

Abstract
Natural killer (NK) cells likely play an important role in immunity to malaria, but the effect of repeated malaria on NK cell responses remains unclear. Here, we comprehensively profiled the NK cell response in a cohort of 264 Ugandan children. Repeated malaria exposure was associated with expansion of an atypical, CD56neg population of NK cells that differed transcriptionally, epigenetically, and phenotypically from CD56dim NK cells, including decreased expression of PLZF and the Fc receptor γ chain, increased histone methylation, and increased protein expression of LAG-3, KIR and LILRB1. CD56neg NK cells were highly functional and displayed greater antibody dependent cellular cytotoxicity than CD56dim NK cells. Higher frequencies of CD56neg NK cells were associated with protection against symptomatic malaria and high parasite densities. Importantly, following marked reductions in malaria transmission, frequencies of these cells rapidly declined, suggesting that continuous exposure to Plasmodium falciparum is required to maintain this modified, adaptive-like NK cell subset.
One Sentence Summary:
Repeated malaria in children is followed by expansion of functional CD56neg NK cells associated with protection from symptomatic malaria.
INTRODUCTION
Plasmodium falciparum (Pf) malaria resulted in an estimated 240 million cases and 624,000 deaths in 2020, with rising morbidity observed in several highly endemic countries exacerbated by the COVID-19 pandemic(1). The burden of disease falls mainly on young African children, with 77% of deaths reported in African children <5 years of age. With increasing age and repeated malaria episodes, children eventually gain protection against severe disease, followed by protection against symptomatic illness. This age-and exposure-dependent clinical immunity is comprised of two distinct components: “anti-parasite” immunity, or partial control of blood-stage parasite densities, and “anti-disease” immunity, or the ability to tolerate higher parasite densities without fever(2). However, clinical immunity is short-lived, and the precise determinants driving these distinct components of naturally acquired anti-malarial immunity remain poorly understood(3).
Although malaria-specific antibodies likely play a key role in clinical immunity through both binding inhibition and Fc-dependent antibody effector functions(4–7), the role of the cellular immune response in clinical immunity is less clear. Innate immune cells can recognize and respond to Plasmodium parasites(8, 9), but pathogen-induced production of inflammatory cytokines by these cells has also been implicated in the pathogenesis of symptomatic and severe malaria(8–10). The development of naturally acquired immunity likely involves the careful regulation of the innate immune response to limit the negative consequences of inflammation while partially controlling parasitemia. Indeed, recent studies have suggested that exposure to malaria may modify several innate immune cells (11–15), potentially through transcriptional and epigenetic reprogramming(13, 16, 17). Whether malaria-induced innate immune modulation plays a role in driving and/or maintaining clinical immunity is unknown.
Natural killer (NK) cells have increasingly been recognized as key players in the host immune response to malaria through both direct parasite recognition(18) and antibody-dependent cellular cytotoxicity(7) (19). These cells have traditionally been divided into subsets based on expression of CD56 and CD16, with CD56dimCD16+ NK cells typically comprising the majority of the NK population in peripheral blood, although the complexity and diversity of NK cell subsets has only recently been interrogated using single cell approaches(20–22). A subset of CD56dim NK cells with low expression of the transcription factor promyelocytic leukemia zinc finger (PLZF) and the signaling Fc receptor γ-chain (FcRγ) were found to have enhanced antibody-dependent cellular cytotoxicity against Pf-infected red blood cells and were associated with resistance to symptomatic malaria in Malian children(23). It remains unclear whether repeated malaria drives expansion of this “adaptive-like” NK cell subset, or whether these cells are maintained stably in the absence of continuous exposure to malaria parasites. Some reports have also described a subset of CD56neg CD16+ NK cells that are expanded in the context of chronic viral infections such as HIV(24), HCV(25), as well as among patients with acute myelogenous lymphoma(26) and malaria-exposed African children with EBV-related Burkitt’s lymphoma(27). Given a reduced capacity of these cells to produce cytokines in response to stimulation, some suggest these cells may represent a dysfunctional and/or exhausted phenotype(26, 28). Whether CD56neg NK cells expand following repeated malaria, have functional capabilities, and/or play a role in clinical immunity to malaria, is unknown.
In this study, we characterized the NK cell response among 264 children followed in the East African International Centers of Excellence in Malaria Research (ICEMR) cohorts in Eastern Uganda. We utilized multiparameter flow cytometry, cellular indexing of transcriptomes and epitopes by sequencing (CITE-Seq), epigenetic profiling using cytometry by time of flight (EpiTOF), and assay for transposase-accessible chromatin using sequencing (ATAC-Seq), along with functional assays, to comprehensively profile NK cell responses. We demonstrate that repeated malaria exposure is associated with expansion of highly functional CD56neg NK cells that strongly correlate with acquisition of clinical immunity to malaria. However, in the absence of continuous malaria exposure, frequencies and function of these cells rapidly decline.
RESULTS
Increasing exposure to malaria is associated with higher frequencies of CD3− CD7+ CD56neg NK cells
We analyzed circulating NK cell populations from PBMC collected in 2013 among 45 children ages 3–7 living in two districts in Eastern Uganda with very different malaria transmission intensities: Jinja, with an estimated annual entomological inoculation rate (aEIR) of 2.8 infectious bites per person per year, and Tororo, with an aEIR of 310 infectious bites per person year(29). Nearly all children were HCMV-positive at the time of sampling (Table S1). CD3−, CD14−, CD19−, CD7+ NK cells were classified by the expression of CD56 and CD16 into three subsets: CD56bright (CD56+ CD16−), CD56dim (CD56dim CD16+) and CD56neg (CD56neg CD16+) (Fig 1A, Fig S1). Children living in highly endemic Tororo district had significantly lower frequencies of CD56dim NK cells (P=5.6e-05) and higher frequencies of CD56neg NK cells (P=1.3e-05), than age-matched Jinja children (Fig 1B, Table S1). Percentages of CD56neg NK cells positively correlated with household-level aEIR, a direct measurement of exposure to Pf infective mosquitoes (P=2.5e-06, Fig 1C). We correlated frequencies of NK cell subsets with age, a surrogate for cumulative malaria exposure in high transmission settings, among a larger Tororo cohort of 242 children ages 6 months to 10 years (Table S2). We found that increasing age was associated with increasing frequencies of CD56neg NK cells (P=1e-06), and decreasing frequencies of CD56dim NK cells (P=1e-04, Fig 1D). Together these data suggest that CD56neg NK cells are increased in children repeatedly exposed to malaria.
Figure 1: Ugandan children living in high transmission settings have high frequencies of atypical CD56neg NK cells.

(A) Representative flow cytometry plots of CD3− CD14− CD19− CD7+ NK cells from three children, as defined by CD56 and CD16 (Fig S1 for full gating). (B) Percentages of CD56dim and CD56neg NK cells from children ages 3–7 years in Tororo (n=35) and Jinja (n=10). Household annual Entomological Inoculation Rate (aEIR) is an estimate of the number of infective bites received in the year prior to sampling across each cohort, as measured via CDC light traps (see methods). (C) Correlation between CD56neg NK cells and measured household aEIR among children assayed in (B). (D) Correlation between NK cell frequencies and age among 242 Tororo children ages 0.6 to 11 years. P-values shown above box plots are calculated using Wilcoxon signed rank test, while the rho (ρ) and p-values associated with the scatterplots are calculated using Spearman’s correlation. Shaded areas represent 95% confidence interval of best fit regression line. All samples were collected in 2013, see Table S1 and S2 for patient characteristics.
Single cell RNA sequencing reveals that CD56neg NK cells have transcriptional features of both “adaptive” and “exhausted” NK cells
To further characterize NK cells at the single cell level, we used Cellular Indexing of Transcriptomes and Epitopes by Sequencing (CITE-Seq), a method of single cell RNA sequencing that simultaneously assesses DNA-barcoded antibodies against cell surface proteins. Peripheral blood mononuclear cells from 10 independent Tororo participant samples were analyzed (n=60,381 total cells, Table S3). Unsupervised clustering of RNA transcripts using uniform manifold approximation and projection (UMAP) revealed that NK cells cluster near T cells, with CD8+ T cells as the closest neighbors (Fig 2A). Using cell surface protein tags for CD56 and CD16, we identified 3 distinct cell subsets within the CD19−, CD3−, CD14− CD7+ NK cell population (Fig 2B) and visualized these cell populations on the RNA UMAP projection. CD56dim and CD56neg NK cells were both identified in the same NK cell RNA cluster, suggesting transcriptional similarities between these subsets (Fig 2C, Fig S2A). However, we also identified gene transcripts differently enriched in each subset (Fig 2D). In comparison to CD56dim NK cells, CD56neg cells had significantly lower expression of ZBTB16 (P=0.001), which encodes the transcription factor PLZF, and FCER1G (P=2.2e-25), which encodes the FcRγ signaling receptor (Fig 2E top row). Conversely, CD56neg NK cells had higher expression of BC11B (P=2.3e-06), a transcription factor shown to repress PLZF(30) (Fig 2E). Expression of PLZF and FcRγ are decreased in adaptive-like NK cells with enhanced capacity for antibody-dependent cellular cytotoxicity (22, 23). CD56neg NK cells had lower expression of genes encoding the cell surface markers KLRB1 (CD161, P=5.7e-15) and KLRF1 (NKp80, P=2.8e-22), and higher expression of CD3G (P=1.6e-11), than CD56dim NK cells, also consistent with an adaptive-like NK cell phenotype(22, 31). Interestingly, CD56neg NK cells had lower expression of IL-18RAP (P=6.1e-07), which encodes the IL-18 receptor accessory protein and mediates IL-18-dependent signal transduction and downstream NF-kB activation and IFNg induction(32). CD56neg NK cells also had higher expression of LAG3 (P=4.2e-07), a checkpoint inhibitor which is induced by type I IFN(33) and chronic in vitro stimulation(34), and is a negative regulator of cytokine production in mature NK cells, suggestive of an exhausted phenotype(34, 35). Expression of killer cell immunoglobulin-like receptor (KIR) genes were similar between CD56dim and CD56neg NK cells, consistent with another published report(36), although KIR genes were not well represented in the dataset (Fig S2B). Together, these data suggest that CD56neg NK cells from malaria-exposed Ugandan children have transcriptional features suggestive of both adaptive-like and exhausted NK cells.
Figure 2: Single-cell RNA Sequencing of NK cells reveal transcriptional similarities and differences between CD56dim and CD56neg NK cells.
