

Abstract
Aliphatic alkylamines are abundant feedstock and versatile building blocks for many organic transformations. While remarkable progress has been made to construct C–N bonds on aliphatic and aromatic carbon centers, the activation and functionalization of C(sp3)–NH2 bonds in primary alkylamines remain a challenging process. In the present work, we discovered an unprecedented method to directly activate the C(sp3)–NH2 bond of primary alkylamines by a high-valent dinuclear CoIII,IV2(μ-O)2 diamond core complex. This reaction results in the installation of other functional groups such as halides and alkene onto the α-carbon center concomitant with the 2-e− oxidation of the nitrogen atom on the amino group to form NH2OH. Our results shed light on future development of catalytic systems enabling versatile functionalization of primary alkylamines based on the dinuclear cobalt system.
Introduction.
Aliphatic alkylamines are abundant feedstock and versatile building blocks in a variety of important biological and synthetic transformations through the construction and activation of their C–N bonds.1 A number of redox and nonredox-based biological processes invoke the cleavage of C–N bonds, such as ubiquitous transamination,2,3 cross exchange of an amino group and a hydrogen atom on vicinal carbon atoms,4 and deaminative conversion of alkylamine to aldehyde.5,6 In particular, deaminative oxidations of primary alkylamines are typically catalyzed by amine oxidases, including the copper-containing amine oxidases (CuAOs) and the flavin-containing monoamine and polyamine oxidases (MAOs and PAOs),5,6 to form aldehydes and ammonia: RCH2NH2 + H2O + O2 → RCHO + NH3 + H2O2. This reaction affords the oxidation of the α-carbon site on the alkyl chain instead of the nitrogen atom of the amino group upon the cleavage of the C–N bond. Interestingly, in contrast, the ammonia-oxidizing enzyme ammonia monooxygenase (AMO) catalyzes the 2-e− oxidation of NH3 to NH2OH with the formation of the N–O bond,7 the first reaction of natural nitrification process that plays key roles in global carbon and nitrogen cycling.8 AMO is a copper-dependent enzyme and a homolog of the methane-oxidizing enzyme particulate methane monooxygenase (pMMO),9–11 although its structure and reaction mechanism are yet to be described.
In synthetic chemistry, remarkable progress has been made to construct C–N bonds on aliphatic and aromatic carbon centers through recent developments of C–H bond amination and C–N bond cross-coupling reactions.12–14 On the other hand, the activation of C–N bonds, particularly the unactivated C(sp3)–NH2 bonds in primary aliphatic alkylamines, has been much less studied.15,16 The conversion of C(sp3)−NH2 bonds to other functionalized moieties is generally a difficult process due to their strong C–N bonds and two active N–H bonds. Available deaminative cross-coupling methods typically require pre-activation of the C(sp3)–NH2 bonds to form imine, ammonium or alkylpyridinium species (Katritzky salts, prepared by the reactions of alkylamines with the pyrylium salts) before the C–N bonds can be cleaved (Scheme 1).15,17–25 Although direct approaches to activate C(sp3)−NH2 bonds are available, they are limited to specific classes of alkylamines such as α-aminoalkylferrocenes and amino acids.26,27
Scheme 1.

Strategies for activating the C(sp3)–NH2 bond of primary aliphatic alkylamines.
Recently, we reported the characterization and reactivity studies of a high-valent bis-μ-oxo CoIII,IV2(μ-O)2 diamond core complex (1), obtained by one-electron oxidation of the CoIII2(μ-O)2 precursor (1a), as the synthetic mimic of the reactive diiron(IV) intermediate Q of soluble methane monooxygenase (sMMO).28,29 1 is able to cleave sp3 C–H bond up to 87 kcal/mol and is much more reactive than the diiron and dicopper analogues.29 More importantly, the full oxidizing power of 1 can be further released upon interacting with Lewis bases to open up its diamond core and generate a terminal CoIV–O moiety, resulting in million-fold rate enhancement and the ability to cleave stronger C–H bonds up to 96 kcal/mol.28 Specifically, when OH− is employed as the Lewis base, a cis-open core species HO–CoIII–O–CoIV–O is generated based on our DFT calculations, where a hydrogen bond is formed between the CoIII–OH and CoIV–O moieties.
In this report, we discovered an unprecedented C(sp3)–NH2 bond activation reactivity of the diamond core complex 1 with primary aliphatic alkylamines to form alkyl chlorides and/or alkenes and hydroxylamine (Scheme 1). This deaminative reaction results in the installation of other functional groups such as chloride and C=C double bond onto the α-carbon center concomitant with the 2-e− oxidation of the nitrogen atom on the amino group (instead of the α-carbon center as in amine oxidases) to form a N–O bond. The formation of alkyl chloride and alkene is strongly affected by the structural and electronic properties of the alkylamine substrates, an approach that can be employed to predict the reaction outcomes. Our work has thus provided an unprecedented method to directly activate and functionalize the C(sp3)–NH2 bond of primary aliphatic alkylamines, a new organic transformation that has yet to be reported in previous discoveries. On the other hand, the oxidation of the amino group/ammonia to hydroxylamine by the high-valent dicobalt species is a synthetic mimic for the enzymatic reaction of AMO, which suggests that a related high-valent copper-oxo species is likely generated as the key intermediate to carry out the ammonia oxidation and N–O bond formation in the enzymatic catalytic cycle.
