

Abstract
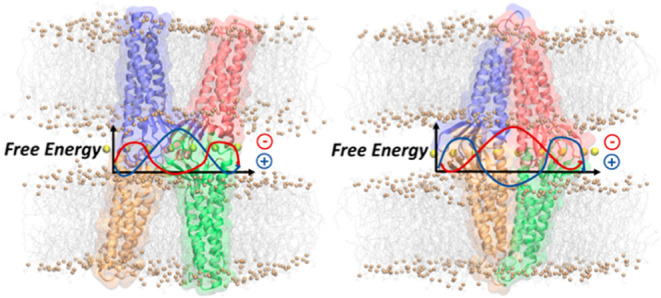
The blood–brain barrier (BBB) strictly regulates the exchange of ions and molecules between the blood and the central nervous system. Tight junctions (TJs) are multimeric structures that control the transport through the paracellular spaces between the adjacent brain endothelial cells of the BBB. Claudin-5 (Cldn5) proteins are essential for TJ formation and assemble into multiprotein complexes via cis-interactions within the same cell membrane and trans-interactions across two contiguous cells. Despite the relevant biological function of Cldn5 proteins and their role as targets of brain drug delivery strategies, the molecular details of their assembly within TJs are still unclear. Two different structural models have been recently introduced, in which Cldn5 dimers belonging to opposite cells join to generate paracellular pores. However, a comparison of these models in terms of ionic transport features is still lacking. In this work, we used molecular dynamics simulations and free energy (FE) calculations to assess the two Cldn5 pore models and investigate the thermodynamic properties of water and physiological ions permeating through them. Despite different FE profiles, both structures present single/multiple FE barriers to ionic permeation, while being permissive to water flux. These results reveal that both models are compatible with the physiological role of Cldn5 TJ strands. By identifying the protein–protein surface at the core of TJ Cldn5 assemblies, our computational investigation provides a basis for the rational design of synthetic peptides and other molecules capable of opening paracellular pores in the BBB.
Keywords: tight junctions, Claudin-5, blood−brain barrier, biological pore models, molecular dynamics, free energy calculations
Introduction
Biological barriers are structures made of layers of tightly bound endothelial/epithelial cells that preserve the characteristics of the body compartments they separate and regulate the exchanges between them. Multimeric protein complexes named tight junctions (TJs)1−6 hold adjacent cells together by forming strands that are visible in freeze-fracture electron microscopy images and seal the paracellular space between cells.7−9
Claudins (Cldns) are the major components of the TJ strands.7,10,11 The Cldn family is composed of 27 tissue-specific homologs with a structure comprising a transmembrane four-helix bundle (TM1-4) embedded in the membrane bilayer (the TM domain). TM helices are joined by two extracellular loops spanning the paracellular space (ECL1-2) and by an intracellular loop in the cytoplasmic region, where the N/C termini are also found.12,13 Cldns are known to assemble into TJs via intermolecular cis-interactions between individual protomers and trans-interactions between proteins of adjacent cells.14 TJ strands regulate the paracellular flux of ions and molecules across the various barriers via highly selective, tissue-specific mechanisms.15,16
The Cldn subtype 5 (Cldn5) is the most abundant TJ protein in the endothelial cells of the blood–brain barrier (BBB), the highly selective interface that preserves the chemical homeostasis of the central nervous system (CNS). In particular, Cldn5 strands are responsible for the very limited BBB paracellular permeability that prevents the uncontrolled permeation of ions and small molecules.17−20 The relevant physiological function of Cldn5 proteins makes them a novel and promising target for strategies to deliver drugs directly to the brain.21−25 However, structure-based approaches are still hampered by a lack of knowledge on the precise assembly of Cldn5 protomers in the BBB TJs.17 Only recently,25 based on prior results of other Cldns,26−28 two structural models of Cldn5 complexes were introduced, both of which display a pore cavity and were named Pore I and Pore II.
The Pore I structure is based on the model originally introduced by Suzuki et al. in ref (29) for the homologous Cldn subtype 15 (Cldn15, PDB ID: 4P79),30 the first member of the family to be crystallized. According to this template, cis-interactions are formed by the ECL1 domains of two neighboring protomers in the same membrane (also named face-to-face interaction7), with opposing β-strands arranged in an antiparallel fashion to generate an extended β-sheet across the two molecules, defining a hydrophilic surface. Moreover, two opposing dimers from adjacent cells create a tetrameric arrangement sustained by trans-interactions between the ECLs of the protomers, resulting in a β-barrel supersecondary structure in the paracellular space that encompasses a pore cavity. After the publication of the Cldn15-based model, this has been refined and validated in several studies using structural modeling and molecular dynamics (MD) simulations,31−38 also for other Cldns. In particular, in ref (37), the authors investigated the mechanism of ion permeation through Cldn5 Pore I by calculating the free energy (FE, or potential of mean force) profiles for various ionic species. Results pointed to the lack of both cation and anion permeation, thus demonstrating that the Pore I conformation properly reproduces the function of barrier to ionic fluxes exerted by BBB TJs.39
On the other hand, Pore II was also introduced by the same group34,35,40 based on previously modeled Cldn5 dimers.34 Although the structure still comprises again two facing Cldn dimers, the cis-arrangement between two protomers in the membrane is characterized by a distinct pattern of interactions involving the TM2 and TM3 helices (also named back-to-back interaction7). More specifically, the authors identified a leucine zipper motif defined by the residues Leu83, Leu90, Leu124, and Leu131 of the two Cldn5 subunits supported by the aromatic interactions between the opposing pairs of Trp138 and Phe127 residues. The presence of this cis-dimerization interface is consistent with the experimental results illustrated in ref (28). Then, similarly to the Pore I configuration,13,40 the Pore II architecture is obtained by joining a couple of these dimers via trans-interactions, although it lacks the cavity-enveloping supersecondary structure of Pore I. The MD simulations presented in ref (40) demonstrate that the Cldn5 Pore II is impermeable to small molecules such as α-D glucose but permissive to water. However, at variance with Pore I, the Pore II model is still limitedly investigated,34,35,40−42 and further studies are required to chart its structural and functional hallmarks. Moreover, a detailed investigation of its ionic permeability has not been performed yet, thus hampering a thorough comparison with Pore I.
The aim of this work is to investigate the two different pore models and to assess their reliability as possible representatives of Cldn5 complexes in the BBB TJs. After building the two tetrameric configurations using Cldn5 protomers modeled from the homologous Cldn15,30 we used all-atom MD simulations to refine their structures in solvated, double-membrane environments and to compute the one-dimensional FE profiles for the permeation of water and ions through both pores. Results show that the Pore I arrangement is structurally more stable, while both are water permeable and present FE barriers of different heights to the passage of ions, consistently with the known role of Cldn5 in increasing the trans-endothelial electrical resistance and reducing the ionic paracellular permeability of the BBB.20 In both the conformations, the FE critical points correlate with the positions of pore-lining charged residues. In particular, barriers for cations are localized in proximity of the Lys65 side chains, while those for Cl– are in correspondence of Glu146 and Asp149. The profiles for the same ions are, however, quite different in the two structures due to distinct arrangements of the residues along the pores. Moreover, the hydration pattern of permeating ions along the pore axis shows a partial depletion of the coordinating water molecules in correspondence with the narrow regions of the pores.33,43
Our findings provide a systematic description of the two Cldn5 tetrameric pore configurations in terms of their structural and permeation properties, indicating that they are both possible Cldn5 assemblies in the TJs of the brain endothelium.
Results and Discussion
The tetrameric structures of Pore I and Pore II are shown in Figures 1–3, which report the arrangements of Cldn5 protomers in dimers (Figure 1), the quaternary structure of the two pores (Figure 2), and the relevant amino acids within their cavities (Figure 3).
Figure 1.

Structural representations of the two equilibrated dimeric structures which prelude to Pore I (A) and Pore II (B). The dimer in panel A is characterized by a face-to-face interaction between the ECL1 domains of two opposite Cldn5 protomers. The dimer in panel B is formed by a back-to-back interaction and stabilized by a leucine zipper pairs in the TM2–TM3 helices of the single Cldn5 protomers.
Figure 3.

Positions of selected residues along Pore I (A) and Pore II (B) models.
Figure 2.

Structural representation of the two equilibrated single-pore models, top and side views. Pore configurations are shown in ribbon cartoon style (A,C) and van Der Waals sphere style (B,D) for Pore I (A,B) and Pore II (C,D) models, respectively.
To construct the Pore I system, two distinct models were set up and simulated. Both the configurations showed a remarkable structural stability of the paracellular domains, evaluated by root-mean-square deviation (rmsd) and cross-distances between facing, pore-lining residues. We then selected the model to be used as the Pore I system based on the pore size and preservation of a hydrogen bond involving the highly conserved Lys157 that was described as structurally relevant in refs (7), (12). Details of the modeling steps and MD simulations setup are provided in the Methods section, while the analysis of the simulations and the assessment of the best Pore I model are reported in the Supporting Information.
FE Calculations
Experimental evidence confirms that Cldn5-based TJs form an efficient barrier to the permeation of small molecules and physiological ions.3,6,39,44−47 Here, to assess the validity of the Cldn5 Pore I and Pore II configurations, we used the umbrella sampling (US) method48 to perform FE calculations for a single water molecule or single Na+, K+, Cl–, Ca2+, and Mg2+ ions permeating across the cavity of the structures.
In all the US simulations, we used the projection of the position of the tagged ion (or water molecule) on the pore axis as collective variable (CV). The FE profiles obtained for the two pore models are reported in Figures 4 and 5, respectively. The errors associated with these calculations were estimated via bootstrapping.49
Figure 4.