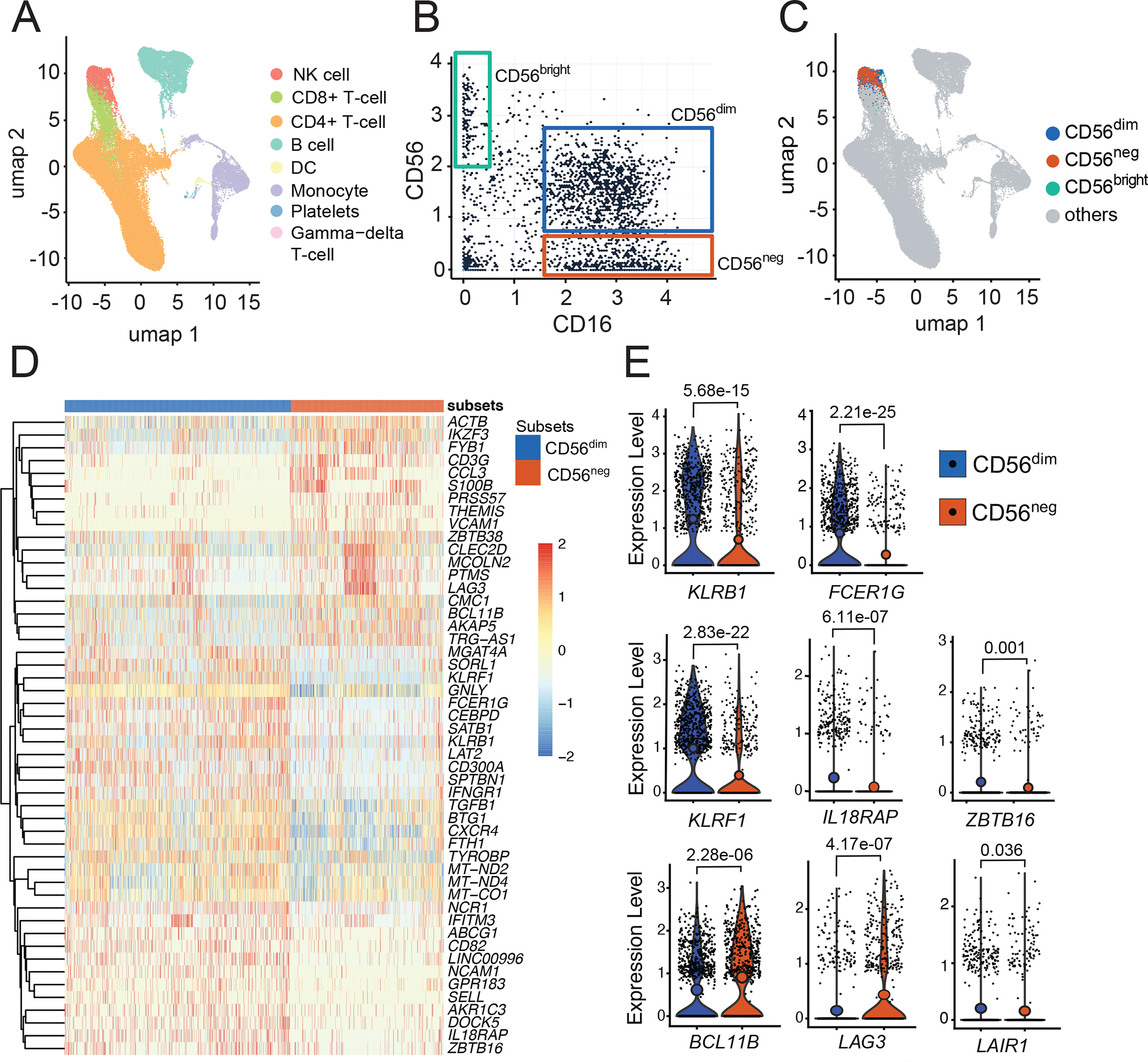
(A) Gene transcripts of n=60381 PBMCs were clustered using Leiden algorithm and visualized in two dimensions with UMAP. (B) Gating of NK cell subsets as defined by surface marker expression of CD16 (x-axis) and CD56 (y-axis). (C) NK cells subsets classified by (B) superimposed with UMAP gene transcript data. CD56dim transcripts are in blue, CD56neg in red, CD56bright in green and non-NK cell transcripts are in grey. (D) Heatmap showing top 50 genes differentially expressed by CD56dim and CD56neg NK cells. (E) Representative violin plots comparing gene transcripts between CD56dim and CD56neg NK cells. P values obtained from likelihood ratio tests as implemented in the Seurat R package. Samples are from 10 individuals, see table S3 for patient characteristics.
CD56neg NK cells from Ugandan children express higher levels of LILRB1 and KIRs and differ epigenetically from CD56dim NK cells
We next compared protein expression between CD56dim and CD56neg subsets from samples collected from 35 Tororo children (Table S4). We initially focused on markers that have been implicated in NK cell differentiation(37) and/or adaptive NK cells(38) using flow cytometry, including cell surface receptors LILRB1 (CD85J), NKG2A, NKG2C, CD57 and NKG2C as well as intracellular signaling receptor FcRy. Consistent with our transcriptomic data, CD56neg NK cells in Tororo children had significantly reduced expression of FcRγ compared with CD56dim NK cells (P=2.9e-05). In addition, CD56neg NK cells also had reduced protein expression of NKG2A (P=4.5e-06), CD57 (P=0.0024), and NKG2C (P=2.2e-04), but higher levels of LILRB1 (P=2.4e-07), compared with CD56dim NK cells (Fig 3A). We examined the combination of these markers, and found that majority of the CD56neg NK cells are LILRB1+, FcRγ−, NKG2A− and CD57−, while CD56dim NK cells are most likely to be FcRγ+ and NKG2A+, but LILRB1− and CD57− (Fig 3B, Figure S3). This phenotypic description of CD56neg NK cells is consistent with similar cell populations within the context of chronic viral infections such as HIV(24). Cell surface expression of KIR2DL1 were similar between CD56neg cells and CD56dim cells, consistent with gene expression data. However, flow cytometric analysis revealed greater cell surface expression of the KIRs KIR3DL1 (P=1.5e-06) and KIR2DL2/L3/S2 (P=0.05) in CD56neg NK cells compared to CD56dim cells (Fig 3C), consistent with other reports which have observed increased KIR protein expression on CD56neg NK cells(24). Additionally, we confirmed other differences seen in our gene expression data, including greater LAG3 expression (P=0.0078) and lower granulysin (P=0.049), among CD56neg compared with CD56dim cells (Fig 3D). Epigenetic differences in NK cells, including both genome-wide DNA hypo- and hypermethylation, have been previously shown to differentiate NK cell subsets, including adaptive NK cells(34, 38). We compared global histone modifications at the single cell level between CD56dim and CD56neg NK cells using epigenetics by time of flight (EpiTOF), as these modifications have previously been shown to differentiate CD56bright and CD56dim NK cells(39). Compared with CD56dim NK cells, CD56neg cells had greater abundance of a number of methylation marks, including di- and trimethylation of histone H3 lysine 4 (H3K4me2 and H3K4me3, (Fig 3E)), marks which are associated with gene expression in mature, aging cells(40). Taken together these data suggest that CD56neg NK cells in malaria-exposed Ugandan children have phenotypic and epigenetic features of mature, adaptive-like NK cells that differentiate them from CD56dim cells.
Figure 3: Cellular phenotyping reveals CD56neg NK cells to have a mature phenotype.

NK cells were gated for LILRB1, FcRγ, NKG2A, CD57 and NKG2C. (A) Frequencies of cells are shown in violin plots. (B) Proportions of cells expressing all four possible marker combinations are shown in dot plots. (C & D) Percentages of NK cells expressing KIRs, granulysin and LAG3. (E) Heatmap comparing distinct histone modifications between CD56dim and CD56neg NK cells as measured by EpiTOF from 36 participant samples. P-values shown above violin and dot plots are calculated using Wilcoxon matched-pairs signed rank test. See Table S4 for number of samples and patient characteristics, Fig S1 and S3 for gating strategies, and Data File S2 for raw data.
CD56neg NK cells display greater antibody-dependent cellular cytotoxicity (ADCC) than CD56dim NK cells
Having demonstrated that CD56neg NK cells were transcriptionally, phenotypically, and epigenetically distinct from CD56dim NK cells, we next compared their functional abilities. Following a short in vitro stimulation with Pf infected red blood cells (iRBCs), neither CD56dim nor CD56neg NK cells produced significant amounts of IFNγ (Fig 4A). However, following IL-12/IL-15/IL-18 stimulation, CD56neg NK cells produced significantly less IFNγ in comparison to CD56dim cells (P=6.6e-09). This result was expected given CITE-Seq data showing decreased expression of IL-18RAP on CD56neg compared to CD56dim NK cells, and consistent with prior reports(28).
Figure 4: CD56neg NK cells have an enhanced ability for antibody-dependent cellular cytotoxicity.

(A) PBMCs were stimulated with iRBCs or cytokines: IL-12 (2ng/ml), IL-15 (10ng/ml) and IL-18 (0.25ug/ml) for 4 hours, after which intracellular IFNγ was measured. (B & C) PBMCs were stimulated with either irbcs alone or opsonized with serum in a ratio of 2 PBMCs to 1 iRBC. (B) IFNγ was measured intracellularly. (C) NK cell degranulation was measured by CD107a expression. (D) Isolated NK cells were stimulated with iRBCs opsonized by either US or Ugandan serum and degranulation measured. (E) Heatplot showing percentages of NK cells expressing cell surface markers degranulating in response to iRBCs opsonized by Ugandan serum. (F) Association between frequencies of CD56neg NK cells and percentage of dead opsonized p815 cells. (G) Isolated NK cells and NK-Depleted PBMCs were stimulated with opsonized p815, after which the percentage of dead cells was measured by 7-AAD staining. P-values for box plots are calculated using Wilcoxon matched-pairs signed rank test, and rho (ρ) and p-value associated with the scatterplot is calculated using Spearman’s correlation. See Table S4 for patient characteristics, and Data File S2 for raw data.
We next tested whether there were differences between CD56dim or CD56neg NK cells in response to antibody-opsonized target cells. We stimulated PBMC obtained from Tororo children with iRBCs alone, iRBC incubated with plasma pooled from either malaria-naïve adults or immune Ugandan adults, or, as a positive control, iRBC incubated with anti-RBC antibodies. In response to iRBC incubated with plasma from immune Ugandan adults, CD56neg NK cells produced significantly more IFNγ (P=4.9e-05) and had greater degranulation (as measured by CD107a, P=2.4e-07) in comparison with CD56dim NK cells, suggesting a heightened capacity to perform ADCC (Fig 4B and C). Following stimulation with iRBCs alone, iRBC treated with plasma from malaria-naïve adults, or uninfected RBCs incubated with plasma from immune Ugandan adults, neither CD56dim nor CD56neg NK produced IFNγ or degranulated (Fig 4B and C, Fig S4), indicating the observed responses were truly antibody-dependent.