Results and Discussion.
We first selected 4-phenylbutylamine (PBA, 2, pKa = 10.21 in aqueous solution) as a probe substrate to investigate the reaction of 1 with alkylamines, with the original goal of evaluating any possible site-selective C–H bond oxidation on the alkyl chain. The introduction of only 1 eq. of PBA into the methanol solution of 1 at −60 °C causes the increase of the self-decay rate of 1 by one order of magnitude from 0.008 to 0.076 s−1. This rate is about 3-fold faster than those observed for 1 in the presence of other simple Lewis bases reported in our previous work.28 The reaction follows first-order kinetics (Figure S1) without the formation of any other intermediate. Surprisingly, this dicobalt-PBA adduct is unable to react with external substrates such as toluene. As shown in Figure 1A, the addition of large excess amounts of toluene does not result in any enhancement of the rate constant, indicating that intermolecular C–H bond oxidations by 1, as we previously observed,28,29 are completely inhibited by the presence of only 1 eq. of PBA. In contrast, a typical second-order rate constant (k2) of 0.23(3) M−1 s−1 is measured for toluene oxidation by 1 in the presence of 1 eq. of triethylamine (pKa = 10.8). These results clearly indicate that primary and tertiary alkylamines have distinct effects on the reactivity of the diamond core complex 1.
Figure 1.
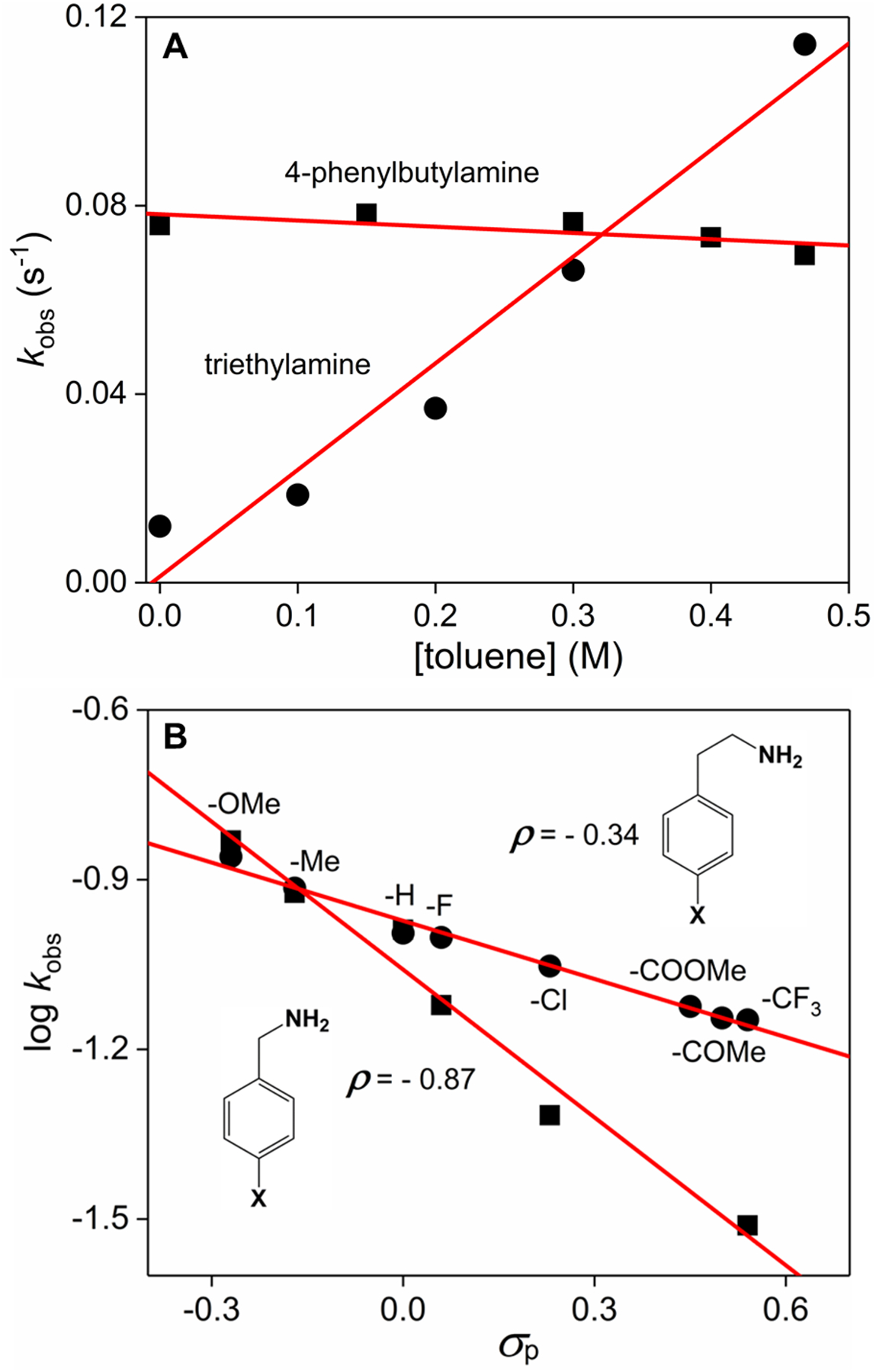
(A) Plots of kobs as a function of the substrate concentration for toluene oxidation by 0.15 mM 1 in the presence of 1 eq. 4-phenylbutylamine (■) and triethylamine (•), respectively, obtained in MeOH at −60 °C. The lines represent the best linear fittings. (B) Hammett correlations for the reaction of 1 with para-X-substituted benzylamine (■) and phenethylamine (•) derivatives.
