

Abstract
Numerous beneficial compounds for treating disease have been discovered in nature. Manumycin-A (Man-A) is a natural, microbially-derived metabolite that inhibits farnesyltransferase activity, reduces cholesterolemia and kills tumors in several experimental disease models. We recently showed that Man-A stimulated Reactive Oxygen Species (ROS) which were upstream of serine dephosphorylation and caspase-dependent cleavage of MEK and Akt in lymphoma apoptosis. Conversely, activation-specific phosphorylation of MEK and Akt proteins on serines was stable in Man-A-resistant tumors suggesting that stimulation of Ser/Thr PPase activity might be required for Man-A tumoricidal activity. We tested this notion and found that indeed, pre-treatment with calyculin-A, and agent that equipotently inhibits PP1 and PP2A, blocked all downstream effects of Man-A whereas, the PP2A-selective inhibitor, Okadaic acid did not. These results suggested that PP1 and not PP2A played a role in Man-A action. CDC2 kinase phosphorylates PP1α on Thr320 and thereby, inhibits PP1α activity. Hence, it was then posited that if PP1α were a critical player in Man-A action, then Man-A treatment might promote dephosphorylation of PP1α on Thr320. Subsequent experiments revealed that indeed, T320 was only dephosphorylated in the tumors that later underwent apoptosis. Lastly, stable over-expression of a constitutively active PP1α mimetic (PP1αT320A mutant), predictably elevated basal ROS levels and enhanced Man-A-stimulated apoptosis. Together, these data suggest that PP1α is an important proximal effector of Man-A mediated lymphoma death and that the mechanisms of Man-A action warrant further investigation because of its potential in treating human diseases.
INTRODUCTION
Many human diseases and conditions have been, and are being treated by natural compounds. These natural compounds include anagelsics and anti-thrombotics, antibiotics, anti-cancer agents, beneficial immunosuppresants and cholesterol-lowering agents1. Manumycin-A (Man-A) is a natural antibiotic which is isolated from the conditioned broth of Streptomyces parvulus 2. Man-A inhibits farnesyltransferases (FTases, 2, 3), a family of enzymes that performs post-translational prenylation of Ras, a process required for the maturation (membrane insertion) of Ras. Man-A is a powerful tumoricide and has been used experimentally to inhibit Ras maturation. Ras dysfunction is observed in over 30% of all known human tumors 4–6 and it was anticipated that targeting Ras maturation would address an appreciable proportion of human cancers. A number of commercial FTAse inhibitors such as R115777, SCH66336, L-778,123 and BMS-214662, were rationally designed and potently inhibited FTases and thereby blocked Ras prenylation. However, in clinical trials, they were surprisingly poor tumoricides against both Ras transformed- and non Ras-transformed cancers 7. Based on our recent report 8 and the data presented herein, we posit that in contrast to the synthetic agents, Man-A is capable of modulating multiple tumoricidal pathways. These include particular ROS, and protein phosphatases and caspases which effect the selective targeting and cleavage of Akt and MEK. Inducing this combination of events makes Man-A superior in effecting a controlled cytotoxic response in both human and murine tumors 8.
We observed that Man-A induced apoptosis involved a cascade that began with a large ROS induction (O2•−, superoxide), followed by dephosphorylation of MEK and Akt, and their caspase-dependent cleavage and finally, apoptosis 8. Interestingly, those and subsequent experiments revealed that tumors containing phospho-stable MEK and Akt, exhibited either a delayed response to Man-A induced apoptosis or did not undergo apoptosis in extended timeframes (herein and Sears et al. 8). Because of the inverse relationship of phospho-Akt and -MEK stability with Man-A sensitivity, herein, we tested the notion that Man-A action might involve modulation of Ser/Thr protein phosphatase activity. Based on our preliminary results (below and data not shown), protein phosphatase 1, and more specifically, protein phosphatase 1 alpha (PP1α), were the most likely candidates for Man-A stimulated dephosphorylation of MEK and Akt.
Protein phosphatase 1 (PP1) is a relatively small (~38 k Da) protein whose specificity and function are controlled by its interactions with an array of regulatory proteins. There are four isoforms of the catalytic subunit: α, γ1, γ2 and δ. The alpha (α) subunit comprises about 67% of the total mass of PP1 expressed in mammalian cells 9. While tyrosine phosphorylation usually initiates growth and proliferative pathways (e.g. EGF receptor, src kinases), Ser/Thr phosphorylation influences many steady state cellular metabolic functions, including for example, but not limited to, growth, regulation of cytoskeletal dynamics, the rates of macromolecular biosynthesis and metabolism, gene transcription and the suppression of apoptotic signals 10, 11. Hence, taken together, it would stand to reason that rapidly-proliferating tumor cells would suppress, or tightly regulate PP1 activation in order to maximize Ser/Thr phosphorylation during cellular growth phases or during cell cycle progression. Therefore, the role of PP1α in Man-A driven Akt and MEK dephosphorylation and cleavage in lymphoma cell apoptosis were explored. The data show that Ser/Thr protein phosphatase 1α (PP1α) is a bona fide effector of Man-A since pharmacological and molecular manipulation of this enzyme modulates the effects of Man-A on ROS, on caspase activation, on MEK and Akt dephosphorylation and on caspase-dependent cleavage of MEK and Akt, and subsequent apoptosis. These data are presented herein.
MATERIALS AND METHODS
Cell Culture
This study utilized the extensively characterized WEHI-231 (IgM+) and A20.2J (IgG+), B cell lymphoma lines 12–14. Some experiments utilized Daudi and Ramos, human IgM+ cell lines 15, and K-562 and Jurkat (human CML and T-cell leukemia models 16.) These cells were kindly provided by Dr. David W. Scott, USUHS, Bethesda, MD. Additional experiments utilized human DU-145 and LNCAP prostate cancer lines 17 (obtained from Dr. Vincent Njar, University of Maryland, Baltimore). Suspension cells were grown in supplemented RPMI 1640 culture medium as previously described 8, 12, 18 or in DMEM as described in the references above. Cell culture supplements were obtained from Sigma-Aldrich (St. Louis, MO), Cellgro (Manassas, VA), Bio-Whittaker (Walkersville, MD), Invitrogen (Carlsbad, CA), Atlanta Biologicals (Lawrenceville, GA) and Gemini Bioproducts (West Sacramento, CA).
Reagents
All reagents were titrated for optimal, sub-toxic dose- and time-responses. Manumycin-A was obtained from A.G. Scientific (San Diego, CA). The broad-spectrum caspase inhibitor, QVD-OPH was obtained from R & D Systems (Minneapolis, MN). N-acetyl cysteine (NAC) was obtained from Sigma-Aldrich. Calyculin A and Okadaic acid, selective inhibitors of protein phosphatases 1 and 2A (PP1 and PP2A), respectively 19, 20, were obtained either from Sigma-Aldrich or from LC Laboratories, (Woburn, MA). Annexin V-FITC or Annexin-V-APC conjugates, mitochondrial probes and ROS detection fluorophores were purchased from BD Biosciences (San Jose, California) and Life Technologies (Grand Island, NY).
ROS and ΔΨm Detection
For 0 – 8h studies, starting culture densities were 0.5 × 106 live cells/ml. Briefly, one ml (1.0 ml) of cells were harvested directly into FACS tubes then stained with dihydroethidium (1 μM DHE final concentration) for superoxide detection 21, or with 2’,7’-dichlorodihydrofluorescein diacetate (DCF, 1 μM final concentration) for detection of H2O2 21. Mitochondrial membrane potential (ΔΨm) was measured with either tetramethylrhodamine (TMR, 22) or with 3,3’-dihexyloxacarbocyanine iodide (DiOC6, 22, 23), each at 50 nM final concentrations. All of the above fluorescing reporter reagents were purchased from Molecular Probes/Life Technologies. Typically, the cells were pulse-stained for the final 20 min of the incubations then processed as previously described 18, 21, 24–26. Cytometric analyses were performed immediately thereafter on Becton Dickinson FACScan or FACScalibur instruments (Becton Dickinson, San Jose, CA). Data analysis was performed with CellQuest® and FlowJo software (Becton Dickinson or TreeStar, Ashland, OR, respectively). Positive oxidation controls included treating cells with up to 500 μM H2O2 (maximal DCF oxidation control). Menadione, a superoxide generator 27, was also used on cells as a positive control for maximal DHE oxidation. Carbonylcyanide 3-chlorophenylhydrazone (CCCP), an uncoupler of mitochondrial respiration 23, was used as a negative control for ΔΨm. Treated and untreated cells were stained and compared by FACS/flow analysis and gate parameters were set to designate DCF+, DHE+ or ΔΨm-low (low mitochondrial potential) cells or as illustrated in the accompanying figure legends. Plasma membrane integrity was determined using Propidium Iodide (PI) exclusion and standard protocols 12.
Apoptosis Assays
Apoptosis was assessed by Annexin-V binding to externalized phosphatidylserine (PS) in cells excluding propidium iodide (Anx V+/PI−) or sub-diploid nuclei determination essentially by previously described and established methods 12, 18, 21, 24. For Anx V staining, briefly, 1.0 ml of cells at 0.5 × 106 /ml were harvested in FACS tubes, washed once with HEPES buffered saline (HBS: 20 mM HEPES, pH 7.4; 120 mM NaCl) and then once in Anx V binding buffer ( HBS supplemented with 2.5 mM CaCl2). For detection of externalized PS, we used Anx V which was directly conjugated to common fluorophores e.g., FITC, PE, PerCP or APC. Anx V APC conjugate was used in triple staining experiments (GFP in FL1, PI in FL3 and AnxV:APC in FL4). The detection conjugates were obtained from Biosource (Camarillo, CA), Life Technologies and Becton Dickinson Biosciences and were used as per standard and manufacturers protocols. Data was collected by flow cytometric analysis and analyzed as described above.
Constructs and Co-transfections
Plasmids bearing relevant cDNAs (AktS473D or PP1αT320A), were used to transfect cells and were prepared using endotoxin-free, plasmid isolation kits from Qiagen (Valencia, CA). Transient co-transfections were performed using the Amaxa Nucleofector System® (Amaxa Inc, U.S.; Gaithersburg, MD). Co-transfections included 1μg of pMaxGFP and 5 μg construct or empty cassette (as noted in Figure Legends). Transfections were optimized as per the manufacturer’s protocols and pMaxGFP expression was monitored by fluorescence microscopy and flow cytometry. Most Man-A treatments were performed 12 to 16h post transfection where the majority (>60%) of the surviving cells were quantitatively observed to express pMaxGFP (see Ref. 8). Constitutively active Akt (AktS473D) constructed in pcDNA3, was a generous gift from Dr. Alexandra Newton (UCSD, CA).
Generation of Stably Transfected Cells Overexpressing PP1αT320A
A20.2J cells were transfected with either 5 μg pcDNA3 or 5 μg PP1αT320A/pcDNA3 using electroporation as previously described 8. The PP1α construct was kindly provided by Dr. Len Neckers, NIH (Bethesda, MD). During the selection period, the transfected cells were subsequently plated, allowed to recover for 24h, washed, then stably selected in the continuous presence of 0.5 mg/ml G-418 (Life Technologies) over 3 weeks. The cells were centrifuged and supplemented with fresh culture medium every 3 to 4 days. For an additional control, mock transfected cells (no cDNA or empty cassette), were grown under similar selection conditions. Under these conditions, no viable cells were detected after 3 weeks. Myc tagged PP1αT320A was detected with anti Myc tag antibody (Cell Signaling) and by western blotting analysis. G418 resistance was also confirmed using anti neomycin phosphotransferase (NPT) antibody (Millipore, Billerica, MA) and western blotting to ascertain expression of NPT in the stably selected cells.
