

Abstract
The majority of human cancers evolve over time through the stepwise accumulation of somatic mutations followed by clonal selection akin to Darwinian evolution. However, the in-depth mechanisms that govern clonal dynamics and selection remain elusive, particularly during the earliest stages of tissue transformation. Cell competition, often referred to as ‘survival of the fittest’ at the cellular level, results in the elimination of less fit cells by their more fit neighbors supporting optimal organism health and function. Alternatively, cell competition may allow an uncontrolled expansion of super-fit cancer cells to outcompete their less fit neighbors thereby fueling tumorigenesis. Recent research discussed herein highlights the various non-cell autonomous principles, including inter-clonal competition and cancer-microenvironment competition supporting the ability of a tumor to progress from the initial stages to tissue colonization. Additionally, we extend current insights from cell competition-mediated clonal interactions and selection in normal tissues to better comprehend those factors that contribute to cancer development.
Keywords: Cell competition, Flower, Clonal selection, p53, Myc
Cell Competition: Key principles and findings
Accumulation of damage and genetic aberrations within a cell may render the cell suboptimal, or less fit, but still viable. Over time, retention of suboptimal cells likely results in tissue abnormalities, disease, or loss of function. Cell competition (CC) is a highly conserved biological mechanism among multicellular organisms that results in the removal of less fit, suboptimal cells by their more fit neighbors. CC is active during tissue development to ensure the contribution of robust cell populations and throughout life as a homeostatic surveillance mechanism to maintain tissue function and health. CC was first experimentally identified in Drosophila when it was found that within mosaic wing discs composed of wildtype (W.T.) cells and heterozygous Minute (M/+) mutant (M.T.) cells that have impaired protein translation and slower growth, W.T. cells grew at the expense of their slower-growing M/+ cellular neighbors, which were markedly eliminated and not detected in the adult wing (1). However, no CC occurred in a homotypic wing disc of M/+cells alone, indicating that relative, not absolute, state of fitness dictates CC; CC did not affect total wing disc size. These observations dictate that homeostatic CC is restrained within tissue compartmentalization and thus acts to maintain tissue integrity. Additional studies revealed that the elimination of suboptimal M/+ cells proceeded through apoptosis and suppression of apoptosis obviated CC-induced expansion of W.T. cells (2). Altogether, these studies demonstrate that expansion of distinct cell populations depends not only on cell-autonomous traits, but the outcome of active selection preceded by communication of cell fitness. We now understand that multiple parameters dictate cell fitness, and differences in growth and proliferation rates alone between interacting cells do not always trigger CC (3). Instead, several complex mechanisms of CC have been identified: mechanical forces (4); cell surface ligand-receptor systems (5); direct communication of fitness status via fitness fingerprints (6); competence in capturing nutrients and signaling cues (an indirect mode of CC similar to trophic theory) (2), and various signaling pathways. Through these mechanisms, CC has been shown to operate via direct cell-cell contacts (6), or longer-range up to several cell diameters apart (4); and by secretion of soluble signaling cues (2). Several CC mechanisms first realized in fruit flies have been identified in advanced multicellular systems whose complex tissue organizations demand multiple regulatory features wherein CC appears to function as a highly-responsive, tunable parameter that integrates environmental cues with inherent fitness-inequalities amongst heterogeneous populations.
Role of Cell Competition in Cancer
Homeostatic CC optimizes the expansion of fit cells but is controlled within normal tissue size and patterning constraints. Thus, it is not surprising that critical genes identified in CC include Myc, which regulates growth and proliferation (7), and also genes within the Hippo signaling pathway, a master regulator of organ size (8,9). Increased expression of the c-Myc proto-oncogene (7,10), hyperactivation of Hippo downstream signaling component Yorkie (Yki, Drosophila homolog of mammalian Yap) (8,9), results in supercompetitors, which are cells that proliferate at the expense of neighboring normal (or W.T.) cells that undergo apoptosis. Additionally, deletions in the tumor suppressor gene, APC (11) is also associated with competitive advantages and elimination of their W.T. neighbors. Notably, the tumor suppressor gene, p53, plays a context-dependent role in CC wherein cells with relatively lower p53 expression outcompete cells with higher p53 in the irradiated hematopoietic stem cell niche (12); and in UV-exposed epidermis (13), or irradiated esophagus (14). M.T. gain-of-function (GOF) p53 exerts a fitness advantage with clonal expansion at the expense of W.T. progenitors fated for terminal differentiation and elimination. These observations of activated oncogenes and inactivated tumor suppressor genes conferring this ‘supercompetitor phenotype’ led to speculation that disrupted CC may result in tumorigenesis. Supercompetitor elimination of W.T. cells is analogous to the phenomena of tumor outgrowth at the expense of normal tissue wherein cancer cells kill normal cells to clear space for their own expansion. Indeed, apoptosis of normal cells in peritumoral tissue has been detected in multiple cancers (6,9,11). In theory, if supercompetitor clonal expansion goes unchecked, this would result in “field cancerization”, which can result in tissue dysplasia and tumors overtime owing to additional mutations. These studies provide intriguing data to suggest that many advantageous cancer-associated mutations are actively selected based on their acquired ability to exploit CC and overcome barriers within heterogeneous multicellular populations required for tumor formation and growth.
CC between cancer and the microenvironment: Friend or foe?
The tumor microenvironment (TME), particularly the tumor-adjacent stroma, plays a crucial role in cancer initiation and progression. The TME can both impede and facilitate tumor development and growth. The tumor-stroma interface is a highly dynamic zone that directly mediates tumor outgrowth and local invasion. In this section, we review recent studies that have attempted to model the initial stages of cancer development and growth and mechanisms by which CC may dictate clonal selection and growth under selective pressures exerted by the tissue microenvironment.
Whether tumor-host CC facilitates the growth of pre-cancerous lesions was experimentally tested using the Drosophila midgut as a model of intestinal adenomas harboring APC−/− resulting in hyperactive Wnt signaling. In this model the tumor cells behaved as supercompetitors and induced apoptosis in the tumor-adjacent tissue and strikingly, inhibition of apoptosis significantly impeded lesion growth (11). One mechanism of tumor-host CC fueling oncogenesis is via direct fitness sensing through Flower transmembrane proteins, which were considered to be fitness fingerprints that allow cells to directly communicate the fitness status of their neighbors. Flower is highly conserved in multicellular organisms, and in humans, Flower is encoded by four isoforms; two isoforms are named Flower-Win and two are named Flower-Lose. Distinct membrane isoform expression marks cells as fit (Flower-Win) or as suboptimal (Flower-Lose) for neighboring cells to recognize. When Flower-Win cells are co-cultured in direct contact with Flower-Lose cells, Flower-Win cells eliminate via apoptosis Flower-Lose cells and undergo compensatory proliferation. Strikingly, several types of human cancers express high levels of Flower-Win, in contrast to the adjacent stroma, which expresses high levels of Flower-Lose, concomitant to increased expression of several pro-apoptotic genes. These results suggest that in human cancers, Flower-Win expressing cancer cells outcompete and eliminate Flower-Lose expressing stromal cells, resulting in tissue attrition, and enabling tumor outgrowth (Fig 1A). Additionally, in vivo mouse models showed that xenografts of Flower-Win expressing cancer cells against a Flower-Lose tissue background resulted in aggressive tumor growth and metastasis, which is in stark contrast to the negligible tumor growth observed from xenografts of Flower-Lose cells against a Flower-Win background. Importantly, inhibition of Flower-mediated competition between tumor and stroma substantially reduced tumor volume and incidence of metastasis and heightened sensitivity toward chemotherapy (6). These results indicate that tumor growth is not the outcome solely of the cancer genome but is mostly a function of fitness inequalities and the elicited CC between the tumor and the host microenvironment.