FE profiles for the permeation of water and physiological ions through the Pore I model. The positions of the most external atoms of the side chains of relevant residues are indicated as dashed lines.
Figure 5.

FE profiles for the permeation of water and physiological ions through the Pore II model. The positions of the most external atoms of the side chains of relevant residues are indicated as dashed lines.
The Pore I configuration is characterized by an hourglass shape with a narrow domain in the middle of the structure, where the Gln57 and Gln63 residues from the four protomers form an uncharged cage (Figure 3A). The pore scaffold is completed by positively charged Lys65 residues, which provide an electrostatic barrier to cations. According to the FE profiles illustrated in Figure 4, the system is permeable to water. In contrast, the profiles of monovalent (Na+ and K+) and divalent (Ca2+ and Mg2+) cations reveal a FE maximum in the constricted region of ∼3 and ∼ 7–8 kcal/mol, respectively, consistent with the pivotal role of electrostatics in controlling the paracellular transport.33,37,43,50−53 Overall, these calculations suggest that the Pore I configuration acts as a seal against the paracellular transport of cations. The FE profile for the Cl– ion shows barriers of about 2 kcal/mol symmetrically positioned at the pore entrances. In these regions, two identical clusters of negatively charged residues, Asp68, Glu146, and Asp149 (Figure 3A), exert a moderate charge repulsion that limits anion access. Our FE profiles are in overall agreement with those calculated by Irudayanathan et al.37 for the same ions permeating through Cldn5 Pore I (there, the authors used the GROMACS code54 and the CHARMM36m force field55 with virtual site parameters for lipids56 and Well-tempered Metadynamics57 for enhanced sampling).
The Pore II system (Figure 5) is also water permeable, but it is characterized by a locally different response to ionic transport. The FE profiles for Na+ and K+ reveal two FE maxima of ∼2 kcal/mol at the two entrances, where the positively charged residues Lys65 and Lys48 are located (Figure 3B), together with Asp68. The profiles for Ca2+ and Mg2+ permeations are characterized by higher barriers, up to 5 kcal/mol. Between the two lateral peaks, a minimum for all cations can be found at the center of the structure correlating with a relevant population of negatively charged residues belonging to the four Cldn5 subunits (Glu146 and Asp149). Because of this cluster of residues, the passage of the Cl– ion is prevented by the presence of a FE barrier reaching 5 kcal/mol, that is only slightly damped in the most central region by the four Arg145 residues.
Pore Size and Hydration of Na+ and Cl– during Permeation
To further investigate the link between the FE profiles and the structure of the pores, because ion permeation can be influenced by a combination of steric and electrostatic effects,33 we calculated the size of the two paracellular cavities. As shown in Figure 6, the two models share the same dimension at the two mouths with a diameter of ∼16–18 Å. On the contrary, the internal radius profile differs between the two models. Indeed, the Pore I structure is characterized by an hourglass shape, with an inner constriction in the central part of ∼ 5–6 Å (Figure 6A), where Gln57, Gln63, and Lys65 residues of the four subunits form a narrow cage. On the contrary, the equilibrated Pore II structure has two constrictions of ∼6 Å (Figure 6B) in each of the two entrances, where the aforementioned residues belonging to two subunits are located.
Figure 6.

Pore cavities of Pore I (A) and Pore II (B) models. Each protomer is represented in different colors, and the pore cavity is shown as a blue surface. In the bottom panels, the pore radius along the pore axis is reported.
We then mapped the hydration pattern of the Na+ and Cl– ions during their permeation across the pore cavity (Figure 7). To this aim, we calculated the average number of coordinating oxygen atoms belonging to the water molecules surrounding the ions in each US window. For this analysis, we adopted a threshold of 3.0 Å for the cation and 3.5 Å for the anion.58
Figure 7.

Average number of ion-coordinating water molecules as a function of the pore–axis coordinate for the Na+ ion through Pore I (A) and Pore II (B) and for the Cl– ion through Pore I (C) and Pore II (D). Error bars represent the standard deviation of the mean.
The ionic hydration profiles correlate with the pore radius and with the FE profiles of the two structures. The Na+ and Cl– ions in the solvent bulk are surrounded by ∼5.5 and ∼6.5 water molecules, respectively, in agreement with the values reported in ref (58).
The Na+ permeating the Pore I cavity (Figure 7A) loses up to one coordinating water molecule in the inner region, where Pore I exhibits the minimal pore radius (Figure 6A), and the pore-lining neutral Gln57 and Gln63 residues are located. The partial depletion of the solvation sphere and the unfavorable electrostatic interactions with the positively charged Lys65 residues add up to generate the energetic barrier displayed in Figure 4. Similarly, the cation permeating the Pore II cavity (Figure 7B) shows two minima in the hydration profile at ∼10 Å and ∼60 Å along the pore axis, which form the narrowest regions (Figure 6B), where the Lys65 residues are located (Figure 3B). This evidence is consistent with the position of the energetic barriers computed with the US calculations (Figure 5). Minor fluctuations of the average number of the coordinating molecules are found between the two peaks, where the pore radius (Figure 6B) is slightly larger than the radius of the Na+ hydration sphere. The hydration profile of the Cl– ion across Pore I (Figure 7C) also correlates with the pore radius and thermodynamics calculations. The FE barriers are found at ∼10 and ∼50 Å corresponding to the positions of Asp149 and Glu146. Here, the pore width allows full hydration of the ion, thus partially screening the interaction with the negatively charged residues. Between these regions, three minima at ∼18, ∼30, and ∼45 Å are observed in the hydration pattern, correlating with the position of the pore-lining residue Lys48 and with the maximal constriction of the cavity. These findings suggest that the main factor responsible for the formation of the Cl– energetic barriers is the electrostatic repulsion exerted by the negatively charged Asp149 and Glu146 residues rather than the steric hindrance of the pore. Indeed, in the inner part of the pore, the anion passes through the narrowest segment experiencing a partial dehydration, which is not associated with a significant thermodynamic barrier. In contrast, the regions where the FE profiles show the highest barriers to passage of the Cl– are wide enough to accommodate the anion with its entire hydration sphere. The antagonistic contributions of the pore shrinkage and the electrostatics justify the lower entity of the barrier found for the anion (∼2 kcal/mol) with respect to the monovalent cations (∼3–3.5 kcal/mol) in the Pore I configuration.
On the other hand, the hydration scheme of the Cl– ion permeating the Pore II model (Figure 7D) reports relevant fluctuations because of electrostatic interactions with the pore-lining charged residues and steric hindrance in the tight regions, where the contact with polar amino acids takes place. The minima at ∼10 and ∼65 Å correlate with the constrictions of the cavity (Figure 6B). Nevertheless, the central section reveals limited fluctuations in the pore radius in the same range of the Cl– hydration sphere. For this reason, the major role to the fluctuations in the coordination pattern of the anion is attributed to the interactions of the ion with the pore-lining residues. To better investigate the mechanisms of the Cl– hydration profiles within Pore II, we analyzed the changes in the coordinating environment of the anion by mapping the interactions of the ion with the pore-lining positively charged residues and the whole protein (Figure 8). Results show that, in the regions at ∼10 and ∼65 Å, the ion interacts not only with Lys65 but also with other protein atoms due to the constriction of the cavity. In the segments centered at ∼22 and ∼46 Å, almost all the interactions with the protein are attributed to Lys48, thus revealing a major role of the residue in coordinating the anion to compensate the partial depletion of its solvation sphere. The central segment of the pore axis reveals a fluctuating pattern where the contacts between the anion and the Arg145 residue are predominant. At the sites around ∼27 and ∼45 Å, corresponding to a pronounced dehydration of the ion, there is substantial interaction with the protein, albeit not with the Arg145 and Lys48 side chains.
Figure 8.

Contributions to the coordination profiles of the Cl– ion in the Pore II cavity. The analysis includes the oxygen atom of the water molecules, the guanidine nitrogen atoms of Arg145, the amine nitrogen atom of both Lys48 and Lys65, the hydroxyl oxygen atom of Ser74, and the non-specific heavy atoms of the protein (also including the above-mentioned residues).
We next analyzed the time evolution of the hydration pattern in specific US windows related to representative hotspots in the hydration profile. We mapped the 20 ns-long trajectories of windows 27, 30, 61, and 63 (corresponding to the same positions, expressed in Å, along the pore axis). In window 27 (Figure 9A), the Cl– ion loses up to two coordinating water molecules (see also Figure 7D). From the analysis of the trajectory, we obtained an average coordination number between the anion and the protein of 1.90 ± 0.24, mainly due to the interaction with the positively charged Arg145 side chain, the polar Ser74 side chain, and, to a minor extent, the Lys48 residue. Conversely, in window 30 (Figure 9B), Cl– is fully hydrated. In this position, we calculated the number of contacts with the negatively charged Glu146 and Asp149, and, as expected, none of them was detected along the entire trajectory. The average coordination number with the protein is only 0.30 ± 0.11, and it is associated with few contacts between the anion and the neighboring positively charged (Arg145) or polar (Ser74) residues. Remarkably, this window corresponds to a high-energy region for the Cl– (Figure 5), revealing a major role of the pore-lining negatively charged residues in blocking the anion permeation. Moreover, we investigated the two windows 61 and 63 (Figure 9C,D), where the Cl– ion loses almost two coordinating water molecules. This region of the cavity is one of the most constricted (Figure 6B), and it also includes the Lys65. In window 61 (Figure 9C), the anion dehydration is mainly due to the stabilizing electrostatic contact with the positively charged Lys65 side chain, with a coordination number of 0.67 ± 0.10, which represents almost the totality of the anion–protein interaction. In contrast, in window 63, the average coordination number of the protein in contact with Cl– is 1.62 ± 0.19, but the contribution of Lys65 is only 0.34 ± 0.10, revealing that the coordination sphere of the anion is completed by multiple contacts with different polar residues such as Ser58, Gln57, and Gln63. Consequently, in the segment spanning between 55 Å and 70 Å (and, symmetrically, between 5 and 20 Å), the stabilizing electrostatic interaction between the anion and Lys65 cooperates with the steric occlusion and the subsequent contact with the polar side chains of other pore-lining residues. The most external regions correspond to relatively low-energy values (Figure 5), confirming that the electrostatic interactions between the anion and the protein control the permeation process.
