

Abstract
Lysine-selective molecular tweezers (MTs) are supramolecular host molecules displaying a remarkably broad spectrum of biologic activities. MTs act as inhibitors of the self-assembly and toxicity of amyloidogenic proteins using a unique mechanism. They destroy viral membranes and inhibit infection by enveloped viruses, such as HIV-1 and SARS-CoV-2, by mechanisms unrelated to their action on protein self-assembly. They also disrupt biofilm of Gram-positive bacteria. The efficacy and safety of MTs have been demonstrated in vitro, in cell culture, and in vivo, suggesting that these versatile compounds are attractive therapeutic candidates for various diseases, infections, and injuries. A lead compound called CLR01 has been shown to inhibit the aggregation of various amyloidogenic proteins, facilitate their clearance in vivo, prevent infection by multiple viruses, display potent anti-biofilm activity, and have a high safety margin in animal models. The inhibitory effect of CLR01 against amyloidogenic proteins is highly specific to abnormal self-assembly of amyloidogenic proteins with no disruption of normal mammalian biologic processes at the doses needed for inhibition. Therapeutic effects of CLR01 have been demonstrated in animal models of proteinopathies, lysosomal-storage diseases, and spinal-cord injury. Here we review the activity and mechanisms of action of these intriguing compounds and discuss future research directions.
Significance Statement
Molecular tweezers are supramolecular host molecules with broad biological applications, including inhibition of abnormal protein aggregation, facilitation of lysosomal clearance of toxic aggregates, disruption of viral membranes, and interference of biofilm formation by Gram-positive bacteria. This review discusses the molecular and cellular mechanisms of action of the molecular tweezers, including the discovery of distinct mechanisms acting in vitro and in vivo, and the application of these compounds in multiple preclinical disease models.
I. Introduction
Over 50 human diseases are known as proteinopathies, in which aberrant protein self-assembly into toxic oligomers and aggregates are causative or play a major role in the pathogenesis (Buxbaum, 1996; Golde et al., 2013; Knowles et al., 2014). Proteinopathies include a wide range of diseases, such as neurodegenerative disorders in which Alzheimer’s disease (AD) and Parkinson’s disease (PD) are the most prominent examples (Hardy and Higgins, 1992; Hardy and Selkoe, 2002). Each disorder can be associated with abnormal self-assembly of one or more amyloidogenic proteins into intra- and/or extracellular characteristic deposits, for example, amyloid plaques and neurofibrillary tangles in AD and Lewy bodies (LBs) in PD. Although the insoluble aggregates long have been thought to cause the associated diseases, smaller, soluble oligomers have been shown to be more toxic and today are believed to be the main culprits causing the pathogenesis in different proteinopathies (Shankar et al., 2007; Outeiro et al., 2008; Karpinar et al., 2009; Nimmrich and Ebert, 2009; Pham et al., 2010; Lasagna-Reeves et al., 2011; Winner et al., 2011; Verma et al., 2015; Delenclos et al., 2019; Kayed et al., 2020). The oligomers, and in some cases also the insoluble deposits, disrupt cellular processes leading to dysfunction and eventually death of the target cells. Despite decades of research, the cause and exact mechanisms of spontaneous misfolding, self-assembly, cytotoxicity, and accumulation of the culprit proteins in each proteinopathy remain elusive (Jucker and Walker, 2013; Marciniuk et al., 2013; Goedert, 2015). Proteinopathies are most common in the central nervous system (CNS), likely due to the high metabolic demands of neurons and their inability to reduce the concentration of the offending proteins through division. Interestingly, in many genetic proteinopathies, for example, polyglutamine diseases, the offending abnormal proteins form toxic assemblies primarily in the CNS even though they are expressed ubiquitously in neuronal and nonneuronal cells in the periphery (Bradford et al., 2010).
Despite tremendous effort to develop therapeutic strategies targeting toxic protein assemblies, to date, most of the available treatments aim only to ease the symptoms, and almost no efficient disease-modifying therapy has been established (Bitan, 2019; Mullard, 2021). Thus, there is an urgent need to develop efficient disease-modifying therapeutic strategies for prevention, treatment, and cure of proteinopathies. Therapeutic approaches including reducing the expression of the offending proteins, enhancing their clearance, inhibiting the self-assembly process, and blocking downstream aggregation-induced cellular toxicity pathways or upstream events triggering protein misfolding and aggregation have been explored. Multiple modalities including gene therapy, biologics, peptides, chaperons, and small molecules have been developed to target common features of amyloidogenic proteins in different proteinopathies (Rahimi et al., 2016; Yadav et al., 2019). However, to date, the field has experienced >99% failure of these attempts in clinical trials (Katsuno et al., 2012; Yiannopoulou et al., 2019; Mullane and Williams, 2020), emphasizing the unique nature of amyloidogenic proteins as molecular pathogens created by our body itself, as opposed to exogenous pathogens or cancer cells, which offer a higher degree of non-self character, facilitating their targeting by various drug modalities.
We have been developing an unusual class of therapeutic small molecules (Fig. 1), molecular tweezers (MTs), which act as broad-spectrum protein-self-assembly inhibitors. MTs use a unique mechanism of action and have been shown to be promising drug candidates for proteinopathies (Attar and Bitan, 2014; Schrader et al., 2016; Malik et al., 2018; Hadrovic et al., 2019). Unlike canonical small-molecule drugs, which typically act as inhibitors of enzymes or receptors and bind their targets with high specificity and nM affinity, MTs do not bind to a specific protein but rather to exposed Lys (and to a lower extent Arg) residues with low µM affinity. As might be expected for such a moderate affinity, the binding is highly labile, as was demonstrated by surface-plasmon resonance experiments (Bier et al., 2013). This mode of binding allows MTs to interfere with weak intermolecular interactions, such as those mediating the formation of aberrant protein oligomers and aggregation seeds, without disrupting the structure or function of normal proteins, where the operating forces have been optimized by millions of years of evolutions and therefore are substantially stronger. An additional important factor contributing to the selectivity of the MTs is the fact that in misfolded or natively unstructured proteins, Lys (and Arg) residues tend to be exposed to the solvent and available for MT binding, as opposed to normal, globular proteins, in which the positively charged residues often are involved in salt bridges and other interactions and therefore are hindered from interacting with the sterically demanding MTs.
Fig. 1.
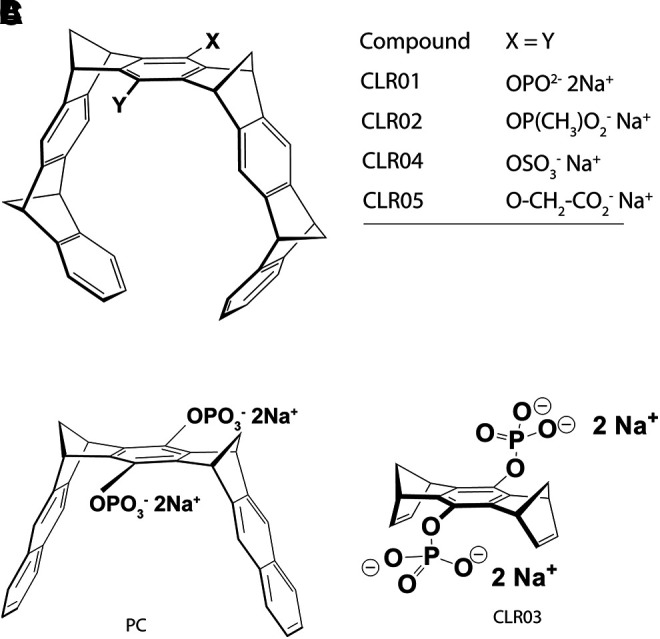
Structures of symmetric MTs. (A) Schematic representation of the torus-shaped MT and the polar head groups in CLR01, CLR02, CLR04, and CLR05. (B) Structure of the molecular clip PC. (C) Structure of the negative-control compound CLR03.
The first two compounds in the MT family tested for their ability to inhibit abnormal protein aggregation were CLR01 and CLR02, which share the same torus-shaped hydrocarbon skeleton and differ in the bridgehead groups—phosphate in CLR01 and methylphosphonate in CLR02 (Fig. 1A). A derivative comprising only the bridgehead component, but lacking the side arms, CLR03 (Fig. 1C), has been used as a negative control in these and subsequent experiments. Both CLR01 and CLR02 were found to inhibit the aggregation of amyloid β-protein (Aβ) in these initial experiments, yet further testing showed that CLR02 was toxic in cell culture. In contrast, CLR01 showed mild toxicity only at concentrations ≥400 μM and therefore became a lead compound used in most of the studies discussed here.
The inhibition of Aβ aggregation was interpreted initially as resulting from disruption of the salt-bridge between Lys28 and Glu22/Asp23 in Aβ, in addition to hydrophobic interactions between the butylene moiety of Lys28 and the sidechain of Val24. These interactions had been hypothesized to stabilize the folding nucleus of Aβ (Lazo et al., 2005) and were found also in early fibril structures of Aβ40 (Antzutkin et al., 2000). However, characterization of the binding sites of CLR01 on Aβ using electron-capture dissociation coupled with mass-spectrometry (ECD-MS) showed that the primary binding site was Lys16, rather than Lys28 (Sinha et al., 2011), likely because Lys16 participates in fewer intramolecular interactions and is exposed to the solvent, ready for interaction with the MT (Sinha et al., 2012b). These findings led us to realize that binding to exposed Lys residues in amyloidogenic proteins, rather than interrupting a particular interaction, might be sufficient for disrupting the aberrant self-assembly of these proteins and prompted testing of CLR01 against multiple other proteins involved in various proteinopathies (Sinha et al., 2011). A few years later, an examination of CLR01 as an inhibitor of the semen amyloid proteins involved in HIV infection (SEVI) (Münch et al., 2007) resulted in the discovery of the direct effect of the compound on viral membranes (Lump et al., 2015) and opened the gate for exploring this separate therapeutic application.
Here we review the multiple studies following up on these initial discoveries, which have involved over 30 research groups around the world. We discuss the different applications of MTs to various disease models, their mechanism of action, currently pending questions, and future directions toward development of these promising compounds for human therapy.
II. Discovery of Lys-/Arg-selective MTs
The molecular skeleton of the MTs was developed by design in a research program dedicated to aromatic interactions. Klärner et al.’s original idea was to create a rigid cavity, which would be at the same time nonpolar and rich in π-electrons. To this end, they combined isolated benzene rings in a convergent arrangement leading to through-space interactions between the separated aromatic systems. The resulting belt-like architecture turned out to be ideally suited for inclusion of cationic guest molecules. It was used as a platform for supramolecular aromatic inclusion events allowing studying inter alia π-cation interactions (Klärner and Kahlert, 2003; Klärner and Schrader, 2013). However, due to the overall nonpolar character of these hydrocarbon compounds, all the initial investigations remained limited to organic solvents. Later, when anionic functionalities were introduced into the central hydroquinone ring, the situation changed profoundly, and binding experiments became possible in buffered aqueous solutions. This led to the discovery of selective Lys and Arg inclusion by a unique mechanism, largely relying on the hydrophobic effect and Coulomb interactions between opposite host and guest charges (Fokkens et al., 2005). The following part details the total synthesis of the parent MT skeleton and subsequent transformations to MTs of successive generations, featuring various anions, linkers, and additional functional elements. It is followed by a general summary of the compounds’ molecular recognition properties with respect to amino acids, peptides, and proteins, which laid the foundation for their various biologic applications.
A. Total Synthesis of MTs
The stereoselective construction of such a rigid aromatic framework with all syn-connections is not trivial. For the synthetic chemist, the target structure invites applying Diels-Alder reactions with varying electron demand, as these lead to six-membered rings in a suprafacial approach with predictable relative stereochemistry.
A retro-synthetic analysis (Scheme 1) of the main transformations necessary for the construction of the belt-like arrangement of aromatic rings indeed reveals that the ideal key step is a repetitive Diels-Alder cycloaddition between two building blocks—5,6-bismethylene-2,3-benzonorbornene 1 as a “diene” and a 1,4,5,8-bismethanotetrahydroanthracene 2 as a “bisdienophile.”
Scheme 1.

Retrosynthetic analysis of the molecular MT skeleton, focusing on the key step between 1 and 2, which represents a neutral Diels-Alder reaction.
The diene is prepared in six steps starting from indene and maleic anhydride (Scheme 2) (Butler and Snow, 1975; Atasoy et al., 1994). At high temperature (200 °C), indene is in an equilibrium with a small, nondetectable amount of isoindene, which is produced by a sigmatropic 1,5-hydrogen shift. This exocyclic diene is trapped in situ by a normal Diels-Alder reaction with maleic anhydride, which serves as the dienophile (Alder et al., 1942). The anhydride adduct is converted subsequently in four conventional steps into trans-5,6-di(chloromethyl)-2,3-benzonorbornene. Base-induced HCl elimination leads to the desired diene 1, often called the “side wall” because it later forms the two cavity walls.
Scheme 2.

Synthesis of diene 1 (5,6-bismethylene-2,3-benzonorbornene), which later forms the two MT side walls from indene.
The bisdienophile 2 substituted by acetoxy groups in the central benzene ring is prepared in four steps (Scheme 3) (Benkhoff et al., 1997). The adduct of the initial Diels-Alder reaction between 1,3-cyclopentadiene and p-benzoquinone is converted by base-induced keto-enol tautomerization to the corresponding hydroquinone, which is oxidized, without isolation, by p-benzoquinone to 5,8-methano-5,8-dihydronaphtho-1,4-quinone. Diels-Alder reaction of this quinone with 1,3-cyclopentadiene at -78°C yields a mixture of the syn and anti adducts in a 65:35 ratio. Fractionating crystallization leads to the pure syn-adduct, which is converted in one step to the desired bisdienophile 2 by a base-catalyzed keto-enol tautomerization and acylation with acetic anhydride. Bisdienophile 2 is also called the “center piece” as it represents the chemically reactive central element of the MT.
Scheme 3.

Synthesis of bisdienophile 2 (9,10-diactoxy-1,4,5,8-octahydro-1,4,5,8-dimethanoanthracene), which later forms the MT’s center piece, from p-benzoquinone.
In the key step, two equivalents of diene 1 are subjected to a double Diels-Alder cycloaddition with one equivalent of bisdienophile 2. The repetitive Diels-Alder reactions between these two building blocks proceed on the exo face of the bisdienophile and the endo face of the diene. This stereoselectivity leads to the bis-adduct in which all four methylene bridges are positioned syn to one another—a prerequisite for creating the MT structure. Oxidative dehydrogenation of both newly formed cyclohexene moieties using 2,3-dichloro-5,6-dicyano-1,4-benzoquinone (DDQ) produces the all-aromatic MT bearing two acetoxy groups on the central benzene ring in an overall yield of 64%. Reduction of the acetoxy groups by LiAlH4 leads to the free hydroquinone MT 3 in a 98% yield (Scheme 4) (Klärner et al., 1996, 1999).
Scheme 4.

Synthesis of the free hydroquinone MT 3 starting from repetitive Diels-Alder reactions between diene 1 and bisdienophile 2.
Hydroquinone MT 3 is the starting material for the synthesis of water-soluble MTs CLR01–CLR05 (Scheme 5). A phosphate or phosphonate function is introduced by reaction with POCl3 or MePOCl2 in the presence of triethylamine. Subsequent hydrolysis using dilute HCl and neutralization by NaOH affords phosphate MT CLR01 (Talbiersky et al., 2008) and phosphonate MT CLR02 (Fokkens et al., 2005). Treatment of hydroquinone MT 3 with a sulfur trioxide pyridinium complex in anhydrous pyridine and subsequent work-up with saturated aqueous NaHCO3 leads to the sulfate MT CLR04 (Dutt et al., 2013a). Finally, the carboxylate analogue is prepared by nucleophilic substitution of the hydroquinone MT on methyl bromoacetate in the presence of potassium carbonate and potassium iodide. Hydrolysis of the ester groups by sodium hydroxide affords the carboxylate MT CLR05 (Dutt et al., 2013a).
Scheme 5.

Synthesis of water-soluble molecular MTs carrying in their central benzene unit two phosphate (CLR01), methanephosphonate (CLR02), sulfate (CLR04), or O-methylenecarboxylate groups (CLR05). Note the different number of charges and pK values.
Due to their low pK values, all MTs are anionic in neutral buffer. It is worth mentioning that each phosphate group of CLR01, which carries two negative charges, is partially protonated at pH 7 so that the negative charge of each group is about –1.5, creating a trianionic species under these conditions.
Further functionalization of the parent MTs proved problematic. In principle, one anionic moiety is sufficient to lock into an ion pair with the included guest cation. However, substitution of one phosphate anion by ether or ester groups blocks the cavity entrance by dispersive and hydrophobic forces (Dutt et al., 2013b). In a systematic investigation, all nonionic aliphatic substituents with more than two carbon atoms in a row at the two central phenolic OH groups were found to lower the MT’s affinity for Lys substantially. Evidence for additional dispersive interactions with the cavity entrance was obtained by crystal structures supported by molecular modeling. Medium and large substituents even showed self-inclusion in the MT’s cavity, further increasing the enthalpic penalty for the decomplexation step. After many attempts, it became clear that both anionic groups must be preserved to keep the cavity open. Additional binding sites may thus be introduced by way of monoesterification of the pendant phosphate anions. This leads to new MTs carrying one or two alkyl or alkynyl phosphate monoesters, which are still water-soluble. Intriguingly, the alkyne moiety prevents self-inclusion inside the MT cavity, because the high local π-electron density of the triple bond leads to strong electrostatic repulsion by the convergent aromatic rings. A good indicator for this is the respective extracellular polymeric substances value.
Two efficient synthetic routes were found for the introduction of additional substituents. The first route utilizes phosphate activation by trichloroacetonitrile, followed by a reaction with the corresponding alcohol and neutralization with NaOH as depicted in Schemes 6 and 7. The second strategy is the milder phosphoramidite method, which is not discussed here (Heid et al., 2018).
Scheme 6.

Synthesis of symmetrical molecular MTs bearing two mono-alkyl or mono-alkynyl phosphate groups on the central benzene unit. CLR01 as a free phosphoric acid is treated with an excess of trichloroacetonitrile followed by a nucleophilic attack of the respective alcohol (TCA method).
Scheme 7.

Synthesis of diphosphate MTs substituted with only one alkyl or alkynyl ester group, starting from the diacetoxy MT.
Careful hydrolysis of the diacetoxy MT with one equivalent of NaOH furnishes the unsymmetrical monoacetoxy MT in 98% yield, which is the starting material for asymmetric MTs. Scheme 7 shows how the free phenol is phosphorylated first and esterified, before the remaining acetoxy group is cleaved and also phosphorylated. This protocol affords, for example, the important key intermediate monobutynyl MT, which can be directly coupled to virtually any azide derivative by way of a click reaction (Heid et al., 2018).
Fluorescence labels, such as TAMRA in CLR16 or fluorescein in CLR18, thus can be subsequently introduced into the terminally alkynylated MT intermediates if they carry a sterically accessible azide group (Scheme 8). Click chemistry employing a Cu(II) source and a reducing agent or Cu(I) halides proceeds smoothly in water/THF mixtures. Intriguingly, none of the reactants needs to carry a protecting group owing to the orthogonal nature of azide and alkyne functionality. The final products are purified by preparative reverse-phase high-performance liquid chromatography, typically on a standard RP-18 stationary phase. These fluorescently labeled MTs were used in cell imaging experiments to track CLR01 derivatives in lysosomes and related cell organelles (Li et al., 2021).
Scheme 8.

