

Abstract
Background:
Elevated rates of mental illness have been reported in clinical studies of sex chromosome aneuploidies (SCAs) but accurate population-based estimates of SCA prevalence, clinical detection rate, and associated risk of psychiatric disorders are lacking.
Methods:
In this study we provide population-valid prevalence estimates of the most common SCA karyotypes (45,X; 47,XXX; 47,XXY; and 47,XYY) and gonadal sex-matched hazard ratios (HR) for schizophrenia spectrum disorder (SSD), bipolar disorder (BPD), major depressive disorder (MDD), autism spectrum disorder (ASD), and attention-deficit hyperactivity disorder (ADHD). Our study leverages the iPSYCH2015 case-cohort dataset including all persons born in Denmark between 1981–2008 with a hospital discharge diagnosis by the end of 2015 of any of the above listed disorders and a random subcohort from the same study population. The assessed sample included 64,533 (54%) gonadal men and 54,948 (46%) gonadal women, and their age during follow-up ranged from 0–34·7 years (mean: 10·9 years). Information on ethnicity is not available.
Findings:
Overall SCA prevalence was 1·5:1000, marginally lower than reported in a previous population-based study (2·1:1000, trend across all four SCA; P=0·046). All four SCAs were associated with increased risk of any psychiatric disorder to a similar level as estimated for several well-known pathogenic copy number variants in a similar population-based setting (HR = 2·2–4·3). Increased risks of ADHD (HR = 2·0–6·2), ASD (HR = 2·7–8·5), and SSD (HR = 1·8–4·6) were also associated with all four SCAs, while increased risks of MDD and BPD were associated with 47,XXY and 47,XYY (HR = 1·9 and 2·7), and 47,XXX (HR = 4·3), respectively. Clinical SCA diagnosis rate was high (93%) for 45,X but much lower (15–22%) for 47,XXX; 47,XXY; and 47,XYY; and did not differ with respect to diagnosis of a psychiatric disorder.
Interpretation:
Increased SCA-associated risk of psychiatric disorders, combined with low clinical SCA diagnosis rates, compromises adequate provision of necessary healthcare and counselling to affected individuals and their families, which might be helped by increased application of genetic testing in clinical settings.
Funding:
The study was funded by the Lundbeck Foundation and the National Institute of Health.
Introduction
Sex chromosome aneuploidies (SCAs) are genetic conditions involving an atypical number of sex chromosomes relative to the typical 46,XY and 46,XX karyotypes. These conditions are usually caused by chromosomal nondisjunction occurring at meiosis or early postzygotic development stages1. The most common SCAs are 45,X; and the three trisomies 47,XXX; 47,XXY; and 47,XYY; with estimated frequencies between 0·5 and 1·3 in 1,000 live births2 - although a high proportion of SCA carriers go undiagnosed1,3. There are no pathognomic features of SCA, and great variability in penetrance and expressivity, but some of the most studied manifestations of SCA include alterations of: growth (short stature in 45,X, tall stature in sex chromosome trisomies)4; gonadal function (ovarian dysgenesis in 45,X5, premature ovarian failure in 47,XXX6 and hypogonadism in 47,XXY7); and neurodevelopment1.
Recent years have seen a growing awareness of association between SCAs and neurodevelopmental and psychiatric impairments8. Still, neuropsychiatric characterization of SCAs has lagged behind that of recurrent autosomal copy number variants (CNVs), despite SCAs involving dosage change of more genes and being more prevalent2 than most recurrent CNVs9,10. The best available research to date has reported high rates of several psychiatric disorders in studies of clinically diagnosed SCA carriers including: psychotic disorders (6–12%), autism (15–30%), and ADHD (30–70%) for the SCA trisomies11, and intellectual disability (ID; 5–10%), ADHD (25%), and anxiety/depression (50%) for 45,X12. Also, a very recent study found clinically significant ADHD symptoms in 24% of 104 children aged 1–6 years with SCA trisomy13.
The few population-based studies that have been published in this field are mostly consistent with the clinical studies, with three of them reporting 2–6 fold risk of psychiatric disorders associated with 45,X14 and 47,XXY15,16, while a fourth such study, of all four SCA karyotypes, only found limited evidence of increased risk of schizophrenia and BPD associated with 47,XYY17.
Despite the high collective prevalence of SCAs and evidence for their association with psychiatric impairments, two obstacles have hindered definitive understanding of the population-based association between mental health outcomes and SCAs – posing a major challenge to adequate healthcare planning and provision for these conditions. First, the most recent prevalence estimates for SCAs from attempted full detection (in a sequential birth series)2, are more than 30 years old. However, several factors that could potentially influence the prevalence of SCAs have changed over this time, including average maternal age and elective abortion in conjunction with prenatal screening18. As such, there is a pressing need for updated population-based prevalence estimates for SCAs to properly assess the scale of clinical need presented by these conditions. Second, to date, there have been no population-based estimates of the penetrance of SCAs for psychiatric outcomes from genetic studies. Securing such estimates is an especially high priority in SCAs, as the rate of psychiatric disorders may differ between the minority of SCAs carriers who are clinically detected, and the majority who remain undetected1,3. Our prior work in recurrent CNVs using the Danish iPSYCH2012 case-cohort study design19 has shown that unbiased population-based samples obtained through nationwide health registers and biobanks are critical for accurate estimation of psychiatric penetrance in under-diagnosed genetic conditions, and can substantially revise the penetrance estimates given by study designs involving traditional case-control samples9 or registry-based studies of clinically diagnosed carriers10.
Here, we address these obstacles by leveraging the Danish iPSYCH2015 case-cohort20 to study the four most prevalent SCAs. We determine and directly contrast prevalence and risk of psychiatric disorders among individuals affected by 45,X; 47,XXX; 47,XXY; or 47,XYY with the corresponding age- and gonadal-sex-matched part of the Danish population. We also harness this study design to provide population-based estimates of the relative reproductive rate in SCA and screen for associations between SCA and several other diagnoses not directly targeted by the iPSYCH2015 case-cohort design. This study follows the STROBE (strengthening the reporting of observational studies in epidemiology)21 guidelines on case-cohort studies22.
