

Abstract
The use of opioids for the treatment of pain, while largely effective, is limited by detrimental side effects including analgesic tolerance, physical dependence, and euphoria, which may lead to opioid abuse. Studies have shown that compounds with a μ-opioid receptor (MOR) agonist/δ-opioid receptor (DOR) antagonist profile reduce or eliminate some of these side effects including the development of tolerance and dependence. Herein we report the synthesis and pharmacological evaluation of a series of tetrahydroquinoline-based peptidomimetics with substitutions at the C-8 position. Relative to our lead peptidomimetic with no C-8 substitution, this series affords an increase in DOR affinity and provides greater balance in MOR and DOR binding affinities. Moreover, compounds with carbonyl moieties at C-8 display the desired MOR agonist/DOR antagonist profile whereas alkyl substitutions elicit modest DOR agonism. Several compounds in this series produce a robust antinociceptive effect in vivo and show antinociceptive activity for greater than 2 h after intraperitoneal administration in mice.
Keywords: Bifunctional ligands, dependence, drug abuse, opioids, peptidomimetics, tolerance
Graphical Abstract
INTRODUCTION
Opioid analgesics are among the most effective drugs for the treatment of moderate to severe pain. Complications from long-term pain management with opioids include gastrointestinal distress, opioid-induced hyperalgesia and the development of analgesic tolerance and physical dependence. Nearly all clinically prescribed opioid analgesics exert both their beneficial and undesirable effects through activation of the μ-opioid receptor (MOR). Evidence suggests that the δ-opioid receptor (DOR) plays a key role in modulating some side effects associated with opioids including analgesic tolerance and physical dependence.1,2 Classic studies using rodent models have demonstrated that by coadministering a DOR antagonist with morphine, or by administering chronic morphine to DOR knockout mice, the development of analgesic tolerance, physical dependence, and drug-seeking behavior are attenuated.3–7 By simultaneously activating MOR and blocking DOR, we aim to develop safer analgesics with reduced tolerance and dependence profiles for the management of a variety of pain conditions.
Current reports and reviews on bifunctional MOR/DOR ligands, which span a wide range of scaffolds and physicochemical properties, have provided support for the development of improved therapeutics that display favorable pharmacodynamic and pharmacokinetic profiles.6–12 Eluxadoline (Viberzi), an orally bioavailable, peripherally acting MOR agonist/DOR antagonist, was FDA approved in 2015 for the treatment of irritable bowel syndrome with diarrhea (IBS-D) and has shown to be effective and well-tolerated in the clinic.13–15 Several laboratories have utilized morphine-like scaffolds to develop MOR agonist/DOR antagonist small molecules, which may show promise as analgesics with reduced antinociceptive tolerance and physical dependence liabilities.10,16,17 An alternative approach by Portoghese and colleagues targets the proposed MOR-DOR heterodimer with a bivalent ligand, whereby the MOR agonist pharmacophore of oxymorphone is linked with the DOR antagonist pharmacophore of naltrindole.18,19 Studies using these bivalent ligands in animal models not only show decreased tolerance and dependence as a function of linker length,18 but also report attenuated conditioned place preference (CPP) and reinstatement, suggesting a lower abuse liability.19 We have recently reported on two MOR agonist/DOR antagonist lead compounds, a glycosylated cyclic peptide7 and a peptidomimetic, 1 (Figure 1),20,21 that produce long-lasting and dose-dependent antinociception in mice after peripheral administration. Elaboration of the peptidomimetic series primarily through modification of the C-6 pendant and at N-1 resulted in MOR agonist/DOR antagonist analogues with a range of relative affinities for MOR and DOR.22–24 Further in vivo evaluation of three such analogs suggested that balanced MOR/DOR affinity is associated with amelioration of tolerance, dependence, and reinforcing properties.25
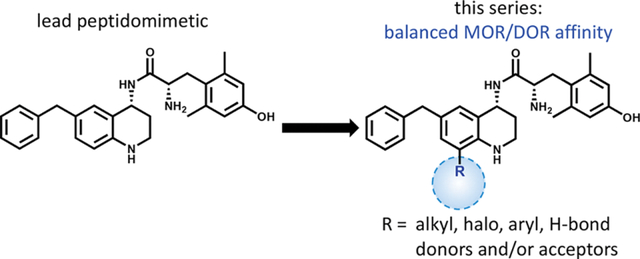
Figure 1.

Lead peptidomimetic 1, featuring the THQ core and numbering convention in blue. Current SAR exploration focuses on substitutions at C-8.
In order to determine what role, if any, balanced MOR/DOR affinities of MOR agonist/DOR antagonist ligands plays in eliminating opioid side effects, we continue to expand the SAR of this series for the development of MOR agonist/DOR antagonist peptidomimetics with a range of relative MOR/DOR affinities. As noted above, our SAR has focused primarily on substitutions at C-6 and N-1.
To further understand and optimize the SAR for our tetrahydroquinoline (THQ) core, we synthesized and evaluated a series of C-8 substitutions while keeping the pharmacophores at C-6 and N-1 constant. We had previously found that ligands with bicyclic C-6 substitutions on the unmodified THQ core preferentially bind MOR over DOR at least 10-fold,22 and N-1 substitutions afford improved balance in MOR and DOR binding affinity, but often elicit partial DOR agonism.24 The C-8 modifications presented in this series probe the spatial and electronic constraints of a binding pocket previously unexplored in this series of peptidomimetics. Here we report the synthesis and evaluation of a series of THQ-based ligands featuring a diverse set of substitutions at C-8 as part of our effort to optimize the MOR agonist/DOR antagonist profile and improve upon our lead peptidomimetic.