Western blots
Cells were treated as indicated in the Figure Legends and processing, SDS-PAGE and blotting procedures were performed essentially as previously described 18. Gradient gels and western blot detection reagents were obtained from Life Technologies and Pierce, respectively. Some PAGE gels were prepared in our lab. For western blots, antibodies included: anti-phospho and total MEK, Akt and anti-cleaved caspase 3 (Asp175, Cat# 9661 from Cell Signaling, Beverly MA. To confirm results, additional anti-caspase 9 and -Akt antibodies were obtained from BD Biosciences and Santa Cruz Biotechnology (Santa Cruz, CA), respectively. Anti PP1α (Santa Cruz catalogue SC-443) and anti-pan PP1 (Santa Cruz, catalogue SC: 7482) antibodies were used to examine either PP1α or total PP1 catalytic unit expression. Anti phospho PP1α (Thr320) antibody was obtained from Cell Signaling Technology. Typical experiments terminating in western blotting required at least 10 million (107) cells. For adhesion culture (Du145 and LNCAP), all floating and adherent cells were recovered after treatments and processed for subsequent analyses.
RESULTS
Effect of PPase Inhibitors on MEK and Akt Processing
MEK activation requires phosphorylation on the Ser220/Ser221 pair, whereas Akt activation requires dual phosphorylation on Thr308 and Ser473 28. It is widely known that PP1 and PP2A dephosphorylate a number of Ser/Thr-phosphorylated targets, including MEK and Akt, both in vitro and in intact cells 28, 29. Our lab recently showed that Man-A treatment initially caused ROS-dependent dephosphorylation of MEK and Akt which was then followed by their caspase-dependent cleavage 8. Since Ser/Thr dephosphorylation appeared to be upstream of MEK and Akt processing and for cytotoxicity downstream of Man-A treatment, we probed the roles of PP1α and PP2A in Man-A action first with a pharmacological approach using the PP1 and PP2A inhibitors, okadaic acid (OA) and Calyculin-A (Cal-A). Importantly, OA inhibits PP2A 100 times more potently than it does PP1 (IC50 = 0.1 nM for PP2A), whereas Cal-A inhibits both enzymes equipotently 19. Cal-A blocked MEK and Akt dephosphorylation and in fact, promoted the accumulation of phospho-MEK and -Akt (Figure 1). Increased phosphorylation of MEK and Akt proteins corresponded with their resistance to being cleaved. On the other hand, 100 nM OA did not prevent MEK and Akt cleavage (Figure 1). Importantly 100 nM OA is 1000 times the IC50 concentration required for inhibiting PP2A 19. Hence, we concluded that PP1 was more likely involved in mediating the effects of Man-A on MEK and Akt.
Figure 1. Effect of PPase Inhibitors on MEK and Akt Phosphorylation and Expression.
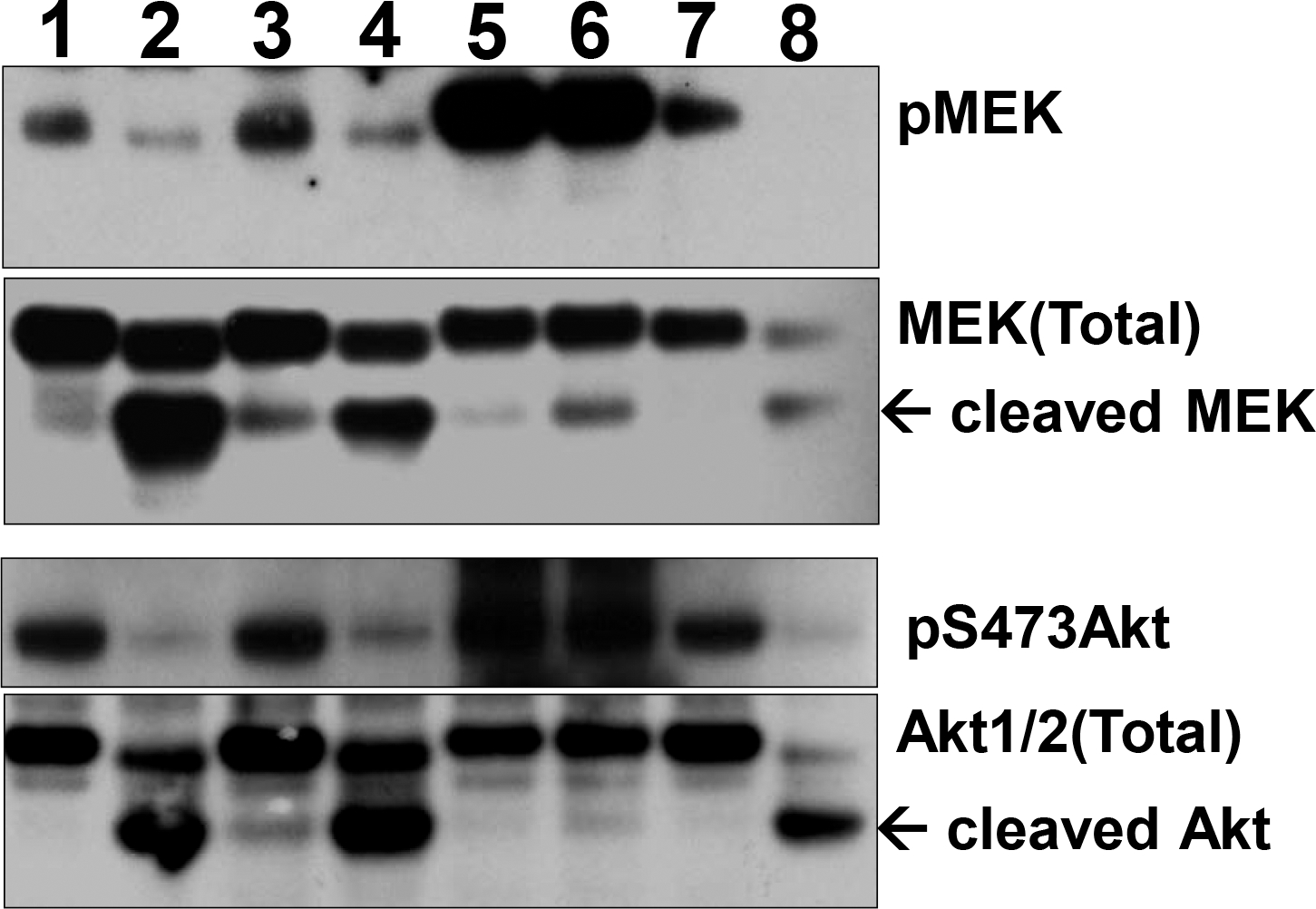
WEHI-231 cells were pre-treated with or without 1 or 10 nM Calyculin-A (Cal-A) or 100 nM Okadaic acid (OA) for 1h. The cells were then treated with or without 4 μM Manumycin-A (Man-A) for 120 min and pMEK/MEK, pAkt/Akt protein expression was determined by western blotting as described in the Materials and Methods. Treatment Lanes: 1, Untreated Control; 2, Man-A; 3, 1 nM Cal-A; 4, 1 nM Cal-A + Man-A; 5, 10 nM Cal-A; 6, 10 nM Cal-A + Man-A; 7, 100 nM OA; 8, 100 nM OA + Man-A. Arrows indicate cleaved MEK and Akt protein products. The results are representative of more than 4 separate, independent experiments.
Man-A Promotes MEK and Akt Cleavage in Murine and Human Hematological Malignancies
We previously showed that Man-A stimulates ROS- and caspase-dependent apoptosis in a panel of human and murine hematological cancer models and that MEK and Akt cleavage occurred fastest in the cells that underwent apoptosis most dramatically in terms of rate and magnitude 20. Next, we wished to quickly examine if the effects of Man-A on MEK and Akt processing were species or cell line specific. Confirming and extending our previous results, treating hematological cancers with Man-A led to Akt cleavage in the tumor lines tested (MEK not shown), regardless of the species of origin of the cells (Figure 2). K-562, a CML model, represents a less differentiated hematological cancer. Interestingly, K-562 was the most resistant to Man-A in terms of Akt processing (Figure 2) and apoptosis (apoptosis data not shown). Conversely, Man-A promoted stronger Akt cleavage in IgM+ murine (WEHI-231) and IgM+ human (Raji, Daudi and Ramos) cells. Jurkat, an acute, T-cell leukemia derived from an adolescent child (see Materials and Methods), was also very responsive to Man-A induced Akt processing (Figure 2). Interestingly, A20.2J, an IgG+ (class switched, murine B cell lymphoma), showed a delayed response to Man-A induced Akt processing (Figure 2) and a delayed response to Man-A mediated apoptosis 20. Based on these results, we concluded that protein processing was independent of the species of origin of the tumor and we would subsequently use WEHI-231 and A20.2J as models of Man-A sensitivity and resistance, respectively.
Figure 2. Man-A Promotes Protein Processing In Both Human and Murine Tumor Lines.
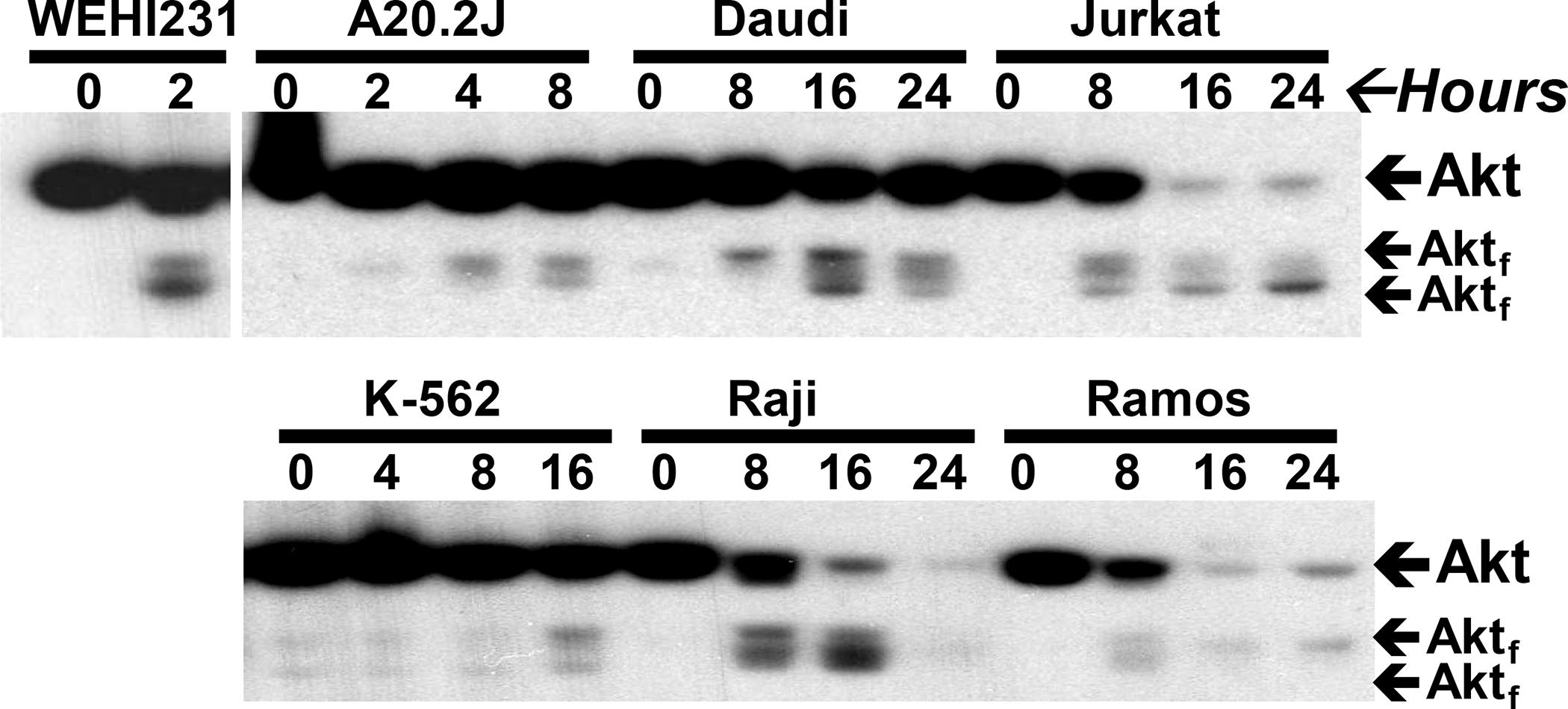
Cells were treated with Man-A as described above. Total protein expression was analyzed by western blotting. WEHI and A202.J are murine lines. Daudi, Jurkat, K-562, Raji and Ramos are human cell lines (Please see Materials and Methods). Subscript ‘f’ indicates fragment. These data are representative of more than four separate independent experiments.
