

Abstract

Metallophilic interactions, which are ubiquitous among d10 metal complexes with linear coordination geometries, can direct one-dimensional assembly. However, the ability of these interactions to manipulate chirality at the hierarchical level largely remains unknown. In this work, we unveiled the role of Au···Cu metallophilic interactions in directing the chirality of multicomponent assemblies. N-heterocyclic carbene–Au(I) complexes bearing amino acid residues formed chiral co-assemblies with [CuI2]− anions via Au···Cu interactions. These metallophilic interactions changed the molecular packing modes of the co-assembled nanoarchitectures from lamellar to columnar chiral packing. This transformation initiated the emergence, inversion, and evolution of supramolecular chirality, thereby affording helical superstructures, depending on the geometry of building units. In addition, the Au···Cu interactions altered the luminescence properties and induced the emergence and amplification of circularly polarized luminescence. This work, for the first time, revealed the role of Au···Cu metallophilic interactions in modulating supramolecular chirality, paving the way for the construction of functional chiroptical materials based on d10 metal complexes.
Keywords: metallophilic interactions, chirality, co-assembly, circularly polarized luminescence, N-heterocyclic carbene−Au(I) complexes
Introduction
Chirality is a widespread phenomenon in nature exhibited by multifarious hierarchical structures ranging from the atomic to the macroscopic scales. Supramolecular chirality, which is defined as the spatially asymmetric packing of building blocks, has witnessed rapid development in fields such as chiroptical materials, sensing, detection, kinetic resolution, and information storage in the last decade.1−4 Unlike the elaborate synthesis of inherently chiral molecular entities,5 generating and controlling the supramolecular chirality of assemblies6−14 based on flexible and dynamically manipulated noncovalent interactions and external fields are more facile and efficient. The methods for modulating the supramolecular chirality of assembled systems are generally divided into kinetic and thermodynamic protocols. These protocols utilize environmental factors (such as the solvent and temperature) and molecular topological design (such as functional groups) to influence the generation and variation of supramolecular chirality.15 Compared with kinetic control, thermodynamic control of the supramolecular chirality of multicomponent assemblies is emerging.16 The evolution of the supramolecular chirality of hierarchical co-assemblies by manipulation of dynamic inter-constituent forces, such as electrostatic, hydrogen bond, halogen bond, π–π stacking, and charge-transfer interactions, has been investigated extensively in recent years.17−21
Metallophilic interactions are based on the propensities of d8 and d10 transition metal ions, such as Pt(II), Au(I), Ag(I), and Cu(I), which are enhanced by the relativistic effect and electron correction of the closed-shell constituents.22−28 Although metallophilic interactions are repulsive in nature, Pauli repulsion is suppressed by the linear coordination geometries of the d10 metal complexes. This leads to a close M···M distance that is shorter than the sum of the van der Waals radius. Thus, metallophilic interactions behave as attractive interactions with an order-of-magnitude strength similar to that of hydrogen bonds.29 By tuning metallophilic interactions, numerous supramolecular materials with tailored properties have been constructed for diverse applications such as optoelectronics and catalysis. Owing to the chemical stability and coordination versatility of N-heterocyclic carbene (NHC) ligands, various chiral NHC–metal complexes have been synthesized.30−32 For instance, Che et al. reported a series of NHC–gold (I) complex salts [Au(NHC)2][MX2] (M = Au or Cu; X = halide, cyanide, or arylacetylide) consisting of alternating cations and anions arranged into one-dimensional (1D) chains. These further generated supramolecular polymers with tunable luminescence properties, in which the synergetic Au···M interactions and Coulombic attraction directed the co-assembly.28,33−37 Among these complex salts, a pair of chiral [Au(NHC)2][Au(CN)2] complex double salts displayed excitation-wavelength-dependent circularly polarized luminescence (CPL).37 The tunable Au···Au distances were proven to be responsible for the tunable photoluminescence (PL). Au···Cu metallophilic interactions assisted by Coulombic attraction not only lead to strong binding energy35 but also facilitate the formation of 1D structures. Thus, such metallophilic interactions are expected to be potential forces in manipulating the supramolecular chirality of nanoarchitectures in multicomponent systems. However, the role of Au···Cu metallophilic interactions in modulating chirality has not yet been revealed.
Herein, we introduced Au···Cu interactions to tune chiral self-assembly and chiroptical properties. Three enantiopure NHC–gold (I) complexes, [Au(NHC)2]+I–, bearing diverse amino acid residues, including alanine (Ala), valine (Val), and phenylalanine (Phe), were synthesized as the model building blocks (Scheme 1a). [CuI2]− ([18-Crown-6]-K+ or [18C6]-K+ as the counter cation)34 was used as the anionic counterpart to co-assemble with the Au(I) complexes through the Au···Cu interactions with auxiliary Coulombic attraction. In solution, the least sterically hindered Au(Ala)2 self-assembled into homochiral helically wound nanofibers. On the other hand, the other two complexes, Au(Val)2 and Au(Phe)2, produced achiral structures on nano- and microscales. Introducing [CuI2]− through a co-precipitation protocol generated distinct co-assembled nanoarchitectures (Scheme 1b). [CuI2]− was exclusively capable of modulating supramolecular chirality compared with the other anions used. Except in the case of the large sterically hindered Au(Phe)2, integrating [CuI2]− into the assemblies induced the formation of helical structures with inverted handedness, consistent with the chiroptical responses. The experimental and theoretical results indicated that the Au···Cu metallophilic interactions in the co-assemblies played a key role in directing the assembly arrays and caused a dramatic change in the molecular packing from lamellar stacking to columnar packing. In addition, the Au··Cu interactions induced luminescent color evolution and the emergence and amplification of CPL (Scheme 1). This work focused on the supramolecular chirality evolution of Au(I)-complex-based chiral assembly materials, exploring a pathway to fabricate chiroptical materials with modifiable handedness based on Au···Cu metallophilic interactions.
Scheme 1. (a) Chemical Structures of Amino Acid-Derived [Au(NHC)2]+I– Complexes and (b) Schematic Representation of Helicity Inversion from Individual Self-Assembly to Co-Assembly with Columnar Chiral Packing.