Methods
Study design
This study was carried out on the iPSYCH2015 case-cohort20, which is an update and expansion of the iPSYCH2012 case-cohort19. The study includes 141,265 individuals selected from all 1,657,449 singleton births in Denmark between May 1st, 1981, and December 31st, 2008, who were alive and residing in Denmark on their first birthday and have a registered mother in the Danish Civil Registration System (CRS)23. The study has a dual design: (1) Cases: All individuals (n=93,608) with one or more of the following psychiatric disorders diagnosed and registered at the Danish Psychiatric Central Research Register (PCRR)24 no later than December 31st, 2015; schizophrenia spectrum disorder (SSD; ICD10 F20–F29, n= 16,008), bipolar disorder (BPD; ICD10 F30–F31, n=3,819), major depressive disorder (MDD; ICD10 F32–F33, and ICD-8 296.09, 296.29, 298.09, 300.49, n=37,555), autism spectrum disorder (ASD; ICD10 F84, n=24,975), or attention-deficit hyperactivity disorder (ADHD; ICD10 F90, n=29,668). (2) Cohort: 50,615 individuals drawn from the source population at random (corresponding to roughly 3%). The index psychiatric disorders were initially selected to represent common complex mental illness with childhood or early adult onset and a common genetic component. An overview of the iPSYCH2015 study design and its application in this study is given in eFigure 1.
As many individuals are diagnosed with more than one of the index disorders the number of samples across diagnosis groups adds up to more than the case total of 93,608. Also, as the randomly drawn population sample naturally includes individuals with diagnosis of mental illness, the numbers of case and subcohort samples add up to more than the total of 141,265 (eFigure 1).
Genotyping & SCA detection
Genotyping was done with Illumina genotyping arrays (Illumina,San Diego, CA, USA) using whole-genome amplified DNA from dried neonatal blood spots extracted from the Danish Neonatal Screening Biobank (DNSB)25. The genotyping of iPSYCH2012 samples is described in detail elsewhere19. Sampling and genotyping of additional samples (iPSYCH2015i, which when combined with iPSYCH2012 constitute the complete iPSYCH2015 case-cohort) differed in several ways20; most importantly, iPSYCH2015i samples were genotyped using the Global Screening Array v2, whereas iPSYCH2012 samples had been genotyped using the PsychArray V1.019. Single nucleotide polymorphism (SNP) genotype calling and quality control were performed using Illumina’s GenTrain software tool for all samples that could be successfully identified and extracted from DNSB. The extraction of B-allele frequency (BAF) as well as probe intensities (in the form of log-R-ratio; LRR) was done with Illumina GenomeStudio. Samples with a genotyping call rate below 95% were excluded from further study19. Access to the data and its use for research purposes was granted by The Danish Scientific Ethics Committee, the Danish Health Data Authority, the Danish data protection agency, and the DNSB Steering Committee.
Carriers of 45,X; 47,XXX; 47,XXY; and 47,XYY were identified by analysing LRR and BAF using a two-stepped approach. First, we identified putative carriers by clustering samples based on mean LRR values for (a) X and Y chromosomes (for samples registered as male), and (b) X and all autosomal chromosomes combined (for samples registered as female). Then, we visually inspected LRR and BAF profiles for X and Y chromosomes for all the putative SCA carriers to determine their sex chromosome carriage status (see the Appendix for more details).
Other available diagnoses and outcomes
We assessed SCA-associated risk for a few other disorders using diagnosis information obtained through PCRR24 and The Danish National Patient Register (DNPR)26 for other iPSYCH2015 studies, see further details on those disorders and the analysis in the Appendix.
Diagnoses for SCAs from DNPR26 were available for a subset of the iPSYCH2015 sample (73,646 of 119,481), and all comparisons between SCA individuals identified from genotype in our study and clinically detected SCA individuals were done in this subset. We considered as clinically detected all individuals with an ICD10 diagnosis of 45,X (Q96.0); 47,XXX (Q97.0); 47,XXY (Q98.0); 47,XYY (Q98.5); or an unspecified Turner syndrome (TS; Q96.9) or Klinefelter syndrome (KS; Q98.4) diagnosis, and a few 47,XXX and 47,XYY individuals with a diagnosis of sex chromosome anomaly not further specified (ICD8: 759.5, or ICD10: Q98.7/Q98.8). We did not have access to records pertaining to pre-natal detection among clinically detected SCA individuals.
Information on the number of offspring of individuals of the study sample by the end of 2015 (including any adopted children) were obtained from the CRS23.
Statistical analysis
Risk of each of the five iPSYCH2015 targeted psychiatric disorders (SSD, BPD, MDD, ASD and ADHD) associated with SCAs was assessed by weighted Cox Proportional Hazard (CPH) models with age at first hospitalisation as outcome. For each CPH model all cases with the index disorder and all individuals from the subcohort were included, without considering case status of other index disorders (eFigure 1). Individuals were censored at the appropriate date if they had; (a) no relevant hospitalisation before 31st December 2015, (b) died, (c) been lost to follow-up or emigrated, whichever occurred first. The SCA status was included as an independent variable. The CPH models were fitted separately for each SCA and in a gonadal sex-specific way reflecting the SCA in question (i.e., 45,X and 47,XXX were tested with 46,XX, and 47,XXY and 47,XYY with 46,XY). Population-unbiased risk estimates were obtained by computing inverse probability of sampling (IPS) weights following Barlow’s procedure27. Regression coefficient’s standard error and 95% confidence intervals (CI95%) were computed using a robust estimator10.
To study the relative reproductive rate, we fitted a Poisson regression type generalised additive model for each SCA karyotype compared to the matching gonadal sex (as explained above) with number of offspring at the end of follow-up as outcome, excluding individuals who were younger than 16 years old at the end of follow-up. SCA status was coded as the independent variable, we adjusted for age by including a smoothed function of age at the end of follow-up as a covariate, allowing for a non-linear fit. Also, all the five iPSYCH2015-targeted psychiatric disorders were included in the model as separate covariates. All statistical analyses were done in R (Version 3.3.1). The survival models were fitted using the survival R package.