Figure 1.

1A: hFwe-mediated tumor-host cell competition regulates tumor outgrowth along the tumor-stroma border. (left) H&E staining of human colon cancer tissue demarcates tumor and stroma. hFweWin-expressing tumor cells compete with hFweLose-expressing stromal cells within the microenvironment for space. Following cell fitness sensing and comparison, hFweWin cells outcompete and eliminate hFweLose-stromal cells via apoptosis, creating space within which tumor cells can take over, resulting in tumor outgrowth at the expense of normal tissue.
1B: c-Myc-mediated cell competition drives cancer growth. (left) H&E staining of human lung cancer tissue demarcates tumor and stroma. Myc-high tumor cells outcompete Myc-low stromal cells, which undergo apoptosis. (right) Staining of human lung cancer sample shows high c-Myc protein in tumor cells and high caspase-3 (Cas3) in stroma cells, indicative of Myc-mediated competition-induced apoptosis.
1C: YAP–mediated cell competition mediates tumor outgrowth in liver cancer. (left) H&E staining of human liver cancer tissue demarcates tumor and stroma. Differential YAP expression between tumor cells (YAP-high) and peritumoral normal hepatocytes (YAP-low) along the tumor-stroma border results in YAP-induced cell competition resulting in expansion of YAP-high tumor cells and apoptosis of YAP-low peritumoral hepatocytes (Created with BioRender.com).
Similarly, in several human cancers, cancer cells with increased expression of c-Myc and/or YAP, appear to outcompete and eliminate via apoptosis adjacent stromal cells with relatively less c-Myc/YAP at the tumor-stroma border (9) (Fig 1B). The ubiquity of cancer-stroma cell competition as mediated by the relative expression of c-Myc and YAP across a wide spectrum of human cancers suggests conservation in mechanisms exploited by oncogenic cells in epithelial cancers.
The beginning stages of tumor initiation remain the most elusive; we still have a nascent and largely theoretical view of the initial interactions between a transformed cell and its surrounding normal neighbors. Accumulating experimental evidence suggests that in some instances, normal epithelial cells can recognize and eliminate a transformed cell through CC. This process is termed EDAC-epithelial defense against cancer and can be imputed as a tumor-suppressive mechanism. In vitro experiments using mammalian cell lines have shown that when oncoprotein RasV12-transformed cells are co-cultured with W.T. epithelial cells, W.T. cells outcompete transformed cells, which are apically extruded from the epithelial monolayer (15). A recent study investigating the role of differential Hippo signaling between liver cancer cells and tumor-adjacent normal hepatocytes in vivo provides compelling evidence that the fitness and competitive advantages of normal cells can protect the epithelia from tumor expansion. Using a mouse model of Notch/Akt-activated driven liver cancer, researchers found that unlike hepatocytes in normal liver, peritumoral hepatocytes displayed accumulation of activated Yap (co-activator for the pro-cancerous transcription factor, TEAD). Deletion of Yap/Taz from peritumoral hepatocytes resulted in accelerated tumor growth (Fig 1C), whereas Yap hyperactivation in peritumoral hepatocytes diminished tumor growth. Secondly, the reduction in tumor growth was a consequence of induction of apoptosis of the cancer cells surrounded by the peritumoral hepatocytes, suggesting increased YAP in the surrounding normal cells exerts a tumor suppressive effect by outcompeting and eliminating cancer cells (8) (Fig 2A). Furthermore, in crypt organoid three-dimensional cultures generated from mouse intestinal epithelial cells, RasV12-transformed cells were extruded into the apical lumen when surrounded by W.T. cells. However, little extrusion occurred when the epithelia was nearly entirely RasV12-transformed (15) (Fig 2B).
Figure 2.
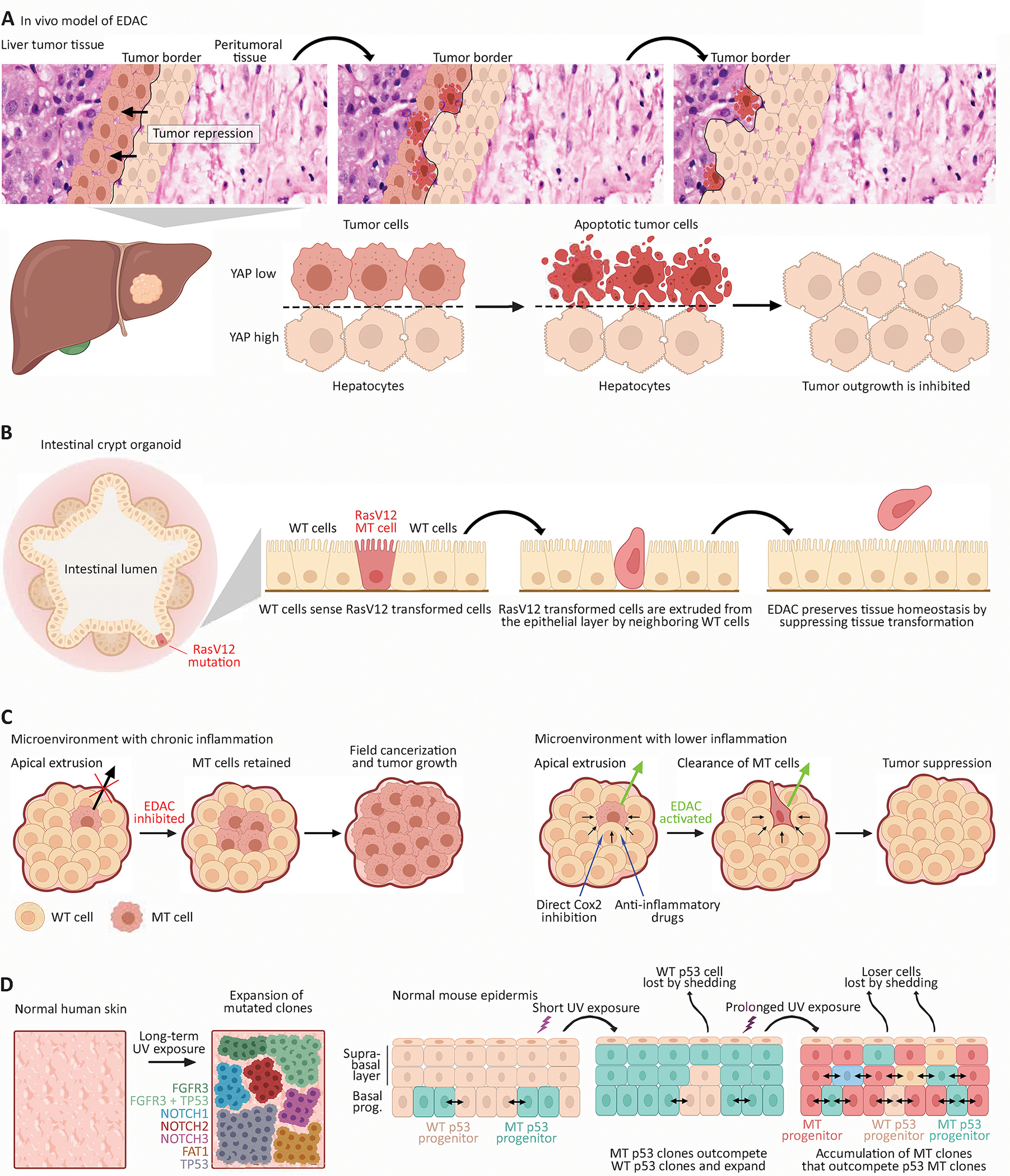
2A. In vivo model of Epithelial Defense against cancer (EDAC). (above) H&E demarcates tumor and stroma of human liver cancer overlaid by an animated depiction of YAP-mediated cell competition between tumor and stromal cells. (below) Within liver tissue, YAP-high peritumoral hepatocytes outcompete YAP-low liver tumor cells, which undergo apoptosis. This results in proliferation of YAP-high hepatocytes and inhibition of tumor outgrowth.