RESULTS AND DISCUSSION
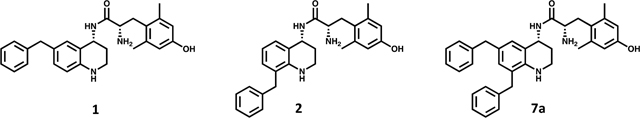
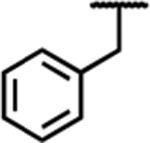
Following the development of 1, we have extensively evaluated the SAR of our peptidomimetic series by incorporating diverse substitutions at the C-6 position,21,22 also referred to as the “pendant.” Prior SAR and computational docking studies have shown that a benzyl pendant at C-6 is well tolerated by a deep binding pocket in both MOR and DOR.20,21,23 To validate the importance of the aryl C-6 pharmacophore in receptor binding and activity, the benzyl pendant was moved to the C-8 position (Table 1, compound 2). Compound 2 showed significantly decreased binding affinities (higher Ki) for all three opioid receptors (MOR, DOR, and κ-opioid receptor (KOR)), and also showed decreased MOR potency (EC50 = 1200 nM) and efficacy (37% stimulation compared to the standard MOR agonist DAMGO), as measured by [35S]-GTPγS binding. Furthermore, compound 2 showed no appreciable activity at DOR or KOR. We then questioned whether this reduction in MOR activity was due to the loss of the C-6 pharmacophore, or to unfavorable ligand–receptor interactions at C-8. To examine this, we combined the C-6 and C-8 benzyl substitutions, giving compound 7a. As shown in Table 1, the binding affinity as well as potency and efficacy of 7a at MOR were restored (Ki = 1 nM; EC50 = 4 nM; 96% stimulation), while the DOR binding affinity improved 10-fold and efficacy 2-fold compared to 1, not only validating the importance of the C-6 pharmacophore for MOR activity, but identifying a key role for the C-8 pharmacophore in modulating DOR affinity and efficacy. This moderate loss in MOR affinity and increase in DOR affinity shifted the MOR/DOR binding ratio (DOR Ki/MOR Ki) from 43 for compound 1 to a more balanced 1.6 for compound 7a (Table 1). Consequently, this 6-, 8-disubstituted THQ analogue established C-8 as a region of interest for future SAR with the principal aim of balancing MOR and DOR binding affinities.
Table 1.
Effects of Benzyl Pendant Position on Binding Affinity, Potency, and Efficacya
| ||||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| binding affinity Ki (nM) | potency EC50 (nM) | efficacy (% stimulation) | ||||||||
|
|
|
|
||||||||
| compd | MOR | DOR | KOR | DOR Ki/MOR Ki | MOR | DOR | KOR | MOR | DOR | KOR |
| 1 | 0.22 (0.02) | 9.4 (0.8) | 68 (2) | 43 | 1.6 (0.3) | 110 (6) | 540 (70) | 81 (2) | 16 (2) | 22 (2) |
| 2 | 48 (9) | 360 (60) | 1500 (400) | 7.5 | 1200 (300) | dns | dns | 37 (4) | dns | dns |
| 7a | 1.0 (0.1) | 1.6 (0.4) | 23 (5) | 1.6 | 4 (2) | 380 (84) | dns | 96 (4) | 42 (7) | dns |
Binding affinities (Ki) were obtained by competitive displacement of radiolabeled [3H]-diprenorphine in membrane preparations. Functional data were obtained using agonist induced stimulation of [35S]-GTPγS binding assay. Potency is represented as EC50 (nM), and efficacy as percent maximal stimulation relative to standard agonist DAMGO (MOR), DPDPE (DOR), or U69,593 (KOR) at 10 μM. All values are expressed as the mean of three separate assays performed in duplicate with standard error of the mean (SEM) in parentheses. dns = does not stimulate (<10%).
In Vitro Structure–Activity Relationships.
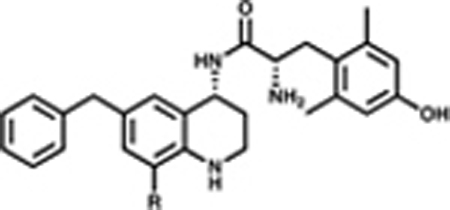
Subsequent compounds in the C-8 series explored the steric environment and depth of the C-8 binding pocket with various alkyl substitutions, ranging from methyl to t-butyl (Table 2). We extended this series to include halogens (F, CF3, Br), which largely fit the same trend as the alkyl set. The alkyl and halogenated series generally showed potent, efficacious agonism at MOR and partial agonism at DOR. Additionally, most alkyl-substituted analogues showed no KOR activation, whereas the halogenated compounds were partial agonists at KOR. In terms of binding, the smallest C-8 substitutions (7b, 7c, 7g, 7h, 7q) maintained high affinity for MOR and increased affinity for DOR relative to the unsubstituted lead peptidomimetic 1; however, these compounds bind MOR over DOR by at least 8:1. Conversely, larger C-8 substitutions (7d, 7e, 7f) slightly decreased MOR affinity and displayed a modest increase in DOR affinity, leading to an improved balance of opioid receptor binding affinities.
Table 2.
Effects of Alkyl and Halogen Substitutions on Affinity, Potency, and Efficacya
|
Binding affinity Ki (nM) | DOR Ki/MOR Ki | Potency, EC50 (nM) | Efficacy, (% stimulation) | |||||||
|---|---|---|---|---|---|---|---|---|---|---|---|
|
|
|
|
|
||||||||
| Cmpd | R2 | MOR | DOR | KOR | MOR | DOR | KOR | MOR | DOR | KOR | |
|
|
|
|
|
||||||||
| 1 |
|
0.22 (0.02) | 9.4 (0.8) | 68 (2) | 43 | 1.6 (0.3) | 110 (6) | 540 (70) | 81 (2) | 16 (2) | 22 (2) |
|
|
|
|
|
||||||||
| 7b |

|
0.24 (0.08) | 1.9 (0.4) | 17 (0.7) | 8 | 4.2 (1.6) | 110 (24) | 760 (140) | 91 (1) | 71 (3) | 52 (2) |
|
|
|
|
|
||||||||
| 7c |
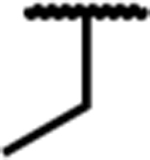
|
0.09 (0.04) | 1.9 (0.4) | 40 (5) | 21 | 6.2 (2.9) | 32 (10) | dns | 74 (2) | 45 (4) | dns |
|
|
|
|
|
||||||||
| 7d |
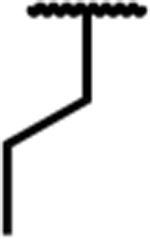
|
0.64 (0.08) | 5.9 (1.5) | 98 (18) | 9 | 23 (7) | 310 (30) | dns* | 90 (6) | 36 (3) | dns* |
|
|
|
|
|
||||||||
| 7e |
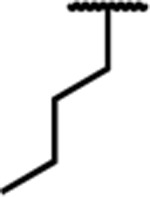
|
0.76 (0.28) | 3.6 (0.5) | 34 (5) | 5 | 17 (4) | 250 (39) | dns* | 85 (2) | 25 (4) | dns* |
|
|
|
|
|
||||||||
| 7f |

|
0.47 (0.18) | 3.8 (0.7) | 48 (7) | 8 | 9.9 (3.6) | 240 (40) | dns* | 83 (5) | 42 (2) | dns* |
|
|
|
|
|
||||||||
| 7g |
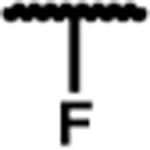
|
0.11 (0.01) | 3.0 (0.3) | 9 (1) | 27 | 1.6 (0.2) | 97 (19) | 370 (8) | 95 (2) | 28 (3) | 40 (1) |
|
|
|
|
|
||||||||
| 7h |

|
0.26 (0.10) | 2.2 (0.7) | 29 (10) | 9 | 1.8 (0.9) | 50 (14) | 580 (14) | 70 (5) | 42 (2) | 18 (4) |
|
|
|
|
|
||||||||
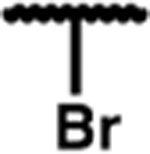
| 7q |
|
0.23 (0.14) | 2.4 (0.8) | 13 (1) | 10 | 1.2 (0.5) | 36 (18) | 310 (95) | 73 (3) | 69 (4) | 29 (1) |
Binding affinities (Ki) were obtained by competitive displacement of radiolabeled [3H]-diprenorphine in membrane preparations. Functional data were obtained using agonist induced stimulation of [35S]-GTPγS binding assay. Potency is represented as EC50 (nM) and efficacy as percent maximal stimulation relative to standard agonist DAMGO (MOR), DPDPE (DOR), or U69,593 (KOR) at 10 μM. All values are expressed as the mean of three separate assays (n = 3) performed in duplicate unless noted otherwise, with SEM in parentheses.