To determine whether malaria-specific NK cell degranulation was independent of other cells, we isolated NK cells from 15 Tororo children by magnetic purification and performed the same ADCC assay. Again, isolated NK cells degranulated more when stimulated with iRBC incubated with pooled plasma from Ugandan adults vs. iRBC incubated with US control plasma (P=2.3e-05, Fig 4D; Fig S1). The majority of degranulating, CD107a+ NK cells following stimulation with Ugandan plasma opsonized parasites were FcRγ−, LILRB1+, NKG2A− and CD57− (Fig 4E), which represent the major fraction of circulating CD56neg NK cells (Fig 3B). Other NK cell populations that degranulated also lacked expression of FcRγ (Fig 4E), including those that were CD56dim, consistent with other studies showing that FcRγ− CD56dim NK cells show enhanced degranulation to antibody-coated targets(23, 41).
As an alternative measure of ADCC, we assessed the ability of NK cells to kill antibody-coated p815 cells (Fig S5a). Following stimulation with antibody-coated p815 cells, we observed a strong positive correlation between frequencies of CD56neg NK cells and p815 killing (P=0.00065, Fig 4F, Fig S5b). We further isolated NK cells and show that these cells are the main cell type that kill antibody opsonized p815 compared to PBMCs that were depleted of NK cells (Fig 4G). Taken together, these results demonstrate that malaria-induced CD56neg NK cells have enhanced ADCC capacity compared to CD56dim NK cells.
Higher frequencies of CD56neg NK cells are associated with protection against subsequent parasitemia and symptoms of malaria
To determine the clinical relevance of these observations, we assessed associations between NK cell percentages and a) parasite densities in the year following the cellular measurement (anti-parasite immunity), and b) the probability of symptoms given Plasmodium infection in the subsequent year (anti-disease immunity). Higher percentages of CD56neg NK cells were associated with significantly lower parasite densities in the year following analysis (P=1e-10, Fig 5A). In contrast, higher percentages of CD56dim NK cells were associated with significantly higher parasite densities (P=1e-08, Fig 5A). When considering the odds of symptoms given Plasmodium infection, higher percentages of CD56neg NK cells were also associated with a lower odds of being symptomatic if infected (Fig 5B, Fig S6). After adjustment for age and household aEIR, children in the highest tertile of CD56neg NK cells had a 71% lower odds of being symptomatic if infected in the subsequent year compared with children in the lowest tertile (adjusted odds ratio (aOR) 0.29, 95% confidence interval 0.13–0.63, P=0.002, Fig 5B). In contrast, higher percentages of CD56dim NK cells were associated with higher odds of symptoms given infection (aOR 3.88 comparing highest vs. lowest tertile of CD56dim NK cells, 95% confidence interval 1.9–8.1, P<0.001, Fig 5B, Fig S6). Together, these data suggest that higher frequencies of CD56neg NK cells, and inversion of the circulating CD56dim to CD56neg ratio, are strongly associated with both anti-parasite and anti-disease immunity to malaria in Ugandan children.
Figure 5: CD56neg NK cells are associated with clinical malaria protection.
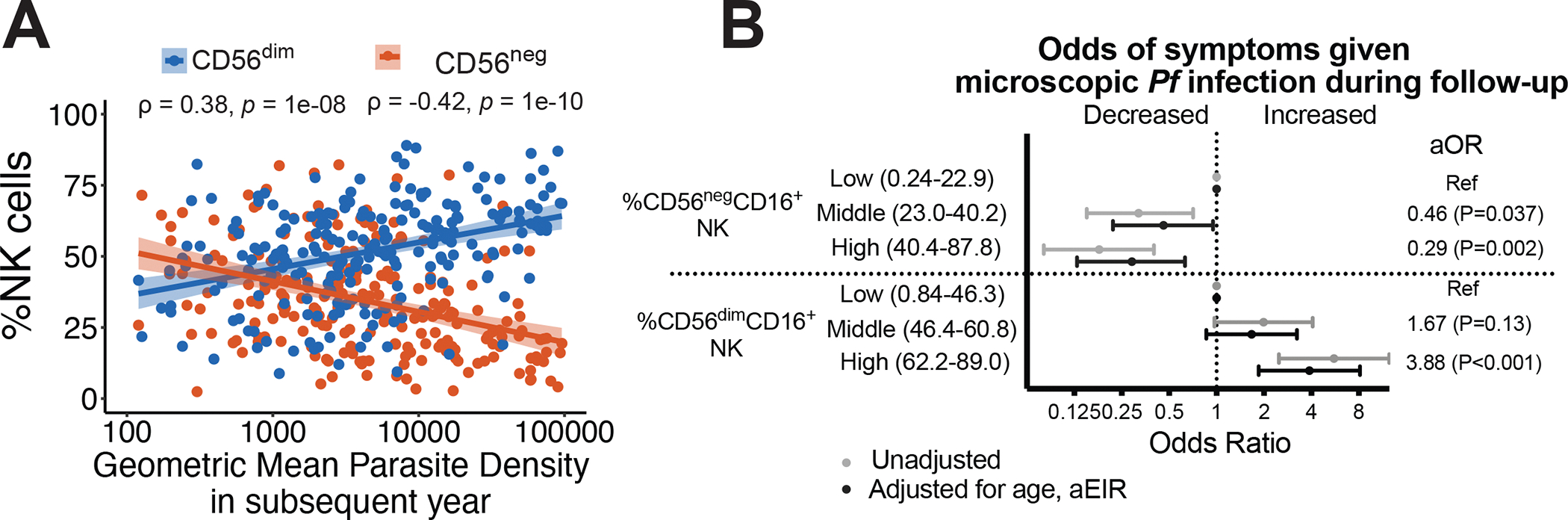
Associations between CD56dim and CD56neg percentages and (A) geometric mean parasite densities in the subsequent year. Rho (ρ) and p-values associated with the scatterplot are calculated using Spearman’s correlation. Shaded areas represent 95% confidence interval of best fit regression line. (B) Associations between CD56dim and CD56neg percentages and probability of symptoms given Pf parasitemia in the subsequent year (see Fig S5 for smoothed relationships). Multilevel mixed effects linear models used to calculate odds of symptoms given microscopic Pf infection (B). Multivariate models adjusted for age and aEIR, and account for clustering by individual and household. Categories represent tertiles of NK response (n=80,81,81 per tertile). aOR: adjusted odds ratio. See Table S2 for patient characteristics.
CD56neg NK cells decline in the setting of reduced malaria transmission
Having shown that functional CD56neg NK cells are increased in children repeatedly exposed to malaria, we wanted to determine whether malaria exposure was required in order to maintain their phenotype and function. We took advantage of a natural experiment in our Tororo cohort where children were followed both before and after initiation of an indoor residual spraying of insecticide (IRS) campaign(42). Prior to initiation of IRS in December 2014, malaria transmission was high and stable in Tororo(29). Following initiation of IRS, malaria transmission, parasite prevalence and malaria incidence markedly declined, and by 2018, the burden of malaria was reduced to very low transmission levels (Fig 6A)(42). We assessed the stability of NK cell phenotypes over time by performing longitudinal assessments in 15 Tororo children before (Aug-Nov 2013) and after (May-Oct 2015) the first round of IRS (Fig S7A), and compared these with a group of 12 similarly aged children sampled in 2018, 4 years after initiation of IRS when transmission was very low. Household aEIR among sampled participants declined from 277 in 2013 to 34 in 2015 to 1in 2018 (Table S5). The percentage of CD56neg NK cells significantly decreased from the pre- to post-IRS period (median 55.2% in 2013 vs. 44.6% in 2015, P=0.0021, Fig 6B), with a concurrent increase of CD56dim cells (Figs 6B and C), and these percentages correlated with household aEIR (Fig S7B). Percentages of CD56neg NK cells were significantly lower in similarly aged children sampled in 2018 (median 10.2% CD56neg NK cells, P=2e-05 comparing 2018 vs. 2015), when transmission and malaria prevalence were markedly lower (Figs 6B and C). Phenotypically, CD56dim NK cells did not significantly change with decreasing malaria transmission (Fig 6D). However, among CD56neg NK populations, we observed a significant decrease of the LILRB1+, FcRγ−, NKG2A+/−, CD57− NK cells that were predominant in 2013 compared with 2018 (P<0.0001), as well as an increase in the LILRB1−, FcRγ+, NKG2A+ CD57− NK cells (P=0.0001, Fig 6D).
Figure 6: Decline of CD56neg NK cells following reductions in malaria transmission.