Synthesis of fluorescent MTs CLR16 and CLR18 by click chemistry starting from the new key intermediate monobutynyl MT. This protocol appears to be generally applicable to almost any given terminal azide and allows the introduction of one additional functional unit onto CLR01.
B. Binding of Guest Molecules by MTs
Though the MT cavity is designed to accommodate cationic guest molecules, there is always competition by undesired self-inclusion of the functionalized phosphate or other “arms” inside the cavity. The inclusion properties of all MTs were assessed by various titration techniques and analytical experiments. In general, inclusion of a guest molecule inside the MT cavity leads to massive upfield shifts of proton NMR signals in the included guest, concomitant with fluorescence quenching in the host molecule. Further evidence was obtained from isothermal calorimetry (ITC) titrations and crystal structures. These techniques allow to separate the desired guest inclusion from the unwanted self-inclusion or dimerization effect and to quantify both processes.
The first evidence for the (unwanted) preferential binding of an alkyl chain inside the MT cavity came from a single-crystal structure and the 1H-NMR spectrum of the MT substituted by two OCH2CO2CH2CH3 groups in the central benzene ring, a precursor of CLR05 (Fig. 2A) (Klärner et al., 1996, 2004). The single-crystal structure showed unambiguously that the ethyl group of one sidechain was bound inside the tweezer cavity whereas the other one was positioned outside (Fig. 2B). In the 1H-NMR spectrum, the signals assigned to the ethyl protons of both sidechains were shifted to smaller δ values by Δδ = 0.8 ppm (CH2) and 1.6 ppm (CH3) compared with the corresponding signals of the bisdienophile lacking the MT sidewalls (Fig. 2A). These shifts certainly are the result of the magnetic anisotropy of the surrounding MT benzene rings around the sidechain. The observation of only one CH2 or CH3 signal for both ethyl groups leads to the conclusion that the inclusion and exclusion of the OCH2CO2CH2CH3 sidechains in the cavity are fast and dynamic processes on the NMR timescale (Fig. 2C) leading to an averaging of chemical shifts of the included and excluded sidechain. Further support for this conclusion came from the 1H-NMR data of the MT substituted by one OCH2CO2CH2CH3 and one OAc group (Fig. 2D). In this case, the complexation-induced 1H NMR shifts Δδ of the sidechain ethyl protons were almost twice as large as those found for the MT disubstituted with two OCH2CO2CH2CH3 groups indicating that in this case the sidechain was completely included inside the MT cavity and did not exchange with the excluded conformation. Very similar effects of self-inclusion were later identified in other asymmetric MTs carrying one phosphate group and an aliphatic substituent (Dutt et al., 2013b).
Fig. 2.

(A) Alkyl sidechain folding of the MT substituted by two OCH2CO2CH2CH3 groups in the central benzene unit detected by its single-crystal structure and its 1H-NMR spectra (CDCl3). (B) 1H-NMR data (CDCl3) of the MT substituted by one OCH2CO2CH2CH3 and one OAc group on the central benzene ring.
Similarly, intermolecular binding of guest molecules inside the MT cavity can be detected by characteristic shifts of the 1H NMR guest signals resulting from the magnetic anisotropy of the surrounding MT benzene rings. In addition, the host–guest complex formation can also be observed by changes in the fluorescence spectra of the MTs during the addition of guest molecules.
Before we begin the discussion of the host–guest complex formation, it is worth mentioning that the 1H-NMR spectra of the phosphate- and sulfate-substituted MTs, CLR01 and CLR04, are themselves concentration-dependent in aqueous buffer (Dutt et al., 2013a). In particular, the 1H NMR signals assigned to the protons attached to the tips of the terminal benzene rings were found to shift in aqueous solution as a function of the MT concentration. The maximum shifts of these 1H NMR signals were determined by dilution titration to be Δδmax = 2.2 ppm for CLR01 and 2.0 ppm for CLR04 compared with the data measured in CD3OD. These findings suggested the formation of the weakly associated dimers, for which the dimerization constants were found to be KDim = 60 M−1 for (CLR01)2 and 370 M−1 for (CLR04)2 in aqueous solution, evidently resulting from nonclassic hydrophobic interactions. Force-field calculations (Fig. 3) suggest intertwined dimer structures in agreement with the observed 1H-NMR signal shifts of the benzene protons at the tips. Notably, this week association is in contrast to an extended MT in which the central aromatic benzene was replaced by a naphthalene carrying two methylphosphonate groups. This naphthalene MT formed a highly stable intertwined dimer (KDim = 2.3·105 M−1) in agreement with the hydrophobic effect, which is expected to be larger for the more extended hydrocarbon skeleton in this case compared with that of the corresponding benzene MT, CLR02 (Klärner et al., 2006). The 1H NMR spectra of the other MTs described here show little to no concentration dependence suggesting that they exist as monomers in dilute aqueous solutions.
Fig. 3.

Structure of a CLR01 dimer calculated using the AMBER*/H2O force field. P atoms are indicated in yellow, O in red, C in gray, and H in white.
All MTs show strong emission bands at λem ≈ 330 nm in their fluorescence spectra upon excitation at λex = 285 nm. Comparison with the fluorescence spectrum of 1,4-dimethoxybenzene (λem = 320 nm) allows the assignment of the MTs’ emission band to the substituted central hydroquinone group (Dutt et al., 2013a). Binding of guest molecules by MTs leads to a partial quenching of these emission bands. Thus, complex formation can be monitored by fluorescence spectroscopy, allowing the respective binding constants, Ka, and hence the dissociation constants Kd (Kd = 1/Ka) to be determined by fluorimetric titration experiments (Fig. 4).
Fig. 4.

Fluorescence titration of CLR01 with Lys or Arg derivatives. All the reactions were carried out in 10 mM phosphate buffer, pH 7.6, and were monitored using λex = 285 nm, λem = 336 nm. (A) Schematic structure of Ac-Lys-OMe. (B) Schematic structure of Ac-Arg-OMe. (C) Fluorescence spectra of CLR01 in the absence or presence of increasing concentrations of Ac-Lys-OMe. (D) Change in fluorescence intensity as a function of increasing Ac-Lys-OMe concentration. (E) Fluorescence spectra of CLR01 in the absence or presence of increasing concentrations of Ac-Arg-OMe. (F) Change in fluorescence intensity as a function of increasing Ac-Arg-OMe concentration.
1. Amino Acid and Peptide Recognition
The complexation behavior of CLR01, CLR02, CLR04, and CLR05 was examined against various Lys and Arg derivatives as well as small, bioactive peptides containing Lys or Arg residues, such as the tripeptide KAA used to build bacterial cell walls (Williams and Bardsley, 1999); KLVFF, which is at the central hydrophobic cluster within the amyloid β-protein (Aβ) sequence (Tjernberg et al., 1997) and considered a nucleation site for pathologic protein aggregation; and KTTKS, a peptide that sends a signal to injured cells to regenerate their own collagen, with potential applications in the anti-aging technology (Tsai et al., 2007).
Representative Kd values from fluorimetric titration experiments are summarized in Table 1. They allow the following conclusions: CLR01, CLR02, and CLR04 are highly selective for Lys and Arg. For example, CLR01 does not bind to a peptide derived from the N-terminus of islet amyloid polypeptide (IAPP2-7), which does not contain either Lys or Arg but binds readily to IAPP1-7, which contains a Lys at position 1 and to IAPP2-14, which contains an Arg at position 11. This confirms earlier results obtained with CLR02, which was shown to complex Lys more strongly than Arg and much more strongly than His. Other amino acids (e.g., Asp, Ser, Phe, Leu, Ala, or Gly) did not bind this MT at all (Fokkens et al., 2005). In contrast to these MTs the O-CH2CO2-substituted MT, CLR05, binds Lys or Arg derivatives significantly weaker (Table 1). Interestingly, despite the weaker binding, this derivative showed strong inhibition of bacterial biofilm by mechanisms that are not completely understood (Malishev et al., 2021).
TABLE 1.
Dissociation constants for MTs with Lys- or Arg-containing amino acids and peptides determined by fluorometric titration experimentsa
| Guest | Kd (µM) | |||
|---|---|---|---|---|
| CLR01 | CLR02 | CLR04 | CLR05 | |
| Ac-Lys-OMe | 17b 9c | 68b | 28b 19c | 226b 643c |
| H-Lys-OH | 21b | 874b | 227c | 1170d |
| KAA | 30b | 905b | 303c | 33333d |
| KLVFF | 20b | 38c | ||
| KKLVFF | 4b | 71b | ||
| KKLVFFAK | 7b | |||
| KKKK | 10b | |||
| Ac-Arg-OMe | 60b 20c | 178b 77d | 882b 281d | |
| H-Arg-OH | 699b,c | 609d | ||
| H-Arg-OMe | 160c | |||
| RGD | 86b | |||
| cRGDfV | 59c | |||
| cGRGDfL | 26d | |||
| IAPP1-7 | 9 | |||
| IAPP2-14 | 104 | |||
| IAPP2-7 | No binding | |||
aDutt et al. (2013a) and Lopes et al. (2015).
bPhosphate buffer 200 mM, pH=7.6.
cPhosphate buffer 10 mM, pH=7.6.
dPhosphate buffer 10 mM, pH=7.2.
Dissociation constants determined independently by 1H-NMR and ITC titration experiments (Dutt et al., 2013a) were in good agreement with fluorescence titrations in Table 1. In addition to the complex stability characterized by these Kd values, the maximum complexation-induced shifts of the 1H-NMR guest signals, Δδmax provided important information about the host–guest complex structures. In the complexes of the phosphate-, phosphonate-, and sulfate-substituted MTs, CLR01, CLR02, and CLR04, respectively, large Δδmax values, up to 6 ppm, were observed for the signals of the guest methylene protons assigned to the Lys or Arg sidechains. These findings suggested threading of these sidechains through the MTs’ cavity. In contrast, in the complexes of the carboxylate-substituted CLR05, the Δδmax values of the corresponding guest protons were substantially smaller, Δδmax < 1 ppm, indicating complex structures different from those of CLR01, CLR02, or CLR04 (Table 2). To gain further structural insight, the structures of the free MTs, free guest molecules, and the corresponding host–guest complexes were optimized using quantum mechanics/molecular mechanics (QM/MM) methods (Fig. 4). The resulting molecular structures were subsequently used for 1H-NMR chemical shift calculations by quantum chemical ab initio methods (11). The comparison of the experimental and calculated 1H-NMR shift data supported the host–guest complex structures. Large theoretical shifts were calculated for the γ-, δ- and ε-methylene protons of the Lys or Arg sidechains in complexes with CLR01, CLR02, and CLR04 in agreement with the experimental data, supporting the threading of these sidechains through the MT cavity. Additional confirmation of the postulated binding mode was obtained by single-crystal structure analyses of the complexes of CLR01 with 14-3-3 proteins (Bier et al., 2013, 2017).
TABLE 2.
Comparison of experimental and calculated (HF/SVP) maximal complexation-induced chemical shifts, Δδmax, for the guest protons in host-guest complexes of MTs with Lys or Arg derivatives
| Δδmaxa (ppm) at position | |||||
|---|---|---|---|---|---|
| Host | Guest | Method | Γ | δ | ε |
| CLR01 | Ac-Lys-OMe | Experimental | 3.91 | ||
| Calculated | 4.62 | 5.51 | 3.62 | ||
| CLR01 | KAA | Experimental | 2.28 | 3.22 | 5.92 |
| Calculated | 2.55 | 5.08 | 5.71 | ||
| CLR02 | Ac-Lys-OMe | Experimental | 1.57, 1.45b | > 4 | |
| Calculated | 1.72 | 3.42, 3.21 | 3.46 | ||
| CLR04 | Ac-Lys-OMe | Experimental | 2.64 | 4.41 | 3.75 |
| Calculated | 1.10 | 3.19 | 4.39 | ||
| CLR05 | Ac-Lys-OMe | Experimental | 0.40 | 0.54 | 0.94 |
| Ac-Lys-OMe in | Calculated | 1.69 | 3.05 | 5.44 | |
| Ac-Lys-OMe out | Calculated | 0.41 | 0.72 | 0.03 | |
| CLR01 | Ac-Arg-OMe | Experimental | 2.54 | 3.75 | |
| Calculated | 2.46 | 5.46 | |||
| CLR02 | Ts-Arg-OMe | Experimental | 4.09, 3.29b | 3.90 | |
| Calculated | 2.51, 1.67b | 4.30 | |||
| CLR04 | Ac-Arg-OMe | Experimental | 2.51 | 3.86 | |
| Calculated | 0.63 | 3.86 | |||
| CLR05 | Ac-Arg-OMe | Experimental | 0.62, 0.48b | 0.96 | |
| Ac-Arg-OMe in | Calculated | 1.39, 1.04b | 3.36 | ||
| Ac-Arg-OMe out | Calculated | 0.38, 0.36b | 0.26 | ||
aΔδmax = δ0-δC; δ0 and δC are the 1H-NMR chemical shifts of the free and complexed guest molecule, respectively. bdiastereotopic H atoms.
As mentioned, the small shifts observed and calculated for the complexes of CLR05 with Lys or Arg guest protons suggested a different binding mode for this MT, in which the sidechains bind outside the cavity (Fig. 5). To test this hypothesis, theoretical Δδmax values were calculated independently for complexes in which the guest sidechain was positioned either inside or outside the cavity. Comparison of the calculated chemical shifts with the experimental ones suggested that although both complexes might exist in a rapid equilibrium, binding outside the cavity is strongly preferred (Table 2). Apparently, the extended OCH2CO2- groups in CLR05 block the MT’s cavity and direct the guest molecule to a position outside the cavity where the major host–guest binding force is electrostatic attraction. QM/MM calculations produced chelate arrangements between both carboxylate anions in CLR05 and the complexed amino acid cation outside the MT’s cavity, made possible by the presence of the additional methylene group in the OCH2CO2- sidechain, which is absent in the other MTs (Fig. 5). The loss of CH-π and hydrophobic interactions in this geometry explains why the complexes of CLR05 with the Lys/Arg sidechains included inside the cavity are significantly less stable than those of CLR01 or CLR04. Evidently, dispersive interactions inside the MTs’ cavities and hydrophobic forces contribute substantially to the stability of the inclusion complexes.
Fig. 5.

Host–guest structures of the complexes of MTs with Lys or Arg derivatives, optimized using QM/MM calculations without counter-ions. Each structure contains a 60 Å water layer (not shown) (Dutt et al., 2013a).
The unique threading binding-mode of Lys and Arg sidechains inside the MT cavity leads to an exceptional selectivity of most MT derivatives for basic amino acids. Hydrophobic forces, dispersive, and electrostatic attractions contribute to the MT’s affinity for these amino acids, which is typically in the low micromolar regimen. These findings are valid not only for the amino acids themselves but also for flexible peptides in which each single basic amino acid is sterically accessible for MT inclusion (Fig. 6). For example, three CLR01 molecules were found to bind to the triply cationic peptides Aβ40 and Aβ42 peptides (Sinha et al., 2011), whereas the dicationic peptide IAPP carried only two CLR01 molecules (Lopes et al., 2015). Importantly, both Aβ and IAPP are natively unstructured peptides in which the cationic sidechains are exposed to the solvent, allowing the MT to bind to all the available positions.
Fig. 6.

CLR01 binding induces a massive upfield shift of Lys sidechain protons inside its cavity. 1H-NMR spectra were recorded for CLR01, IAPP(1 − 7), and their 1:1 or 1:3 complexes in 10 mM phosphate-buffered D2O, pH 7.2. The Lys1 side-chain methylene signals are assigned as α, β, γ, δ, and ε. All the methylene resonances, most notably the δ- and ε-protons, are shifted upfield by 4.4 and 4.5 ppm, respectively, indicating strong binding of CLR01 to Lys1.
2. Protein Surface Recognition
On natively folded, compact protein surfaces, titration experiments with MTs showed binding of multiple MT molecules, but, in these cases, the number of the molecules was found by crystallography to be substantially lower than the number of available binding sites because only those Lys and Arg residues that were exposed allowed binding of the sterically demanding MTs (Bier et al., 2013). QM/MM calculations predicted a less efficient binding mode for the MTs on protein areas with several basic amino acids in close proximity. In these cases, the MTs may prefer to form electrostatic clusters employing multiple Coulomb interactions in a chelate type, without inclusion of sidechains inside their cavities (QM/MM) (Bier et al., 2013).
A study addressing the binding mode and binding sites of supramolecular host molecules known to bind cationic amino acid residues compared side-by-side MTs, sulfonated calixarenes, pyrenes, and porphyrins using ubiquitin as a model protein target. The direct comparison revealed that these compounds differed greatly in their preference for different target areas on the protein surface. Thus, whereas CLR01 preferentially bound to Lys residues in the unstructured, flexible C-terminal tail of the protein, whereas the other compounds tended to occupy folded surface patches rich in Arg residues (Mallon et al., 2016). The study, which used 2D-NMR experiments and molecular modeling, suggested that MT ligands prefer binding to accessible Lys residues in conformationally flexible protein regions, supporting their ability to interfere selectively with misfolding and self-assembly of amyloidogenic proteins while leaving normal processes mediated by folded proteins undisturbed. Similarly, recognition of the flexible Lys-rich C-terminus of the effector protein SpHtp3 by CLR01 prevented the protein’s interaction with its cognate receptor protein, gp96, and inhibited cell entry of parasites (Trusch et al., 2018).
Several crystal structures between CLR01 and different proteins illustrated the exquisite selectivity for Lys and Arg and confirmed the postulated unique threading binding mode involving ion pair formation (Bier et al., 2013, 2017). They also pointed to a combination of Coulomb attraction with the hydrophobic effect and a strong preference of the nonpolar MT skeleton for nonpolar patches on the protein surface (Fig. 7). The same noncovalent interactions are relevant for aggregation processes and form the basis of our postulated process-specific inhibition of such processes, as opposed to protein-specific binding, which was found to be very weak, namely in the high micromolar regimen (Talbiersky et al., 2008). NMR evidence also showed multiple incidences of MTs binding well accessible Lys or Arg residues at exposed locations, for example, at the edge of α-helices or at isolated basic sites (Trusch et al., 2018). These experimental findings were accompanied by QM/MM calculations and MD simulations and led to empirical rules for preferred Lys surroundings for tweezer docking. QM/MM calculations also helped to distinguish among ambiguous NMR effects so that neighboring Lys residues with similar heteronuclear single quantum coherence cross peaks could be precisely assigned (Fokkens et al., 2005; Trusch et al., 2018). Fluorescence polarization titrations revealed the ability of MTs to interfere with protein–protein interactions despite their weak binding (Bier et al., 2013, 2017; Trusch et al., 2018), a prerequisite for redirecting the path of protein aggregation. Beuck et al. studied MT binding to proteins containing uniformly 13C,15N-labeled Lys and Arg and employed 2D H2(C)N spectra for the precise analysis of Lys/Arg complexation on protein surfaces (Fig. 8). This method now allows to determine the exact binding order on all preferred binding sites (Hogeweg et al., 2017). All of these findings are of fundamental importance for understanding and describing the complexation behavior of MTs on naturally occurring peptides and proteins. They also form the basis for explaining and predicting the powerful effects of MTs as modulators/inhibitors of aberrant protein aggregation.
Fig. 7.

Crystal structures of CLR01 docked onto Lys (A, B) or Arg residues (C, D) on 14-3-3 protein surfaces. In both cases, the postulated binding mode was corroborated by experimental evidence, and the threading mode of the cationic sidechain together with the new ion pair between the MT phosphate and the included cation were demonstrated.
Fig. 8.