Analysis of the reaction products of PBA by GC-MS showed the formation of 1-chloro-4-phenylbutane (12% yield) and 4-phenyl-1-butene (36% yield, all yields are referred to 1), accounting for ~50% of the oxidizing equivalents used (Figure S2). The formation of ketone and aldehyde products is not detected even if the reaction is carried out in air. Together these observations suggest that a) there is an intramolecular reaction pathway of the dicobalt-PBA adduct that outcompetes intermolecular C–H bond cleavage of toluene, and b) this intramolecular reaction affords the deamination of the alkyl chain to form the chlorinated and desaturated products, both of which require the activation of the C–NH2 bond presumably in a heterolytic manner. The lack of ketone and aldehyde products in the presence of O2 is strong evidence against the formation of a carbon radical as the result of homolytic C–NH2 bond cleavage. We carried out careful control experiments to exclude any background oxidation of PBA by residual CAN in the solution (See SI for more information). We also measured the oxidation potentials of both PBA and CAN using cyclic voltammetry, showing that CAN is unable to oxidize PBA under our experimental conditions (Figure S3).
The source of chloride appears to be the starting Co(II) complex Co(II)(TPA)Cl2. Only the desaturated but not chlorinated product was observed if the perchlorate form Co(II)(TPA)(ClO4)2 is used as the starting complex for the same experiment (Figure S4). We then optimized experimental conditions for the formation of 1-chloro-4-phenylbutane using tetrabutylammonium chloride (TBACl) as the Cl− source, and found that its highest yield (24%) can be obtained with a ratio of 1 : PBA: Cl− = 1 : 1 : 30 at a Cl− concentration of ~0.1 M (Figure S5A, Tables S1 and S2). The addition of too much chloride above the optimized ratio inhibits the formation of both the alkyl chloride and alkene, presumably because the direct reaction of Cl− with the dicobalt-PBA adduct becomes significant at high Cl− concentrations. On the other hand, increasing the amount of PBA increases the formation of the alkene (up to 32% yield) but decreases the yield of the alkyl chloride (Figure S5B). We hypothesize that this is because the excess amount of PBA acts as a Lewis base to promote the deprotonation necessary to form the alkene (see more details in later discussions). On the other hand, when Cl− is replaced by Br− in the solution, we observed the formation of 1-bromo-4-phenylbutane in a yield (26%) similar to that of 1-chloro-4-phenylbutane (Figure S6). Furthermore, no halogenation product was identified if F− or I− is used as the nucleophile.
We expanded the substrate scope (Table 1) to investigate the effects of the steric and electronic properties of the alkylamines on their reactions with 1 under the optimized conditions for the alkyl chloride formation (30 eq. Cl-). For each amine substrate studied, we measured the rate constant and quantified the reaction product(s) (Table S1). For amines that afford low product yields (such as 5f, 9, 15 and 16), we typically recovered >50% of the starting substrates. Our results show that, for alkylamines (2–4a) that have a linear alkyl chain between a phenyl ring and the terminal amino group and are able to form both the alkyl chloride and alkene, the reaction appears to favor slightly the formation of alkyl chloride as the alkyl chain becomes shorter, while the typical combined product formation yields are 40–50% (Figures S7–S9). For benzylamine (5a), the simplest substrate in this category where it is impossible to form the alkene product, we observed the formation of benzyl chloride in 65% yield.
Table 1.
Substrate scope of C(sp3)–NH2 bond activation reactions by 1 in methanol at −60 °C with 30 eq. TBACl (optimized condition for alkyl chloride formation).a

|
All product yields are determined by GC-MS and normalized to the formation yield of complex 1. See SI for more details.
The overall C–NH2 bond cleavage is strongly affected by the electronic properties of the amine substrates. We employed benzylamine and its derivatives with a variety of para-substituents as a set of test substrates (5a–f), and found that the substrates with electron-donating groups (EDG) react faster with 1 and afford the corresponding alkyl chlorides in higher yields compared to those with electron-withdrawing groups (EWG) (Figures S10–S22). The Hammett plot (Figure 1B) clearly shows that the logarithm of the reaction rate constant kobs correlates linearly as a function of the Hammett parameter σpara of the aromatic substituents with a slope of ρ = −0.87. The negative ρ value is a clear indication that the reaction builds partial positive charges on the benzylic carbon center. Interestingly, the Hammett plot of phenethylamine and derivatives (4a–h), where a CH2 spacer is placed between the benzylic carbon site and the amino group, affords also a linear correlation with a much smaller slope of ρ = −0.34 (Figure 1B). This result indicates that the para-substituents on the phenyl ring have a reduced effect on the α-carbon center as the alkyl chain becomes longer. This group of substrates (4a–h) affords both the alkyl chloride and alkene in typical yields of 20–35% and 10–20%, respectively (Figures S23–S36). Notably, substrates derived from two bioactive alkaloids, including serotonin and dopamine derivatives having related arylethylamino moieties (5-methoxytryptamine, 6 and 3,4-dimethoxy-phenethylamine, 7) are converted to the corresponding alkyl chloride and alkene in moderate yields (Figures S37–S39). These results demonstrate that our reaction is well tolerated to a broad range of functional groups at a remote position, including aryl and alkyl halides, ether, ester, ketone, and unprotected indole ring.