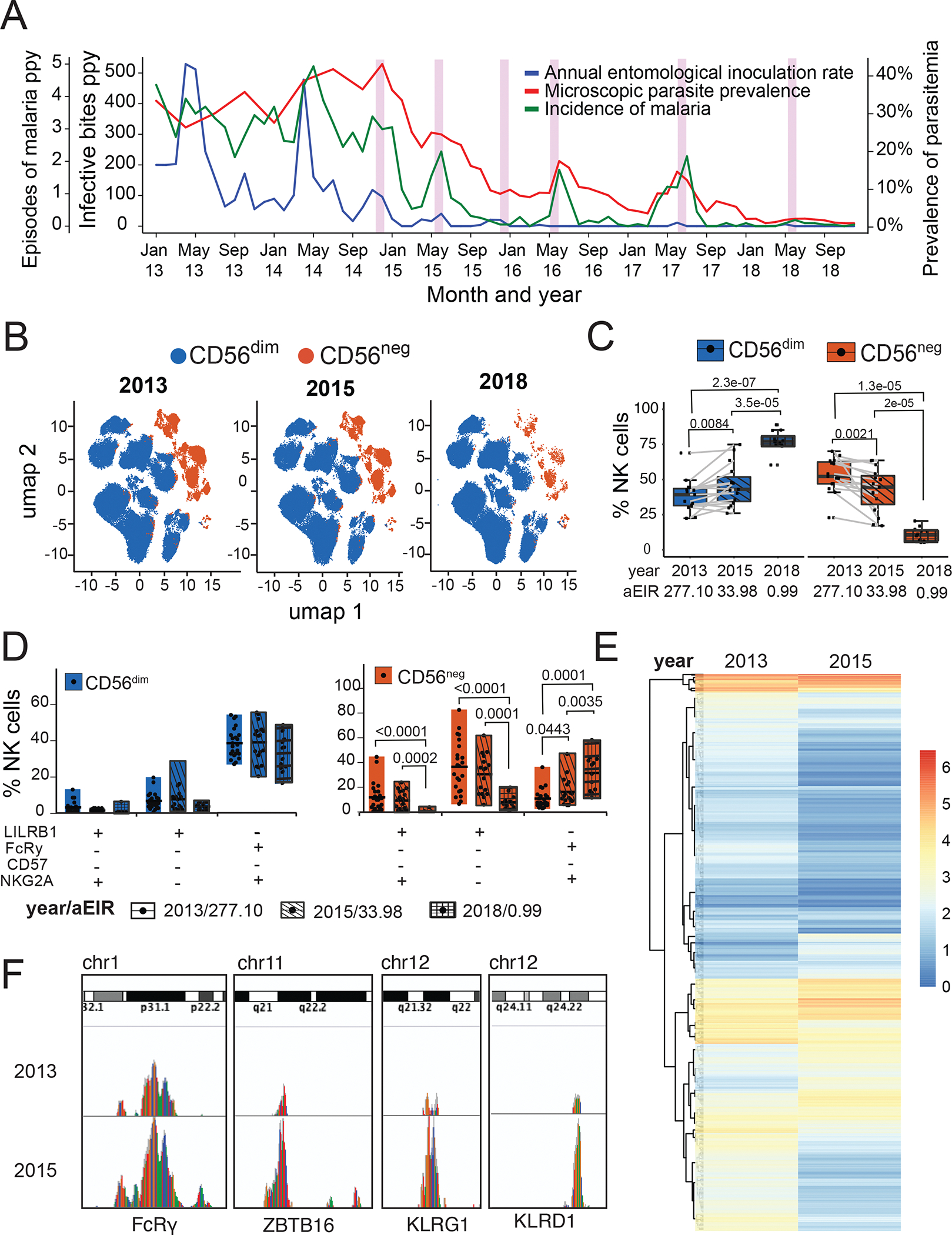
(A) Measures of malaria burden in Tororo from 2013 to 2018: Annual entomological inoculation rate (aEIR, blue, middle y-axis), malaria incidence (green, left y-axis), and parasitemia (red, right y-axis). Pink bars show rounds of indoor residual spraying with insecticides. (B) Unsupervised clustering of CD56dim (blue) and CD56neg (red) NK cells within individuals sampled in 2013 and 2015 (paired) and 2018 (unpaired, age-matched), visualized by UMAP projections. (C) Percentages of CD56dim and CD56neg NK cells over time as aEIR decreases. (D) Proportion of CD56dim (blue) and CD56neg (red) NK cells expressing all possible marker combinations of CD85j, FcRγ, NKG2A and CD57 plotted over time as aEIR decreases. Clear box indicates 2013, diagonal lined box indicates 2015, and hatched box indicates 2018. (E) ATAC-Seq heatmap showing differential accessibility of genes from FACS-sorted CD3−CD14−CD19−CD7+ NK cells obtained from a Tororo child sampled in 2013 and 2015 (F) ATAC-Seq genome tracks showing chromatin accessibility (peaks) at the FcRy, ZBTB16, KLRG1 and KLRD1 loci for 2013 and 2015 paired samples. P-values for box plots are calculated using Wilcoxon matched-pairs signed rank test. See Table S5 for patient characteristics, and Data File S2 for raw data.
Interestingly, the CD56neg populations that decreased were the two most predominant populations that degranulated when stimulated with antibody-opsonized parasites. To determine whether reductions in malaria transmission also altered epigenetic stability of NK cells, we measured chromatin accessibility of sorted NK cells collected in 2013 and 2015 using ATAC-Seq. We found that reductions in malaria transmission were associated with global changes in chromatin accessibility (Fig 6E). Specifically, accessibility to the promoter regions of the FcRγ, ZBTB16, KLRG1 and KLRD1 increased as malaria transmission declined (Fig 6F). These data suggest that loss of malaria exposure leads to changes in NK cell chromatin accessibility involving genes implicated in NK cell function.
Antibody-mediated cellular cytotoxicity function decline in CD56neg NK cell after decrease in malaria transmission
Finally, since we observed changes phenotypically and epigenetically in NK cells following declines in malaria transmission, we evaluated whether there were concomitant changes in NK cell function. We observed a significant decrease of total NK cell degranulation in response to Ugandan plasma opsonized iRBCs with decreasing malaria transmission (P=0.0014 comparing 2018 vs. 2013, Fig 7A, Fig S7B), with a stable response to a positive control, anti-RBC opsonized iRBC (Fig 7A). Looking more closely at cellular phenotypic subsets, we saw that CD56neg NK cells, but not CD56dim NK cells, exhibited significant reduction of degranulation in response to Ugandan plasma opsonized iRBCs with decreasing malaria transmission (P=8.5e-05 in CD56neg NK cells vs. P=0.32 in CD56dim NK comparing 2018 vs. 2013, Fig 7B). Surprisingly, we observed a similar decline in CD56neg, but not CD56dim, degranulation in response to anti-RBC opsonized iRBC (Fig 7C, Fig S7C). Furthermore, we observed a reduced ability to kill antibody-covered p815 cells post-IRS (Fig 7D). Collectively, these data are consistent with the hypothesis that ongoing malaria transmission is required to maintain both the frequency and effector function of CD56neg NK cells. Furthermore, the changing population of CD56neg NK cells in the setting of declining malaria transmission resulted in reduced ADCC activity of NK cells, including both degranulation and killing of target cells.
Figure 7: Reduction of ADCC by CD56neg NK cells following decline of malaria transmission.

(A) PBMCs were stimulated either with iRBCs alone or opsonized with serum. Degranulation was measured through the different years with decreasing aEIR. (B & C) PBMCs stimulated with iRBCs opsonized with Ugandan serum analyzed by NK cell subset. Degranulation by (B) CD56dim and (C) CD56neg NK cells was measured over time. (D) Killing of opsonized p815 cells by PBMCs collected from 2013 and 2015. Clear box indicates 2013 with an aEIR of 258.3, diagonal lined box indicates 2015 with an EIR of 25.8 and hatched box indicates 2018 with an aEIR of 0.43. P-values for box plots are calculated using Wilcoxon matched-pairs signed rank test. See Table S5 for number of samples and patient characteristics, and Data File S2 for raw data.
Discussion
In this study, we characterized the NK cell response among children followed longitudinally across a range of malaria transmission intensities in Uganda. Children repeatedly exposed to malaria had increased frequencies of CD56neg NK cells that resembled CD56dim NK cells transcriptionally, but with several key differences, including decreased expression of PLZF and FcRγ, global histone modifications, and increased protein expression of KIR3DL1 and KIR2DL2/L3/S2, LILRB1, and LAG-3. These CD56neg NK cells were highly functional, displaying greater malaria parasite-induced ADCC than CD56dim NK cells, and higher percentages of these cells were associated with both lower parasite densities and a lower probability of symptomatic disease upon subsequent Plasmodium infections. Importantly, following marked reductions in malaria transmission due to indoor residual spraying of insecticides, frequencies – and function - of CD56neg NK cells rapidly declined, suggesting that continuous exposure to malaria is required to maintain this modified, adaptive-like NK cell subset.
Although CD56 is the prototypical identifier of circulating human NK cells(43), our data suggest that CD56neg NK cells make up a significant fraction – and in many cases the majority – of circulating NK cells among children living in a high malaria transmission setting. CD56neg NK cells expressed CD7, which has previously been shown to differentiate NK cells from monocyte/DC like myeloid cells(44, 45), and transcriptional profiling of PBMC by CITE-Seq confirmed that CD56neg NK cells cluster similarly with CD56dim NK cells. Prior literature has suggested that CD56neg NK cells are dysfunctional and likely represent an exhausted phenotype, given that they have a reduced ability to produce cytokines in response to stimulation(26, 28). We similarly observed that CD56neg cells in malaria-exposed children produce less IFN-γ in response to IL-12/IL-15/IL-18 stimulation than CD56dim NK cells. CD56neg NK cells expressed lower levels of IL-18RAP, which likely limits IL-18-mediated cellular activation(32), and higher levels of LAG-3, which likely negatively regulates cytokine production in these cells similar to its role in other NK cells(34). Intriguingly, we also observed that CD56neg cells express high levels of the inhibitory receptor LILRB1. Recent studies have illuminated that a subset of Pf RIFINs, a family of variant surface proteins expressed on the surface of infected red blood cells, are able to directly bind LILRB1(46, 47). K562 cells transfected with a LILRB1 binding RIFIN were more resistant to NK cell-mediated lysis than those transfected with a RIFIN unable to bind LILRB1, suggesting that Pf RIFIN expression may play a role in immune evasion43. Whether CD56neg NK cells bind Pf-RIFIN, and whether this regulates their subsequent activation, remains to be determined.
Along with a reduced response to cytokine stimulation, higher frequencies of CD56neg NK cells were associated with a higher probability of being asymptomatic when subsequently infected with malaria parasites, independent of age and household level transmission intensity. In contrast, CD56dim NK cells were much more inflammatory in vitro, and higher frequencies of these cells were associated with a higher probability of symptoms upon infection. Given the age dependent inversion of the circulating CD56dim to CD56neg ratio among children living in a high malaria transmission setting, these data are consistent with the hypothesis that epigenetic and transcriptional regulation of the NK cell response is important in limiting the negative consequences of inflammation due to repeated blood-stage malaria.
Despite this reduced response to cytokine stimulation, we observed that CD56neg NK cells potently degranulate in response to opsonized iRBC and were able to mediate killing of opsonized p815 cells, consistent with a role for these cells having a specialized capacity to perform ADCC. Higher percentages of these cells were also associated with lower parasite densities upon subsequent infection, suggesting an important role of these cells in vivo in mediating anti-parasite immunity. Our data are similar to recently described CD56dim adaptive NK cells, also characterized by low PLZF and FcRγ expression, which were found to have enhanced ADCC against Pf infected red blood cells and were associated with protection against incident malaria(23). That study identified NK cells based on CD56 expression and therefore excluded CD56neg NK cells. Given their similar phenotype and function to the CD56neg cells we describe, we suspect significant overlap between adaptive, PLZFlow/FcRγlow CD56dim and CD56neg NK cell subsets. Several recent reports have also highlighted specialized antibody-dependent functions of other innate immune cells, including ADCC(14) and antibody dependent phagocytosis (ADP)(48) from CD16-expressing γδT cells, as well as ADP from monocytes(5, 49) and neutrophils(50). Together, these data suggest that multiple innate immune subsets may be involved in antibody-dependent protection against parasitemia. However, the relative contributions from each subset towards protection – and whether repeated malaria drives epigenetic, transcriptional, and functional remodeling of these cells - remain to be determined.