Figure 9.

Analysis of the coordination environment of the Cl– ion along the 20 ns-long trajectories in windows (A) 27, (B) 30, (C) 61, and (D) 63 of the US scheme.
Discussion
TJs are complex intercellular systems observed in both epithelial and endothelial cells, responsible for the control of the paracellular diffusion processes. Among the various tissue-specific TJs, a major interest is devoted to those located in the BBB. Although it is well known that Cldn5 proteins are the backbone of TJ strands of the BBB, we still miss a complete understanding of how they seal the paracellular spaces by oligomerization within the same cell (via cis interactions) and between adjacent cells (via trans interactions).
Here, we refined two structural models of pore-forming Cldn5 complexes that have been recently introduced.40 These models, proposed for Cldn5 and other Cldns, are only partly consistent with experimental results, so that their validation has not already been concluded.37,40 Pore I is in agreement with the structural model proposed by Suzuki and collaborators for the homologous Cldn15,29 in which two protomers belonging to the same cell interact with their ECL1 domains to form a cis-dimer. A couple of these dimers from two opposite cells are then supposed to interact in trans forming a Cldn tetramer characterized by a β-barrel-like paracellular cavity.29 After the publication of this model, criticisms were expressed about its validity,12 regarding steric hindrances at the paracellular interface and inconsistency with the experimentally demonstrated interactions between TM helices of cis-protomers.27,28,42 On the contrary, in the Pore II structure, Cldn5 cis-dimers are formed via a leucine zipper pattern belonging to TM2/TM3. In particular, TM2/TM3-mediated interactions have been previously described by experiments of Cldn5-based systems.28 Several successive works revised the two models.
Various research groups successfully refined the Pore I configuration for Cldn15 and other Cldns,31−38 showing that non-overlapping conformations for the ECLs are possible and that the resulting tetramer is stable. Moreover, experimental results based on electron microscopy techniques42,59 indicated an arrangement compatible with the Pore I configuration for Cldn3, Cldn10b, and Cldn11. Additional experiments for Cldn3, Cldn10b, and Cldn15 showed that the palmitoyl groups are located in proximity of the TM domains.42,60,61 This last occurrence raises the possibility that palmitoylation could perturb the tight packing of cis-interactions at the TM level, thus favoring the Pore I configuration. On the contrary, the multiscale molecular simulations described in ref (62) reported that Cldn5 palmitoylation enhances the probability of the dimeric arrangement that characterizes Pore II over the other possible cis-configurations occurring at the TM level. Nevertheless, in the absence of a conclusive experimental result able to discriminate between the two models, both the configurations remain worthy of refinement and study, also in the hypothesis of a heterogeneous distribution of the two pore arrangements. Indeed, computational works indicate the potential coexistence of the two pores in the same TJ strands. Coarse-grained MD simulations of self-assembly of Cldn protomers suggest diverse possible cis-dimers arrangements in the strand, all consistent with the units that construct the two pore configurations.34,35,40 Furthermore, the same authors confirmed the formation of similar dimers using the PANEL software to obtain millions of Cldn–Cldn conformations and to analyze the amino acid contact maps.41,63 The results showed the presence of both the Pore I and Pore II structures investigated in this work for Cldn5.
In this framework, we used MD simulations and FE calculations to quantify the thermodynamic features of ionic permeation events through the pore cavities of the two Cldn5 models. The study of ionic processes across biological channels has been an important topic in molecular modeling to look into the details of protein models.64 In our previous work, we used the same approach to refine the configuration of the Cldn15 pore based on the original structure of ref (29). Our efforts contributed to the validation of this structure, confirming the role of the investigation of ion permeation processes for structural validations.
In this work, we extended our analysis to the two structural models built for Cldn5 subunits. The HOLE profiles revealed a different pore shape between the two models. The Pore I structure is characterized by an hourglass shape, with the inner constriction in correspondence of residues Gln57, Gln63, and Lys65 of the four subunits, measuring ∼ 5–6 Å in diameter. On the contrary, the Pore II structure has two constrictions of ∼6 Å in diameter each in proximity of one of the two entrances, where the same residues Gln57, Gln63, and Lys65, now from two subunits, are located. Indeed, it is remarkable that, despite their different topologies, the narrowest pore regions in both models are related to the same set of residues. Our FE calculations reveal that both the pores are water permeable, a feature not yet fully clarified experimentally but consistent with previous computational results37,40 and postulated by some authors.65 On the contrary, the pores show FE barriers to cations. Interestingly, in both the models, the position of the cation barriers corresponds to the narrowest regions. This is in line with the fact that the minimum pore diameters are close to the size of a hydrated Na+ ion and slightly smaller than the diameter of the Cl– hydration shell.
This observation pushed us to investigate the details of ion hydration during Na+ and Cl– permeation by US simulations. The passage of the Na+ ion through the constrictions induces a partial dehydration of its shell. This contributes to generate the FE barrier, together with the unfavorable electrostatic repulsions between the cation and the Lys65 residues of the different Cldn5 subunits. The coupling of steric and electrostatic effects has been already observed in another Cldn-based paracellular system,33 and it is also relevant for the study of other, more conventional, narrow protein channels such as gramicidin66 or in the selectivity filter of K+67,68 and Na+69 channels.
As for the hydration pattern of Cl–, both steric and electrostatic effects induced pronounced fluctuations in the number of water molecules coordinating the anion passing through the pore axis. The smaller solvation energy of Cl– with respect to Na+ (−6.4 and −17.2 kcal/mol, respectively70,71) provides a more labile coordinating shell to the anion, and the effects are particularly evident in the regions of the cavities where the pore radius is comparable to the size of the Cl– hydration sphere. The analysis of the coordinating environment of the anion permeating the Pore II cavity revealed that the energetics of the barriers are mainly driven by the unfavorable electrostatic interactions with the pore-lining acidic residues, while the depletion of the solvation sphere due to steric hindrance is not correlated with high-energy regions. Conversely, a stabilizing interacting network with positively charged and polar amino acids able to fill the solvation sphere of the ion is observed in the regions of maximal constriction. For both the models, the FE profiles suggest that the permeation of Cl– is limited by the presence of the negatively charged Asp149 and Glu146 residues. These data indicate the absence of preference for cation versus anion selectivity for both the pore models in agreement with the well-known characteristics of the BBB.20,46,47 Our results complement those exposed for Pore I in ref (37), extending the validation of the two models in terms of ionic permeation features.
In recent years, peptides and peptidomimetics have attracted interest as potentially useful tools in therapeutic approaches for a variety of pathologies.72 So far, however, the lack of detailed structural information has limited the design of specific inhibitors of Cldn5 polymerization.25 The overarching goal of the present work is to contribute to the understanding of Cldn5 assemblies’ structural details, so as to facilitate the future design of new generations of peptides. Still, moving from structure-based, computational design to in vitro testing and then effective in vivo treatments presents significant challenges. As for the BBB, new sophisticated experimental platforms are designed to overcome the limitations of the most commonly used Transwell system. A 3D human multicellular assembloid model of the BBB with more advanced barrier properties has been recently developed and tested for its functionality.73,74 The assembloid includes endothelial cells, astrocytes, and pericytes, spontaneously organizing in a layered spherical structure around an internal brain-like core to mimic the in vivo barrier architecture and the synergistic interactions between the three cellular components. The development of BBB assembloids together with the recently reported choroid plexus organoids75 adds unprecedented opportunities to study the BBB’s physiology and its transient, non-disruptive modulation by the peptidomimetic approach that we are currently pursuing under the guide of the MD structural predictions. As for in vivo, the most successful approach to date provided encouraging results in early-stage clinical trials, showing non-invasive, reversible permeabilization of the BBB via transient opening of TJs by focused ultrasound.76 At present, however, there are very few in vivo approaches to safely alter the BBB permeability by targeting specific molecules,77 making proof-of-concept studies crucial to the development of potentially effective treatments.
Conclusions
The BBB plays a pivotal role in controlling the brain homeostasis, thanks to its high selectivity that prevents the passage of harmful molecules from the blood. As a consequence, it is a significant obstacle to effective brain drug delivery in the treatment of CNS diseases.15−17,20,78−80 To overcome this limitation, strategies are emerging to enhance the BBB permeability by modulating the passive transport across the TJs in the paracellular space.21−25 This approach has already provided promising results from in vitro experiments of drug-enhancer peptides.81,82 Although it is well known that the TJ scaffold is essentially formed by Cldn5 protein complexes, the fine structural details of Cldn5 multimeric arrangement are still missing.7,12,83 Recently, two tetrameric pore-forming models have been introduced after computational investigations based on coarse-grained MD simulations.40 Despite the different topological configurations, both the structures, originally named Pore I and Pore II, recapitulate various features from experimental results,29,30,84 but a systematic comparison of these systems is still missing. In this work, we refined the structures of the two Cldn5 pore configurations in solvated double-bilayer environment by all-atom MD simulation. Then, we calculated the FE profiles for single water molecule/ion translocation across the two pores. Both the structures fit the typical barrier-like behavior of Cldn5 in the BBB TJs. The findings illustrated in this work extend our knowledge of Cldn5 TJ structures and, although in the simplest case of single-pore systems, offer a molecular description of the BBB Cldn5 role. Furthermore, by identifying Cldn5 homomeric interaction surfaces in the TJs, our results can contribute to develop experimental strategies to enhance the drug delivery process across the BBB by modulating the paracellular permeability.