2B. In vitro intestinal organoid model of EDAC. Within the crypt domain of intestinal organoids, RasV12-transformed cells (red cell) surrounded by neighboring W.T. cells are sensed by W.T. cells and undergo apical extrusion. This results in the elimination of M.T. cells and prevention of tumor development.
2C. Inflammation suppresses CC and EDAC. (left) A tissue micro-environment burdened with chronic inflammation inhibits homeostatic cell competition between W.T. and M.T. cells, resulting in suppression of EDAC and thereby retention of M.T. cells. This leads to field cancerization and potentially tumor growth. (right) Targeting inflammation by COX2 inhibition or anti-inflammatory drugs restores cell competition and activates EDAC; W.T. cells eliminate M.T. cells, and tumor growth is suppressed.
2D. Mutant clone dynamics within normal human skin and mouse epidermis. (left) DNA sequencing of aged normal human skin reveals a high degree of genetic mutations resulting in a patchwork of M.T. clones, most likely due to lifetime of UV-exposure. Most frequent mutations were found in FGFR3, NOTCH1–3, FAT1, TP53. Also detected was a subclone containing FGFR3+TP53 mutations (size of clones relative to each other not drawn to scale). (right) Mouse epidermis exposed to U.V. U.V. exposure is highly mutagenic and generates a patchwork of M.T. clones. Short-term U.V. exposure results in GOF p53-M.T. epidermal progenitors that outcompete p53-W.T. progenitors and rapidly expand. Long-term U.V. exposure results in the out competition of p53-M.T. clones by M.T. clones with presumed increased fitness and competitive strength (Created with BioRender.com).
The microenvironment acts as a selective pressure that heavily dictates the relative fitness status of interacting cells and thus, the outcomes of CC-mediated clonal selection. Indeed, inflammation has been found to attenuate CC-mediated elimination of transformed cells. For example, obesity-induced chronic inflammation and metabolic alterations suppressed apical extrusion of RasV12-transformed cells by W.T. cells in mouse pancreatic and small intestine tissue; however, aspirin treatment rescued CC-driven extrusion of RasV12-expressing cells (16). Further investigations into the role of inflammation in CC subversion found normal cells surrounding RasV12-transformed cells in vitro display upregulated cyclooxygenase (COX)-2, a key mediator of inflammation, and Prostaglandin (PG) E2, a downstream mediator of COX-2. Induction of pancreatitis, a state of chronic pancreatic inflammation, in mice with RasV12-transformed pancreatic epithelia drastically suppressed extrusion of RasV12-transformed cells. Importantly, and similar to obesity-driven suppression of CC, reduction of inflammation by COX-2 inhibition promoted apical extrusion of RasV12 cells by W.T. cells (17). These results indicate that inflammation can directly impede the anti-cancer properties of CC resulting in higher retention of transformed cells (Fig 2C). Another aspect of the tumor microenvironment that selects for M.T. cells is the immune system. Tumors induce an immune inflammatory response that contribute to selection of aggressive clonal populations. During early tumor development, cytotoxic immune cells such as natural killer cells and macrophages recognize and eliminate immunogenic cancer cells. The remaining neoplastic tissue that is less immunogenic evades the immune system and progresses towards a clinically detectable tumor. Over time, cytokines from tumor and stromal cells elicit alterations in macrophage activation and function (18–20). These tumor associated macrophages that originally selected against M.T. cells now accelerate tumor growth by promoting angiogenesis (21), immune suppression, and metastasis (18,22).
A recent investigation into the stem cell dynamics within mosaic mouse intestinal crypts attempted to quantify the competitive advantage of distinct M.T. clonal populations (oncogenic Kras M.T., Apc−/−, p53-M.T.) and their potential in tumor initiation. M.T. Kras and Apc−/− stem cells outcompeted W.T. cells, but W.T. cells overtime replaced M.T. cells and prevented field cancerization effects (23). These findings are reminiscent of clonal dynamics observed from lineage tracing of p53-M.T clones in normal epidermis and esophagus, in which over time, the tissue was able to re-achieve homeostasis (13,14). The ability of epithelial tissues to return to homeostasis following initial M.T. clonal expansion is puzzling, and future investigations into how CC regulates tissue plasticity long-term are crucial. Interestingly, in the intestinal crypt, p53-M.T. had a selective advantage and outcompeted W.T. stem cells in colitis-affected intestines, in which the surrounding cells exhibited reduced fitness due to colitis-induced chronic inflammation (23). Collectively, the results of these studies suggest an inflammatory microenvironment impacts the fitness status of normal, non-transformed cells, which likely play crucial roles in determining outcomes of CC in early stages of tumor development. Total stem cell number within the crypt itself may also modulate CC outcomes: increased total stem cell population enhanced CC-removal of M.T. stem cells by W.T. cells (24).
Advanced sequencing technologies have revealed that normal aged human tissue contains a remarkable degree of genomic mutations and other aberrations, despite the tissue maintaining structural and functional integrity with no clinically detectable cancer (25,26). Revelations into the degree of mutations found in normal tissue postulate what mechanisms dictate expansion versus contraction of M.T. clones and their progression into malignancy. Two recent studies, which sought to trace M.T. clonal dynamics underlying CC in normal tissue, found that altering the exposure to specific stressors or altering fitness of W.T. cells greatly impacted CC outcomes (13,14). Two model tissues were investigated- the epidermis and the esophagus, which are both squamous epithelial tissues, wherein tissue homeostasis is maintained by a pool of proliferating progenitor cells that terminally differentiate as they progress toward the tissue surface and are shed. Clonal lineage tracing in the normal murine esophagus found that p53-M.T. progenitors outcompeted their W.T. neighbors following low-dose ionizing radiation, but competition was obviated by antioxidant pretreatment. This indicates that alteration of the microenvironment affects CC outcomes (14) and that enhancement of relative fitness status of W.T. cells tilts CC in favor of tumor suppression. These concepts are supported from findings of a murine model of the mutagen-exposed esophagus that manifested a heavily mutated mosaic epithelium composed of Notch and TP53 M.T. clones, wherein the growth of M.T. clones was directly impacted by competition with their neighbors, and the proliferative advantage of M.T. clones diminished when faced with cells with an equivalent fitness status (27). Mapping of M.T. clones in the normal human esophageal epithelium revealed a strong selection of clones mutated in various cancer genes, including TP53 and most notably, NOTCH1, arguing these clones harbored a competitive advantage over their neighboring cells (25). In mouse esophageal epithelium, single-cell induction of Notch-inhibiting mutations in mice with sporadic p53 M.T. clones resulted in rare double-M.T. clones that grew larger than p53-M.T. clones (28), thus illustrating how cooperative mutations that advance competitive potential impacts clonal selection and perhaps eventual field effects.
