

Summary
Prader-Willi syndrome (PWS) is a rare genetic disorder due to lack of genes expression inherited from the paternal chromosome 15q11-q13 region usually from paternal deletions, maternal uniparental disomy 15 or imprinting defect. There are two different nutritional stages reported in an individual with PWS; first stage during infancy marked by feeding and growth difficulties and second stage where hyperphagia starts and leads to development of obesity. However, the exact mechanism of hyperphagia development, from having difficulties in feeding during early years to insatiable appetite after they grow is still unknown and is the focused in this review. The keywords used for literature search such as "Prader-Willi syndrome", "hyperphagia", "obesity", and "treatment" were used to create the search strings by using synonyms in order to retrieve the relevant records from PubMed, Scopus and Science Direct. The possible mechanism of hyperphagia can be classed into hormonal abnormalities such as increase in ghrelin and leptin from infancy to adulthood. Low level of hormones was observed in the thyroid, insulin and peptide YY at certain ages. Neuronal abnormalities contributed by Orexin A and brain structure alteration was documented at 4-30 years old. Treatment in the form of drugs such as livoletide, topiramate, and diazoxide could potentially alleviate these abnormalities and make hyperphagia less prominent in PWS. The approaches are important to regulate the hormonal changes and neuronal involvement as potentially controlling hyperphagia and obesity.
Keywords: appetite, genetic disorders, hormones, neurodevelopment, overeating
1. Introduction
Prader-Wi l l i syndrome (PWS ) i s a genet ic neurodevelopmental disorder (1). The prevalence of this syndrome is 1/10,000-1/30,000 with approximately 350,000-400,000 individuals worldwide reported with this condition that occurs equally regardless of gender and race (2). PWS is due to loss of gene function in the region of chromosome 15 located at 15q11.2-q13 that is expressed in the paternal gene while the genes on the maternal copy are turned off or inactive in a condition known as genomic imprinting (3,4). Three different causes can lead to PWS. In most cases of PWS, about 70 percent of the cases occur when a paternal chromosome 15 segment is deleted. In another 25 percent of cases, individuals with PWS have two copies of chromosome 15 inherited from the maternal side instead of one copy from each parent, known as maternal uniparental disomy. Another 5 per cent is caused by a chromosome rearrangement called translocation or by a mutation or other defect that abnormally deactivates a gene on paternal chromosome 15. Thus, individuals with these chromosomal changes lose specific critical genes in this segment that leads to PWS.
PWS is characterized by severe hypotonia with difficulty in sucking and breastfeeding during infancy before progressing to overeating and the gradual development of morbid obesity from childhood to adulthood. They also have poor motor and language development as well as cognitive disabilities. PWS individuals have been reported with behavioural problems such as skin picking, difficulty with a change in routine, temper tantrums, obsessive, compulsive behaviours and mood fluctuations (5). In addition, they also suffer from endocrine abnormalities that are present in the form of genital hypoplasia, incomplete pubertal development and infertility in addition to a short physique related to growth hormone deficiency (2).
There are two different nutritional stages reported in an individual with PWS; the first stage is marked by feeding and growth difficulties during infancy and the second stage is when the hyperphagia starts and leads to the development of obesity (6). Hyperphagia is the constant pathologic urge to consume food and constant hunger that can advance into dangerous food-seeking behaviour (7). This leads to the central feature of PWS individuals in which obesity is a primary phenotypic component (8).
PWS is the most commonly known cause of morbid obesity in humans. The annual mortality rate is estimated at 1-4% primarily due to complications of hyperphagia and obesity-related causes (7). The contribution of hyperphagia and obesity as a cause of death in PWS is often discussed as a consequence of cardiorespiratory failure. As this clinical manifestation is mainly presented in PWS, this review sought to summarize the different types of mechanisms in the manifestation of hyperphagia from infancy to adulthood and possible pharmacological treatment approaches for PWS individuals with hyperphagia.
2. Literature search
The keywords and synonyms were used to generate the search strings to retrieve relevant records from PubMed, Scopus and Science Direct (Supplemental Table S1). The search strings were combined using Boolean operators (AND, OR, NOT) by using the advanced search in the database to screen the relevant articles from 2016 to 2022. To limit the records retrieved to research papers, titles and abstracts were screened to eliminate books, reviews, conference papers, and other non-research publications. The titles and abstracts of the chosen original research in English were selected in this review. Then, relevant primary research articles were screened for relevant content to hyperphagia and obesity in PWS individuals in English. The references are classified based on the mechanism and treatment.