Methods
Pore I
The Pore I configuration was assembled with four Cldn5 protomers matching the quaternary structure published by Suzuki et al.29 The Cldn5 protomers were modeled from the Cldn15 homologs using structures from two different works,30,32 obtaining two putative models for Pore I. The first model (named Model1) was built starting from the tetrameric configuration of Cldn15 simulated by Alberini et al.32 The second one (named Model2) was assembled starting from the configuration of the Cldn15 pore published by Suzuki et al.29 In this case, a single Cldn5 structure was modeled adopting the crystal structure of the mouse Cldn15 protomer as a template (PDB ID: 4P79).30
Model1
The pore simulated by Alberini et al.32 was disassembled in four separated Cldn15 monomers, which have adopted slightly different conformations after the simulated trajectories of 250 ns described in ref (32). Each of these four protomers was used for the homology modeling of four Cldn5 monomers via the SWISS-MODEL85 program. The four raw models of Cldn5 were then refined with the ModRefiner86 server. The resulting protomers of Cldn5 were superimposed on the Cldn15 template32 with the UCSF Chimera87 Matchmaker tool. Afterward, the tetrameric system was refined with the GalaxyRefineComplex tool,88,89 and the configuration with highest score was selected for MD simulations. The complex was then oriented with the pore axis parallel to the Cartesian y-axis and embedded in a double bilayer of pure 1-palmitoyl-2-oleoyl-sn-glycero-3-phosphocholine (POPC), solvated with explicit three-point (TIP3P)90 water molecules and charge-neutralized with counterions using VMD 1.9.3.91 The fully hydrogenated pdb file of the protein complex was generated with the CHARMM-GUI PDB manipulator tool.92,93 Two hexagonal membranes were generated using the membrane builder tool of the same platform93,94 and equilibrated separately for 10 ns with the NAMD 3.0 software95 and the CHARMM36m force field55 using hexagonal periodic boundary conditions. The final simulation box is a hexagonal prism with a base inscribed in a square of approximately 120.0 × 120.0 Å2 and a height of around 160.0 Å. The topology file was built with the psfgen tool of VMD 1.9.391 with the parameters of the CHARMM36m force field55 and the four disulfide bridges were preserved between residues Cys54 and Cys64 found in the ECL1 of each protomer.
Model2
The Cldn5 protomer for Model2 was built starting from the crystal structure of the isolated Cldn15 published by Suzuki et al. (PDB ID: 4P79).30 The crystal lacks a segment of eight residues (34–41) in ECL1 that is automatically built by SWISS-MODEL85 during the homology modeling of Cldn5. The resulting structure was refined with a ModRefiner,86 consistently with the workflow illustrated for the Model1 and replicated in four identical copies. Following the same protocol illustrated for Model1, the four Cldn5 protomers were assembled to form the tetrameric arrangement. Analogously, the optimal system was embedded in a hexagonal double POPC bilayer solvated with water and charge-neutralized with counterions.
Equilibration and Unbiased MD Simulations
Both Model1 and Model2 systems contain about 200000 atoms. They were equilibrated with a multistep protocol where, after a first energy minimization, they were heated up to 310 K and simulated for 30 ns with a progressive release of positional restraints on the heavy atoms. Each model was then simulated for 1 μs. To avoid any rigid body rotational or translational displacement of the protein, the coordinates of the Cα atoms of the residues 6, 9, 20, 23, 79, 82, 97, 100, 117, 120, 138, 141, 166, 169, 177, and 180, all belonging to the most external residues on the TM α helices, were restrained to their initial values by harmonic potentials. The use of restraints on TM and/or ECLs backbone atoms in the isolated pore conformations can be justified by the fact that this model structure misses the neighbor protomers of the strands, which in the physiological TJ architecture form a scaffold that constraints the pore, limiting the fluctuations of its domains. Notably, in all of our extended MD simulations of Pore I, the ECL domains were stable, preserving the β-barrel structure, and so we limited the restraints to few atoms of the TM helices. The systems were simulated in the NPT ensemble at P = 1 atm and T = 310 K, maintained by a Langevin thermostat and Nosé–Hoover Langevin piston.96,97 Long-range electrostatic interactions were computed using the Particle Mesh Ewald algorithm.98 Chemical bonds between hydrogen atoms and protein heavy atoms were constrained with SHAKE,99 while those of the water molecules with SETTLE.100 The NAMD 3.0 program95 with the CHARMM36m force field55 was used to perform the simulations. Based on the pore size and the presence of a hydrogen bond deemed structurally relevant in refs (7)(12), we selected Model2 to represent Pore I and continue with the FE calculations. The comparative analysis of the simulations is reported in the Supporting Information.
Pore II
The Pore II configuration was described in ref (40). In this architecture, the cis-interface originates from the interaction of two neighboring protomers at the level of the TM helices arranging in a leucine zipper composed by the residues Leu83, Leu90, Leu124, and Leu131 on TM2 and TM3, supported by two homophilic π–π interactions between Phe127 and Trp138 on the opposing TM domains (Figure 1B). To build this structure, we first simulated a Cldn5 protomer, again homology-modeled from the Cldn15 template. The protein was embedded in a rectangular pure POPC membrane bilayer and equilibrated with a 110 ns-long all-atom MD simulation in an explicit solvent. The trajectory was analyzed to assess the structural stability of the protein (see the Supporting Information). An equilibrated configuration of the Cldn5 protomer was extracted from the trajectory and used to reproduce the cis-interface via a docking protocol. The leucine zipper TM interaction between two copies of the protein was predicted by the MEMDOCK server,101 which includes a specific algorithm for docking α-helical membrane proteins. The dimer selected by MEMDOCK101 was further refined with DOCKING2,102−104 and the structure finely reproducing the leucine zipper was embedded in a pure POPC membrane, solvated with explicit water, and equilibrated with ∼100 ns of all-atom MD simulation in the presence of charge-neutralizing counterions. The final dimer complex was replicated, and the two copies were used to assemble the Pore II configuration with a further docking approach. Following the protocol suggested in ref (40), the Pore II complex was generated using ClusPro105−109 to reproduce the trans-interactions occurring between two opposing dimers at the level of the paracellular domains. Afterward, the tetrameric structure was refined using GalaxyRefine,88,89 oriented with the pore axis parallel to the Cartesian y-axis, and embedded in a hexagonal double bilayer of pure POPC, solvated with water and charge-neutralized with counterions. The topology file was built with the psfgen tool of VMD 1.9.391 with CHARMM36m parameters,55 and the four disulfide bridges were preserved between residues Cys54 and Cys64 found in the ECL1 of each protomer.
Equilibration and Unbiased MD Simulation
The Pore II simulation setup (∼200,000 atoms) followed the same protocol described for the two putative models of Pore I. Additionally, further harmonic restraints were applied on the Cα atoms of the residues 11, 14, 25, 28, 78, 81, 99, 102, 116, 119, 143, 146, 166, 169, 183, and 186 in the ECLs.
Pore Size Analysis
The size of the paracellular pores was monitored along the trajectory with the HOLE program.110,111 The algorithm maps the radius of a protein cavity along a given axis (here, the y-axis) by fitting a spherical probe with the van der Waals radii of the pore-lining atoms. For all the models, a 15 Å threshold was chosen for the pore radius, and representative structures spaced by 10 ns along the production trajectory were selected and analyzed (see the Supporting Information).
FE Calculations
The FE profiles for the permeation of a water molecule and single ions were calculated using the US method.48 A restraining term is added to the MD potential to confine a CV (function of the Cartesian coordinates of the system) in selected regions, named windows, allowing proper sampling even in high-energy regions of the landscape. As CV, we chose the coordinate of the tagged permeating ion along the pore axis, previously aligned with the Cartesian y-axis, and the restraining potential Vi(y) in each window i is
where yi0 indicates the value in Å at which the CV is restrained in the window (called center) and k is a constant that is appropriately chosen in order to ensure a sufficient overlap of the CV distributions from adjacent windows (in this work, we used k = 2.0 kcal/(molÅ2) for all the simulations). In each window, the displacement of the ion orthogonal to the pore axis is confined within a disk of radius r0 + δ, where r0 is the pore radius as determined by the HOLE program110,111 and δ = 2 Å. The equilibrated conformation of the system was used as the starting structure of all the US windows, and the ion was manually positioned at each center yi.
The Pore I cavity was split into 60 windows. Each window was minimized and simulated for 16 ns with the same setup described for the standard MD simulation, adding the bias to the force field112 via the colvars module.113 Positional restraints on selected Cα atoms were applied as described for the unbiased simulations. The first nanosecond of production was excluded from the statistics. Due to the elongated shape of the Pore II cavity, 75 windows were required to sample the entire cavity. The simulation followed the same protocol adopted for Pore I, except that 20 ns of production per window were carried out in order to achieve a proper convergence. The FE profiles are obtained by combining the CV distributions of all windows using the weighted histogram analysis method.49,114,115 We employed the code from the Grossfield group available at http://membrane.urmc.rochester.edu/content/wham. The block error analysis was also implemented to the calculated FE profiles (see the Supporting Information).