Importantly, we show that higher percentages of CD56neg NK cells were observed in children with greater Pf exposure, and that, following a marked reduction in malaria transmission, both the frequency and function of these cells declines rapidly. Together, these data strongly suggest that continuous exposure to malaria is required to maintain circulating frequencies of CD56neg NK cells. It remains to be determined how malaria drives expansion of these cells, and whether these cells represent a clonal, “terminally differentiated” population of NK cells analogous to other effector populations that have been described(51) vs. expansion of a unique NK cell subset. In contrast to CMV infection, where virally encoded peptides induce expansion of CMV-specific, NKG2C-expressing adaptive NK cells(52), our in vitro data do not suggest that CD56neg NK cells respond directly to Pf stimulation. Nonetheless, their heightened malaria-specific and non-specific (e.g., p815 cells) antibody-dependent functionality suggests that these cells play a critical role in innate/adaptive immune bridging.
There were limitations in this study. Not all assays could be performed on all subjects due to sample availability and cost. However, this remains a comprehensive study, leveraging the infrastructure of the East African ICEMR with longitudinal sampling in multiple subjects. Despite their phenotypic, transcriptional, and functional similarity to other NK cells, the ontology of these cells remains unclear, and it is possible that CD56neg NK cells may be better classified as an alternative innate lymphoid cell (ILC) subset(53). Additional functional characterization of these cells is ongoing. Epidemiological analyses are limited to associations with measure of clinical immunity conditional on participants being infected (i.e. microscopic parasite densities or symptoms if infected). This study is unable to determine whether NK cell subsets are associated developing a blood stage infection in the first place, nor whether these subsets are associated with protection against sub-microscopic parasitemia. Although our data strongly suggest that repeated exposure to malaria results in increased frequencies of CD56neg NK cells, we cannot exclude the possibility that other infections may drive expansion of these cells in this setting. None of the children in this cohort were HIV-infected, and nearly all children were CMV seropositive at the time of sampling, precluding comparisons between CMV seropositive and CMV seronegative children. Although subjects were not tested for EBV, the prevalence of EBV in similar East African settings has been noted to be very high early in life(54). Furthermore, associations between aEIR and NK cell phenotypes, and declining percentages of CD56neg NK cells in children following interruptions in malaria transmission, strongly implicate the need for repeated malaria exposure to drive higher frequencies of these cells. Absolute cell counts of NK cell subsets were not available for this cohort. While it is recognized that absolute numbers of peripheral blood lymphocytes and major lymphoid subsets change over the first few years of life, it is unknown whether these changes in cell concentrations are linked to function and/or protection from disease. Finally, given the low number of infections following IRS initiation, we were not able to determine whether this immunologic change was associated with a higher propensity for either symptomatic and/or high parasite density infections.
By showing that adaptive-like, functional CD56neg NK cells are associated with anti-disease and anti-parasite protection against malaria, we have identified a clear role of CD56neg NK cells in malaria-endemic settings. Understanding factors that drive programming of this unique NK cell subset will help guide therapeutic translation, including enhancing vaccine-elicited protection. Our findings that these cells are lost in the absence of continuous malaria exposure has important implications regarding maintenance of clinical immunity following interruptions in malaria transmission. It is generally presumed that clinical immunity to malaria is short-lived and wanes rapidly(3), due in part to selective and rapid loss of malaria-specific antibodies(55). Understanding whether the loss of this cellular immune response plays a role in the resurgence and/or delay of malaria following withdrawal of effective malaria control interventions remains to be determined(56). Finally, as higher frequencies of CD56neg NK cells have been implicated in the pathogenesis of malignancies including Burkitt’s Lymphoma(27) and AML(26), understanding the impact of these malaria-induced NK cell changes on immune surveillance of malignancy, and responses to other infectious diseases and/or vaccinations, remains critically important.
MATERIALS AND METHODS
Study Design
Samples were obtained from children followed longitudinally through the East African International Centres of Excellence in Malaria Research cohorts in Tororo and Jinja districts(29). In these settings, malaria transmission is year-round, with two seasonal peaks and varied transmission intensities(29). In Tororo District, before December 2014, malaria control was limited to the distribution of LLINs and case management. IRS with the carbamate bendiocarb was first initiated in December 2014, with additional rounds administered in June 2015 and November 2015. In June 2016, IRS was administered with the organophosphate pirimiphos-methyl (Actellic), with repeated rounds in June 2017 and June 2018. IRS was not initiated in Jinja District.
In both sites, all children from 100 randomly selected households were enrolled into the cohort study if they met the following eligibility criteria: 1) documented age between 6 months and less than 10 years, 2) full-time resident of the household, 3) no intention to move out of the subcounty for the next 2 years, 4) agreement to come to a dedicated study clinic located within the subcounty for any febrile illness, 5) agreement to avoid antimalarial medications administered outside the study, and 6) provision of written informed consent from parent or guardian(29). At enrollment, study participants and their parents/guardians were given a LLIN and underwent a standardized evaluation, including evaluation for any chronic medical conditions. No children were found to be HIV-infected.
Children were followed for all care at dedicated study clinics. Those who presented with a fever (tympanic temperature >38.0 °C) or history of fever in the previous 24 hours had blood obtained by finger prick for a thick smear. If the thick smear was positive for Plasmodium parasites, the patient was diagnosed with malaria regardless of parasite density, and was treated with artemether-lumefantrine. Routine assessments, including blood smears to detect for microscopic Pf parasitemia, were performed monthly. Participants with asymptomatic parasitemia were not treated with antimalarial drugs in accordance with local guidelines. Household annualized entomologic inoculation rates (aEIR) in the year prior to sampling were estimated from monthly Centers for Disease Control and Prevention (CDC) light trap mosquito collections. The aEIR was the product of the annual human biting rate (total number of female Anopheles mosquitoes captured/number of house nights of collection X 365 days/year) and the sporozoite rate (number of mosquitoes testing positive for Pf sporozoites/number of mosquitoes tested). CMV serostatus was measured using Human Anti-cytomegalovirus IgG ELISA Kit (CMV) ab108724. Regarding KIR genotypes, KIR2DL1 and 3DL1 are present in >98% of Ugandan ICEMR participants(57).
Written informed consent was obtained from the parent or guardian of all study participants. The study protocols were approved by the Uganda National Council of Science and Technology (HS 1019), the Makerere University School of Medicine Research and Ethics Committee (2011–167), the University of California, San Francisco Committee on Human Research (11–05995), and the Institutional Review Board at Stanford University (41197).
PBMC and Plasma Isolation
At select study visits, 3 to 10 mls of blood were obtained in acid citrate dextrose tubes and/or heparin tubes. Plasma was removed and peripheral blood mononuclear cells (PBMC) isolated by density gradient centrifugation (Ficoll-Histopaque; GE Life Sciences) were counted and cryopreserved in liquid nitrogen prior to shipping to Stanford for downstream analyses. Analysis of cell viability using Guava Viacount (Millipore) consistently demonstrated >90% viability after thaw. Plasma pooled from 12 adults followed in the high transmission Tororo district or from deidentified plasma from adult US donors was used in ADCC assays. Participant characteristics of samples analyzed in each figure are provided in Tables S1–5. Data from participants may be represented in more than one figure or table.
NK cell phenotyping
Thawed cells were rested at 37°C for 5 hours, washed with 1XPBS, and incubated with cell surface antibodies at room temperature for 30 mins. Cells were washed with FACS buffer (1XPBS with 0.5% BSA and 0.5M EDTA), fixed for 15 min (Fix A, Thermo Fisher), and then permeabilized (Perm B, Thermo Fisher) prior to incubation with intracellular antibodies (Table S6) for 20 min at room temperature. Cells were washed by FACS buffer, then resuspended with 1x PBS before analysis on an Attune NXT flow cytometer. Data was analyzed with FlowJo X software (Tree Star).
CITE-Seq
The 10X Chromium platform was used to perform CITE-Seq. All protocols, including sample preparation, library preparation, and instrument and sequencing settings, are available at: https://support.10xgenomics.com/single-cell-gene-expression. Approximately 1,000,000 cells from 10 patient samples were stained with Human TruStain FcX Fc Blocking Reagent (BioLegend, 422302) for 10 min at room temperature, then stained for 30 min at 4 °C. Antibodies and clones used for CITE-Seq were anti-human TotalSeq B reagents (Biolegend): TBNK Cocktail, CD7 (CD7-6B7); TCR.Vd2 (B6); CD279 (EH12.2H7); HLADR (L243); CD123 (6H6); CD38 (HB-7); CD57 (QA17A04); CD370(8F9);CD1c (L161). Cells were then washed twice with PBS supplemented with 2% FCS and 2 mM EDTA (Sigma) before resuspending in PBS and counting. Approximately 10,000 cells per sample were loaded onto the 10x Chromium controller. Gene expression libraries were prepared for each sample according to the manufacturer’s protocol (10x Genomics). Cell-surface protein libraries were subjected to double the manufacturer’s recommended primer concentration and seven to eight amplification cycles during the sample index PCR to reduce the likelihood of daisy chains forming. All libraries were sequenced using a NovaSeq 6000 (Illumina) to achieve a minimum of 50,000 paired-end reads per cell for gene expression and 20,000 paired-end reads per cell for cell-surface protein libraries.
Epigenetic landscape profiling using cytometry by time of flight (EpiTOF)
EpiTOF analysis using cytometry by time of flight was performed as previously described (39). Briefly, cryopreserved PBMCs were thawed and incubated in RPMI 1640 media (ThermoFisher) containing 10% FBS (ATCC) at 37°C for 1 hour prior to processing. Cisplatin (ENZO Life Sciences) was added to 10 mM final concentration for viability staining for 5 minutes before quenching with CyTOF Buffer (PBS (ThermoFisher) with 1% BSA (Sigma), 2mM EDTA (Fisher), 0.05% sodium azide). Cells were centrifuged at 400 g for 8 minutes and stained with lanthanide-labeled antibodies (Table S7) against immunophenotypic markers in CyTOF buffer containing Fc receptor blocker (BioLegend) for 30 minutes at room temperature (RT). Following extracellular marker staining, cells were washed 3 times with CyTOF buffer and fixed in 1.6% PFA (Electron Microscopy Sciences) at 1×106 cells/ml for 15 minutes at RT. Cells were centrifuged at 600 g for 5 minutes post-fixation and permeabilized with 1 mL ice-cold methanol (Fisher Scientific) for 20 minutes at 4C. 4 mL of CyTOF buffer was added to stop permeabilization followed. Mass-tag sample barcoding was performed following the manufacturer’s protocol (Fluidigm). Individual samples were then combined and stained with intracellular antibodies in CyTOF buffer containing Fc receptor blocker (BioLegend) overnight at 4C. The following day, cells were washed twice in CyTOF buffer and stained with 250 nM 191/193Ir DNA intercalator (Fluidigm) in PBS with 1.6% PFA for 30 minutes at RT. Cells were washed twice with CyTOF buffer and once with double-deionized water (ddH2O) (ThermoFisher) followed by filtering through 35 mm strainer to remove aggregates. Cells were resuspended in ddH2O containing four element calibration beads (Fluidigm) and analyzed on CyTOF2 (Fluidigm).