To compare SCA population prevalence with that of the largest previous study2, we considered live-born individuals with the corresponding karyotype (including mosaicism) in the previous study2 with identified SCA carriers in our study and tested the difference for each SCA and the trend across all SCAs with a two-sided Fishe’s exact test and Mantel-Haenzel test, respectively.
Role of the funding source
The sponsors of the study had no role in the study design; the collection, analysis, and interpretation of data; the writing of the report; nor in the decision to publish the results.
Results
After quality control 119,481 genotyped samples of the iPSYCH2015 case-cohort remained (eFigure 1 & eTable 1), including 78,726 case individuals diagnosed with one or more of the ascertained psychiatric disorders (SSD, BPD, MDD, ASD or ADHD), and 43,326 cohort individuals (3%) randomly drawn from the source population. The assessed sample included 64,533 (54%) gonadal men and 54,948 (46%) gonadal women, and their age during follow-up ranged from 0–34·7 years (mean: 10·9 years). We identified 387 carriers across the four SCA karyotypes under study (45,X; 47,XXX; 47,XYY; and 47,XYY) among the 119,481 samples.
Hospital records regarding chromosomal conditions, including any clinical SCA diagnoses, were available for 73,646 samples, and out of the 240 SCA carriers identified by our study in this subset, 55 (23%) also had a clinical SCA diagnosis (see Methods). The clinical diagnosis rate was high for 45,X (93%), but low for 47,XXX (15%), 47,XXY (22%), and 47,XYY (15%). Mean age (in years) at clinical SCA diagnosis was as follows: 45,X (10·6), 47,XXX (10·1), 47,XXY (14·9), and 47,XYY (12·1). Also, a minority (24%) of clinically diagnosed SCA individuals, mostly involving TS and KS, were not indicated as carriers with our SNP array-based approach, with evidence that those not identified were likely enriched for non-canonical dosage alterations (see Appendix).
Population-valid risk associated with each of the five iPSYCH2015 index psychiatric disorders was estimated by contrasting individuals in the respective case group with individuals in the cohort (restricting to gonadal sex corresponding to the SCA), using age at first admission with the disorder as outcome and appropriate IPS weights (see Methods and eFigure 1). The risk associated with diagnosis of one (or more) of the five psychiatric disorders was significantly increased for all four SCAs, with HR range 2·1–4·3, highest for 47,XYY (Figure 1 upper left panel & eTable 2). The combined rate of the five psychiatric disorders did not differ significantly between SCA carriers with and without a hospital SCA diagnosis (P = 0·65). All SCAs were associated with increased risk of the two primarily childhood-onset disorders, ASD (HR = 2·7–8·5) and ADHD (HR = 2·0–6·2), lowest for 47,XXY and highest for 45,X (Figure 1 & eTable 2). All four SCAs also associated with increased risk of SCZ (HR = 1·8–4·6), 47,XXY and 47,XYY with that of MDD (HR = 1·9 and 2·6), and 47,XXX with that of BPD (HR = 4·3) (Figure 1 & eTable 2).
Figure 1. Risk of psychiatric disorders associated with SCA karyotypes.
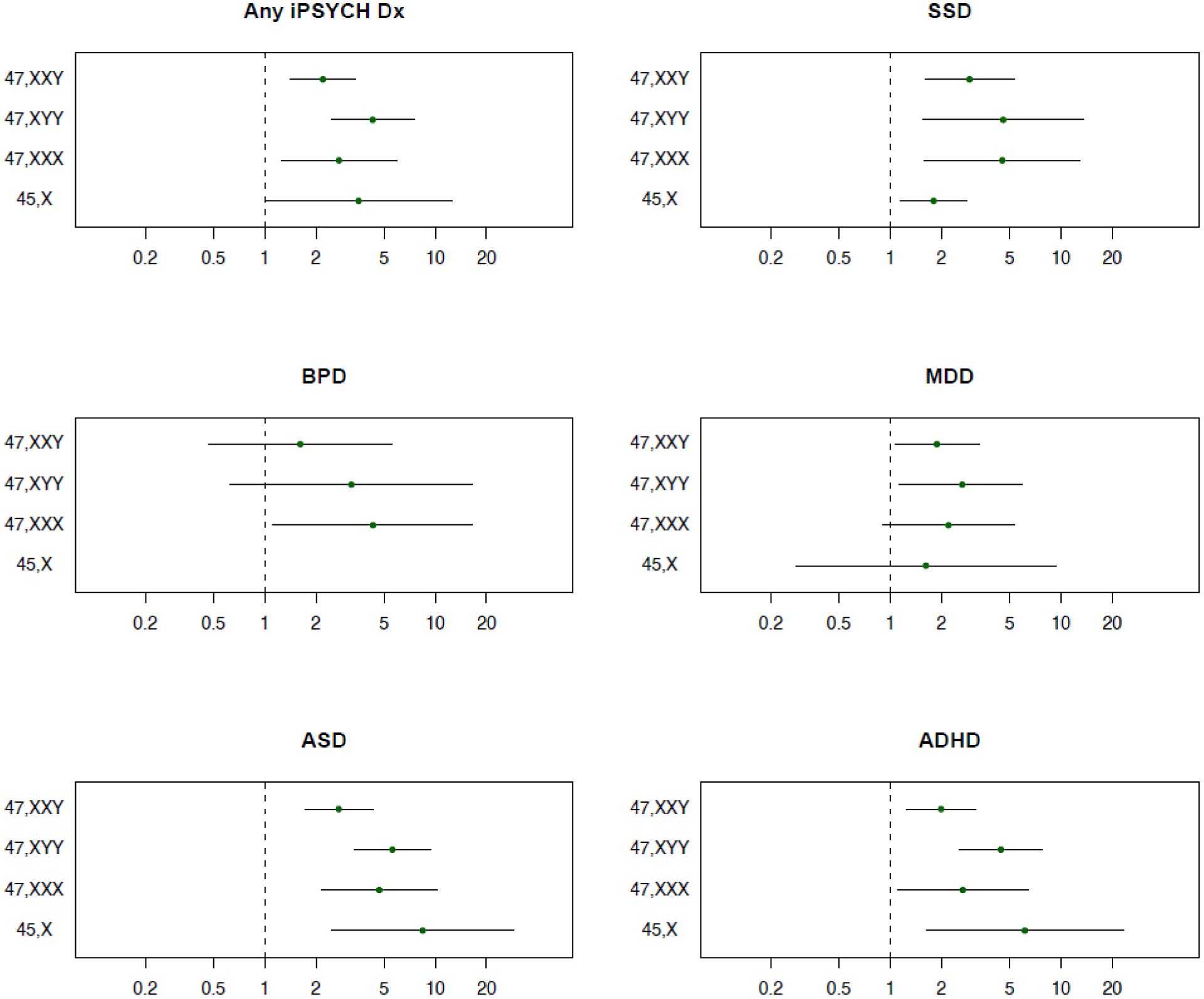
Hazard ratios (HR) and 95% confidence intervals (CI95%) were estimated with a population-weighted Cox proportional hazard model. SSD; schizophrenia spectrum disorder. BPD; bipolar disorder. MDD; major depressive disorder. ASD; autism spectrum disorder. ADHD; attention-deficit hyperactivity disorder. Any iPSYCH Dx; individuals diagnosed with one or more of the above specified disorders. Higher HR indicates that SCA individuals are more likely to develop the disorder than individuals of the same sex without SCA.