Acknowledgments
We thank Jörg Piontek and Matteo Ceccarelli for useful discussions; and Sergio Decherchi, Diego Moruzzo, and Andrea L. Benfenati for valuable help. Computing time allocations were granted by the CINECA supercomputing center under the ISCRA initiative. We also gratefully acknowledge the HPC infrastructure and the Support Team at Fondazione Istituto Italiano di Tecnologia.
Glossary
Abbreviations
- BBB
Blood–brain barrier
- CLDN
Claudin
- CNS
Central nervous system
- CV
Collective variable
- ECL
Extracellular loop
- FE
Free energy
- MD
Molecular dynamics
- PMF
Potential of mean force
- POPC
1-palmitoyl-2-oleoyl-sn-glycero-3-phosphocholine
- RMSD
Root-mean-square deviation
- TJ
Tight junction
- TM
Trans-membrane
- US
Umbrella sampling
- WHAM
Weighted histogram analysis method.
Supporting Information Available
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/acschemneuro.2c00139.
All-atom MD simulation results and analysis of the Pore I models, free energy profiles with the block error analysis, and all-atom MD simulation results and analysis of the Cldn5 protomer (PDF)
Author Contributions
⊥ A.B. and G.A. equally contributed to perform the simulations, analyze the data, discuss the results, and write the manuscript. L.M. and F.B. supervised the research project, discussed the results, and revised the manuscript.
The research was supported by IRCCS Ospedale Policlinico San Martino (Ricerca Corrente and 5 × 1000 grants to FB and LM) and by Telethon/Glut-1 Onlus Foundations (GSP19002_PAsGlut009 and GSA22A002 projects to FB).
The authors declare no competing financial interest.
Supplementary Material
References
- Furuse M.; Sasaki H.; Fujimoto K.; Tsukita S. A Single Gene Product, Claudin-1 or -2, Reconstitutes Tight Junction Strands and Recruits Occludin in Fibroblasts. J. Cell Biol. 1998, 143, 391–401. 10.1083/jcb.143.2.391. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morita K.; Sasaki H.; Furuse M.; Tsukita S. Endothelial Claudin. J. Cell Biol. 1999, 147, 185–194. 10.1083/jcb.147.1.185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Anderson J. M. Molecular Structure of Tight Junctions and Their Role in Epithelial Transport. Physiology 2001, 16, 126–130. 10.1152/physiologyonline.2001.16.3.126. [DOI] [PubMed] [Google Scholar]
- Farquhar M. G.; Palade G. E. Junctional Complexes in Various Epithelia. J. Cell Biol. 1963, 17, 375–412. 10.1083/jcb.17.2.375. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gumbiner B. Structure, Biochemistry, and Assembly of Epithelial Tight Junctions. Am. J. Physiol. Cell Physiol. 1987, 253, C749–C758. 10.1152/ajpcell.1987.253.6.C749. [DOI] [PubMed] [Google Scholar]
- Krause G.; Winkler L.; Mueller S. L.; Haseloff R. F.; Piontek J.; Blasig I. E. Structure and Function of Claudins. Biochim. Biophys. Acta Biomembr. 2008, 1778, 631–645. 10.1016/j.bbamem.2007.10.018. [DOI] [PubMed] [Google Scholar]
- Piontek J.; Krug S. M.; Protze J.; Krause G.; Fromm M. Molecular Architecture and Assembly of the Tight Junction Backbone. Biochim. Biophys. Acta Biomembr. 2020, 1862, 183279. 10.1016/j.bbamem.2020.183279. [DOI] [PubMed] [Google Scholar]
- Claude P.; Goodenough D. A. Fracture Faces of Zonulae Occludentes from “Tight” and “Leaky” Epithelia. J. Cell Biol. 1973, 58, 390–400. 10.1083/jcb.58.2.390. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Claude P. Morphological Factors Influencing Transepithelial Permeability: A Model for the Resistance of TheZonula Occludens. J. Membr. Biol. 1978, 39, 219–232. 10.1007/BF01870332. [DOI] [PubMed] [Google Scholar]
- Bonetta L. Endothelial Tight Junctions Form the Blood–Brain Barrier. J. Cell Biol. 2005, 169, 378–379. 10.1083/jcb1693fta1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Günzel D.; Yu A. S. L. Claudins and the Modulation of Tight Junction Permeability. Physiol. Rev. 2013, 93, 525–569. 10.1152/physrev.00019.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krause G.; Protze J.; Piontek J. Assembly and Function of Claudins: Structure-Function Relationships Based on Homology Models and Crystal Structures. Semin. Cell Dev. Biol. 2015, 42, 3–12. 10.1016/j.semcdb.2015.04.010. [DOI] [PubMed] [Google Scholar]
- Piorntek J.; Winkler L.; Wolburg H.; Müller S. L.; Zuleger N.; Piehl C.; Wiesner B.; Krause G.; Blasig I. E. Formation of Tight Junction: Determinants of Homophilic Interaction between Classic Claudins. FASEB J. 2008, 22, 146–158. 10.1096/fj.07-8319com. [DOI] [PubMed] [Google Scholar]
- Angelow S.; Ahlstrom R.; Yu A. S. L. Biology of Claudins. Am. J. Physiol. Ren. Physiol. 2008, 295, F867–F876. 10.1152/ajprenal.90264.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Powell D. W. Barrier Function of Epithelia. Am. J. Physiol. 1981, 241, G275–G288. 10.1152/ajpgi.1981.241.4.G275. [DOI] [PubMed] [Google Scholar]
- Tang V. W.; Goodenough D. A. Paracellular Ion Channel at the Tight Junction. Biophys. J. 2003, 84, 1660–1673. 10.1016/S0006-3495(03)74975-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bauer H.-C.; Krizbai I. n. A.; Bauer H.; Traweger A. You Shall Not Pass”—Tight Junctions of the Blood Brain Barrier. Front. Neurosci. 2014, 8, 392. 10.3389/fnins.2014.00392. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Abbott N. J.; Patabendige A. A. K.; Dolman D. E. M.; Yusof S. R.; Begley D. J. Structure and Function of the Blood-Brain Barrier. Neurobiol. Dis. 2010, 37, 13–25. 10.1016/j.nbd.2009.07.030. [DOI] [PubMed] [Google Scholar]
- Daneman R.; Prat A. The Blood-Brain Barrier. Cold Spring Harbor Perspect. Biol. 2015, 7, a020412. 10.1101/cshperspect.a020412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pardridge W. M. The Blood-Brain Barrier: Bottleneck in Brain Drug Development. NeuroRX 2005, 2, 3–14. 10.1602/neurorx.2.1.3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hashimoto Y.; Campbell M. Tight Junction Modulation at the Blood-Brain Barrier: Current and Future Perspectives. Biochim. Biophys. Acta Biomembr. 2020, 1862, 183298. 10.1016/j.bbamem.2020.183298. [DOI] [PubMed] [Google Scholar]
- Staat C.; Coisne C.; Dabrowski S.; Stamatovic S. M.; Andjelkovic A. V.; Wolburg H.; Engelhardt B.; Blasig I. E. Mode of Action of Claudin Peptidomimetics in the Transient Opening of Cellular Tight Junction Barriers. Biomaterials 2015, 54, 9–20. 10.1016/j.biomaterials.2015.03.007. [DOI] [PubMed] [Google Scholar]
- Neuhaus W.; Piontek A.; Protze J.; Eichner M.; Mahringer A.; Subileau E.-A.; Lee I.-F. M.; Schulzke J. D.; Krause G.; Piontek J. Reversible Opening of the Blood-Brain Barrier by Claudin-5-Binding Variants of Clostridium Perfringens Enterotoxin’s Claudin-Binding Domain. Biomaterials 2018, 161, 129–143. 10.1016/j.biomaterials.2018.01.028. [DOI] [PubMed] [Google Scholar]
- Zwanziger D.; Hackel D.; Staat C.; Böcker A.; Brack A.; Beyermann M.; Rittner H.; Blasig I. E. A Peptidomimetic Tight Junction Modulator to Improve Regional Analgesia. Mol. Pharm. 2012, 9, 1785–1794. 10.1021/mp3000937. [DOI] [PubMed] [Google Scholar]
- Dithmer S.; Staat C.; Müller C.; Ku M.-C.; Pohlmann A.; Niendorf T.; Gehne N.; Fallier-Becker P.; Kittel Á.; Walter F. R.; Veszelka S.; Deli M. A.; Blasig R.; Haseloff R. F.; Blasig I. E.; Winkler L. Claudin Peptidomimetics Modulate Tissue Barriers for Enhanced Drug Delivery. Ann. N.Y. Acad. Sci. 2017, 1397, 169–184. 10.1111/nyas.13359. [DOI] [PubMed] [Google Scholar]