Results and Discussion
Three amino-acid-derived NHC–Au(I) complexes ([Au(NHC)2]+I–) formed co-assemblies with [18C6]-K+[CuI2]− via solution-processed bottom-up assembly. Aggregates of the three [Au(NHC)2]+I– complexes were obtained by evaporating the respective acetonitrile solutions. The co-assembly of the [Au(NHC)2]+I– complexes and [18C6]-K+[CuI2]− was triggered by co-precipitation in acetonitrile, followed by incubation and aging, under ambient conditions. The presence of cationic [Au(NHC)2]+ and anionic [CuI2]− in the co-assemblies was confirmed by electrospray ionization–mass spectrometry in the positive and negative modes, respectively (Figures S1–S3). Both self- and co-assemblies were adequately studied by electron microscopy, circular dichroism (CD), thin-film powder X-ray diffraction (XRD), PL, and CPL.
Because it has the least sterically hindered methyl group at the α-carbon position of the amino acid residue, Au(Ala)2 was prioritized in the study. The self-assembly of Au(Ala)2 produced twisted nanofibrous structures with a mean diameter of approximately 100 nm, as observed by scanning electron microscopy (SEM). As shown in Figures 1a and S4, the Au(l-Ala)2 self-assembly displays M-handed homochirality, whereas P-chiral fibers are observed exclusively for the Au(d-Ala)2 self-assembly (Figures 1c and S4). When Au(Ala)2 co-assembled with [CuI2]−, similar fibrous structures were observed, albeit with reversed helicity. The M-type Au(l-Ala)2 self-assembly changed to P-type fibers, and the P-chiral Au(d-Ala)2 self-assembly converted to M-chiral helical fibers after co-assembly with [CuI2]− (Figures 1b,d and S5).
Figure 1.

SEM images of (a,c) Au(l-Ala)2 and Au(d-Ala)2 self-assemblies and (b,d) their respective co-assemblies with [CuI2]−. CD spectral comparisons of (e) Au(l-Ala)2 and Au(d-Ala)2 self-assemblies and (f) their respective co-assemblies with [CuI2]−. (g) XRD pattern comparison of the individual self-assemblies and co-assemblies. (h) Schematic models of lamellar and cubic packing arrangements based on XRD results.
This unique supramolecular chirality inversion
was further
supported
by the CD spectra. The Au(Ala)2 self-assembly featured
a well-defined mirror Cotton effect at 230 nm resulting from the absolute
chirality of the Ala residues. Moreover, a couple of exciton-type
Cotton effects centered at approximately 310 nm were attributed to
the aromatic groups (NHC). This result implies a chirality transfer
from the amino acid residue to NHC and a shift from molecular chirality
to supramolecular chirality (Figure 1e). This chirality transfer boosted the degree of polarization.
The dissymmetry g-factor (gabs), which reflects the polarization of absorbance, was calculated
as gabs =  = θ (mdeg)/(32,980
× Abs). In
contrast to the negligible gabs of Au(Ala)2 in the solution state, a dramatic increase from 4.0 ×
10–4 to 1.1 × 10–2 was observed
after self-assembly. This was magnified 27.5-fold because of the effective
chirality transfer to the supramolecular scale (Figure S6). Introducing [CuI2]− into the Au(Ala)2 self-assembly clearly reversed the
Cotton effects. The Au(l-Ala)2/CuI2 co-assembly exhibited a positive extended exciton band at approximately
320 nm, in contrast to the negative-then-positive exciton Cotton effects
shown by the Au(l-Ala)2 self-assembly. The same
condition was observed for the Au(d-Ala)2/CuI2 co-assembly, which showed a negative band, in contrast to
the positive-then-negative exciton band exhibited by the Au(d-Ala)2 self-assembly (Figure 1f). Such inversion in the CD spectra corresponds
well to the helical morphology transformation, confirming the supramolecular
chirality inversion after the integration of [CuI2]− into the Au(Ala)2 self-assembly.
= θ (mdeg)/(32,980
× Abs). In
contrast to the negligible gabs of Au(Ala)2 in the solution state, a dramatic increase from 4.0 ×
10–4 to 1.1 × 10–2 was observed
after self-assembly. This was magnified 27.5-fold because of the effective
chirality transfer to the supramolecular scale (Figure S6). Introducing [CuI2]− into the Au(Ala)2 self-assembly clearly reversed the
Cotton effects. The Au(l-Ala)2/CuI2 co-assembly exhibited a positive extended exciton band at approximately
320 nm, in contrast to the negative-then-positive exciton Cotton effects
shown by the Au(l-Ala)2 self-assembly. The same
condition was observed for the Au(d-Ala)2/CuI2 co-assembly, which showed a negative band, in contrast to
the positive-then-negative exciton band exhibited by the Au(d-Ala)2 self-assembly (Figure 1f). Such inversion in the CD spectra corresponds
well to the helical morphology transformation, confirming the supramolecular
chirality inversion after the integration of [CuI2]− into the Au(Ala)2 self-assembly.
A physical insight into the effect of [CuI2]− on the tuning of supramolecular chirality was further investigated.
Based on the XRD pattern comparison (Figure 1g), the molecular packing of the aggregates
evolved after the incorporation of [CuI2]−. The Au(Ala)2 self-assembly showed strong diffraction
peaks at 7.8, 15.0, and 22.2° with an approximate distance ratio
of 1:1/2:1/3, corresponding to a lamellar structure with (100), (200),
and (300) planes and a d-spacing of 1.13 nm. This d-spacing is approximately equal to the length of the Au(Ala)2 molecule (1.30 nm), suggesting that the molecules are stacked
in a layer-by-layer motif. However, the Au(Ala)2/CuI2 co-assembly showed a different XRD pattern than the Au(Ala)2 self-assembly, implying that [CuI2]− induced a different molecular packing model. For the Au(Ala)2/CuI2 co-assembly, the diffraction peaks at 6.92,
7.76, 9.62, and 11.32° with a ratio of  3:
3: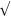 4:
4: :
: corresponded to bicontinuous
cubic packing
with a d-spacing of 1.54 nm, which was larger than
that of the lamellar structure of the Au(Ala)2 self-assembly.
