | 8 | 0.5 | 16 | 0.312 | (83) |
The MIC is given as μg/mL, except when specified by “*”, which indicates that the MIC is given as μM.
The synergy was determined by the time-killing method; N.D., not determined; N.A., not available. OM, outer membrane; IM, inner membrane.
Scheme 2. Chemical Structures of Outer Membrane Permeabilizers.

Antimicrobial peptides (AMPs) with cationic and amphiphilic properties have been widely evaluated as antibiotic adjuvants.57,58 The most widely studied antimicrobial peptide is colistin (Table 2b, Scheme 2b). Colistin initially interacts with the negatively charged lipid A components in LPS. Because the binding of colistin to LPS is much stronger than that of the divalent cations Mg2+ or Ca2+, it competitively replaces these divalent ions to weaken and release the LPS molecules, forming permeabilized pores in the outer membrane. Because colistin effectively forms physical pores in the outer membrane, it synergizes with various classes of antibiotics such as rifampicin and carbapenems.59,60 However, the spread of the plasmid-mediated colistin resistance gene mcr-1 has resulted in an increase in resistance to colistin.61,62Mcr-1 encodes a phosphoethanolamine transferase that modifies LPS via addition of a cationic phosphoethanolamine moiety to one phosphate group in lipid A, resulting in a reduction of the net anionicity of the bacterial membrane. Interestingly, a recent study found that despite clinical resistance, colistin retained sufficient membrane-permeabilizing ability to sensitize mcr-1-positive Enterobacteriaceae to several Gram-positive-specific antibiotics.63 This property may be linked to the presence of one remaining anionic phosphate group in lipid A of mcr-1-positive Enterobacteriaceae that promotes residual colistin binding. Colistin is an older drug that was previously withdrawn from use due to its nephrotoxicity and neurotoxicity.64 It resurfaced as a last-resort treatment option for carbapenem-resistant Gram-negative bacterial infections. However, the toxicity concern remains. Clinical prescription of colistin requires close monitoring of vital biomarkers during treatment.65 Use of a low dose of colistin as a potentiator may leverage the toxicity concern. However, the use of colistin as an antibiotic adjuvant should be avoided as much as possible to limit resistance to this last resort Gram-negative antibiotic.
Polymyxin B nonapeptide (PMBN) (Table 2c, Scheme 2c) is a shorter, fatty acid lipid-tail-deficient derivative of polymyxin B that retains the outer membrane-permeabilizing activity of polymyxin B. PMBN also binds to LPS,66 resulting in the release of divalent cross-linkers, which increases outer membrane permeability. PMBN increases the sensitivity of E. coli to hydrophobic antibiotics by increasing outer membrane permeability.67 The enantiomer of PMBN is not active, indicating that the stereochemical configuration is important.68 However, although it is less toxic than colistin, PMBN’s nephrotoxicity was still observed in preclinical studies, preventing its further development as a therapeutic adjuvant.69 SPR741 (Table 2d, Scheme 2d), another polymyxin B derivative, possesses modification of the N-terminus of polymyxin to reduce both its hydrophobicity and cationicity. SPR741 potentiates the action of large-molecular-weight antibiotics, such as rifampicin, against clinically relevant extensively drug-resistant A. baumannii by increasing outer membrane permeability via a similar mechanism to that of colistin and PMBN.70,71 This drug candidate entered clinical trials in 2016 and has since cleared two Phase I clinical trials in healthy adults, supporting its further clinical development.72
Recently, Zhu et al. discovered a short linear antimicrobial peptide, SLAP-S25 (Table 2e, Scheme 2e), that restores the efficacy of colistin against multiple mcr-1-mediated colistin-resistant Gram-negative pathogens.73 However, it is not effective against K. pneumoniae, which has closely packed LPSs due to the relatively long acyl lipid chain length of lipid A (Figure 3C). Mechanistic studies have indicated that SLAP-S25 disrupts both the outer and cytoplasmic membranes, enhancing antibiotic penetration. SLAP-S25 displaces the Mg2+ ions that hold the LPS together,73 resulting in increased outer membrane permeability. However, because SLAP-S25 also targets the lipid phosphatidylglycerol, which is also present in pulmonary surfactants,74 certain toxicity concerns exist for treating lung infections with SLAP-S25.
The guanidine group of thanatin not only sequesters the Zn2+ ions from β-lactamase to inhibit MBLs, it also displaces Ca2+ ions to induce LPS release in Gram-negative bacteria (Table 2f, Scheme 1k),42 leading to outer membrane disruption. This dual mechanism of action may account for the excellent potentiating effect of thanatin with β-lactam antibiotics against many NDM-1-producing E. coli and K. pneumoniae isolates.
Recently, Rutherford et al. found that the inner membrane protein PbgA plays an important role in the control of LPS biogenesis and outer membrane integrity.75 The LABv2.1 peptide (YPMXFRRFLEKWGLLR) (Table 2g, Scheme 2f), derived from the interfacial binding domain of PbgA, shows high affinity for lipid A via targeting of 1-phospho-GlcNAc, which decreases LPS density, thereby increasing outer membrane permeability.75 As a result, LABv2.1 potentiates the action of the outer membrane-impermeable antibiotics rifampicin, erythromycin, vancomycin, and colistin against E. coli, with a fold decrease in the minimum inhibitory concentration (MIC) ranging from 32 to >1024.
The combined application of AMPs and antibiotics represents a natural method to exploit the therapeutic values of AMPs. Indeed, they are a part of the innate immune system, which works in concert with other defense mechanisms such as neutrophil extracellular traps (NETs) to fight infection.55,76 NETs are networks of extracellular fibers composed of DNA and granule proteins that can trap and kill extracellular bacteria.77,78 Siryaporn et al. found that AMP LL-37 (Table 2h) and magainin-2 potentiated the antimicrobial potency of extracellular histone by promoting penetration of bacterial outer membrane barriers.79 The colocalization of histones and AMPs in NETs is necessary for their antibacterial effects. Without AMPs, extracellular histones do not have antibacterial effects under physiological conditions. AMPs form transient pores in bacterial membranes that allow histones to condense the intracellular chromosomal DNA, resulting in inhibition of transcription and bacterial cell death. Thus, the coexistence of histones and AMPs in many immune cells,77,80,81 such as macrophages and dendritic cells, represents a natural synergistic method to clear host infection.
Many AMPs are synergistic when combined due to their capacity to increase outer membrane permeability. However, few in vitro active AMPs are potent in animal models due to their rapid degradation by host enzymes.57 Recently, Chan-Park and coll. invented a new peptidomimetic based on the β-peptide backbone, PAS8-b-PDM12 (Table 2i, Scheme 2g), that sensitizes all antibiotic-resistant ESKAPE Gram-negative bacteria, including mcr-1 colistin-resistant clinical isolates, to rifampicin by increasing their outer membrane permeability.82 The extra methylene in the backbone of β-peptides compared to α-peptides, which are not recognized and targeted by the host enzymes, makes them intrinsically resistant to proteolysis. This property facilitates in vivo potency in animal models of sepsis caused by all ESKAPE Gram-negative bacteria. The design of PAS8-b-PDM12 represents a significant improvement over previous antimicrobial α-peptide-sensitizing agents. Chan-Park et al. also developed another chitosan derivative (2,6-DAC) (Table 2j, Scheme 2h) that potentiates the action of various antibiotics by enhancing their penetration.83 This in vitro synergistic drug combination was also validated in mice; the combination of 2,6-DAC with novobiocin/rifampicin resulted in a marked reduction in bacterial load in murine intraperitoneal and lung multidrug-resistant A. baumannii infection models.
The intrinsic resistance of Gram-negative bacteria to antibiotics provides an opportunity to broaden the applications of many antibiotics that are only effective against Gram-positive bacteria by coadministering them with outer membrane permeabilizers. This strategy is critically important given the difficulty of discovering novel antibiotic classes for Gram-negative bacteria. The so-called modern approaches focusing on antibiotic discovery, such as combinatorial chemistry and high-throughput screening, have produced several possible targets, but Gram-negative outer membrane impermeability remains a challenge for many drug discovery programs. An example is illustrated in a report from the pharmaceutical company GlaxoSmithKline (GSK). Scientists at GSK identified more than 160 target genes and performed high-throughput chemical screening several times during a 7-year period.84,85 However, only actinonin-targeting peptide deformylase progressed to Phase I clinical trials.84,86 The most attractive aspect of the development of outer membrane permeabilizer adjuvants is that it is possible to potentiate the action of numerous Gram-positive antibiotics as well as new preclinical drug candidates. One of the most promising outer membrane permeabilizers is pentamidine. This drug has been in clinical use since 1937 to treat trypanosomiasis, leishmaniasis, and babesiosis; therefore, its safety profile and pharmacology are well-known, and a drug repurposing approach could be cost- and time-effective for its translation into therapeutic application.87 Although other compounds such as SPR741 and PAS8-b-PDM12 also showed promising in vitro and in vivo efficacy and safety profiles as new chemical entities, they will need to progress through clinical trials. Indirect approaches to outer membrane disruption, such as inhibition of LPS biosynthesis or transport, may also lead to the development of outer membrane permeabilizers88,89 and are worthy of future studies.