In a similar study, it was shown that within the UV-exposed epidermis, progenitors accumulate GOF p53 mutations, which outcompete and proliferate at the expense of their W.T. neighbors. However, over time, the epidermis can achieve homeostasis and M.T.-progenitors no longer outcompete W.T. neighbors. Notably, long-term U.V. exposure decreases p53-M.T. clonal expansion, which is conceivably due to UV-induced accumulation of additional mutations, which generates additional aggressive M.T. clones that can outcompete p53-M.T. clones (13). This is similar to what has been observed in normal aged human skin subjected to a lifetime of UV-exposure, which is heavily burdened with many cancer-associated somatic mutations, including those in TP53 (Fig 2D). It is also possible that if increased density of M.T. clones persist, this may continue to promote W.T. cell loss by differentiation- this would help achieve homeostasis and fate balance between proliferation and differentiation, thus maintaining tissue integrity. However, if exposure to a specific mutagen or inflammation persists, this would tilt the fate of positively selected M.T. clones toward uncontrolled expansion, and overtime, accrual of additional mutations, and subsequent tumor formation.
CC in clonal expansion of normal and premalignant human epithelium
Epithelial homeostasis relies on appropriate stem cell behavior to maintain a population of differentiated daughter cells that form a functional epithelium. This process is tightly regulated but is susceptible to changes that can result in tumorigenesis. The risk of oncogenic progression within tissues such as the human colon is thought to be based on stem cells acquiring sufficient mutations in critical genes that become fixed within the epithelial unit which itself clonally expands forming a cancerized field effect from which tumors may arise. To achieve this, a M.T. stem cell must outcompete its W.T. neighbors resulting in all future progeny being similarly mutated (29). Within epithelial units such as the intestinal crypt, there are several stem cells that each compete with their neighbors for space within the stem cell niche. Within the niche there is an optimum microenvironment that maintains stem cell behavior and the longer a stem cell remains mitotically active at this location, the greater the likelihood that its stem cell progeny will outcompete its neighbors; a process often called niche succession (30). This ultimately results in the formation of a clone where all cells are derived from a single parental stem cell. Indeed, Corominas-Murtra et al. (31) were able to mathematically demonstrate that ‘stemness’ arises from the random neutral competition of cells within the niche using the mammary gland, kidney development and the intestinal crypt as their models and that competition for space is the limiting factor determining the number of stem cells within the niche. Competition in the normal epithelium is not solely restricted to neighboring stem cells; clonal units that comprise many stem cells may form within the epithelium, and the degree to which they expand is determined by selection or the lack thereof when competing for space in the presence of other clones (32).
In the absence of positive selection, clonal competition and expansion is thought to occur via a neutral process where clones stochastically appear and disappear over time. In humans, tissue stem cell behavior can be visualized through the use of lineage tracing methodologies to map clonal fate (33,34). Naturally occurring mutations that result in biochemical defects is a common method to measure stem cell dynamics within human epithelial tissues. Using mutations within the mitochondrial genome that result in a biochemical deficiency of cytochrome c oxidase (CCO) it has been possible to both observe and measure stem cell dynamics within the normal human intestinal crypt, skin, pancreas, and breast (35,36). CCO-deficiency can be easily identified histologically (Fig 3A), and microdissection sequencing of multiple CCO-deficient cells can demonstrate they are clones, each sharing the same mitochondrial DNA mutation as their neighbors. As new M.T. stem cells arise, they compete with their W.T. neighbors resulting in either losing or winning the right to seed the entire stem cell niche with their progeny. To determine the rate at which they do this, we can take advantage of the fact that daughter cells inherit their parent’s stem cell genotype and in the intestinal crypt move in a conveyer belt manner (35,37). This effectively means that cells located further up the crypt represent an older cell population than those at the crypt base. The proportion of CCO-deficient to CCO-normal cells at each physical level within the crypt represents the state of cell competition at that particular time point. Generating en face serial sections all the way through partially CCO-deficient crypts allows us to trace the expansion and contraction of CCO-deficient clones over time; this has been referred colloquially as the ‘wiggle’ (Fig 3B). Determining the change in the size of CCO-deficiency between each serial section in the reconstructed crypt allows us to calculate the rate at which clonal expansion and competition between clones occur. In the normal adult human crypt, deviations in clone size relative to sequential sections were found to be balanced, and therefore CCO-deficiency represented a neutral marker of clonal expansion, suggesting that niche succession in morphologically normal crypts is based on stochastic stem cell divisions (37). Interestingly, neutral drift also occurs in partially CCO-deficient adenomatous crypts with the important distinction that if we view niche succession as a game, the game (the loss or replacement of stem cells) is played out at a faster pace yet the outcome of whether an adenomatous crypt becomes entirely CCO-deficient or CCO-normal, remains stochastic. Therefore, neutral stem cell drift occurs even in APC-mutated crypts if all stem cells possess the same genotype.
Figure 3.
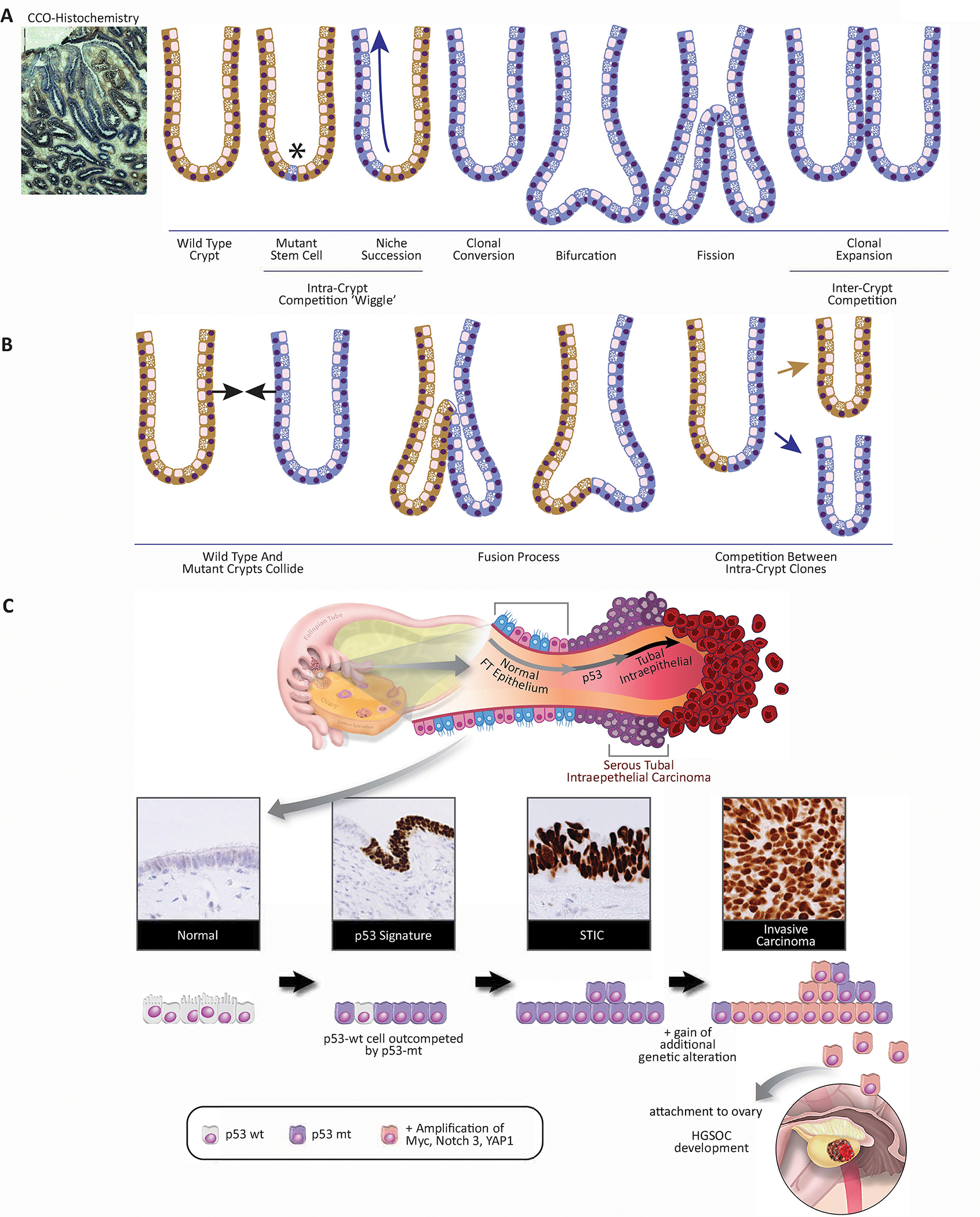
3A. CCO-deficiency as a result of mitochondrial DNA mutation. This process begins with a CCO-M.T. stem cell arising at the base of a CCO-W.T. colon crypt. Neutral stem cell competition results in niche succession and clonal conversion to a CCO-deficient crypt. M.T. crypts divide by fission creating a M.T. patch that competes for space with the colon mucosa.