2D H2(C)N spectra specific for Arg (A) and Lys (B) of the 13C/15N-labeled, small protein domain hpin1-WW at increasing CLR01 concentrations. The superimposed titration curves (C) and (D) reveal a strict order in preferred binding sites for the MT on the same protein. (E, F) Successive recovery of Lys (E) and Arg (F) protons upon back titration of a 1:1 hpin1-WW MTs complex with unlabeled Lys. (G) MT binding mode of the Lys sidechain.Schematic structure of the small protein domain hpin1-WW.
3. Membrane Recognition
Similar considerations are important for the inclusion of the trimethylammonium choline headgroup of membrane-forming phospholipids. Intriguingly, direct 1H-NMR titrations of sphingomyelin and related phospholipids with CLR01 produced marked upfield shifts of the N-Me signal of up to 3 ppm (Weil et al., 2020). Clearly, the cationic NMe3+ choline head group can be accommodated inside the tweezer cavity. This new, unique binding mode was proposed based on extensive MD simulations and QM/MM calculations of CLR01 docked onto model membranes. As a result of the calculations, the tweezer body was drawn even further into the lipid bilayer, offering a straightforward explanation for the increased surface tension of viral membranes (see below) (Weil et al., 2020).
Further mechanistic elucidation came from fluorescence microscopy imaging of liposome models (GUVs), which imitated ordinary cell membranes with 2-dioleoyl-sn-glycero-3-phosphocholine (DOPC) as their only lipid component (Lump et al., 2015; Weil et al., 2020). Viral membranes were simulated with DOPC and 30% additional lipid rafts, that is, cholesterol and sphingomyelin. After CLR01 addition, pure DOPC liposomes remained unchanged, but liposomes containing lipid rafts broke exactly at the border between fluorescently labeled DOPC and the rafts, leading to complete leakage of an included blue dye out of the liposomes. The data suggest that the disruption of the lipid-raft containing membranes was due to the MT-induced increase in surface tension at the boundary between different lipid phases. A direct proof for the surface disruption was obtained using extensive electron microscopy measurements, which documented surface perturbation and in some cases massive deformation of spherical viral particles and a total loss of peplomers (Weil et al., 2020).
These findings prompted introduction of lipid anchors to the phosphate anions in the form of aliphatic or aromatic esters. Cell culture and mouse experiments demonstrated a significant, up to 100-fold improvement in IC50 values of antiviral activity into the nanomolar regimen, demonstrating that the additional ester arms had a powerful destabilizing effect on the viral membranes. Subsequent structure-activity relationship studies pointed to certain linear alkyl and benzyl esters as new lead structures, which are currently being examined against a broad variety of viruses.
The experiments discussed here led to a detailed understanding of the forces underlying membrane disruption of viral envelopes and the application of MTs as broad antivirals (Le et al., 2022).
III. Biologic Applications
The following sections discuss the effect of MTs on different diseases and pathologic conditions and their potential as therapeutic agents for these conditions.
A. Synucleinopathies and Related Conditions
The primary pathology in synucleinopathies, including PD, dementia with Lewy bodies (DLB), and multiple system atrophy (MSA), is the abnormal intracellular accumulation and deposition of α-synuclein (α-syn), a 140-residue protein whose structure and function are active areas of investigation (Ghiglieri et al., 2018; Sulzer and Edwards, 2019; Runwal and Edwards, 2021). Interestingly, α-syn deposits are in neurons in PD and DLB, where they are called LBs and Lewy neurites, and primarily in oligodendrocytes in MSA, in which the deposits are called glial cytoplasmic inclusions (Dickson, 2001). Although a small fraction of α-syn is thought to form a native, α-helix-rich tetramer (Bartels et al., 2011; Wang et al., 2011), the protein is mostly intrinsically disordered. Upon oligomerization and aggregation, α-syn acquires prion-like properties, facilitating propagation of these toxic assemblies from cell to cell (Desplats et al., 2009; Bartels et al., 2011; Fauvet et al., 2012). Different studies have suggested that α-syn oligomers are the most toxic species contributing to the pathology of different synucleinopathies (Outeiro et al., 2008; Winner et al., 2011; Martin et al., 2012) and to the propagation of the misfolded, toxic structures in a prion-like manner (Lee et al., 2010; Luk et al., 2012; Jucker and Walker, 2013; Clavaguera et al., 2015; Hijaz and Volpicelli-Daley, 2020). Therefore, inhibition of α-syn oligomerization and aggregation and dissociation of existing fibrillar deposits may limit disease progression and inhibit the neurotoxic effects of α-syn. With regard to the latter point, however, it is crucial to ensure that dissociation of fibrillar aggregates does not lead to the formation of toxic oligomers.
Initial examination of the impact of CLR01 on α-syn aggregation was done using thioflavin-T (ThT) fluorescence and electron microscopy. These experiments showed that CLR01 prevented α-syn’s fibril formation in a dose-dependent manner (Fig. 9A, B). A complete inhibition of β-sheet formation was observed at a 1:1 α-syn:CLR01 concentration ratio and a partial inhibition at a 10:1 α-syn:CLR01 ratio (Prabhudesai et al., 2012). CLR01 not only inhibited α-syn aggregation but also was found to disaggregate preformed α-syn fibrils (Prabhudesai et al., 2012). The gradual decrease in ThT signal and changes in morphology (Fig. 9C) suggested that CLR01 halted α-syn fibril formation and dissociated the existing fibrils. This was an important finding because it suggested that CLR01 could be used not only for prevention of α-syn self-assembly and toxicity in synucleinopathies but also for treatment of the disease after aggregated α-syn already had been deposited in the brain. In the same study, CLR01 protected differentiated rat pheochromocytoma, PC-12 cells from the toxicity of 20 μM α-syn oligomers added to the cell-culture medium with half-maximal inhibition (IC50) = 4 ± 1 μM. The substoichiometric inhibition was attributed to the high number of potential binding sites for CLR01, 15 Lys residues, in α-syn. CLR01 also inhibited completely the toxicity of α-syn expressed in human embryonic kidney, HEK293, cells when added at 1 μM (Prabhudesai et al., 2012). In a more recent study, a dose-response analysis showed that CLR01 inhibited the intracellular accumulation of oligomeric α-syn in human neuroblastoma, SH-SY5Y cells, with IC50 = 85.4 nM (Bengoa-Vergniory et al., 2020).
Fig. 9.

CLR01 inhibits α-syn assembly and disaggregates α-syn in vitro. (A) β-Sheet formation in α-syn in the absence or presence of different concentrations of CLR01 or CLR03 was followed by measuring ThT fluorescence. (B) Electron micrographs of α-syn in the absence or presence of 10-fold molar excess of CLR01 or CLR03 at the end of the reactions shown in panel A. Scale bars denote 100 nm. (C) Disaggregation of α-syn fibrils by CLR01 was initiated at two time points: On day 8 (dissociation reaction D1) or on day 24 (D2), and the reactions were monitored using ThT fluorescence. Electron micrographs were obtained periodically and show the morphology of α-syn at the indicated time points. Scale bars denote 100 nm. The data are an average of three independent experiments. Reprinted by permission from Springer Nature Customer Service Centre GmbH: Springer Nature, Neurotherapeutics, A novel “molecular tweezer” inhibitor of α-synuclein neurotoxicity in vitro and in vivo, Prabhudesai, S., Sinha, S., Attar, A., Kotagiri, A., Fitzmaurice, A. G., Lakshmanan, R. Ivanova, M. I., Loo, J. A., Klärner, F. G., Schrader, T., Stahl, M., Bitan, G., and Bronstein, J. M. (2012).
In the latter study, the effect of CLR01 on α-syn aggregation and toxicity was examined in induced pluripotent stem-cell (iPSC)-derived dopaminergic neurons treated with LB extracts from postmortem, human PD-brain samples. The LB-treated cells displayed shortened neuronal processes and increased blebbing compared with cells treated with control brain extracts that did not contain LBs. CLR01 treatment improved the neurite length and reduced the blebbing back to control level (Bengoa-Vergniory et al., 2020). In addition, treatment of LB-treated cells with CLR01 reduced α-syn-oligomer puncta, measured using an α-syn proximity ligation assay (AS-PLA) (Roberts et al., 2015) and rescued α-syn-mediated cytotoxicity and aggregation in LB-treated iPSC-derived dopaminergic neurons in a dose-dependent manner (Bengoa-Vergniory et al., 2020).
The first in vivo testing of CLR01 was in a zebrafish (ZF) embryo model of α-syn toxicity, which expresses human wild-type α-syn under control of the neuronal HuC promoter (Prabhudesai et al., 2012). Expression of α-syn in the ZF embryos caused severe toxicity. Morphologically, the embryos were deformed, ranging from a modest bend in the tail region to gross deformation. A few embryos had normal morphology but were partially or completely paralyzed. Most of these embryos died within 48 to 72 hours post-fertilization (hpf). Addition of 1 or 10 μM CLR01 to the water in which the embryos developed led to a dramatic improvement in both their phenotype and survival. The protective effect was both dose and time dependent. Thus, at the high dose, survival improved threefold at 72 hpf and 13-fold at 240 hpf compared with untreated fish. CLR01 was found to rescue α-syn-induced apoptosis of the fish neurons and to facilitate degradation of α-syn by the ubiquitin-proteasome system (UPS) (Prabhudesai et al., 2012).
In a subsequent study examining the toxicity of the pesticide Ziram (zinc dithiocarbamate), which had been found to be strongly linked to an increased prevalence of PD (Fitzmaurice et al., 2013, 2014), a surprising finding was that part of the mechanism by which Ziram caused strong toxicity in ZF embryos was through accumulation and aggregation of the endogenous ZF γ-synuclein, the closest ortholog to human α-syn. Therefore, CLR01 was tested for its ability to inhibit Ziram toxicity and was found to reduce significantly the Ziram-induced neurotoxicity in the ZF, which was measured by quantifying the number of neurons expressing green-fluorescent protein (GFP)-conjugated vesicular monoamine transporter 2 (VMAT2) (Lulla et al., 2016). VMAT2 is an integral membrane protein involved in the transport of monoamines, particularly neurotransmitters, including dopamine. The GFP-VMAT2 conjugate thus was used to visualize the ZF monoaminergic, including dopaminergic, neurons. The results of these ZF studies have been summarized previously in Fig. 7 of Schrader et al. (2016) and therefore are not reproduced here.
A different fish study used a lamprey model of spinal-cord injury (SCI). The background for the study was a report by Busch and Morgan who found that SCI caused accumulation of the endogenous lamprey synuclein in a subset of giant reticulospinal neurons, which correlated with poor survival of these neurons, whereas neurons that showed good survival after SCI did not accumulate synuclein (Busch and Morgan, 2012). To test whether the accumulation of synuclein reflected formation of toxic oligomers and/or aggregates, and if CLR01 could protect the fish against the toxicity of these species, Fogerson et al. applied a single dose of 1-mM CLR01 or buffer to the spinal-cord transection site via a small Gelfoam piece, which yields an effective concentration of CLR01 in the spinal cord in the 1 to 100 μM range. Eleven weeks post-transection, punctate synuclein accumulation was found in poor-survivor neurons of vehicle-treated animals, whereas CLR01 treatment resulted in significant reduction of synuclein accumulation within the giant reticulospinal neurons, and the staining pattern was diffuse as opposed to punctate in the CLR01-treated fish. In correlation with the reduced synuclein accumulation and change in the morphology from aggregated to diffuse, CLR01 treatment improved significantly the survival of the neurons (Fogerson et al., 2016).
CLR01 then was tested in a mouse model expressing wild-type human α-syn under the Thy1 promoter, which leads to a broad expression of α-syn in the brain (Chesselet et al., 2012). First, to avoid the blood-brain barrier and obtain proof of concept, CLR01 was administered intracerebroventricularly (i.c.v.) for 28 days via osmotic minipumps. The treatment was found to improve motor deficits assessed using the challenge-beam test by 29% compared with mice treated with vehicle. After treatment completion, the brain of the mice was extracted using a three-step protocol, fractionating the proteins into (1) a buffer-soluble fraction containing cytoplasmic and water-soluble extracellular proteins; (2) detergent-soluble, membrane-associated proteins; and (3) insoluble proteins from inclusion bodies and aggregates. A significant reduction in α-syn was observed using western blots in the buffer-soluble fraction of the striatum region of CLR01-treated animals compared with those treated with vehicle, suggesting that the treatment reduced toxic, soluble oligomers. Importantly, the study suggested that CLR01 could improve motor deficits in the mice by lowering soluble α-syn in the striatum, even though the treatment did not change the level of aggregated α-syn in the substantia nigra (SN) (Richter et al., 2017), supporting the notion that the soluble/oligomeric form was the main culprit (Conway et al., 2000, 2001).
The impact of CLR01 administered subcutaneously at 0.4 mg/Kg per day via osmotic minipumps then was tested in the same mice for 28 days. A 14% improvement was found in the challenge-beam test, smaller than the 29% improvement found using i.c.v.-administered CLR01, yet using this route of administration, a significant improvement also was found in the pole test, in which mice are placed face-up on a vertical pole and the time they require to turn and descend is measured. Significant improvement was found both in the time it took the CLR01-treated mice to turn and in the time it took them to descend from the pole compared with vehicle-treated mice. Perhaps counterintuitively, following i.c.v. administration, there was no improvement in the pole test, but this likely was due to limitation in the ability of the mice to maneuver themselves on the pole due to the canulae implanted in their brains, as compared with the less demanding movement across the challenge beam.
In another proof-of-concept study, the effect of i.c.v.-administered CLR01 was tested in a mouse model of the rare synucleinopathy MSA (Herrera-Vaquero et al., 2019). In this model, overexpression of wild-type, human α-syn in oligodendrocytes is driven using the proteolipid protein (PLP) promoter (PLP-α-syn mice) (Stefanova and Wenning, 2015). Animals were administered 0.3 or 1.0 mg/Kg per day CLR01 for 32 days using osmotic minipumps. In an open-field behavioral test, vehicle-treated PLP-α-syn mice showed a significant anxiety-like behavior, reflected by a lower fraction of time spent in the center of the field relative to the periphery. CLR01 improved the anxiety-like behavior dose dependently. Immunohistochemistry (IHC) analysis showed that the treatment reduced glial cytoplasmic inclusions (GCIs) in the brain of the treated mice dose dependently. Moreover, the reduction in GCI density in various brain regions, including the prefrontal cortex, basolateral amygdala, and commissura anterior was found to correlate strongly with the improvement in the anxiety-like behavior. Western-blot analysis of α-syn phosphorylated at serine 129 (pS129-α-syn) was performed following brain extraction into buffer-soluble and insoluble fractions, as described earlier. In the soluble fraction, only monomeric pS129-α-syn was observed, and its abundance was not affected by CLR01 treatment. In contrast, in the insoluble fraction, both pS129-α-syn monomer and a high-molecular-weight (HMW) band were observed, and both were reduced dose-dependently by the treatment. The decrease in the HMW pS129-α-syn band intensity correlated strongly (r = –0.453, p = 0.039) with the improvement in the anxiety-like behavior, suggesting that this band represented a toxic form of α-syn.
Neurodegeneration in the substantia nigra pars compacta (SNc) at 6 months of age is a prominent neuropathologic feature of the PLP-α-syn mouse model. CLR01 treatment inhibited SNc neuronal loss, assessed by the number of tyrosine-hydroxylase (TH)-positive neurons, dose-dependently and the inhibition correlated with a decrease in the abundance of HMW α-syn oligomers and an increase in the abundance of monomers measured using native-PAGE/western blots. In addition, the treatment reduced prion-like seeding of α-syn measured using biosensor HEK293T cells (Fig. 10) (Holmes et al., 2014). These cells express the familial-PD-causing variant, A53T-α-syn, conjugated to yellow fluorescent protein (YFP) or cyan fluorescent protein (CFP), both in the same cells. Introduction of α-syn oligomers or small fibrils, which act as seeds, to the biosensor cells leads to co-aggregation of the endogenous YFP- and CFP-conjugated α-syn. The aggregation can be visualized by fluorescence microscopy as bright puncta and quantified with high sensitivity using flow cytometry, measuring the fluorescence-resonance energy transfer (FRET) signal between CFP and YFP (Fig. 10A) (Holmes et al., 2014; Yamasaki et al., 2019). When the soluble fraction of brain lysates from PLP-α-syn mice were added to the cells, a significant, dose-dependent reduction in the seeding activity was observed in CLR01- compared with vehicle-treated mice (Fig. 10B), which correlated strongly with the improvement in anxiety-like behavior (Fig. 10C). Thus, the study suggested that CLR01 treatment decreased dose dependently the presence of several neurotoxic forms of α-syn, including HMW oligomers, pS129-α-syn, and seeding-competent α-syn, all of which correlated with the improvement in the behavioral phenotype.
Fig. 10.