As shown in Figure 1g, at around 25° of the co-assembly XRD patterns, it should
be attributed to the regular arrangement of atomic levels in the co-assembled
fibers, which corresponds to a Au···Cu distance of
about 3.5 Å. Similar fine features have been previously observed
in XRD patterns of the orderly packed Au25 cluster assembled
nanoribbon.38 Based on the above-mentioned
results, the proposed packing motif evolution from the Au(Ala)2 self-assembly to the Au(Ala)2/CuI2 co-assembly
is shown in Figure 1h. The Au(Ala)2 molecules self-assemble into a lamellar
microstructure and then become homochiral helical fibers based on
the absolute chirality of the Ala residues. Previous studies of Au(NHC)2/CuI2 co-assemblies showed that consecutive Au···Cu
interactions benefit the formation of 1D packing with alternating
cationic [Au(NHC)2]+ and anionic [CuI2]−.33,34 In the present Au(Ala)2/CuI2 co-assembly system, cationic [Au(Ala)2]+ and anionic [CuI2]− units
tended to pack alternately along the 1D direction on the periphery
of the co-assembled arrays through Au···Cu interactions
and then formed cubic sub-column structures accompanied by supramolecular
chirality inversion compared with the Au(Ala)2 self-assembly.
corresponded to bicontinuous
cubic packing
with a d-spacing of 1.54 nm, which was larger than
that of the lamellar structure of the Au(Ala)2 self-assembly.
As shown in Figure 1g, at around 25° of the co-assembly XRD patterns, it should
be attributed to the regular arrangement of atomic levels in the co-assembled
fibers, which corresponds to a Au···Cu distance of
about 3.5 Å. Similar fine features have been previously observed
in XRD patterns of the orderly packed Au25 cluster assembled
nanoribbon.38 Based on the above-mentioned
results, the proposed packing motif evolution from the Au(Ala)2 self-assembly to the Au(Ala)2/CuI2 co-assembly
is shown in Figure 1h. The Au(Ala)2 molecules self-assemble into a lamellar
microstructure and then become homochiral helical fibers based on
the absolute chirality of the Ala residues. Previous studies of Au(NHC)2/CuI2 co-assemblies showed that consecutive Au···Cu
interactions benefit the formation of 1D packing with alternating
cationic [Au(NHC)2]+ and anionic [CuI2]−.33,34 In the present Au(Ala)2/CuI2 co-assembly system, cationic [Au(Ala)2]+ and anionic [CuI2]− units
tended to pack alternately along the 1D direction on the periphery
of the co-assembled arrays through Au···Cu interactions
and then formed cubic sub-column structures accompanied by supramolecular
chirality inversion compared with the Au(Ala)2 self-assembly.
We further evaluated the influence of amino acid residues on the individual self-assemblies and co-assemblies. Unlike Au(Ala)2, Au(Val)2 possesses an isopropyl group at the α-carbon position of the amino acid residue, which may result in different self- and co-assembly behaviors. In contrast to the helical twisted nanofiber aggregates of Au(Ala)2, stacked nanosheets on the microscale (Figures 2a and S7) were generated by the crystallization-induced self-assembly of Au(l-Val)2. After the addition of 1 equiv of [CuI2]− to the nanosheets, a gel phase was generated, transforming the co-assembly into twisted nanoribbons with a mean diameter of 50 nm, as observed by SEM and transmission electron microscopy (TEM) (Figures 2b,c and S7). M-handedness was observed for the Au(l-Val)2/CuI2 co-assembly. The morphological transformation between the Au(d-Val)2 self-assembly and Au(d-Val)2/CuI2 co-assembly was similar to that of the corresponding l-enantiomers (Figure S8). Supramolecular chirality emerged through the co-assembly with [CuI2]−.
Figure 2.

(a) SEM images of the Au(l-Val)2 self-assembly. (b,c) SEM and TEM images of the Au(l-Val)2 co-assembly with [CuI2]−. (d) Comparison of the gabs values of the Au(l-Val)2 and Au(d-Val)2 self-assemblies and their co-assemblies with [CuI2]−. (e) Schematic representation of nanoscale chirality induced by [CuI2]−. (f) Comparison of XRD patterns simulated from the single-crystal data of the Au(l-Val)2, individual self-assembly, and co-assembly. (g) Intermolecular interactions based on the X-ray crystal structure of Au(l-Val)2. Arrows indicate the C–H···O interactions. (h) Packing viewed along the b axis of the crystal structure. (i) Simulated packing transformation induced by [CuI2]− in the self-assembly scenario.
The generation and modulation of supramolecular chirality through the introduction of [CuI2]− during the assembly process were investigated by CD characterization. A pair of well-defined mirror curves were obtained for the Au(Val)2 self-assembly with an almost negligible gabs of approximately 3 × 10–4, indicative of the low degree of polarization of the absorption (Figure S9a). After incorporating [CuI2]−, strong Cotton effects centered at approximately 314 nm were observed (Figure S9b). The gabs also increased up to 6 × 10–3, which was 20-fold higher than that of the Au(Val)2 self-assembly. This signifies that the emergence of supramolecular chirality is beneficial to the chiroptical properties (Figure 2d). Furthermore, similar to the Au(Ala)2 self-assembly, introducing [CuI2]− into the Au(Val)2 self-assembly also achieved chirality inversion independent of the amino acid residue. As shown in Figure S9a, the Au(l-Val)2 self-assembly features negative and positive exciton-type Cotton effects at approximately 314 nm, which are assigned to the NHC groups. When [CuI2]− was integrated into the Au(l-Val)2 self-assembly, the Cotton effect at 314 nm was inversed as a positive signal (Figure S9b). By varying the configuration from the l-to the d-enantiomer, the same CD signal variation occurred, confirming the negligible artifacts and validating the decisive effect of the absolute chirality on the guidance of the supramolecular chirality. However, in the solid states, the anisotropic effects are basically inevitable. In order to rule out the artifacts, we actually performed multiple tests to confirm the accuracy for both CD and CPL spectroscopy. Based on the above-mentioned results, incorporating [CuI2]− into the Au(Val)2 self-assembly enables the generation of supramolecular chirality to achieve chirality amplification and inversion.