Based on the three chosen databases in this scoping review including Pubmed, Science Direct and Scopus, there were a total of 778 records retrieved which were Pubmed (n = 54), Science Direct (n = 424) and Scopus (n = 300). Then, 430 duplicates were removed. After the initial screening, there were 348 articles, 9 records were removed due to the articles detected were without titles along with authors and abstract in the EndNote system. Then, the total of 339 records, complete with title, names and abstract in the EndNote system were identified, 49 records were removed because it included books and unrelated articles. Finally, a total of 290 records were selected as research articles that had undergone secondary selection. A total of 258 articles were excluded due to hyperphagia or obesity that are not related to PWS and 11 articles were published in a language other than English. Finally, 21 studies including 10 mechanism studies and 11 treatment studies were included into the literature synthesis. The screening process was done by more than one person to avoid bias and contradiction in the selection of studies.
3. Potential mechanisms causing hyperphagia
PWS is a genetic neurological disorder due to loss of function on the long arm (q11-q13) of the paternal chromosome 15. The paternally expressed PWS region located on chromosome 15 contains genes encoding polypeptides such as MKRN3, MAGEL2, NECDIN and snoRNA (2). The critical gene for most PWS phenotypes involves the snoRNA gene, SNORD116 are affected (9). PWS baby could survive until adulthood, however they tend to develop into obesity resulted from hyperphagia. The prevalence of overweight and obesity in PWS is around 40% in children and adolescents (10), while this percentage tends to increase between 80% and 90% in adulthood (11,12). Although PWS individuals have poor nutrition and appetite during infancy, they experience uncontrolled appetite leading to weight gain after the age of 4 (6). While the exact mechanism has not yet been fully explained, the development of obesity is mainly related to dysfunction in the feeding centre of the hypothalamus and its hormones that lead to uncontrol food intake and energy expenditure (13). Disruption in the hypothalamic pathway of satiety control results in persistent and unsatiated appetite, hyperphagia and hunger-related eating behaviours. It is closely associated with hormones and neuronal abnormalities that can cause body composition to change and stimulate hyperphagia.
Hypothalamic neurons sense both neural and physiological signals and respond by releasing neurotransmitters and peptide neuromodulators into the brain (14). Hypothalamic neurons also regulate puberty, reproduction, stress, circadian rhythms, immune function, and more complex behaviours such as social behaviour. Furthermore, hormones are released from the endocrine glands to regulate physiology and behaviour. The dysfunction of human hypothalamic neurons and hormones have been linked to obesity, hypertension, mood disorders and sleep disorders (15). For instance, it helps to regulate body composition, that is composed of fat, bone and muscle in the body. However, an alteration in a high percentage of fat with less muscle mass is present in PWS instead of a normal body composition that consists of less fat and more muscle mass (16).
A deficiency of thyroid hormone levels in PWS was observed as early as infancy (17). About 20-30% of PWS patients have thyroid hormone deficiency (2,19). Low T4, T3 and thyroid stimulating hormone (TSH) levels are among the cause of the floppy baby syndrome and hypotonia. Apart from that, the reduction of thyroid hormone leads to a change in metabolic rate and the reduction of energy consumption (17). This makes PWS individuals more prone to develop obesity later as they age.
Next, an increase in ghrelin has been seen in PWS individuals since they were 5 weeks old (18). Ghrelin is a powerful orexigenic hormone, an appetite stimulant hormone that increases appetite and food intake. The stomach secretes ghrelin during fasting or when hungry. The individual will feel hungrier and the increment of hormone level will gradually decrease after eating. However, the ghrelin level will remain high even after food consumption in PWS. Hyperghrelinemia was also experienced as early as one year old, who was still in nutritional phase 1a, characterized by poor appetite and feeding (19). Ghrelin also is associated with playing an essential role in the adaptation of the fetus to intrauterine malnutrition as well as the growth of infants who are smaller than the average fetus, known as small for gestational age (SGA) (20). High levels of ghrelin have been observed in SGA and premature infants leading to the hypothesis that intrauterine growth restriction and low birth weight are the possible physiological effects of excessive ghrelin secretion (20,21). Therefore, it is possible that the high ghrelin levels observed in PWS infants are a physiological response to their birth weight being on average about 15% less than expected (22). Increased ghrelin leads to two different results according to age which is an adaptation of the fetus to malnutrition as well as the growth in small-sized babies, but the hormone acts as an appetite stimulant later in life (21).