3B. Crypts are able to fuse. Two phenotypically distinct crypts collide and begin to fuse in a process the reverse of fission. This results in two independent clones competing for the stem cell niche. The winner (in this scenario) is random but results in the removal of the loser from the colon.
3C. Mutations in TP53 drive cell competition in early fallopian tube precursors of ovarian cancer. The majority of high-grade serous carcinomas arise in the distal fallopian tube. Histology: Normal tubal epithelium is composed of ciliated and secretory cells. Acquisition of TP53 mutations, through the repetitive release of inflammatory follicular fluid during ovulation, confirms an advantage to secretory cells. Their expansion leads to the formation of p53 signatures. These cells stain strongly for p53 by immunohistochemistry. Subsequent alterations in oncogenes and tumor suppressors, like MYC, PTEN, RB, YAP1, or NOTCH1, among others, cooperate with M.T. TP53 to drive the transformation of the p53 signatures into serous tubal intraepithelial carcinoma (STIC lesions). STIC lesions retain strong staining for p53. STIC cells outcompete neighboring W.T. cells and eventually invade through the basement membrane to establish an invasive high-grade serous carcinoma (Created with BioRender.com).
Crypts themselves compete for space within the colon and are able to divide (by a process known as crypt fission) resulting in clonal expansion and can also fuse with neighbor crypts resulting in potential clonal contraction (38,39). In the normal human colon, a lifetime of clonal expansion can result in a significant patch size of related crypts. Novelli et al. (38), have shown that random x-inactivation of a germline inherited M.T. allele for the G6PD gene early in development can result in the formation of large patches of clonally related crypts late in adult life. However, if M.T. crypts with no selective advantage arise later in life (such as CCO-deficiency) patch size is very small (33,40). Not only does this show that crypt fission is rare in the normal adult colon, but it is difficult for field cancerization to develop to any significant extent if there is no selective advantage over W.T. crypts. Additionally, clonal expansion is countered by an apparent similar rate of crypt fusion to the extent that overall crypt numbers are maintained in the human colon (39). If a crypt fusion event occurs, there is an immediate increase in the number of stem cells in the newly-formed crypt where each parent crypt has contributed 50% of the new fused crypt stem cells. The competition between these two distinct clones begins as if it were a normal crypt and niche succession eventually results in a clonal crypt with a similar number of stem cells to its neighbors with no remaining evidence of the loser half of the crypt.
While it is likely that the vast majority of somatic mutations that occur in human epithelial cells are neutral, there is evidence on how those with selective advantages behave. As discussed earlier in this review, in animal models, mutations in apc, kras and p53 can result in outcompeting W.T. intestinal stem cells via biased niche succession and crypts M.T. in these genes preferentially expanding via increased crypt fission as they confer proliferative and survival advantages to the individual crypt (23,34). Using somatically acquired mutations as a means to lineage trace stem cell clones in the human colonic crypt, Nicholson et al. (34) have shown a marked difference in the rate of niche succession and clonal expansion between neutral and pro-oncogenic mutations in morphologically normal colon. The average patch size of M.T. tumor suppressor STAG2 crypts is significantly larger than either the patch size of CCO-deficient (33) or M.T. period acid Schiff crypts (34) that have no selective advantage. It is highly unlikely that in truly normal human epithelium, even under conditions of natural selection that M.T. clones can expand through the entire tissue and it is therefore highly unlikely that individual clones are able to undergo clonal sweeps where the entire epithelium is clonal.
One of the most surprising observations of human clonal expansion in recent years was the discovery that using next-generation sequencing (NGS) technologies, normal epithelial cells particularly those taken from the eyelid (26) and esophagus (25) of aged individuals display a significant mutation burden and diversity of pro-oncogenic genes in the absence of cancer. Within the normal esophagus, the size of individual clones appeared to be affected through smoking and in the eyelid by sun U.V. exposure but time appears to be the predominant factor of clone size. This observation is not uniform across all human epithelial tissues. In the normal human bladder, while there is a similar diversity of mutations, however, most under positive selection are detected in chromatin remodeling genes whereas comparison genes associated with bladder cancer are relatively rare (41). Overall, it is now clear that the normal epithelium represents a mosaic of competing clones that may or may not determine a patient’s cancer risk.
With the advent of NGS and genome atlas projects, tumor and normal tissue sequencing has established that many somatic mutations do not reliably result in cancer development and that most genetic changes within a particular cancer subtype are not shared across patients. While gradual, stepwise somatic mutations are essential to tumorigenesis, cancer sequencing studies have demonstrated that punctuated bursts of genetic change at the karyotype level are necessary for cancer progression (42–44). Chromosomal instability (CIN), classified as either structural or numerical, has typically been known to diminish cellular fitness. However, studies investigating chemotherapy resistance have demonstrated that CIN-driven macroevolution provides cancer cells with additional mechanisms to thrive under stressful environments (45). In particular, aneuploidy has been demonstrated to induce chemotherapy resistance in patient derived xenografts (46) and promote metastatic progression (47) through increased epithelial-to-mesenchymal transition (EMT) activity (48). Understanding the role of CIN in cell fitness will play a crucial role in evaluating cancer risk, as higher levels of CIN are typically associated with poorer prognosis (48).