Asterisk (*) indicates n = 2; dns = does not stimulate (<10% stimulation).
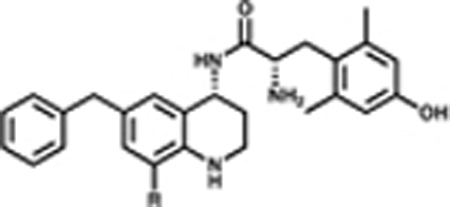
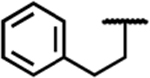
Expanding upon the alkyl and halogen subsets, we synthesized a series of analogues featuring aryl, carbonyl, and amine substitutions, summarized in Table 3. The phenethyl (7l) and ethyl ester (7n) C-8 substituted compounds both improved the MOR/DOR binding affinity ratio relative to 1 (DOR Ki/MOR Ki = 4.1 and 3.7, respectively), however, they were not as balanced as the benzyl C-8 substituted compound 7a (DOR Ki/MOR Ki = 1.6). Compounds 7l and 7n maintained appreciable MOR efficacy (>70% stimulation), yet only modestly stimulated DOR (≤15% stimulation), whereas 7a produced partial agonism at DOR (EC50 = 380 nM; 42% stimulation). Evaluation of three amide-linked C-8 substituted compounds (ethyl amide 7m, phenyl amide 7o, and benzyl amide 7p) resulted in binding affinities that favored MOR over DOR approximately 10:1. Functionally, these compounds produced the desired MOR agonist/DOR antagonist profile. Additionally, this series of carbonyl-containing compounds increased KOR affinity relative to 1 and displayed KOR antagonism. The carboxylic acid-substituted compound (7r) was the outlier in the carbonyl series, showing a significant loss of KOR binding affinity (Ki = 210 nM), however affinity and activity at MOR and DOR were comparable to the other compounds in the carbonyl series. The amine substitutions examined in this series were all cyclic, tertiary amines (e.g., piperidine (7i), morpholine (7j), and piperazine (7k). These C-8 substitutions elicited minor differences at MOR, with affinities and efficacies comparable to 1, however, these compounds diverged markedly at DOR and KOR. Compound 7k displayed the weakest DOR affinity in this series by a considerable margin (Ki = 15 nM), while 7i and 7j both displayed high affinity and partial agonism for DOR. Furthermore, while 7i and 7k showed a large increase in KOR binding affinity (Ki = 0.93 and 1.9 nM, respectively) and produced partial KOR agonism, 7j showed a more modest increase in KOR affinity (Ki = 7.3 nM) and did not stimulate KOR.
Table 3.
Effects of Aryl, Carbonyl, and Amino Substitutions on Affinity, Potency, and Efficacya
|
Binding affinity Ki (nM) | DOR Ki/MOR Ki | Potency, EC50 (nM) | Efficacy, (% stimulation) | |||||||
|---|---|---|---|---|---|---|---|---|---|---|---|
|
|
|
|
|
||||||||
| Cmpd | R2 | MOR | DOR | KOR | MOR | DOR | KOR | MOR | DOR | KOR | |
|
|
|
|
|
||||||||
| 7a |
|
1.0 (0.1) | 1.6 (0.4) | 23 (5) | 2 | 4 (2) | 380 (84) | dns | 96 (4) | 42 (7) | dns |
|
|
|
|
|
||||||||
| 7l |
|
0.37 (0.07) | 1.4 (0.7) | 27 (8) | 4 | 44 (17) | 33 (13) | dns* | 78 (1) | 15 (2) | dns* |
|
|
|
|
|
||||||||
| 7m |
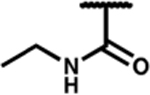
|
0.20 (0.06) | 1.8 (0.4) | 25 (4) | 9 | 2.1 (0.3) | dns | dns | 56 (5) | dns | dns |
|
|
|
|
|
||||||||
| 7n |
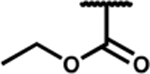
|
1.0 (0.3) | 3.7 (0.3) | 47 (2) | 4 | 4.9 (0.3) | dns | dns* | 71 (3) | dns | dns* |
|
|
|
|
|
||||||||
| 7o |
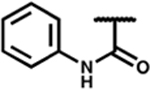
|
0.32 (0.08) | 5.4 (0.5) | 29 (5) | 17 | 1.2 (0.7) | dns | dns* | 49 (4) | dns | dns* |
|
|
|
|
|
||||||||
| 7p |
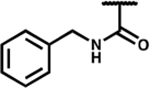
|
0.17 (0.03) | 3.0 (0.4) | 30 (2) | 18 | 3.4 (1.4) | dns | dns | 73 (5) | dns | dns |
|
|
|
|
|
||||||||
| 7r |
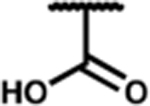
|
0.47 (0.16) | 2.4 (0.4) | 210 (6) | 5 | 4.4 (1.7) | dns | dns | 67 (3) | dns | dns |
|
|
|
|
|
||||||||
| 7i |
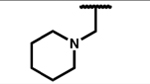
|
0.07 (0.03) | 4.4 (1.0) | 0.93 (0.18) | 63 | 2.3 (0.2) | 27 (2) | 100 (33) | 93 (2) | 31 (4) | 30 (6) |
|
|
|
|
|
||||||||
| 7j |
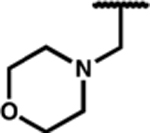
|
0.15 (0.04) | 2.3 (0.7) | 7.3 (1.4) | 15 | 1.8 (0.4) | 180 (47) | dns | 96 (2) | 29 (5) | dns |
|
|
|
|
|
||||||||
| 7k |
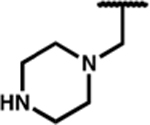
|
0.35 (0.18) | 15 (3) | 1.9 (0.5) | 43 | 8.2 (3.5) | 290 (100) | 170 (67) | 60 (2) | 18 (1) | 17 (1) |
Binding affinities (Ki) were obtained by competitive displacement of radiolabeled [3H]-diprenorphine in membrane preparations. Functional data were obtained using agonist induced stimulation of [35S]-GTPγS binding assay. Potency is represented as EC50 (nM) and efficacy as percent maximal stimulation relative to standard agonist DAMGO (MOR), DPDPE (DOR), or U69,593 (KOR) at 10 μM. All values are expressed as the mean of three separate assays (n = 3) performed in duplicate unless noted otherwise, with SEM in parentheses.