For all the substrates studied, the formation of the alkyl chloride occurs only on the α-carbon site adjacent to the amino group. No other regio-isomers are observed. The chlorination appears to be highly sensitive to the crowdedness of the α-carbon center. For example, comparison of the results obtained from phenethylamine (4a) and α-methylbenzylamine (8) shows that the formation of (2-chloroethyl)benzene (34%) is in a higher yield than (1-chloroethyl)benzene (15%, Table 1, Figure S40), consistent with that the α-carbon site in phenethylamine is less sterically hindered than that in α-methylbenzylamine. Furthermore, increasing the steric hindrance of benzylamine by adding one and two phenyl rings onto the α-carbon center (benzhydrylamine, 9 and triphenylmethylamine, 10) results in decreased reaction rates (Table S1) and significantly reduced alkyl chloride formation yields for this series of substrates (Table 1, Figure S41). Specifically, the formation of the alkyl chloride is undetectable for triphenylmethylamine. Similar results (no alkyl chloride formation) are obtained also for sterically hindered substrates such as 1-adamantylamine (11), cumylamine (15) and 1,1-diphenylethan-1-amine (16), where the amino group is adjacent to a tertiary α-carbon center, indicating that these α-carbon sites are inaccessible by Cl-. Interestingly, moving the steric crowdedness slightly away from the α-carbon center of 1-adamantylamine by adding a CH2 spacer between the adamantyl and the amino groups (1-adamantanemethylamine, 12) restored the chlorination reactivity with the formation of 1-adamantanemethyl chloride in 40% yield (Figure S42). Taken together, these results clearly indicate that the chlorination occurs through an SN2 nucleophilic attack of the α-carbon center of the amine substrate by Cl− in the solution.
On the other hand, the formation of alkene appears to follow an E2 pathway, where the alkylamine also functions as a Lewis base to deprotonate the b-carbon (if applicable) and form a C=C bond in the same transition state as the C–NH2 bond cleavage. We employed phenethylamine deuterated at the b-carbon site to probe this mechanism. A competition experiment with 1:1 mixed non-deuterated and deuterated phenethylamine as the substrate afforded both non-deuterated and deuterated styrene products in a ratio of ~20:1, indicating a H/D kinetic isotope effect (KIE) of ~20. This large KIE strongly suggests that the cleavage of the b-C–H bond is a significant component of the rate-determining step. In addition, substrates with a tertiary α- or β-carbon site afforded alkenes with much reduced yields. For example, only trace amounts of alkene products are observed for β-methyl-phenethylamine (13) and 2,2-diphenylethylamine (14), likely due to the steric inaccessibility of their bprotons by the Lewis base in the course of the reactions. Furthermore, cumylamine (15) and 1,1-diphenylethylamine (16) (regio-isomers of 13 and 14, respectively) afforded the formation of alkenes in only ~10% yields (Figures S43 and S44), much lower than those substrates with α-CH–NH2 or α-CH2–NH2 moieties. The steric hindrance of the α-carbon site in these two alkylamines likely makes it difficult for them to access the substrate β-protons in an E2 reaction as a Lewis base. Notably, these sterically hindered alkylamines (10, 11, 15 and 16) all react with 1 in a rate that is typically 3–5 folds slower than those of other alkylamine substrates (Table S1), indicating that this subset of substrates have weaker binding abilities to the cobalt center due to the sterically hindered α-carbon center. This E2 process is also consistent with our observations that the alkene formation yield is dependent on the amine concentration in the reaction solutions (Figures S5 and S45), as well as substrates 2 and 3 afford the corresponding alkenes with the C=C bond at the terminal position of the alkyl chain without the formation of other regio-isomers, which otherwise is expected for an E1 process.
The lack of formation of ketone or aldehyde product in our reactions strongly indicates that the alkyl chain is not oxidized. Therefore, we hypothesize that the amino group is oxidized by the high-valent dicobalt species to generate a 2e− oxidized species, likely NH2OH. Indeed, product analysis of the post-reaction solution of 1 with a representative alkylamine substrate (such as benzylamine) by ESI-MS showed the formation of a mono-cationic species with m/z = 34, which is assignable to [NH3OH]+. This signal shifted by one mass unit to m/z = 35 if 15N enriched substrates are employed (Figure S46). Quantification of the NH2OH formation yield is carried out using 15N NMR spectroscopy. As shown in Figure S47, the 15N NMR spectrum of the reaction solution of 1 with 15N-labeled benzylamine (chemical shift ~29 ppm, 15N-ammonia as the reference) affords a signal at ~107 ppm. Addition of a known amount of the authentic 15NH2OH sample into the reaction solution causes the increase of the 107 ppm signal, indicating that this signal is originated from 15NH2OH formed from the reaction of 1 with benzylamine (see SI for more details). Integrations of the 107 ppm signal further showed that NH2OH is formed in a yield of ~60%, consistent with the formation yield of 65% for benzyl chloride obtained by GC-MS. Moreover, in order to further verify that complex 1 is able to oxidize the nitrogen atom of an amino group without an alkyl chain, we employed 15N-ammonia as the substrate to react with 1. As shown in Figure S48, the 15N NMR spectrum of the reaction solution clearly shows the formation of 15NH2OH as the only product in ~60% yield. Therefore, our reaction mimics the one catalyzed by the enzyme AMO.