The following mechanism is the increase of leptin which can be seen in two different studies; in seven-month-old subjects (before hyperphagia starts) and in adulthood (after the hyperphagia phase starts) (1,23). The function of leptin is to control the long-term balance between food intake and energy use (18). The small infant size at birth was associated with higher leptin levels in umbilical cord blood and in turn, was associated with higher body mass index weight gain at 4 years of age (24). Besides, the effect of high leptin in adults is associated with low Brain-Derived Neurotrophic Factor (BDNF). BDNF acts as a satiety signal guided by leptin-melanocortin signalling. In PWS, the neural circuits in this area become dysfunctional and cause a decrease in local BDNF levels. Low BDNF level causes less peripheral density and this in turn causes adiposity to secrete more leptin. A prolonged increase in leptin causes leptin resistance and subsequently lack of satiety, overeating and increased weight.
Low peptide YY (PYY) and insulin levels are also involved in hyperphagia and obesity of PWS. The function of PYY and insulin are to stimulate pro-opiomelanocortin (POMC) neurons and inhibit neuropeptide Y (NPY) follows by activation of melanocortin receptor 4 (MC4R) to induce satiety (13). As PWS individuals have low levels of PYY and insulin, NPY is released and prevents MC4R activation that leads to increased food intake (26,27). This causes failure of satiety control as α and β-MSH or MC4R fail to be activated (28).
In addition to hormones, other mechanisms from the study's findings involved neuronal abnormality via changes in brain structure. Changes in several brain areas (hypothalamus, amygdala, hippocampus, orbitofrontal and medial prefrontal cortex) play an important role in regulating abnormal food intake in PWS. Functional magnetic resonance imaging showed higher activity in reward/limbic regions (nucleus accumbens, amygdala) in subjects with PWS (29). Mainly, subjects with PWS exhibited greater food activation in limbic and paralimbic regions (hypothalamus, amygdala, hippocampus) and lower activation in cortical inhibitory regions (orbitofrontal cortex, medial prefrontal cortex) (29,30). In addition, reduced functional connectivity between the ventral striatum and limbic structures (hypothalamus and amygdala) was reported in subjects with PWS and it was associated with obsessive eating behaviour (30). These brain function studies show that hypothalamic control disorders, dysfunction in food reward-related circuit areas and impairments in inhibitory control areas contribute to hyperphagia and extreme obesity in PWS.
Low levels of Orexin A could be seen in subjects aged 5 to 11 years, a phase where hyperphagia has already started. The function of orexin is to stimulate appetite and increase food intake (31). These findings are consistent with the association of orexin with serious neurological dysfunction involving food-addictive behaviour (32,33). The dopamine-rich ventral tegmental area (VTA) and the nucleus accumbens (NA) function as behavioural regulators, behaviour driven by food reward and addiction. Both structures are heavily innervated by orexin neurons and express high levels of orexin receptors (34). Excessive orexin stimulation in the hypothalamus, as well as the VTA and NA contribute to hyperphagia by increasing the reward value of food in patients with PWS. Thus, the insatiable appetite and unusual eating-related problems exhibited by PWS patients indicate abnormalities in the orexin system. The findings are summarized in Table 1 and tabulated in Figure 1 according to nutritional phase and age in PWS.
Table 1. Mapping of evidence regarding the potential mechanisms of hyperphagia.
| Abnormality | Subject's age | Level of abnormality | Details | Ref. |
|---|---|---|---|---|
| Thyroid hormone | 1 week-3 years | Decreased | ―Function: Regulate whole body metabolism. ―In PWS: ↓ in PWS resulting in altered metabolic rate and energy expenditure. |
(17) |
| Ghrelin | 5 weeks-36 years old | Elevated | ―Function: Regulates short-term food intake, ↑ in hunger, ↓ after food intake. ―In PWS: Persistently ↑ ghrelin even after food intake leads to weight gain. ↑ body fat. |