Oncogenic mutations alone are not necessarily sufficient to generate large-scale field cancerization, and microenvironment factors such as inflammation play an essential role in clonal expansion. Inflammation appears to strongly select for p53-M.T.crypts, and this is reflected in human cancers associated with inflammation such as ulcerative colitis and Crohn’s disease (49). The evolution of cancer begins long before the emergence of a detectable malignancy. As described above, ostensibly healthy tissues accrue mutations throughout life, and a subset of the mutation bearing cells have an altered phenotype that experiences evolutionary selection leading to an expansion of the fitter clones. Sometimes the clonal expansion is the first step towards cancer. Studies in Barrett’s esophagus and Inflammatory Bowel Disease (IBD) – two chronic inflammatory conditions of the respective ends of the gastrointestinal tract – are excellent examples of this pre-cancer evolution. In both conditions, M.T. cells, often carrying alterations in critical cancer-associated genes including CDKN2A (p16) and TP53 (p53), experience positive selection and expand over huge extents. For example, in Barrett’s, alterations to CDKN2A are often present throughout the entire Barrett’s lesion (50), and in general, the expansion of M.T. clones is positively correlated with cancer risk (51). In IBD, we documented in one remarkable Crohn’s colitis patient the occurrence of multiple TP53 clones, one of which grew along the entire colon length and crossed into the small intestine during four years of surveillance (52). Large clonal expansions are found more commonly in people with IBD who progress to colorectal cancer (53).
However, in many examples, cells bearing cancer-associated mutations do not progress to cancer in a person’s lifetime. For example, in Barrett’s esophagus mutations of many cancer-associated genes are also found in never-dysplastic Barrett’s; only TP53 and SMAD4 mutations are stage-specific to cancer (54). In the colon, epithelial cells within crypts contain thousands of somatic mutations by middle age and 1% of crypts reportedly carry a driver alteration (55), yet less than 10% of people will develop colorectal cancer in their lifetime (56). Thus, it appears that the initial stages of cancer evolution involve many ‘false starts’ where the acquisition of a so-called cancer mutation is not, in fact, the first step along the evolutionary trajectory to cancer – or at least not the first step upon a trajectory that is rapid enough to lead to cancer in a patient’s lifetime. Moreover, whether ‘false starts’ are a result of the wrong combination of genes being simultaneously activated or a consequence of induction of counteracting genomic changes remains a possibility to be explored.
CC could play a role in preventing cells with a cancer-associated mutation from forming cancer. As described above, the neutral competition to retain a place in the stem cell niche is commonplace across tissues. Competition between stem cells for a place in the intestinal crypt niche is a clear example. While a cancer-associated mutation may give an advantage in the competition for a place in the niche, it may be insufficient to lead to the growth of the niche itself, or alternatively be insufficient to allow the M.T. cells to grow without reliance on the stem cell niche. In the intestine, for example, KRAS mutations are positively selected in the intestinal crypt (23,40), but alone are insufficient to produce neoplasia. In other words, CC could conceivably act as a ‘red herring’ for somatic evolution, providing selective pressure for a phenotype that is competitive for initial expansion (e.g., within the intestinal crypt), but not necessarily adapted for subsequent development beyond the confines of the initial growth (e.g., to drive the growth of the crypt population).
Similarly, in ovarian cancer, mutations in TP53 are obligatory (57), occurring in nearly 100% of cases, and are detected in the earliest recognized precursor lesions in the fallopian tube called ‘p53 signatures’ (58). These early precursors are composed of stretches of contiguous non-proliferative secretory cells that maintain a near-normal morphology yet harbor somatic TP53 mutation and accumulation of DNA damage (Fig 3C). The development of these precursors is epidemiologically and experimentally linked to ovulation and the associated inflammation triggered by the repetitive release of monthly follicular fluid that contains cytokines, chemokines, and reactive oxygen species (59,60). These p53 signature cells are flanked by benign W.T. secretory cells. Next-generation whole-exome sequencing shows that p53 signatures are not only the precursors to high-grade serous carcinoma (HGSC) (61), the most common subtype of ovarian cancer, they can also persist for decades before acquiring additional alterations that lead to an in-situ carcinoma (61,62). This suggests that M.T. p53-expressing secretory cells have a transient fitness advantage that enables the M.T. cells to expand and develop into a p53 signature. However, in the absence of additional mutations, these cells can persist but are kept in check by neighboring W.T. cells. This is relevant since nearly a third to 50% of all women have ‘p53 signatures’ in their fallopian tubes regardless of risk (e.g., BRCA1/2 mutation status) yet ovarian cancer is a relatively rare disease.
Interestingly, these molecular studies also showed that multiple tubal precursors could arise in the fallopian tube, and they are not always related. Specifically, in a fallopian tube with multiple ‘p53 signatures’, each signature may harbor a different TP53 mutation, presumably due to the stochastic effects of ovulation, and only one of these signatures may ultimately develop into an in situ lesion and invasive carcinoma. Presumably, the TP53 mutations that were unsuccessful in progressing to carcinoma were limited by competition with a more aggressive TP53 mutation that is detected in the subsequent in situ lesion and invasive carcinoma. It also suggests that not all TP53 mutations are created equal and that W.T. cells can often impose a restraint on their impact, an example of EDAC.
The ability of some ‘p53 signatures’ to progress into in situ lesions and HGSC is often accompanied by the acquisition of MYC, NOTCH3, or YAP1 amplification. As seen in other organs, these oncogenes enable the nascent p53 signature cells to outcompete and likely eliminate their adjacent W.T. cells. Although the acquisition of these activated oncogenes may take decades, once acquired, the progression from a tubal cancer to an ovarian cancer with widespread dissemination to the peritoneal cavity can occur rapidly within six years (61,62).
Conclusion
Advanced sequencing technologies that allow identification of extensive cellular heterogeneity coupled with intricate clonal lineage tracing techniques to analyze the temporal and spatial regulation of clonal behavior is rapidly advancing our understanding of mechanisms underlying normal tissue growth versus aberrant growth, i.e., cancer. Cancer clonal evolution is dependent on cell-extrinsic interactions between other cells as well as cues from the microenvironment. Observational findings from various human tissues and experimental models suggest CC may be a crucial mediator of clonal dynamics in normal tissues, and throughout tumorigenesis, as it has been implicated in initial tissue transformation (both permissive and suppressive to tumor formation), field cancerization, tumor outgrowth at the expense of normal tissue, and offers a distinct lens into the evolution of premalignant lesions into malignant cancers. As our understanding of CC increases, new potential therapeutic targets will be uncovered and specific genomic changes allowing more accurate staging of premalignant and metastatic cancers will lead too enhanced prognostication of therapeutic responses.
Acknowledgements
ERC to E.M., SNSF, Swiss Cancer League, LB692, and FCT LB506 to R.G. This work was also supported by NIH SPORE P50 CA228991 (R.D.), NIH/NCI R01 CA244993 (P.B.F.), NIH/NCI R01 CA259599 (P.B.F.), the U.S. Department of Defense (OC170094, OC180420), a grant from Virginia’s Commonwealth Health Research Board (P.B.F.), the VCU Massey Cancer Center core grant NIH-NCI Cancer Center Support Grant P30 CA016059 (J.R.T., R.A.W., P.B.F., R.G.), the Dr. Miriam and Sheldon G. Adelson Medical Research Foundation (R.D.), the Honorable Tina Brozman Foundation for Ovarian Cancer Research (R.D.), The Penn Medicine Abramson Cancer Center - Ovarian Cancer Translational Center of Excellence (R.D.), The Basser Center for BRCA (R.D.), and the Claneil Foundation (R.D.). T.A.G. acknowledges support from Cancer Research U.K. (A19771). S.A.M. also acknowledges support from Cancer Research U.K. (A21446 & A29072). Support from the Thelma Newmeyer Corman Endowment (P.B.F.) is also acknowledged. All figures are original or references; figures were prepared using BioRender.