Targeting of Akt by Man-A Promotes Lymphoma Apoptosis
Since phosphorylated/activated MEK and Akt are found in virtually all tumors, MEK and Akt are considered as ‘high value’ therapeutic targets and are the foci of several ongoing clinical trials (See www.clinicaltrials.gov). It was also considered that processing of MEK and Akt could represent latter stages of the apoptotic process in response to Man-A. To examine the relationship of MEK or Akt deactivation to apoptosis, lymphoma cells were pre-treated with or without the MEK inhibitors PD98059 and U-0126 or the PI-3K/Akt inhibitor, LY294002 30, 31, followed by Man-A treatment. Caspase inhibitors and antioxidant treatment blocked Man-A induced apoptosis (Figure 3A), confirming our previous results and the results of others 8. Additional experiments showed that MEK inhibition and Man-A co-treatment dramatically reduced proliferation in some of the lymphomas tested, which is consistent with the results of Richards et al. (Data not shown and ref. 31). In contrast, the PI-3K/Akt inhibitor, LY294002, enhanced Man-A-driven apoptosis (Figure 3A, MEK inhibitor data not shown). Hence, since MEK appeared to be upstream of growth-enhancement, but PI-3K/Akt seemed to play a more powerful role in tumor survival, we co-transfected WEHI-231 cells with pMaxGFP® with or without empty vector (pcDNA3®) or a constitutively-active mimetic of Akt constructed in the pcDNA3® vector (AktS473D/pcDNA3® 32, 33) and then examined the effect of Man-A on apoptosis in those cells. (The co-transfection methods were previously described in reference 8). The experiments herein showed that compared to transfection with pcDNA3® vector alone, transfection with the AktS473D/pcDNA3 construct had a modest effect on DHE-sensitive ROS, suggesting that Akt loss is not upstream of ROS generation. On the other hand, Man-A induced apoptosis (as measured by Anx V positivity in GFP+/PI− cells), was dramatically reversed in those cells (Figure 3B). Thus, these new data establish Akt dephosphorylation and processing as important steps in Man-A driven cytotoxicity.
Figure 3. Panel A, Targeting of Akt by Man-A Promotes Lymphoma Apoptosis.
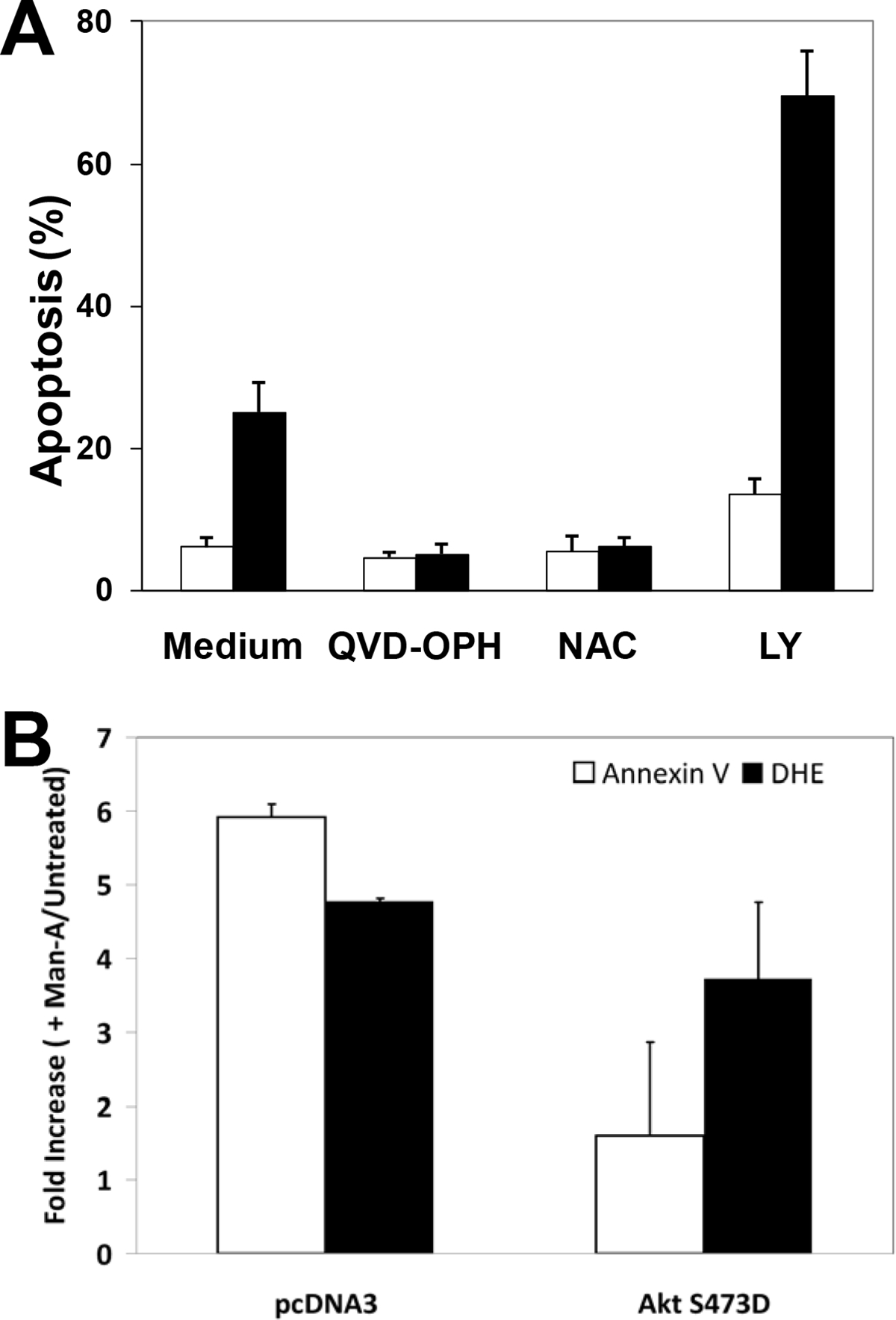
WEHI-231 cells were pre-treated with or without 10 μM QVD-OPH, 5 mM N-Acetyl Cysteine (NAC) or 10 μM LY294002 for 1 hour, followed by treatment with (■) or without (□) 4 μM Man-A for 2 hours. The label “Apoptosis (%)” denotes the percentage of sub-diploid nuclei measured after performing DNA content analysis as described in the Materials and Methods. Data are expressed as per cent Apoptosis and are representative of more than four separate, independent experiments. Panel B, Effects of Transiently Transfected AktS473D on Man-A Induced Apoptosis and Superoxide Production. WEHI-231 cells were transiently transfected with AktS473D/pcDNA3 using the Nucleofector method (Amaxa, See Materials and Methods) for 16h with the pMaxGFP cDNA in combinations with or without pcDNA3 vector alone or AktS473D constructed in pcDNA3. The cells were treated for 120 min with 4 μM Man-A. Apoptosis was assessed by measuring Annexin V binding to externalized phosphatidyl serine on GFP+/PI− cells, exactly as previously described by Sears et al. (Ref. 8). DHE probe was used to detect superoxide production as described in Materials and Methods and previously 8. Treatments were performed in triplicate and the data are expressed as the average fold increase (Treated / Untreated) in mean Annexin V-APC or DHE fluorescence ± SEM. The data are representative of at least two separate, independent experiments.
Differential Effects of Ser/Thr Protein Phosphatase Inhibitors on Events Downstream of Man-A
The data presented above show that Cal-A inhibits Man-A-stimulated MEK and Akt dephosphorylation and cleavage (Figure 1). Next, it was important to test the impact of Ser/Thr PPase inhibition on the Man-A initiated death cascade, i.e., ROS induction and apoptosis, as previously described 8. These additional experiments revealed that Cal-A and not OA blocked ROS induction and apoptosis (Figure 4 Panels, A and B, respectively). Since PP1 is the only known target of Cal-A to date, together, the data strongly suggested that PP1α plays an important role in the Man-A mediated apoptotic response in this lymphoma.
Figure 4. Differential Effects of Serine/Threonine Protein Phosphatase Inhibitors on Man-A Stimulated ROS and Apoptosis.