(18) |
| Leptin | 7 months-5 years old | Elevated | ―Function: Help regulate the long-term balance between the body's food intake and energy use. | (1) |
| Insulin | Children (median age 11.35 years old) | Decreased | ―Function: Stimulate POMC and inhibit NPY neurons leading to stimulation of MC4R to induce satiety ―In PWS: ↓ PWS leads to MC4R not being stimulated. |
(26) |
| Peptide YY | 19-42 years old | Decreased | ―Function: Induce satiety by stimulating POMC and inhibiting NPY resulting in activation of α and β-MSH and reducing gastric emptying. ―In PWS: ↓ PYY in PWS causes loss of stimulating signal to POMC, fails to stimulate α and β-MSH. |
(27) |
| Brain-Derived Neurotrophic Factor (BDNF) and leptin | 30 adults | BDNF: Decreased Leptin: Elevated | ―Function BDNF: act as a satiety signal. ―Function Leptin: helps regulate the long-term balance between the body's food intake and energy use. ―In PWS: BDNF signalling is compromised, ↓ and local BDNF levels, ↑ leptin, resulting in leptin resistance, leading to hyperphagia and obesity. |
(24) |
| Ghrelin and Glucagon-like peptide-1 | Adults, median age 27.5 years old | Ghrelin: Elevated GLP-1: Decreased | ―Function ghrelin: Regulates short-term food intake, ↑ in hunger, ↓ after food intake. ―Function GLP-1: is an appetite suppressor hormone. Elevated satiety signal concentrations could be a compensatory response to higher ghrelin levels. ―In PWS: Persistently ↑ ghrelin even after food intake leads to weight gain. ↓ in GLP-1 secretion, resulting increase in gastric emptying rates. |
(23) |
| Altered brain structure | Children, median age 7.2 years old | Cortical volume: Decrease White matter integrity: Reduced fractional anisotropy (FA) Grey matter volume: Decrease | ―Cortisol volume = decreased cortical volume in the bilateral frontal, medial prefrontal cortex and anterior cingulate lead to dysfunctions in regulations of appetite, increased self-reported hunger and increased risk of overeating through an imbalance between cognitive and emotional processing. ―White matter integrity and Gray matter volume: ↓ FA indicates ↓ white matter health. Grey and white matter damage were present in the brain regions associated with food intake in PWS. |
(30) |
| Orexin A | 5-11 years old | Elevated | ―Function Orexin A: stimulates appetite and increases food consumption. ―In PWS: Overstimulation of orexin signalling in the hypothalamus contribute to hyperphagia by increasing the food addiction. |
(31) |
Figure 1.
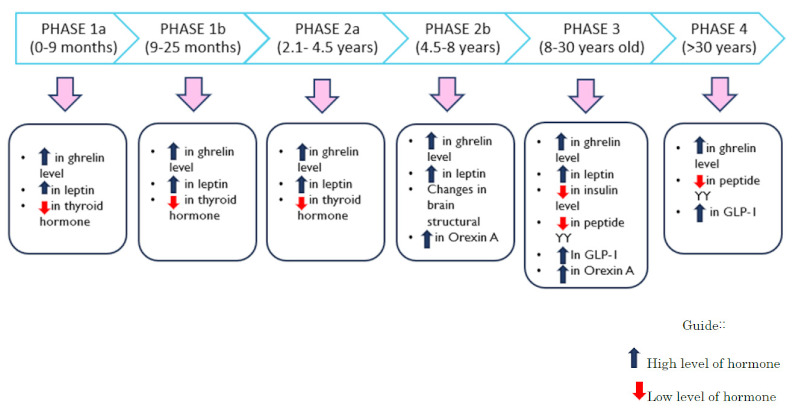
Summary of the hormonal and physiological changes involved in the development of hyperphagia from infants to adults in Prader-Willi syndrome. The changes are tabulated according to nutritional phase and age in Prader Willi syndrome.
4. Potential treatment to reduce hyperphagia
Several pharmaceutical companies are developing drugs to target the mechanisms (Table 2). Among the potential treatments to correct ghrelin abnormalities are AZP-531 and RM-853. Two primary forms of ghrelin are found in circulation, acylated ghrelin (AG) and unacylated ghrelin (UAG). Studies show that the ratio of AG to UAG is relevant to hyperphagia in PWS (35). A hypothesis has been made that when UAG levels are too low, it will cause a higher AG/UAG ratio, which then results and leads to the development of hyperphagia and obesity (36). Therefore, an approach to treat hyperphagia and obesity in PWS is via pharmacological alteration of the AG/ UAG ratio, through an increase in UAG concentration. AZP-531, an amino acid peptide, is a stable UAG analogue (36). AZP-531 (livoletide) treatment reduced waist circumference and fat mass, but no significant changes were detected in weight. Besides, no serious side effects were observed during the study, indicating that AZP-531 is well tolerated in PWS individuals (37). In addition, RM-853 [Ghrelin O-acyltransferase (GOAT)] is an enzyme that catalyzes the octanoylation of ghrelin (38). Inhibition of GOAT will inhibit the production of AG and block orexigenic and adipogenic effects. This restriction will also increase UAG levels. Ghrelin signalling through GOAT inhibition provides a potential therapeutic opportunity to treat hyperphagia and obesity (39).