Combined population prevalence of the SCAs was 1·45 per 1,000 (estimated in the full cohort) and ranged from <0·23 for 45,X to 0·65 for 47,XXX; 0·81 for 47,XYY; and 1·23 for 47,XXY, relative to their respective gonadal sex (Figure 2 & eTable 1 bottom line). While none of the prevalence estimates differed significantly from those previously reported2 (P>0·05 for each of the four SCAs), combined they trended towards lower prevalence (P=0·046).
Figure 2. SCA Prevalence in Denmark.
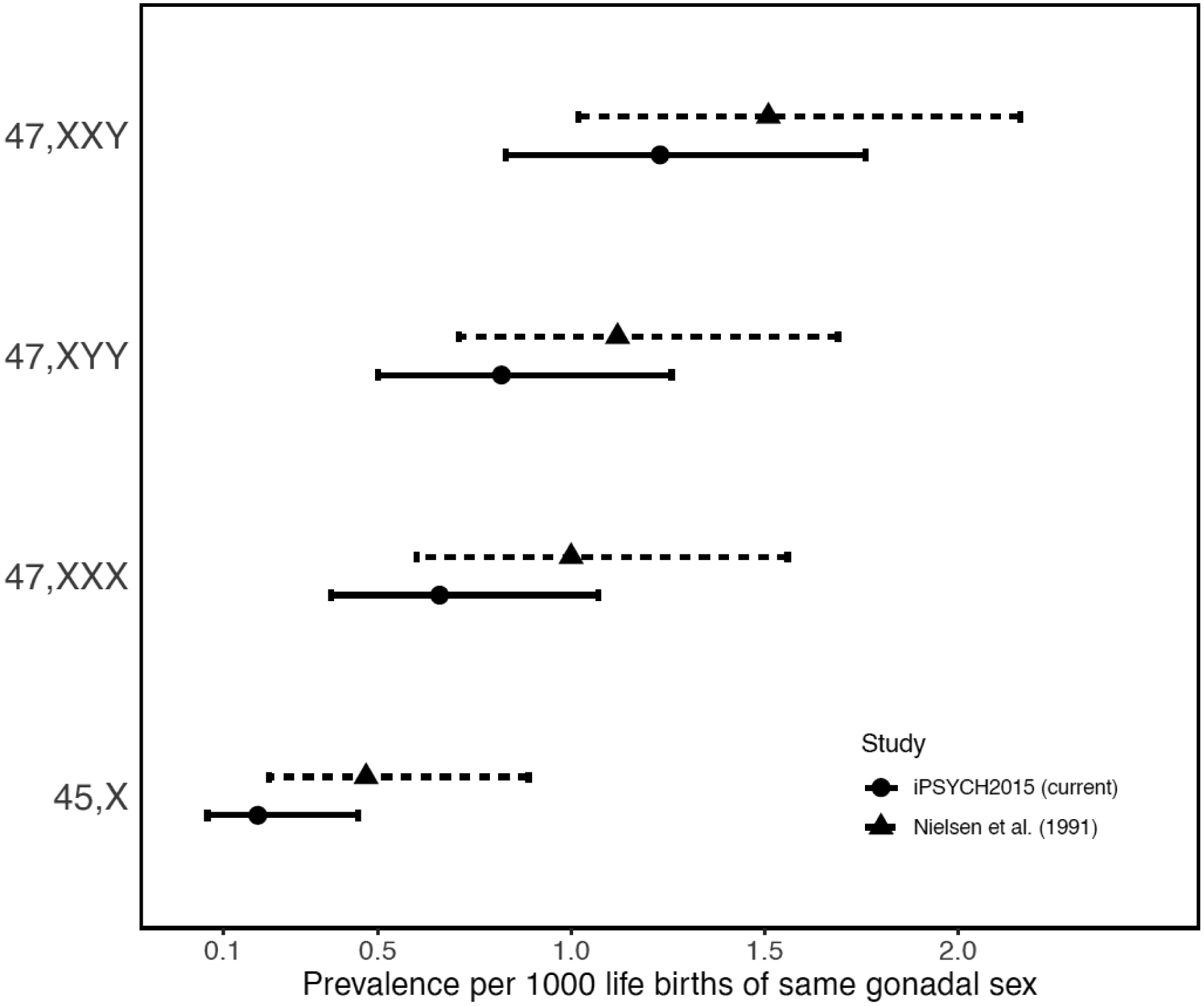
Prevalence estimates per 1000 individuals of matching gonadal sex of the four herein studied SCA karyotypes in the iPSYCH2015 random population cohort compared to those of a previous population-based study2 (with 95% confidence intervals).