We carried out electron paramagnetic resonance (EPR) studies to further examine the fate of the dicobalt complex in the aforementioned reactions. The starting CoIII2(μ-O)2 complex 1a is diamagnetic and EPR silent (Figure 2A). The oxidation of 1a by CAN afforded the mixed-valent CoIII,IV2(μ-O)2 complex 1 as an S = 1/2 species with an EPR signal centered at g ~ 2 (Figure 2B). Spin quantification of this signal suggests that the formation yield of complex 1 is ~60–70%. These results are consistent with the ones that we reported previously for 1.29 The addition of NH3 to the solution of 1 resulted in complete decay of the S = 1/2 species and the concomitant formation of a new, broad EPR signal with observed g resonances at 6.85, 4.80, 3.82, and 2.73 (Figure 2C), which is assignable to an S = 3/2 Co(II) species (1b). The observed EPR signal is originated from the MS = ± 1/2 Kramers doublet of an S = 3/2 spin state. The temperature dependent EPR measurements suggested that this MS = ± 1/2 Kramers doublet should be the ground doublet (Figure S49), thus the axial zero field splitting (D) of 1b should be positive. Simulation and quantification of this signal (See Figure 2 and the figure caption) indicated that the formation yield of 1b is 80±20% (average of six samples, see Table S3 for more details) from complex 1, suggesting that 1 is a 2-e− oxidant in the course of reaction with NH3. Given the basic reaction environment with added amine/ammonia, we propose that 1b is mixed-valent dicobalt(II,III) species.
Figure 2.

X-band EPR spectra of 1a, 1 and 1b. (A) 1a; (B) a sample containing 1 obtained by treating 1a with CAN; (C) a sample containing 1b by treating 1 mM 1a with 3 mM CAN then treating it with 0.2 M NH3. The black line shows the corresponding simulation for 1b. The simulation parameters are: g = [2.50, 2.38, 2.87], A(59Co) = [100, 240, 390] MHz, D = +50 cm−1, E/D = 0.15, σ(E/D) = 0.04. The data collection conditions are: microwave frequency 9.63 GHz, microwave power 2 mW, modulation frequency 100 KHz, modulation amplitude 0.5 mT, temperature 17 K. See the SI for discussion on the simulation.
Mechanistic considerations.
We propose a mechanistic sketch (Scheme 2) that highlights key fundamental steps necessary for this novel transformation. Detailed computational investigation of the reaction mechanism is underway and will be the subject of another publication. Coordination of the alkylamine (Scheme 2, step A, using PBA as a representative example) first opens up the diamond core of 1 to generate an open core species 1c. As we have demonstrated in a previous publication,28 the binding of a Lewis base (pKa in a range of 4–16) to 1 is weak (Keq = 0.31 M−1) and the equilibrium strongly disfavors the formation of the open core. Therefore, 1c should be a minor species in the reaction solution. Given that the amino group (−NH2) is also a hydrogen bond donor similar to hydroxide, we hypothesize that 1c is a cis-open core adduct (instead of trans-open core) with a hydrogen bound N–H•••O–Co(IV) moiety. We turned to DFT calculations to obtain a geometry-optimized structure of this key intermediate in order to better understand it. As shown in Figure 3A, ethylamine-coordinated dicobalt complex cis-EtNH2CoIII–O–CoIV–O has a long Co•••Co distance of 3.5 Å characteristic of an open-core configuration. Interestingly, the NH•••O interatomic distance between the N–H bond and O–Co(IV) moiety of this complex is as short as 1.45 Å, which, together with a linear arrangement of the N-H•••O atoms, suggests a strong intramolecular hydrogen bonding. The unpaired electron in cis-EtNH2-CoIII–O–CoIV–O is primarily localized on the terminal oxygen atom (+0.38 a.u.) and Co(IV) (+0.48 a.u.), exhibiting a significant radical character on the terminal oxygen atom. By contrast, tertiary alkylamines such as triethylamine are unable to form the hydrogen bound cis-open core configuration but rather generate a trans-open core upon coordinating to the cobalt center. This structural difference appears to have a significant impact on the reaction outcomes---intramolecular C–N bond cleavage with PBA vs. intermolecular C–H bond activation in the presence of triethylamine (Figure 1A).
Scheme 2.

Schematic illustration for the reaction pathway of 1 with PBA.
Figure 3.

Geometry optimized structures of (A) 1c and (B) 1d obtained at the BP86/6–31G(D)+PCM(methanol) level of theory. Cobalt: pink, oxygen: red, nitrogen: blue, carbon: gray, hydrogen: white.