Efflux Pump Inhibitors
Efflux pumps contribute to another type of intrinsic drug resistance in both Gram-negative and Gram-positive bacteria.10,11,90,91 Efflux pumps are protein complexes that can “pump out” exotoxins such as antimicrobial agents and toxins from bacteria, thereby reducing their intracellular concentration. Clinical resistance to several antibiotics has been associated with the action of efflux pumps in all ESKAPE pathogens.92,93 Efflux pump protein complexes are classified into five groups based on their sequence homology, substrate specificity, supramolecular characteristics, and energy source (Figure 4A),12 namely (i) ATP-binding cassette (ABC) efflux pumps, (ii) multi-drug and toxic compound extrusion (MATE) efflux pumps, (iii) major facilitator superfamily (MFS) efflux pumps, (iv) small multi-drug resistance (SMR) efflux pumps, and (v) resistance nodulation-cell division (RND) efflux pumps, which are the most prominent and clinically relevant efflux pumps. The ABC, MATE, MFS, and SMR pumps are cytoplasmic membrane-bound efflux pumps that exist in both Gram-negative and Gram-positive bacteria for the removal of substrate molecules from the cytosol. RND efflux pumps are only present in Gram-negative bacteria and remove substrate molecules from both the cytosol and periplasmic spaces.94 Although ABC efflux systems are powered by ATP hydrolysis, the other four efflux pump classes use the proton motive force (PMF) directly as the energy source to export substrates. The RND efflux pumps are unique in that they have tripartite components consisting of a transporter located in the cytoplasmic membrane, an exit duct located in the outer membrane, and a periplasmic adaptor channel that connects the other two components.95
Figure 4.

(A) Schematic representation of the five main types of bacterial efflux systems and their energy sources. (B) Representative tripartite complex of two RND efflux pumps: AcrA-AcrB-TolC and MexA-MexB-OprM. (C) Schematic illustration of the three-step rotating substrate efflux mechanism: access, binding, and extrusion.
The most well-studied and characteristic efflux pumps are the AcrA-AcrB-TolC and MexA-MexB-OprM pumps (Figure 4B) found in P. aeruginosa and E. coli. In both systems, the cytoplasmic membrane-bound transporters AcrB and MexB are responsible for recognizing and extruding substrates. Because the cytoplasmic membrane transporters AcrB and MexB share more than 86% structural similarity, they operate via a similar three-step rotating substrate efflux mechanism: access, binding, and extrusion (Figure 4C).96 First, in the access state, the vestibule (entrance) of the transporter is open to allow the potential drugs access to the vestibule. Second, the substrates bind to the binding pockets via either hydrophobic or aromatic–aromatic interaction with the phenylalanine acid residues or form hydrogen bonds with the asparagine and glutamine acid residues. Third, the movement of the central helix valve, which is the component powered by the PMF, shrinks the binding pocket to push the substrate through the extrusion parts to the exit channel. Most antibiotic classes are efflux pump substrates.97−101 Several pharmacological strategies are being developed to inhibit efflux pumps. Pyridopyrimidine and pyranopyridine derivatives, β-thujaplicin analogs, aminoglycoside hybrids, and cationic polymers were designed to mitigate efflux pump action via either binding to the affinity site of the efflux pump,102 reducing efflux pump expression,103 or dissipating the energy source essential for efflux.104
Efflux pump function can be inhibited via competition with antibiotics for binding to efflux pump complexes. This is the mechanism of action of phenylalanine-arginine β-naphthylamide (PAβN) (Table 3a, Scheme 3a),102 which competitively binds to the substrate pocket used by efflux pumps to target antibiotics. PAβN inhibits clinically relevant RND pumps in several Gram-negative bacteria, including P. aeruginosa, and sensitizes them to quinolones, piperacillin, cefotaxime, ceftazidime, ciprofloxacin, tetracycline, and chloramphenicol.105−107 Although PAβN has been known for decades, it has not been translated into clinical use due to its toxicity, which may arise from its cationic moiety.108,109 Pyridopyrimidine derivatives, which lack net cationic charges, do not have an antimicrobial ability but may avoid the toxicity issue of PAβN. D13-9001(Table 3b, Scheme 3b) is the most promising pyridopyrimidine derivative due to its high efficiency of binding to the efflux pump transporter and its water solubility.109,110 Crystal structure analysis has shown that the compound is deeply inserted into a hydrophobic trap rich in phenylalanine residues in the AcrB and MexB transporters but not in the MexY transporter.109 This is the primary cause of the failure of D13-9001 to inhibit efflux in bacteria overexpressing MexX-MexY-OprM efflux pumps. Unlike PAβN, D13-9001 does not directly bind to the same substrate pocket as the exported drugs but prevents the conformational change necessary for the functional rotation drug-efflux mechanism. Therefore, it potentiates the action of a broader spectrum of antibiotics than that of PAβN. Recently, Flipo, Hartkoorn, Pos and collaborators identified a series of pyridylpiperazine-based RND efflux pump inhibitors that sensitized E. coli to antibiotics via binding to the unique site on the transmembrane domain of the AcrB transporter, resulting in inhibition of antibiotic efflux.91 The chemically optimized compound BDM88855 (Table 3c, Scheme 3c) potentiated all antibiotics substrates for AcrB in wild-type E. coli to a similar extend compared to an AcrB efflux pump deficient mutant.
Table 3. Summary of the Potentiating Effects of Efflux Pump Inhibitors.
| no. | OM permeabilizers | molecular structure | combined drug | microbial species | proposed mechanisms | MICa of drug alone | MICa in combination | fold decrease | FIC | ref |
|---|---|---|---|---|---|---|---|---|---|---|
| a | PAβN | Scheme 3a | ciprofloxacin | P. aeruginosa | bind to the drug-binding pocket | 0.125 | 0.032 | 4 | N.A. | (105) |
| b | D13-9001 | Scheme 3b | carbenicillin | P. aeruginosa | jam the machinery of efflux | 64/128 | 4 | 16/32 | N.A. | (110) |
| c | BDM88855 | Scheme 3c | oxacillin | E. coli | jam the machinery of efflux | >100* | 3.0* | >33.3 | N.A. | (91) |
| d | MBX3132 | Scheme 3d | minocycline | E. coli | bind to the drug-binding pocket | 1 | 0.25 | 4 | N.A. | (111) |
| e | MBX3132 | Scheme 3e | levofloxacin | K. pneumoniae | bind to the drug-binding pocket | 1 | 0.25 | 4 | N.A. | (111) |
| f | MBX4191 | Scheme 3f | minocycline | E. coli, K. pneumoniae | bind to the drug-binding pocket | 32–128 | 4 | 8–32 | N.A. | (112) |
| g | β-thujaplicin analogs | Scheme 3g | doxycycline | E. coli | downregulate the expression of efflux pumps | 156* | 122* | 1.3 | N.A. | (103) |
| h | Loperamide | Scheme 3h | minocycline | P. aeruginosa | PMF collapse | 32 | 1 | 32 | <0.28 | (104) |
| i | IITR08027 | Scheme 3i | norfloxacin | E. coli | PMF collapse | 0.3215 | 0.0098 | 32 | N.A. | (116) |
| j | econazole | Scheme 3j | colistin | E. coli | PMF collapse | >64 | 0.5 | 128 | <0.5 | (118) |
| k | tobramycin-efflux pump inhibitor hybrid | Scheme 3k | minocycline | P. aeruginosa | PMF collapse | 8 | 0.5 | 16 | 0.19 | (120) |
| l | tobramycin-lysine hybrid | Scheme 3l | novobiocin | P. aeruginosa | PMF collapse | 1024 | 8 | 128 | 0.04 | (122) |
| m | C12(ω7) K-β12 | Scheme 3m | erythromycin | E. coli | PMF collapse | 512 | 0.5 | 1024 | ≤0.20 | (124) |
| n | PAS8-b-PDM12 | Scheme 2g | novobiocin | E. coli | PMF collapse | 100 | 0.195 | 512 | 0.25 | (82) |
The MIC is given as μg/mL, except when specified by “*”, which indicates that the MIC is given as μM. PMF, proton motive force; N.A., not available.
Scheme 3. Chemical Structures of Efflux Pump Inhibitors.