- Angelow S.; Yu A. S. L. Structure-Function Studies of Claudin Extracellular Domains by Cysteine-Scanning Mutagenesis. J. Biol. Chem. 2009, 284, 29205–29217. 10.1074/jbc.M109.043752. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Van Itallie C. M.; Mitic L. L.; Anderson J. M. Claudin-2 Forms Homodimers and Is a Component of a High Molecular Weight Protein Complex. J. Biol. Chem. 2011, 286, 3442–3450. 10.1074/jbc.M110.195578. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rossa J.; Ploeger C.; Vorreiter F.; Saleh T.; Protze J.; Günzel D.; Wolburg H.; Krause G.; Piontek J. Claudin-3 and Claudin-5 Protein Folding and Assembly into the Tight Junction Are Controlled by Non-Conserved Residues in the Transmembrane 3 (TM3) and Extracellular Loop 2 (ECL2) Segments. J. Biol. Chem. 2014, 289, 7641–7653. 10.1074/jbc.M113.531012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Suzuki H.; Tani K.; Tamura A.; Tsukita S.; Fujiyoshi Y. Model for the Architecture of Claudin-Based Paracellular Ion Channels through Tight Junctions. J. Mol. Biol. 2015, 427, 291–297. 10.1016/j.jmb.2014.10.020. [DOI] [PubMed] [Google Scholar]
- Suzuki H.; Nishizawa T.; Tani K.; Yamazaki Y.; Tamura A.; Ishitani R.; Dohmae N.; Tsukita S.; Nureki O.; Fujiyoshi Y. Crystal Structure of a Claudin Provides Insight into the Architecture of Tight Junctions. Science 2014, 344, 304–307. 10.1126/science.1248571. [DOI] [PubMed] [Google Scholar]
- Fuladi S.; Jannat R.-W.; Shen L.; Weber C. R.; Khalili-Araghi F. Computational Modeling of Claudin Structure and Function. Int. J. Mol. Sci. 2020, 21, 742. 10.3390/ijms21030742. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alberini G.; Benfenati F.; Maragliano L. A Refined Model of Claudin-15 Tight Junction Paracellular Architecture by Molecular Dynamics Simulations. PLOS ONE 2017, 12, e0184190 10.1371/journal.pone.0184190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alberini G.; Benfenati F.; Maragliano L. Molecular Dynamics Simulations of Ion Selectivity in a Claudin-15 Paracellular Channel. J. Phys. Chem. B 2018, 122, 10783–10792. 10.1021/acs.jpcb.8b06484. [DOI] [PubMed] [Google Scholar]
- Irudayanathan F. J.; Trasatti J. P.; Karande P.; Nangia S. Molecular Architecture of the Blood Brain Barrier Tight Junction Proteins--A Synergistic Computational and In Vitro Approach. J. Phys. Chem. B 2016, 120, 77–88. 10.1021/acs.jpcb.5b09977. [DOI] [PubMed] [Google Scholar]
- Irudayanathan F. J.; Wang X.; Wang N.; Willsey S. R.; Seddon I. A.; Nangia S. Self-Assembly Simulations of Classic Claudins-Insights into the Pore Structure, Selectivity, and Higher Order Complexes. J. Phys. Chem. B 2018, 122, 7463–7474. 10.1021/acs.jpcb.8b03842. [DOI] [PubMed] [Google Scholar]
- Samanta P.; Wang Y.; Fuladi S.; Zou J.; Li Y.; Shen L.; Weber C.; Khalili-Araghi F. Molecular Determination of Claudin-15 Organization and Channel Selectivity. J. Gen. Physiol. 2018, 150, 949–968. 10.1085/jgp.201711868. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Irudayanathan F. J.; Nangia S. What Does It Take for an Ion to Pass Through a Tight Junction Pore?. Langmuir 2020, 36, 6757–6764. 10.1021/acs.langmuir.0c00877. [DOI] [PubMed] [Google Scholar]
- Zhao J.; Krystofiak E. S.; Ballesteros A.; Cui R.; Van Itallie C. M.; Anderson J. M.; Fenollar-Ferrer C.; Kachar B. Multiple Claudin-Claudin Cis Interfaces Are Required for Tight Junction Strand Formation and Inherent Flexibility. Commun. Biol. 2018, 1, 50. 10.1038/s42003-018-0051-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Günzel D.; Fromm M. Claudins and Other Tight Junction Proteins. Compr. Physiol. 2012, 2, 1819–1852. 10.1002/cphy.c110045. [DOI] [PubMed] [Google Scholar]
- Irudayanathan F. J.; Wang N.; Wang X.; Nangia S. Architecture of the Paracellular Channels Formed by Claudins of the Blood-Brain Barrier Tight Junctions. Ann. N.Y. Acad. Sci. 2017, 1405, 131–146. 10.1111/nyas.13378. [DOI] [PubMed] [Google Scholar]
- Rajagopal N.; Durand A. J.; Nangia S. Predicting Selectivity of Paracellular Pores for Biomimetic Applications. Mol. Syst. Des. Eng. 2020, 5, 686–696. 10.1039/C9ME00177H. [DOI] [Google Scholar]
- Hempel C.; Protze J.; Altun E.; Riebe B.; Piontek A.; Fromm A.; Lee I. M.; Saleh T.; Günzel D.; Krause G.; Piontek J. Assembly of Tight Junction Strands: Claudin-10b and Claudin-3 Form Homo-Tetrameric Building Blocks That Polymerise in a Channel-Independent Manner. J. Mol. Biol. 2020, 432, 2405–2427. 10.1016/j.jmb.2020.02.034. [DOI] [PubMed] [Google Scholar]
- Yu A. S. L.; Cheng M. H.; Angelow S.; Günzel D.; Kanzawa S. A.; Schneeberger E. E.; Fromm M.; Coalson R. D. Molecular Basis for Cation Selectivity in Claudin-2–Based Paracellular Pores: Identification of an Electrostatic Interaction Site. J. Gen. Physiol. 2008, 133, 111–127. 10.1085/jgp.200810154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Colegio O. R.; Itallie C. V.; Rahner C.; Anderson J. M. Claudin Extracellular Domains Determine Paracellular Charge Selectivity and Resistance but Not Tight Junction Fibril Architecture. Am. J. Physiol. Cell Physiol. 2003, 284, C1346–C1354. 10.1152/ajpcell.00547.2002. [DOI] [PubMed] [Google Scholar]
- Van Itallie C. M.; Anderson J. M. Claudins and Epithelial Paracellular Transport. Annu. Rev. Physiol. 2006, 68, 403–429. 10.1146/annurev.physiol.68.040104.131404. [DOI] [PubMed] [Google Scholar]
- Wen H.; Watry D. D.; Marcondes M. C. G.; Fox H. S. Selective Decrease in Paracellular Conductance of Tight Junctions: Role of the First Extracellular Domain of Claudin-5. Mol. Cell. Biol. 2004, 24, 8408–8417. 10.1128/MCB.24.19.8408-8417.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nitta T.; Hata M.; Gotoh S.; Seo Y.; Sasaki H.; Hashimoto N.; Furuse M.; Tsukita S. Size-Selective Loosening of the Blood-Brain Barrier in Claudin-5-Deficient Mice. J. Cell Biol. 2003, 161, 653–660. 10.1083/jcb.200302070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Torrie G. M.; Valleau J. P. Nonphysical Sampling Distributions in Monte Carlo Free-Energy Estimation: Umbrella Sampling. J. Comput. Phys. 1977, 23, 187–199. 10.1016/0021-9991(77)90121-8. [DOI] [Google Scholar]
- Kumar S.; Rosenberg J. M.; Bouzida D.; Swendsen R. H.; Kollman P. A. The Weighted Histogram Analysis Method for Free-Energy Calculations on Biomolecules. I. The Method. J. Comput. Chem. 1992, 13, 1011–1021. 10.1002/jcc.540130812. [DOI] [Google Scholar]
- Krug S. M.; Günzel D.; Conrad M. P.; Lee I.-F. M.; Amasheh S.; Fromm M.; Yu A. S. L. Charge-Selective Claudin Channels. Ann. N.Y. Acad. Sci. 2012, 1257, 20–28. 10.1111/j.1749-6632.2012.06555.x. [DOI] [PubMed] [Google Scholar]
- Hou J.; Renigunta A.; Yang J.; Waldegger S. Claudin-4 Forms Paracellular Chloride Channel in the Kidney and Requires Claudin-8 for Tight Junction Localization. Proc. Natl. Acad. Sci. U. S. A. 2010, 107, 18010–18015. 10.1073/pnas.1009399107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Van Itallie C. M.; Fanning A. S.; Anderson J. M. Reversal of Charge Selectivity in Cation or Anion-Selective Epithelial Lines by Expression of Different Claudins. Am. J. Physiol. Ren. Physiol. 2003, 285, F1078–F1084. 10.1152/ajprenal.00116.2003. [DOI] [PubMed] [Google Scholar]
- Colegio O. R.; Van Itallie C. M.; McCrea H. J.; Rahner C.; Anderson J. M. Claudins Create Charge-Selective Channels in the Paracellular Pathway between Epithelial Cells. Am. J. Physiol. Cell Physiol. 2002, 283, C142–C147. 10.1152/ajpcell.00038.2002. [DOI] [PubMed] [Google Scholar]
- Abraham M. J.; Murtola T.; Schulz R.; Páll S.; Smith J. C.; Hess B.; Lindahl E. GROMACS: High Performance Molecular Simulations through Multi-Level Parallelism from Laptops to Supercomputers. SoftwareX 2015, 1-2, 19–25. 10.1016/j.softx.2015.06.001. [DOI] [Google Scholar]
- Huang J.; Rauscher S.; Nawrocki G.; Ran T.; Feig M.; de Groot B. L.; Grubmüller H.; MacKerell A. D. CHARMM36m: An Improved Force Field for Folded and Intrinsically Disordered Proteins. Nat. Methods 2017, 14, 71–73. 10.1038/nmeth.4067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Loubet B.; Kopec W.; Khandelia H. Accelerating All-Atom MD Simulations of Lipids Using a Modified Virtual-Sites Technique. J. Chem. Theory Comput. 2014, 10, 5690–5695. 10.1021/ct500100f. [DOI] [PubMed] [Google Scholar]
- Barducci A.; Bussi G.; Parrinello M. Well-Tempered Metadynamics: A Smoothly Converging and Tunable Free-Energy Method. Phys. Rev. Lett. 2008, 100, 020603. 10.1103/PhysRevLett.100.020603. [DOI] [PubMed] [Google Scholar]