Asterisk (*) indicates n = 2; dns = does not stimulate (<10% stimulation).
In Vivo Antinociceptive Activity.
All final compounds excluding 7k and 7o were evaluated in vivo for antinociceptive activity using the mouse warm water tail withdrawal (WWTW) assay (Table 4). In the alkyl series, compounds 7b, c, and e were fully efficacious, showing dose dependent antinociception and reaching the cutoff latency of 20 s at 10 mg/kg after intraperitoneal (ip) administration, whereas 7d showed no significant antinociceptive effect at the same dose. The t-butyl analogue 7f was partially active in vivo, with a latency of 10 s at 10 mg/kg. In the carbonyl series, only the ethyl ester analogue 7n also showed full efficacy. Larger substitutions including the cyclic amines 7i and j, aryl rings 7a and l, and amides 7m, o, and p, produced no antinociception at the doses tested. Additionally, the relatively small carboxylic acid 7r and halogenated analogues 7g, h, and q produced no antinociceptive effect at the doses tested. Results of the in vivo screening are summarized in Table 4. Of the bioactive analogues 7b, c, e, and n, the duration of action for 7e and n proved to be the longest at 2.5 h. This is a modest improvement over the lead 1 (2 h).
Table 4.
Antinociceptive Activity of Compounds 7a–r in Mouse WWTW Assaya
| C-8 substitution | antinociceptive activity at 10 mg/kg | duration of action | C-8 substitution | antinociceptive activity at 10 mg/kg | duration of action | ||
|---|---|---|---|---|---|---|---|
| 7a | benzyl | no activity | 7j | methylmorpholine | no activity | ||
| 7b | methyl | fully efficacious | 1.5 h | 7k | methylpiperazine | did not test | |
| 7c | ethyl | fully efficacious | 1.0 h | 7l | phenethyl | no activity | |
| 7d | n-propyl | no activity | 7m | ethyl amide | no activity | ||
| 7e | n-butyl | fully efficacious | 2.5 h | 7n | ethyl ester | fully efficacious | 2.5 h |
| 7f | t-butyl | partially active | 7o | phenyl amide | did not test | ||
| 7g | fluoro | no activity | 7p | benzyl amide | no activity | ||
| 7h | trifluoromethyl | no activity | 7q | bromo | no activity | ||
| 7i | methylpiperidine | no activity | 7r | carboxylic acid | no activity |
Results from the mouse WWTW assay after ip administration of compound 7a–r at 10 mg/kg. Antinociceptive activities defined as fully efficacious for 20 s latency to tail withdrawal, partially active for 10 s above baseline, or no activity for no significant difference from baseline. Duration of action is defined here as the time it takes to return to baseline after a 10 mg/kg bolus injection of test compound.
All compounds reported in this series maintained a high binding affinity at MOR and demonstrated partial to full MOR agonism in vitro compared to the standard full agonist DAMGO. With compound 7k as the sole exception, all compounds had improved DOR affinity relative to the lead compound 1. Accordingly, both lipophilic and polar C-8 substitutions provided compounds with a greater balance in MOR/DOR receptor binding affinity. While most compounds displayed DOR agonist activity, those with carbonyl C-8 substitutions (7m, n, o, p, and r) were consistently found to lack efficacy in the GTPγS binding assay. We have previously shown that peptidomimetics that do not produce GTPγS binding are in fact functional antagonists, as demonstrated by a shift in the concentration response curve of a standard agonist. Using SNC80 as a standard DOR agonist, we calculated the Ke values of 7m, n, o, p, and r to be 25.4, 42.5, 31.5, 16.7, and 15.7 nM, respectively, confirming their DOR antagonist proerties.
Based on our computational models,26 we predict the C-8 substitutions to primarily interact with extracellular loop 2 and the N-terminal tail of MOR and DOR, and to a lesser extent with transmembrane helices 3, 4, and 5. Due to the flexibility in these regions of the receptor, accurately correlating computationally predicted ligand–receptor interactions with the SAR data was not feasible. Comprehensive evaluation of this series of compounds suggests that C-8 carbonyl moieties block DOR activation quite effectively and bulky alkyl and aryl groups, such as n-butyl and phenethyl, respectively, attenuate DOR activation relative to smaller alkyl, aryl, and halogen-containing groups. We have previously shown that a bulky C-6 pendant interacts favorably with the active-state MOR binding pocket, yet there is a steric clash between a large C-6 pendant and the analogous amino acid residues in the active-state DOR.21,26 We propose from our SAR analysis that the active-state binding pockets of MOR and DOR likely interact with the C-8 substitutions in a similar manner. For preferentially binding the MOR active-state pocket and the DOR inactive-state pocket with a high affinity, a bulky C-6 pendant and carbonyl moiety at the C-8 position are key pharmacophore elements that produce the desired MOR agonist/DOR antagonist profile with an improved receptor binding affinity balance.