Opperman et al. prepared a series of pyranopyridines that was several orders of magnitude more potent than PAβN to sensitize Enterobacteriaceae to antibiotics.111 The two pyranopyridine compounds, MBX3132 (Table 3d, Scheme 3d) and MBX3135 (Table 3e, Scheme 3e), exhibited potency at a concentration as low as 0.1 μM, which was 500-fold lower than that of PAβN. Due to the large degree of steric hindrance, MBX3132 and MBX3135 bound more tightly to the binding pocket of the AcrB transporter in E. coli than did PAβN. Therefore, MBX3132 and MBX3135 achieved effective inhibition at extremely low concentrations. To enhance solubility and biocompatibility, the terminal acetyl group was changed to a polar tetrazole.112 The resulting compound, MBX4191 (Table 3f, Scheme 3f), had high water solubility (100 M) and limited cytotoxicity (IC50 = 47 M). An in vivo mouse study also supported the high biocompatibility of MBX4191, which was well-tolerated after a single intravenous (IV) injection of 200 mg/kg and multiple IV injections of 50 mg/kg, twice a day for 4 days. Most importantly, MBX4191 rescued the antibiotic activity of minocycline against minocycline-resistant K. pneumoniae in a murine sepsis infection model. These excellent in vitro and in vivo efficacy and safety profiles highlight the potential of MBX4191 for development for clinical application.113
Substrate pocket blocking or other methods of “jamming” the machinery of efflux comprise a direct approach that can be highly effective and fast acting. An indirect approach involves targeting genes that regulate efflux pump protein expression. Kishony et al. found that β-thujaplicin analogs (Table 3g, Scheme 3g) downregulated the tetracycline resistance efflux pump,103 enabling an effective second-phase treatment with doxycycline.
Because efflux pumps are powered by the PMF,114 compounds that target the PMF in bacteria can inhibit efflux pump activity. Loperamide (Table 3h, Scheme 3h), an antidiarrheal drug, was found to potentiate the efficacy of tetracycline antibiotics by dissipating the electrical component of the PMF.104 Because of the well-characterized toxicology and pharmacology of loperamide, it is a promising agent for adjuvant therapy. However, because loperamide is toxic when given by IV administration at a low dose (1 mg/kg),115 it cannot be used in combination therapy to treat systemic infections. By screening over 8000 synthetic molecules, Pathania et al. identified the small molecule IITR08027 (Table 3i, Scheme 3i), which potentiates the efficacy of ciprofloxacin by disruption of the PMF.116 IITR08027 is a hydrophobic weak acid that can shuttle H+ through the impermeable cytoplasmic membrane into the cytosol, resulting in distortion of the PMF. The strong hydrophobicity of IITR08027 may help it penetrate the hydrophobic lipid bilayer of the cytoplasmic membrane and move within the cytoplasm.117 Chen et al. found that econazole (Table 3j, Scheme 3j), in combination with colistin, achieved synergistic killing of colistin-resistant Gram-negative bacteria by collapsing the PMF.118 Although colistin binds to LPS and replaces the divalent ions to increase the outer membrane permeability, it also affects the electron transport chain after interaction with the cytoplasmic membrane, resulting in a reduction of the PMF.118 Econazole reduces the PMF by functioning as an ionophore to transport protons from the periplasm to the cytosol.118 The combination of the two drugs synergistically damages the bacterial cytoplasmic membrane and collapses the PMF.
A series of aminoglycoside hybrids, including a tobramycin-efflux pump inhibitor hybrid (Table 3k, Scheme 3k) and a tobramycin-lysine hybrid (Table 3l, Scheme 3l), was recently synthesized. The molecules synergistically eradicated P. aeruginosa when used in combination with antibiotics that are efflux pump substrates.119−122 The potentiating effect of these hybrids is mediated by an increase in outer membrane permeability and depolarization of the cytoplasmic membrane.120 The tobramycin moiety interacts with LPS to replace ionic cross-linkers, and the cationic amphiphilic part of the hybrids causes cytoplasmic membrane depolarization. In contrast to some efflux pump inhibitors such as PAβN and D13-9001, which cannot penetrate outer membranes with low permeability such as that of P. aeruginosa, aminoglycoside hybrids can penetrate membranes via a self-promoted uptake mechanism.123
Cationic peptides that target the bacterial cytoplasmic membrane also have the potential to dissipate the transmembrane PMF; however, only a few molecules have been reported. One example is the small cationic lipo-peptide C12(ω7) K-β12 (Table 3m, Scheme 3m), which potentiates the activity of tetracycline and erythromycin against E. coli by targeting the electric potential (Δψ) of the PMF.124 Because C12(ω7) K-β12 disrupts the cytoplasmic membrane,125 it triggers an influx of Na+ and K+, resulting in dissipation of the electric potential. Furthermore, the newly identified β-peptide PAS8-b-PDM12 (Table 3n, Scheme 2g) deactivates efflux pump systems by dissipating the PMF while also increasing the outer membrane permeability.82 The ideal alternation of the cationic (−CH2NH2) and hydrophobic ((−CH3)2) moieties at the β2 and β3 positions, together with their similar lengths in PAS8-b-PDM12, results in its strong membrane deformation ability. The two major intrinsic resistance mechanisms in Gram-negative bacteria, specifically, impermeability of the outer membrane barrier and multidrug efflux pumps, act in concert, leading to high levels of multidrug resistance. By overcoming both the outer membrane barrier and efflux pumps, PAS8-b-PDM12 potentiated the activity of novobiocin against all ESKAPE Gram-negative bacteria, which is a unique attribute unmatched by any other reported molecules.
Although many molecules have been identified as potential efflux pump inhibitors, their translation into clinical application is still lacking. This may be due to their toxicity in mammalian cells and preclinical animal models. The introduction of hydrophilic groups onto molecules improves biocompatibility.126−128 Resolution of the three-dimensional structure of the substrate binding pocket of efflux pump transporters may help to design efflux pump inhibitors with enhanced efficacy. This approach allowed the identification of a promising compound, MBX4191, which showed excellent in vivo biocompatibility and in vivo potency.113 Compounds that target the energy source of efflux pump systems have also shown encouraging results. For instance, loperamide, an FDA-approved drug, deactivates efflux pump systems by dissipating the PMF.104 It is worth noting that overexpression of efflux pump systems is frequently associated with other resistance mechanisms in ESKAPE pathogens, such as reduction of outer membrane permeability and/or enzymatic degradation.129 Therefore, compounds that can interfere with multiple resistance mechanisms, such as PAS8-b-PDM12 and aminoglycoside hybrids, are particularly promising for potentiation of the action of multiple antibiotics against ESKAPE pathogens.82,120
Summary and Outlook
The widespread antibiotic resistance of ESKAPE pathogens, together with the dwindling antibiotic pipeline, has resulted in an antimicrobial resistance crisis that requires new antibiotic classes or infection treatment strategies.130−132 Traditional approaches to discovering new antibiotic classes are uncertain. For instance, the discovery of a novel natural antibiotic class was estimated to require screening of at least 107 isolates.28 Natural resistance mechanisms such as low penetration barrier, drug efflux, or enzymatic degradation explain the difficulty of discovering promising new antibiotic classes. In this context, combination therapy using approved or experimental antibiotics and their adjuvants can disable those mechanisms and provide an alternative strategy to combat the antimicrobial resistance crisis.
Our review summarized the chemical structures of novel antimicrobial adjuvants that can sensitize drug-resistant bacteria by disrupting their resistance pathways and the mechanisms of action underlying them, specifically the β-lactamase inhibitors, outer membrane permeabilizers, and efflux pump inhibitors. Combination therapies may provide new approaches to impart or restore efficacy to older drugs against drug-resistant ESKAPE pathogens.133 It is interesting to note that a few molecules, e.g., thanatin, aminoglycoside hybrids, and PAS8-b-PDM12, possess dual mechanisms of inhibition and show excellent in vivo synergy against multidrug-resistant pathogens. Moreover, many drug-resistant pathogens possess multiple resistance mechanisms, contributing to highly resistant phenotypes. For example, carbapenem-resistant P. aeruginosa not only contains plasmid-mediated carbapenemases but also shows increased expression of efflux pumps and reduced porin expression, which decreases its outer membrane permeability.134 Thus, multifunctional antibacterial adjuvants that can overcome multiple resistance mechanisms would be a valuable direction for future studies. The use of combination systems with three or more components to treat multidrug-resistant bacteria also deserves consideration. Moreover, several antituberculars are prodrugs that need to be bioactivated into active metabolites by specific mycobacterial enzymes. A combination of antibiotic prodrug with the compound that boosts the production of activating enzymes may be a feasible strategy against M. tuberculosis. Recently, Willand, Baulard, et al. discovered a small molecule SMARt751 that potentiated the efficacy of ethionamide (ETH) against a panel of resistant M. tuberculosis both in vitro and in vivo. Elegant mode of action studies demonstrated that SMARt751 promoted the production of MymA via inhibition of a cryptic negative transcriptional regulator.135 To date, these systems have received only limited attention. With the advent of artificial intelligence, which may be applied to virtual combination screening and data analysis, the discovery of promising drug cocktails and their underpinning rules may be accelerated.136,137
Acknowledgments
This work was funded and supported by the Singapore MOE Tier 3 grant (MOE2018-T3-1-003).
The authors declare no competing financial interest.