47,XXX; 47,XXY; and 47,XYY associated with reduced relative reproductive rate (estimated as number of offspring in a Poisson regression model, see Methods), with Poisson regression coefficients ranging from 0·15 for 47,XXY; to 0·32 and 0·38 for 47,XYY and 47,XXX; respectively (Figure 3 & eTable 3). No offspring were recorded among the 19 identified 45,X carriers, which is consistent with the well-characterised ovarian dysgenesis in 45,X5.
Figure 3. SCA association with relative reproductive rate.
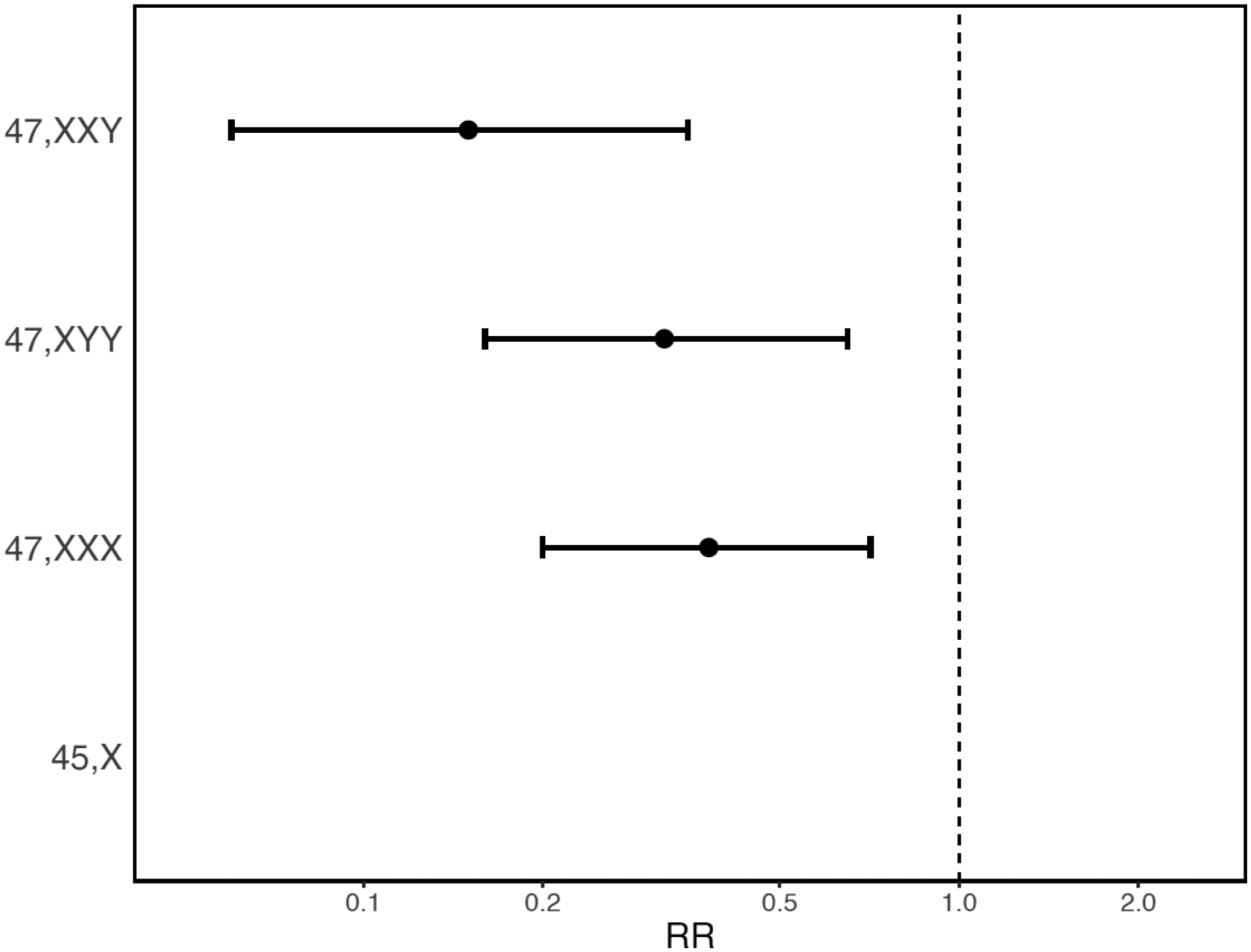
Risk Ratio (RR) and a corresponding 95% confidence interval is estimated as the relative change in number of offspring compared to individuals of the same gonadal sex without SCA, obtained by fitting a Poisson regression model, accounting for age at end of follow-up and psychiatric case status. A model could not be fitted for 45,X as no offspring were observed among carriers. Lower RR indicates that individuals with the SCA karyotype have fewer offspring than individuals of the same sex without SCA.
From available hospital records of eight disorders (other than the five iPSYCH2015 index psychiatric disorders) present in two or more carriers of a given SCA (see Appendix) we observed a very high risk of congenital malformations of the circulatory system associated with 45,X (HR = 30·6) and moderate (HR 1·8–3·7) increases in risk of ID, syncope, febrile seizures, asthma, hernia, and migraine associated with one or more SCA karyotypes (eFigure 2 & eTable 4).
Discussion
This study extends understanding of SCA karyotypes in several important ways. Most fundamentally, it is the first study to provide population-based estimates of SCA associated risk of psychiatric disorders that simultaneously considers the four most common SCA karyotypes detected through genotyping. Prior estimates of enrichment of psychiatric diagnoses in SCA have mainly derived from diagnostic surveys in groups of individuals with clinical SCA diagnosis11–13, or registry-based research14–17, (also relying solely on clinically detected SCA carriers). Our prevalence estimates of each SCA karyotype are consistent with those reported earlier for the Danish population2, although a marginally significant trend for lower prevalence was observed.
The most important finding of this study is that of an overall increased risk of psychiatric disorders associated with all four SCA karyotypes. These findings are largely in keeping with previous SCA studies11–17 but add context in several important aspects. For instance, the associated HRs are similar or even higher in magnitude than those observed in many rarer gene dosage disorders that have a longer-standing history as models of genetic risk in psychiatric research (e.g., 22q11.2 deletion and CNVs at 16p11.2)9,10. This observation highlights the relative impact of SCAs as genomic disorders contributing significantly to risk of psychiatric disorders at population level.