Table 2. Mapping of evidence on potential treatments to reduce hyperphagia.
| Treatment | Mechanism of action | Advantages | Ref. |
|---|---|---|---|
| AZP-531 (Livoletide) | Decreases the appetite-stimulating effects of ghrelin. | Potential to address PWS-specific increase in ghrelin. | (37) |
| RM-853 (ghrelin o-acyltransferase (GOAT) inhibitor) | Inhibitor of an enzyme that catalyzes ghrelin octanolycation, thus resulting in reduced production of ghrelin. | Possible to modify food intake and prevent weight gain. | (39) |
| Topiramate | In the hypothalamus, topiramate increased mRNA for neuropeptide Y. | Possible reduced food intake acutely and increased metabolic rate. There were also significant reduction in leptin. | (40) |
| Diazoxide | K+ ATP channel agonist that may exert therapeutic effects through the down-regulation of insulin secretion, modulation of hypothalamic neuropeptide Y concentrations, increased GABAnergic neuronal excitability, and activation of KATP channels in adipocytes. | FDA-approved drug for the treatment of hyperinsulinemia and hypoglycemia. Potential to treat hyperphagia. | (41) |
| Carbetocin | Carbetocin is an oxytocin analogue that has the exact mechanism and outcomes as oxytocin. | Have a longer half-life than oxytocin. | (44) |
| Beloranib | inhibits an enzyme methionine aminopeptidase 2 (MetAP2) that reduces hunger while stimulating the use of stored fat as an energy source | FDA-approved drug for the treatment of hyperphagia and weight loss. | (45) |
| Setmelanotide | Activates MC4R and this causes the inhibition of food intake. | Possibility to address an underlying defect in hunger circuits. | (46) |
| Oxytocin | Binds to G protein-coupled receptor and this leads to activation of several systems that regulate appetite. | Potential to replace the insufficiency of oxytocin in patients with PWS and may have positive effects on hyperphagia. | (47) |
| Tesofensine | serotonin-noradrenaline-dopamine reuptake inhibitor acts primarily as an appetite suppressant with related effects on fat oxidation and resting energy expenditure. | Potential treatment to reduce appetite, decrease food craving. | (48) |
| JD5037 (antiobesity drug candidate) | Restricted cannabinoid-1 receptor (CB1R) an antagonist that targets the overstimulated endocannabinoid system in PWS to reduce appetite. | Potential to treat obesity-related metabolic disorders without producing adverse nervous system effects. | (49) |
| CBDA-O-methyl ester (EPM301) | Enhance serotonergic 5-HT1A receptor activation following agonist binding. | Weight loss, increased ambulation, and improved glycemic and lipid profiles. | (50) |
Potential treatments to correct PYY and insulin abnormalities are topiramate, diazoxide and setmelanotide. Topiramate reduced the mRNA for NPY. As NPY stimulates food intake, increases motivation to eat and delays satiety, reducing its level could reduce food intake and increase metabolic rate (40). The next approach is via Diazoxide, a K+-ATP channel agonist approved by the FDA for treating hypoglycemia, hyperinsulinemia and acute hypertension. Diazoxide has a therapeutic effect on PWS through insulin secretion from pancreatic β-cells, modulation of hypothalamic NPY, and activation of KATP channels in adipocytes (41). This study showed that oral diazoxide administration for 12 weeks reduced fat mass, lowered blood glucose, and increased endurance capacity. Although the effects of diazoxide on PWS-related hyperphagia are not yet well understood, current evidence suggests that diazoxide deserves further research attention. PWS individuals may also be responsive to the therapeutic activation of MC4R, which provides a rationale for treating obesity with setmelanotide. Setmelanotide is a potent and selective MC4R agonist for treating genetic disorders of obesity. It binds with high affinity to human MC4R, resulting in efficient MC4R activation that may potentially reduce hyperphagia associated with PWS (42).
Another study finds that targeting oxytocin, a hormone produced in the hypothalamic paraventricular nucleus (PVN) and supraoptic nucleus, could better regulate food intake and satiety (43). Thus, the oxytocin analogue, carbetocin also has been introduced intranasally to PWS and shows positive effects because it could improve hyperphagia (44). The following approach is via appetite suppressant. Tesofensine, a serotonin-noradrenaline-dopamine monoamine reuptake inhibitor was co-administered with metoprolol (to reduce blood pressure) and showed that it could inhibit hunger feeling and food cravings by boosting the three neurotransmitters' activity. Meanwhile, Beloranib inhibits an enzyme methionine aminopeptidase 2 (MetAP2) that reduces hunger and induces weight loss (45). While JD5037, a cannabinoid-1 receptor blocker reacts on CB receptors function in inhibiting food intake and increasing satiety.
5. Conclusion
Based on this review, the mechanism of development of hyperphagia and obesity could be seen from hormonal changes (ghrelin, leptin, insulin, thyroid, PYY), Orexin A alteration and changes in brain structure. Several drugs have been shown to potentially ameliorate abnormalities of hormones, body composition and eating behaviour. However, these treatments need to be studied further because most still have no record of long-term safety and effectiveness in PWS individuals.