References
- McKenna M. The antibiotic paradox: why companies can’t afford to create life-saving drugs. Nature 2020, 584, 338–341. 10.1038/d41586-020-02418-x. [DOI] [PubMed] [Google Scholar]
- Blair J. M.; Webber M. A.; Baylay A. J.; Ogbolu D. O.; Piddock L. J. Molecular mechanisms of antibiotic resistance. Nature reviews microbiology 2015, 13 (1), 42–51. 10.1038/nrmicro3380. [DOI] [PubMed] [Google Scholar]
- Cook M. A.; Wright G. D. The past, present, and future of antibiotics. Science Translational Medicine 2022, 14 (657), eabo7793 10.1126/scitranslmed.abo7793. [DOI] [PubMed] [Google Scholar]
- Vena A.; Castaldo N.; Bassetti M. The role of new β-lactamase inhibitors in gram-negative infections. Current Opinion in Infectious Diseases 2019, 32 (6), 638–646. 10.1097/QCO.0000000000000600. [DOI] [PubMed] [Google Scholar]
- Wright G. D. Bacterial resistance to antibiotics: enzymatic degradation and modification. Advanced drug delivery reviews 2005, 57 (10), 1451–1470. 10.1016/j.addr.2005.04.002. [DOI] [PubMed] [Google Scholar]
- Ramirez M. S.; Tolmasky M. E. Aminoglycoside modifying enzymes. Drug resistance updates 2010, 13 (6), 151–171. 10.1016/j.drup.2010.08.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Labby K. J.; Garneau-Tsodikova S. Strategies to overcome the action of aminoglycoside-modifying enzymes for treating resistant bacterial infections. Future medicinal chemistry 2013, 5 (11), 1285–1309. 10.4155/fmc.13.80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang N.; Luo J.; Deng F.; Huang Y.; Zhou H. Antibiotic Combination Therapy: A Strategy to Overcome Bacterial Resistance to Aminoglycoside Antibiotics. Frontiers in Pharmacology 2022, 10.3389/fphar.2022.839808. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zgurskaya H. I.; López C. A.; Gnanakaran S. Permeability barrier of Gram-negative cell envelopes and approaches to bypass it. ACS infectious diseases 2015, 1 (11), 512–522. 10.1021/acsinfecdis.5b00097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li X.-Z.; Plésiat P.; Nikaido H. The challenge of efflux-mediated antibiotic resistance in Gram-negative bacteria. Clin. Microbiol. Rev. 2015, 28 (2), 337–418. 10.1128/CMR.00117-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schindler B. D.; Kaatz G. W. Multidrug efflux pumps of Gram-positive bacteria. Drug Resistance Updates 2016, 27, 1–13. 10.1016/j.drup.2016.04.003. [DOI] [PubMed] [Google Scholar]
- Henderson P. J.; Maher C.; Elbourne L. D.; Eijkelkamp B. A.; Paulsen I. T.; Hassan K. A. Physiological Functions of Bacterial “Multidrug” Efflux Pumps. Chem. Rev. 2021, 121, 5417. 10.1021/acs.chemrev.0c01226. [DOI] [PubMed] [Google Scholar]
- Masi M.; Réfregiers M.; Pos K. M.; Pagès J.-M. Mechanisms of envelope permeability and antibiotic influx and efflux in Gram-negative bacteria. Nature microbiology 2017, 2 (3), 1–7. 10.1038/nmicrobiol.2017.1. [DOI] [PubMed] [Google Scholar]
- Fernández L.; Hancock R. E. Adaptive and mutational resistance: role of porins and efflux pumps in drug resistance. Clin. Microbiol. Rev. 2012, 25 (4), 661–681. 10.1128/CMR.00043-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mulani M. S.; Kamble E. E.; Kumkar S. N.; Tawre M. S.; Pardesi K. R. Emerging strategies to combat ESKAPE pathogens in the era of antimicrobial resistance: a review. Frontiers in microbiology 2019, 10.3389/fmicb.2019.00539. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tyers M.; Wright G. D. Drug combinations: a strategy to extend the life of antibiotics in the 21st century. Nature Reviews Microbiology 2019, 17 (3), 141–155. 10.1038/s41579-018-0141-x. [DOI] [PubMed] [Google Scholar]
- Chang M.; Mahasenan K. V.; Hermoso J. A.; Mobashery S. Unconventional Antibacterials and Adjuvants. Acc. Chem. Res. 2021, 54, 917–929. 10.1021/acs.accounts.0c00776. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Finlay J.; Miller L.; Poupard J. A. A review of the antimicrobial activity of clavulanate. J. Antimicrob. Chemother. 2003, 52 (1), 18–23. 10.1093/jac/dkg286. [DOI] [PubMed] [Google Scholar]
- Blair J. M.; Webber M. A.; Baylay A. J.; Ogbolu D. O.; Piddock L. J. Molecular mechanisms of antibiotic resistance. Nature reviews microbiology 2015, 13 (1), 42. 10.1038/nrmicro3380. [DOI] [PubMed] [Google Scholar]
- Bush K.; Bradford P. A. Interplay between β-lactamases and new β-lactamase inhibitors. Nature Reviews Microbiology 2019, 17 (5), 295–306. 10.1038/s41579-019-0159-8. [DOI] [PubMed] [Google Scholar]
- Meletis G. Carbapenem resistance: overview of the problem and future perspectives. Therapeutic advances in infectious disease 2016, 3 (1), 15–21. 10.1177/2049936115621709. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Control C. f. D.; Prevention, Antibiotic Resistance Threats in the United States 2019, US Department of Health and Human Services, CDC, Atlanta, GA: 2019. [Google Scholar]
- Tooke C. L.; Hinchliffe P.; Bragginton E. C.; Colenso C. K.; Hirvonen V. H.; Takebayashi Y.; Spencer J. β-Lactamases and β-Lactamase Inhibitors in the 21st Century. Journal of molecular biology 2019, 431 (18), 3472–3500. 10.1016/j.jmb.2019.04.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tehrani K. H.; Martin N. I. β-lactam/β-lactamase inhibitor combinations: an update. MedChemComm 2018, 9 (9), 1439–1456. 10.1039/C8MD00342D. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Higashitani F.; Hyodo A.; Ishida N.; Inoue M.; Mitsuhashi S. Inhibition of β-lactamases by tazobactam and in-vitro antibacterial activity of tazobactam combined with piperacillin. J. Antimicrob. Chemother. 1990, 25 (4), 567–574. 10.1093/jac/25.4.567. [DOI] [PubMed] [Google Scholar]
- Docquier J.-D.; Mangani S. An update on β-lactamase inhibitor discovery and development. Drug Resistance Updates 2018, 36, 13–29. 10.1016/j.drup.2017.11.002. [DOI] [PubMed] [Google Scholar]
- Bush K.; Jacoby G. A. Updated functional classification of β-lactamases. Antimicrob. Agents Chemother. 2010, 54 (3), 969–976. 10.1128/AAC.01009-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lewis K. The Science of Antibiotic Discovery. Cell 2020, 181 (1), 29–45. 10.1016/j.cell.2020.02.056. [DOI] [PubMed] [Google Scholar]
- Crandon J. L.; Nicolau D. P. In vitro activity of cefepime/AAI101 and comparators against cefepime non-susceptible Enterobacteriaceae. Pathogens 2015, 4 (3), 620–625. 10.3390/pathogens4030620. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Papp-Wallace K. M.; Bethel C. R.; Caillon J.; Barnes M. D.; Potel G.; Bajaksouzian S.; Rutter J. D.; Reghal A.; Shapiro S.; Taracila M. A. Beyond piperacillin-tazobactam: cefepime and AAI101 as a potent β-lactam- β-lactamase inhibitor combination. Antimicrob. Agents Chemother. 2019, 10.1128/AAC.00105-19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vallejo J. A.; Martínez-Guitián M.; Vázquez-Ucha J. C.; González-Bello C.; Poza M.; Buynak J. D.; Bethel C. R.; Bonomo R. A.; Bou G.; Beceiro A. LN-1–255, a penicillanic acid sulfone able to inhibit the class D carbapenemase OXA-48. J. Antimicrob. Chemother. 2016, 71 (8), 2171–2180. 10.1093/jac/dkw105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sader H. S.; Castanheira M.; Flamm R. K. Antimicrobial activity of ceftazidime-avibactam against Gram-negative bacteria isolated from patients hospitalized with pneumonia in US medical centers, 2011 to 2015. Antimicrob. Agents Chemother. 2017, 10.1128/AAC.02083-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ehmann D. E.; Jahić H.; Ross P. L.; Gu R.-F.; Hu J.; Kern G.; Walkup G. K.; Fisher S. L. Avibactam is a covalent, reversible, non-β-lactam β-lactamase inhibitor. Proc. Natl. Acad. Sci. U. S. A. 2012, 109 (29), 11663–11668. 