CLR01 treatment reduces α-syn seeds in MSA mouse brain. (A) Examples of unseeded (top) and seeded (bottom) biosensor cells. (B) FRET analysis of seed content in the brain extracts’ soluble fraction. The error bars indicate SD. **p < 0.01, ****p < 0.0001 one-way ANOVA with post hoc Tukey test. (C) Linear regression analysis of the correlation between the seeding activity and the center/periphery ratio. This figure was published originally in BBA—Molecular Basis of Disease, 1865, M. Herrera-Vaquero, D. Bouquio, M. Kallab, K. Biggs, G. Nair, J. Ochoa, A. Heras-Garvina, C. Heid, I. Hadrovic, W. Poewe, G. K. Wenning, F.-G. Klärner, T. Schrader, G. Bitan, and N. Stefanova. The molecular tweezer CLR01 reduces aggregated, pathologic, and seeding-competent α-synuclein in experimental multiple system atrophy, 165513, Copyright Elsevier (2019).
Recently, CLR01 was tested in another PD mouse model, called SNCA-OVX, which overexpresses wild-type human α-syn on a mouse snca-null background. This model shows localized, early, and selective deficits in dopamine neurotransmission in the nigrostriatal pathway followed by loss of dopaminergic neurons, reduced SNc dopamine neuron firing rates, and motor impairments in aged animals. This constellation of PD-like phenotypes caused by elevated human α-syn is associated with soluble HMW α-syn species observed beginning at 3 months of age. SNCA-OVX mice do not develop Lewy-body pathology, suggesting that they model early-stage PD (Janezic et al., 2013).
Twelve-month-old SNCA-OVX mice showed rescued motor deficits in their latency to fall in the Rotarod test (Dunham and Miya, 1957) and their speed in CatWalk [a gait-analysis system (Vrinten and Hamers, 2003)] after treatment with 0.14 mg/Kg CLR01 administered subcutaneously twice a week for 2 months, concomitant with a significant reduction in α-syn oligomers measured using AS-PLA in the dopaminergic neurons (Bengoa-Vergniory et al., 2020). Microglia in the SN are preferentially ramified (homeostatic) in WT animals, whereas the microglia of SNCA-OVX mice adopt a more ameboid morphology, suggesting an activated state, which is part of an inflammatory response to the α-syn pathology. CLR01 treatment shifted the microglial morphology back to the ramified appearance in the SN of the mice. Another component of the neuroinflammatory response is activation of the astrocytes, which can be visualized and quantified using the astrocytic marker glial fibrillary acidic protein (GFAP). CLR01 treatment reduced GFAP immunostaining in the brain of the treated mice compared with mice receiving vehicle (Bengoa-Vergniory et al., 2020), demonstrating that CLR01 inhibited not only motor deficits and oligomerization of α-syn in this mouse model but also the neuroinflammation resulting from α-syn-mediated insults.
Because OVX-SNCA mice do not develop Lewy-body-like pathology, CLR01 was tested also in models in which fibrillar α-syn seeds are introduced intracranially into a mouse brain, leading to spread of Lewy-body-like fibrillar deposits in the brain and representing a relatively late stage of PD pathology (Luk et al., 2012). Recombinant mouse α-syn pre-formed fibrils (PFFs) were injected into the striatum of 3-month-old, C57Bl/6, wild-type mice. In this model, small seeds of nonphosphorylated PFFs induce the transformation of the endogenous mouse α-syn into pathologic aggregates. Subcutaneous administration of 0.14 mg/Kg CLR01 twice weekly for 1 month reduced pS129-α-syn aggregates in the SNc and improved both general neuronal viability, assessed by Nissl staining, and dopaminergic-neuron viability measured by immunostaining of dopamine transporter, compared with animals treated similarly with PBS. In a similar experiment, LB extracts from a postmortem PD brain were injected into the striatum of 4-month-old, C57Bl/6 mice, which then were aged for additional 3 months. This model mimics an advanced and severe PD pathology. At 7 months of age, the mice were treated for 2 months with 0.04 mg/Kg per day CLR01 administered subcutaenously via osmotic minipumps. The treatment improved significantly dopaminergic neurodegeneration measured by tyrosine-hydroxylase immunostaining and reduced proteinase-K resistant α-syn aggregates. Importantly, these data demonstrated that CLR01 treatment not only prevents α-syn aggregation in the brain but also can disrupt PFFs in the mouse brain when administered peripherally at a low dose. Taken together, the in vitro, cell culture, and animal studies demonstrate that CLR01 is a highly effective and safe drug candidate for synucleinopathies.
B. Alzheimer’s Disease and Other Tauopathies
Neurodegenerative diseases associated with aberrant post-translational modification and self-assembly of the microtubule-association protein tau into pathologic oligomers and aggregates are known as tauopathies, of which the most common is Alzheimer diseases (AD). Unlike other tauopathies, in which tau is the primary neuropathologic protein, in AD, tau self-assembly and neurotoxicity occur downstream of neurotoxic insults by Aβ. Both proteins are thought to form neurotoxic oligomers first, which later may transform into amyloid fibrils and deposit in the brain. Thus, extracellular amyloid plaques composed primarily of Aβ and intracellular neurofibrillary tangles comprising hyperphosphorylated tau (p-tau) are the main pathologic hallmarks of AD (Arriagada et al., 1992). The monoclonal antibody aducanumab, which has been shown to reduce amyloid plaques in the brain of patients with AD in clinical trials, recently has been approved by the Food and Drug Administration, raising hope that it will be the first disease-modifying therapy for AD. However, contradicting clinical trial results (Knopman et al., 2021) also have raised concerns about the ability of this drug to improve cognitive deficits (Mullard, 2021). There are no current disease-modifying therapies targeting tau, and, unfortunately, two clinical trials testing immunotherapy against the N-terminal region of tau recently have failed (Jabbari and Duff, 2021). In this section, we discuss the inhibitory effect of CLR01 on both Aβ and tau.
1. Amyloid β-Protein
Effective therapy for AD would target the early pathology before devastating neurodegeneration occurs. Formation of toxic Aβ oligomers is thought to be one of the earliest, potentially the earliest, pathologic event in AD (Kirkitadze et al., 2002; Selkoe, 2002, 2008) and therefore preventing the formation of these oligomers and/or facilitating their clearance are expected to be highly effective ways to prevent AD and treat the diseases in its prodromal and early stages. CLR01 has been shown to interact with Aβ40 and Aβ42 rapidly and modulate their self-assembly into formation of nontoxic oligomers that are similar in size or smaller than the toxic oligomers formed in the absence of CLR01 (Sinha et al., 2011). Top-down mass spectrometry and solution-state NMR studies showed that CLR01 binds primarily at Lys16 in Aβ40 and Aβ42 leading to formation of nontoxic assemblies, which, unlike oligomers formed in the absence of CLR01, are not detectable by the oligomer-specific antibody A11 and do not form fibrils (Sinha et al., 2011).
Replica exchange molecular dynamics (REMD) simulations and QM/MM calculations were carried out to further investigate the binding sites and effect of CLR01 on the aggregation of two Aβ42 monomers. The results showed that the MT could encapsulate both Lys residues of the Aβ42 monomers (Mittal et al., 2018), in agreement with ion mobility spectroscopy–mass spectrometry (IM–MS) experiments indicating a 1:2 stoichiometry for Aβ1–42: CLR01 binding (Zheng et al., 2015). Contrarily, only labile interactions were found in the simulations with Arg5. In agreement with experimental data (Sinha et al., 2011), the results indicated that Lys16 was favored for CLR01 binding compared with Lys28, which was related to additional stabilizing interactions between the MT encapsulating Lys16 and the neighbor residue Gln15. Furthermore, the simulations allowed proposing a molecular mechanism for the effect of CLR01 on Aβ42, characterized by an aggregation pathway different than the one acting in the absence of the MT. In the presence of CLR01, aggregation is driven by the encapsulation of Lys residues by the MT and by secondary intermolecular interactions between MT molecules as well as secondary interactions of CLR01 with other residues of Aβ42 (Mittal et al., 2018).
Similar to α-syn (Fig. 9C), incubation of pre-formed fibrils of either Aβ40 or Aβ42 with 10-fold excess CLR01 led to a slow dissociation of the fibrils, suggesting that the compound could be effective in vivo not only for prevention but also for treatment of Aβ self-assembly and pathology. Further mechanistic investigation using ion-mobility spectroscopy-coupled mass spectrometry (IM-MS) showed that CLR01 stabilized Aβ42 monomers, dimers, and tetramers and prevented formation of larger oligomers. Interestingly, the cross-section of CLR01-bound Aβ42 dimers and tetramers was significantly smaller than that of Aβ42 dimers or tetramers formed in the absence of CLR01, suggesting that CLR01 induces compaction of the structure, which inhibits formation of larger oligomers or conversion into amyloid fibrils (Zheng et al., 2015).
CLR01 was found to protect differentiated PC-12 cells, primary rat hippocampal neurons, and mixed primary neuronal/glial cultures against Aβ-induced cell death (Sinha et al., 2011, 2012a). The compound also rescued primary neurons from Aβ42-induced synaptotoxicity (Attar et al., 2012). Incubation of primary rat hippocampal neurons with 3 μM Aβ42 showed substantial depletion of dendritic spines and abundant axonal swelling, in which endosomes and lysosomes are thought to accumulate. Ten-fold excess CLR01, but not the negative-control MT derivative, CLR03, rescued this phenotype to approximately 80% of the level of neuron not exposed to Aβ42. Moreover, CLR01 also rescued primary hippocampal mouse neurons from Aβ42-induced reduction of spontaneous synaptic firing (miniature excitatory postsynaptic currents), induced synaptic firing (excitatory postsynaptic currents), and long-term potentiation, a cellular correlate of learning and memory (Attar et al., 2012).
Metal cations, such as Zn2+, have been reported to be important modulators of Aβ self-assembly (Lovell et al., 1998; Bush, 2002; Tõugu et al., 2009). A high concentration of Zn2+ (about 1 mM) was found in amyloid plaques in the AD brain along with increased levels in patients’ serum (Watt et al., 2010). Zn2+ ions have been found to associate with Aβ and induce the formation of neurotoxic Aβ assemblies depending on the specific conditions, including the concentration of Aβ and Zn2+, the ratio between the concentrations, and the solution conditions (Bush, 2003; Ali et al., 2004; Townsend et al., 2006; Lacor et al., 2007). Therefore, testing the activity of aggregation and toxicity inhibitors not only on Aβ itself but also on Aβ42-Zn2+ complexes has been postulated to be important. Zn2+ has been shown previously to accelerate formation of atypical, β-sheet rich, nonfibrillar aggregates of Aβ40 and Aβ42, and the toxicity of Aβ40-Zn2+ and Aβ42-Zn2+ complexes in cell culture was found to depend on the method of addition (Solomonov et al., 2012; Mason et al., 2020). When the complexes were added all at once, they rapidly formed nontoxic, larger aggregates, yet when the same total amount was divided into four parts, to maintain the concentration low and prevent rapid aggregation, the Aβ-Zn2+ complexes were potently toxic (Solomonov et al., 2012; Mason et al., 2020).
CLR01 has been found to have a relatively high affinity for Zn2+ (Fig. 11), Kd = 5 µM, compared with other metal ions (Wilch et al., 2017), and therefore when applied to Aβ-Zn2+ complexes, it can target both Aβ42 and Zn2+ simultaneously. ThT fluorescence analysis at different CLR01 concentrations added to Aβ42 in the presence of Zn2+ showed a rapid increase in fluorescence in the presence of CLR01, which correlated with formation of nonfibrillar structures similar to those observed when CLR01 was added to other amyloidogenic proteins (Sinha et al., 2011; Gessel et al., 2012; Attar and Bitan, 2014; Herzog et al., 2015; Schrader et al., 2016; Wilch et al., 2017; Hadrovic et al., 2019; Mason et al., 2020). Electron microscopy analysis of Aβ42- Zn2+ in the presence of 10 µM CLR01 showed a mixture of short, thin fibrils and oligomer-like structures, whereas in the presence of 30 µM or 100 µM CLR01, mostly oligomers and rarely fibrillar structures were observed. The effect of CLR01 on Aβ42 toxicity in the presence of Zn2+ then was tested in differentiated PC-12 cells using the MTT assay. These experiments showed that CLR01 inhibited the toxicity of Aβ42-Zn2+ with IC50 = 20 µM, which surprisingly was about 2.5 times lower than the IC50 for CLR01 inhibition of Aβ42 in the absence Zn2+ (Sinha et al., 2011), suggesting that the ability of CLR01 to bind to Zn2+ may act cooperatively with binding to Lys to inhibit Aβ42-mediated toxicity (Mason et al., 2020).
Fig. 11.

Inclusion complex of CLR01 and Zn2+. P atoms are indicated in tan, O in red, C in cyan, and H in white. The structure of the complex was calculated with the DFTB3-D3H5 method (Řezáč, 2017) implemented in the DFTB+ program (Hourahine et al., 2020) in implicit solvent (Onufriev and Case, 2019). The optimized structure resembles the experimentally determined single-crystal structure of a Cs+ atom with a MT containing one OH group and one O-CH2-CO2- group at the central hydroquinone ring (Klärner et al., 2000). π-cation interactions between four of the five aromatic MT rings and the metal cation, as well as the ionic interactions between Zn2+ and both OP(OH)O2- side chains, lead to the binding of the metal cation inside the MT cavity.
After establishing the inhibitory effect of CLR01 on Aβ self-assembly in vitro and Aβ-induced toxicity in cell-culture models, the compound was tested in a triple-transgenic mouse model of AD, which overexpresses mutant forms of the human genes encoding presenilin 1 (PS1) containing the M146V substitution; amyloid β-protein precursor (APP) containing the double substitution KM670/671NL, each of which causes early-onset familial AD; and tau containing the substitution P301L, which causes familial frontotemporal dementia. This model exhibits both amyloid plaques and neurofibrillary tangles (Oddo et al., 2003). Animals were administered 0.04 mg/Kg per day CLR01 or vehicle (sterile saline), subcutaneously using osmotic minipumps for 28 days. Mice treated with CLR01 showed a significant, 33% decrease in plaque burden in their brain. A similar reduction in p-tau was observed in CLR01-treated mice. Moreover, the CLR01-treated mice showed a 46% reduction in the number of microglia in the hippocampus compared with vehicle-treated triple-transgenic mice, suggesting reduced neuroinflammation in the mice receiving CLR01 (Attar et al., 2012). Importantly, all of these beneficial effects were observed in 14- to 15-month-old mice, well after the onset of plaque and tangle pathology, demonstrating that despite the low dose and short duration of the study, the treatment not only halted but also reversed the buildup of the toxic protein aggregates in the brain.
In another small study, CLR01 was tested in a transgenic rat model of AD expressing familial AD-linked mutated forms of human APP (K670N/M671L/V717I) and PS1 (M146V). The animals were treated with 0.1 or 0.3 mg/Kg per day, subcutaneously using osmotic minipumps for 28 days at the age of 9 months, in which this model has moderate plaque pathology. The treatment resulted in 45% and 52% reduction in the plaque pathology, respectively, when compared with the vehicle-treated animals (Malik et al., 2018), further supporting the therapeutic potential of CLR01.
2. Tau
It is estimated that >30 million people currently are suffering from tauopathies, including AD, frontotemporal dementia (FTD), progressive supranuclear palsy (PSP), corticobasal degeneration (CBD), and chronic traumatic encephalopathy (CTE) (Spires-Jones et al., 2017; Chang et al., 2018). Thus, modulating tau accumulation, post-translational modification, and aggregation, and inhibiting tau toxicity are central strategies in developing disease-modifying therapy for tauopathies. Unlike most amyloidogenic proteins, tau does not aggregate spontaneously and aberrant phosphorylation, as well as other post-translational modifications, are key factors inducing its self-assembly. In vitro aggregation of tau typically is triggered by addition of polyanions, such as heparin or arachidonic acid (Goedert et al., 1996; Wilson and Binder, 1997).
The first demonstration that CLR01 could inhibit tau aggregation was in experiments in which tau was induced to aggregate by arachidonic acid. ThT-fluorescence measurements showed that CLR01 inhibited tau β-sheet formation completely at a 1:1 tau:CLR01 concentration ratio (Sinha et al., 2011). The strong inhibition was rationalized by the high number of binding sites for CLR01 on tau. Similar to α-syn, whose aggregation also was inhibited by 1 molar equivalent of CLR01, approximately 10% of the amino acid residues in tau are Lys.
To mimic more closely the pathologic tau found in patients’ brain, which contains multiple post-translational modifications including hyperphosphorylation, a later study used in vitro phosphorylated tau, which was produced by incubation either with activated extracellular signal-regulated kinase 2 (ERK2), p-tauERK, or with a whole rat brain extract (RBE), p-tau(S262A)RBE. The S262A substitution in the latter form was included to promote aggregation (Despres et al., 2019). These in vitro phosphorylated forms of tau were compared with unphosphorylated tau induced to aggregate by heparin. The study characterized the oligomerization and aggregation properties of these forms and the effect of CLR01 on their aggregation and seeding. p-TauERK did not form fibrils and therefore was used only in oligomerization but not in fibrillation experiments. Seeding was studied using a HEK293 biosensor cell line (Holmes et al., 2014), similar to the one described previously for α-syn. Both cell lines were created by the Diamond group and are similar in their design and function. Unlike the α-syn biosensor cells, which express a variant of full-length α-syn, in the tau-biosensor cells, it is not the full-length protein but rather the aggregation-prone repeat-domain of tau, containing the FTD-associated substitution P301S, which is conjugated to CFP or YFP. Addition of tau seeds, but not α-syn seeds, to the tau-biosensor cells leads to intracellular tau aggregation that can be observed as bright puncta by a fluorescence microscope and quantified with high sensitivity using FRET-based flow cytometry (Holmes et al., 2014; Maina et al., 2022).
Because heparin and CLR01 are both negatively charged, they may compete for binding to positively charged residues in tau. To address whether this is the case, SPR was used to assess the competition between the two compounds. As would be expected for CLR01’s labile binding mode, it was found to be a weak competitor for heparin’s more avid binding to tau. Thus, sixfold excess CLR01 was needed for inhibiting heparin-induced tau aggregation whereas aggregation of p-tau(S262A)RBE was inhibited completely by two equivalents of CLR01, suggesting that heparin indeed interfered with the binding of CLR01 to tau. Using the biosensor cells, treatment with CLR01 was found to inhibit intracellular aggregation of tau seeded by fibrils formed either by unphosphorylated tau in the presence of heparin or by p-tau(S262A)RBE. The effect of CLR01 on tau fibrils formed using heparin-induced tau fibrils was atypical, increasing seeding at CLR01 concentrations <0.3 μM and inhibiting the seeding dose dependently at higher concentrations, likely reflecting perturbation of the interaction between CLR01 and tau by heparin. In contrast, inhibition of p-tau(S262A)RBE aggregation followed a typical sigmoidal behavior and was effected by CLR01 with IC50 = 660 ± 140 nM (Despres et al., 2019).
ECD-MS investigation of the binding sites of CLR01 using the longest of the six human isoforms of tau, in which 44 out of 441 residues are Lys and 14 are Arg, found preferential binding to Lys residues located in the repeat domain, which is known to be the main region driving tau self-assembly (Nshanian et al., 2019). These observations were corroborated by a solution-state NMR investigation, which showed CLR01 binding to multiple Lys residues in tau’s repeat domain, primarily Lys321 and Lys331, in the third and fourth repeats, respectively. Tau phosphorylation either by ERK or by a whole rat-brain extract had little impact on CLR01 binding (Despres et al., 2019).
The experiments described above using the triple-transgenic mouse model of AD had shown a robust clearance of p-tau pathology. However, tau-pathology in AD and in this mouse model is believed to be a downstream consequence of Aβ insults (Stancu et al., 2014). Thus, the question whether the observed reduction in p-tau in this model was a direct effect of CLR01 treatment on tau or a secondary effect resulting from inhibition in Aβ toxicity remained unanswered. To address this question, a pure tauopathy mouse model expressing the FTD-linked P301S-tau variant (PS19 line) (Yoshiyama et al., 2007) was treated with 0.3 or 1.0 mg/Kg per day CLR01 or vehicle, administered subcutaneously for 35 days using osmotic minipumps.
A prominent feature of the behavioral variant of FTD (Lindau et al., 2000), which often presents also in patients with AD (Moretti et al., 2001), is disinhibition. Supporting the relevance of the PS19 mouse model to studying human tauopathy, an early-onset, progressive, disinhibition-like behavior has been reported in these mice (Przybyla et al., 2016). CLR01 protected the P301S-tau mice from the disinhibition-like behavior, which was assessed by the number of freezing episodes in the open-field test, normalizing the number of these episodes to the level of wild-type mice. The maximal effect was observed already in the low-dose group (Di et al., 2021). The treatment also prevented deterioration in muscle strength, a phenotype starting at 6 to 7-months of age in these mice. Muscle strength was measured using the grip-strength test, in which the animals are placed hanging upside down on a wire mesh and the latency to fall is measured. Again, both treatment groups showed complete rescue of this phenotype compared with vehicle-treated P301S-tau mice (Di et al., 2021). The improvement in these behavioral phenotypes correlated with reduced toxic p-tau aggregates in the hippocampus, measured by immunostaining with monoclonal antibody AT8, which binds specifically to tau phosphorylated at Ser202 and Thr205 (Mercken et al., 1992). In the vehicle-treated group, 11.2 ± 6.7% of the hippocampus was AT8-positive. CLR01 treatment reduced the hippocampal AT8-positive area to 5.1 ± 3.7% (p = 0.0156 compared with vehicle) in the 0.3 mg/Kg per day group and 5.0 ± 3.8% (P = 0.0127) in the 1.0 mg/Kg per day group. Similarly, reduction of aggregated intracellular tau measured using Gallyas silver-staining (Gallyas, 1971) (Fig. 12A–C) was maximal already at 0.3 mg/Kg per day CLR01 (Fig. 12D). The number of cells containing tau aggregates in the CA3 region of the hippocampus was reduced from 477± 244 cells per mm2 in the vehicle-treated transgenic mice to 126 ± 62 (P = 0.0001) and 171 ± 68 (P = 0.0005) cells per mm2. CLR01 treatment also was found to reduce tau-oligomers measured using dot blots in the soluble fraction of brain extract using monoclonal antibody tau oligomeric complex-1 (TOC1) (Ward et al., 2013), and neuroinflammation assessed as the area covered by microglia in the hippocampal CA3 region (Di et al., 2021).
Fig. 12.