Acknowledgements
We thank Persatuan Sindrom Prader-Willi Malaysia and Prof. Thong Meow Keong for their support in completing this manuscript
Funding:
None.
Conflict of Interest
The authors have no conflicts of interest to disclose.
References
- 1. Goldstone AP, Holland AJ, Hauffa BP, Hokken-Koelega AC, Tauber M; speakers contributors at the Second Expert Meeting of the Comprehensive Care of Patients with PWS. Recommendations for the diagnosis and management of Prader-Willi syndrome. J Clin Endocrinol Metab. 2008; 93:4183-4193. [DOI] [PubMed] [Google Scholar]
- 2. Cassidy SB, Schwartz S, Miller JL, Driscoll DJ. Prader- Willi syndrome. Genet Med. 2012; 4:10-26. [DOI] [PubMed] [Google Scholar]
- 3. Butler MG. Genomic imprinting disorders in humans: a mini-review. J Assist Reprod Genet. 2009; 26:477-486. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Stevenson DA, Heinemann J, Angulo M, Butler MG, Loker J, Rupe N, Kendell P, Cassidy SB, Scheimann A. Gastric rupture and necrosis in Prader-Willi syndrome. J Pediatr Gastroenterol Nutr. 2007; 45:272-274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Angulo MA, Butler MG, Cataletto ME. Prader-Willi syndrome: a review of clinical, genetic, and endocrine findings. J Endocrinol Invest. 2015; 38:1249-1263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Bueno Díez M, Caixàs Pedragós A. Prader-Willi syndrome and hyperphagia: a challenge to investigate. Endocrinol Nutr. 2014; 61:121-122. [DOI] [PubMed] [Google Scholar]
- 7. Butler MG, Manzardo AM, Heinemann J, Loker C, Loker J. Causes of death in Prader-Willi Syndrome: Prader-Willi Syndrome Association (USA) 40-year mortality survey. Genet Med. 2017; 19:635-642. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Heymsfield SB, Avena NM, Baier L, et al. Hyperphagia: current concepts and future directions proceedings of the 2nd International Conference on Hyperphagia. Obesity. 2014; 22:S1-S17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Buiting K. Prader-Willi syndrome and Angelman syndrome. Am J Med Genet C Semin Med Genet. 2010; 154C:365-376. [DOI] [PubMed] [Google Scholar]
- 10. Diene G, Mimoun E, Feigerlova E, Caula S, Molinas C, Grandjean H, Tauber M; French Reference Centre for PWS. Endocrine disorders in children with Prader- Willi syndrome - data from 142 children of the French database. Horm Res Paediatr. 2010; 74:121-128. [DOI] [PubMed] [Google Scholar]
- 11. Grugni G, Crino A, Bosio L, et al. The Italian National Survey for Prader-Willi syndrome: an epidemiologic study. Am J Med Genet A. 2008; 146A:861-872. [DOI] [PubMed] [Google Scholar]
- 12. Sinnema M, Einfeld SL, Schrander-Stumpel CT, Maaskant MA, Boer H, Curfs LM. Behavioral phenotype in adults with Prader-Willi syndrome. Res Dev Disabil. 2011; 32:604-612. [DOI] [PubMed] [Google Scholar]
- 13. Khan MJ, Gerasimidis K, Edwards CA, Shaikh MG. Mechanisms of obesity in Prader-Willi Syndrome. Pediatr Obes. 2018; 13:3-13. [DOI] [PubMed] [Google Scholar]
- 14. Merkle FT, Maroof A, Wataya T, Sasai Y, Studer L, Eggan K, Schier AF. Generation of neuropeptidergic hypothalamic neurons from human pluripotent stem cells. Development. 2015; 142:633-643. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Kim JH, Choi JH. Pathophysiology and clinical characteristics of hypothalamic obesity in children and adolescents. Ann Pediatr Endocrinol Metab. 2013; 18:161-167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Marzullo P, Mele C, Minocci A, Mai S, Scacchi M, Sartorio A, Aimaretti G, Grugni G. Fat-free mass is better related to serum uric acid than metabolic homeostasis in Prader-Willi syndrome. Nutrients. 2020; 12:2583. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Vaiani E, Herzovich V, Chaler E, Chertkoff L, Rivarola MA, Torrado M, Belgorosky A. Thyroid axis dysfunction in patients with Prader-Willi syndrome during the first 2 years of life. Clin Endocrinol (Oxf). 2010; 73:546-550. [DOI] [PubMed] [Google Scholar]