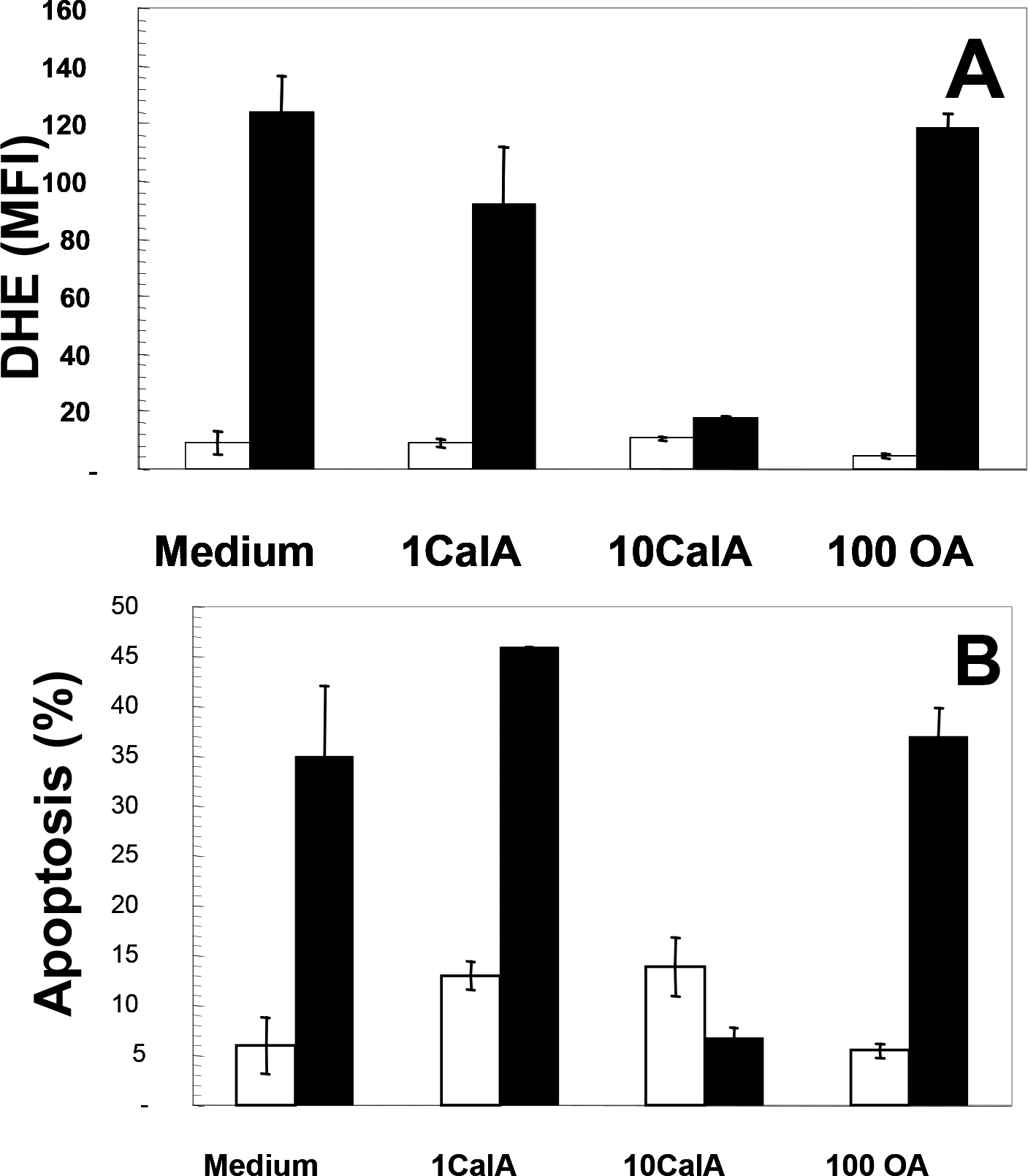
Panel A, WEHI-231 cells were treated with or without 1 or 10 nM Cal-A (Labeled as 1 or 10CalA, respectively) or with or without 100 nM Okadaic acid (Labeled as 100 OA) for 1h. The cells were then treated with (■) or without (□) 4 μM Man-A for 60 min and superoxide production measured with the dihydroethidium (DHE ) probe as described in Materials and Methods. Panel B, Apoptosis was determined by the DNA content and propidium iodide staining method as described above (In the legend of Figure 2 and in the Materials and Methods). Data are expressed as per cent sub-diploid nuclei. Treatments were performed in triplicate and data are expressed as means ± SEM. The data are representative of at least 4 separate, independent experiments.
Stability of Thr320 Phosphorylation on PP1α Reflects Relative Resistance to Man-A
The data above implicated PP1 as a potential target of Man-A. In addition, several groups have shown that inhibitory phosphorylation of PP1α on Thr320 is mediated by CDC2 11, 34–36. Therefore, we then examined if the response to Man-A involved correlative changes in Thr320 phosphorylation on PP1α. If PP1α were important for Man-A mediated apoptosis, we anticipated that Man-A would stimulate dephosphorylation of Thr320 in the cells that later underwent apoptosis. Indeed, the results showed that treatment with Man-A for 60 min resulted in complete dephosphorylation of the inhibitory site on PP1α in the WEHI-231 but not in A20.2J or prostate (Du145 or LNCAP cancer cells (Figure 5). Importantly, failure to induce PP1α dephosphorylation coincided with failure of Man-A to induce caspase 3 cleavage and apoptosis in the treated cells (Figure 5 and ref. 8. Apoptosis data not shown). Hence, sensitivity to Man-A-mediated apoptosis correlated positively with its ability to both induce ROS and to cause both Akt and PP1α dephosphorylation (Figures 1 and 5). In addition, in consideration with the data presented in Figure 2 above, it might appear that Man-A might selectively target hematological malignancies depending on their stages of activation and/or differentiation.
Figure 5.
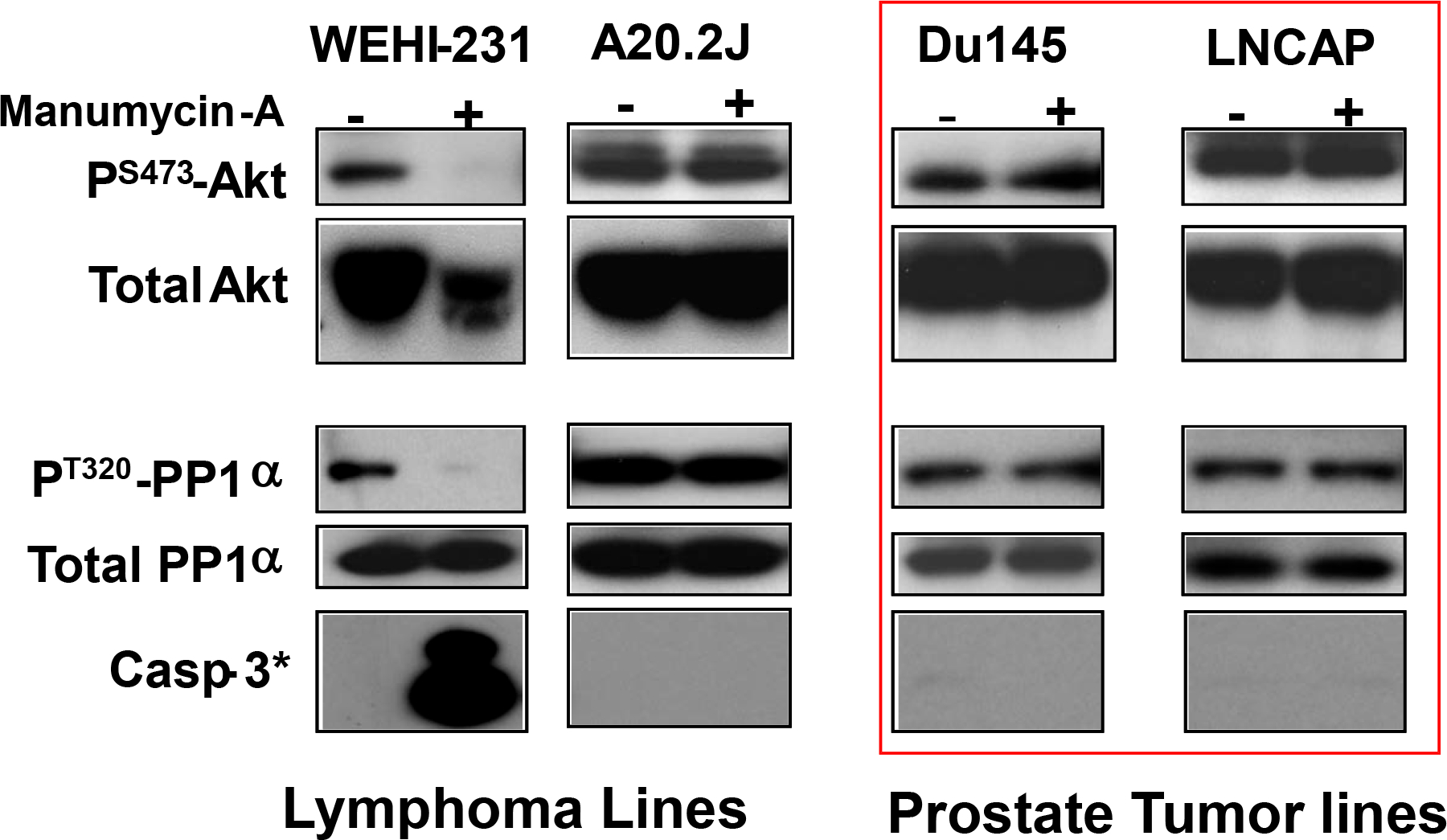
Effect of Man on P-Akt, total Akt, P-PP1α/PP1α, and cleaved (activated) Caspase 3. Human prostate cancer (Du145 and LNCAP) and murine lymphoma (WEHI-231 and A20.2J) cell lines were maintained in exponential growth phase and were treated with or without 4 μM Manumycin-A for 0 (−) or 2h (+). Proteins were analyzed by western blot as described above. Note1: cells producing the data shown in rectangle with thick lines are more resistant to Man-A. Note2: Akt loss occurs only in WEHI-231. The data are representative of at least 3 separate, independent experiments.
Effects of Stable over-expression of Constitutively-Activated PP1α Mimetic on Man-A Action
In consideration of the time to effect 50% apoptosis, A20.2J lymphoma cells can resist the effects of Man-A as much as 8 times longer than their WEHI-231 counterparts (above and ref. 8) and PP1α remains stably phosphorylated in Man-A resistant A20.2J and prostate cancer cells (Figure 5). In addition, whereas the PP2A-specific inhibitor OA had modest effects on Man-A modulated endpoints, the PP1 and PP2A inhibitor, Cal-A, blocked both ROS induction and apoptosis (Figure 4). Therefore, to further explore the role of PP1α in Man-A action, the constitutively-active PP1α mimetic (PP1αThr320A) was stably over-expressed in A20.2J cells, which we decided to use as a ‘resistance model.’ Importantly, mutation of T320 on PP1α to alanine (T320A) renders PP1α constitutively-active since CDC2 cannot phosphorylate the intended inhibitory site 11, 34–36. It was posited that if PP1α plays a major role in Man-A action, then expression of PP1αT320A should elevate ROS and sensitize A20.2J cells to Man-A mediated apoptosis. Indeed, stable transfection with PP1αT320A/pcDNA3 alone enhanced basal superoxide production in A20.2J (Figure 6A, solid bars) and amplified the effect of Man-A on A20.2J apoptosis 5-fold (from 5% to ~26%, hatched bars). Parallel results were observed in experiments conducted on human Du145, LNCAP and MCF7 (prostate and breast cancer cell lines) transfected with PP1α Thr320A (Data not shown). Hence, overall, these data strongly support our working hypothesis that PP1α is a critical intermediate in Man-A-mediated cellular apoptosis.
Figure 6. Stable Transfection With Constitutively- Active PP1αT320A cDNA Sensitizes A20.2J To Man-A Mediated Apoptosis.