CLR01 treatment reduces tau aggregation in the CA3 region of P301S-tau mice. Brain sections from P301S-tau mice were stained with Gallyas silver-stain. (A–C) Representative images of the hippocampus (top) and zooming on the CA3 region (bottom) are presented for the mice treated with 0 (A), 0.3 (B), or 1.0 (C) mg/Kg CLR01 and show black/dark brown cells containing aggregated tau. (D) The data were quantified as the number of tau-aggregate-containing cells per mm2 in the CA3. The data are presented as mean ± SD. P values were calculated using a one-way ANOVA with post hoc Tukey test. This figure was published originally in BMC—Alzheimer’s Research and Therapy, 2021, J. Di, I. Siddique, Z. Li, G. Malki, S. Hornung, S. Dutta, I. Hurst, E. Eshaaya, A. Wang, S. Tu, A. Boghos, I. Ericsson, F.-G. Klärner, T. Schrader, and G. Bitan. The molecular tweezer CLR01 improves behavioral deficits and reduces tau pathology in P301S- tau transgenic mice, Creative Commons Attribution 4.0 International License.
The seeding capability of brain lysates from vehicle- and CLR01-treated P301S-tau mice also was tested using the tau-biosensor cell line described earlier. The integrated FRET density was 133 ± 114 in the vehicle-treated mice and was reduced to 59 ± 90 (p = 0.053) and 45 ± 48 (p = 0.016) in the low- and high-dose treatment groups, respectively, in agreement with the cell-culture findings showing that CLR01 inhibits seeded intracellular tau aggregation, as described previously. The inhibition of seeded aggregation in the brain of the transgenic mice correlated well (r = –0.314, P = 0.028) with the improvement in grip strength (Di et al., 2021). Taken together, the data demonstrated clearly that CLR01 inhibited tau self-assembly, facilitated p-tau clearance, and reduced tau pathology in the brain of the mice by its direct action on tau in vivo, rather than as a secondary effect through inhibition of Aβ toxicity. This demonstrated ability of CLR01 to ameliorate simultaneously, yet independently, both Aβ and tau pathology in the brain makes it a particularly attractive candidate for treatment of AD, where clinical trials of experimental therapies addressing only one of the offending proteins have repeatedly failed to produce a meaningful clinical improvement despite clear evidence of target engagement.
C. Amyotrophic Lateral Sclerosis
ALS is a rare neuromuscular disorder characterized by progressive degeneration of motor neurons in the spinal cord, brain stem, and motor cortex (Arora and Khan, 2021). Most ALS cases are sporadic, whereas 5% to 10% are familial (Chiò et al., 2013). The first gene linked to familial ALS was superoxide dismutase 1 (SOD1) (Deng et al., 1993; Rosen et al., 1993), in which over 160 mutations cause 15% to 20% of the familial ALS cases (Sreedharan and Brown, 2013). The resulting amino acid substitutions in the protein lead to SOD1 destabilization, misfolding, and accumulation of neurotoxic oligomers and aggregates (Sreedharan and Brown, 2013). To date, only two drugs have been approved for ALS treatment, riluzole (Martin et al., 1993) and edaravone (Sawada, 2017), which increase survival by 2 to 3 months but do not address the cause of the disease. Inhibition of SOD1 self-assembly can lead to development of promising therapy for familial ALS cases involving mutations in the SOD1 gene. Moreover, aggregation of other proteins, such as transactive response DNA binding protein 43 kDa (TDP-43), fused in sarcoma (FUS), ubiquilin 2, and repeat-associated non-AUG (RAN) translation products of C9ORF72 have been reported to cause, or contribute to, the pathology of both familial and sporadic cases of ALS. Therefore, aggregation modulators, especially broad-spectrum inhibitors such as the MTs, could be useful in these cases and provide wide-range therapy for ALS. However, to date, only a few small-molecule aggregation inhibitors have been tested in the context of ALS, primarily against SOD1 aggregation (Xia et al., 2011; Trippier et al., 2014; Brown et al., 2020), and secondarily against TDP-43 (Martínez-González et al., 2020).
To begin assessing the potential utility of MTs for ALS, the effect of CLR01 on the aggregation of the wild-type and disease-associated variants of SOD1, including E21K, H46R, D76Y, and G93A (Deng et al., 1993; Rosen et al., 1993; Wong et al., 1995), was evaluated using the ThT assay. CLR01 was found to inhibit the aggregation of all of these variants in a dose-dependent manner, yet with different kinetics (Malik et al., 2019). A complete or near-complete inhibition was observed at fivefold excess CLR01 in all cases.
CLR01 also was tested for its ability inhibit the aggregation of TDP-43. Although ThT arguably is the most common assay for evaluation of amyloidogenic protein aggregation, some amyloid fibrils resist ThT binding, precluding the use of this assay for measurement of their aggregation kinetics. Sinha et al. reported previously that this was the case for transthyretin and the peptide PrP(106–126) and in both cases used turbidity measurements instead to assess CLR01’s ability to inhibit the aggregation of these proteins (Sinha et al., 2011). Similarly, in the case of TDP-43, ThT did not show a change in fluorescence during the aggregation process and therefore we evaluated the aggregation process of the protein using a turbidity assay. In addition, we analyzed the morphology of the protein at the end of the aggregation reaction using transmission electron microscopy (TEM). The aggregation of TDP-43 is a very fast process, making it difficult to maintain the protein in an unaggregated state in vitro and initiate the aggregation reaction reproducibly. To overcome this difficulty, TPD-43 was expressed and purified as a conjugate with small ubiquitin-like modifier (SUMO), keeping it in an unaggregated form. The aggregation then was initiated by addition of ubiquitin-like protease 1 (Ubl 1), which cleaves the SUMO tag and releases the highly aggregation-prone TDP-43.
The reaction mixtures of 3 μM TDP-43 in the absence or presence of 1, 5, or 10 equivalents CLR01 were shaken at 1400 rpm and absorbance was measured at λ = 395 nm to evaluate the turbidity of the solution. In the absence of CLR01, an increase in absorbance was observed up to about 10 h and then reached a plateau (Fig. 13A). CLR01 inhibited the aggregation of TDP-43 dose dependently, reaching near-complete inhibition at a 1:10 TDP-43:CLR01 concentration ratio. Morphologic analysis at the end of the reactions showed that TDP-43 formed abundant amorphous aggregates, which were inhibited dose dependently by CLR01 (Fig. 13B).
Fig. 13.

CLR01 inhibits TDP-43 aggregation. (A) Three μM SUMO-conjugated TDP-43 were incubated in the absence or presence of 1, 5, or 10 equivalents of CLR01. The aggregation reaction was initiated by cleavage of the SUMO tag and monitored using turbidity. (B) Electron micrographs taken at the end of the aggregation reactions. Scale bars denote 100 nm.
C9ORF72-derived RAN translation of the aberrant repeat-expansion domain forms five RAN polypeptides—poly-(PR), poly-(PA), poly-(GP), poly-(GA), and poly-(GR), of which the Arg-containing peptides, poly-(GR) and poly-(PR) were reported to be the most neurotoxic (Gupta et al., 2017; Shi et al., 2017). To test whether CLR01 could protect cells from the toxicity of these two RAN polypeptides, PC12 cells were treated with 20 μM each of synthetic (GR)20 or (PR)20 conjugated to a human influenza hemagglutinin tag for 48 h in the absence or presence of CLR01 and the viability of the cells then was measured using the MTT assay. Both peptides decreased the cell viability to 40-50% and their toxic activity was inhibited dose dependently by CLR01 (Fig. 14A). To further examine the protective activity of CLR01 in a system that is closer to the actual disease, the effect of CLR01 was tested on the viability of induced pluripotent stem-cell (iPSC)-derived motor neurons from a patient with ALS carrying an approximately 6kb C9ORF72 expansion. The cells were labeled with an Isl1-tomato knock-in reporter to identify the motor neurons and were followed by longitudinal tracking (daily robotic imaging) to measure their survival. A significant increase in cell viability (Kaplan-Meier analysis) in the presence of 10 μM CLR01 showed that the compound could protect human motor neurons from the toxic effects of Arg-containing RAN peptides (Fig. 14B).
Fig. 14.