Moreover, in addition to balancing binding affinty at MOR and DOR and demonstrating that C-8 carbonyl substitutions produce DOR antagonism in vitro, this series afforded several compounds with promising in vivo antinociceptive activities. We have shown that four compounds (7b, c, e, and n) produced a full antinociceptive effect in the WWTW assay in mice after peripheral administration. Contrary to expectations, 7d, which incorporated the n-propyl substitution, showed no effect in vivo, whereas 7e, having the n-butyl substitution, produced full antinociceptive activity. 7d has only a single carbon difference from the bioactive 7c and 7e, so it is surprising that its in vivo effects would be drastically different. Among the carbonyl subset, only the ethyl ester (7n) was fully efficacious.
As shown in Table 4, most compounds in this series demonstrated no antinociceptive activity at the doses tested in vivo. While we cannot definitively attribute a loss of activity to any individual factor for all compounds in the series, typical physicochemical properties such as high molecular weight, lipophilicity, polar surface area, and hydrogen bond partners are likely to inhibit membrane permeability and access to the CNS. Accordingly, our reported in vivo SAR was constrained to small C-8 modifications. Small alkyl substitutions (7b–f) showed the best correlation with in vivo antinociceptive activity. However, the relatively low molecular weight fluoro-substituted compounds (7g and h) showed no activity in vivo at the doses tested, indicating pharmacokinetic obstacles besides molecular weight. Another pair of relatively low molecular weight analogues with differing in vivo effects, 7m and n, suggest new parameters affecting bioavailability. Although 7m and n are comparable in size, 7m features an hydrogen bond donating amide moiety, whereas 7n bears a more lipophilic, hydrogen bond accepting ester functionality. The added hydrogen bond donating amide may affect specific interactions with proteins that impact CNS access (e.g., active transporters, efflux proteins, metabolizing enzymes), or nonspecific parameters, including polarity, and by extension, passive membrane permeability. Unsurprisingly, the carboxylic acid substituted compound (7r) showed no in vivo activity despite its relatively low molecular weight, likely due to the poor blood-brain barrier permeability of carboxylic acid moieties. Lastly, the relatively large aryl rings (7a and l), aryl amides (7o and p), and amine heterocycles (7i and k) evoked no response in vivo at the doses tested, likely due to a combination of unfavorable physicochemical parameters.
In summary, we have shown that modifications at the C-8 position of the THQ scaffold of our peptidomimetic series maintain high MOR affinity while improving DOR affinity, thus achieving our goal of developing more balanced receptor binding affinity compounds. Additionally, we have identified the C-8 carbonyl moiety as a key pharmacophore element for blocking DOR activation, thereby achieving the pharmacologically favorable MOR agonist/DOR antagonist profile. Although it is not known that a 1:1 MOR/DOR binding affinity ratio is optimal, preliminary in vivo data for a set of related compounds in our peptidomimetic series show the greatest reduction in tolerance and dependence when MOR/DOR affinity is relatively balanced. Confirmation of this trend will require evaluation of tolerance and dependence liabilities of additional analogues with a range of MOR and DOR affinities. The compounds reported to have in vivo activity here (7b, c, e, and n) encompass a range of MOR/DOR affinity ratios (7.9, 21, 4.7, and 3.7, respectively) and therefore should provide valuable data supporting or refuting this trend. In conclusion, these bioavailable peptidomimetics serve as promising leads in the search for future MOR agonist/DOR antagonist analgesics.
METHODS
Synthesis.
All reagents and solvents were obtained commercially and were used without further purification. Intermediates were purified by flash chromatography using a Biotage Isolera One instrument. Most purification methods utilized a hexanes/ethyl acetate solvent system, with a linear gradient between 20 and 100% ethyl acetate, though dichloromethane/methanol/triethylamine solvent systems were utilized for some polar, amine-containing intermedites. Purification of final compounds was performed using a Waters semipreparative HPLC with a Vydac protein and peptide C18 reverse phase column, using a linear gradient of 0% solvent B (0.1% TFA in acetonitrile) in solvent A (0.1% TFA in water) to 100% solvent B in solvent A at a rate 1% per minute, monitoring UV absorbance at 230 nm. The purity of final compounds was assessed using a Waters Alliance 2690 analytical HPLC instrument with a Vydac protein and peptide C18 reverse phase column. A linear gradient (gradient A) of 0% solvent B in solvent A to 70% solvent B in solvent A in 70 min, measuring UV absorbance at 230 nm was used to determine purity. All final compounds used for testing were ≥95% pure, as determined by analytical HPLC. 1H NMR and 13C NMR data were obtained on a 500 MHz Varian spectrometer using CDCl3 or CD3OD as solvents. The identities of final compounds were verified by mass spectrometry using an Agilent 6130 LC–MS mass spectrometer in positive ion mode.
Compounds presented in this series were synthesized as described in Schemes 1–5, starting with a commercially available, appropriately substituted aniline. In Scheme 1, anilines 1a–h were first coupled with 3-bromopropionyl chloride (i), yielding intermediates 2a–h. These intermediates then underwent (ii) a base-catalyzed β-lactam formation (3a–h). This was followed by a triflic acid-catalyzed Fries rearrangement (iii) to produce the THQ core (4c, 4g). In order to achieve the desired aryl bromide substitution pattern for further functionalization, compounds denoted as following Scheme 1 were treated with N-bromosuccinimide to install a bromine at either C-8 (4a) or C-6 (4b–h). Compounds 4i–k, shown in Scheme 2, were derived from 3b. After cyclization and aromatic bromination, the THQ amine was trifluoroacetyl-protected, then the methyl group at C-8 underwent benzylic bromination with N-bromosuccinimide and benzoyl peroxide. The benzylic bromide was then substituted under basic conditions with potassium carbonate and the specified amine (along with partial loss of the trifluoroacetyl protecting group), leaving the C-8 substituted aryl bromides 4i–k. Complete TFA loss observed in step (vi).
Scheme 1a.

a(i) 3-Bromopropionyl chloride, K2CO3. (ii) NaOtBu. (iii) TfOH. (iv) NBS. (v) see Scheme 2. (vi) benzyl (5a–k) or phenethyl (5l) boronic acid, Pd(dppf)Cl2, K2CO3. (vii, viii) see Schemes 3 and 4.