ATAC-Seq
ATAC-Seq analysis was performed as previously described using FAST-ATAC(58). In brief, live CD3−CD14−CD19−CD7+ NK cells were sorted into FACS buffer and pelleted by centrifugation at 500g RCF at 4°C in a pre-cooled fixed-angle centrifuge. The supernatant was carefully removed, and 50 microliters of transposase mixture (25 ul of 2x TD buffer, 2.5 ul TDE1, 0.25 ul 2% digitonin, 22.5 ul nuclease-free water) added to the cells. After mixing, samples were incubated at 37°C for 30 min in an Eppendorf ThermoMixer with agitation at 300 rpm. Transposed DNA was then purified using a QIAGEN MinElute Reaction Cleanup kit (28204) and purified DNA was eluted in 10 ul elution buffer (10mM Tris-HCl, pH 8). Transposed fragments were amplified and purified and libraries quantified using qPCR before sequencing. Fast-ATAC libraries were sequenced using paired-end, dual-index library sequencing on an Illumina NextSeq instrument (75bp kit, 36 base pair reads, ~20–30 million reads/sample).
Functional assays
Parasite Culture and Isolation
P. falciparum 3D7 asexual stage parasites were cultured in RPMI 1640 with 25mM HEPES, 25mM sodium bicarbonate, 1% gentamycin, and enriched with 0.5% Albumax (pH 6.75) and 250uM hypoxanthine, under atmospheric conditions (5% oxygen, 5% carbon dioxide, and 95% nitrogen). To retain synchronous cultures, parasite growth was treated with 5% D-sorbitol. Schizont isolation was performed using MACS cell separation LD columns (Miltenyi Biotec), and stored in −80°C. For experiments, schizonts were thawed, washed and resuspended in RPMI before addition to cells. Mycoplasma contamination was assayed using the MycoAlert Mycoplasma Detection Kit (Lonza).
NK cell isolation
We used the Human NK Cell Isolation Kit (Miltenyi Biotec) to negatively isolate NK cells from freshly thawed PBMCs. First, cells were resuspended with buffer and mixed with NK cell biotin-antibody cocktail for 5 mins at 4°C. Then, the provided NK Cell Microbead cocktail was added and incubated for 10mins at 4°C. This was followed by magnetic cell separation using an LS column (Miltenyi Biotec). Flowthrough was collected, washed and resuspended in R10 (RPMI 1610 (Sigma) with 10% FBS, 1% L-Glutamine (Thermo Fisher), 1% Penicillin and Gentamycin (Corning)).
Stimulation Assay
Thawed PBMC were rested for 1 hour at 37°C before the addition of either uninfected RBCs, schizonts or cytokine cocktail containing 2.5ng/ml of recombinant human IL-12 (R&D systems), 10ng/ml of IL-15 (PeproTech) and 0.25ug of IL-18 (R&D systems). Cultures were incubated for 1 hour at 37°C before addition of Protein Transport Inhibitor Brefeldin A (BD). Cells were kept at 37°C for an additional 3 hours before antibody staining (Table S6) and analysis on an Attune NXT flow cytometer.
ADCC – Degranulation Assay
Freshly thawed schizonts were washed and incubated with RPMI, 10% pooled US plasma, 10% pooled immune Ugandan plasma or 1:100 anti-RBC (Abcam) at a concentration of 5×107 for 1 hour at 37°C. Following opsonization, cells were washed and resuspended in R10 (see NK Isolation) before mixing with PBMCs or isolated NK cells. CD107a at a dilution of 1:250 (Clone H4A3, BioLegend) was immediately added with ER inhibitors – Monensin (BD Biosciences) and Protein Transport Inhibitor containing brefeldin A (BD Biosciences). Cells were spun for 3 mins at 100×g before incubating for 5 hours, after which cells were stained and analyzed using the Attune NXT flow cytometer. Data was analyzed with FlowJo X software (Tree Star).
ADCC - p815 Killing Assay
Killing assay performed as previously described(59) with some modifications. Briefly, p815 cells (a mouse leukemia line) were stained with CellTrace Violet Cell Proliferation Kit (ThermoFisher) at 37°C for 20 mins. Reaction was stopped by addition of RPMI (Corning) with 10% FBS at 37C for 5 mins. Stained p815 cells were either opsonized with 10 μg/ml p815 rat anti-mouse antibody (Clone: 2.4G2, BD Biosciences) or left uncoated for 30 mins at 37°C. After incubation, PBMCs were cocultured with opsonized and uncoated target cells at an effector:target ratio of 10:1. Co-cultures were incubated at 37°C for 5 hours, before staining for dead and dying cells using 7-AAD Viability staining solution (1:25, Biolegend) and PE Annexin V (1:20, Biosciences) in 1x BD Annexin V Binding Buffer for 15 mins at room temperature. Volume was brought up to 200 μl and analyzed on the Attune NXT Flow Cytometer within the hour and analyzed with FlowJo X (Tree Star) software. Percentage of killed target cells was calculated by subtracting the percentages of dead uncoated target cells from the percentages of dead opsonized target cells.
Statistical analysis
All statistical analyses were performed using STATA version 16 (College Station), SPICE v.5.3 (NIAID), Prism 9.0 (GraphPad), or R version 4.2.0. Comparisons of cellular percentages between groups were performed using the Wilcoxon rank sum and/or t test, and the Wilcoxon signed-rank and/or paired t test was used to compare paired data. Associations between continuous variables were assessed using Spearman’s rank correlation (ρ). Dimensionality reduction was performed with UMAP(60). Associations between NK cell tertiles and the monthly probability of symptoms if parasitemic (detected by microscopy), and geometric mean parasite densities if parasitemic, were evaluated using multilevel mixed-effects models. These analyses accounted for repeated measures within individuals and were clustered on household to account for multiple children living in the same household. In multivariate analyses, odds ratios for symptoms when infected were adjusted for age and aEIR. Two-sided p-values were calculated for all test statistics and P < 0.05 was considered significant.
CITE-Seq Analysis
Droplet libraries were processed using Cell Ranger v4.0. Reads were aligned to the GRCh38 human genome using STAR (v2.5.1b) and unique molecular identifiers (UMIs) deduplicated. Antibody-derived tag (ADT) counts for each protein were first normalized using counts per million and log transformed, with a pseudocount of +1. To estimate the background signal for each protein, a two-component Gaussian mixture model, implemented in the R package function ‘mclust’ (v5.4.7), was fit across the droplets with a total UMI count of >10 and <100 from each experimental sample separately. The mean of the first Gaussian mixture model component for each protein was then subtracted from the log counts per million from the QC-passed droplets in the respective experimental sample. Raw data was filtered to remove cells that expressed fewer than 200 genes and >10% mitochondrial reads. We used the variable stabilization transformation (VST) from the Seurat R package(61) to integrate and transform the count data into normalized and scaled data that can be used for downstream analysis.
We used UMAP for dimensionality reduction and data visualization, the Louvain algorithm to identify cell clusters, and the singleR package to annotate cell clusters by comparing transcriptomic profiles between cell clusters and reference data from the Human Primary Cell Atlas(62). We manually gated protein-level expression data to identify three NK cell subsets, including CD56neg (CD19− CD3− CD14− CD7+ CD16+ CD56−), CD56dim (CD19− CD3− CD14− CD7+ CD16+ CD56dim) and CD56bright NK cells (CD19− CD3− CD14− CD7+ CD16− CD56+), and these subsets were mapped to the UMAP projection. We performed likelihood ratio tests (implemented as the FindMarkers function in the Seurat R package(61)) to identify differentially expressed genes between CD56− NK cells and CD56dim NK cells.
EpiTOF Analysis
Raw data were pre-processed using FlowJo to identify cell events from individual samples by palladium-based mass tags, and to segregate specific immune cell populations by immunophenotypic markers (Gating hierarchy in Fig S3). Single-cell data for various immune cell subtypes from individual subjects were exported from FlowJo for downstream computational analyses. The exported Flowjo data were then analyzed following previously reported methods(39). In brief, the value of each histone mark was regressed against the total amount of histones, represented by measured values of H3 and H4. For sample level analyses, the values of each histone mark were averaged for each cell type in each sample. Distances of epigenetic profiles of NK cell types were obtained by computing Euclidean distances from the centers of the epigenetic profiles for each cell type. Statistical significance of the differences between groups at the sample level was assessed by computing an effect size with Hedges’ g formula, and p-values corrected for multiple comparisons with the Benjamini-Hochberg method.
ATAC-Seq Analysis
We used ChrAccR, an R package for comprehensive analysis of chromatin accessibility data (https://greenleaflab.github.io/ChrAccR/). We identified open chromatin peaks that varied across the different timepoints from the same child and that are differentially enriched for transcription factor binding motifs in regulatory elements (e.g. gene promoters and distal enhancers). Specific sites of interest visualized with Integrative Genome Viewer (Broad Institute, Cambridge, MA).
Supplementary Material
Acknowledgments:
We are grateful to all the parents and guardians for giving their consent and to the study participants for their cooperation. We thank all the members of the study team for their tireless effort and excellent work.
Funding:
Support for this work was provided by the following grants: National Institutes of Health grant U01AI150741 (to BG and PJ), National Institutes of Health grant R01AI093615 (to MEF), National Institutes of Health grant U19AI089674 (to GD and MK), Australian National Health and Medical Research Council Ideas Grant CIA GNT1181932 and Career Development Fellowship 1 GNT1141632 (to MB), and Bill and Melinda Gates Foundation/Stanford Center for Human Systems Immunology (OPP 1113682, Pilot Project to PJ).
Footnotes
Competing interests: Authors declare that they have no competing interests.
Clinical data from both study cohorts are fully accessible online via the ClinepiDB platform (clinepidb.org, “PRISM” studies), at the level of the cohort, household, individual, and visit.