Also, in concordance with previous reports1,3 we find that only a small fraction of SCA carriers had been clinically detected (except for 45,X). Importantly, a test across all four SCA types showed no indication that the clinically detected subset had higher rates of psychiatric disorders.
When considering the case-cohort design of iPSYCH2015 and its unique ability to perform a genetic assessment of SCA karyotypes in a well-powered population-representative sample of individuals with and without psychiatric disorders, these findings become very important. Previously, due to the known low clinical detection rate of SCA karyotypes (particularly SCA trisomies), it has not been clear whether the increased rates of psychiatric disorders observed in clinically detected SCA carriers would also apply among undetected carriers.
We also observe an increased risk of BPD associated with 47,XXX. To the best of our knowledge this is the first reported association of this kind, although a recent study of 74 individuals with 47,XXX reported increased rates of affective and psychotic disorders, including BPD28. This finding also draws attention to the fact that most studies of SCA have focussed on children and adolescents, and hence, risk estimates for most typically adult-onset disorders is limited.
Taken together, our findings highlight a uniform increased risk of major psychiatric disorders associated with SCA karyotypes, and an unmet opportunity of early diagnosis and treatment intervention. This is especially important in the case of 47,XXX and 47,XYY with both a low clinical detection rate and a relatively high associated risk (4–5 fold) of major psychiatric disorders such as ADHD, ASD and SSD.
Despite limited power to study SCA associated changes in relative reproductive rate given the comparatively young age of the case-cohort during follow-up, we observed substantial reduction in the number of offspring associated with all three SCA trisomies (no offspring were recorded for 45,X carriers). Although other factors than fertility influence the reproductive rate, this large relative reduction in number of offspring is consistent with the late or compromised gonadal development and inadequate hormonal levels commonly noted in SCA carriers.1,5–7
In our analysis of other outcomes available for the iPSYCH2015 sample, we replicated several previously reported associations, including with congenital heart defects in 45,X5, ID in 47,XXX6, and seizure-related disorders and asthma in 47,XYY;29 and observed indications of increased risk in a few other disorders. However, these disorders were not part of the iPSYCH2015 case-cohort design, and as we only assessed risk for disorders presenting in at least two carriers for a given SCA, the associated risk estimates can be inflated and may only be considered as suggestive.
In the CPH analysis each hazard function was estimated separately and only in the subset of samples with the gonadal sex corresponding to the SCA karyotype. While this means that we cannot directly test differences in psychiatric risk patterns across SCA karyotypes in our sample, a few karyotype-based features are noteworthy, and could potentially carry both clinical and basic science implications if confirmed in a formal comparative analysis.
For instance, we observe a nominally greater increase in risk of psychiatric disorders in 47,XXX than in 47,XXY. The biological basis for this differential impact of X-chromosome gain in females vs. males is unclear and warrants further research. Paradoxically, studies of gene expression from peripheral tissues in SCA suggest that X-chromosome gain induces a substantially greater transcriptomic disruption in male vs. female cells30, which is counter to the observed risk levels for psychiatric outcomes. However, we lack functional genomic studies of SCA effects on human brain tissue, which would arguably be more relevant for understanding potential differences in psychiatric penetrance between SCA subtypes. In conducting such future studies, it would be important to examine mechanisms beyond cis-regulatory effects through which SCAs could impact cellular functions, such as altered dosage of non-genic regulatory elements31, broader disruptions of nuclear architecture32, and changes in cell division rates33.
We also observe nominally greater risks of psychiatric outcomes associated with 47,XYY than with 47,XXY, which is in keeping with earlier studies of clinical samples34,35. While such a risk pattern could seem unlikely, given that X-chromosome gain leads to substantially greater changes in gene dosage and brain anatomy36 than Y-chromosome gain, other potential sources of divergence in psychiatric risk profiles across the SCA trisomies could explain this. These include impaired gonadal function (seen in 47,XXY, and less so in 47,XXX, but not in 47,XYY), and interactions between X-chromosome dosage and different biological contexts in males (XY background and testes) vs. females (XX background and ovaries).
Notwithstanding the disparate direction and effect size magnitude of X- and Y- chromosome dosage effects on total brain size37, neuroanatomical studies have revealed that supernumerary X- and Y-chromosomes exert spatially overlapping effects on regional brain anatomy which could theoretically underpin the shared increases in psychiatric risk reported here. Specifically, increases in both X- and Y-chromosome dosage are associated with contraction of fronto-temporal cortices, and increases in parieto-occipital cortices37–39 as well as reductions in the relative size of several cerebellar36 and subcortical40 regions.
While the population-representative design of the iPSYCH2015 case-cohort and the leverage of nationwide health-registers has many advantages, it also has limitations. For example, while the bloodspot-based genotyping enables powerful population-based detection of genetic conditions such as SCA carriage, it has limited ability to detect mosaic aneuploidies and partial sex chromosome alterations. Around one third and one quarter of clinically diagnosed TS and KS subjects, respectively, were not indicated as SCA carriers in our analysis. In the case of TS these had a significantly higher average age at diagnosis than those also found with 45,X by our analysis, and most had only an unspecified TS diagnosis (ICD-10: Q96.9), suggesting that they may not be karyotype 45,X carriers. For KS we found no such difference in KS diagnosis subtype or age at diagnosis, suggesting that at least some could be true 47,XXY not detected by our analysis.
Although, the design of the iPSYCH2015 study optimises the power to assess risk conferred of the index psychiatric disorders, in a population-representative manner, the power to estimate associated risks of other outcomes, including those mental disorders that are not specifically targeted in the design, is limited. Also, the relatively young age during follow-up limits the study power of disorders (including index disorders such as BPD) that have a late typical age at onset.