Footnotes
All authors have no conflicts of interest to declare.
References
- 1.Morata G, Ripoll P. Minutes: mutants of drosophila autonomously affecting cell division rate. Dev Biol 1975;42:211–21 [DOI] [PubMed] [Google Scholar]
- 2.Moreno E, Basler K, Morata G. Cells compete for decapentaplegic survival factor to prevent apoptosis in Drosophila wing development. Nature 2002;416:755–9 [DOI] [PubMed] [Google Scholar]
- 3.de la Cova C, Abril M, Bellosta P, Gallant P, Johnston LA. Drosophila myc regulates organ size by inducing cell competition. Cell 2004;117:107–16 [DOI] [PubMed] [Google Scholar]
- 4.Matamoro-Vidal A, Levayer R. Multiple Influences of Mechanical Forces on Cell Competition. Curr Biol 2019;29:R762–R74 [DOI] [PubMed] [Google Scholar]
- 5.Yamamoto M, Ohsawa S, Kunimasa K, Igaki T. The ligand Sas and its receptor PTP10D drive tumour-suppressive cell competition. Nature 2017;542:246–50 [DOI] [PubMed] [Google Scholar]
- 6.Madan E, Pelham CJ, Nagane M, Parker TM, Canas-Marques R, Fazio K, et al. Flower isoforms promote competitive growth in cancer. Nature 2019;572:260–4 [DOI] [PubMed] [Google Scholar]
- 7.Claveria C, Giovinazzo G, Sierra R, Torres M. Myc-driven endogenous cell competition in the early mammalian embryo. Nature 2013;500:39–44 [DOI] [PubMed] [Google Scholar]
- 8.Moya IM, Castaldo SA, Van den Mooter L, Soheily S, Sansores-Garcia L, Jacobs J, et al. Peritumoral activation of the Hippo pathway effectors YAP and TAZ suppresses liver cancer in mice. Science 2019;366:1029–34 [DOI] [PubMed] [Google Scholar]
- 9.Di Giacomo S, Sollazzo M, de Biase D, Ragazzi M, Bellosta P, Pession A, et al. Human Cancer Cells Signal Their Competitive Fitness Through MYC Activity. Sci Rep 2017;7:12568. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Moreno E, Basler K. dMyc transforms cells into super-competitors. Cell 2004;117:117–29 [DOI] [PubMed] [Google Scholar]
- 11.Suijkerbuijk SJ, Kolahgar G, Kucinski I, Piddini E. Cell Competition Drives the Growth of Intestinal Adenomas in Drosophila. Curr Biol 2016;26:428–38 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Bondar T, Medzhitov R. p53-mediated hematopoietic stem and progenitor cell competition. Cell Stem Cell 2010;6:309–22 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Murai K, Skrupskelyte G, Piedrafita G, Hall M, Kostiou V, Ong SH, et al. Epidermal Tissue Adapts to Restrain Progenitors Carrying Clonal p53 Mutations. Cell Stem Cell 2018;23:687–99 e8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Fernandez-Antoran D, Piedrafita G, Murai K, Ong SH, Herms A, Frezza C, et al. Outcompeting p53-Mutant Cells in the Normal Esophagus by Redox Manipulation. Cell Stem Cell 2019;25:329–41 e6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Kon S, Ishibashi K, Katoh H, Kitamoto S, Shirai T, Tanaka S, et al. Cell competition with normal epithelial cells promotes apical extrusion of transformed cells through metabolic changes. Nat Cell Biol 2017;19:530–41 [DOI] [PubMed] [Google Scholar]
- 16.Sasaki A, Nagatake T, Egami R, Gu G, Takigawa I, Ikeda W, et al. Obesity Suppresses Cell-Competition-Mediated Apical Elimination of RasV12-Transformed Cells from Epithelial Tissues. Cell Rep 2018;23:974–82 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Sato N, Yako Y, Maruyama T, Ishikawa S, Kuromiya K, Tokuoka SM, et al. The COX-2/PGE2 pathway suppresses apical elimination of RasV12-transformed cells from epithelia. Commun Biol 2020;3:132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Gonzalez H, Hagerling C, Werb Z. Roles of the immune system in cancer: from tumor initiation to metastatic progression. Genes Dev 2018;32:1267–84 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Teng MW, Galon J, Fridman WH, Smyth MJ. From mice to humans: developments in cancer immunoediting. J Clin Invest 2015;125:3338–46 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Mantovani A, Marchesi F, Malesci A, Laghi L, Allavena P. Tumour-associated macrophages as treatment targets in oncology. Nat Rev Clin Oncol 2017;14:399–416 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Shojaei F, Zhong C, Wu X, Yu L, Ferrara N. Role of myeloid cells in tumor angiogenesis and growth. Trends Cell Biol 2008;18:372–8 [DOI] [PubMed] [Google Scholar]
- 22.Qian BZ, Zhang H, Li J, He T, Yeo EJ, Soong DY, et al. FLT1 signaling in metastasis-associated macrophages activates an inflammatory signature that promotes breast cancer metastasis. J Exp Med 2015;212:1433–48 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Vermeulen L, Morrissey E, van der Heijden M, Nicholson AM, Sottoriva A, Buczacki S, et al. Defining stem cell dynamics in models of intestinal tumor initiation. Science 2013;342:995–8 [DOI] [PubMed] [Google Scholar]
- 24.Bruens L, Ellenbroek SIJ, Suijkerbuijk SJE, Azkanaz M, Hale AJ, Toonen P, et al. Calorie Restriction Increases the Number of Competing Stem Cells and Decreases Mutation Retention in the Intestine. Cell Rep 2020;32:107937. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Martincorena I, Fowler JC, Wabik A, Lawson ARJ, Abascal F, Hall MWJ, et al. Somatic mutant clones colonize the human esophagus with age. Science 2018;362:911–7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Martincorena I, Jones PH, Campbell PJ. Constrained positive selection on cancer mutations in normal skin. Proc Natl Acad Sci U S A 2016;113:E1128–9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Colom B, Alcolea MP, Piedrafita G, Hall MWJ, Wabik A, Dentro SC, et al. Spatial competition shapes the dynamic mutational landscape of normal esophageal epithelium. Nat Genet 2020;52:604–14 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Alcolea MP, Jones PH. Cell competition: winning out by losing notch. Cell Cycle 2015;14:9–17 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Leedham SJ, Wright NA. Expansion of a mutated clone: from stem cell to tumour. J Clin Pathol 2008;61:164–71 [DOI] [PubMed] [Google Scholar]
- 30.Baker AM, Graham TA, Wright NA. Pre-tumour clones, periodic selection and clonal interference in the origin and progression of gastrointestinal cancer: potential for biomarker development. J Pathol 2013;229:502–14 [DOI] [PubMed] [Google Scholar]
- 31.Corominas-Murtra B, Scheele C, Kishi K, Ellenbroek SIJ, Simons BD, van Rheenen J, et al. Stem cell lineage survival as a noisy competition for niche access. Proc Natl Acad Sci U S A 2020;117:16969–75 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Vermeulen L, Snippert HJ. Stem cell dynamics in homeostasis and cancer of the intestine. Nat Rev Cancer 2014;14:468–80 [DOI] [PubMed] [Google Scholar]