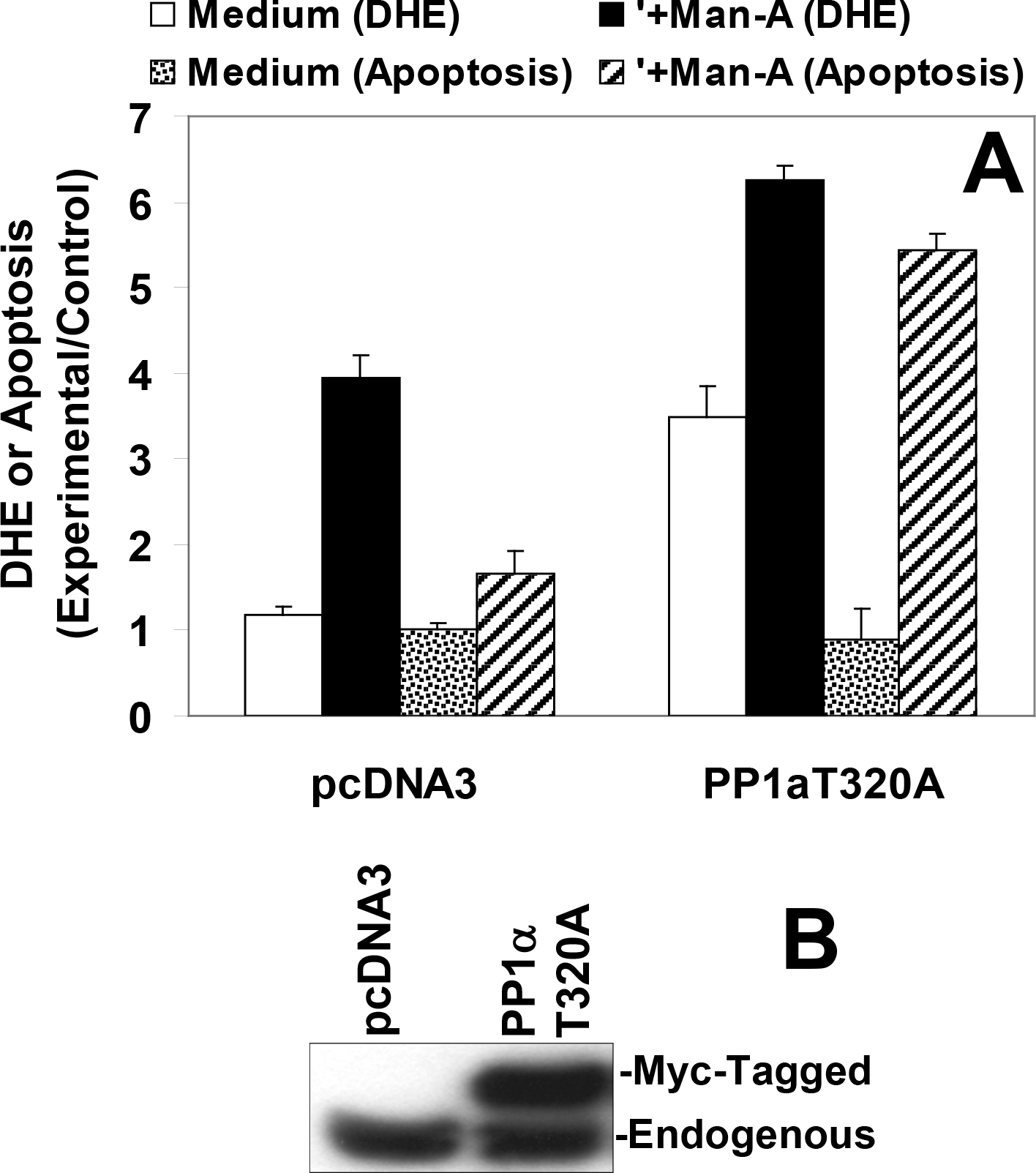
A20.2J were transfected with either 5 μg pcDNA3 or 5 μg PP1αT320A/pcDNA3 then stably selected with G-418. Stably-selected cells were treated with or without 4 μM Man-A for 6h. Superoxide production and apoptosis were measured using DHE and propidium iodide fluorescent reporters as described above. The data are expressed as Experimental/Control. Apoptosis in the control, untreated cells was 4.0%.
Inhibition of PP1α Restores Cellular Hydrogen Peroxide Tone
We had previously observed that sensitivity to Man-A induced apoptosis corresponded with excess superoxide (O2•−) generation, relative to H2O2 8. The data presented herein show that Cal-A blocked Man-A induced DHE oxidation (O2•− generation, Figure 4, Above). In addition, over-expression of PP1α increased DHE oxidation (O2•− generation, Figure 6A), leading us to conclude early that active PP1α was upstream of the production of ROS. Therefore, we initially posited that Cal-A-mediated protection from Man-A driven apoptosis, most likely involved reduction of ROS (both O2•− and H2O2). Since we had not yet examined H2O2 production in the presence of PP1 inhibitors, cells were then treated with or without Cal-A, followed by treatment with or without Man-A and ROS (H2O2) was measured using the RedOx sensitive dye DCFDA. Surprisingly, treatment with Cal-A resulted in increased DCFDA oxidation (increased H2O2 production, Figure 7A) under the same conditions where Cal-A decreased DHE oxidation (O2•−, Figure 4A).
Figure 7. Effects of PP1α Inhibitor on H2O2 Production and Apoptosis.
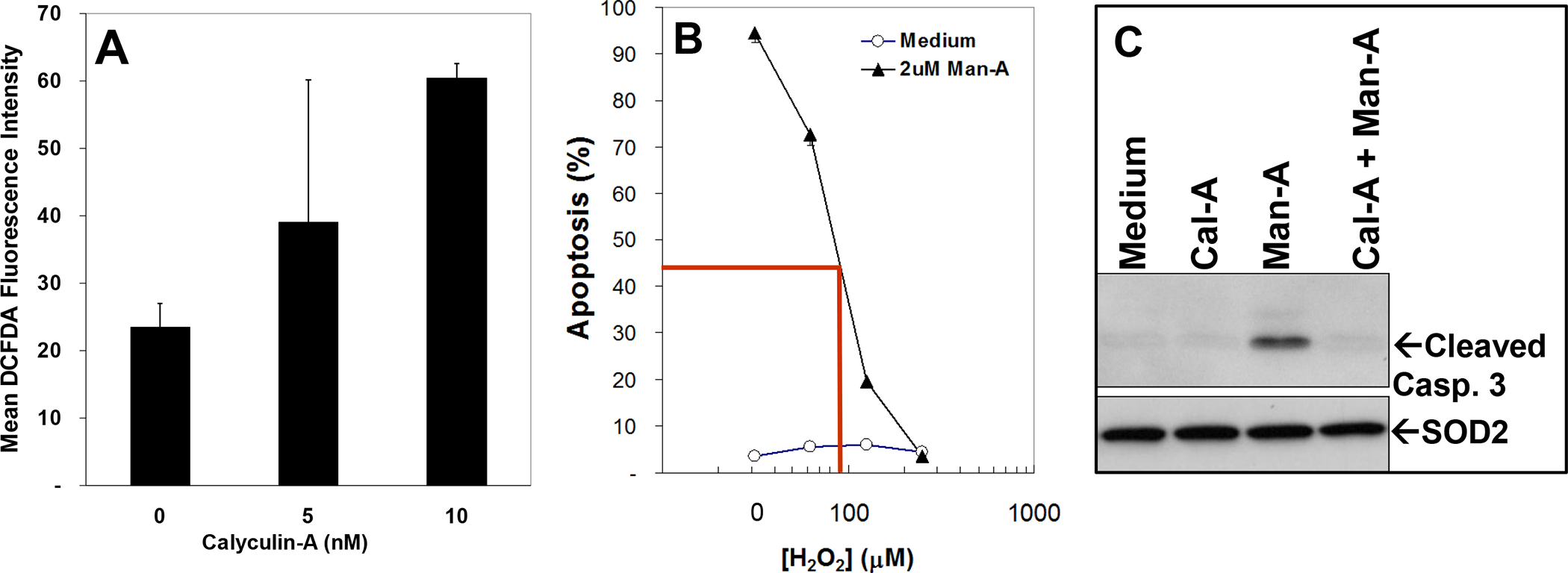
Panel A, 2 ml of WEHI-231 cells at 0.25 × 106 /ml were plated on 24 well plates and pretreated treated with or without Calyculin-A in triplicate for 1h. The cells were then pulse-labeled with DCFDA, harvested, washed and DCFDA fluorescence measured as described in Materials and Methods and above. The data are presented as Mean DCFDA fluorescence ± SEM and represent at least 3 separate, independent experiments. Panel B, WEHI-231 cells were treated with or without varying concentrations of H2O2 for 1h. The cells were washed and re-plated in fresh medium, pre-equilibrated at standard incubating conditions for the cells (37 °C, 95% air/5% CO2), then treated with or without 2 μM Man-A for 4 hrs. Apoptosis was assessed using the DNA-content and propidium iodide method as described above. Panel C, WEH-231 cells were treated as described above and processed for protein detection by Western blotting as described above and in the Materials and Methods. Blots were probed for activated (cleaved) caspase 3 as described in the legend of Figure 5. SOD2 protein expression under the same conditions was examined by western blotting. No changes in SOD2 protein expression were observed.
Conversely, it was possible that increased H2O2 production could be an unrelated or a secondary event in Cal-A-mediated protection from Man-A driven apoptosis. To test this notion, WEHI-231 lymphoma cells were pre-treated with or without varying, physiologically-relevant 37 doses of H2O2. The cells were then washed and re-suspended in fresh, pre-equilibrated RPMI-1640 (37°C, 5% CO2, 95% air. See Materials and Methods) and treated with or without Man-A for 2 hrs. These resulting data showed that indeed, pre-treatment with μM (physiological) amounts of H2O2, resulted in dose-dependent inhibition of Man-A driven apoptosis (Figure 7B). In addition, Cal-A treatment blocked Man-A stimulated caspase 3 activation (Figure 7C, Compare to Figure 5). Hence, we concluded that alteration of specific PP1α-associated ROS is critical for Man-A-driven outcomes in tumor cells. More specifically, it appears that Man-A drives the collapse of H2O2 tone which in turn, leads to un-repressed PPases and proteases, which then drive protein dephosphorylation and apoptosis. SOD2 protein expression did not change both indicating equal protein loading under these conditions and that the Man-A-driven collapse of H2O2 does not involve depletion of SOD2 enzyme (Figure 7C).
Effect of PPase Inhibitor Cal-A on PP1Phosphorylation and Expression
We were intrigued by the effects of Cal-A on activation-related phosphorylation of MEK and Akt and the high level of PP1α phosphorylation on T320 in Man-A resistant tumor cells. So, we next explored the effect of Cal-A treatment on PP1α phosphorylation, anticipating that Cal-A treatment would promote phosphorylation of PP1α on the Thr320 inhibitory site of PP1α. The results showed that both low serum and Man-A treatment reduced PP1α phosphorylation on Thr320 (Figure 8, top). Indeed, treatment with Cal-A (+) but not vanadate (tyrosine phosphatase inhibitor, 38) dramatically increased PP1α phosphorylation on Thr320 which would be consistent with Cal-A increasing inhibition (reducing activation) of PP1α. Subsequent treatment with Man-A could not reverse the effect of Cal-A on PP1α which is consistent with the notion that by failing to engage PP1α dephosphorylation or activation, Man-A then fails to promote MEK and Akt processing and then apoptosis. That Man-A tumoricidal activity utilizes PP1α is supported by two key pieces of evidence above: 1), expression of active PP1α sensitized otherwise resistant cells to Man-A (Figure 6) and 2), hyper-phosphorylation of PP1α made otherwise Man-A sensitive lymphoma, resistant to the apoptotic effects of the compound (Figure 8). Further analysis of resolved cellular phosphoproteins under the conditions presented in Figure 8 showed that vanadate increased total cellular phosphorylation on tyrosine (data not shown).
Figure 8. Effect of PPase Inhibitor Cal-A on PP1Phosphorylation and Expression.