10.1073/pnas.1205073109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Russ D.; Glaser F.; Tamar E. S.; Yelin I.; Baym M.; Kelsic E. D.; Zampaloni C.; Haldimann A.; Kishony R. Escape mutations circumvent a tradeoff between resistance to a beta-lactam and resistance to a beta-lactamase inhibitor. Nat. Commun. 2020, 11 (1), 1–9. 10.1038/s41467-020-15666-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Papp-Wallace K. M.; Bonomo R. A. New β-lactamase inhibitors in the clinic. Infectious Disease Clinics 2016, 30 (2), 441–464. 10.1016/j.idc.2016.02.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Livermore D. M.; Warner M.; Mushtaq S.; Woodford N. Interactions of OP0595, a novel triple-action diazabicyclooctane, with β-lactams against OP0595-resistant Enterobacteriaceae mutants. Antimicrob. Agents Chemother. 2016, 60 (1), 554–560. 10.1128/AAC.02184-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morinaka A.; Tsutsumi Y.; Yamada M.; Suzuki K.; Watanabe T.; Abe T.; Furuuchi T.; Inamura S.; Sakamaki Y.; Mitsuhashi N. OP0595, a new diazabicyclooctane: mode of action as a serine β-lactamase inhibitor, antibiotic and β-lactam ‘enhancer’. J. Antimicrob. Chemother. 2015, 70 (10), 2779–2786. 10.1093/jac/dkv166. [DOI] [PubMed] [Google Scholar]
- Durand-Réville T. F.; Guler S.; Comita-Prevoir J.; Chen B.; Bifulco N.; Huynh H.; Lahiri S.; Shapiro A. B.; McLeod S. M.; Carter N. M. ETX2514 is a broad-spectrum β-lactamase inhibitor for the treatment of drug-resistant Gram-negative bacteria including Acinetobacter baumannii. Nature microbiology 2017, 2 (9), 17104. 10.1038/nmicrobiol.2017.104. [DOI] [PubMed] [Google Scholar]
- González M. M.; Kosmopoulou M.; Mojica M. F.; Castillo V.; Hinchliffe P.; Pettinati I.; Brem J. r.; Schofield C. J.; Mahler G.; Bonomo R. A. Bisthiazolidines: a substrate-mimicking scaffold as an inhibitor of the NDM-1 carbapenemase. ACS infectious diseases 2015, 1 (11), 544–554. 10.1021/acsinfecdis.5b00046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hinchliffe P.; González M. M.; Mojica M. F.; González J. M.; Castillo V.; Saiz C.; Kosmopoulou M.; Tooke C. L.; Llarrull L. I.; Mahler G. Cross-class metallo-β-lactamase inhibition by bisthiazolidines reveals multiple binding modes. Proc. Natl. Acad. Sci. U. S. A. 2016, 113 (26), E3745-E3754 10.1073/pnas.1601368113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rossi M.-A.; Martinez V.; Hinchliffe P.; Mojica M. F.; Castillo V.; Moreno D. M.; Smith R.; Spellberg B.; Drusano G. L.; Banchio C., 2-Mercaptomethyl-thiazolidines use conserved aromatic-S interactions to achieve broad-range inhibition of metallo-β-lactamases. Chemical Science 2021.122898. 10.1039/D0SC05172A [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ma B.; Fang C.; Lu L.; Wang M.; Xue X.; Zhou Y.; Li M.; Hu Y.; Luo X.; Hou Z. The antimicrobial peptide thanatin disrupts the bacterial outer membrane and inactivates the NDM-1 metallo-β-lactamase. Nat. Commun. 2019, 10 (1), 1–11. 10.1038/s41467-019-11503-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chiou J.; Wan S.; Chan K.-F.; So P.-K.; He D.; Chan E. W.-c.; Chan T.-h.; Wong K.-y.; Tao J.; Chen S. Ebselen as a potent covalent inhibitor of New Delhi metallo-β-lactamase (NDM-1). Chem. Commun. 2015, 51 (46), 9543–9546. 10.1039/C5CC02594J. [DOI] [PubMed] [Google Scholar]
- Sun H.; Zhang Q.; Wang R.; Wang H.; Wong Y.-T.; Wang M.; Hao Q.; Yan A.; Kao R. Y.-T.; Ho P.-L. Resensitizing carbapenem-and colistin-resistant bacteria to antibiotics using auranofin. Nat. Commun. 2020, 11 (1), 1–13. 10.1038/s41467-020-18939-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Patel T. S.; Pogue J. M.; Mills J. P.; Kaye K. S. Meropenem-vaborbactam: a new weapon in the war against infections due to resistant Gram-negative bacteria. Future microbiology 2018, 13 (09), 971–983. 10.2217/fmb-2018-0054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hecker S. J.; Reddy K. R.; Lomovskaya O.; Griffith D. C.; Rubio-Aparicio D.; Nelson K.; Tsivkovski R.; Sun D.; Sabet M.; Tarazi Z. Discovery of cyclic boronic acid QPX7728, an ultrabroad-spectrum inhibitor of serine and metallo-β-lactamases. J. Med. Chem. 2020, 63 (14), 7491–7507. 10.1021/acs.jmedchem.9b01976. [DOI] [PubMed] [Google Scholar]
- Zhang J.; Chen Y. P.; Miller K. P.; Ganewatta M. S.; Bam M.; Yan Y.; Nagarkatti M.; Decho A. W.; Tang C. Antimicrobial metallopolymers and their bioconjugates with conventional antibiotics against multidrug-resistant bacteria. J. Am. Chem. Soc. 2014, 136 (13), 4873–4876. 10.1021/ja5011338. [DOI] [PubMed] [Google Scholar]
- Thappeta K. R. V.; Vikhe Y. S.; Yong A. M. H.; Chan-Park M. B.; Kline K. Combined efficacy of an antimicrobial cationic peptide polymer with conventional antibiotics to combat multi-drug resistant pathogens. ACS Infectious Diseases 2020, 6, 1228. 10.1021/acsinfecdis.0c00016. [DOI] [PubMed] [Google Scholar]
- Lewis K. New approaches to antimicrobial discovery. Biochemical pharmacology 2017, 134, 87–98. 10.1016/j.bcp.2016.11.002. [DOI] [PubMed] [Google Scholar]
- Domalaon R.; Idowu T.; Zhanel G. G.; Schweizer F. Antibiotic hybrids: the next generation of agents and adjuvants against gram-negative pathogens?. Clin. Microbiol. Rev. 2018, 31 (2), e00077-17 10.1128/CMR.00077-17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Molinaro A.; Holst O.; Di Lorenzo F.; Callaghan M.; Nurisso A.; D’Errico G.; Zamyatina A.; Peri F.; Berisio R.; Jerala R. Chemistry of lipid A: at the heart of innate immunity. Chem.—Eur. J. 2015, 21 (2), 500–519. 10.1002/chem.201403923. [DOI] [PubMed] [Google Scholar]
- Kabanov D.; Prokhorenko I. Structural analysis of lipopolysaccharides from Gram-negative bacteria. Biochemistry (Moscow) 2010, 75 (4), 383–404. 10.1134/S0006297910040012. [DOI] [PubMed] [Google Scholar]
- Clifton L. A.; Skoda M. W.; Le Brun A. P.; Ciesielski F.; Kuzmenko I.; Holt S. A.; Lakey J. H. Effect of divalent cation removal on the structure of gram-negative bacterial outer membrane models. Langmuir 2015, 31 (1), 404–412. 10.1021/la504407v. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Breidenstein E. B.; de la Fuente-Núñez C.; Hancock R. E. Pseudomonas aeruginosa: all roads lead to resistance. Trends in microbiology 2011, 19 (8), 419–426. 10.1016/j.tim.2011.04.005. [DOI] [PubMed] [Google Scholar]
- Lam S. J.; O’Brien-Simpson N. M.; Pantarat N.; Sulistio A.; Wong E. H.; Chen Y.-Y.; Lenzo J. C.; Holden J. A.; Blencowe A.; Reynolds E. C. Combating multidrug-resistant Gram-negative bacteria with structurally nanoengineered antimicrobial peptide polymers. Nature microbiology 2016, 1 (11), 1–11. 10.1038/nmicrobiol.2016.162. [DOI] [PubMed] [Google Scholar]
- Stokes J. M.; MacNair C. R.; Ilyas B.; French S.; Côté J.-P.; Bouwman C.; Farha M. A.; Sieron A. O.; Whitfield C.; Coombes B. K. Pentamidine sensitizes Gram-negative pathogens to antibiotics and overcomes acquired colistin resistance. Nature microbiology 2017, 2 (5), 17028. 10.1038/nmicrobiol.2017.28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cassone M.; Otvos Jr L. Synergy among antibacterial peptides and between peptides and small-molecule antibiotics. Expert review of anti-infective therapy 2010, 8 (6), 703–716. 10.1586/eri.10.38. [DOI] [PubMed] [Google Scholar]
- Lazzaro B. P.; Zasloff M.; Rolff J.. Antimicrobial peptides: Application informed by evolution. Science 2020, 368 ( (6490), ). 10.1126/science.aau5480 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tascini C.; Gemignani G.; Ferranti S.; Tagliaferri E.; Leonildi A.; Lucarini A.; Menichetti F. Microbiological activity and clinical efficacy of a colistin and rifampin combination in multidrug-resistant Pseudomonas aeruginosa infections. Journal of chemotherapy 2004, 16 (3), 282–287. 10.1179/joc.2004.16.3.282. [DOI] [PubMed] [Google Scholar]