Scheme 5a.

a(ix) (R)-(+)-2-methyl-2-propanesulfinamide, then NaBH4. (x) HCl, then diBocDmt, PyBOP, 6-Cl HOBt, DIPEA, then TFA. (xi) LiOH (prior to TFA deprotection).
Scheme 2a.

a(iii) TfOH. (iv) NBS. (v) TFAA. (v′) NBS, benzoyl peroxide (v″) amine, K2CO3.
To convert intermediates 4 to 5, the aryl bromide was functionalized via either Suzuki coupling (step (vi), Scheme 1) to give 5a–l, or carbonylation (step (vii), Scheme 3) to give 5n and 4a′ followed by amide coupling (step (viii), Scheme 4) for 5m, o, and p.
Scheme 3a.

a(vii) CO, Pd(dppf)Cl2, MeOH (5n) or H2O (4a′).
Scheme 4a.

a(viii) Amine, PyBOP, DIPEA.
In Scheme 5, intermediates 5a–p (and 4a) were carried forward through a reductive amination step that utilized Ti(OEt)4 and a chiral Ellman sulfinamide, followed by NaBH4 to yield the desired R stereochemistry at C-4. During this step, the methyl ester of 5n converted to an ethyl ester, likely due to nucleophilic attack by ethoxide ions liberated from the titanium complex. Addition of concentrated HCl (x) cleaved the sulfinamide, leaving a primary amine HCl salt. This amine then underwent amide coupling with N-,O-diBoc 2′,6′-dimethyl-L-tyrosine (diBocDmt), followed by Boc deprotection with trifluoroacetic acid, yielding final compounds 7a–q. Compound 7r was produced by hydrolysis (xi) of the ester from 6n, prior to Boc deprotection. All final compounds were purified by semipreparative reverse-phase HPLC.
In Vitro Pharmacology.
Cell Lines and Membrane Preparations.
All tissue culture reagents were purchased from Gibco Life Sciences (Grand Island, NY, U.S.). C6-rat glioma cells stably transfected with a rat MOR (C6-MOR) or rat DOR (C6-DOR) and Chinese hamster ovary (CHO) cells stably expressing a human KOR (CHO-KOR) were used for all in vitro assays. Cells were grown to confluence at 37 °C in 5% CO2 in Dulbecco’s modified Eagle medium (DMEM) containing 10% fetal bovine serum and 5% penicillin/streptomycin. Membranes were prepared by washing confluent cells three times with ice cold phosphate buffered saline (0.9% NaCl, 0.61 mM Na2HPO4, 0.38 mM KH2PO4, pH 7.4). Cells were detached from the plates by incubation in warm harvesting buffer (20 mM HEPES, 150 mM NaCl, 0.68 mM EDTA, pH 7.4) and pelleted by centrifugation at 1600 rpm for 3 min. The cell pellet was suspended in ice-cold 50 mM Tris- HCl buffer, pH 7.4, and homogenized with a Tissue Tearor (Biospec Products, Inc., Bartlesville, OK) for 20 s. The homogenate was centrifuged at 15 000 rpm for 20 min at 4 °C. The pellet was rehomogenized in 50 mM Tris-HCl with a Tissue Tearor for 10 s, followed by recentrifugation. The final pellet was resuspended in 50 mM Tris-HCl and frozen in aliquots at 80 °C. Protein concentration was determined via a BCA protein assay (Thermo Scientific Pierce, Waltham, MA) using bovine serum albumin as the standard.
Radioligand Competition Binding Assays.
Radiolabeled compounds were purchased from PerkinElmer (Waltham, MA). Opioid ligand binding assays were performed by competitive displacement of 0.2 nM [3H]-diprenorphine (250 μCi, 1.85 TBq/mmol) by the peptidomimetic from membrane preparations containing opioid receptors as described above. The assay mixture, containing membranes (20 μg protein/tube) in 50 mM Tris-HCl buffer (pH 7.4), [3H]-diprenorphine, and various concentrations of test peptidomimetic, was incubated at room temperature on a shaker for 1 h to allow binding to reach equilibrium. For several peptidomimetics, radioligand competition binding assays were performed in high Na+ buffer (50 mM Tris-HCl, 100 mM NaCl, 5 mM MgCl2, 1 mM EDTA, pH 7.4) and samples were incubated at room temperature for 75 min to allow binding to reach equilibrium. Samples were rapidly filtered through Whatman GF/C filters using a Brandel harvester (Brandel, Gaithersburg, MD) and washed five times with 50 mM Tris-HCl buffer. Bound radioactivity on dried filters was determined by liquid scintillation counting, after saturation with EcoLume liquid scintillation cocktail, in a Wallac 1450 MicroBeta (PerkinElmer, Waltham, MA). Nonspecific binding was determined using 10 μM naloxone. The results presented are the mean ± standard error (SEM) from at least three separate assays performed in duplicate. Ki (nM) values were calculated using nonlinear regression analysis to fit a logistic equation to the competition data using GraphPad Prism, version 6.0c, for Mac OS X (GraphPad Software Inc., La Jolla, CA).
[35S]-GTPγS Binding Assays.
Agonist stimulation of [35S]guanosine 5′-O-[γ-thio]triphosphate [35S]-GTPγS, 1250 Ci, 46.2 TBq/mmol) binding to G protein was measured as described previously.27 Briefly, membranes (10–20 μg of protein/tube) were incubated for 1 h at 25 °C in GTPγS buffer (50 mM Tris-HCl, 100 mM NaCl, 5 mM MgCl2, pH 7.4) containing 0.1 nM [35S]-GTPγS, 30 μM guanosine diphosphate (GDP), and varying concentrations of test peptidomimetic. G protein activation following receptor activation with peptidomimetic was compared with 10 μM of the standard compounds [d-Ala2,N-MePhe4,Gly-ol]enkephalin (DAMGO) at MOR, d-Pen2,5-enkephalin (DPDPE) at DOR, or U69,593 at KOR. The reaction was terminated by vacuum filtration of GF/C filters that were washed 10 times with GTPγS buffer. Bound radioactivity was measured as previously described. The results are presented as the mean ± standard error (S.E.M.) from at least three separate assays performed in duplicate; potency (EC50 (nM)) and percent stimulation were determined using nonlinear regression analysis with GraphPad Prism, as above.
Ke Determination.