- 33.Greaves LC, Preston SL, Tadrous PJ, Taylor RW, Barron MJ, Oukrif D, et al. Mitochondrial DNA mutations are established in human colonic stem cells, and mutated clones expand by crypt fission. Proc Natl Acad Sci U S A 2006;103:714–9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Nicholson AM, Olpe C, Hoyle A, Thorsen AS, Rus T, Colombe M, et al. Fixation and Spread of Somatic Mutations in Adult Human Colonic Epithelium. Cell Stem Cell 2018;22:909–18 e8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Fellous TG, McDonald SA, Burkert J, Humphries A, Islam S, De-Alwis NM, et al. A methodological approach to tracing cell lineage in human epithelial tissues. Stem Cells 2009;27:1410–20 [DOI] [PubMed] [Google Scholar]
- 36.Baker AM, Cereser B, Melton S, Fletcher AG, Rodriguez-Justo M, Tadrous PJ, et al. Quantification of Crypt and Stem Cell Evolution in the Normal and Neoplastic Human Colon. Cell Rep 2019;27:2524. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Park HS, Goodlad RA, Wright NA. Crypt fission in the small intestine and colon. A mechanism for the emergence of G6PD locus-mutated crypts after treatment with mutagens. Am J Pathol 1995;147:1416–27 [PMC free article] [PubMed] [Google Scholar]
- 38.Novelli M, Cossu A, Oukrif D, Quaglia A, Lakhani S, Poulsom R, et al. X-inactivation patch size in human female tissue confounds the assessment of tumor clonality. Proc Natl Acad Sci U S A 2003;100:3311–4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Greaves LC, Nooteboom M, Elson JL, Tuppen HA, Taylor GA, Commane DM, et al. Clonal expansion of early to mid-life mitochondrial DNA point mutations drives mitochondrial dysfunction during human ageing. PLoS Genet 2014;10:e1004620. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Snippert HJ, Schepers AG, van Es JH, Simons BD, Clevers H. Biased competition between Lgr5 intestinal stem cells driven by oncogenic mutation induces clonal expansion. EMBO Rep 2014;15:62–9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Lawson ARJ, Abascal F, Coorens THH, Hooks Y, O’Neill L, Latimer C, et al. Extensive heterogeneity in somatic mutation and selection in the human bladder. Science 2020;370:75–82 [DOI] [PubMed] [Google Scholar]
- 42.Heng HH, Bremer SW, Stevens JB, Horne SD, Liu G, Abdallah BY, et al. Chromosomal instability (CIN): what it is and why it is crucial to cancer evolution. Cancer Metastasis Rev 2013;32:325–40 [DOI] [PubMed] [Google Scholar]
- 43.Heng J, Heng HH. Genome Chaos, Information Creation, and Cancer Emergence: Searching for New Frameworks on the 50th Anniversary of the “War on Cancer”. Genes (Basel) 2021;13 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Vendramin R, Litchfield K, Swanton C. Cancer evolution: Darwin and beyond. EMBO J 2021;40:e108389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Lukow DA, Sausville EL, Suri P, Chunduri NK, Wieland A, Leu J, et al. Chromosomal instability accelerates the evolution of resistance to anti-cancer therapies. Dev Cell 2021;56:2427–39 e4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Gao H, Korn JM, Ferretti S, Monahan JE, Wang Y, Singh M, et al. High-throughput screening using patient-derived tumor xenografts to predict clinical trial drug response. Nat Med 2015;21:1318–25 [DOI] [PubMed] [Google Scholar]
- 47.Turajlic S, Swanton C. Metastasis as an evolutionary process. Science 2016;352:169–75 [DOI] [PubMed] [Google Scholar]
- 48.Vasudevan A, Baruah PS, Smith JC, Wang Z, Sayles NM, Andrews P, et al. Single-Chromosomal Gains Can Function as Metastasis Suppressors and Promoters in Colon Cancer. Dev Cell 2020;52:413–28 e6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Baker AM, Cross W, Curtius K, Al Bakir I, Choi CR, Davis HL, et al. Evolutionary history of human colitis-associated colorectal cancer. Gut 2019;68:985–95 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Martinez P, Mallo D, Paulson TG, Li X, Sanchez CA, Reid BJ, et al. Evolution of Barrett’s esophagus through space and time at single-crypt and whole-biopsy levels. Nat Commun 2018;9:794. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Maley CC, Galipeau PC, Li X, Sanchez CA, Paulson TG, Blount PL, et al. The combination of genetic instability and clonal expansion predicts progression to esophageal adenocarcinoma. Cancer Res 2004;64:7629–33 [DOI] [PubMed] [Google Scholar]
- 52.Galandiuk S, Rodriguez-Justo M, Jeffery R, Nicholson AM, Cheng Y, Oukrif D, et al. Field cancerization in the intestinal epithelium of patients with Crohn’s ileocolitis. Gastroenterology 2012;142:855–64 e8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Salk JJ, Salipante SJ, Risques RA, Crispin DA, Li L, Bronner MP, et al. Clonal expansions in ulcerative colitis identify patients with neoplasia. Proc Natl Acad Sci U S A 2009;106:20871–6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Weaver JMJ, Ross-Innes CS, Shannon N, Lynch AG, Forshew T, Barbera M, et al. Ordering of mutations in preinvasive disease stages of esophageal carcinogenesis. Nat Genet 2014;46:837–43 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Lee-Six H, Olafsson S, Ellis P, Osborne RJ, Sanders MA, Moore L, et al. The landscape of somatic mutation in normal colorectal epithelial cells. Nature 2019;574:532–7 [DOI] [PubMed] [Google Scholar]
- 56.Sasieni PD, Shelton J, Ormiston-Smith N, Thomson CS, Silcocks PB. What is the lifetime risk of developing cancer?: the effect of adjusting for multiple primaries. Br J Cancer 2011;105:460–5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Cancer Genome Atlas Research N. Integrated genomic analyses of ovarian carcinoma. Nature 2011;474:609–15 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Lee Y, Miron A, Drapkin R, Nucci MR, Medeiros F, Saleemuddin A, et al. A candidate precursor to serous carcinoma that originates in the distal fallopian tube. J Pathol 2007;211:26–35 [DOI] [PubMed] [Google Scholar]
- 59.Emori MM, Drapkin R. The hormonal composition of follicular fluid and its implications for ovarian cancer pathogenesis. Reprod Biol Endocrinol 2014;12:60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Reid BM, Permuth JB, Sellers TA. Epidemiology of ovarian cancer: a review. Cancer Biol Med 2017;14:9–32 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Labidi-Galy SI, Papp E, Hallberg D, Niknafs N, Adleff V, Noe M, et al. High grade serous ovarian carcinomas originate in the fallopian tube. Nat Commun 2017;8:1093. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Wu RC, Wang P, Lin SF, Zhang M, Song Q, Chu T, et al. Genomic landscape and evolutionary trajectories of ovarian cancer precursor lesions. J Pathol 2019;248:41–50 [DOI] [PMC free article] [PubMed] [Google Scholar]