Data and materials availability:
All data generated or analyzed in this study are available in the main text or the supplementary materials. Raw data from figures with n<25 observations is in data file S2. RNA and ATAC sequencing data have been deposited at NCBI Gene Expression Omnibus under accession number GSE210943 and are publicly available. (https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE210943)
References
- 1.“World malaria report 2021,” (World Health Organization, Geneva, Switzerland, 2021). [Google Scholar]
- 2.Rodriguez-Barraquer I, Arinaitwe E, Jagannathan P, Kamya MR, Rosenthal PJ, Rek J, Dorsey G, Nankabirwa J, Staedke SG, Kilama M, Drakeley C, Ssewanyana I, Smith DL, Greenhouse B, Quantification of anti-parasite and anti-disease immunity to malaria as a function of age and exposure. Elife 7, (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Langhorne J, Ndungu FM, Sponaas AM, Marsh K, Immunity to malaria: more questions than answers. Nat Immunol 9, 725–732 (2008). [DOI] [PubMed] [Google Scholar]
- 4.Tan J, Piccoli L, Lanzavecchia A, The Antibody Response to Plasmodium falciparum: Cues for Vaccine Design and the Discovery of Receptor-Based Antibodies. Annu Rev Immunol, (2018). [DOI] [PubMed] [Google Scholar]
- 5.Osier FH, Feng G, Boyle MJ, Langer C, Zhou J, Richards JS, McCallum FJ, Reiling L, Jaworowski A, Anders RF, Marsh K, Beeson JG, Opsonic phagocytosis of Plasmodium falciparum merozoites: mechanism in human immunity and a correlate of protection against malaria. BMC Med 12, 108 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Boyle MJ, Reiling L, Feng G, Langer C, Osier FH, Aspeling-Jones H, Cheng YS, Stubbs J, Tetteh KK, Conway DJ, McCarthy JS, Muller I, Marsh K, Anders RF, Beeson JG, Human antibodies fix complement to inhibit Plasmodium falciparum invasion of erythrocytes and are associated with protection against malaria. Immunity 42, 580–590 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Arora G, Hart GT, Manzella-Lapeira J, Doritchamou JY, Narum DL, Thomas LM, Brzostowski J, Rajagopalan S, Doumbo OK, Traore B, Miller LH, Pierce SK, Duffy PE, Crompton PD, Desai SA, Long EO, NK cells inhibit Plasmodium falciparum growth in red blood cells via antibody-dependent cellular cytotoxicity. Elife 7, (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Stevenson MM, Riley EM, Innate immunity to malaria. Nat Rev Immunol 4, 169–180 (2004). [DOI] [PubMed] [Google Scholar]
- 9.Chua CL, Brown G, Hamilton JA, Rogerson S, Boeuf P, Monocytes and macrophages in malaria: protection or pathology? Trends Parasitol 29, 26–34 (2013). [DOI] [PubMed] [Google Scholar]
- 10.Riley EM, Jakobsen PH, Allen SJ, Wheeler JG, Bennett S, Jepsen S, Greenwood BM, Immune response to soluble exoantigens of Plasmodium falciparum may contribute to both pathogenesis and protection in clinical malaria: evidence from a longitudinal, prospective study of semi-immune African children. Eur J Immunol 21, 1019–1025 (1991). [DOI] [PubMed] [Google Scholar]
- 11.Jagannathan P, Kim CC, Greenhouse B, Nankya F, Bowen K, Eccles-James I, Muhindo MK, Arinaitwe E, Tappero JW, Kamya MR, Dorsey G, Feeney ME, Loss and dysfunction of Vdelta2(+) gammadelta T cells are associated with clinical tolerance to malaria. Sci Transl Med 6, 251ra117 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Jagannathan P, Lutwama F, Boyle MJ, Nankya F, Farrington LA, McIntyre TI, Bowen K, Naluwu K, Nalubega M, Musinguzi K, Sikyomu E, Budker R, Katureebe A, Rek J, Greenhouse B, Dorsey G, Kamya MR, Feeney ME, Vdelta2+ T cell response to malaria correlates with protection from infection but is attenuated with repeated exposure. Sci Rep 7, 11487 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Guha R, Mathioudaki A, Doumbo S, Doumtabe D, Skinner J, Arora G, Siddiqui S, Li S, Kayentao K, Ongoiba A, Zaugg J, Traore B, Crompton PD, Plasmodium falciparum malaria drives epigenetic reprogramming of human monocytes toward a regulatory phenotype. PLoS Pathog 17, e1009430 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Farrington LA, Callaway PC, Vance HM, Baskevitch K, Lutz E, Warrier L, McIntyre TI, Budker R, Jagannathan P, Nankya F, Musinguzi K, Nalubega M, Sikyomu E, Naluwu K, Arinaitwe E, Dorsey G, Kamya MR, Feeney ME, Opsonized antigen activates Vdelta2+ T cells via CD16/FCgammaRIIIa in individuals with chronic malaria exposure. PLoS Pathog 16, e1008997 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Walk J, de Bree LCJ, Graumans W, Stoter R, van Gemert GJ, van de Vegte-Bolmer M, Teelen K, Hermsen CC, Arts RJW, Behet MC, Keramati F, Moorlag S, Yang ASP, van Crevel R, Aaby P, de Mast Q, van der Ven A, Stabell Benn C, Netea MG, Sauerwein RW, Outcomes of controlled human malaria infection after BCG vaccination. Nat. Commun. 10, 874 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Dobbs KR, Embury P, Vulule J, Odada PS, Rosa BA, Mitreva M, Kazura JW, Dent AE, Monocyte dysregulation and systemic inflammation during pediatric falciparum malaria. JCI Insight 2, (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Crabtree JN, Caffrey DR, de Souza Silva L, Kurt-Jones EA, Dobbs K, Dent A, Fitzgerald KA, Golenbock DT, Lymphocyte crosstalk is required for monocyte-intrinsic trained immunity to Plasmodium falciparum. J Clin Invest 132, (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Artavanis-Tsakonas K, Riley EM, Innate immune response to malaria: rapid induction of IFN-gamma from human NK cells by live Plasmodium falciparum-infected erythrocytes. J Immunol 169, 2956–2963 (2002). [DOI] [PubMed] [Google Scholar]
- 19.Wolf AS, Sherratt S, Riley EM, NK Cells: Uncertain Allies against Malaria. Front Immunol 8, 212 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Horowitz A, Strauss-Albee DM, Leipold M, Kubo J, Nemat-Gorgani N, Dogan OC, Dekker CL, Mackey S, Maecker H, Swan GE, Davis MM, Norman PJ, Guethlein LA, Desai M, Parham P, Blish CA, Genetic and environmental determinants of human NK cell diversity revealed by mass cytometry. Sci Transl Med 5, 208ra145 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Strauss-Albee DM, Fukuyama J, Liang EC, Yao Y, Jarrell JA, Drake AL, Kinuthia J, Montgomery RR, John-Stewart G, Holmes S, Blish CA, Human NK cell repertoire diversity reflects immune experience and correlates with viral susceptibility. Sci Transl Med 7, 297ra115 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Yang C, Siebert JR, Burns R, Gerbec ZJ, Bonacci B, Rymaszewski A, Rau M, Riese MJ, Rao S, Carlson KS, Routes JM, Verbsky JW, Thakar MS, Malarkannan S, Heterogeneity of human bone marrow and blood natural killer cells defined by single-cell transcriptome. Nat. Commun. 10, 3931 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Hart GT, Tran TM, Theorell J, Schlums H, Arora G, Rajagopalan S, Sangala ADJ, Welsh KJ, Traore B, Pierce SK, Crompton PD, Bryceson YT, Long EO, Adaptive NK cells in people exposed to Plasmodium falciparum correlate with protection from malaria. J Exp Med 216, 1280–1290 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Mavilio D, Lombardo G, Benjamin J, Kim D, Follman D, Marcenaro E, O’Shea MA, Kinter A, Kovacs C, Moretta A, Fauci AS, Characterization of CD56−/CD16+ natural killer (NK) cells: a highly dysfunctional NK subset expanded in HIV-infected viremic individuals. Proc Natl Acad Sci U S A 102, 2886–2891 (2005). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Gonzalez VD, Falconer K, Bjorkstrom NK, Blom KG, Weiland O, Ljunggren HG, Alaeus A, Sandberg JK, Expansion of functionally skewed CD56-negative NK cells in chronic hepatitis C virus infection: correlation with outcome of pegylated IFN-alpha and ribavirin treatment. J Immunol 183, 6612–6618 (2009). [DOI] [PubMed] [Google Scholar]
- 26.Chretien AS, Devillier R, Granjeaud S, Cordier C, Demerle C, Salem N, Wlosik J, Orlanducci F, Gorvel L, Fattori S, Hospital MA, Pakradouni J, Gregori E, Paul M, Rochigneux P, Pagliardini T, Morey M, Fauriat C, Dulphy N, Toubert A, Luche H, Malissen M, Blaise D, Nunes JA, Vey N, Olive D, High-dimensional mass cytometry analysis of NK cell alterations in AML identifies a subgroup with adverse clinical outcome. Proc Natl Acad Sci U S A 118, (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Forconi CS, Cosgrove CP, Saikumar-Lakshmi P, Nixon CE, Foley J, Ong’echa JM, Otieno JA, Alter G, Munz C, Moormann AM, Poorly cytotoxic terminally differentiated CD56(neg)CD16(pos) NK cells accumulate in Kenyan children with Burkitt lymphomas. Blood Adv 2, 1101–1114 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Bjorkstrom NK, Ljunggren HG, Sandberg JK, CD56 negative NK cells: origin, function, and role in chronic viral disease. Trends Immunol 31, 401–406 (2010). [DOI] [PubMed] [Google Scholar]
- 29.Kamya MR, Arinaitwe E, Wanzira H, Katureebe A, Barusya C, Kigozi SP, Kilama M, Tatem AJ, Rosenthal PJ, Drakeley C, Lindsay SW, Staedke SG, Smith DL, Greenhouse B, Dorsey G, Malaria transmission, infection, and disease at three sites with varied transmission intensity in Uganda: implications for malaria control. Am J Trop Med Hyg 92, 903–912 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Holmes TD, Pandey RV, Helm EY, Schlums H, Han H, Campbell TM, Drashansky TT, Chiang S, Wu CY, Tao C, Shoukier M, Tolosa E, Von Hardenberg S, Sun M, Klemann C, Marsh RA, Lau CM, Lin Y, Sun JC, Mansson R, Cichocki F, Avram D, Bryceson YT, The transcription factor Bcl11b promotes both canonical and adaptive NK cell differentiation. Sci Immunol 6, (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Brownlie D, Scharenberg M, Mold JE, Hard J, Kekalainen E, Buggert M, Nguyen S, Wilson JN, Al-Ameri M, Ljunggren HG, Marquardt N, Michaelsson J, Expansions of adaptive-like NK cells with a tissue-resident phenotype in human lung and blood. Proc Natl Acad Sci U S A 118, (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Born TL, Thomassen E, Bird TA, Sims JE, Cloning of a novel receptor subunit, AcPL, required for interleukin-18 signaling. J Biol Chem 273, 29445–29450 (1998). [DOI] [PubMed] [Google Scholar]