The spontaneous self-assembly of Au(l-Val)2 produced
achiral nanosheets, while the introduction of [CuI2]− generated supramolecular chirality with M-handedness (Figure 2e). To gain insights into the assembly mechanism of Au(Val)2 and Au(Val)2/CuI2, single crystals
of Au(l-Val)2 were cultured through a liquid-phase
diffusion protocol (dimethyl formamide vs diethyl ether), and XRD
analysis was performed. The single-crystal structures exhibited C–H···O
interactions between the Au(l-Val)2 molecules,
with distances of 2.365–2.702 Å, which are in the range
of the reported hydrogen bond lengths (Figure 2g). The structure of the assembly supported
by C–H···O interactions was characterized by
a lamellar array along the b axis with alternately
stacked [Au(NHC)2]+ units and amino acid residues
(colored green and purple, respectively, shown in Figure 2h). Similar noncovalent C–H···O
interactions might be present during the spontaneous aggregation,
and the XRD pattern of the Au(Val)2 self-assembly was different
from that simulated using Au(Val)2 single crystals (Figure 2f). This indicates
that the solution-processed self-assembly adopts a distinct arrangement
from that produced by thermodynamic crystallization. The XRD pattern
of the Au(Val)2 self-assembly showed two sets of peaks
at 5.72, 10.78, and 15.84° (1, 2, 3) and 6.7, 12.8, and 18.82°
(1′, 2′, 3′). This indicates the co-existence
of two lamellar structures along two different directions with d-spacing values of 1.54 and 1.32 nm, in accordance with
the size of the Au(Val)2 molecules (1.60 and 1.31 nm from
different directions). After co-assembly with [CuI2]−, an XRD pattern different from that of the individual
self-assembly appeared. The main diffraction peaks of the Au(Val)2/CuI2 co-assembly were located at 4.78, 5.38, and
6.98° (approximate ratio of  :
: :
: ), corresponding to the (110),
(200), and
(210) planes of hexagonal columnar packing, respectively, with a d-spacing of 3.2 nm calculated from the Bragg equation.
A relatively weaker peak at around 27° should be assigned as
Au···Cu distance with about 3.3 Å. According to
the above-mentioned results, the Au(Val)2 self-assembly
formed lamellar structures along two directions (Figure 2i). After incorporating [CuI2]− into the assembly, the resulting co-assembly
exhibited hexagonal packing, which further facilitated the assembly
of a helical nanofibrous structure.
), corresponding to the (110),
(200), and
(210) planes of hexagonal columnar packing, respectively, with a d-spacing of 3.2 nm calculated from the Bragg equation.
A relatively weaker peak at around 27° should be assigned as
Au···Cu distance with about 3.3 Å. According to
the above-mentioned results, the Au(Val)2 self-assembly
formed lamellar structures along two directions (Figure 2i). After incorporating [CuI2]− into the assembly, the resulting co-assembly
exhibited hexagonal packing, which further facilitated the assembly
of a helical nanofibrous structure.
Owing to a larger sterically hindered benzyl group at the α-position, Au(Phe)2 exhibited different self- and co-assembly behaviors compared with Au(Ala)2 and Au(Val)2 (Figure 3a). As shown in Figure 3b, Au(d-Phe)2 self-assembles into slender fibrous structures, which then twist into thick fibers with an average diameter of approximately 250 nm without evident nanoscale chirality. Integrating [CuI2]− into the assembly led to a morphological transformation from flexible fibers to rigid rods with a diameter of approximately 80 nm but without the generation of morphological supramolecular chirality (Figure 3c,d). The same situation also occurred in the Au(l-Phe)2 self-assembly and Au(l-Phe)2/CuI2 co-assembly (Figure S10).
Figure 3.

(a) Schematic representation of the morphological transformation of Au(Phe)2 induced by [CuI2]−. SEM images of the (b) Au(d-Phe)2 self-assembly and (c) Au(d-Phe)2/CuI2 co-assembly. (d) TEM images of the Au(l-Phe)2/CuI2 co-assembly. CD spectral comparisons of (e) Au(l-Phe)2 and Au(d-Phe)2 self-assemblies and (f) their respective co-assemblies with [CuI2]−. (g) XRD pattern comparison of the individual self-assemblies and co-assemblies.
The CD signals of the Au(Phe)2 and Au(Phe)2/CuI2 assemblies were also reversed, similar to those of the other two systems. As shown in Figure 3e, the CD spectra of the Au(Phe)2 self-assembly with different configurations appear as well-defined mirror curves. The pair of extended bands centered at approximately 325 nm were positive for Au(l-Phe)2 and negative for Au(d-Phe)2. Incorporating [CuI2]− into the Au(Phe)2 self-assembly noticeably changed the Cotton effects (Figure 3f). The resultant Au(Phe)2/CuI2 co-assembly exhibited a couple of extended exciton-type Cotton effects at approximately 330 nm, with positive and negative bands for Au(d-Phe)2/CuI2, unlike the totally negative band for the Au(d-Phe)2 self-assembly. Such behavior suggests that chirality inversion occurs after co-assembly with [CuI2]−.
The thin-film XRD patterns of
the Au(Phe)2 and Au(Phe)2/CuI2 assemblies
were recorded to probe the molecular
packing modes before and after the integration of [CuI2]− (Figure 3g). The Au(Phe)2 self-assembly displayed clear
diffraction peaks at 6.88, 12.3, 13.64, 18.4, and 21.56° with
an approximate ratio of 1: :
: :
: :
: , corresponding to hexagonal
columnar packing
with a d-spacing of 1.28 nm. For the Au(Phe)2/CuI2 co-assembly, the XRD pattern showed a set
of peaks at 3.88, 6.66, 7.58, 10.1, and 11.4° with the same approximate
ratio, indicating the same hexagonal columnar packing, albeit with
a larger d-spacing of 2.26 nm. Unlike the Au(Phe)2 self-assembly, the co-assembly displayed a distinct peak
at around 27°, which corresponded to a Au···Cu
distance of about 3.3 Å. The above-mentioned phenomenon reveals
that the introduction of [CuI2]− into
the Au(Phe)2 self-assembly does not change the hexagonal
columnar arrangement but stretches the assembled structure. Unlike
the Au(Ala)2/CuI2 and Au(Val)2/CuI2 co-assemblies, the Au(Phe)2/CuI2 co-assembly
possessing a large sterically hindered amino acid residue failed to
generate morphological supramolecular chirality, indicating the effect
of the substituents on the handedness of the assemblies. However,
the integration of [CuI2]− into the assembly
also resulted in chirality inversion reflected in the chiroptical
properties, further confirming the vital function of Au···Cu
metallophilic interactions in manipulating chirality evolution.
, corresponding to hexagonal
columnar packing
with a d-spacing of 1.28 nm. For the Au(Phe)2/CuI2 co-assembly, the XRD pattern showed a set
of peaks at 3.88, 6.66, 7.58, 10.1, and 11.4° with the same approximate
ratio, indicating the same hexagonal columnar packing, albeit with
a larger d-spacing of 2.26 nm. Unlike the Au(Phe)2 self-assembly, the co-assembly displayed a distinct peak
at around 27°, which corresponded to a Au···Cu
distance of about 3.3 Å. The above-mentioned phenomenon reveals
that the introduction of [CuI2]− into
the Au(Phe)2 self-assembly does not change the hexagonal
columnar arrangement but stretches the assembled structure. Unlike
the Au(Ala)2/CuI2 and Au(Val)2/CuI2 co-assemblies, the Au(Phe)2/CuI2 co-assembly
possessing a large sterically hindered amino acid residue failed to
generate morphological supramolecular chirality, indicating the effect
of the substituents on the handedness of the assemblies. However,
the integration of [CuI2]− into the assembly
also resulted in chirality inversion reflected in the chiroptical
properties, further confirming the vital function of Au···Cu
metallophilic interactions in manipulating chirality evolution.