Finally, while the study relies on diagnoses form a nationwide in- and out-patient based hospital based register25, which does not include information on individuals who are diagnosed and treated for psychiatric illness solely by general practitioners or privately practising psychiatrists. This means that we are likely missing a subset of milder instances of mental illness (severe form of illness most often will be diagnosed and treated in a hospital). This also means that we are unable to address any dimensional psychopathology that may manifest across diagnostic domains also in undiagnosed SCA individuals.
In conclusion, the combination of relatively high associated risk of psychiatric disorders, and low clinical detection rates for three out of four SCA karyotypes (with average age of clinical diagnosis ranging from 9–14 years for those few who are clinically detected) suggests that most SCA carriers may lack appropriate clinical care provision. Increased use of genetic testing in clinical settings would likely promote early diagnosis, inform on risk of SCA-associated illness and complications, and increase likelihood of positive treatment outcome for psychiatric disorders.
Supplementary Material
Research in context.
Evidence before this study
Elevated rates of mental illness have been reported in clinical studies of sex chromosome aneuploidies (SCAs) but few population-based studies of SCA associated risk of psychiatric disorders have been published. We searched MEDLINE, until October 30, 2022, for studies published in English, containing combinations of the following terms: psychiatric, schizophrenia, bipolar disorder, major depression, autism or ADHD; with association or risk; and 45,X, Turner syndrome, 47,XXX, Trisomy X, 47,XXY, Klinefelter syndrome, 47,XYY or Jacob’s syndrome; and register or population.
We identified only four population-based studies addressing risk of psychiatric disorders for one or more SCA karyotype. Three out of four studies reported increased risk of psychiatric disorders. All four studies relied on register-based clinical diagnoses of SCA individuals and therefore did not include the majority of SCA individuals that are assumed to be clinically undetected.
Added value of this study
To the best of our knowledge, this is the first study combining data from nation-wide biobanks and health registers to estimate the association of genotype-based SCA exposure with the risk of psychiatric outcomes in a population-representative manner. This design differs from that of previous population-based studies in a very important way, as it includes the large fraction of SCA carriers that usually are not clinically detected (and hence not included in studies based on clinical SCA diagnoses).
We found all four tested SCA karyotypes to be associated with increased risk of psychiatric disorders, and that the risk was similarly increased in the large fraction of SCA carriers who were without clinical SCA diagnosis, as in SCA carriers with a clinical SCA diagnosis.
Implications of all the available evidence
Our results corroborate the increased rates and risks of psychiatric disorders reported for SCA individuals in most studies of selected clinical samples and register-based population studies of clinically detected SCA individuals. They also show that the many individuals with SCAs who remain clinically undetected with respect to their SCA carriage are at an equally increased risk of developing psychiatric disorders. Increased use of genetic testing in clinical settings could provide earlier diagnosis of emerging psychiatric complications in these individuals and hence increase chances of a favourable treatment outcome.
Acknowledgements
The iPSYCH Initiative was approved by the Danish Scientific-Ethical Committee. This work was supported by grants form the Lundbeck Foundation (R165-2013-15320, R155-2014-1724, R248-2017-2003, and R335-2019-2318) and the NIH (R01 MH124789-01 and R01 MH130581).
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Declaration of interests
The authors declare no potential conflict of interests.
Data sharing statement
Regarding access to study data, please contact the corresponding authors.
References
- 1.Skuse D, Printzlau F, Wolstencroft J. Sex chromosome aneuploidies. Handb Clin Neurol 2018; 147: 355–76. [DOI] [PubMed] [Google Scholar]
- 2.Nielsen J, Wohlert M. Chromosome abnormalities found among 34,910 newborn children: results from a 13-year incidence study in Arhus, Denmark. Hum Genet 1991; 87: 81–3. [DOI] [PubMed] [Google Scholar]
- 3.Gravholt CH, Chang S, Wallentin M, Fedder J, Moore P, Skakkebæk A. Klinefelter Syndrome: Integrating Genetics, Neuropsychology, and Endocrinology. Endocr Rev 2018; 39: 389–423. [DOI] [PubMed] [Google Scholar]
- 4.Ottesen AM, Aksglaede L, Garn I, et al. Increased number of sex chromosomes affects height in a nonlinear fashion: a study of 305 patients with sex chromosome aneuploidy. Am J Med Genet A 2010; 152A: 1206–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Gravholt CH, Viuff MH, Brun S, Stochholm K, Andersen NH. Turner syndrome: mechanisms and management. Nat Rev Endocrinol 2019; 15: 601–14. [DOI] [PubMed] [Google Scholar]
- 6.Tartaglia NR, Howell S, Sutherland A, Wilson R, Wilson L. A review of trisomy X (47,XXX). Orphanet J Rare Dis 2010; 5: 8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Lizarazo AH, McLoughlin M, Vogiatzi MG. Endocrine aspects of Klinefelter syndrome. Curr Opin Endocrinol Diabetes Obes 2019; 26: 60–5. [DOI] [PubMed] [Google Scholar]