Agonist stimulation of [35S]-GTPγS binding by the known standard agonist SNC80 at DOR was measured as described above. This was then compared to [35S]-GTPγS binding stimulated by SNC80 in the presence of test compound (1000 nM). Both conditions produced 100% stimulation relative to SNC80. The difference between the EC50 of SNC80 alone and in the presence of test antagonist is the shift in concentration response. The Ke was then calculated as Ke = (concentration of compound)/ (concentration response shift – 1). The results presented are the mean from at three separate assays performed in duplicate.
In Vivo Characterization of Compounds.
Drug Preparation.
All compounds were administered by intraperitoneal (ip) injection in a volume of 10 mL/kg of body weight. Test compounds were dissolved in 5% DMSO (v/v) in sterile saline (0.9% NaCl w/v).
Animals.
Male C57BL/6 wild type mice (stock number 000664, Jackson Laboratory, Sacramento CA) bred in-house from breeding pairs and weighing between 20 and 30 g at 8–16 weeks old, were used for behavioral experiments. Mice were group-housed with free access to food and water at all times. Experiments were conducted in the housing room, maintained on a 12 h light/dark cycle with lights on at 7:00 am; all experiments were conducted during the light cycle. Studies were performed in accordance with the University of Michigan Committee on the Use and Care of Animals and the Guide for the Care and Use of Laboratory Animals (National Research Council, 2011 publication).
Antinociception.
Antinociceptive effects were evaluated in the mouse WWTW assay. Withdrawal latencies were determined by briefly placing a mouse into a cylindrical plastic restrainer and immersing 2–3 cm of the tail tip into a water bath maintained at 50 °C. The latency to tail withdrawal or rapidly flicking the tail back and forth was recorded with a maximum cutoff time of 20 s (50 °C) to prevent tissue damage. Antinociceptive effects were determined using a cumulative dosing procedure. Each mouse received an injection of saline ip and then 30 min later baseline withdrawal latencies were recorded. Following baseline determinations, cumulative doses of the test compound (1, 3.2, and 10 mg/kg) were given ip at 30 min intervals. At 30 min after each injection, the tail withdrawal latency was measured as described above. To determine the duration of antinociceptive action, baseline latencies were determined as described above. Thirty minutes after baseline determination, animals were given a 10 mg/kg bolus injection of test compound ip. Latency to tail withdrawal was then determined at 5, 15, and 30 min after injections, and every 30 min thereafter until latencies returned to baseline values.
Supplementary Material
Funding
This study was supported by NIH Grant DA003910 (H.I.M., E.M.J., and J.R.T.). J.P.A. was supported by the Substance Abuse Interdisciplinary Training Program administered by NIDA, 5 T32 DA007267, and N.W.G. by the Pharmacological Sciences Training Program, T32-GM007767.
ABBREVIATIONS
- MOR
μ-opioid receptor
- DOR
δ-opioid receptor
- KOR
κ-opioid receptor
- DAMGO
[d-Ala2, N-MePhe4, Gly-ol]-enkephalin
- DPDPE
[d-Pen2,d-Pen5] encephalin
- THQ
tetrahydroquinoline
- WWTW
warm water tail withdrawal
- di-Boc-Dmt
N,O-Boc 2′,6′-dimethyl-L-tyrosine
- DIPEA
N,N-diisopropylethylamine
- PyBOP
benzotriazol-1-yloxytripyrroli-dinophosphonium hexafluorophosphate
- 6-Cl HOBt
1-hydroxy-6-chloro-benzotriazole
- CPP
conditioned place preference
- MIDA
N-methyliminodiacetic acid
Footnotes
The authors declare no competing financial interest.
ASSOCIATED CONTENT
Supporting Information
The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/acschemneur-o.8b00139.
Molecular formula strings (XLSX)
REFERENCES
- (1).Porreca F, Takemori AE, Sultana M, Portoghese PS, Bowen WD, and Mosberg HI (1992) Modulation of mu-mediated antinociception in the mouse involves opioid delta-2 receptors. J. Pharmacol. Exp. Ther. 263, 147–152. [PubMed] [Google Scholar]
- (2).Zhu Y, King M. a., Schuller AGP, Nitsche JF, Reidl M, Elde RP, Unterwald E, Pasternak GW, and Pintar JE (1999) Retention of supraspinal delta-like analgesia and loss of morphine tolerance in δ opioid receptor knockout mice. Neuron 24, 243–252. [DOI] [PubMed] [Google Scholar]
- (3).Abdelhamid EE, Sultana M, Portoghese PS, and Takemori AE (1991) Selective blockage of delta opioid receptors prevents the development of morphine tolerance and dependence in mice. J. Pharmacol. Exp. Ther. 258, 299–303. [PubMed] [Google Scholar]
- (4).Fundytus ME, Schiller PW, Shapiro M, Weltrowska G, and Coderre TJ (1995) Attenuation of morphine tolerance and dependence with the highly selective δ-opioid receptor antagonist TIPP[ψ]. Eur. J. Pharmacol. 286, 105–108. [DOI] [PubMed] [Google Scholar]
- (5).Hepburn MJ, Little PJ, Gingras J, and Kuhn CM (1997) Differential effects of naltrindole on morphine-induced tolerance and physical dependence in rats. J. Pharmacol. Exp. Ther. 281, 1350–1356. [PubMed] [Google Scholar]
- (6).Schiller PW, Fundytus ME, Merovitz L, Weltrowska G, Nguyen TM, Lemieux C, Chung NN, and Coderre TJ (1999) The opioid mu agonist/delta antagonist DIPP-NH(2)[ψ] produces a potent analgesic effect, no physical dependence, and less tolerance than morphine in rats. J. Med. Chem. 42, 3520–3526. [DOI] [PubMed] [Google Scholar]
- (7).Mosberg HI, Yeomans L, Anand JP, Porter V, Sobczyk-Kojiro K, Traynor JR, and Jutkiewicz EM (2014) Development of a bioavailable μ opioid receptor (MOPr) agonist, δ opioid receptor (DOPr) antagonist peptide that evokes antinociception without development of acute tolerance. J. Med. Chem. 57, 3148–3153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (8).Dietis N, Guerrini R, Calo G, Salvadori S, Rowbotham DJ, and Lambert DG (2009) Simultaneous targeting of multiple opioid receptors: A strategy to improve side-effect profile. Br. J. Anaesth. 103, 38–49. [DOI] [PubMed] [Google Scholar]