- Lev B.; Roux B.; Noskov S. Y. Relative Free Energies for Hydration of Monovalent Ions from QM and QM/MM Simulations. J. Chem. Theory Comput. 2013, 9, 4165–4175. 10.1021/ct400296w. [DOI] [PubMed] [Google Scholar]
- Krystofiak E. S.; Heymann J. B.; Kachar B. Carbon Replicas Reveal Double Stranded Structure of Tight Junctions in Phase-Contrast Electron Microscopy. Commun. Biol. 2019, 2, 98. 10.1038/s42003-019-0319-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rodenburg R. N. P.; Snijder J.; van de Waterbeemd M.; Schouten A.; Granneman J.; Heck A. J. R.; Gros P. Stochastic Palmitoylation of Accessible Cysteines in Membrane Proteins Revealed by Native Mass Spectrometry. Nat. Commun. 2017, 8, 1280. 10.1038/s41467-017-01461-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Van Itallie C. M.; Gambling T. M.; Carson J. L.; Anderson J. M. Palmitoylation of Claudins Is Required for Efficient Tight-Junction Localization. J. Cell Sci. 2005, 118, 1427–1436. 10.1242/jcs.01735. [DOI] [PubMed] [Google Scholar]
- Rajagopal N.; Irudayanathan F. J.; Nangia S. Palmitoylation of Claudin-5 Proteins Influences Their Lipid Domain Affinity and Tight Junction Assembly at the Blood-Brain Barrier Interface. J. Phys. Chem. B 2019, 123, 983–993. 10.1021/acs.jpcb.8b09535. [DOI] [PubMed] [Google Scholar]
- Rajagopal N.; Nangia S. Obtaining Protein Association Energy Landscape for Integral Membrane Proteins. J. Chem. Theory Comput. 2019, 15, 6444–6455. 10.1021/acs.jctc.9b00626. [DOI] [PubMed] [Google Scholar]
- Khalili-Araghi F.; Tajkhorshid E.; Roux B.; Schulten K. Molecular Dynamics Investigation of the ω-Current in the Kv1.2 Voltage Sensor Domains. Biophys. J. 2012, 102, 258–267. 10.1016/j.bpj.2011.10.057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fisher D.; Mentor S. Are Claudin-5 Tight-Junction Proteins in the Blood-Brain Barrier Porous?. Neural Regener. Res. 2020, 15, 1838–1839. 10.4103/1673-5374.280308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Allen T. W.; Andersen O. S.; Roux B. Energetics of Ion Conduction through the Gramicidin Channel. Proc. Natl. Acad. Sci. U.S.A. 2004, 101, 117–122. 10.1073/pnas.2635314100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bostick D. L.; Brooks C. L. Selectivity in K+ Channels Is Due to Topological Control of the Permeant Ion’s Coordinated State. Proc. Natl. Acad. Sci. U. S. A. 2007, 104, 9260–9265. 10.1073/pnas.0700554104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Noskov S. Yu.; Roux B. Importance of Hydration and Dynamics on the Selectivity of the KcsA and NaK Channels. J. Gen. Physiol. 2007, 129, 135–143. 10.1085/jgp.200609633. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Corry B.; Thomas M. Mechanism of Ion Permeation and Selectivity in a Voltage Gated Sodium Channel. J. Am. Chem. Soc. 2012, 134, 1840–1846. 10.1021/ja210020h. [DOI] [PubMed] [Google Scholar]
- Palascak M. W.; Shields G. C. Accurate Experimental Values for the Free Energies of Hydration of H+, OH-, and H3O+. J. Phys. Chem. A 2004, 108, 3692–3694. 10.1021/jp049914o. [DOI] [PubMed] [Google Scholar]
- Lamoureux G.; Roux B. Absolute Hydration Free Energy Scale for Alkali and Halide Ions Established from Simulations with a Polarizable Force Field. J. Phys. Chem. B 2006, 110, 3308–3322. 10.1021/jp056043p. [DOI] [PubMed] [Google Scholar]
- Kaspar A. A.; Reichert J. M. Future Directions for Peptide Therapeutics Development. Drug Discovery Today 2013, 18, 807–817. 10.1016/j.drudis.2013.05.011. [DOI] [PubMed] [Google Scholar]
- Bergmann S.; Lawler S. E.; Qu Y.; Fadzen C. M.; Wolfe J. M.; Regan M. S.; Pentelute B. L.; Agar N. Y. R.; Cho C.-F. Blood-Brain-Barrier Organoids for Investigating the Permeability of CNS Therapeutics. Nat. Protoc. 2018, 13, 2827–2843. 10.1038/s41596-018-0066-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pisani F.; Castagnola V.; Simone L.; Loiacono F.; Svelto M.; Benfenati F. Tunneling Nanotubes Connect Blood-Brain Barrier Cells: Role of Pericytes in Barrier Preservation During Ischemia. Cell Death Dis. 2022, 13, 582. 10.1101/2022.03.02.482668. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parichha A.; Suresh V.; Chatterjee M.; Kshirsagar A.; Ben-Reuven L.; Olender T.; Taketo M. M.; Radosevic V.; Bobic-Rasonja M.; Trnski S.; Holtzman M. J.; Jovanov-Milosevic N.; Reiner O.; Tole S. Constitutive Activation of Canonical Wnt Signaling Disrupts Choroid Plexus Epithelial Fate. Nat. Commun. 2022, 13, 633. 10.1038/s41467-021-27602-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rezai A. R.; Ranjan M.; D’Haese P.-F.; Haut M. W.; Carpenter J.; Najib U.; Mehta R. I.; Chazen J. L.; Zibly Z.; Yates J. R.; Hodder S. L.; Kaplitt M. Noninvasive Hippocampal Blood-Brain Barrier Opening in Alzheimer’s Disease with Focused Ultrasound. Proc. Natl. Acad. Sci. U.S.A. 2020, 117, 9180–9182. 10.1073/pnas.2002571117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li X.; Vemireddy V.; Cai Q.; Xiong H.; Kang P.; Li X.; Giannotta M.; Hayenga H. N.; Pan E.; Sirsi S. R.; Mateo C.; Kleinfeld D.; Greene C.; Campbell M.; Dejana E.; Bachoo R.; Qin Z. Reversibly Modulating the Blood-Brain Barrier by Laser Stimulation of Molecular-Targeted Nanoparticles. Nano Lett. 2021, 21, 9805–9815. 10.1021/acs.nanolett.1c02996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pardridge W. M. Drug Transport across the Blood–Brain Barrier. J. Cereb. Blood Flow Metab. 2012, 32, 1959–1972. 10.1038/jcbfm.2012.126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lochhead J. J.; Yang J.; Ronaldson P. T.; Davis T. P. Structure, Function, and Regulation of the Blood-Brain Barrier Tight Junction in Central Nervous System Disorders. Front. Physiol. 2020, 11, 914. 10.3389/fphys.2020.00914. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nation D. A.; Sweeney M. D.; Montagne A.; Sagare A. P.; D’Orazio L. M.; Pachicano M.; Sepehrband F.; Nelson A. R.; Buennagel D. P.; Harrington M. G.; Benzinger T. L. S.; Fagan A. M.; Ringman J. M.; Schneider L. S.; Morris J. C.; Chui H. C.; Law M.; Toga A. W.; Zlokovic B. V. Blood–Brain Barrier Breakdown Is an Early Biomarker of Human Cognitive Dysfunction. Nat. Med. 2019, 25, 270–276. 10.1038/s41591-018-0297-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bocsik A.; Walter F. R.; Gyebrovszki A.; Fülöp L.; Blasig I.; Dabrowski S.; Ötvös F.; Tóth A.; Rákhely G.; Veszelka S.; Vastag M.; Szabó-Révész P.; Deli M. A. Reversible Opening of Intercellular Junctions of Intestinal Epithelial and Brain Endothelial Cells With Tight Junction Modulator Peptides. J. Pharm. Sci. 2016, 105, 754–765. 10.1016/j.xphs.2015.11.018. [DOI] [PubMed] [Google Scholar]
- Liao Z.; Yang Z.; Piontek A.; Eichner M.; Krause G.; Li L.; Piontek J.; Zhang J. Specific Binding of a Mutated Fragment of Clostridium Perfringens Enterotoxin to Endothelial Claudin-5 and Its Modulation of Cerebral Vascular Permeability. Neuroscience 2016, 327, 53–63. 10.1016/j.neuroscience.2016.04.013. [DOI] [PubMed] [Google Scholar]
- Piontek J.; Fritzsche S.; Cording J.; Richter S.; Hartwig J.; Walter M.; Yu D.; Turner J. R.; Gehring C.; Rahn H.-P.; Wolburg H.; Blasig I. E. Elucidating the Principles of the Molecular Organization of Heteropolymeric Tight Junction Strands. Cell. Mol. Life Sci. 2011, 68, 3903–3918. 10.1007/s00018-011-0680-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rossa J.; Ploeger C.; Vorreiter F.; Saleh T.; Protze J.; Günzel D.; Wolburg H.; Krause G.; Piontek J. Claudin-3 and Claudin-5 Protein Folding and Assembly into the Tight Junction Are Controlled by Non-Conserved Residues in the Transmembrane 3 (TM3) and Extracellular Loop 2 (ECL2) Segments. J. Biol. Chem. 2014, 289, 7641–7653. 10.1074/jbc.M113.531012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Waterhouse A.; Bertoni M.; Bienert S.; Studer G.; Tauriello G.; Gumienny R.; Heer F. T.; de Beer T. A. P.; Rempfer C.; Bordoli L.; Lepore R.; Schwede T. SWISS-MODEL: Homology Modelling of Protein Structures and Complexes. Nucleic Acids Res. 2018, 46, W296–W303. 10.1093/nar/gky427. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu D.; Zhang Y. Improving the Physical Realism and Structural Accuracy of Protein Models by a Two-Step Atomic-Level Energy Minimization. Biophys. J. 2011, 101, 2525–2534. 10.1016/j.bpj.2011.10.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pettersen E. F.; Goddard T. D.; Huang C. C.; Couch G. S.; Greenblatt D. M.; Meng E. C.; Ferrin T. E. UCSF Chimera--a Visualization System for Exploratory Research and Analysis. J. Comput. Chem. 2004, 25, 1605–1612. 10.1002/jcc.20084. [DOI] [PubMed] [Google Scholar]