To explore the Au···Cu metallophilic interactions within a co-assembly, density functional theory (DFT) calculations were performed on the Au(l-Val)2/CuI2 complex using the geometry extracted from the crystal structure. As shown in Figure 4a, the interaction energy (ΔE) between Au(l-Val)2 and [CuI2]− is −112.84/2 = −56.42 kcal/mol, which is comparable to the energy of an anion–cation d10···d10 interaction, such as the Au···Au interaction.35 Such a large binding energy indicates the strong binding affinity between [Au(Val)2]+ and [CuI2]− due to the combined effects of the Au···Cu metallophilicity and the anion–cation and hydrogen bond interactions. The optimized structure of the Au(l-Val)2/CuI2 complex showed a sandwich construct with alternately stacked cationic Au(l-Val)2 and anionic [CuI2]−. This arrangement is believed to be more favorable for supramolecular chiral packing along 1D growth. The Cu···Au···Cu torsion angle was 170.88°, which, being close to 180°, was beneficial for forming a 1D assembled structure (Figure 4b). The Au···Cu distances were 3.458 and 3.295 Å, which were comparable to the distances of d10···d10 metallophilic interactions (especially Au···Cu interactions) reported in the literature.33,34 Also, the Au···Cu distances obtained from the DFT calculations were consistent with the observations from the XRD patterns of the co-assemblies. In addition to the Au···Cu interactions, N–H···I hydrogen bonds were observed in the complex structure with bond lengths of 2.806 and 2.949 Å.
Figure 4.

(a) Energy diagram of Au(Val)2/CuI2 co-assembly formation. (b) DFT-optimized structures of Au(Val)2/CuI2 at the B3LYP/def2-SVP level of theory. (c,f) DFT-optimized stacked structures and their corresponding calculated ECD spectra of the Au(Val)2/CuI2 co-assembly at the B3LYP/def2-SVP level of theory.
To investigate the correlation between supramolecular geometry and CD activity, electronic circular dichroism (ECD) calculations were performed. The Au(l-Val)2/CuI2 complex with optimized clockwise or anticlockwise screw sense was applied for the ECD calculations (Figure 4c–f). The Au(l-Val)2/CuI2 complex with a clockwise screw sense (M-handedness) displayed a positive Cotton effect in the range of 250–350 nm (Figure 4c,e) while that with the anticlockwise screw sense (P-handedness) afforded a positive-then-negative exciton Cotton effect at a wavelength of 300 nm (Figure 4d,f). The positive Cotton effect presented by the M-handed structure was consistent with the experimental CD spectra of the Au(l-Val)2/CuI2 co-assembly prepared by co-precipitation. This result indicates that the supramolecular chirality of the Au(l-Val)2/CuI2 co-assembly follows a clockwise M-handedness, which is in accordance with the observed morphology. Thus, the pivotal anion–cation Au···Cu interactions, assisted by the hydrogen bonds between Au(l-Val)2 and [CuI2]−, guide the co-assembly structure and influence the helical direction of the supramolecular chirality.
The emergence of Au···Cu metallophilic interactions dramatically changed the PL properties of the Au(I) complexes. The fluorescence spectra of the self- and co-assemblies were measured, as shown in Figure 5a–c. The [Au(NHC)2]+I– complexes (NHC = Ala, Val, and Phe) displayed cyan emissions with major peaks centered at 456, 466, and 486 nm. After integration with [CuI2]−, the Au(Ala)2/CuI2 and Au(Val)2/CuI2 co-assemblies exhibited large emission shifts of up to 140 nm, while the emission shift of the Au(Phe)2/CuI2 co-assembly was 60 nm. The emission peaks of the Au(NHC)2/CuI2 (NHC = Ala, Val, and Phe) co-assemblies were 597, 612, and 546 nm, contributing to orange, red, and yellow luminescence, respectively, which was in good agreement with the luminescence images shown in Figure 5a–c. The decay profiles (Figure S11) showed that the emission lifetime of the self- and co-assemblies lasted for hundreds to thousands of nanoseconds, indicating phosphorescent emission.
Figure 5.

(a,b) Fluorescence spectra of the self- and co-assemblies. Insets show the luminescence colors of the corresponding systems observed by fluorescence microscopy. CPL spectra of the (c) Au(Ala)2/CuI2 co-assembly, (d) Au(Val)2 self-assembly, and (e) Au(Val)2/CuI2 co-assembly. (f) HOMO–LUMO energy levels and the corresponding electron distribution maps of the Au(Val)2 and Au(Val)2/CuI2 assemblies.
The synergistic Au···Cu metallophilic interactions in the co-assemblies were assumed to facilitate supramolecular chirality and thus favor CPL emission. Samples of the self- and co-assemblies in the film state were used in the CPL tests, and the linear dichroism effect was excluded by repeated experiments. The CPL spectra of the Au(Ala)2 and Au(Phe)2 self-assemblies and Au(Phe)2/CuI2 co-assembly could not be obtained, possibly due to negligible luminescence dissymmetry factors (glum). In Figure 5d, Au(l-Ala)2/CuI2 and Au(d-Ala)2/CuI2 show right and left CPL, respectively. As the CPL spectrum represents the chirality in the photoexcited state while the CD spectrum reflects the chirality in the ground state, it is understandable that they are not consistent with each other owing to structural relaxation. The glum values were determined using the equation glum = 2 × [ellipticity/(32,980/ln 10)]/total fluorescence intensity at the CPL maximum peak.39 In addition, the glum of both the Au(l-Ala)2/CuI2 and Au(d-Ala)2/CuI2 co-assemblies was ± 7 × 10–3 (Figure S12). For the Au(l-Val)2/CuI2 and Au(d-Val)2/CuI2 co-assemblies, CPL with a glum of ± 0.01 was obtained in contrast to the negligible CPL of the Au(l-Val)2 and Au(d-Val)2 self-assemblies (Figures 5e and S12). The chiroptical properties including CD and CPL originate from the differential absorption and emission of left- and right-handed circularly polarized light, which are determined by the screw sense of electronic transition dipole moments. In the self-assemblies, the point chirality of amino acids shall induce the helical orientation or packing of NHC-based chromophores, leading to the helical packing of transition dipole moments to afford chiroptical properties. With respect to this fact, the emergence of chiroptical properties is hardly associated with the scattering effect of light by the nanoscale assemblies. The CD and CPL extremum wavelengths correspond to the absorption and emission peaks of chromophores. The helical packing of chromophores in most cases is consistent with the helical sense observed using microscopic techniques such as SEM. The evolution behavior of the luminescence and chiroptical properties from self-assembly to co-assembly was caused by the Au···Cu metallophilic interactions. These interactions not only influenced the luminescence, leading to different degrees of bathochromic shift of the emission peaks, but also amplified the CPL to induce supramolecular chirality.