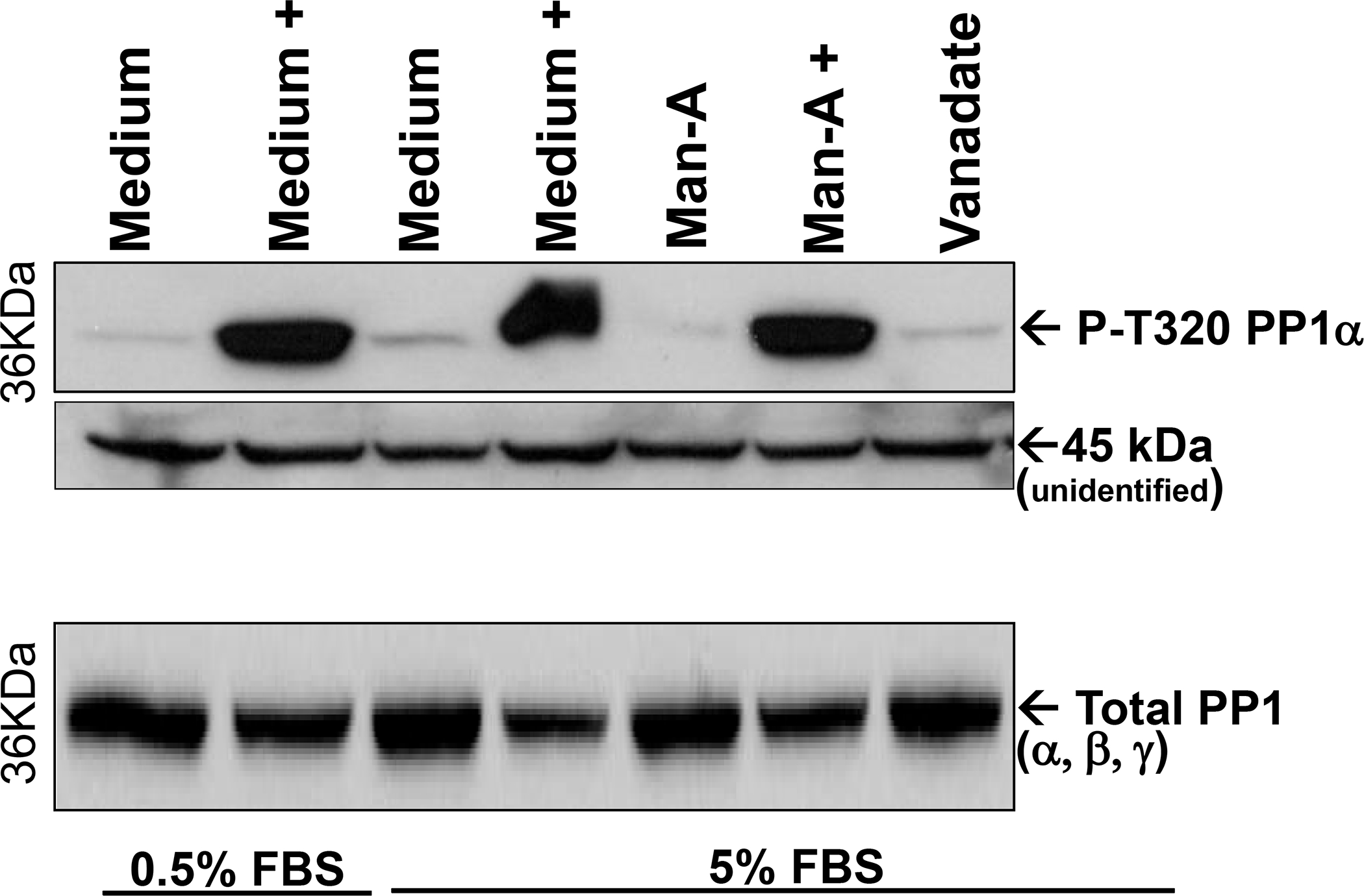
WEHI-231 cells were cultured overnight in the presence of either 0.5% FBS (low serum) or 5% FBS (normal serum) conditions. Prior to adding Man-A (4 μM for 2hrs), or Sodium Vanadate (200 mM for 2hrs), the cells were treated without or with (+) 7 .5 nM Calyculin A for 15 min. The cells were processed for analysis of proteins by western blotting as described above. Phospho PP1α or total PP1 (Using Pan PP1 Ab) were detected by western blotting. An unidentified band at 45 kDa serves as a loading control. The results are representative of more than 3 (three), separate, independent experiments.
Calyculin-A Stimulates Generalized Ser/Thr Phosphorylation in WEHI-231 Lymphoma
Because of the effects of Man-A on PP1α phosphorylation and the effects of Cal-A on MEK, Akt and PP1α phosphorylation, we then examined general Ser/Thr phosphorylation in the presence or absence of Cal-A, Man-A or vanadate. These experiments were conducted using the anti phospho Ser/Thr-Phe motif antibody (CST-9631) from Cell Signaling Technology. The Phospho-Ser/Thr-Phe antibody detects this common motif in Ser/Thr kinase targets such as PKA, CAK, PKC, p90RSK, CDK2 and others (See Cell Signaling Technology Website). As expected, treatment with 7.5 nM Cal-A caused strong overall Ser/Thr phosphorylation in the treated cells which could not be reversed with Man-A treatment (Figure 9, Panel A). Also as anticipated, treatment with the tyrosine phosphatase inhibitor vanadate treatment did not increase the signals detected with CST-9631 antibody(Figure 9A). Due to the intensity of the signals from the lanes where cells had been treated with Cal-A, we re-ran the SDS-PAGE gels and omitted the extracts from Cal-A treated cells and then re-examined P-Ser/Thr-Phe signals using western blot analysis. Contrast was enhanced using Adobe Photoshop® software (Adobe). These results clearly showed that compared to cells grown in 5% FBS, Man-A but not vanadate promoted the general loss of Ser/Thr phosphorylation signals (Figure 9, PanelB). Man-A also promoted the loss of prominent phosphoprotein signals at around 72, 55 42, 38 and 30 kD. Future and ongoing experiments include co-immunoprecipitations followed by analysis by Mass Spec to determine the identities of these proteins. Following these experiments, we concluded that that Man-A-mediated tumoricidal activity involves induction of ser/thr protein phosphatase activity since blockade of this process by a ser/thr protein phosphatase inhibitor such as Cal-A, blocks the downstream events that leads to tumor apoptosis.
Figure 9. Effects of Calyculin A, Man-A and Sodium Orthovanadate on Phosphoproteins in WEHI-231 Lymphoma Cells.
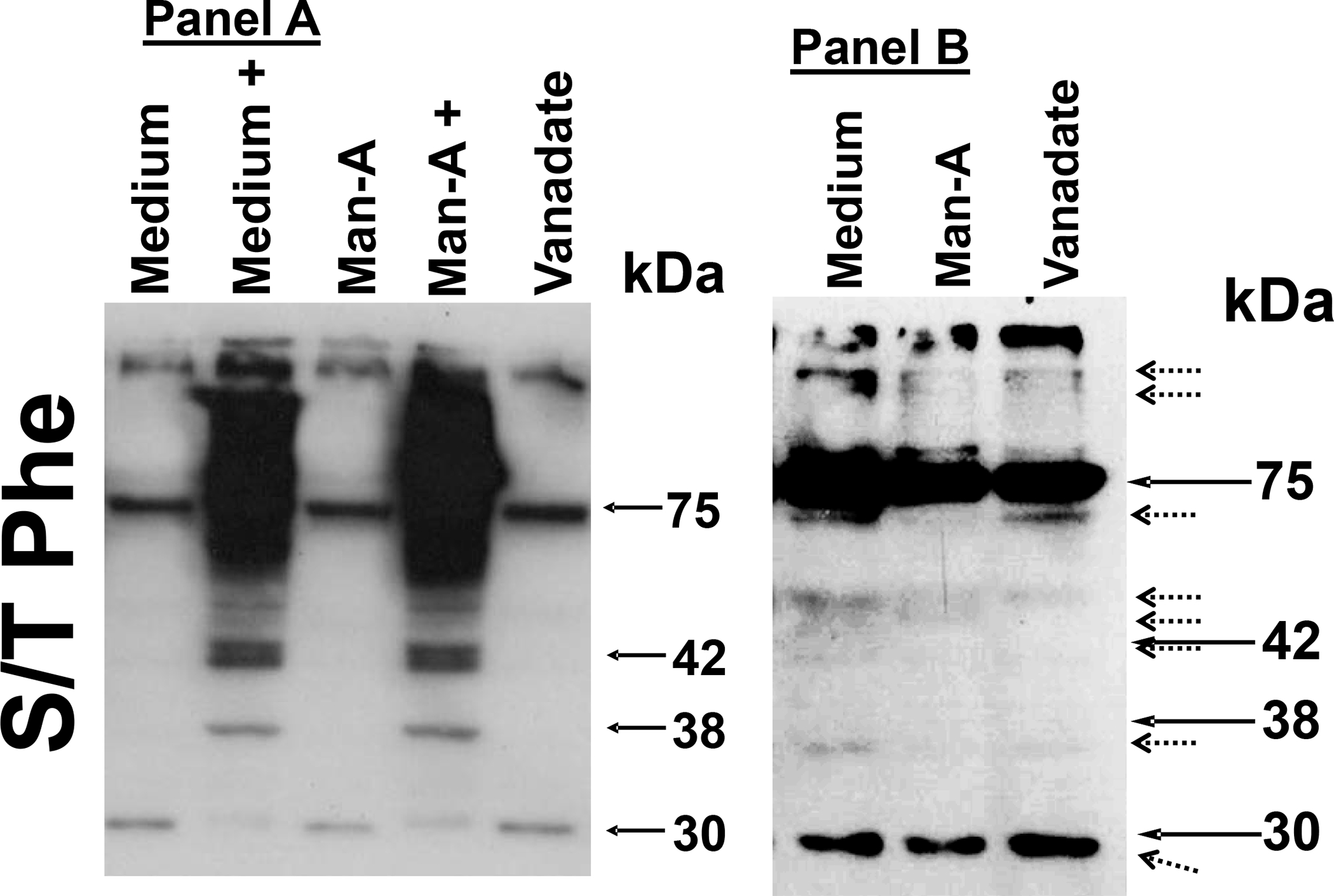
Log growth phase WEHI-231 cells were pre-treated without or with 7.5 nM Calyculin-A (+), followed by treatment without or with 4 μM Man-A or 200 μM sodium orthovanadate. Cells were processed for analysis of phosphoproteins by western blotting as described above. Filters were probed with the Cell Signaling Technology catalogue CST-9631 antibody which detects the Ser/Thr-Phe motif common in Ser/Thr kinase targets such as PKA, CAK, PKC, p90RSK, CDK2 and others (See Cell Signaling Technology Website). Panel 9A, Cal-A But Not Vanadate Stimulates Ser/Thr Phosphorylation of Multiple Proteins in WEHI-231 Cells. Panel 9B, Man-A and Not Vanadate Reduces Ser/Thr Phosphorylation of Multiple Protein Targets in WEHI-231 Cells. Because of the intensity of the phospho protein signal in samples treated with Cal-A, those samples were omitted from the analysis in Panel 9B. Contrast of the phosphoprotein bands in the original autoradiograph was enhanced (Right) using Adobe Photoshop®.
DISCUSSION
We have provided compelling evidence above that Man-A activates or targets protein phosphatase 1 α (PP1α.) It is well established that increased protein phosphorylation plays a critical role in increased anabolism in normal and cancer cells. In fact, conventional and new-line therapeutics target tyrosine kinases (e.g. ibrutinib39) and Trametinib and MK2206 for Ser/Thr protein kinases Akt, MEK40, 41. Intriguingly, whereas many tumors have been successfully treated using targeted kinase therapy, Man-A and possibly other natural agents may allow the selective targeting of protein phosphatases to kill tumors. By further studying the effects of these compounds, we might understand how they might be utilized to activate protein phosphatases and treat human diseases involving activation or Ser/Thr-phosphorylation-mediated dysplasia. Furthermore, as Ser/Thr PPase-inactivating agents such as Cal-A, okadaic acid and SV40 are potent tumor inducing agents 20, conversely, compounds that activate PPases and suppress tumor growth, would be welcome additions to the arsenal array of tools to use against cancer.