- 18. Kweh FA, Miller JL, Sulsona CR, Wasserfall C, Atkinson M, Shuster JJ, Goldstone AP, Driscoll DJ. Hyperghrelinemia in Prader-Willi syndrome begins in early infancy long before the onset of hyperphagia. Am J Med Genet A. 2016; 167A:69-79. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Feigerlova E, Diene G, Conte-Auriol F, Molinas C, Gennero I, Salles JP, Arnaud C, Tauber M. Hyperghrelinemia precedes obesity in Prader-Willi syndrome. J Clin Endocrinol Metab. 2008; 93:2800-2805. [DOI] [PubMed] [Google Scholar]
- 20. Sahin H, Erener T, Erginoz E, Vural M, Ilikkan B, Kavuncuoglu S, Yildiz H, Perk Y. The relationship of active ghrelin levels and intrauterine growth in preterm infants. Eur J Endocrinol. 2012; 166:399-405. [DOI] [PubMed] [Google Scholar]
- 21. Fidanci K, Meral C, Suleymanoglu S, Pirgon Ö, Karademir F, Aydınöz S, Özkaya H, Gültepe M, Göçmen İ. Ghrelin levels and postnatal growth in healthy infants 0-3 months of age. J Clin Res Pediatr Endocrinol. 2010; 2:34-38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Miller JA, Lynn CH, Driscoll DC, Goldstone AP, Gold JA, Kimonis V, Dykens E, Butler MG, Shuster JJ, Driscoll DJ. Nutritional phases in Prader-Willi syndrome. Am J Med Genet A. 2011; 155A:1040-1049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Bueno M, Esteba-Castillo S, Novell R, Giménez-Palop O, Coronas R, Gabau E, Corripio R, Baena N, Viñas-Jornet M, Guitart M, Torrents-Rodas D, Deus J, Pujol J, Rigla M, Caixàs A. Lack of postprandial peak in brain-derived neurotrophic factor in adults with Prader-Willi syndrome. PLoS One. 2016; 11:e0163468. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Kaar JL, Brinton JT, Crume T, Hamman RF, Glueck DH, Dabelea D. Leptin levels at birth and infant growth: The EPOCH study. J Dev Orig Health Dis. 2014; 5:214-218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Obradovic M, Sudar-Milovanovic E, Soskic S, Essack M, Arya S, Stewart AJ, Gojobori T, Isenovic ER. Leptin and obesity: role and clinical implication. Front Endocrinol (Lausanne). 2021; 12:585887. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Haqq AM, Muehlbauer MJ, Newgard CB, Grambow S, Freemark M. The metabolic phenotype of Prader- Willi syndrome (PWS) in childhood: heightened insulin sensitivity relative to body mass index. J Clin Endocrinol Metab. 2011; 96:E225-E232. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Rigamonti AE, Bini S, Piscitelli F, Lauritano A, Di Marzo V, Vanetti C, Agosti F, De Col A, Lucchetti E, Sartoria A. Hedonic eating in Prader-Willi syndrome is associated with blunted PYY secretion. Food Nutr Res. 2017; 61:1297553. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Doulla M, McIntyre AD, Hegele RA, Gallego PH. A novel MC4R mutation associated with childhood-onset obesity: A case report. Paediatr Child Health. 2014; 19:515-518. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Holsen LM, Zarcone JR, Brooks WM, Butler MG, Thompson TI, Ahluwalia JS, Nollen NL, Savage CR. Neural mechanisms underlying hyperphagia in Prader- Willi syndrome. Obesity (Silver Spring). 2006; 14:1028-1037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Xu M, Zhang Y, Von Deneen KM, Zhu H, Gao JH. Brain structural alterations in obese children with and without Prader-Willi syndrome. Hum Brain Mapp. 2017; 38:4228-4238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Manzardo AM, Johnson L, Miller JL, Driscoll DJ, Butler MG. Higher plasma orexin a levels in children with Prader-Willi syndrome compared with healthy unrelated sibling controls. Am J Med Genet A. 2016; 170:2328-2333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Harris GC, Wimmer M, Aston-Jones G. A role for lateral hypothalamic orexin neurons in reward seeking. Nature. 2005; 437:556-559. [DOI] [PubMed] [Google Scholar]
- 33. Sakurai T. The role of orexin in motivated behaviours. Nat Rev Neurosci. 2014; 15:719-731. [DOI] [PubMed] [Google Scholar]