The cis-open core species 1c then likely undergoes a non-rate-determining step (Scheme 2, step B) to oxidize the amino nitrogen forming an N-O bond and consume the Co(IV)–O oxidant. This hypothesis is consistent with our observations that 1) external substrates such as toluene are unable to intercept the Co(IV)–O oxidant, and 2) weak C-H bonds in the alkylamine substrates such as the benzylic C-H bonds in benzylamine are not oxidized. We propose that this transformation affords a two-electron reduced dicobalt(II,III) species 1d. Geometry-optimized structure of 1d by DFT (Figure 3B) shows that it is bridged by a hydroxyl ligand and a NH-O moiety, forming a five-member ring with a N-O bond length of 1.436 Å. This structure is in accordance with a number of Co(μ-OH)(μ-OO)Co complexes reported in literature.30–32 Our calculations show that 1d is ~2.4 kcal/mol more stable than 1c, suggesting that the conversion of 1c to 1d is thermodynamically favorable. The formation of an N-O bond can be accomplished by the Co(IV)–O moiety in 1c via a direct oxygen atom transfer (OAT) or following classical hydrogen atom transfer (HAT) and radical rebound processes. We favor the OAT pathway because the N-H bonds of primary alkylamines are typically strong (>100 kcal/mol) and the upper limit of the C-H bond that the Co(IV)–O oxidant can cleave is only 96 kcal/mol.28 Although recent work from other groups has shown that the coordination of NH3 to a transition metal center induces weakening of the N-H bond, the degree of perturbation is expected to be small for cobalt.33,34
The rate-determining step (Scheme 2, step C) appears to be the C-N bond cleavage, as clearly demonstrated by the Hammett relationship. This is a non-redox step that affords the formation of organic product(s) via SN2 or E2 pathway discussed above. We further employed CH3NH2 and CD3ND2 as probe substrates and measured their rate constants with 1. As shown in Figure S50, these measurements afforded a H/D kinetic isotope effect (KIE) of 1.37. This value is much smaller than those observed in our previous work for CH/C-D bond activation by the open core complex,28 suggesting that it is not a primary KIE. Instead, we assign it to a asecondary KIE consistent with the change of the α-carbon hybridization from sp3 to sp2 in the SN2 transition state.35
Furthermore, the C-N bond cleavage (step C) cannot precede the N-O bond formation (step B). Our control experiments show that no benzyl chloride is formed if benzylamine is added to a mononuclear Co(III)-TPA complex under otherwise identical conditions, indicating that the binding of the alkylamine to the cobalt center alone is insufficient to trigger C-N bond cleavage. In addition, no reaction was observed when acetamide (CH3C(O)NH2) and benzamide (PhC(O)NH2) are employed as substrates, likely because the formation of N-O bond for these two substrates is difficult due to the electron-withdrawing carbonyl group adjacent to NH2, even though their α-carbon is more acidic and susceptible for nucleophilic attack.
The diamond core complex 1 thus represents the first example of a high-valent metal-oxo complex that mediates direct activation of the C(sp3)–NH2 bond of aliphatic primary alkylamines. This reaction takes advantages of the unique features of the dicobalt-alkylamine adduct, where the alkylamine is both a strong Lewis base to open the CoIII,IV2(μ-O)2 diamond core and a hydrogen bond donor to “lock” the cisopen core configuration between the CoIII–NH2 and CoIV–O moieties. Both the open core structure and the cis-configuration are critical to ensure a successful intramolecular C(sp3)–NH2 bond cleavage and N–O bond formation. By contrast, the diamond core itself is unable to activate the C(sp3)–NH2 bond. For example, a closely related CuIII2(μ-O)2 species reported recently oxidizes primary alkylamines to nitriles, a desaturation reactivity instead of heterolytic cleavage of the CH2–NH2 bond.36 The formation of the C–Cl (or C–Br) bond, on the other hand, is the result of Cl− (or Br) being both a weak Lewis base incapable to compete with the alkylamine for binding the cobalt center and a strong nucleophile in methanol for attacking the α-carbon site. The employment of such a protic solvent further decreases the nucleophilicity (not basicity) of the primary alkylamine substrates so that no formation of secondary alkylamine is observed as the reaction product under optimized chlorination conditions.
Conclusion.
In conclusion, we have discovered a novel organic transformation that converts primary aliphatic alkylamines to functionalized product(s) via direct activation of the inert C(sp3)–NH2 bond by a high-valent CoIII,IV2(μ-O)2 complex. The merits of our work are multi-folds. First, the direct method for converting aliphatic primary alkylamines to other functionalities such as alkyl halides and alkenes without any pre-activation of the C(sp3)–NH2 bond is unprecedented. Furthermore, our mechanistic studies have shown that the formation of C–Cl and C=C bonds proceeds through two separate pathways (SN2 and E2, respectively), each of which can be modulated by the structural and electronic properties of the alkylamine substrates, as well as the amount of Cl-/amine added into the reaction solution. These conditions can thus be employed to predict the reaction outcomes and enable more complex experimental designs. At the current stage, this prototype reaction is stoichiometric and fundamental in nature. However, we envision that, in the future development of this work, nucleophiles other than Cl−/Br− can be employed to design other C–X (X = N, O, C) bond coupling reactions and catalytic systems can be further developed using primary alkylamines as the substrates to enable novel synthetic utilities.
On the other hand, the formation of NH2OH as the oxidation product of alkylamines and ammonia is a synthetic mimic of the enzymatic 2-e− oxidation of ammonia by AMO. Our work thus suggests that a high-valent copper-oxo intermediate is likely generated in the AMO catalytic cycle as the key oxidant. While a related enzymatic intermediate has yet to be characterized, our work has demonstrated the powerful abilities of synthetic model complexes to provide insights about enzymatic reactions.