To gain insights into the correlation between the Au···Cu metallophilic interactions and lower-energy transitions of the co-assemblies, we performed time-dependent DFT calculations on the Au(l-Val)2 and Au(l-Val)2/CuI2 assemblies using geometries extracted from the crystal structures. The resulting frontier orbitals are depicted in Figure 5f. The highest occupied molecular orbital (HOMO) and lowest unoccupied molecular orbital (LUMO) energy levels of Au(l-Val)2 were −10.61 and −3.27 eV, respectively. The HOMO of the Au(l-Val)2/CuI2 complex was localized on the [CuI2]− anions, with contribution from the Au···Cu interactions, with an energy of −4.92 eV. The LUMO was localized on the [Au(NHC)]+ cations with an energy of 1.50 eV. Therefore, the low-lying electronic transitions can be described as ligand (anion)-to-ligand (cation) charge transfer mixed with Au···Cu interactions.34 The narrower HOMO–LUMO energy gap (5.42 eV) of the co-assembly compared with that of the individual self-assembly (7.34 eV), which corresponds to the bathochromic emission, indicates the favorability of the co-assembly directed by Au···Cu metallophilic interactions with the assistance of Coulombic attraction. Thus, the theoretical calculations and experimental results confirm that Au···Cu metallophilic interactions play an important role in regulating the luminescence and chiroptical properties and the amplification of CPL emission.
Conclusions
In summary, we illustrated the modulation of supramolecular chirality via Au···Cu metallophilic interactions in co-assemblies of cationic NHC–Au(I) complexes and anionic [CuI2]−. The intrinsic aggregation of the [Au(NHC)2]+I– complexes varied with the steric size of the amino acid residue (Ala, Val, and Phe), producing different nanostructures such as helical nanofibers, stacked nanosheets, and thick fibers. After introducing [CuI2]−, the self-assembly structures transformed into helical packing, exhibiting supramolecular chirality for the Au(Ala)2 and Au(Val)2 self-assemblies. However, for the large sterically hindered Au(Phe)2 self-assembly, morphological supramolecular chirality was not observed, although the chiroptical property was also inverted after co-assembly with [CuI2]−. The experimental and theoretical results indicated that the chiroptical and supramolecular chirality inversion and evolution were due to the changed molecular packing geometry directed by the Au···Cu metallophilic interactions. These interactions, assisted by Coulombic attraction, favored co-assembly and allowed the transformation to columnar chiral packing, which induced the inversion and emergence of a supramolecular chiral nanoarchitecture. Moreover, the Au···Cu metallophilic interactions also played an important role in the regulation of the luminescence properties and amplification of the CPL emission. Knowledge of the crucial function of Au···Cu metallophilic interactions in controlling supramolecular chirality in Au(NHC)2/CuI2 co-assemblies would guide the establishment of supramolecular chiral structures based on a reasonable choice of substituents. Thus, the findings of this work would facilitate the design and construction of functional chiroptical materials based on d10 metal complexes.
Methods
Assembly Methods
Self-assembly was carried out by evaporating the solvent slowly. For example, 8.75 mg of Au(Ala)2 was dispersed in 1 mL of acetonitrile and then spread on a quartz plate to form a film assembly. The co-assembly was prepared by a co-precipitation protocol. For the Au(Ala)2/CuI2 co-assembly system, Au(Ala)2 (8.75 mg, 10 mmol) dispersed in acetonitrile (1 mL) and 18-Crown-6 K [CuI2] (6.21 mg, 10 mmol) dispersed in acetonitrile (0.5 mL) were mixed and then co-precipitated to give a solid phase. For Au(Val)2/CuI2 and Au(Phe)2/CuI2 co-assembly systems, similar procedures were performed to afford gel and solid phases, respectively. All samples were aged under ambient conditions for at least 5 h before testing.
DFT Calculation Details
In order to gain insights into the metallophilic interactions in the co-assembly systems, the Au(l-Val)2/CuI2 system was selected as an example to be investigated according to the DFT calculations performed with the B3LYP-D3 (B3LYP with Grimme’s DFT-D3 correction using zero-damping) functional using the Gaussian 16 suite of programs. A geometry of Au(l-Val)2 extracted from the single-crystal XRD data combined with two optimized geometries of [CuI2]− was used to construct a co-assembled structure and optimized using the B3LYP function and the def2-SVP basis set for all atoms. Coordinates were obtained from the above-mentioned optimized configuration for constructing molecular electrostatic potential maps (isodensity = 0.001 au). The single-point energy of Au(Val)2 and [CuI2]− and their counterpoise-corrected energy were calculated at the B3LYP/def2-TZVP level of theory. The interaction energy was computed as the difference in energy between the co-assembly and the sum of the energies of the individual components (equation below) and was corrected for basis set superposition error using the counterpoise technique. Interaction energy: 2Eint = ECO – EAu – 2ECu, where ECO = energy of the Au(Val)2/CuI2 co-assembly, EAu = single-point energy of Au(Val)2, and ECu = single-point energy of [CuI2]−.
Acknowledgments
We gratefully acknowledge the financial support from the National Key R&D Program of China (2021YFA1200301), the National Natural Science Foundation of China (Nos. 92061201, 21825106, and 22205211), the Postdoctoral Research Grant in Henan Province (No. 202101001), and Zhengzhou University.
Supporting Information Available
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/jacsau.2c00653.
Author Contributions
CRediT: Ya-Jie Wang conceptualization, formal analysis, funding acquisition, investigation, writing-original draft, writing-review & editing; Xiao-Yan Shi formal analysis, investigation; Pengyao Xing conceptualization, formal analysis, writing-review & editing; Shuang-Quan Zang conceptualization, funding acquisition, project administration, supervision, writing-review & editing.
The authors declare no competing financial interest.