CLR01 inhibits the toxicity of C9ORF72 dipeptide repeats. (A) Twenty μM of synthetic peptides (GR)20 or (PR)20 were incubated with differentiated PC12 cells for 48 hours in the absence or presence of 5- or 10-fold excess CLR01 and cell viability was measured using the MTT assay. P values were calculated using a one-way ANOVA. (B) iPSC-derived motor neurons from a patient with C9ORF72 repeat expansion were incubated in the absence or presence of 10 μM CLR01 and cell viability was measured using longitudinal tracking (daily robotic imaging).
To test the effect of CLR01 on SOD1 aggregation and toxicity in vivo, the compound was administered by a daily subcutaneous injection to G93A-SOD1 mice (Malik et al., 2019). This model overexpresses relatively high levels of G93A-SOD1 and develops neurodegeneration of spinal motor neurons starting at about 60 days of age, followed by rapidly progressing motor deficits leading to paralysis and death at 100 to 120 days of age (Gurney et al., 1994). Mice were treated with 0.5 or 5.0 mg/Kg CLR01 or with vehicle starting at 50 days of age until they met criteria for euthanasia due to advanced weakness or weight loss. IHC of the spinal cord showed little change in total SOD1 in mice treated with CLR01, whereas the misfolded pathologic form of SOD1 staining, detected using antibody 10C12 (Grad et al., 2014; Atlasi et al., 2018), was decreased significantly with a maximal response of about threefold decrease already at 0.5 mg/Kg. Similar results were obtained also by immunostaining with antibody C4F6, which is less specific for pathologic SOD1 (Atlasi et al., 2018).
Surprisingly, despite the marked reduction in the immunostaining for misfolded SOD1, the treatment did not improve survival, motor function (Rotarod test), or muscle strength deterioration [grip-strength test, respiration volume (Brooks and Dunnett, 2009)]. This is the only case to date in which the reduction of the toxic form of an offending amyloidogenic protein did not correlate with improvement in disease phenotype in an animal model, raising questions regarding the causative role of the misfolded SOD1 detected by antibody 10C12 in this model. A potential explanation for this unexpected result is the high copy number of the SOD1 transgene in this particular mouse model, which reduces the concentration of the available co-factors Zn2+ and Cu2+ in the brain. As mentioned, when the binding of CLR01 to a series of common metal ions was examined, the only one that showed high affinity (low μM range) was Zn2+ (Wilch et al., 2017). Thus, the lack of therapeutic effect by CLR01 in the G93A-SOD1 mouse model might not have resulted from an insufficient impact of CLR01 on misfolded SOD1 but rather reflected a model-specific depletion of brain Zn2+, which would not be expected in familial ALS.
D. Other CNS Diseases
1. Huntington's Disease
In Huntington’s diseases (HD), misfolding and aggregation is associated with the expansion of a polyglutamine (polyQ) tract in the region of the protein corresponding to exon 1 of the cognate Htt gene (Htte1). The expanded polyQ sequence forms the core of the amyloid fibrils (Sivanandam et al., 2011; Bugg et al., 2012; Hoop et al., 2014; Lin et al., 2017). Because CLR01 binds specifically to Lys residues, we did not expect it to affect the aggregation of Htte1, where no Lys residues are in the fibril core (Hoop et al., 2014; Adegbuyiro et al., 2017). However, the 17-residue N-terminal region (N17) immediately adjacent to the polyQ domain of Htt has been reported to be an important initiator and mediator of the aggregation (Thakur et al., 2009; Jayaraman et al., 2012). This region includes three Lys residues, which have been shown to be important for Htt aggregation (Arndt et al., 2015). Therefore, Vöpel et al. tested whether, through binding to these Lys residues, CLR01 could modulate or inhibit the aggregation of Htte1. CLR01 was found to inhibit the aggregation of htte1 not only in test-tube assays but also when the protein was induced to aggregate intracellularly in HeLa cells expressing Htte1 containing a 72Q expansion. REMD simulations and ECD-MS measurements showed that the inhibition was achieved through binding of CLR01 to Lys residues in the N17 domain and remodeling the secondary structure of this region (Vöpel et al., 2017). Lys9 in the N17 sequence was identified using QM/MM calculations and ECD-MS measurements as the primary binding site of CLR01 in Htte1.
2. Mucopolysaccharidosis Type IIIA
Lysosomal storage diseases (LSDs) consist of over 70 diseases characterized by lysosomal dysfunction due to mutations in genes encoding lysosomal and certain non-lysosomal enzymes. Most LSDs are inherited in an autosomal recessive manner (Filocamo and Morrone, 2011). LSDs typically occur in infants and children and often are related to neurodegeneration due to the failure of the autophagy-lysosome pathway (ALP). Over the last several years, options for the treatment available for LSDs have increased, including bone marrow transplantation, enzyme replacement therapy, substrate reduction, chaperones-based therapies, and gene therapies. Despite these encouraging advances, the availability of therapy for many LSDs is limited or nonexistent (Beck, 2018).
Mucopolysaccharidosis type III (MPS III, Sanfilippo syndrome) is a type of LSD, caused by a deficiency in one of the four enzymes involved in the catabolism of the glycosaminoglycan heparan sulfate. MPS III is characterized by a progressive cognitive decline and severe hyperactivity. It is the one of the most common types of MPS, with an estimated prevalence between 0.3 and 4.1 cases for every 100,000 live births, depending on the subtype—A, B, C, or D (Andrade et al., 2015).
A mouse model of the more common subtype, MPS IIIA, shows in addition to the primary lysosomal storage of undegraded heparan sulfate, progressive accumulation of multiple amyloid protein aggregates as a secondary storage in the CNS neurons, including α-syn, Aβ, tau, and prion protein (Sambri et al., 2017; Monaco and Fraldi, 2020). Concomitantly, the autophagy-lysosome system becomes defective resulting in the accumulation of enlarged autolysosomes at about 6 months of age. The mice develop severe and progressive neurologic defects reminiscent of the human disease, including synaptic dysfunction, cognitive and memory deficits, and neuroinflammation (Bhaumik et al., 1999; Sorrentino et al., 2019).
Because multiple amyloidogenic proteins were found to accumulate in the lysosomes of MPS IIIA mice, CLR01 was tested for its ability to reduce the amyloid burden and potentially improve the disease phenotype in these mice. Although such treatment does not fix the defective enzyme, targeting the secondary storage of amyloid is a novel therapeutic approach for MPS III that could substantially delay symptom onset, ameliorate symptoms, and expand the time window for approaches, such as enzyme-replacement therapy and gene therapy.
One mg/Kg CLR01 was administered to the mice by a daily subcutenous injection between 4.5 and 9 months of age. The treatment was found to fully rescue the mice from the memory deficits measured in the fear-conditioning test compared with vehicle-treated mice (Monaco et al., 2020). To visualize the amyloid pathology in the brain, brain sections were stained with thioflavin-S (ThS) a common dye for histologic staining of amyloid (Sigurdsson, 2005). CLR01 treatment resulted in a marked reduction in ThS staining in brain sections of CLR01-treated mice compared with those from vehicle-treated mice. In agreement with the decrease in accumulation of amyloid proteins, CLR01 treatment reduced the lysosome diameter in the neurons of the treated mice from an abnormal size of >800 nm to a normal value of approximately 400 nm.
The microtubule-associated protein 1A/1B-light chain 3 (LC3) has two forms: a cytoplasmic form, LC3-I, which is converted to a membrane-associated form LC3-II found in autophagosomes. Punctate appearance of LC3-II in immunofluorescence analysis is a common method for labeling autophagosomes (Tanida et al., 2008). Immunofluorescence analysis of the MPS IIIA mice showed a striking increase in the overall number of LC3-positive puncta compared with wild-type mice, representing the halt in the autophagic flux in MPS-IIIA mouse brain. This phenotype was decreased dramatically after CLR01 treatment, suggesting that the treatment relieved the blockage of the autophagic flux, resulting in clearance of the protein aggregates. CLR01 treatment also reduced significantly neuroinflammation in the mouse brains, measured by immunostaining of microglial and astrocytic markers, and protected the neurons against loss of synaptic vesicles, which was assessed by TEM (Monaco et al., 2020).
E. Non-CNS Amyloidoses
Systemic (non-CNS) amyloidoses are diseases associated with abnormal protein misfolding and formation of toxic amyloids in various organs outside the CNS, including heart, liver, kidney, skin, eyes, lungs, and others (Cohen, 1967). Some amyloidoses affect multiple organs, e.g., light-chain (AL) amyloidosis and systemic (reactive) AA amyloidosis, whereas others are organ-specific including cardiac amyloidosis, type 2 diabetes in which amyloid accumulates in the pancreas, and conjunctival amyloidosis (Desai et al., 2010).
1. Transthyretin Amyloidosis
Several diseases including, senile systemic amyloidosis, familial amyloid polyneuropathy, and familial amyloid cardiomyopathy, are associated with misfolding and aggregation of transthyretin (TTR) (Westermark et al., 2003; Ruberg and Berk, 2012; Çakar et al., 2019). TTR is a homotetramer protein that serves as a transporter of thyroxin and retinol in the serum and cerebrospinal fluid (Blake, 1978; Larsson et al., 1985). More than 100-point mutations in the TTR gene lead to misfolding of the protein and its abnormal self-assembly (Saraiva, 1995; 2001). In these cases, TTR tetramers dissociate into monomers that misfold, form toxic oligomers, and accumulate as amyloid fibrils (Ruberg and Berk, 2012). The TTR amyloid deposits extracellularly in various organs including the heart, kidneys, gastrointestinal system, peripheral nervous system, and vascular system. Thus, we tested if CLR01 could modulate TTR aggregation and toxicity, starting with simple test-tube and cell-culture experiments (Cardoso et al., 2007; Ferreira et al., 2014).
Initial demonstration of the inhibitory effect of CLR01 on wild-type TTR aggregation was achieved using a turbidity assay (Sinha et al., 2011). CLR01 also prevented the toxicity of wild-type TTR oligomers added to differentiated PC-12 cells and inhibited the aggregation of mutant TTR in a rat Schwannoma cell line, RN22, transfected with TTR-L55P, the least stable and the most pathogenic variant, which leads to the earliest onset of disease in mutation carriers (McCutchen et al., 1995; Lashuel et al., 1998; Cardoso et al., 2007). Immunodetection of TTR in an immunofiltration assay showed that cells incubated in the presence of CLR01 displayed significantly fewer aggregates compared with those incubated in the absence of the MT (Ferreira et al., 2014). In addition, CLR01 protected RN22 cells against TTR-Y78F-mediated toxicity. The TTR-Y78F variant was designed to destabilize the native structure and stabilize an intermediate structure in the fibrillogenesis pathway, facilitating the demonstration of TTR misfolding, self-assembly, and toxicity (Redondo et al., 2000; Terazaki et al., 2006). Pretreatment of TTR-Y78F with CLR01 rescued RN22 cells from the cytotoxicity of the protein’s oligomers in a dose-dependent manner (Ferreira et al., 2014).
Following up on these experiments, the therapeutic potential of CLR01 was tested in an animal model of TTR amyloidosis. A transgenic mouse model (hTTR V30M/HSF) expressing the amyloidogenic human TTR V30M variant on a mouse TTR-null background and heterozygous for the heat shock transcription factor 1 (HSF1) was used in these experiments. These genetic manipulations lead to an extensive, early accumulation of non-fibrillar TTR starting at the age of 1 month, evolving later into fibrillar deposits in distinct organs including the peripheral nervous system and gastrointestinal tract (Santos et al., 2010). Animals were treated for 5 weeks at the age of 4 months with 1.2 mg/Kg per day CLR01 administered subcutaneously using osmotic minipumps. TTR deposition measured by IHC was found to be significantly reduced in the treated mice compared with mice receiving vehicle in all the target organs, including 33% reduction in the stomach, 51% in the colon, and 47% in the dorsal root ganglion (Ferreira et al., 2014).
Extracellular deposition of TTR on the cell membrane causes permeabilization of the membrane and consequently perturbation of intracellular calcium homeostasis. This mechanism has been proposed as a common primary pathogenic process responsible for initiating several pathogenic signaling pathways. Assessing these pathways by IHC, CLR01 treatment was found to reduce tissue injury indicated by decreased staining for the endoplasmic-reticulum-stress marker BiP (34% in the stomach and 67% in both the colon and DRG), the apoptosis marker Fas/CD95 death receptor (39% in the stomach, 56% in the colon, and 68% in the DRG), and the protein nitrosylation marker 3-nitrotyrosine (78% in the stomach, 76% in the colon, and 68% in the DRG).
2. Desmin-Related Cardiomyopathy
Mutations in desmin or its chaperone αB-crystallin, a small heat-shock-like protein, can cause a disease called desmin-related myopathy (DRM), characterized by formation of toxic oligomers and insoluble, intracellular proteinaceous deposits of the mutant protein and its interaction partners, ultimately resulting in muscle weakness and dilated cardiomyopathy (Pattison and Robbins, 2008). There is no effective treatment available for this disease, though some complications can be prevented by implanting a pacemaker or by a heart transplantation. CLR01 was tested both in cell culture and in a mouse model of DRM. In primary, neonatal, rat ventricular myocytes expressing mutant αB-crystalline leading to a R120G substitution (CryABR120G) CLR01 treatment decreased CryABR120G-induced cytotoxicity in a dose-dependent manner (Xu et al., 2017). In a transgenic mouse model in which CryABR120G overexpression under the α-myosin heavy-chain promoter is restricted to cardiomyocytes, CLR01 was administered by a daily subcutaneous injection at 1, 3, or 6 mg/Kg for 5 weeks beginning at 4 weeks of age and then the mice were sacrificed for IHC analysis of the heart. Similar to the cell-culture results, CryABR120G‐positive aggregates were found to decrease in a dose‐dependent manner and were significantly lower by about threefold at the highest dose compared with vehicle-treated mice. In addition, sensitive mRNA markers of hypertrophy and stress, atrial- and brain-specific natriuretic peptides, respectively, were found to be highly increased in CryABR120G animals relative to their wild-type counterparts and were reduced significantly by the CLR01 treatment (Xu et al., 2017).
3. Type 2 Diabetes
Abnormal self-association of islet amyloid polypeptide (IAPP) into toxic oligomers and aggregates is an important deleterious mechanism thought to kill insulin-producing pancreatic islet β-cells in type 2 diabetes, a disease affecting >400 million people worldwide (Sharma et al., 2021). IAPP is one of the most amyloidogenic proteins known. It has only one Lys residue at position 1 in its 37-amino-acids sequence, and therefore CLR01 was not expected to affect its assembly or toxicity. Surprisingly, using the ThT fluorescence assay, CLR01 was found to inhibit IAPP aggregation at a 1:10 ratio, respectively (Sinha et al., 2011). Further investigation using ThT fluorescence and TEM showed that CLR01 inhibited IAPP aggregation at even lower concentration ratios (Lopes et al., 2015). Circular dichroism spectroscopy showed that CLR01 facilitated the formation of α-helical structures already at [CLR01] ≤ [IAPP] and excess CLR01 stabilized these structures. Solution-state NMR showed that CLR01 predominantly bound to Lys1 and to a lower extent to Arg11. Cell-culture experiments using the MTT-reduction assay showed that CLR01 protected rat insulinoma RIN5fm cells from IAPP-induced toxicity with IC50 = 6 ± 3 μM (Sinha et al., 2011) and inhibited IAPP-induced apoptosis in these cells (Lopes et al., 2015). In agreement with the NMR findings, REMD simulations showed that both Lys1 and Arg11 sidechains contributed to the binding of CLR01 to IAPP (see also the Fig. 6), yet the precise mechanism allowing CLR01 to inhibit IAPP aggregation at low substoichoimetric ratios remains to be elucidated.
4. p53 Aggregation
p53 is a key transcription factor and cell cycle regulator whose proper function prevents cancer development. The functional unit of p53 is a homotetramer of 393-amino-acid polypeptide monomers. The DNA binding domain, p53-DBD, is intrinsically unstable and unfolds rapidly at body temperature (Bullock et al., 2000). Certain mutations in the cognate TP53 gene that lead to destabilizing substitutions in the protein cause conformational changes that promote dissociation of the tetramer, misfolding, and aggregation of p53 (Friedler et al., 2003). G245S and R249S within the p53DBD are two such substitutions, which are among the six most frequent “conformational mutations” in human cancers (Joerger and Fersht, 2007).
CLR01 was tested for its ability to prevent the aggregation of G245S-p53 and R249S-p53 using several biophysical assays. First, the effect of CLR01 on G245S-p53 aggregation was evaluated using ThT fluorescence, CD spectroscopy, and static light scattering and showed that CLR01 induced formation of β-sheet-rich structures at a 1:5 G245S-p53:CLR01 concentration ratio (Herzog et al., 2015). Dot blot probed with polyclonal antibody OC, which is selective for fibrillar amyloid structures, showed increased OC binding with increasing CLR01 concentrations, supporting the formation of β-sheet-rich G245S-p53 structures, although these structures did not have the typical morphology of amyloid fibrils [similar to the β-sheet-rich structures formed by Aβ in the presence of Zn2+ (Solomonov et al., 2012; Mason et al., 2020)]. CLR01 induced similar β-sheet-rich structures in the R249S-p53 variant. However, turbidity measurements showed that fivefold excess CLR01, the same concentration ratio that induced the β-sheet-rich structure in both variants, also inhibited their aggregation. TEM analysis showed that in presence of CLR01, the p53DBD variants formed smaller, disperse aggregates compared with larger aggregates in the absence of the MT (Herzog et al., 2015). Toxicity was assessed using the XTT cell-viability assay in human H1299 lung carcinoma cells and showed that CLR01 protected the cells from G245S-p53DBD- or R249S-p53DBD-induced toxicity at 5:1 and 2:1 concentration ratios, respectively. CLR01 had a hormetic effect on the toxicity of the p53 variants. It protected the cells from the toxicity of G245S-p53DBD or R249S-p53DBD back to the level of untreated cells at fivefold and twofold excess, but its protective effect declined at higher concentration ratios for reasons that currently are not well understood (Herzog et al., 2015).
F. Semen Amyloids
Human seminal fluid naturally contains amyloid fibrils formed by fragments derived from the abundant semen proteins prostatic acid phosphatase (PAP) and semenogelins 1 and 2 (SEM 1 and 2) (Münch et al., 2007; Arnold et al., 2012; Roan et al., 2014; Röcker et al., 2018a). These fibrils play important roles in reproduction and innate immunity as they bind to apoptotic sperm cells and bacterial pathogens, promoting their phagocytotic clearance in the female reproductive tract (Easterhoff et al., 2013; Roan et al., 2017). Semen amyloids were identified in 2007 as agents that promote the infectivity of the human immunodeficiency virus 1 (HIV-1), a mainly sexually transmitted pathogen (Münch et al., 2007). The mechanism underlying infectivity enhancement is a direct interaction of the positively charged fibrils with the negatively charged viral and cellular membranes, which results in orders-of-magnitude increased rates of viral attachment and infection (Roan et al., 2009). Further studies showed that semen itself enhances HIV-1 infection and semen amyloids are the responsible factor (Roan et al., 2014). Thus, antagonizing the HIV-1-promoting activity of semen could reduce rates of sexual viral transmission, and several compounds interfering with formation and function of semen amyloids have been described (reviewed in Röcker et al., 2018a). Since amyloidogenic peptides in semen are rich in Lys and Arg residues, CLR01 was hypothesized to modulate their HIV-1 enhancing activity (Lump et al., 2015). Indeed, CLR01 (but not CLR03, which lacks the hydrophobic side walls) not only inhibited the formation of PAP and SEM fibrils but also remodeled preformed semen amyloids, thereby abrogating their ability to interact with virions and to promote infection (Lump et al., 2015). Unexpectedly, CLR01 also exhibited a direct antiviral effect by disrupting the membrane of HIV-1 and other enveloped viruses (see below). This combined anti-amyloid and anti-HIV-1 activity renders CLR01 an interesting microbicide candidate for preventing or reducing sexual HIV-1 transmission (Lump et al., 2015).
G. Antiviral Activity of MTs
The finding that CLR01 had direct anti-HIV-1 activity was surprising and stimulated further research. It was shown that CLR01, but not CLR03, inhibited HIV-1 infection independently of the viral coreceptor tropism or HIV-1 strain, with IC50 values in the µM range, by directly targeting the virion but not the cell (Lump et al., 2015). Atomic force microscopy showed that CLR01 caused a physical disruption of the retroviral particles confirming a direct antiviral effect (Lump et al., 2015). Further biophysical experiments using giant unilamellar vesicles (GUVs) demonstrated that CLR01 preferentially destroys lipid raft-enriched membranes, which are typical for viral envelopes derived from the plasma membrane (Lump et al., 2015). Thus, CLR01’s ability to exert broad antiviral activity against other enveloped and nonenveloped viruses was investigated. In agreement with the mechanism proposed here, CLR01 was found to suppress infection by all analyzed enveloped viruses, including members of the herpesviruses (Herpes Simplex Virus 1 and 2, Cytomegalovirus), flaviviruses (Zika Virus), filoviruses (Ebola Virus), hepaciviruses (Hepatitis C Virus), paramyxoviruses (Measles Virus), orthomyxoviruses (Influenza A Virus), pneumoviruses (Respiratory Syncytial Virus), and coronaviruses (SARS-CoV-2) (Lump et al., 2015; Röcker et al., 2018b; Weil et al., 2020; Brenner et al., 2021). In contrast, CLR01 did not abrogate infectivity of nonenveloped adenoviruses or the encephalomyocarditis virus, further confirming that the compound targets the viral envelope (Lump et al., 2015; Röcker et al., 2018b; Weil et al., 2020; Brenner et al., 2021). These and the mechanistic data discussed later show that CLR01 attacks and destabilizes the viral membrane, ultimately leading to a loss of viral infectivity. The observed IC50 values against the different viruses ranged between the low to median µM range. These different activities could be explained by differences in the composition of the viral lipid membrane, the accessibility of the membrane due to tightly packed glycoproteins, or the effects of membrane curvature and tension of virus particles of different sizes. Another explanation that might affect the MTs antiviral activity is the total number of infectious particles and/or the presence of enveloped but noninfectious particles in a given viral stock solution.
In studies aimed to clarify the mechanism underlying membrane disruption by MTs, the antiviral effects of CLR05 and the molecular clip PC were investigated, together with CLR01 (Fig. 1) (Weil et al., 2020). PC features planar naphthalene side walls, which result in a different cavity with respect to that of CLR01. As discussed later, only CLR01 and CLR05, but not PC, formed closed inclusion complexes with lipid head groups of viral membranes, thereby altering lipid orientation and increasing surface tension. Consequently, CLR01 and CLR05, but not CLR03 or PC, showed broad antiviral activity against enveloped viruses, suggesting that introducing aliphatic ester arms into each phosphate group of CLR01 might act as lipid anchors that promote membrane targeting (Weil et al., 2020). Indeed, the best antiviral MT candidate was found to be the one containing n-pentylphosphate groups as side arms (Scheme 6). This derivative was approximately one order of magnitude more effective against HIV-1 than CLR01 (Weil et al., 2020). These findings open the path for structural optimization toward generation of even more potent MTs that represent promising leads for a novel class of broad-spectrum antivirals. Advanced MTs might be particularly useful for prevention and treatment of viruses where currently no effective therapy exists, as was the case for the first 1.5 years of the SARS-CoV-2 pandemic, or other newly emerging enveloped human pathogens.
More recently, it also became clear that CLR01 not only inhibits cell-free virus infection but also cell-to-cell viral spread (Brenner et al., 2021). Many enveloped viruses, including several herpesviruses, have evolved a cell-to-cell mode of spread involving direct cell–cell contacts, which most likely also accounts for viral dissemination in vivo (Sattentau, 2008). Clinical isolates of the human cytomegalovirus (HCMV) grow mainly in a cell-associated manner without releasing infectious progeny virus into the extracellular environment (Jackson and Sparer, 2018). Interestingly, this direct cell-to-cell transmission of HCMV could be blocked by CLR01 but not by antibodies that neutralize cell-free HCMV infection (Brenner et al., 2021). Similar results were obtained for HSV-1 and HSV-2, suggesting that MTs may be particularly useful in the prevention or treatment of herpesviral infections. However, the exact mechanism by which CLR01 restricts herpesvirus cell-to-cell spread remains to be determined, and it would be interesting to investigate whether CLR01 and newer MTs also block cell-associated spread of other viruses.
H. Bacterial Biofilm
Bacterial infections often involve surface-associated bacterial communities embedded in a polymeric matrix called biofilm (Flemming et al., 2016). The composition of the biofilm matrix varies among bacterial strains and includes polysaccharides, extracellular DNA, and proteins constructing solid layers supporting bacterial cell growth and development (Limoli et al., 2015; Schwartz et al., 2016). Biofilm proteins often are self-assembled into functional amyloids, which provide high stability to the structure and resemble amyloids involved in proteinopathies (Blanco et al., 2012; Zaman and Andreasen, 2020). The biofilm functional amyloids contribute to bacterial persistence in the host cells and promote their resistance to antimicrobial drugs. Thus, inhibiting biofilm formation or disassembling biofilm by targeting the functional amyloids involved in these structures is a promising strategy for tackling bacterial infection and drug resistance.
Among gramicidin-positive bacteria, Staphylococcus aureus (S. aureus) infections are one of the five most common causes of hospitalizations in the United States and have an estimated >25% mortality rate (Bridier et al., 2011; Schlecht et al., 2015). S. aureus bacteria form a rigid, impenetrable biofilm containing functional amyloid proteins named phenol-soluble modulins (PSMs) that contribute to the pathogenicity and drug resistance of these bacteria (Cheung et al., 2014). In a recent study (Malishev et al., 2021), CLR01, CLR05, and CLR03 as a negative control were tested for their impact on S. aureus biofilm formation.
Both CLR01 and CLR05 inhibited S. aureus growth at concentrations >50 µM with IC50 values of about 130 µM and 90 µM respectively, whereas CLR03 was inactive, as expected. Therefore, in subsequent experiments, the anti-biofilm activity of these compounds was studied at concentrations ≤50 μM, which allowed the bacteria to survive. Confocal fluorescence microscopy images recorded after 24-h incubation of S. aureus with 50 µM of each compound at 37°C showed pronounced disruption of the biofilm assembly (Fig. 15A) (Malishev et al., 2021). The ability of the MTs to inhibit biofilm formation also was evaluated using scanning electron microscopy in 10% mouse serum to test whether inhibition could still be observed in the presence of biomolecules, including competition by Lys and Arg residues in the environment. This analysis revealed a significant reduction in S. aureus biofilm mass in the presence of both MTs, though, interestingly, under these conditions CLR01 was more effective than CLR05.
Fig. 15.

CLR01 and CLR05 disrupt S. aureus biofilm in distinct manners. (A) 50 µ M CLR01, CLR03, or CLR05 were added to S. aureus cultures at t = 0. Biofilm images show viable cells stained in green using the LIVE/DEAD BacLight Bacterial Viability Kit. The images were recorded after 24 h of incubation using confocal fluorescence microscopy (excitation/emission: 485/498 nm). Scale bars, 50 mm. (B) TEM micrographs of PSMα1 aggregated in the absence or presence of equimolar MT concentrations results in short scarce fibrils in the presence of CLR01 and quasi-oval multiple fibril bundles in the case of CLR05. (C) TEM micrographs testing potential disaggregation of preformed PSMα1 fibrils by CLR01 or CLR05. The MTs were added at a 1:1 concentration ratio and incubated for 24 hours at 37°C. Scale bars denote 300 nm. Reprinted by permission from Elsevier License: Elsevier, Cell Chemical Biology, Inhibition of Staphylococcus aureus biofilm-forming functional amyloid by molecular tweezers, Malishev R, Salinas N, Gibson J, Eden A, Mieres-Perez J, Ruiz-Blanco Y, Malka O, Kolusheva S, Klärner F, Schrader T, Sanchez-Garcia E, Wang C, Landau M, Bitan G, Jelinek R (2021).
PSMα is a member of the PSM peptide family. Some, though not all, the peptides in this family form amyloid in the context of biofilm formation (Salinas et al., 2018; Malishev et al., 2021). One of the peptides known to form amyloid is PSMα1, and therefore this peptide was used as a model to further investigate the mechanism by which the MTs disrupt biofilm using experimental and computational approaches. A ThT-fluorescence assay showed that CLR01 inhibited PSMα1 β-sheet formation completely at a 1:1 concentration ratio, whereas CLR05 was a weaker inhibitor of PSMα1 β-sheet formation, requiring fivefold excess for complete inhibition. TEM analysis revealed that in the presence of CLR01, PSMα1 formed relatively scarce, short, and thin fibrils, as opposed to abundant long and thick fibrils in the absence of MTs. Interestingly, in the presence of equimolar CLR05, PSMα1 formed quasi-oval “bundles” of short fibrils, distinct from the abundant, long fibrils formed by PSMα1 on its own (Fig. 15B). Equimolar concentrations of CLR01, and to a lesser extent CLR05, dissociated PSMα1 fibrils upon incubation for 24 hours (Fig. 15C). The stark difference between the kinetics of these dissociation reactions and those of Aβ (Sinha et al., 2011), α-syn (Prabhudesai et al., 2012), and SOD1 (Malik et al., 2019), which required excess CLR01 and took weeks, demonstrates the substantially lower intrinsic stability of functional amyloids, which are designed to be assembled and disassembled according to the needs of the organism, compared with disease-related amyloids forming under pathologic conditions.
Solution-state NMR experiments demonstrated that CLR01 binding to PSMα1 involved inclusion of Lys sidechains inside the cavity of the MT, as has been demonstrated previously (Fig. 16) (Talbiersky et al., 2008; Lopes et al., 2015), whereas CLR05 binds more weakly and uses a distinct binding mode (Fig. 5). The weaker binding of CLR05 compared with CLR01 indicated that the stronger anti-biofilm activity of this compound likely is mediated via mechanisms other than its direct binding to PSMα1. Molecular-dynamics simulations and free-energy calculations supported the different binding modes of the two MTs observed by NMR yet deciphering the mechanisms by which each compound disrupts S. aureus biofilm will require further study.
Fig. 16.