The antibiotic properties of Manumycin-A (Man-A) were discovered in the 1960s and its farnesyltransferase inhibitor (FTI) properties were discovered in the early 1990s 2. Although it is becoming clearer that the targets of Man-A action are not limited to FTases 4, the mechanism(s) of Man-A action are still not fully understood. Man-A is an effective, experimental tumoricide in both cell culture and in animal studies. It is highly effective against both solid (pancreas, thyroid and brain) and disseminated (e.g. hematological tumor models) when used alone or in combination with agents such as paclitaxel 42–46.
Despite the apparent diversity in mechanisms of Man-A mediated cytotoxicity, we and at least two other groups have shown that ROS induction is common and is absolutely essential for downstream effects of Man-A 8, 47, 48. Our group specifically showed that the Man-A induced ROS were upstream of a cascade that included rapid dephosphorylation of MEK and Akt, caspsase activation, MEK and Akt cleavage, then apoptosis 8. We also observed that tumors containing phospho-stable AktS473 and MEKSer220/Ser221 were highly resistant to Man-A induced apoptosis 8. Hence, we asked if Ser/Thr protein phosphatase inhibitors would protect MEK and Akt from dephosphorylation and cleavage and if they would prevent Man-A stimulated apoptosis. Using first pharmacological then molecular approaches, the results show that targeting or manipulation of the serine/threonine phosphatase PP1α, modulates all of the effects of Man-A, including ROS induction and the dephosphorylation of MEK and Akt and their subsequent cleavage. Hence, these results establish PP1α as a proximal target and effector of the Man-A-driven apoptotic response.
Recently, several studies have shown that CDC2 phosphorylates Thr320 on PP1α resulting in a dramatic reduction in its PPase activity 35, 36. Our data show that Man-A indeed causes dephosphorylation of PP1α on Thr320 (relieves inhibition). Moreover, forced over-expression of a constitutively active mimetic of PP1α (PP1αThr320A) in a tumor cell line typically more resistant to Man-A, resulted in elevated basal ROS (O2•−) and enhanced Man-A stimulated apoptosis. Furthermore, inhibition of PP1α and pharmacological depletion of PP1α and no other PP1 catalytic isoforms, conferred resistance to Man-A-mediated cytoxicity in an otherwise Man-A sensitive lymphoma. Together, these data strongly support that PP1α is a bona fide target and effector of the natural tumoricide, Man-A.
Since inhibition of PP1 reduced DHE oxidation (O2•−) and over-expressing PP1αThr320A resulted in elevated DHE oxidation, we initially reasoned that suppression of ROS provided protection from Man-A driven apoptosis. On the other hand, our data support the notion that loss of H2O2 and not necessarily induction of O2•− might account for sensitivity to Man-A driven tumor cytotoxicity. Indeed, transient pre-treatment of the cells with micromolar (μM) amounts of exogenously-added H2O2 prevented Man-A driven cell death. If Man-A collapses ‘essential’ H2O2 and provokes protein dephosphorylation and caspase activation, then the observations above are consistent with evidence showing that H2O2 oxidizes and thereby inhibits the thiol active sites of both protein phosphatases and caspases 26, 49, 50. Importantly, treatment with the Ser/Thr protein phosphatase inhibitor, Cal-A, restored H2O2 (DCF) levels, blocked the rise in O2•− (DHE), prevented caspase activation and blocked apoptosis. Hence, from these data, it seems possible that the Man-A driven collapse of H2O2 might allow opportunistic activation of PP1α and amplification of pro-apoptotic loops that culminate in threshold PP1α activation and the activation of caspases. Importantly too, we attempted to examine the effect of Man-A on PP1α phosphatase activity in vitro after PP1α immunoprecipitation followed by the widely used malachite green and p-nitrophenol assays. These data showed little detectable changes in PP1α activity in vitro. We then realized that all the protocols we tested required the addition of dithiothreitol (DTT), a thiol reducing agent. Without DTT, there was no detectable changes in low phosphatase activity (data not shown). Overall, firstly, these results support the notion that care must be taken in extrapolating in vitro results to intracellular phenomena. Secondly, if oxidation terminates PP1α activity in vitro, then it is possible that PP1α can be reversibly oxidized in intact cells. This notion would be supported by the data presented above. Lastly, that addition of a Ser/Thr PPase inhibitor increases H2O2 production raises the possibility that either a single key target or a complex network of interconnected players regulated by Ser/Thr phosphorylation, may be involved in this process. These ideas are currently under investigation in our laboratory.
Our working hypothesis of Man-A driven tumor death is presented in figure 10.
Figure 10. Proposed Working Model Of Mechanism(s) of Man-A Action:
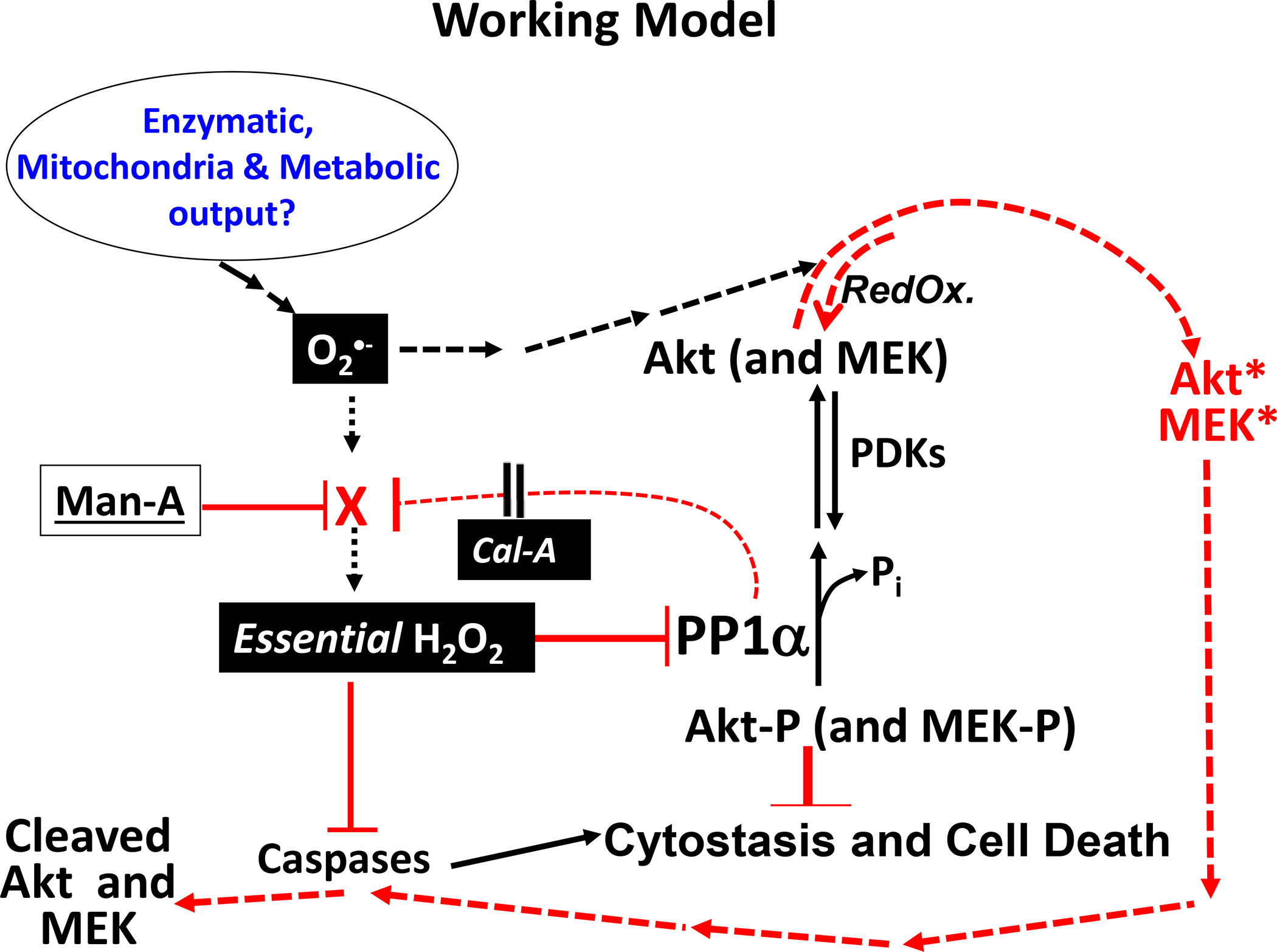
Man-A might antagonize a protein or pathway which is responsible for converting superoxide (O2•−) hydrogen peroxide (to H2O2). This would result in accumulation of O2•− and loss of to H2O2 as are observed. Collapse of inhibitory (essential) H2O2 would result in loss of inhibition of PP1α and of caspases which are proteins that have been demonstrated to contain reversibly oxidizable catalytic sulfhdryls. Active PP1α would dephosphorylate Akt and MEK (Akt-P and MEK-P) as observed. Perhaps increased levels of highly reactive O2•− terminally oxidizes Akt and MEK, which targets the deactivated and now RedOx damaged proteins for caspase-mediated cleavage. We and others have observed reversibly oxidized Akt (and MEK) under normal, rapid growth conditions. Hence, it is possible that initial oxidation can be counteracted by RedOx enzymes (esp. reductases, illustrated). However, once oxidation proceeds past a point (e.g. thiol oxidation to sulfinic and sulfonic acid moieties), the proteins advance from deactivated to damaged and then terminally deactivated (cleaved). Then the cells capitulate to apoptosis as is observed. Man-A could also either directly stimulate PP1α or activate CDC2 (neither is illustrated or elaborated). Solid lines ( ___ ), established mechanisms. Dotted lines (----), speculated and reasonable mechanisms.
Acknowledgments
Supported by grants CA-94027, CA-122882 (NCI) and GC00432490 (U-MD Grant In Aid) to G.B.C.