- Heo L.; Lee H.; Seok C. GalaxyRefineComplex: Refinement of Protein-Protein Complex Model Structures Driven by Interface Repacking. Sci. Rep. 2016, 6, 32153. 10.1038/srep32153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heo L.; Park H.; Seok C. GalaxyRefine: Protein Structure Refinement Driven by Side-Chain Repacking. Nucleic Acids Res. 2013, 41, W384–W388. Web Server issue) 10.1093/nar/gkt458. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jorgensen W. L.; Chandrasekhar J.; Madura J. D.; Impey R. W.; Klein M. L.. Comparison of Simple Potential Functions for Simulating Liquid Water. J. Chem. Phys. 1983, 79. 10.1063/1.445869. [DOI] [Google Scholar]
- Humphrey W.; Dalke A.; Schulten K. VMD: Visual Molecular Dynamics. J. Mol. Graphics 1996, 14, 33–38. 10.1016/0263-7855(96)00018-5. [DOI] [PubMed] [Google Scholar]
- Jo S.; Cheng X.; Islam S. M.; Huang L.; Rui H.; Zhu A.; Lee H. S.; Qi Y.; Han W.; Vanommeslaeghe K.; MacKerell A. D.; Roux B.; Im W. CHARMM-GUI PDB Manipulator for Advanced Modeling and Simulations of Proteins Containing Nonstandard Residues. Adv. Protein Chem. Struct. Biol. 2014, 96, 235–265. 10.1016/bs.apcsb.2014.06.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jo S.; Kim T.; Iyer V. G.; Im W. CHARMM-GUI: A Web-Based Graphical User Interface for CHARMM. J. Comput. Chem. 2008, 29, 1859–1865. 10.1002/jcc.20945. [DOI] [PubMed] [Google Scholar]
- Wu E. L.; Cheng X.; Jo S.; Rui H.; Song K. C.; Dávila-Contreras E. M.; Qi Y.; Lee J.; Monje-Galvan V.; Venable R. M.; Klauda J. B.; Im W. CHARMM-GUI Membrane Builder toward Realistic Biological Membrane Simulations. J. Comput. Chem. 2014, 35, 1997–2004. 10.1002/jcc.23702. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Phillips J. C.; Hardy D. J.; Maia J. D. C.; Stone J. E.; Ribeiro J. V.; Bernardi R. C.; Buch R.; Fiorin G.; Hénin J.; Jiang W.; McGreevy R.; Melo M. C. R.; Radak B. K.; Skeel R. D.; Singharoy A.; Wang Y.; Roux B.; Aksimentiev A.; Luthey-Schulten Z.; Kalé L. V.; Schulten K.; Chipot C.; Tajkhorshid E. Scalable Molecular Dynamics on CPU and GPU Architectures with NAMD. J. Chem. Phys. 2020, 153, 044130. 10.1063/5.0014475. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Feller S. E.; Zhang Y.; Pastor R. W.; Brooks B. R. Constant Pressure Molecular Dynamics Simulation: The Langevin Piston Method. J. Chem. Phys. 1995, 103, 4613–4621. 10.1063/1.470648. [DOI] [Google Scholar]
- Martyna G. J.; Tobias D. J.; Klein M. L. Constant Pressure Molecular Dynamics Algorithms. J. Chem. Phys. 1994, 101, 4177–4189. 10.1063/1.467468. [DOI] [Google Scholar]
- Darden T.; York D.; Pedersen L. Particle Mesh Ewald: An N·log(N) Method for Ewald Sums in Large Systems. J. Chem. Phys. 1993, 98, 10089–10092. 10.1063/1.464397. [DOI] [Google Scholar]
- Ryckaert J.-P.; Ciccotti G.; Berendsen H. J. C. Numerical Integration of the Cartesian Equations of Motion of a System with Constraints: Molecular Dynamics of n-Alkanes. J. Comput. Phys. 1977, 23, 327–341. 10.1016/0021-9991(77)90098-5. [DOI] [Google Scholar]
- Miyamoto S.; Kollman P. A. SETTLE: An Analytical Version of the SHAKE and RATTLE Algorithm for Rigid Water Models. J. Comput. Chem. 1992, 13, 952–962. 10.1002/jcc.540130805. [DOI] [Google Scholar]
- Hurwitz N.; Schneidman-Duhovny D.; Wolfson H. J. Memdock: an α-helical membrane protein docking algorithm. Bioinf. 2016, 32, 2444–2450. 10.1093/bioinformatics/btw184. [DOI] [PubMed] [Google Scholar]
- Lyskov S.; Gray J. J. The RosettaDock Server for Local Protein-Protein Docking. Nucleic Acids Res. 2008, 36, W233–W238. Web Server issue) 10.1093/nar/gkn216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chaudhury S.; Berrondo M.; Weitzner B. D.; Muthu P.; Bergman H.; Gray J. J. Benchmarking and Analysis of Protein Docking Performance in Rosetta v3.2. PLoS One 2011, 6, e22477 10.1371/journal.pone.0022477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lyskov S.; Chou F.-C.; Conchúir S. Ó.; Der B. S.; Drew K.; Kuroda D.; Xu J.; Weitzner B. D.; Renfrew P. D.; Sripakdeevong P.; Borgo B.; Havranek J. J.; Kuhlman B.; Kortemme T.; Bonneau R.; Gray J. J.; Das R. Serverification of Molecular Modeling Applications: The Rosetta Online Server That Includes Everyone (ROSIE). PLoS One 2013, 8, e63906 10.1371/journal.pone.0063906. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Desta I. T.; Porter K. A.; Xia B.; Kozakov D.; Vajda S. Performance and Its Limits in Rigid Body Protein-Protein Docking. Struct. Lond. Engl. 2020, 28, 1071–1081.e3. 10.1016/j.str.2020.06.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vajda S.; Yueh C.; Beglov D.; Bohnuud T.; Mottarella S. E.; Xia B.; Hall D. R.; Kozakov D. New Additions to the ClusPro Server Motivated by CAPRI. Proteins 2017, 85, 435–444. 10.1002/prot.25219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kozakov D.; Hall D. R.; Xia B.; Porter K. A.; Padhorny D.; Yueh C.; Beglov D.; Vajda S. The ClusPro Web Server for Protein-Protein Docking. Nat. Protoc. 2017, 12, 255–278. 10.1038/nprot.2016.169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kozakov D.; Beglov D.; Bohnuud T.; Mottarella S. E.; Xia B.; Hall D. R.; Vajda S. How Good Is Automated Protein Docking?. Proteins 2013, 81, 2159–2166. 10.1002/prot.24403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xia B.; Vajda S.; Kozakov D. Accounting for Pairwise Distance Restraints in FFT-Based Protein–Protein Docking. Bioinformatics 2016, 32, 3342–3344. 10.1093/bioinformatics/btw306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smart O. S.; Breed J.; Smith G. R.; Sansom M. S. A Novel Method for Structure-Based Prediction of Ion Channel Conductance Properties. Biophys. J. 1997, 72, 1109–1126. 10.1016/S0006-3495(97)78760-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smart O. S.; Neduvelil J. G.; Wang X.; Wallace B. A.; Sansom M. S. P. HOLE: A Program for the Analysis of the Pore Dimensions of Ion Channel Structural Models. J. Mol. Graphics 1996, 14, 354–360. 10.1016/S0263-7855(97)00009-X. [DOI] [PubMed] [Google Scholar]
- Klauda J. B.; Venable R. M.; Freites J. A.; O’Connor J. W.; Tobias D. J.; Mondragon-Ramirez C.; Vorobyov I.; MacKerell A. D.; Pastor R. W. Update of the CHARMM All-Atom Additive Force Field for Lipids: Validation on Six Lipid Types. J. Phys. Chem. B 2010, 114, 7830–7843. 10.1021/jp101759q. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fiorin G.; Klein M. L.; Hénin J. Using Collective Variables to Drive Molecular Dynamics Simulations. Mol. Phys. 2013, 111, 3345–3362. 10.1080/00268976.2013.813594. [DOI] [Google Scholar]
- Souaille M.; Roux B. Extension to the Weighted Histogram Analysis Method: Combining Umbrella Sampling with Free Energy Calculations. Comput. Phys. Commun. 2001, 135, 40–57. 10.1016/S0010-4655(00)00215-0. [DOI] [Google Scholar]
- Smith L. G.; Tan Z.; Spasic A.; Dutta D.; Salas-Estrada L. A.; Grossfield A.; Mathews D. H. Chemically Accurate Relative Folding Stability of RNA Hairpins from Molecular Simulations. J. Chem. Theory Comput. 2018, 14, 6598–6612. 10.1021/acs.jctc.8b00633. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.