- 33.Narayanan S, Ahl PJ, Bijin VA, Kaliaperumal N, Lim SG, Wang C-I, Fairhurst A-M, Connolly JE, LAG3 is a Central Regulator of NK Cell Cytokine Production. bioRxiv, 2020.2001.2031.928200 (2020). [Google Scholar]
- 34.Merino A, Zhang B, Dougherty P, Luo X, Wang J, Blazar BR, Miller JS, Cichocki F, Chronic stimulation drives human NK cell dysfunction and epigenetic reprograming. J Clin Invest 129, 3770–3785 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Anderson AC, Joller N, Kuchroo VK, Lag-3, Tim-3, and TIGIT: Co-inhibitory Receptors with Specialized Functions in Immune Regulation. Immunity 44, 989–1004 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Smith SL, Kennedy PR, Stacey KB, Worboys JD, Yarwood A, Seo S, Solloa EH, Mistretta B, Chatterjee SS, Gunaratne P, Allette K, Wang YC, Smith ML, Sebra R, Mace EM, Horowitz A, Thomson W, Martin P, Eyre S, Davis DM, Diversity of peripheral blood human NK cells identified by single-cell RNA sequencing. Blood Adv 4, 1388–1406 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Bjorkstrom NK, Riese P, Heuts F, Andersson S, Fauriat C, Ivarsson MA, Bjorklund AT, Flodstrom-Tullberg M, Michaelsson J, Rottenberg ME, Guzman CA, Ljunggren HG, Malmberg KJ, Expression patterns of NKG2A, KIR, and CD57 define a process of CD56dim NK-cell differentiation uncoupled from NK-cell education. Blood 116, 3853–3864 (2010). [DOI] [PubMed] [Google Scholar]
- 38.Schlums H, Cichocki F, Tesi B, Theorell J, Beziat V, Holmes TD, Han H, Chiang SC, Foley B, Mattsson K, Larsson S, Schaffer M, Malmberg KJ, Ljunggren HG, Miller JS, Bryceson YT, Cytomegalovirus infection drives adaptive epigenetic diversification of NK cells with altered signaling and effector function. Immunity 42, 443–456 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Cheung P, Vallania F, Warsinske HC, Donato M, Schaffert S, Chang SE, Dvorak M, Dekker CL, Davis MM, Utz PJ, Khatri P, Kuo AJ, Single-Cell Chromatin Modification Profiling Reveals Increased Epigenetic Variations with Aging. Cell 173, 1385–1397 e1314 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Cruz C, Della Rosa M, Krueger C, Gao Q, Horkai D, King M, Field L, Houseley J, Tri-methylation of histone H3 lysine 4 facilitates gene expression in ageing cells. Elife 7, (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Hwang I, Zhang T, Scott JM, Kim AR, Lee T, Kakarla T, Kim A, Sunwoo JB, Kim S, Identification of human NK cells that are deficient for signaling adaptor FcRgamma and specialized for antibody-dependent immune functions. Int Immunol 24, 793–802 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Nankabirwa JI, Arinaitwe E, Rek J, Kilama M, Kizza T, Staedke SG, Rosenthal PJ, Rodriguez-Barraquer I, Briggs J, Greenhouse B, Bousema T, Drakeley C, Roos DS, Tomko SS, Smith DL, Kamya MR, Dorsey G, Malaria Transmission, Infection, and Disease following Sustained Indoor Residual Spraying of Insecticide in Tororo, Uganda. Am J Trop Med Hyg 103, 1525–1533 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Caligiuri MA, Human natural killer cells. Blood 112, 461–469 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Milush JM, Long BR, Snyder-Cappione JE, Cappione AJ 3rd, York VA, Ndhlovu LC, Lanier LL, Michaelsson J, Nixon DF, Functionally distinct subsets of human NK cells and monocyte/DC-like cells identified by coexpression of CD56, CD7, and CD4. Blood 114, 4823–4831 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Milush JM, Lopez-Verges S, York VA, Deeks SG, Martin JN, Hecht FM, Lanier LL, Nixon DF, CD56negCD16(+) NK cells are activated mature NK cells with impaired effector function during HIV-1 infection. Retrovirology 10, 158 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Saito F, Hirayasu K, Satoh T, Wang CW, Lusingu J, Arimori T, Shida K, Palacpac NMQ, Itagaki S, Iwanaga S, Takashima E, Tsuboi T, Kohyama M, Suenaga T, Colonna M, Takagi J, Lavstsen T, Horii T, Arase H, Immune evasion of Plasmodium falciparum by RIFIN via inhibitory receptors. Nature 552, 101–105 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Chen Y, Xu K, Piccoli L, Foglierini M, Tan J, Jin W, Gorman J, Tsybovsky Y, Zhang B, Traore B, Silacci-Fregni C, Daubenberger C, Crompton PD, Geiger R, Sallusto F, Kwong PD, Lanzavecchia A, Structural basis of malaria RIFIN binding by LILRB1-containing antibodies. Nature 592, 639–643 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Junqueira C, Polidoro RB, Castro G, Absalon S, Liang Z, Sen Santara S, Crespo A, Pereira DB, Gazzinelli RT, Dvorin JD, Lieberman J, gammadelta T cells suppress Plasmodium falciparum blood-stage infection by direct killing and phagocytosis. Nat Immunol 22, 347–357 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Musasia FK, Nkumama IN, Frank R, Kipkemboi V, Schneider M, Mwai K, Odera DO, Rosenkranz M, Furle K, Kimani D, Tuju J, Njuguna P, Hamaluba M, Kapulu MC, Wardemann H, Team C-SS, Osier FHA, Phagocytosis of Plasmodium falciparum ring-stage parasites predicts protection against malaria. Nat. Commun. 13, 4098 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Garcia-Senosiain A, Kana IH, Singh S, Das MK, Dziegiel MH, Hertegonne S, Adu B, Theisen M, Neutrophils dominate in opsonic phagocytosis of P. falciparum blood-stage merozoites and protect against febrile malaria. Commun Biol 4, 984 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Huot N, Rascle P, Petitdemange C, Contreras V, Sturzel CM, Baquero E, Harper JL, Passaes C, Legendre R, Varet H, Madec Y, Sauermann U, Stahl-Hennig C, Nattermann J, Saez-Cirion A, Le Grand R, Keith Reeves R, Paiardini M, Kirchhoff F, Jacquelin B, Muller-Trutwin M, SIV-induced terminally differentiated adaptive NK cells in lymph nodes associated with enhanced MHC-E restricted activity. Nat. Commun. 12, 1282 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Hammer Q, Ruckert T, Borst EM, Dunst J, Haubner A, Durek P, Heinrich F, Gasparoni G, Babic M, Tomic A, Pietra G, Nienen M, Blau IW, Hofmann J, Na IK, Prinz I, Koenecke C, Hemmati P, Babel N, Arnold R, Walter J, Thurley K, Mashreghi MF, Messerle M, Romagnani C, Peptide-specific recognition of human cytomegalovirus strains controls adaptive natural killer cells. Nat Immunol 19, 453–463 (2018). [DOI] [PubMed] [Google Scholar]
- 53.Simoni Y, Fehlings M, Kloverpris HN, McGovern N, Koo SL, Loh CY, Lim S, Kurioka A, Fergusson JR, Tang CL, Kam MH, Dennis K, Lim TKH, Fui ACY, Hoong CW, Chan JKY, Curotto de Lafaille M, Narayanan S, Baig S, Shabeer M, Toh SES, Tan HKK, Anicete R, Tan EH, Takano A, Klenerman P, Leslie A, Tan DSW, Tan IB, Ginhoux F, Newell EW, Human Innate Lymphoid Cell Subsets Possess Tissue-Type Based Heterogeneity in Phenotype and Frequency. Immunity 46, 148–161 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Piriou E, Asito AS, Sumba PO, Fiore N, Middeldorp JM, Moormann AM, Ploutz-Snyder R, Rochford R, Early age at time of primary Epstein-Barr virus infection results in poorly controlled viral infection in infants from Western Kenya: clues to the etiology of endemic Burkitt lymphoma. J Infect Dis 205, 906–913 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Ssewanyana I, Rek J, Rodriguez I, Wu L, Arinaitwe E, Nankabirwa JI, Beeson JG, Mayanja-Kizza H, Rosenthal PJ, Dorsey G, Kamya MR, Drakeley C, Greenhouse B, Tetteh KKA, Impact of a Rapid Decline in Malaria Transmission on Antimalarial IgG Subclasses and Avidity. Front Immunol 11, 576663 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Greenwood B, Zongo I, Dicko A, Chandramohan D, Snow RW, Ockenhouse C, Resurgent and delayed malaria. Malar J 21, 77 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Digitale JC, Callaway PC, Martin M, Nelson G, Viard M, Rek J, Arinaitwe E, Dorsey G, Kamya M, Carrington M, Rodriguez-Barraquer I, Feeney ME, Association of Inhibitory Killer Cell Immunoglobulin-like Receptor Ligands With Higher Plasmodium falciparum Parasite Prevalence. J Infect Dis 224, 175–183 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Corces MR, Buenrostro JD, Wu B, Greenside PG, Chan SM, Koenig JL, Snyder MP, Pritchard JK, Kundaje A, Greenleaf WJ, Majeti R, Chang HY, Lineage-specific and single-cell chromatin accessibility charts human hematopoiesis and leukemia evolution. Nat. Genet. 48, 1193–1203 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Roy Chowdhury R, Vallania F, Yang Q, Lopez Angel CJ, Darboe F, Penn-Nicholson A, Rozot V, Nemes E, Malherbe ST, Ronacher K, Walzl G, Hanekom W, Davis MM, Winter J, Chen X, Scriba TJ, Khatri P, Chien YH, A multi-cohort study of the immune factors associated with M. tuberculosis infection outcomes. Nature 560, 644–648 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.McInnes L, Healy J, Melville J, UMAP: uniform manifold approximation and projection for dimension reduction. (2020). [Google Scholar]
- 61.Hao Y, Hao S, Andersen-Nissen E, Mauck WM 3rd, Zheng S, Butler A, Lee MJ, Wilk AJ, Darby C, Zager M, Hoffman P, Stoeckius M, Papalexi E, Mimitou EP, Jain J, Srivastava A, Stuart T, Fleming LM, Yeung B, Rogers AJ, McElrath JM, Blish CA, Gottardo R, Smibert P, Satija R, Integrated analysis of multimodal single-cell data. Cell 184, 3573–3587 e3529 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Mabbott NA, Baillie JK, Brown H, Freeman TC, Hume DA, An expression atlas of human primary cells: inference of gene function from coexpression networks. BMC Genomics 14, 632 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
All data generated or analyzed in this study are available in the main text or the supplementary materials. Raw data from figures with n<25 observations is in data file S2. RNA and ATAC sequencing data have been deposited at NCBI Gene Expression Omnibus under accession number GSE210943 and are publicly available. (https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE210943)