References
- 1.Demain AL, Sanchez S. Microbial drug discovery: 80 years of progress. The Journal of antibiotics 2009;62: 5–16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Hara M, Akasaka K, Akinaga S, Okabe M, Nakano H, Gomez R, Wood D, Uh M, Tamanoi F. Identification of Ras farnesyltransferase inhibitors by microbial screening. Proc Natl Acad Sci U S A 1993;90: 2281–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Sattler I, Thiericke R, Zeeck A. The manumycin-group metabolites. Nat Prod Rep 1998;15: 221–40. [DOI] [PubMed] [Google Scholar]
- 4.Sebti SM, Adjei AA. Farnesyltransferase inhibitors. Semin Oncol 2004;31: 28–39. [DOI] [PubMed] [Google Scholar]
- 5.Shaw RJ, Cantley LC. Ras, PI(3)K and mTOR signalling controls tumour cell growth. Nature 2006;441: 424–30. [DOI] [PubMed] [Google Scholar]
- 6.Basso AD, Kirschmeier P, Bishop WR. Lipid posttranslational modifications. Farnesyl transferase inhibitors. J Lipid Res 2006;47: 15–31. [DOI] [PubMed] [Google Scholar]
- 7.Steelman LS, Pohnert SC, Shelton JG, Franklin RA, Bertrand FE, McCubrey JA. JAK/STAT, Raf/MEK/ERK, PI3K/Akt and BCR-ABL in cell cycle progression and leukemogenesis. Leukemia 2004;18: 189–218. [DOI] [PubMed] [Google Scholar]
- 8.Sears KT, Daino H, Carey GB. Reactive oxygen species-dependent destruction of MEK and Akt in Manumycin stimulated death of lymphoid tumor and myeloma cell lines. Int J Cancer 2008;122: 1496–505. [DOI] [PubMed] [Google Scholar]
- 9.Okada T, Fujii T, Tanuma N, Mitsuhashi S, Urano T, Araki Y, Shima H, Kikuchi K. Analysis of isoform specific function of PP1 catalytic subunits in mammalian cells using siRNA. Int J Oncol 2004;25: 1383–8. [PubMed] [Google Scholar]
- 10.Cohen PT. Protein phosphatase 1--targeted in many directions. J Cell Sci 2002;115: 241–56. [DOI] [PubMed] [Google Scholar]
- 11.Berndt N. Protein dephosphorylation and the intracellular control of the cell number. Front Biosci 1999;4: D22–42. [DOI] [PubMed] [Google Scholar]
- 12.Mueller CM, Scott DW. Distinct molecular mechanisms of Fas resistance in murine B lymphoma cells. J Immunol 2000;165: 1854–62. [DOI] [PubMed] [Google Scholar]
- 13.Oi VT, Morrison SL, Herzenberg LA, Berg P. Immunoglobulin gene expression in transformed lymphoid cells. Proc Natl Acad Sci U S A 1983;80: 825–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Gururajan M, Chui R, Karuppannan AK, Ke J, Jennings CD, Bondada S. c-Jun N-terminal kinase (JNK) is required for survival and proliferation of B-lymphoma cells. Blood 2005;106: 1382–91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Kaptein JS, Lin CK, Wang CL, Nguyen TT, Kalunta CI, Park E, Chen FS, Lad PM. Anti-IgM-mediated regulation of c-myc and its possible relationship to apoptosis. The Journal of biological chemistry 1996;271: 18875–84. [DOI] [PubMed] [Google Scholar]
- 16.Wu J, Suzuki H, Zhou YW, Liu W, Yoshihara M, Kato M, Akhand AA, Hayakawa A, Takeuchi K, Hossain K, Kurosawa M, Nakashima I. Cepharanthine activates caspases and induces apoptosis in Jurkat and K562 human leukemia cell lines. J Cell Biochem 2001;82: 200–14. [DOI] [PubMed] [Google Scholar]
- 17.van Duijn PW, Trapman J. PI3K/Akt signaling regulates p27(kip1) expression via Skp2 in PC3 and DU145 prostate cancer cells, but is not a major factor in p27(kip1) regulation in LNCaP and PC346 cells. Prostate 2006;66: 749–60. [DOI] [PubMed] [Google Scholar]
- 18.Carey GB, Scott DW. Role of phosphatidylinositol 3-kinase in anti-IgM- and anti-IgD-induced apoptosis in B cell lymphomas. J Immunol 2001;166: 1618–26. [DOI] [PubMed] [Google Scholar]
- 19.Chatfield K, Eastman A. Inhibitors of protein phosphatases 1 and 2A differentially prevent intrinsic and extrinsic apoptosis pathways. Biochem Biophys Res Commun 2004;323: 1313–20. [DOI] [PubMed] [Google Scholar]
- 20.McConnell JL, Wadzinski BE. Targeting protein serine/threonine phosphatases for drug development. Mol Pharmacol 2009;75: 1249–61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Devadas S, Hinshaw JA, Zaritskaya L, Williams MS. Fas-stimulated generation of reactive oxygen species or exogenous oxidative stress sensitize cells to Fas-mediated apoptosis. Free Radic Biol Med 2003;35: 648–61. [DOI] [PubMed] [Google Scholar]
- 22.Scaduto RC Jr., Grotyohann LW. Measurement of mitochondrial membrane potential using fluorescent rhodamine derivatives. Biophys J 1999;76: 469–77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Rottenberg H, Wu S. Quantitative assay by flow cytometry of the mitochondrial membrane potential in intact cells. Biochim Biophys Acta 1998;1404: 393–404. [DOI] [PubMed] [Google Scholar]
- 24.Hinshaw JA, Mueller CM, Scott DW, Williams MS. B cell receptor signaling mediates immediate protection from Fas-induced apoptosis upstream of caspase activation through an atypical protein kinase C isozyme and de novo protein synthesis. Eur J Immunol 2003;33: 2490–500. [DOI] [PubMed] [Google Scholar]
- 25.Jackson SH, Devadas S, Kwon J, Pinto LA, Williams MS. T cells express a phagocyte-type NADPH oxidase that is activated after T cell receptor stimulation. Nature immunology 2004;5: 818–27. [DOI] [PubMed] [Google Scholar]
- 26.Kwon J, Qu CK, Maeng JS, Falahati R, Lee C, Williams MS. Receptor-stimulated oxidation of SHP-2 promotes T-cell adhesion through SLP-76-ADAP. Embo J 2005;24: 2331–41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Castedo M, Hirsch T, Susin SA, Zamzami N, Marchetti P, Macho A, Kroemer G. Sequential acquisition of mitochondrial and plasma membrane alterations during early lymphocyte apoptosis. J Immunol 1996;157: 512–21. [PubMed] [Google Scholar]
- 28.Li L, Ren CH, Tahir SA, Ren C, Thompson TC. Caveolin-1 maintains activated Akt in prostate cancer cells through scaffolding domain binding site interactions with and inhibition of serine/threonine protein phosphatases PP1 and PP2A. Mol Cell Biol 2003;23: 9389–404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Mao L, Yang L, Arora A, Choe ES, Zhang G, Liu Z, Fibuch EE, Wang JQ. Role of protein phosphatase 2A in mGluR5-regulated MEK/ERK phosphorylation in neurons. The Journal of biological chemistry 2005;280: 12602–10. [DOI] [PubMed] [Google Scholar]
- 30.Vlahos CJ, Matter WF, Hui KY, Brown RF. A specific inhibitor of phosphatidylinositol 3-kinase, 2-(4- morpholinyl)-8-phenyl-4H-1-benzopyran-4-one (LY294002). The Journal of biological chemistry 1994;269: 5241–8. [PubMed] [Google Scholar]
- 31.Richards JD, Dave SH, Chou CH, Mamchak AA, DeFranco AL. Inhibition of the MEK/ERK signaling pathway blocks a subset of B cell responses to antigen. J Immunol 2001;166: 3855–64. [DOI] [PubMed] [Google Scholar]
- 32.Gao T, Furnari F, Newton AC. PHLPP: a phosphatase that directly dephosphorylates Akt, promotes apoptosis, and suppresses tumor growth. Mol Cell 2005;18: 13–24. [DOI] [PubMed] [Google Scholar]
- 33.Brognard J, Sierecki E, Gao T, Newton AC. PHLPP and a second isoform, PHLPP2, differentially attenuate the amplitude of Akt signaling by regulating distinct Akt isoforms. Mol Cell 2007;25: 917–31. [DOI] [PubMed] [Google Scholar]
- 34.Liu CW, Wang RH, Berndt N. Protein phosphatase 1alpha activity prevents oncogenic transformation. Mol Carcinog 2006;45: 648–56. [DOI] [PubMed] [Google Scholar]
- 35.Xu W, Yuan X, Jung YJ, Yang Y, Basso A, Rosen N, Chung EJ, Trepel J, Neckers L. The heat shock protein 90 inhibitor geldanamycin and the ErbB inhibitor ZD1839 promote rapid PP1 phosphatase-dependent inactivation of AKT in ErbB2 overexpressing breast cancer cells. Cancer Res 2003;63: 7777–84. [PubMed] [Google Scholar]
- 36.Liu CW, Wang RH, Dohadwala M, Schonthal AH, Villa-Moruzzi E, Berndt N. Inhibitory phosphorylation of PP1alpha catalytic subunit during the G(1)/S transition. The Journal of biological chemistry 1999;274: 29470–5. [DOI] [PubMed] [Google Scholar]
- 37.Whisler RL, Goyette MA, Grants IS, Newhouse YG. Sublethal levels of oxidant stress stimulate multiple serine/threonine kinases and suppress protein phosphatases in Jurkat T cells. Arch Biochem Biophys 1995;319: 23–35. [DOI] [PubMed] [Google Scholar]
- 38.Secrist JP, Burns LA, Karnitz L, Koretzky GA, Abraham RT. Stimulatory effects of the protein tyrosine phosphatase inhibitor, pervanadate, on T-cell activation events. The Journal of biological chemistry 1993;268: 5886–93. [PubMed] [Google Scholar]
- 39.Byrd JC, Furman RR, Coutre SE, Flinn IW, Burger JA, Blum KA, Grant B, Sharman JP, Coleman M, Wierda WG, Jones JA, Zhao W, et al. Targeting BTK with ibrutinib in relapsed chronic lymphocytic leukemia. The New England journal of medicine 2013;369: 32–42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Wilson JM, Kunnimalaiyaan S, Gamblin TC, Kunnimalaiyaan M. MK2206 inhibits hepatocellular carcinoma cellular proliferation via induction of apoptosis and cell cycle arrest. The Journal of surgical research 2014;191: 280–5. [DOI] [PubMed] [Google Scholar]
- 41.Wright CJ, McCormack PL. Trametinib: first global approval. Drugs 2013;73: 1245–54. [DOI] [PubMed] [Google Scholar]
- 42.Mazzocca A, Giusti S, Hamilton AD, Sebti SM, Pantaleo P, Carloni V. Growth inhibition by the farnesyltransferase inhibitor FTI-277 involves Bcl-2 expression and defective association with Raf-1 in liver cancer cell lines. Mol Pharmacol 2003;63: 159–66. [DOI] [PubMed] [Google Scholar]
- 43.Zhou JM, Zhu XF, Pan QC, Liao DF, Li ZM, Liu ZC. Manumycin induces apoptosis in human hepatocellular carcinoma HepG2 cells. Int J Mol Med 2003;12: 955–9. [PubMed] [Google Scholar]
- 44.Frassanito MA, Cusmai A, Piccoli C, Dammacco F. Manumycin inhibits farnesyltransferase and induces apoptosis of drug-resistant interleukin 6-producing myeloma cells. Br J Haematol 2002;118: 157–65. [DOI] [PubMed] [Google Scholar]
- 45.Yeung SC, Xu G, Pan J, Christgen M, Bamiagis A. Manumycin enhances the cytotoxic effect of paclitaxel on anaplastic thyroid carcinoma cells. Cancer Res 2000;60: 650–6. [PubMed] [Google Scholar]
- 46.Kainuma O, Asano T, Hasegawa M, Kenmochi T, Nakagohri T, Tokoro Y, Isono K. Inhibition of growth and invasive activity of human pancreatic cancer cells by a farnesyltransferase inhibitor, manumycin. Pancreas 1997;15: 379–83. [DOI] [PubMed] [Google Scholar]
- 47.Zhang Q, Tsukahara F, Maru Y. N-acetyl-cysteine enhances growth in BCR-ABL-transformed cells. Cancer Sci 2005;96: 240–4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Pan J, She M, Xu ZX, Sun L, Yeung SC. Farnesyltransferase inhibitors induce DNA damage via reactive oxygen species in human cancer cells. Cancer Res 2005;65: 3671–81. [DOI] [PubMed] [Google Scholar]
- 49.Kwon J, Lee SR, Yang KS, Ahn Y, Kim YJ, Stadtman ER, Rhee SG. Reversible oxidation and inactivation of the tumor suppressor PTEN in cells stimulated with peptide growth factors. Proc Natl Acad Sci U S A 2004;101: 16419–24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Hampton MB, Orrenius S. Dual regulation of caspase activity by hydrogen peroxide: implications for apoptosis. FEBS Lett 1997;414: 552–6. [DOI] [PubMed] [Google Scholar]