- Petrosillo N.; Ioannidou E.; Falagas M. Colistin monotherapy vs. combination therapy: evidence from microbiological, animal and clinical studies. Clinical Microbiology and Infection 2008, 14 (9), 816–827. 10.1111/j.1469-0691.2008.02061.x. [DOI] [PubMed] [Google Scholar]
- Poirel L.; Kieffer N.; Liassine N.; Thanh D.; Nordmann P. Plasmid-mediated carbapenem and colistin resistance in a clinical isolate of Escherichia coli. Lancet Infectious Diseases 2016, 16 (3), 281. 10.1016/S1473-3099(16)00006-2. [DOI] [PubMed] [Google Scholar]
- Zhang Q.; Wang R.; Wang M.; Liu C.; Koohi-Moghadam M.; Wang H.; Ho P.-L.; Li H.; Sun H. Re-sensitization of mcr carrying multidrug resistant bacteria to colistin by silver. Proc. Natl. Acad. Sci. U. S. A. 2022, 119 (11), e2119417119 10.1073/pnas.2119417119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- MacNair C. R.; Stokes J. M.; Carfrae L. A.; Fiebig-Comyn A. A.; Coombes B. K.; Mulvey M. R.; Brown E. D. Overcoming mcr-1 mediated colistin resistance with colistin in combination with other antibiotics. Nat. Commun. 2018, 9 (1), 458. 10.1038/s41467-018-02875-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lim L. M.; Ly N.; Anderson D.; Yang J. C.; Macander L.; Jarkowski III A.; Forrest A.; Bulitta J. B.; Tsuji B. T. Resurgence of colistin: a review of resistance, toxicity, pharmacodynamics, and dosing. Pharmacotherapy: The Journal of Human Pharmacology and Drug Therapy 2010, 30 (12), 1279–1291. 10.1592/phco.30.12.1279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Biswas S.; Brunel J.-M.; Dubus J.-C.; Reynaud-Gaubert M.; Rolain J.-M. Colistin: an update on the antibiotic of the 21st century. Expert review of anti-infective therapy 2012, 10 (8), 917–934. 10.1586/eri.12.78. [DOI] [PubMed] [Google Scholar]
- Tsubery H.; Ofek I.; Cohen S.; Fridkin M. Structure- function studies of polymyxin B nonapeptide: implications to sensitization of gram-negative bacteria. Journal of medicinal chemistry 2000, 43 (16), 3085–3092. 10.1021/jm0000057. [DOI] [PubMed] [Google Scholar]
- Vaara M.; Vaara T. Sensitization of Gram-negative bacteria to antibiotics and complement by a nontoxic oligopeptide. Nature 1983, 303 (5917), 526–528. 10.1038/303526a0. [DOI] [PubMed] [Google Scholar]
- Tsubery H.; Ofek I.; Cohen S.; Fridkin M. The functional association of polymyxin B with bacterial lipopolysaccharide is stereospecific: studies on polymyxin B nonapeptide. Biochemistry 2000, 39 (39), 11837–11844. 10.1021/bi000386q. [DOI] [PubMed] [Google Scholar]
- Vaara M. Novel derivatives of polymyxins. J. Antimicrob. Chemother. 2013, 68 (6), 1213–1219. 10.1093/jac/dkt039. [DOI] [PubMed] [Google Scholar]
- French S.; Farha M.; Ellis M. J.; Sameer Z.; Côté J.-P.; Cotroneo N.; Lister T.; Rubio A.; Brown E. D. Potentiation of Antibiotics against Gram-Negative Bacteria by Polymyxin B Analogue SPR741 from Unique Perturbation of the Outer Membrane. ACS infectious diseases 2020, 6 (6), 1405–1412. 10.1021/acsinfecdis.9b00159. [DOI] [PubMed] [Google Scholar]
- Zurawski D. V.; Reinhart A. A.; Alamneh Y. A.; Pucci M. J.; Si Y.; Abu-Taleb R.; Shearer J. P.; Demons S. T.; Tyner S. D.; Lister T. SPR741, an antibiotic adjuvant, potentiates the in vitro and in vivo activity of rifampin against clinically relevant extensively drug-resistant Acinetobacter baumannii. Antimicrob. Agents Chemother. 2017, 10.1128/AAC.01239-17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eckburg P. B.; Lister T.; Walpole S.; Keutzer T.; Utley L.; Tomayko J.; Kopp E.; Farinola N.; Coleman S. Safety, tolerability, pharmacokinetics, and drug interaction potential of SPR741, an intravenous potentiator, after single and multiple ascending doses and when combined with β-lactam antibiotics in healthy subjects. Antimicrob. Agents Chemother. 2019, 63 (9), e00892-19 10.1128/AAC.00892-19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Song M.; Liu Y.; Huang X.; Ding S.; Wang Y.; Shen J.; Zhu K. A broad-spectrum antibiotic adjuvant reverses multidrug-resistant Gram-negative pathogens. Nature Microbiology 2020, 5 (8), 1040–1050. 10.1038/s41564-020-0723-z. [DOI] [PubMed] [Google Scholar]
- Veldhuizen R.; Nag K.; Orgeig S.; Possmayer F. The role of lipids in pulmonary surfactant. Biochimica et Biophysica Acta (BBA)-Molecular Basis of Disease 1998, 1408 (2–3), 90–108. 10.1016/S0925-4439(98)00061-1. [DOI] [PubMed] [Google Scholar]
- Clairfeuille T.; Buchholz K. R.; Li Q.; Verschueren E.; Liu P.; Sangaraju D.; Park S.; Noland C. L.; Storek K. M.; Nickerson N. N.; Martin L.; Dela Vega T.; Miu A.; Reeder J.; Ruiz-Gonzalez M.; Swem D.; Han G.; DePonte D. P.; Hunter M. S.; Gati C.; Shahidi-Latham S.; Xu M.; Skelton N.; Sellers B. D.; Skippington E.; Sandoval W.; Hanan E. J.; Payandeh J.; Rutherford S. T. Structure of the essential inner membrane lipopolysaccharide-PbgA complex. Nature 2020, 584, 479. 10.1038/s41586-020-2597-x. [DOI] [PubMed] [Google Scholar]
- Mookherjee N.; Anderson M. A.; Haagsman H. P.; Davidson D. J. Antimicrobial host defence peptides: Functions and clinical potential. Nat. Rev. Drug Discovery 2020, 19 (5), 311–332. 10.1038/s41573-019-0058-8. [DOI] [PubMed] [Google Scholar]
- Brinkmann V.; Reichard U.; Goosmann C.; Fauler B.; Uhlemann Y.; Weiss D. S.; Weinrauch Y.; Zychlinsky A. Neutrophil extracellular traps kill bacteria. science 2004, 303 (5663), 1532–1535. 10.1126/science.1092385. [DOI] [PubMed] [Google Scholar]
- Papayannopoulos V. Neutrophil extracellular traps in immunity and disease. Nature Reviews Immunology 2018, 18 (2), 134. 10.1038/nri.2017.105. [DOI] [PubMed] [Google Scholar]
- Doolin T.; Amir H. M.; Duong L.; Rosenzweig R.; Urban L. A.; Bosch M.; Pol A.; Gross S. P.; Siryaporn A. Mammalian histones facilitate antimicrobial synergy by disrupting the bacterial proton gradient and chromosome organization. Nat. Commun. 2020, 11 (1), 1–16. 10.1038/s41467-020-17699-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Doster R. S.; Rogers L. M.; Gaddy J. A.; Aronoff D. M. Macrophage extracellular traps: a scoping review. Journal of innate immunity 2018, 10 (1), 3–13. 10.1159/000480373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Loures F. V.; Röhm M.; Lee C. K.; Santos E.; Wang J. P.; Specht C. A.; Calich V. L.; Urban C. F.; Levitz S. M. Recognition of Aspergillus fumigatus hyphae by human plasmacytoid dendritic cells is mediated by dectin-2 and results in formation of extracellular traps. PLoS Pathog 2015, 11 (2), e1004643 10.1371/journal.ppat.1004643. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Si Z.; Lim H. W.; Tay M. Y.; Du Y.; Ruan L.; Qiu H.; Reghu S.; Chen Y.; Tiong W. S.; Marimuthu K. A glycosylated cationic block poly (beta-peptide) reverses intrinsic antibiotic resistance in all ESKAPE Gram-negative bacteria. Angew. Chem., Int. Ed. 2020, 132, 6886. 10.1002/ange.201914304. [DOI] [PubMed] [Google Scholar]
- Si Z.; Hou Z.; Vikhe Y. S.; Thappeta K. R. V.; Marimuthu K.; De P. P.; Ng O. T.; Li P.; Zhu Y.; Pethe K. Antimicrobial Effect of a Novel Chitosan Derivative and Its Synergistic Effect with Antibiotics. ACS Appl. Mater. Interfaces 2021, 13, 3237. 10.1021/acsami.0c20881. [DOI] [PubMed] [Google Scholar]
- Payne D. J.; Gwynn M. N.; Holmes D. J.; Pompliano D. L. Drugs for bad bugs: confronting the challenges of antibacterial discovery. Nat. Rev. Drug Discovery 2007, 6 (1), 29–40. 10.1038/nrd2201. [DOI] [PubMed] [Google Scholar]