Supplementary Material
References
- Liu M.; Zhang L.; Wang T. Supramolecular Chirality in Self-Assembled Systems. Chem. Rev. 2015, 115, 7304–7397. 10.1021/cr500671p. [DOI] [PubMed] [Google Scholar]
- Yashima E.; Ousaka N.; Taura D.; Shimomura K.; Ikai T.; Maeda K. Supramolecular Helical Systems: Helical Assemblies of Small Molecules, Foldamers, and Polymers with Chiral Amplification and Their Functions. Chem. Rev. 2016, 116, 13752–13990. 10.1021/acs.chemrev.6b00354. [DOI] [PubMed] [Google Scholar]
- Dong J.; Liu Y.; Cui Y. Supramolecular Chirality in Metal–Organic Complexes. Acc. Chem. Res. 2021, 54, 194–206. 10.1021/acs.accounts.0c00604. [DOI] [PubMed] [Google Scholar]
- Gong Z.-L.; Li Z.-Q.; Zhong Y.-W. Circularly Polarized Luminescence of Coordination Aggregates. Aggregate 2022, 3, e177 10.1002/agt2.177. [DOI] [Google Scholar]
- Mori T. Chiroptical Properties of Symmetric Double, Triple, and Multiple Helicenes. Chem. Rev. 2021, 121, 2373–2412. 10.1021/acs.chemrev.0c01017. [DOI] [PubMed] [Google Scholar]
- Zhang L.; Qin L.; Wang X.; Cao H.; Liu M. Supramolecular Chirality in Self-Assembled Soft Materials: Regulation of Chiral Nanostructures and Chiral Functions. Adv. Mater. 2014, 26, 6959–6964. 10.1002/adma.201305422. [DOI] [PubMed] [Google Scholar]
- Huang S.; Yu H.; Li Q. Supramolecular Chirality Transfer toward Chiral Aggregation: Asymmetric Hierarchical Self-Assembly. Adv. Sci. 2021, 8, 2002132. 10.1002/advs.202002132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peng Q. C.; Luo X. M.; Qin Y. J.; Wang T.; Bai B.; Wei X. L.; Li K.; Zang S. Q. Influence of Wallach’s Rule on Chiral AIE Systems and Its Application in Cryptographic Information Storage. CCS Chem. 2022, 4, 3686–3692. 10.31635/ccschem.022.202201998. [DOI] [Google Scholar]
- Peng Q.; Shuai Z. Molecular Mechanism of Aggregation-Induced Emission. Aggregate 2021, 2, e91 10.1002/agt2.91. [DOI] [Google Scholar]
- Ma S.; Du S.; Pan G.; Dai S.; Xu B.; Tian W. Organic Molecular Aggregates: From Aggregation Structure to Emission Property. Aggregate 2021, 2, e96 10.1002/agt2.96. [DOI] [Google Scholar]
- Zhang X.; Xu Y.; Valenzuela C.; Zhang X.; Wang L.; Feng W.; Li Q. Liquid Crystal-Templated Chiral Nanomaterials: From Chiral Plasmonics to Circularly Polarized Luminescence. Light Sci. Appl. 2022, 11, 233. 10.1038/s41377-022-00913-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang L.; Urbas A. M.; Li Q. Nature-Inspired Emerging Chiral Liquid Crystal Nanostructures: From Molecular Self-Assembly to DNA Mesophase and Nanocolloids. Adv. Mater. 2020, 32, 1801335. 10.1002/adma.201801335. [DOI] [PubMed] [Google Scholar]
- Chen X.-M.; Zhang S.; Chen X.; Li Q. Tunable Circularly Polarized Luminescent Supramolecular Systems: Approaches and Applications. ChemPhotoChem 2022, 6, e202100256 10.1002/cptc.202100256. [DOI] [Google Scholar]
- He Y.; Lin S.; Guo J.; Li Q. Circularly Polarized Luminescent Self-organized Helical Superstructures: From Materials and Stimulus-Responsiveness to Applications. Aggregate 2021, 2, e141 10.1002/agt2.141. [DOI] [Google Scholar]
- Ye Q.; Zheng F.; Zhang E.; Bisoyi H. K.; Zheng S.; Zhu D.; Lu Q.; Zhang H.; Li Q. Solvent Polarity Driven Helicity Inversion and Circularly Polarized Luminescence in Chiral Aggregation Induced Emission Fluorophores. Chem. Sci. 2020, 11, 9989–9993. 10.1039/d0sc04179c. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dou X.; Mehwish N.; Zhao C.; Liu J.; Xing C.; Feng C. Supramolecular Hydrogels with Tunable Chirality for Promising Biomedical Applications. Acc. Chem. Res. 2020, 53, 852–862. 10.1021/acs.accounts.0c00012. [DOI] [PubMed] [Google Scholar]
- Xing P.; Zhao Y. Controlling Supramolecular Chirality in Multicomponent Self-Assembled Systems. Acc. Chem. Res. 2018, 51, 2324–2334. 10.1021/acs.accounts.8b00312. [DOI] [PubMed] [Google Scholar]
- Wang F.; Ji W.; Yang P.; Feng C.-L. Inversion of Circularly Polarized Luminescence of Nanofibrous Hydrogels through Co-assembly with Achiral Coumarin Derivatives. ACS Nano 2019, 13, 7281–7290. 10.1021/acsnano.9b03255. [DOI] [PubMed] [Google Scholar]
- An S.; Hao P.; Xing P. Halogen Bonding Mediated Hierarchical Supramolecular Chirality. ACS Nano 2021, 15, 15306–15315. 10.1021/acsnano.1c06178. [DOI] [PubMed] [Google Scholar]
- Liu G.; Sheng J.; Teo W. L.; Yang G.; Wu H.; Li Y.; Zhao Y. Control on Dimensions and Supramolecular Chirality of Self-Assemblies through Light and Metal Ions. J. Am. Chem. Soc. 2018, 140, 16275–16283. 10.1021/jacs.8b10024. [DOI] [PubMed] [Google Scholar]
- Martial B.; Lefèvre T.; Buffeteau T.; Auger M. Vibrational Circular Dichroism Reveals Supramolecular Chirality Inversion of α-Synuclein Peptide Assemblies upon Interactions with Anionic Membranes. ACS Nano 2019, 13, 3232–3242. 10.1021/acsnano.8b08932. [DOI] [PubMed] [Google Scholar]