Supplementary Material
ACKNOWLEDGMENT
Support of this work for Y.L. and D.W. was provided by the University of Montana, Montana INBRE (IDeA Networks of Biomedical Research Excellence, grant NIGMS P20GM103474) and National Science Foundation (grant CHE-2102339). S.H. and M.R.T. were supported by an Institutional Development Award (IDeA) from the National Institute of General Medical Sciences (grant P20GM103451). The DFT calculations were performed using computational resources awarded by the Extreme Science and Engineering Discovery Environment (XSEDE) TGCHE170004. J.X. and Y.G. thank the support from NIGMS grants P01GM125924 and P01GM127588.
Footnotes
Supporting Information. Experimental methods, Figures S1–S50, Tables S1–S4. The Supporting Information is available free of charge on the ACS Publications website.
REFERENCES
- (1).Froidevaux V; Negrell C; Caillol S; Pascault J-P; Boutevin B Biobased Amines: From Synthesis to Polymers; Present and Future. Chem. Rev 2016, 116, 14181–14224. [DOI] [PubMed] [Google Scholar]
- (2).Cai W; Qiao X; Zhang H; Li B; Guo J; Zhang L; Chen W-W; Zhao B Asymmetric Biomimetic Transamination of α-keto Amides to Peptides. Nat. Commun 2021, 12, 5174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (3).Mayer RJ; Kaur H; Rauscher SA; Moran J Mechanistic Insight into Metal Ion-Catalyzed Transamination. J. Am. Chem. Soc 2021, 143, 19099–19111. [DOI] [PubMed] [Google Scholar]
- (4).Wu B; Szymanski W; Heberling MM; Feringa BL; Janssen DB Aminomutases: Mechanistic Diversity, Biotechnological Applications and Future Perspectives. Trends Biotechnol. 2011, 29, 352–362. [DOI] [PubMed] [Google Scholar]
- (5).Klema VJ; Wilmot CM The Role of Protein Crystallography in Defining the Mechanisms of Biogenesis and Catalysis in Copper Amine Oxidase. Int. J. Mol. Sci 2012, 13, 5375–5405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (6).Cona A; Rea G; Angelini R; Federico R; Tavladoraki P Functions of Amine Oxidases in Plant Development and Defence. Trends Plant Sci. 2006, 11, 80–88. [DOI] [PubMed] [Google Scholar]
- (7).Norton JM; Alzerreca JJ; Suwa Y; Klotz MG Diversity of Ammonia Monooxygenase Operon in Autotrophic Ammonia-oxidizing Bacteria. Arch. Microbiol 2002, 177, 139–149. [DOI] [PubMed] [Google Scholar]
- (8).Heil J; Vereecken H; Bruggemann N A Review of Chemical Reactions of Nitrification Intermediates and Their Role in Nitrogen Cycling and Nitrogen Trace Gas Formation in Soil. Eur. J. Soil Sci 2016, 67, 23–39. [Google Scholar]
- (9).Fisher OS; Kenney GE; Ross MO; Ro SY; Lemma BE; Batelu S; Thomas PM; Sosnowski VC; DeHart CJ; Kelleher NL; Stemmler TL; Hoffman BM; Rosenzweig AC Characterization of a Long Overlooked Copper Protein from Methane- and Ammonia-oxidizing Bacteria. Nat. Commun 2018, 9, 4276. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (10).Musiani F; Broll V; Evangelisti E; Ciurli S The Model Structure of the Copper-dependent Ammonia Monooxygenase. J. Biol. Inorg. Chem 2020, 25, 995–1007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (11).Ross MO; MacMillan F; Wang J; Nisthal A; Lawton TJ; Olafson BD; Mayo SL; Rosenzweig AC; Hoffman BM Particulate Methane Monooxygenase Contains only Mononuclear Copper Centers. Science 2019, 364, 566–570. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (12).Park Y; Kim Y; Chang S Transition Metal-Catalyzed C–H Amination: Scope, Mechanism, and Applications. Chem. Rev 2017, 117, 9247–9301. [DOI] [PubMed] [Google Scholar]
- (13).Clark JR; Feng K; Sookezian A; White MC Manganesecatalysed Benzylic C(sp3)–H Amination for Late-stage Functionalization. Nat. Chem 2018, 10, 583–591. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (14).Ruiz-Castillo P; Buchwald SL Applications of PalladiumCatalyzed C–N Cross-Coupling Reactions. Chem. Rev 2016, 116, 1256412649. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (15).Ouyang K; Hao W; Zhang W-X; Xi Z Transition-Metal-Catalyzed Cleavage of C–N Single Bonds. Chem. Rev 2015, 115, 12045–12090. [DOI] [PubMed] [Google Scholar]
- (16).Wang Q; Su Y; Li L; Huang H Transition-metal Catalysed C–N Bond Activation. Chem. Soc. Rev 2016, 45, 1257–1272. [DOI] [PubMed] [Google Scholar]
- (17).Katritzky AR; Marson CM Pyrylium Mediated Transformations of Primary Amino Groups into Other Functional Groups. New Synthetic Methods (41). Angew. Chem. Int. Ed 1984, 23, 420–429. [Google Scholar]