- Tommasi R.; Brown D. G.; Walkup G. K.; Manchester J. I.; Miller A. A. ESKAPEing the labyrinth of antibacterial discovery. Nat. Rev. Drug Discovery 2015, 14 (8), 529–542. 10.1038/nrd4572. [DOI] [PubMed] [Google Scholar]
- Butler M. S.; Blaskovich M. A.; Cooper M. A. Antibiotics in the clinical pipeline in 2013. Journal of antibiotics 2013, 66 (10), 571–591. 10.1038/ja.2013.86. [DOI] [PubMed] [Google Scholar]
- Miró-Canturri A.; Ayerbe-Algaba R.; Smani Y. Drug repurposing for the treatment of bacterial and fungal infections. Frontiers in microbiology 2019, 10, 41. 10.3389/fmicb.2019.00041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mandler M. D.; Baidin V.; Lee J.; Pahil K. S.; Owens T. W.; Kahne D. Novobiocin enhances polymyxin activity by stimulating lipopolysaccharide transport. J. Am. Chem. Soc. 2018, 140 (22), 6749–6753. 10.1021/jacs.8b02283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- MacNair C. R.; Tsai C. N.; Brown E. D. Creative targeting of the Gram-negative outer membrane in antibiotic discovery. Ann. N.Y. Acad. Sci. 2020, 1459 (1), 69–85. 10.1111/nyas.14280. [DOI] [PubMed] [Google Scholar]
- Brawley D. N.; Sauer D. B.; Li J.; Zheng X.; Koide A.; Jedhe G. S.; Suwatthee T.; Song J.; Liu Z.; Arora P. S. Structural basis for inhibition of the drug efflux pump NorA from Staphylococcus aureus. Nat. Chem. Biol. 2022, 18, 706. 10.1038/s41589-022-00994-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Plé C.; Tam H.-K.; Vieira Da Cruz A.; Compagne N.; Jiménez-Castellanos J.-C.; Müller R. T.; Pradel E.; Foong W. E.; Malloci G.; Ballée A. Pyridylpiperazine-based allosteric inhibitors of RND-type multidrug efflux pumps. Nat. Commun. 2022, 13 (1), 1–11. 10.1038/s41467-021-27726-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Webber M.; Piddock L. The importance of efflux pumps in bacterial antibiotic resistance. J. Antimicrob. Chemother. 2003, 51 (1), 9–11. 10.1093/jac/dkg050. [DOI] [PubMed] [Google Scholar]
- Sun J.; Deng Z.; Yan A. Bacterial multidrug efflux pumps: mechanisms, physiology and pharmacological exploitations. Biochemical and biophysical research communications 2014, 453 (2), 254–267. 10.1016/j.bbrc.2014.05.090. [DOI] [PubMed] [Google Scholar]
- Blanco P.; Hernando-Amado S.; Reales-Calderon J. A.; Corona F.; Lira F.; Alcalde-Rico M.; Bernardini A.; Sanchez M. B.; Martinez J. L. Bacterial multidrug efflux pumps: much more than antibiotic resistance determinants. Microorganisms 2016, 4 (1), 14. 10.3390/microorganisms4010014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Symmons M. F.; Bokma E.; Koronakis E.; Hughes C.; Koronakis V. The assembled structure of a complete tripartite bacterial multidrug efflux pump. Proc. Natl. Acad. Sci. U. S. A. 2009, 106 (17), 7173–7178. 10.1073/pnas.0900693106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murakami S.; Nakashima R.; Yamashita E.; Matsumoto T.; Yamaguchi A. Crystal structures of a multidrug transporter reveal a functionally rotating mechanism. Nature 2006, 443 (7108), 173–179. 10.1038/nature05076. [DOI] [PubMed] [Google Scholar]
- Nakae T.; Nakajima A.; Ono T.; Saito K.; Yoneyama H. Resistance to β-Lactam Antibiotics inPseudomonas aeruginosa Due to Interplay between the MexAB-OprM Efflux Pump and β-Lactamase. Antimicrob. Agents Chemother. 1999, 43 (5), 1301–1303. 10.1128/AAC.43.5.1301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhong P.; Shortridge V. D. The role of efflux in macrolide resistance. Drug Resistance Updates 2000, 3 (6), 325–329. 10.1054/drup.2000.0175. [DOI] [PubMed] [Google Scholar]
- Li X.-Z.; Livermore D. M.; Nikaido H. Role of efflux pump (s) in intrinsic resistance of Pseudomonas aeruginosa: resistance to tetracycline, chloramphenicol, and norfloxacin. Antimicrob. Agents Chemother. 1994, 38 (8), 1732–1741. 10.1128/AAC.38.8.1732. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mahamoud A.; Chevalier J.; Alibert-Franco S.; Kern W. V.; Pages J.-M. Antibiotic efflux pumps in Gram-negative bacteria: the inhibitor response strategy. J. Antimicrob. Chemother. 2007, 59 (6), 1223–1229. 10.1093/jac/dkl493. [DOI] [PubMed] [Google Scholar]
- Köhler T.; Kok M.; Michea-Hamzehpour M.; Plesiat P.; Gotoh N.; Nishino T.; Curty L. K.; Pechere J.-C. Multidrug efflux in intrinsic resistance to trimethoprim and sulfamethoxazole in Pseudomonas aeruginosa. Antimicrob. Agents Chemother. 1996, 40 (10), 2288–2290. 10.1128/AAC.40.10.2288. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jamshidi S.; Sutton J. M.; Rahman K. M. Computational study reveals the molecular mechanism of the interaction between the efflux inhibitor PAβN and the AdeB transporter from Acinetobacter baumannii. ACS omega 2017, 2 (6), 3002–3016. 10.1021/acsomega.7b00131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stone L. K.; Baym M.; Lieberman T. D.; Chait R.; Clardy J.; Kishony R. Compounds that select against the tetracycline-resistance efflux pump. Nat. Chem. Biol. 2016, 12 (11), 902. 10.1038/nchembio.2176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ejim L.; Farha M. A.; Falconer S. B.; Wildenhain J.; Coombes B. K.; Tyers M.; Brown E. D.; Wright G. D. Combinations of antibiotics and nonantibiotic drugs enhance antimicrobial efficacy. Nat. Chem. Biol. 2011, 7 (6), 348–350. 10.1038/nchembio.559. [DOI] [PubMed] [Google Scholar]
- Lamers R. P.; Cavallari J. F.; Burrows L. L. The efflux inhibitor phenylalanine-arginine beta-naphthylamide (PAβN) permeabilizes the outer membrane of gram-negative bacteria. PloS one 2013, 8 (3), e60666 10.1371/journal.pone.0060666. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gill E. E.; Franco O. L.; Hancock R. E. Antibiotic adjuvants: diverse strategies for controlling drug-resistant pathogens. Chemical biology & drug design 2015, 85 (1), 56–78. 10.1111/cbdd.12478. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reading E.; Ahdash Z.; Fais C.; Ricci V.; Wang-Kan X.; Grimsey E.; Stone J.; Malloci G.; Lau A. M.; Findlay H. Perturbed structural dynamics underlie inhibition and altered efflux of the multidrug resistance pump AcrB. Nat. Commun. 2020, 11 (1), 1–11. 10.1038/s41467-020-19397-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fleeman R. M.; Debevec G.; Antonen K.; Adams J. L.; Santos R. G.; Welmaker G. S.; Houghten R. A.; Giulianotti M. A.; Shaw L. N. Identification of a novel polyamine scaffold with potent efflux pump inhibition activity toward multi-drug resistant bacterial pathogens. Frontiers in microbiology 2018, 9, 1301. 10.3389/fmicb.2018.01301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nakashima R.; Sakurai K.; Yamasaki S.; Hayashi K.; Nagata C.; Hoshino K.; Onodera Y.; Nishino K.; Yamaguchi A. Structural basis for the inhibition of bacterial multidrug exporters. Nature 2013, 500 (7460), 102. 10.1038/nature12300. [DOI] [PubMed] [Google Scholar]
- Ranjitkar S.; Jones A. K.; Mostafavi M.; Zwirko Z.; Iartchouk O.; Barnes S. W.; Walker J. R.; Willis T. W.; Lee P. S.; Dean C. R. Target (MexB)-and efflux-based mechanisms decreasing the effectiveness of the efflux pump inhibitor D13–9001 in Pseudomonas aeruginosa PAO1: uncovering a new role for MexMN-OprM in efflux of β-lactams and a novel regulatory circuit (MmnRS) controlling MexMN expression. Antimicrob. Agents Chemother. 2019, 10.1128/AAC.01718-18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sjuts H.; Vargiu A. V.; Kwasny S. M.; Nguyen S. T.; Kim H.-S.; Ding X.; Ornik A. R.; Ruggerone P.; Bowlin T. L.; Nikaido H. Molecular basis for inhibition of AcrB multidrug efflux pump by novel and powerful pyranopyridine derivatives. Proc. Natl. Acad. Sci. U. S. A. 2016, 113 (13), 3509–3514. 10.1073/pnas.1602472113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Laws M.; Shaaban A.; Rahman K. M. Antibiotic resistance breakers: current approaches and future directions. FEMS Microbiology Reviews 2019, 43 (5), 490–516. 10.1093/femsre/fuz014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moir D. T.; Opperman T. J.; Aron Z. D.; Bowlin T. L., Adjunctive therapy for multidrug-resistant bacterial infections: type III secretion system and efflux inhibitors. Drug discovery today 2021.262173. 10.1016/j.drudis.2021.03.031 [DOI] [PubMed] [Google Scholar]