- 34. Harris GC, Wimmer M, Randall-Thompson JF, Aston- Jones G. Lateral hypothalamic orexin neurons are critically involved in learning to associate an environment with morphine reward. Behav Brain Res. 2007; 183:43-51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Beauloye V, Diene G, Kuppens R, Zech F, Winandy C, Molinas C, Faye S, Kieffer I, Beckers D, Nergårdh R, Hauffa B, Derycke C, Delhanty P, Hokken-Koelega A, Tauber M. High unacylated ghrelin levels support the concept of anorexia in infants with Prader-Willi syndrome. Orphanet J Rare Dis. 2016; 11:56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Kuppens RJ, Diene G, Bakker NE, Molinas C, Faye S, Nicolino M, Bernoux D, Delhanty PJ, van der Lely AJ, Allas S, Julien M, Delale T, Tauber M, Hokken-Koelega AC. Elevated ratio of acylated to unacylated ghrelin in children and young adults with Prader-Willi syndrome. Endocrine. 2015; 50:633-642. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Delhanty PJ, Sun Y, Visser JA, van Kerkwijk A, Huisman M, van Ijcken WF, Swagemakers S, Smith RG, Themmen AP, van der Lely AJ. Unacylated ghrelin rapidly modulates lipogenic and insulin signaling pathway gene expression in metabolically active tissues of GHSR deleted mice. PLoS One. 2020; 5:e11749. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Hougland JL. Ghrelin octanoylation by ghrelin O-acyltransferase: unique protein biochemistry underlying metabolic signaling. Biochem Soc Trans. 2019; 47:169-178. [DOI] [PubMed] [Google Scholar]
- 39. Cleverdon ER, McGovern-Gooch KR, Hougland JL. The octanoylated energy regulating hormone ghrelin: an expanded view of ghrelin's biological interactions and avenues for controlling ghrelin signaling. Mol Membr Biol. 2016; 33:111-124. [DOI] [PubMed] [Google Scholar]
- 40. Consoli A, Çabal Berthoumieu S, Raffin M, Thuilleauxet D, Poitou C, Coupaye M, Pinto G, Lebbah S, Zahr N, Tauber M, Cohen D, Bonnot O. Effect of Topiramate on eating behaviours in Prader-Willi syndrome: toprader double-blind randomised placebo-controlled study. Transl Psychiatry. 2019; 9:274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Kimonis V, Surampalli A, Wencel M, Gold JA, Cowen NM. A randomized pilot efficacy and safety trial of diazoxide choline controlled- release in patients with Prader-Willi syndrome. PLoS One. 2019; 14:e0221615. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Collet TH, Dubern B, Mokrosinski J, et al. Evaluation of a melanocortin-4 receptor (MC4R) agonist (Setmelanotide) in MC4R deficiency. Mol Metab. 2017; 6:1321-1329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Miller J. The potential of oxytocin for the treatment of hyperphagia in Prader-Willi syndrome. Expert Opin Orphan Drugs. 2018; 6:247-251. [Google Scholar]
- 44. Dykens EM, Miller J, Angulo M, Roof E, Reidy M, Hatoum HT, Willey R, Bolton G, Korner P. Intranasal carbetocin reduces hyperphagia in individuals with Prader-Willi syndrome. JCI Insight. 2018; 3:98333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Pottorf TS, Fagan MP, Burkey BF, Cho DJ, Vath JE, Tran PV. MetAP2 inhibition reduces food intake and body weight in a ciliopathy mouse model of obesity. JCI Insight. 2020; 5:e134278. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Falls BA, Zhang Y. Insights into the allosteric mechanism of setmelanotide (RM-493) as a potent and first-in-class melanocortin-4 receptor (MC4R) agonist to treat rare genetic disorders of obesity through an in silico approach. ACS Chem Nerosci. 2019; 10:1055-1065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Miller JL, Tamura R, Butler MG, Kimonis V, Sulsona C, Gold JA, Driscoll DJ. Oxytocin treatment in children with Prader-Willi syndrome: a double-blind, placebo-controlled, crossover study. Am J Med Genet A. 2017; 173:1243-1250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Bentzen BH, Grunnet M, Hyveled-Nielsen L, Sundgreen C, Lassen JB, Hansen HH. Anti-hypertensive treatment preserves appetite suppression while preventing cardiovascular adverse effects of tesofensine in rats. Obesity (Silver Spring). 2013; 21:985-992. [DOI] [PubMed] [Google Scholar]
- 49. Nagappan A, Shin J, Jung MH. Role of cannabinoid receptor type 1 in insulin resistance and its biological implications. Int J Mol Sci. 2019; 20:2109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Ben-Cnaan E, Permyakova A, Azar S, Hirsch S, Baraghithy S, Hinden L, Tam J. The metabolic efficacy of a cannabidiolic acid (CBDA) derivative in treating diet-and genetic-induced obesity. Int J Mol Sci. 2022; 23:5610. [DOI] [PMC free article] [PubMed] [Google Scholar]