- (9).Schiller PW (2010) Bi- or multifunctional opioid peptide drugs. Life Sci. 86, 598–603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (10).Ananthan S, Saini SK, Dersch CM, Xu H, McGlinchey N, Giuvelis D, Bilsky EJ, and Rothman RB (2012) 14-Alkoxy- and 14-acyloxypyridomorphinans: μ agonist/ δ antagonist opioid analgesics with diminished tolerance and dependence side effects. J. Med. Chem. 55, 8350–8363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (11).Salvadori S, Trapella C, Fiorini S, Negri L, Lattanzi R, Bryant SD, Jinsmaa Y, Lazarus LH, and Balboni G (2007) A new opioid designed multiple ligand derived from the μ opioid agonist endomorphin-2 and the δ opioid antagonist pharmacophore Dmt-Tic. Bioorg. Med. Chem. 15, 6876–6881. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (12).Turnaturi R, Aricò G, Ronsisvalle G, Parenti C, and Pasquinucci L (2016) Multitarget opioid ligands in pain relief: new players in an old game. Eur. J. Med. Chem. 108, 211–228. [DOI] [PubMed] [Google Scholar]
- (13).Dove LS, Lembo A, Randall CW, Fogel R, Andrae D, Davenport JM, McIntyre G, Almenoff JS, and Covington PS (2013) Eluxadoline benefits patients with irritable bowel syndrome with diarrhea in a phase 2 study. Gastroenterology 145, 329–338.e1. [DOI] [PubMed] [Google Scholar]
- (14).Sobolewska-Włodarczyk A, Włodarczyk M, Storr M, and Fichna J (2016) Clinical potential of eluxadoline in the treatment of diarrhea-predominant irritable bowel syndrome. Ther. Clin. Risk Manage. 12, 771–775. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (15).Lembo AJ, Lacy BE, Zuckerman MJ, Schey R, Dove LS, Andrae DA, Davenport JM, McIntyre G, Lopez R, Turner L, and Covington PS (2016) Eluxadoline for irritable bowel syndrome with diarrhea. N. Engl. J. Med. 374, 242–253. [DOI] [PubMed] [Google Scholar]
- (16).Ananthan S, Kezar HS, Carter RL, Saini SK, Rice KC, Wells JL, Davis P, Xu H, Dersch CM, Bilsky EJ, Porreca F, and Rothman RB (1999) Synthesis, opioid receptor binding, and biological activities of naltrexone-derived pyrido- and pyrimidomorphinans. J. Med. Chem. 42, 3527–3538. [DOI] [PubMed] [Google Scholar]
- (17).Wells JL, Bartlett JL, Ananthan S, and Bilsky EJ (2001) In vivo pharmacological characterization of SoRI 9409, a nonpeptidic opioid mu-agonist/delta-antagonist that produces limited antinociceptive tolerance and attenuates morphine physical dependence. J. Pharmacol. Exp. Ther. 297, 597–605. [PubMed] [Google Scholar]
- (18).Daniels DJ, Lenard NR, Etienne CL, Law P-Y, Roerig SC, and Portoghese PS (2005) Opioid-induced tolerance and dependence in mice is modulated by the distance between pharmacophores in a bivalent ligand series. Proc. Natl. Acad. Sci. U. S. A. 102, 19208–19213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (19).Lenard NR, Daniels DJ, Portoghese PS, and Roerig SC (2007) Absence of conditioned place preference or reinstatement with bivalent ligands containing mu-opioid receptor agonist and deltaopioid receptor antagonist pharmacophores. Eur. J. Pharmacol. 566, 75–82. [DOI] [PubMed] [Google Scholar]
- (20).Wang C, McFadyen I, Traynor JR, and Mosberg HI (1998) Design of a high affinity peptidomimetic opioid agonist from peptide pharmacophore models. Bioorg. Med. Chem. Lett. 8, 2685–2688. [DOI] [PubMed] [Google Scholar]
- (21).Mosberg HI, Yeomans L, Harland A. a., Bender AM, Sobczyk-Kojiro K, Anand JP, Clark MJ, Jutkiewicz EM, and Traynor JR (2013) Opioid peptidomimetics: Leads for the design of bioavailable mixed efficacy μ opioid receptor (MOR) agonist/δ opioid receptor (DOR) antagonist ligands. J. Med. Chem. 56, 2139–2149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (22).Bender AM, Griggs NW, Anand JP, Traynor JR, Jutkiewicz EM, and Mosberg HI (2015) Asymmetric synthesis and in vitro and in vivo activity of tetrahydroquinolines featuring a diverse set of polar substitutions at the 6 position as mixed efficacy μ opioid receptor/δ opioid receptor ligands. ACS Chem. Neurosci. 6, 1428–1435. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (23).Harland AA, Yeomans L, Griggs NW, Anand JP, Pogozheva ID, Jutkiewicz EM, Traynor JR, and Mosberg HI (2015) Further optimization and evaluation of bioavailable, mixed-efficacy μ-opioid receptor (mor) agonists/ δ -opioid receptor (dor) antagonists: balancing mor and dor affinities. J. Med. Chem. 58, 8952–8969. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (24).Harland AA, Bender AM, Griggs NW, Gao C, Anand JP, Pogozheva ID, Traynor JR, Jutkiewicz EM, and Mosberg HI (2016) Effects of n-substitutions on the tetrahydroquinoline (thq) core of mixed-efficacy μ-opioid receptor (mor)/δ-opioid receptor (dor) ligands. J. Med. Chem. 59, 4985–4998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (25).Anand JP, Kochan KE, Nastase AF, Montgomery D, Griggs NW, Traynor JR, Mosberg HI, and Jutkiewicz EM (2018) In vivo effects of μ opioid receptor agonist/δ opioid receptor antagonist peptidomimetics following acute and repeated administration. Br. J. Pharmacol, DOI: 10.1111/bph.14148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (26).Anand JP, Purington LC, Pogozheva ID, Traynor JR, and Mosberg HI (2012) Modulation of opioid receptor ligand affinity and efficacy using active and inactive state receptor models. Chem. Biol. Drug Des. 80, 763–770. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (27).Traynor JR, and Nahorski SR (1995) Modulation by mu-opioid agonists of guanosine-5′-O-(3-[35S]thio)triphosphate binding to membranes from human neuroblastoma SH-SY5Y cells. Mol. Pharmacol. 47, 848–854. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.