- 8.Green T, Flash S, Reiss AL. Sex differences in psychiatric disorders: what we can learn from sex chromosome aneuploidies. Neuropsychopharmacology 2019; 44: 9–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Sánchez XC, Helenius D, Bybjerg-Grauholm J, et al. Comparing Copy Number Variations in a Danish Case Cohort of Individuals With Psychiatric Disorders. JAMA Psychiatry 2022; 79: 59–69. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Olsen L, Sparsø T, Weinsheimer SM, et al. Prevalence of rearrangements in the 22q11. 2 region and population-based risk of neuropsychiatric and developmental disorders in a Danish population: a case-cohort study. Lancet Psychiatry 2018; 5: 573–80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.van Rijn S A review of neurocognitive functioning and risk for psychopathology in sex chromosome trisomy (47,XXY, 47,XXX, 47, XYY). Curr Opin Psychiatry 2019; 32: 79–84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Hutaff-Lee C, Bennett E, Howell S, Tartaglia N. Clinical developmental, neuropsychological, and social–emotional features of Turner syndrome. Am J Med Genet C Semin Med Genet 2019; 181: 126–34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Kuiper K, Swaab H, Tartaglia N, van Rijn S. Early developmental impact of sex chromosome trisomies on attention deficit-hyperactivity disorder symptomology in young children. Am J Med Genet A 2021; 185: 3664–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Björlin Avdic H, Butwicka A, Nordenström A, et al. Neurodevelopmental and psychiatric disorders in females with Turner syndrome: a population-based study. J Neurodev Disord 2021; 13: 51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Cederlöf M, Ohlsson GA, Larsson H, et al. Klinefelter syndrome and risk of psychosis, autism and ADHD. J Psychiatr Res 2014; 48: 128–30. [DOI] [PubMed] [Google Scholar]
- 16.Bojesen A, Juul S, Birkebaek NH, Gravholt CH. Morbidity in Klinefelter syndrome: a Danish register study based on hospital discharge diagnoses. J Clin Endocrinol Metab 2006; 91: 1254–60. [DOI] [PubMed] [Google Scholar]
- 17.Mors O, Mortensen PB, Ewald H. No evidence of increased risk for schizophrenia or bipolar affective disorder in persons with aneuploidies of the sex chromosomes. Psychol Med 2001; 31: 425–30. [DOI] [PubMed] [Google Scholar]
- 18.Viuff MH, Stochholm K, Uldbjerg N, Nielsen BB, Danish Fetal Medicine Study Group, Gravholt CH. Only a minority of sex chromosome abnormalities are detected by a national prenatal screening program for Down syndrome. Hum Reprod 2015; 30: 2419–26. [DOI] [PubMed] [Google Scholar]
- 19.Pedersen CB, Bybjerg-Grauholm J, Pedersen MG, et al. The iPSYCH2012 case–cohort sample: new directions for unravelling genetic and environmental architectures of severe mental disorders. Mol Psychiatry 2017; 23: 6–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.[Preprint] Bybjerg-Grauholm J, Pedersen CB, Bækvad-Hansen M, et al. The iPSYCH2015 Case-Cohort sample: updated directions for unravelling genetic and environmental architectures of severe mental disorders. medRxiv 2020; doi: 10.1101/2020.11.30.20237768 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Vandenbroucke JP, von Elm E, Altman DG, et al. Strengthening the Reporting of Observational Studies in Epidemiology (STROBE): explanation and elaboration. PLoS Med 2007; 4: e297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Sharp SJ, Poulaliou M, Thompson SG, White IR, Wood AM. A review of published analyses of case-cohort studies and recommendations for future reporting. PLoS One 2014; 9: e101176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Pedersen CB. The Danish Civil Registration System. Scand J Public Health 2011; 39: 22–25. [DOI] [PubMed] [Google Scholar]
- 24.Mors O, Perto GP, Mortensen PB. The Danish Psychiatric Central Research Register. Scand J Public Health 2011; 39: 54–7. [DOI] [PubMed] [Google Scholar]
- 25.Hollegaard MV, Grove J, Grauholm J, et al. Robustness of genome-wide scanning using archived dried blood spot samples as a DNA source. BMC Genet 2011; 12: 58. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Schmidt M, Schmidt SAJ, Sandegaard JL, Ehrenstein V, Pedersen L, Sørensen HT. The Danish National Patient Registry: a review of content, data quality, and research potential. Clin Epidemiol 2015; 7: 449–90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Barlow WE, Ichikawa L, Rosner D, Izumi S. Analysis of case-cohort designs. J Clin Epidemiol 1999; 52: 1165–72. [DOI] [PubMed] [Google Scholar]
- 28.Wigby K, D’Epagnier C, Howell S, et al. Expanding the phenotype of Triple X syndrome: A comparison of prenatal versus postnatal diagnosis. Am J Med Genet A 2016; 170: 2870–81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Bardsley MZ, Kowal K, Levy C, et al. 47,XYY syndrome: clinical phenotype and timing of ascertainment. J Pediatr 2013; 163: 1085–94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Raznahan A, Parikshak NN, Chandran V, et al. Sex-chromosome dosage effects on gene expression in humans. Proc Natl Acad Sci U S A 2018; 115: 7398–403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Raznahan A, Won H, Glahn DC, Jacquemont S. Convergence and Divergence of Rare Genetic Disorders on Brain Phenotypes: A Review. JAMA Psychiatry 2022; 79: 818–828. [DOI] [PubMed] [Google Scholar]
- 32.Jowhar Z, Shachar S, Gudla PR, et al. Effects of human sex chromosome dosage on spatial chromosome organization. Mol Biol Cell 2018; 29: 2458–69. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Otter M, Schrander-Stumpel CTRM, Curfs LM. Triple X syndrome: a review of the literature. Eur J Hum Genet 2010; 18: 265–71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Rau S, Whitman ET, Schauder K, et al. Patterns of psychopathology and cognition in sex chromosome aneuploidy. J Neurodev Disord 2021; 13: 61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Ross JL, Roeltgen DP, Kushner H, et al. Behavioral and social phenotypes in boys with 47,XYY syndrome or 47,XXY Klinefelter syndrome. Pediatrics 2012; 129: 769–78. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Mankiw C, Park MTM, Reardon PK, et al. Allometric Analysis Detects Brain Size-Independent Effects of Sex and Sex Chromosome Complement on Human Cerebellar Organization. J Neurosci 2017; 37: 5221–31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Raznahan A, Lee NR, Greenstein D, et al. Globally Divergent but Locally Convergent X- and Y-Chromosome Influences on Cortical Development. Cereb Cortex 2016; 26: 70–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Lepage JF, Hong DS, Raman M, et al. Brain morphology in children with 47, XYY syndrome: a voxel- and surface-based morphometric study. Genes Brain Behav 2014; 13: 127–34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Bryant DM, Hoeft F, Lai S, et al. Neuroanatomical phenotype of Klinefelter syndrome in childhood: a voxel-based morphometry study. J Neurosci 2011; 31: 6654–60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Reardon PK, Clasen L, Giedd JN, et al. An Allometric Analysis of Sex and Sex Chromosome Dosage Effects on Subcortical Anatomy in Humans. J Neurosci 2016; 36: 2438–48. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
Regarding access to study data, please contact the corresponding authors.