- Nikaido H. Prevention of drug access to bacterial targets: permeability barriers and active efflux. Science 1994, 264 (5157), 382–388. 10.1126/science.8153625. [DOI] [PubMed] [Google Scholar]
- Hurwitz A.; Sztern M.; Looney G.; Ben-Zvi Z. Loperamide effects on hepatobiliary function, intestinal transit and analgesia in mice. Life sciences 1994, 54 (22), 1687–1698. 10.1016/0024-3205(94)00609-1. [DOI] [PubMed] [Google Scholar]
- Bhattacharyya T.; Sharma A.; Akhter J.; Pathania R. The small molecule IITR08027 restores the antibacterial activity of fluoroquinolones against multidrug-resistant Acinetobacter baumannii by efflux inhibition. International journal of antimicrobial agents 2017, 50 (2), 219–226. 10.1016/j.ijantimicag.2017.03.005. [DOI] [PubMed] [Google Scholar]
- Valderrama K.; Pradel E.; Firsov A. M.; Drobecq H.; Bauderlique-le Roy H.; Villemagne B.; Antonenko Y. N.; Hartkoorn R. C. Pyrrolomycins are potent natural protonophores. Antimicrob. Agents Chemother. 2019, 63 (10), e01450-19 10.1128/AAC.01450-19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu C.; Chen K.; Chan K. F.; Chan E. W. C.; Guo X.; Chow H. Y.; Zhao G.; Zeng P.; Wang M.; Zhu Y., Imidazole Type Antifungal Drugs Are Effective Colistin Adjuvants That Resensitize Colistin-Resistant Enterobacteriaceae. Advanced Therapeutics, 202032000084. 10.1002/adtp.202000084 [DOI] [Google Scholar]
- Gorityala B. K.; Guchhait G.; Goswami S.; Fernando D. M.; Kumar A.; Zhanel G. G.; Schweizer F. Hybrid antibiotic overcomes resistance in P. aeruginosa by enhancing outer membrane penetration and reducing efflux. Journal of medicinal chemistry 2016, 59 (18), 8441–8455. 10.1021/acs.jmedchem.6b00867. [DOI] [PubMed] [Google Scholar]
- Yang X.; Goswami S.; Gorityala B. K.; Domalaon R.; Lyu Y.; Kumar A.; Zhanel G. G.; Schweizer F. A tobramycin vector enhances synergy and efficacy of efflux pump inhibitors against multidrug-resistant Gram-negative bacteria. Journal of medicinal chemistry 2017, 60 (9), 3913–3932. 10.1021/acs.jmedchem.7b00156. [DOI] [PubMed] [Google Scholar]
- Gorityala B. K.; Guchhait G.; Fernando D. M.; Deo S.; McKenna S. A.; Zhanel G. G.; Kumar A.; Schweizer F. Adjuvants based on hybrid antibiotics overcome resistance in Pseudomonas aeruginosa and enhance fluoroquinolone efficacy. Angew. Chem., Int. Ed. 2016, 55 (2), 555–559. 10.1002/anie.201508330. [DOI] [PubMed] [Google Scholar]
- Lyu Y.; Yang X.; Goswami S.; Gorityala B. K.; Idowu T.; Domalaon R.; Zhanel G. G.; Shan A.; Schweizer F. Amphiphilic tobramycin-lysine conjugates sensitize multidrug resistant gram-negative bacteria to rifampicin and minocycline. Journal of medicinal chemistry 2017, 60 (9), 3684–3702. 10.1021/acs.jmedchem.6b01742. [DOI] [PubMed] [Google Scholar]
- Mingeot-Leclercq M.-P.; Glupczynski Y.; Tulkens P. M. Aminoglycosides: activity and resistance. Antimicrob. Agents Chemother. 1999, 43 (4), 727–737. 10.1128/AAC.43.4.727. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goldberg K.; Sarig H.; Zaknoon F.; Epand R. F.; Epand R. M.; Mor A. Sensitization of gram-negative bacteria by targeting the membrane potential. FASEB J. 2013, 27 (9), 3818–3826. 10.1096/fj.13-227942. [DOI] [PubMed] [Google Scholar]
- Sarig H.; Livne L.; Held-Kuznetsov V.; Zaknoon F.; Ivankin A.; Gidalevitz D.; Mor A. A miniature mimic of host defense peptides with systemic antibacterial efficacy. FASEB J. 2010, 24 (6), 1904–1913. 10.1096/fj.09-149427. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhong W.; Shi Z.; Mahadevegowda S. H.; Liu B.; Zhang K.; Koh C. H.; Ruan L.; Chen Y.; Zeden M. S.; Pee C. J. Designer broad-spectrum polyimidazolium antibiotics. Proc. Natl. Acad. Sci. U. S. A. 2020, 117 (49), 31376–31385. 10.1073/pnas.2011024117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang K.; Du Y.; Si Z.; Liu Y.; Turvey M. E.; Raju C.; Keogh D.; Ruan L.; Jothy S. L.; Reghu S. Enantiomeric glycosylated cationic block co-beta-peptides eradicate Staphylococcus aureus biofilms and antibiotic-tolerant persisters. Nat. Commun. 2019, 10 (1), 1–14. 10.1038/s41467-019-12702-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li J.; Zhang K.; Ruan L.; Chin S. F.; Wickramasinghe N.; Liu H.; Ravikumar V.; Ren J.; Duan H.; Yang L. Block copolymer nanoparticles remove biofilms of drug-resistant Gram-positive bacteria by nanoscale bacterial debridement. Nano Lett. 2018, 18 (7), 4180–4187. 10.1021/acs.nanolett.8b01000. [DOI] [PubMed] [Google Scholar]
- Zgurskaya H. I.; Walker J. K.; Parks J. M.; Rybenkov V. V. Multidrug Efflux Pumps and the Two-Faced Janus of Substrates and Inhibitors. Acc. Chem. Res. 2021, 54, 930–939. 10.1021/acs.accounts.0c00843. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Micoli F.; Bagnoli F.; Rappuoli R.; Serruto D., The role of vaccines in combatting antimicrobial resistance. Nature Reviews Microbiology 2021.19287. 10.1038/s41579-020-00506-3 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Si Z.; Zheng W.; Prananty D.; Li J.; Koh C. H.; Kang E.-T.; Pethe K.; Chan-Park M. B. Polymers as advanced antibacterial and antibiofilm agents for direct and combination therapies. Chemical Science 2022, 13 (2), 345–364. 10.1039/D1SC05835E. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roemhild R.; Bollenbach T.; Andersson D. I. The physiology and genetics of bacterial responses to antibiotic combinations. Nature Reviews Microbiology 2022, 20, 478. 10.1038/s41579-022-00700-5. [DOI] [PubMed] [Google Scholar]
- Hobson C.; Chan A. N.; Wright G. D. The Antibiotic Resistome: A Guide for the Discovery of Natural Products as Antimicrobial Agents. Chem. Rev. 2021, 121, 3464. 10.1021/acs.chemrev.0c01214. [DOI] [PubMed] [Google Scholar]
- Meletis G.; Exindari M.; Vavatsi N.; Sofianou D.; Diza E. Mechanisms responsible for the emergence of carbapenem resistance in Pseudomonas aeruginosa. Hippokratia 2012, 16 (4), 303. [PMC free article] [PubMed] [Google Scholar]
- Flipo M.; Frita R.; Bourotte M.; Martínez-Martínez M. S.; Boesche M.; Boyle G. W.; Derimanov G.; Drewes G.; Gamallo P.; Ghidelli-Disse S. The small-molecule SMARt751 reverses Mycobacterium tuberculosis resistance to ethionamide in acute and chronic mouse models of tuberculosis. Science Translational Medicine 2022, 14 (643), eaaz6280 10.1126/scitranslmed.aaz6280. [DOI] [PubMed] [Google Scholar]
- Stokes J. M.; Yang K.; Swanson K.; Jin W.; Cubillos-Ruiz A.; Donghia N. M.; MacNair C. R.; French S.; Carfrae L. A.; Bloom-Ackermann Z. A deep learning approach to antibiotic discovery. Cell 2020, 180 (4), 688–702. 10.1016/j.cell.2020.01.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pascucci M.; Royer G.; Adamek J.; Asmar M. A.; Aristizabal D.; Blanche L.; Bezzarga A.; Boniface-Chang G.; Brunner A.; Curel C.; Dulac-Arnold G.; Fakhri R. M.; Malou N.; Nordon C.; Runge V.; Samson F.; Sebastian E.; Soukieh D.; Vert J.-P.; Ambroise C.; Madoui M.-A. AI-based mobile application to fight antibiotic resistance. Nat. Commun. 2021, 12 (1), 1173. 10.1038/s41467-021-21187-3. [DOI] [PMC free article] [PubMed] [Google Scholar]