- Ji J.; Wei X.; Wu W.; Fan C.; Zhou D.; Kanagaraj K.; Cheng G.; Luo K.; Meng X.-G.; Yang C. The More the Slower: Self-Inhibition in Supramolecular Chirality Induction, Memory, Erasure, and Reversion. J. Am. Chem. Soc. 2022, 144, 1455–1463. 10.1021/jacs.1c13210. [DOI] [PubMed] [Google Scholar]
- Yam V. W.-W.; Au V. K.-M.; Leung S. Y.-L. Light-Emitting Self-Assembled Materials Based on d8 and d10 Transition Metal Complexes. Chem. Rev. 2015, 115, 7589–7728. 10.1021/acs.chemrev.5b00074. [DOI] [PubMed] [Google Scholar]
- Katz M. J.; Sakai K.; Leznoff D. B. The Use of Aurophilic and Other Metal–Metal Interactions as Crystal Engineering Design Elements to Increase Structural Dimensionality. Chem. Soc. Rev. 2008, 37, 1884–1895. 10.1039/b709061g. [DOI] [PubMed] [Google Scholar]
- Pyykkö P. Strong Closed-Shell Interactions in Inorganic Chemistry. Chem. Rev. 1997, 97, 597–636. 10.1021/cr940396v. [DOI] [PubMed] [Google Scholar]
- Schmidbaur H.; Schier A. Aurophilic Interactions as a Subject of Current Research: An Up-Date. Chem. Soc. Rev. 2012, 41, 370–412. 10.1039/c1cs15182g. [DOI] [PubMed] [Google Scholar]
- Schmidbaur H.; Schier A. Argentophilic Interactions. Angew. Chem., Int. Ed. 2015, 54, 746–784. 10.1002/anie.201405936. [DOI] [PubMed] [Google Scholar]
- Harisomayajula N. V. S.; Makovetskyi S.; Tsai Y.-C. Cuprophilic Interactions in and between Molecular Entities. Chem.—Eur. J. 2019, 25, 8936–8954. 10.1002/chem.201900332. [DOI] [PubMed] [Google Scholar]
- Chen Y.; Cheng G.; Li K.; Shelar D. P.; Lu W.; Che C.-M. Phosphorescent Polymeric Nanomaterials with Metallophilic d10···d10 Interactions Self-Assembled from [Au(NHC)2]+ and [M(CN)2]−. Chem. Sci. 2014, 5, 1348–1353. 10.1039/c3sc52989d. [DOI] [Google Scholar]
- Wan Q.; Yang J.; To W.-P.; Che C.-M. Strong Metal–Metal Pauli Repulsion Leads to Repulsive Metallophilicity in Closed-Shell d8 and d10 Organometallic Complexes. Proc. Natl. Acad. Sci. U.S.A. 2021, 118, e2019265118 10.1073/pnas.2019265118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang F.; Liu L.; Wang W.; Li S.; Shi M. Chiral NHC–Metal-Based Asymmetric Catalysis. Coord. Chem. Rev. 2012, 256, 804–853. 10.1016/j.ccr.2011.11.013. [DOI] [Google Scholar]
- Deng M.; Mukthar N. F. M.; Schley N. D.; Ung G. Yellow Circularly Polarized Luminescence from C1-Symmetrical Copper(I) Complexes. Angew. Chem., Int. Ed. 2020, 59, 1228–1231. 10.1002/anie.201913672. [DOI] [PubMed] [Google Scholar]
- Ying A.; Ai Y.; Yang C.; Gong S. Aggregation-Dependent Circularly Polarized Luminescence and Thermally Activated Delayed Fluorescence from Chiral Carbene-CuI-Amide Enantiomers. Angew. Chem., Int. Ed. 2022, 61, e202210490 10.1002/anie.202210490. [DOI] [PubMed] [Google Scholar]
- Cao Y.; Yang J.-G.; Deng Y.; Wang S.; Liu Q.; Shen C.; Lu W.; Che C.-M.; Chen Y.; He L. Amine-Responsive Disassembly of AuI–CuI Double Salts for Oxidative Carbonylation. Angew. Chem., Int. Ed. 2020, 59, 2080–2084. 10.1002/anie.201914089. [DOI] [PubMed] [Google Scholar]
- Liu Q.; Xie M.; Chang X.; Cao S.; Zou C.; Fu W.-F.; Che C.-M.; Chen Y.; Lu W. Tunable Multicolor Phosphorescence of Crystalline Polymeric Complex Salts with Metallophilic Backbones. Angew. Chem., Int. Ed. 2018, 57, 6279–6283. 10.1002/anie.201803965. [DOI] [PubMed] [Google Scholar]
- Liu Q.; Xie M.; Chang X.; Gao Q.; Chen Y.; Lu W. Correlating Thermochromic and Mechanochromic Phosphorescence with Polymorphs of a Complex Gold(I) Double Salt with Infinite Aurophilicity. Chem. Commun. 2018, 54, 12844–12847. 10.1039/c8cc05210g. [DOI] [PubMed] [Google Scholar]
- Da X.; Yu F.-H.; Zhang C.; Wang Z.; Jian Y.; Hou Y.; Chen Y.; Wang X.; Zhou Q. A Bioorthogonal Assembly Based on Metallophilic Interactions for Selective Imaging and PDT Treatment of Cancer Cells. Inorg. Chem. Front. 2022, 9, 2290–2297. 10.1039/d2qi00147k. [DOI] [Google Scholar]
- Yang J.-G.; Li K.; Wang J.; Sun S.; Chi W.; Wang C.; Chang X.; Zou C.; To W.-P.; Li M.-D.; Liu X.; Lu W.; Zhang H.-X.; Che C.-M.; Chen Y. Controlling Metallophilic Interactions in Chiral Gold(I) Double Salts towards Excitation Wavelength-Tunable Circularly Polarized Luminescence. Angew. Chem., Int. Ed. 2020, 59, 6915–6922. 10.1002/anie.202000792. [DOI] [PubMed] [Google Scholar]
- Wu Z.; Du Y.; Liu J.; Yao Q.; Chen T.; Cao Y.; Zhang H.; Xie J. Aurophilic Interactions in the Self-Assembly of Gold Nanoclusters into Nanoribbons with Enhanced Luminescence. Angew. Chem., Int. Ed. 2019, 58, 8139–8144. 10.1002/anie.201903584. [DOI] [PubMed] [Google Scholar]
- Yang D.; Duan P.; Zhang L.; Liu M. Chirality and Energy Transfer Amplified Circularly Polarized Luminescence in Composite Nanohelix. Nat. Commun. 2017, 8, 15727. 10.1038/ncomms15727. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.