- (18).Ni S; Li C-X; Mao Y; Han J; Wang Y; Yan H; Pan Y Ni-catalyzed deaminative cross-electrophile coupling of Katritzky salts with halides via C─N bond activation. Sci. Adv 2019, 5, eaaw9516. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (19).Zeng X; Yan W; Zacate SB; Cai A; Wang Y; Yang D; Yang K; Liu W Copper-Catalyzed Deaminative Difluoromethylation. Angew. Chem. Int. Ed 2020, 59, 16398–16403. [DOI] [PubMed] [Google Scholar]
- (20).Baker KM; Baca DL; Plunkett S; Daneker ME; Watson MP Engaging Alkenes and Alkynes in Deaminative Alkyl–Alkyl and Alkyl–Vinyl Cross-Couplings of Alkylpyridinium Salts. Org. Lett 2019, 21, 9738–9741. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (21).Scharfbier J; Gross BM; Oestreich M Stereospecific and Chemoselective Copper-Catalyzed Deaminative Silylation of Benzylic Ammonium Triflates. Angew. Chem. Int. Ed 2020, 59, 1577–1580. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (22).Zhang X; Qi D; Jiao C; Liu X; Zhang G Nickel-catalyzed Deaminative Sonogashira Coupling of Alkylpyridinium Salts Enabled by NN2 Pincer Ligand. Nat. Commun 2021, 12, 4904. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (23).Plunkett S; Basch CH; Santana SO; Watson MP Harnessing Alkylpyridinium Salts as Electrophiles in Deaminative Alkyl–Alkyl Cross-Couplings. J. Am. Chem. Soc 2019, 141, 2257–2262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (24).Ashley MA; Rovis T Photoredox-Catalyzed Deaminative Alkylation via C–N Bond Activation of Primary Amines. J. Am. Chem. Soc 2020, 142, 18310–18316. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (25).Dorsheimer JR; Ashley MA; Rovis T Dual Nickel/PhotoredoxCatalyzed Deaminative Cross-Coupling of Sterically Hindered Primary Amines. J. Am. Chem. Soc 2021, 143, 19294–19299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (26).Zhou M-G; Zhang W-Z; Tian S-K Direct Enantiospecific Substitution of Primary a-aminoalkylferrocenes via Lewis Acidcatalyzed C–N Bond Cleavage. Chem. Commun 2014, 50, 14531–14534. [DOI] [PubMed] [Google Scholar]
- (27).Kalutharage N; Yi CS Deaminative and Decarboxylative Catalytic Alkylation of Amino Acids with Ketones. Angew. Chem. Int. Ed 2013, 52, 13651–13655. [DOI] [PubMed] [Google Scholar]
- (28).Li Y; Handunneththige S; Xiong J; Guo Y; Talipov MR; Wang D Opening the CoIII,IV2(μ-O)2 Diamond Core by Lewis Bases Leads to Enhanced C-H Bond Cleaving Reactivity. J. Am. Chem. Soc 2020, 142, 21670–21678. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (29).Li Y; Handunneththige S; Farquhar ER; Guo Y; Talipov MR; Li F; Wang D Highly Reactive CoIII,IV2(m-O)2 Diamond Core Complex that Cleaves C-H Bonds. J. Am. Chem. Soc 2019, 141, 20127–20136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (30).Cho YI; Joseph DM; Rose MJ “Criss-Crossed” Dinucleating Behavior of an N4 Schiff Base Ligand: Formation of a μ-OH,μ-O2 Dicobalt(III) Core via O2 Activation. Inorg. Chem 2013, 52, 1329813300. [DOI] [PubMed] [Google Scholar]
- (31).Kotani H; Hong D; Satonaka K; Ishizuka T; Kojima T Mechanistic Insight into Dioxygen Evolution from Diastereomeric mPeroxo Dinuclear Co(III) Complexes Based on Stoichiometric ElectronTransfer Oxidation. Inorg. Chem 2019, 58, 3676–3682. [DOI] [PubMed] [Google Scholar]
- (32).Wang H-Y; Mijangos E; Ott S; Thapper A Water Oxidation Catalyzed by a Dinuclear Cobalt–Polypyridine Complex. Angew. Chem. Int. Ed 2014, 53, 14499–14502. [DOI] [PubMed] [Google Scholar]
- (33).Bezdek MJ; Guo S; Chirik PJ Coordination-induced Weakening of Ammonia, Water, and Hydrazine X–H Bonds in a Molybdenum Complex. Science 2016, 354, 730–733. [DOI] [PubMed] [Google Scholar]
- (34).Bhattacharya P; Heiden ZM; Wiedner ES; Raugei S; Piro NA; Kassel WS; Bullock RM; Mock MT Ammonia Oxidation by Abstraction of Three Hydrogen Atoms from a Mo−NH3 Complex. J. Am. Chem. Soc 2017, 139, 2916–2919. [DOI] [PubMed] [Google Scholar]
- (35).Anslyn EV; Dougherty DA Modern Physical Organic Chemistry; University Science, 2005. [Google Scholar]
- (36).Large TAG; Mahadevan V; Keown W; Stack TDP Selective Oxidation of Exogenous Substrates by a bis-Cu(III) bis-oxide Complex: Mechanism and Scope. Inorg. Chim. Acta 2019, 486, 782–792. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.