CLR01 binding to Lys and to PSMα1. (A) A model of the interaction between CLR01 and Lys. P atoms are in green, O atoms in red, C atoms in gray, and N atoms in blue (MD simulation). A HPO4- group at the bridgehead of CLR01 (partially protonated at pH 7.4) forms an ion pair with the ε-NH3+ of Lys. The ammonium end group of the included butylene sidechain is engaged in π-cation interactions with the surrounding aromatic side walls of CLR01 (dotted lines) and further stabilized by the hydrophobic effect. (B) A model of a PSMα1 fibril (surface) with CLR01 (licorice representation) encapsulating a Lys residue. Lys patches are indicated in red over the overall protein surface (blue). The inset shows in more detail the encapsulation of Lys by CLR01 and how the MT establishes additional interactions with a neighbor Lys residue, which also interacts with the complexed Lys.
IV. Mechanism of Action Against Amyloidogenic Proteins
A. Anti-amyloid Activity In Vitro
Unlike many inhibitors of protein aggregation (Liu and Bitan, 2012; Rahimi et al., 2016), MTs do not target the cross-β structures of amyloid fibrils but rather interfere with a combination of hydrophobic and electrostatic interactions involving Lys and, to a lower extent, Arg residues, which are key to the early self-assembly processes before formation of the cross-β structures. As this unique mode of inhibitory activity allows MTs to interfere with the abnormal self-assembly of virtually any amyloid protein as long as Lys or Arg residues are available; we call this mode of activity “process-specific” to distinguish it from the typical specificity of small-molecule inhibitors of a particular protein target. Thus, MTs can be used as broad-spectrum inhibitors of the oligomerization, aggregation, and toxicity of most amyloidogenic proteins as has been shown for CLR01, originally for 10 different proteins (Sinha et al., 2011) and subsequently for additional proteins (Table 3). The only other example of a similar approach we are aware of is a substantially larger curcubit[7]uril that inhibits Aβ aggregation and toxicity by targeting a particular amino acid, in that case Tyr (Lee et al., 2014), albeit with considerably lower efficacy than CLR01. In vitro, binding of at least 1 MT molecule to at least 1 Lys (or Arg) residue is necessary for inhibition, and, therefore, the MTs act stoichiometrically (though sometimes the concentration ratios are sub-stoichiometric and other times super-stoichiometric, depending on the specific protein). Importantly, this mode of action is distinct from the catalytic action of MTs in vivo (see later discussion).
TABLE 3.
Proteins against which CLR01 has been tested and the main related diseases
| Protein/mutant | Inhibition of aggregation | Inhibition of toxicity | Associated disease |
|---|---|---|---|
| Aβ40, Aβ42 | √ | √ | Alzheimer’s disease |
| Tau | √ | √ | Tauopathies |
| α-Synuclein | √ | √ | Synucleinopathies |
| IAPP | √ | √ | Type 2 diabetes |
| Calcitonin | √ | √ | Medullary Carcinoma of the thyroid |
| Insulin | √ | √ | Injection-related amyloidosis |
| β2-Microglobulin | √ | √ | Dialysis-related amyloidosis |
| TTR | √ | √ | Senile systemic amyloidosis, familial amyloid polyneuropathy, familial amyloid cardiomyopathy |
| SOD1 | √ | √ | ALS |
| IgG light chain | √ | NA* | Light-chain amyloidosis |
| TDP43 | √ | NA* | ALS, FTD |
| C9ORF72 RAN translation products containing Arg | √ | √ | ALS, others |
| SEVI | √ | √ | HIV infection |
| P53 | √ | √ | Cancer |
| Huntingtin | √ | √ | Huntington’s disease |
| PMSα1 | √ | √ | Bacterial biofilm |
| Ataxin 3 | √ | √ | Spinocerebellar ataxia type 3 |
| αB-Crystallin | √ | √ | Desmin-related cardiomyopathy |
*NA – not available.
Unlike with CLR01, computational and experimental studies indicated no inhibition of seminal amyloid formation by CLR05 or PC, nor remodeling of the fibrils (Weil et al., 2020). Further testing the effect of the MTs and PC on infectivity enhancement mediated by SEVI fibrils revealed complete inhibition of HIV-1 by CLR01 but not by CLR05 or PC (Weil et al., 2020). However, CLR05 reduced amyloid-infectivity enhancement, whereas PC had no effect at all. Free energy calculations indicated that with respect to CLR01, CLR05 displayed in general a reduced ability to form inclusion complexes with Lys or Arg residues of PAP248-286, an amyloidogenic peptide forming the SEVI fibrils (Lump et al., 2015; Weil et al., 2020). In addition, CLR05 did not interact effectively with Lys281 or Lys282. These two Lys residues, located at the C-terminal region of PAP248–286, form part of the stable cross-β SEVI fibril core. The differences between the activity of CLR01 and CLR05 were related to the fact that the latter forms both chelates and inclusion complexes with Lys/Arg residues in solution via its carboxylate groups. Comparison between the two MTs suggests that the nature of the negatively charged group attached to the central hydroquinone is the key to the difference in activity. Thus, the anti-seminal-amyloid activity of CLR01 results not only from inclusion of Lys/Arg in its torus-shaped cavity but also from the hydrogen phosphate groups adorning its periphery, which favor the formation of supramolecular complexes with Lys/Arg. The formation of the inclusion complexes between CLR01 and Lys/Arg residues in seminal peptides such as PAP248-286 leads to a neutralized zeta potential of the fibrils and thus to elimination of the fibrils’ ability to carry virions to the cell membrane (Lump et al., 2015).
CLR01 binds to Lys, simple Lys derivatives, and small Lys-containing peptides with approximately 10 μM affinity (Sinha et al., 2011; Dutt et al., 2013a). It binds Arg derivatives with 5 to 10-times lower affinity and does not recognize other amino acids or small co-factors (Fokkens et al., 2005; Talbiersky et al., 2008). Importantly, the binding of CLR01 is kinetically labile and has a high on-off rate, which was demonstrated by surface-plasmon resonance (Bier et al., 2013) and 1H-NMR spectroscopy (Dutt et al., 2013a). The moderate affinity and high on-off rate are crucial for the selectivity of MTs for the abnormal assemblies of amyloid proteins. Stable proteins, whose structure was optimized by millions of years of evolution, are unaffected by such gentle binding, whereas the weak forces mediating formation of metastable oligomers and nuclei, which (mis)fold considerably slower and through a rougher energy landscape (Yu et al., 2015), are disrupted efficiently by MT binding. Moreover, there is a profound difference between freely accessible Lys residues on unstructured protein domains, which are easily accessible to MTs, and folded proteins whose surface often does not allow the sterically demanding MTs to approach a Lys residue close enough for binding. This was evidenced by x-ray crystallography, isothermal titration calorimetry, and molecular dynamics simulations combined with QM/MM calculations of the complex between CLR01 and 14-3-3, in which only 5 of 17 surface Lys residues were available for CLR01 binding (Bier et al., 2013, 2017). Thus, the mode by which MTs target the abnormal self-assembly process itself, rather than a specific protein, is based on the fundamental difference in the binding energies holding polypeptide chains together in metastable oligomers versus normal proteins.
The binding sites of CLR01 on various amyloidogenic proteins have been studied using ECD-MS and/or solution-state NMR and the findings are summarized in Table 4. As discussed above, determining the primary binding site for CLR01 on Aβ was particularly informative in the early days of studying the anti-aggregation activity of MTs and in fact, opened the gate to discovering their broad-spectrum activity. Aβ has two Lys residues at positions 16 and 28, in addition to one Arg residue at position 5. The first publication describing water-soluble, Lys-selective MTs (Fokkens et al., 2005) appeared around the same time a central turn structure in monomeric Aβ was discovered (Lazo et al., 2005). The turn was stabilized by both hydrophobic and electrostatic interactions involving Lys28, leading us to postulate that MTs might bind to Lys28, disrupt these interactions, and thereby inhibit Aβ self-assembly. Though the experimental evidence indeed showed that CLR01 was a potent inhibitor of both Aβ40 and Aβ42 oligomerization and aggregation (Sinha et al., 2011; Zheng et al., 2015), we were surprised when the main binding site for the compound was found by both ECD-MS and solution-state NMR to be Lys16 (Sinha et al., 2011). These findings suggested that disruption of the central turn in Aβ might not be necessary for inhibition of its self-assembly, which prompted experiments testing the inhibitory activity of CLR01 against multiple other amyloidogenic proteins and the discovery of its broad-spectrum activity (Table 3).
TABLE 4.
CLR01 binding sites on different amyloidogenic proteins
| Protein | Binding Sites | Method | Reference |
|---|---|---|---|
| Aβ40, Aβ42 | Lys16 primary, Lys28 secondary, Arg5 tertiary | ECD-MS/MS and solution-state NMR | (Sinha et al., 2011) |
| Huntingtin exon 1 | Lys9 | ECD-MS/MS | (Vöpel et al., 2017) |
| PSMα1 | Lys9, Lys12, Lys21 | Solution-state NMR | (Malishev et al., 2021) |
| SOD1 | Lys70 and/or Lys75 | ECD-MS/MS | (Malik et al., 2019) |
| Tau | Multiple Lys residues, mainly in the MTBD, Lys331 primary, Lys321 secondary | ECD-MS/MS and solution-state NMR | (Despres et al., 2019; Nshanian et al., 2019) |
| α-Synuclein | Lys10 and/or Lys12 | ECD-MS/MS | (Acharya et al., 2014) |
Using the same methods in combination with molecular dynamics simulations, QM/MM, and free-energy calculations, CLR01’s primary binding sites were found to be at Lys 10 and/or Lys 12 on α-syn (Acharya et al., 2014), mainly in the MTBD of tau, in which Lys331 had the highest affinity followed by Lys321 (Despres et al., 2019; Nshanian et al., 2019), and Lys9 of Htte1 (Vöpel et al., 2017). In addition, recent NMR experiments showed that all three Lys residues in PSMα1 bound CLR01 (Malishev et al., 2021). The identification of these multiple binding sites reveals important mechanistic aspects of CLR01’s inhibitory activity. First, the main binding sites of the compound on different proteins do not occur at particular consensus motifs but rather appear to be determined by the unique amino-acid sequence and conformation in each case. MT binding can be favored especially when charged groups such as hydrogen phosphates and carboxylates are adorning the central hydroquinone, by formation of additional salt bridges and hydrogen bond interactions between the MT’s charged groups and residues neighboring the encapsulated Lys or Arg (Bier et al., 2013). Second, the main binding sites do not have to be at a region important for the protein’s aggregation to achieve efficient inhibition. The first point is demonstrated in Table 5, showing the flanking residues around each Lys residue recognized as a main binding site for CLR01. It stands out in particular when one considers the seven-repeat motifs at the N-terminus of α-syn and four-repeat domain comprising the microtubule-binding region of tau. In each of these cases, certain Lys residues in specific repeats have been identified as primary binding sites for CLR01, whereas Lys residues experiencing similar environments in nearby repeats are not. The second point supports the unique mechanism of action of MTs, which is distinct from typical inhibitors binding to specific binding sites of receptors or active sites of enzymes. MTs bind Lys residues with moderate affinity and high on-off rates. A major factor determining their preferred binding sites is simply the exposure of the Lys residue to the environment, implying that residues involved in intramolecular interactions, such as salt-bridges, would be less available for the MTs’ binding. Thus, the inhibition of self-assembly is mediated by disruption of intermolecular interactions involving Lys (and nearby residues) and by the temporary reversal of the positive charge at the site of binding to a negative charge for the duration of the MT binding.
TABLE 5.
CLR01 binding sites
| Protein | Binding Site |
|---|---|
| Aβ | H13HQKLVF19 |
| Htte1 | K6LMKAFS12 |
| SOD1 | L67SRKHGG73 |
| G72GPKDEE78 | |
| Tau | C291GSKDNI297 |
| D295NIKSVP301 | |
| I308VYKPVD314 | |
| D314LSKVTS320 | |
| V318TSKCGS324 | |
| I328HHKPGG334 | |
| α-Synuclein | G7LSKAKE13 |
| S9KAKEGV15 |
Providing additional insight into the mechanism of action of MTs, the impact of CLR01 binding was shown to modulate the “reconfiguration rate” of α-syn, that is, the rate by which the monomer conformation changes, affecting both intra- and intermolecular interactions. This rate can be measured by introducing a fluorophore-quencher system, e.g., Trp and Cys, into the sequence of the protein (Ahmad et al., 2012; Ahmad and Lapidus, 2012). A slow reconfiguration rate typically indicates strong intramolecular interactions and a stable structure, whereas a fast reconfiguration rate suggests the lack of a stable structure. When the reconfiguration rate is moderate, the structural stability is low and intermolecular interactions may be favorable, leading to self-assembly, whereas a fast reconfiguration rate decreases the stability of intermolecular interactions and the probability of self-association. Using this system, CLR01 was shown to increase the reconfiguration rate of α-syn, providing a plausible mechanistic explanation for the strong inhibitory action of the MT on α-syn aggregation (Acharya et al., 2014).
In addition to inhibiting formation of toxic assemblies, CLR01 also has been demonstrated to dissociate existing amyloid fibrils of various amyloidogenic proteins, including Aβ40 and Aβ42 (Sinha et al., 2011) α-syn (; Prabhudesai et al., 2012; Bengoa-Vergniory et al., 2020), SOD1 (Malik et al., 2019), SEVI and PAP(85-120) (Lump et al., 2015), and PSMα1 (Malishev et al., 2021). Incubation of CLR01 with fibrils of IAPP, a particularly amyloidogenic protein bearing only two binding sites for CLR01 at Lys1 and Arg11, did not dissociate the fibrils but led to an arrest of fibril growth (Lopes et al., 2015). In view of the gentle binding of CLR01 described earlier, its ability to dissociate pre-existing amyloid fibrils, where the forces holding the polypeptide chains together are particularly strong, might be surprising. Because these forces are weaker in functional amyloids, such as those formed by SEVI, PAP(85–120), and PSMα1, the dissociation of these fibrils was achieved within hours using low protein:CLR01 concentration ratios, as opposed to the pathologic amyloid fibrils, which required excess CLR01 and several weeks for dissociation. In these latter cases, binding of CLR01 to exposed Lys residues on the surface of the fibril likely does not contribute to fibril dissociation. Rather, when CLR01 binds to the minute amount of soluble protein that is in equilibrium with the fibril, it does not allow the protein to re-bind to the fibril, thereby slowly shifting the equilibrium toward fibril dissociation (Fig. 17).
Fig. 17.

Putative schematic mechanism of fibril dissociation by MTs. Time-dependent EM analysis allow the following conclusions: Binding of MTs to the fibrils of disease-associated proteins, such as Aβ, does not disrupt the fibrils. However, in the presence of excess MT, binding to the small fraction of soluble protein molecules does not allow them to re-bind to the fibrils, slowly shifting the equilibrium toward dissociation.
B. Anti-amyloid Activity In Cellulo and In Vivo
Despite the in-depth insight into the mechanisms by which MTs inhibit abnormal protein self-assembly and dissociate existing aggregates in vitro, until recently, little was known about the way they interact with cells, distribute within cells, and affect amyloidogenic proteins in the context of a cellular environment. A major impediment to answering these questions is that the intrinsic fluorescence of most MTs, although sufficient for measurement of binding in simple buffers (Fig. 4), is too weak to observe on the background of a cell. Therefore, to address these questions, recently two fluorescent MT derivatives were prepared in which fluorophore beacons were attached to one of the phosphate groups of CLR01 (Li et al., 2021). In a derivative called CLR16, the fluorophore is 5-carboxytetramethylrhodamine (TAMRA), whereas in CLR18 it is 6-fluorescein amidite (FAM). Most of the work to date has been done using the red-fluorescent CLR16 to allow counterstaining with various cellular markers labeled by GFP, whereas the green CLR18 was used for validation of key experiments (Li et al., 2021).
In the first study using CLR16, the compound was shown to be internalized in human oligodendroglioma cells (Herrera-Vaquero et al., 2019). Subsequent studies showed that this internalization is a general phenomenon as CLR16 was found also internalize into mouse primary neurons and primary astrocytes, SH-SY5Y cells, and HEK293 cells, primarily via dynamin-mediated endocytosis (Li et al., 2021). Inspection of cells treated with either CLR16 or CLR18 under a fluorescence microscope revealed that most of the intracellular fluorescence appeared to be punctate, rather than diffuse (Fig. 18). Possible explanations included intracellular aggregation of the MTs or their concentration in cellular compartments. As discussed earlier, MTs have been found to form dimers only at high concentrations (Fig. 3). In addition, CLR01 was found not to form colloids or micelles despite its amphipathic nature, likely due to its rigid structure (Lump et al., 2015), suggesting against intracellular aggregation of the fluorescent MTs. Therefore, the bright puncta were hypothesized to reflect concentration of the MTs in particular cellular organelles, and subsequent experiments were performed to decipher the identity of these organelles.
Fig. 18.
MTs are internalized in SH-SY5Y cells and concentrate in puncta. SH-SY5Y cells were incubated with fluorescent MTs for 24 hours and visualized by fluorescence microscopy. (A) Cells were transiently transfected with GFP-actin (green), incubated with 5 µM CLR16 (red), and nuclei were stained with Hoechst (blue). (B) Cells were incubated with 10 µM CLR18 (green) and nuclei were stained with Hoechst (blue). This figure was published originally in Communications Biology, Li, Z., Siddique, I., Hadrovic, I., Kirupakaran, A., Li, J., Zhang, Y., Klärner, F-G., Schrader, T., Bitan, G. Lysine-selective molecular tweezers are cell penetrant and concentrate in lysosomes, 165513, Copyright Nature Portfolio (2021).
Cells were treated with the fluorescent MT derivatives and counterstained with dyes or markers for specific organelles. These experiments showed that the MTs did not co-localize with mitochondria or nuclei but rather overlapped weakly with early endosomes, moderately with late endosomes and autophagosomes, and most strongly with lysosomes (Li et al., 2021). These data revealed that MTs tend to concentrate in acidic compartments, which are the very compartments to which cells direct misfolded and aggregated proteins for degradation. Though the reason for the concentration in acidic compartments currently is not well understood, the data helped explain the strong therapeutic effects of low-dose CLR01 found in multiple animal models, suggesting that the compound concentrates right where it is most needed, achieving a high effective concentration in the small volume of these organelles and acting catalytically to facilitate enzymatic degradation of misfolded, aberrantly assembled proteins by lysosomal cathepsins. This putative mechanism helps explain the high efficacy of CLR01 observed in the mouse model of MPS IIIA described earlier, in which different amyloidogenic proteins form secondary lysosomal storage (Monaco et al., 2020).
It might be argued that the relatively large fluorophores changed the chemical characteristics of CLR01 and therefore their internalization and cellular distribution do not reflect the behavior of the unlabeled MT. To address this concern, we incubated the cells with CLR16 in the presence of increasing concentrations of CLR01 and found that CLR01 competed with the internalization and lysosome concentration of CLR16, suggesting that the behavior of both compounds in a cellular environment was similar (Li et al., 2021).
A crucial demand from drugs aimed at treating CNS diseases is penetration through the blood–brain barrier (BBB). The brain-to-blood ratio of CLR01 has been tested using 3H-CLR01 initially using intravenous administration and was found to be 2% to 3% after 20 minutes in wild-type mice and mouse models of AD and PD (Attar et al., 2014; Richter et al., 2017). Although this first-pass penetration of CLR01 into the brain was relatively low, it was more than might be expected for a compound with a molecular mass of 722 g/mole and up to four negative charges. Interestingly, we also found that although the plasma half-life of CLR01 was approximately 2.5 hours, the small percentage that passed through the BBB persisted in the brain 72 hours later (Attar et al., 2014), providing the first clue for the strong efficacy of CLR01 administered peripherally in the multiple mouse models of CNS proteinopathies discussed earlier.
A second clue was discovered recently, as part of the study testing CLR01 in the SNCA-OVX mouse model of PD. The study re-evaluated the BBB penetration of the compound, which this time was administered subcutaneously in the pharmacokinetics experiments, to match the efficacy experiments. Surprisingly, when CLR01 was administered subcutaneously in these mice, its brain-to-blood ratio was an order of magnitude higher than following intravenous administration and appeared to saturate at approximately 40% (Bengoa-Vergniory et al., 2020) (Fig. 19). This substantial increase in brain-to-blood ratio was not related to the mouse model itself because two mice included in the study were wild-type littermates (red dots in Fig. 19). Rather, the increase in brain-to-blood ratio reflected a substantially lower concentration of CLR01 in the blood and a three- to fourfold increase in the brain concentration following subcutaneous administration compared with intravenous administration, leading to the hypothesis that using this route of administration, CLR01 is temporarily deposed in the subcutaneous adipose tissue, from which it is released slowly into the bloodstream. Indeed, initial tissue-distribution analysis showed that a large fraction of CLR01 was found in the adipose tissue at the injection site 5 minutes after injection (manuscript in preparation), supporting this proposed mechanism.
Fig. 19.

Brain-to-blood ratio of CLR01 following SC administration. OVX-SNCA mice were administered increasing doses of CLR01 spiked with 10% 3H-CLR01 (3.92 μCi/g body weight in the highest dose). Blood and brain were harvested at 1, 8, or 24 hours, digested in Solvable (Perkin-Elmer) and the radioactivity counted by scintillation. Brain-to-blood ratio (%) was calculated as cpm/g brain divided by cpm/mL blood ×100. Two wild-type littermates were included in the 8-hour group and are marked by red dots.
In the SNCA-OVX Parkinson’s disease mouse model, a dose of 0.14 mg/kg administered twice a week for 2 months was found to have a significant beneficial therapeutic effect. Using computer modeling, the intracellular average drug concentration was calculated to be 240 ng/mL, with a predicted maximum concentration of 510 ng/mL, corresponding to cells exposed to concentrations above the calculated EC50 of 85.4 nM 93% of the time. Moreover, the model predicted a rapid intracellular accumulation of CLR01. The extracellular average concentration was calculated to be 100 ng/mL, the maximum concentration was 230 ng/mL, and time above the EC50 in the extracellular space was 81%. These numbers support the findings that administration of the same dose of CLR01 using osmotic minipumps led to effective clearance of amyloid plaques and neurofibrillary tangles in the triple-transgenic mouse model of AD (Attar et al., 2012). The concentrations of the major amyloidogenic proteins in the brain are: Aβ42 approximately 7.5 ng/mL (Wang et al., 1999), tau approximately 120 μg/mL (Iqbal et al., 2010), and α-syn 1.4 to 14 μg/mL (Bernal-Conde et al., 2020). Thus, the calculated intracellular concentration of CLR01 allowed it to disrupt formation of toxic oligomers, the concentration of which would be predicted to be substantially lower than that of the total protein. However, as MTs bind to exposed Lys residues on any protein and to free Lys, the percentage of CLR01 available for interaction with the amyloidogenic proteins might be considerably lower than the total concentration measured in the brain. How then is the high efficacy of the compound achieved?
The recent study using fluorescent MT derivatives provided a third major clue. The study revealed that upon endocytosis, the MTs concentrate rapidly in acidic organelles, primarily in lysosomes, and to a lower extent in late endosomes and autophagosomes, which together comprise about 2% of the total volume of a cell. These organelles are exactly those to which the cells direct misfolded and aggregated amyloidogenic proteins, making the effective concentration of CLR01 one to two orders of magnitude higher than the concentration calculated based on total brain volume and reducing substantially the number of competing Lys/Arg residues in the remaining volume of the cell. Together with the catalytic activity in the lysosomes, as opposed to the stoichiometric inhibition of aggregation in vitro, these new pieces of the puzzle explain how CLR01 achieves high efficacy at substantially lower doses in vivo than would be predicted based on in vitro data.
C. Disruption of Viral Membranes
CLR01 and CLR05 exerted broad antiviral activity against all analyzed enveloped, but not naked viruses (Lump et al., 2015; Röcker et al., 2018b; Weil et al., 2020; Brenner et al., 2021), suggesting that the viral lipid bilayer is the target of the tweezers. In fact, experiments using synthetic GUVs and liposomes confirmed a direct membrane disrupting activity (Lump et al., 2015; Weil et al., 2020). Importantly, the potency in liposome destruction correlated strongly with the antiviral activity, providing further evidence that the tweezers exert antiviral activity by destroying the viral envelope. To obtain additional mechanistic insights, molecular dynamics simulations and NMR studies were performed, which showed that CLR01, CLR05, and PC encapsulated the lipid head groups inside their cavities (Fig. 20), yet PC did not form “closed” inclusion complexes. Unlike PC, CLR01 and CLR05 induced changes in the orientation of the head groups of the lipids, which raised surface tension resulting in destabilization of the viral membrane.
Fig. 20.

CLR01 binding to lipid headgroups. (A) Model viral membrane (surface representation: cholesterol in pink, DOPC in gray, SM in cyan) with CLR01 molecules (licorice representation) encapsulating SM and DOPC lipids. (B) A SM lipid headgroup inside CLR01’s cavity.
In addition, QM/MM calculations indicated that CLR01-sphingomyelin (SM) complexes are more stable than CLR01-DOPC complexes. As viral membranes derived from the plasma membrane are enriched in cholesterol and SM (Lorizate et al., 2013), the preferred destabilization of viral membranes in comparison with cellular membranes can be rationalized by the difference in stability of these complexes. The ability of MTs to disrupt viral membranes also results from the increased surface tension due to the higher curvature of these membranes compared with cellular membranes making the latter more resistant to disruption by MTs. Membrane repair mechanisms of cells, which are absent in viruses, might further help prevent MT-mediated damage to the cells.
Importantly, free-energy calculations allowed establishing that CLR01 is unlikely to penetrate deep inside the membrane or cross the bilayers. Thus, the key to the antiviral activity is related to the MTs’ effect on the lipid orientation and/or the preference of the MTs toward lipids, such as SM.
In addition, biomolecular simulations of potentiated MTs featuring novel aliphatic or aromatic ester groups and displaying improved antiviral activity against SARS-CoV-2 were carried out. The results indicated that the presence of MTs’ substituents that can act as “inserted side arms” into the membrane increases the surface tension at a local site of the viral membrane, in addition to the changes in lipid orientation discussed previously. Therefore, the insertion of lipophilic arms, which increase the MT affinity for the lipid membrane provides a rationale for the enhanced and highly effective antiviral activity of these novel MTs.
V. Drug-like Properties and Toxicology
A. CLR01’s Chemistry and Pharmacokinetics
CLR01 is soluble in aqueous solutions at 10 to 15 mM. It complies with three of the four Lipinski rules-of-5 (Lipinski et al., 2001). It has two H-bond donors and eight H-bond acceptors, and its cLogP = 2.64. CLR01’s polar surface area of 139.18 Å2 also supports its druggability. The one violated rule is the size. The molecular mass of CLR01 is 722, above the recommended 500-Da cutoff. In addition, the negatively charged phosphate groups of CLR01 would raise caution for traditional medicinal chemists or pharmacologists. However, as discussed earlier, substantial empirical data demonstrate internalization into cells, BBB permeability following subcutaneous administration well above expectation, and a putative catalytic mechanism in vivo, explaining the efficacy in multiple animal models of CNS proteinopathies upon peripheral administration.
Pharmacologic characterization of CLR01 began with standard in vitro experiments and yielded the following results (Attar et al., 2014):
CLR01 was stable in human (99 ± 9%) and mouse (97 ± 2%) liver microsome preparations for 60 minutes, whereas testosterone, which was used as a positive control, was degraded down to 29% and 1%, respectively.
CLR01 was stable (100 ± 5%) in both human and mouse plasma for 60 minutes at 37°C.
As would be expected based on its mechanism of action, CLR01 was 96 ± 3% and 99.0 ± 0.2% bound to human and mouse plasma proteins, respectively. These results were comparable to testosterone (98.6 ± 0.2% and 94 ± 2%, respectively).
CLR01’s IC50 for inhibition of five major cytochrome P (CYP) 450 isoforms were all above 1 μM (Table 6), suggesting against significant drug–drug interactions (Obach et al., 2006). In addition, CYP450 34A induction by CLR01 was moderate relative to the antibiotic rifampicin used as a positive control (Attar et al., 2012).
TABLE 6.
CYP450 inhibition
| CYP450 Isoform | Standard IC50 (μM) | CLR01 IC50 (μM) |
|---|---|---|
| 1A2 | 0.51 (naphthoflavone) | >20 |
| 2D6 | 0.03 (quinidine) | 3.6 |
| 2C9 | 0.33 (sulphaphenazole) | 2.2 |
| 2C19 | 3.6 (troglitazone) | 1.5 |
| 3A4 | 0.03 (ketaconazole) | 1.7 |
The plasma concentration of CLR01 following a 1-mg/kg I.V. injection initially (20 minutes post-injection) was five times higher than upon subcutaneous administration, yet clearance was faster in the intravenous route so the area under the curve was only about two times higher for intravenously administered CLR01 compared with the subcutaneous route. The half-life in both routes was determined to be about 2.5 hours (Attar et al., 2014) but was quite variable in each group. These data suggest that mechanisms other than those tested in plasma in vitro are responsible for CLR01 clearance. Continuous subcutaneous administration of 1 mg/kg per day CLR01 via osmotic minipumps for 21 days showed a plasma steady-state concentration of about 140 nM (Lopes et al., 2015). In contrast, oral bioavailability by gavage following administration at 10 mg/kg was low (Attar et al., 2014). In addition to the size and negative charge of CLR01, which likely have restricted its oral absorption, two additional factors might have lowered oral bioavailability. First, the mice in those initial experiments were fed ad libitum and the proteins in the food acted as a sink for CLR01. Second, CLR01 might have been metabolized in the GI tract. We hypothesized that the most likely metabolism of CLR01 would be dephosphorylation. To begin testing this hypothesis, the compound was incubated in vitro with alkaline phosphatase or with whole mouse brain extracts for 60 minutes. The release of free phosphate was measured, using p-nitrophenylphosphate (PNP) as a positive control. The measurements showed quantitative dephosphorylation of PNP, but no release of free phosphate from CLR01 in either condition (Attar et al., 2014), likely because its rigid structure confers metabolic stability, in agreement with the plasma and liver-microsome experiments. These results suggested that the low oral bioavailability of CLR01 might result from inability to be absorbed by the gut and/or from its binding to food proteins.
B. CLR01’s Specificity In Vitro
To test the hypothesis that CLR01 could disrupt aberrant protein self-assembly while sparing normal assembly or other physiologic functions, the protein:CLR01 concentration ratio needed for inhibiting aggregation of amyloid proteins was compared with that required for interfering with a normal assembly process—tubulin polymerization. The protein:CLR01 concentration ratios needed for half-maximal inhibition (IC50) of different amyloid proteins were mostly in the range 1:0.1 to 1:5, respectively (Sinha et al., 2011; Schrader et al., 2016). These concentration ratios had no effect on tubulin polymerization. Only at a tubulin:CLR01 concentration ratio of 1:55, respectively, partial disruption of the process was observed (Attar et al., 2014).
CLR01 also was tested as an inhibitor of enzymes containing key Lys residues at or near the active site. The enzyme:CLR01 ratios needed for inhibiting the activity of alcohol dehydrogenase or PARP-1 were 1:865 (Talbiersky et al., 2008) and 1:1,435 (Wilch et al., 2017), respectively. These data support the mechanism of action of CLR01 and demonstrate that despite its ability to bind exposed Lys residues essentially on any protein, its gentle, labile binding affects amyloid proteins’ self-assembly at concentrations orders of magnitude lower than those needed for disruption of normal biologic processes.
C. CLR01’s Specificity and Toxicity in Cell Culture
Because in most of the assays used to measure the toxicity of amyloidogenic proteins the proteins were added to cells exogenously, the concentrations of CLR01 needed for inhibition depended on the concentration of the protein used and, as discussed earlier, generally were in the high nM to low μM range. Inhibition of α-syn oligomerization when expressed endogenously in SH-SY5Y cells by CLR01 was achieved with IC50 = 85.4 nM (Bengoa-Vergniory et al., 2020) and inhibition of seeded intracellular tau aggregation in HEK293 biosensor cells with IC50 = 660 nM (Despres et al., 2019). Toxicity of CLR01 itself in cell culture was observed at about three orders of magnitude higher concentrations and depended on the cell type used. In differentiated PC-12 cells, no toxicity was observed up to 200 μM, and approximately 15% reduced viability (MTT assay) occurred at 400 μM (Sinha et al., 2011). No reduction in viability (XTT assay) was found up to 500 μM in the HIV reporter cell line TZM-bl (Lump et al., 2015).
D. CLR01’s Safety In Vivo
To test the safety of CLR01 in mice, it was administered intraperitoneally to 2-month-old wild-type mice either as an acute bolus at 10 or 100 mg/kg with subsequent analysis at 24 hours or chronically for 30 days (Attar et al., 2014). In the acute setting, administration of 100 mg/kg CLR01 initially caused obvious signs of distress to the mice, mainly freezing and hunching, which resolved completely within 2 hours. No mouse died until the time point of euthanasia at 24 hours. Histologic and serological analyses showed liver injury but no damage to other major organs, including heart, lungs, kidneys, pancreas, spleen, or brain (Attar et al., 2014). In the 10-mg/kg group there were no signs of distress and no significant serological findings. In one out of eight mice in this group there were mild signs of liver degeneration. Therefore, 10 mg/kg per day was used as the high dose in the subsequent chronic administration experiments, whereas a low dose was 3 mg/kg per day. There were no behavioral or histologic findings in either of the chronic-treatment groups. The only significant serological change was an approximately 40% decrease in blood cholesterol in the 10-mg/kg per day group (Attar et al., 2014). These findings demonstrate that CLR01 has a high safety margin, though the formal therapeutic index could not be determined because lethal doses were not reached. In further support of the safety of CLR01 and its mechanism of action, in all the efficacy in vivo studies performed to date, dose range 0.04 to 6.0 mg/kg per day, no treatment-related side effects were observed, including behavioral changes, morbidity, mortality, weight loss, or any indication that CLR01 treatment caused distress to the treated animals. Overall, the chemistry, pharmacokinetics, and safety data suggest that CLR01 administered subcutaneously is an attractive drug candidate for multiple proteinopathies and LSDs for which currently disease-modifying therapy is not available and support further development of orally bioavailable derivatives or pro-drugs.
VI. Pending Questions and Future Directions
The body of evidence supporting the therapeutic efficacy, high safety, and pharmacokinetic profile of MTs, particularly CLR01, has been growing over the last decade. To date, positive therapeutic effects have been demonstrated in more than 10 in vivo studies using four different species—zebrafish, lamprey, mouse, and rat, some of which are highlighted in Fig. 21. Inhibition of pathologic protein self-assembly was found for nearly 20 proteins involved in diverse diseases, including AD, other tauopathies, PD, ALS, MSA, TTR amyloidosis, desmin-related myopathy, and MPS IIIA, as well as in spinal cord injury, viral infection, and bacterial-biofilm formation. MTs act by remodeling toxic protein assemblies into benign structures prone to degradation and, importantly, facilitate cellular clearance mechanisms, such as the UPS and ALP, acting catalytically in vivo, which yields far greater impact than would be expected based on their quasi-stoichiometric inhibitory activity in vitro. This difference in mechanism between in vitro and in vivo environments is what allows CLR01 administered peripherally at doses as low as 0.14 mg/kg twice weekly or 0.04 mg/kg daily, yielding brain concentrations in the nanomolar range, to achieve high efficacy (Attar et al., 2012; Bengoa-Vergniory et al., 2020) even though inhibition of the corresponding proteins (Aβ, tau, α-syn) in vitro requires micromolar concentrations of CLR01. Despite the progress made over the last decade, several questions remain to be answered and additional data will support paving the path into clinical trials and the eventual goal of using MTs as disease-modifying therapy in humans.
Fig. 21.

Highlighted efficacy studies of CLR01 in different animal models.
First, although the BBB permeability of CLR01 has been shown to be sufficient for achieving strong therapeutic effects and the brain-to-blood ratio can be increased by an order of magnitude by administering the compound subcutaneously, rather than intravenously, the first-pass penetration of the compound is limited and likely can be improved. Ongoing studies are testing new MT derivatives and CLR01 pro-drugs and initial results suggest that compounds with similar activity and greater BBB penetration can be obtained. An important and currently unanswered question is the mechanism by which MTs enter through the BBB. As their size and charge would be expected to keep these compounds outside the brain, a likely mechanism by which they manage to get through the barrier is opportunistic “hitchhiking,” bound to Lys residues on protein transporters, such as those importing glucose or amino acids into the brain or the transferrin receptor. Another currently unexplained phenomenon is the trapping of MTs in the brain (Attar et al., 2014). A plausible explanation for this phenomenon is the concentration of the compounds intracellularly in acidic organelles, which prevents their diffusion back into the extracellular space and the systemic circulation.
Two additional suboptimal pharmacokinetic characteristics of CLR01 are its relatively short plasma half-life (about 2.5 hours) and low oral bioavailability. The relatively short half-life could reflect either metabolic modifications or excretion of the compound. Based on the structure of CLR01, the most likely metabolic alteration is dephosphorylation. However, incubation of the compound either with alkaline phosphatase or a whole mouse brain extract yielded no detectable phosphate release, whereas a positive control compound, 4-nitrophenylphosphate, was dephosphorylated quantitatively under the same conditions (Attar et al., 2014). These findings do not rule out other modifications, but they suggest against dephosphorylation, the most likely modification, as the cause for the moderate plasma half-life. Formal metabolite analysis and measurement of the kinetics of CLR01 excretion remain to be done. The low oral bioavailability of CLR01 (about 1%) may be due to the size and charge of the compound, which makes passing through the gut–blood barrier difficult. An alternative explanation is binding to proteins in food, which would interfere with absorption of orally administered CLR01 into the blood. The initial pharmacokinetic experiments measuring oral absorption were done in mice fed ad libitum. Future testing in fasting animals will allow distinguishing between these two hypotheses. Encouragingly, initial testing of new MT derivatives and CLR01 pro-drugs suggests that the plasma half-life can be extended, and the oral bioavailability can be increased without compromising the activity or safety of the compounds.
The broad-spectrum activity of MTs against abnormal protein aggregation suggests that they can be effective against multiple additional amyloidogenic proteins and may have more applications than those explored to date. For example, MTs may be effective drug candidates for prionoses. Another example is eye diseases involving protein aggregation, such as retinal ganglion cell degeneration in glaucoma, where an extended exposure of proteins to reduced sugars leads to the nonenzymatic glycation of amino groups by Maillard reaction and alters the biologic activity and degradation of these proteins (Tezel et al., 2007). Other proteinopathies predicted to be good candidates for MT therapy are Aβ-related diseases other than Alzheimer’s disease, such as cerebral amyloid angiopathy and inclusion-body myositis, systemic diseases, including primary (AL) amyloidosis and reactive (AA) amyloidosis, and rare genetic diseases, such as Finnish hereditary systemic amyloidosis caused by GSN (gelsolin) mutations, or Familial British dementia and Familial Danish dementia caused by mutations in BRI2. A particularly interesting case are poly-Q expansion diseases, in which the core of the amyloid consists of the poly-Q tract and thus is inaccessible to the MTs, yet nearby Lys residues may allow them to inhibit the aggregation of the mutant protein, as has been shown for Htte1 (Vöpel et al., 2017).
Among LSDs, it will be interesting to test if MTs can help reduce not only the secondary storage of amyloidogenic proteins, as has been shown in a mouse model of MPS IIIA (Monaco et al., 2020) but also lipid storage, particularly of sphingomyelin, in view of the recent discovery of the affinity of various MTs for the headgroups of these lipids (Weil et al., 2020). In Niemann-Pick disease type-C (NPC), lysosomal hydrolases along with the mutated transporter enzymes (NPC1 and NPC2) restrict breakdown and recycling of various cellular products (Evans and Hendriksz, 2017), which may result in deposition of the APP C-terminal fragment C99 and Aβ42 in the late endosomes (Yamazaki et al., 2001; Jin et al., 2004). In Krabbe disease, mutations in the gene encoding galactocerebrosidase lead to a dysfunctional enzyme, which is responsible for the hydrolysis of the galactosyl moiety from several galactolipids (Wenger et al., 1997). Aggregated forms of α-syn have been reported to accumulate as secondary storage in this disease in neuronal lysosomes (Smith et al., 2014). In Gaucher disease, deficiency of the lysosomal enzyme glucocerebrosidase leads to the accumulation of glucosylceramide and glucosylsphingosine (Platt, 2014; Breiden and Sandhoff, 2019), leading to deposition of α-syn and amyloidogenic APP fragments. The failure to break down sphingosines and ceramides in this disease may lead to accumulation of sphingomyelin clusters, which potentially can be disrupted by MT binding to their head groups, as has been reported previously (Lump et al., 2015; Weil et al., 2020). As MTs may disrupt both the secondary storage of amyloidogenic proteins and lipid clusters, particularly of sphingomyelin, they are attractive therapeutic agents for these and potentially other LSDs, especially in combination with enzyme replacement therapy or gene therapy correcting the genetic defect while addressing also the existing accumulation of proteins and lipids at the time of the treatment initiation.
MTs also could be useful against lipofuscinoses, such as Batten and Stargardt diseases, based on their ability to interfere with abnormal protein aggregation and protein–lipid interactions (Bitan, 2019). An additional potential application of MTs is CNS injury, including spinal-cord injury, different types of traumatic brain injury, and stroke, in which amyloidogenic proteins, such as tau or α-syn, are elevated (Acosta et al., 2015; Fogerson et al., 2016; Bi et al., 2017; Wang et al., 2017, 2020; Hayden et al., 2019; Shahim et al., 2020) and may form toxic aggregates that harm the cells surrounding the injury site.
Although proteinopathies are characterized by abnormal protein aggregation, they are complex diseases in which many deleterious mechanisms act in concert. Therefore, inhibition of aggregation and clearance of existing aggregates may not be enough for achieving the desired therapeutic effects. It is thus plausible that in some cases MTs could be used in combination with other drugs, ideally other disease-modifying drugs. For example, antisense-oligonucleotide and siRNA therapies are promising approaches for treatment of diseases in which the main mechanism is a toxic gain of function by an amyloidogenic protein, such as poly-Q diseases (Silva et al., 2020) and other rare proteinopathies (Kluve-Beckerman et al., 2011; Daoutsali et al., 2021), but at the time of treatment initiation, substantial aggregation might have already occurred and facilitation of clearance by MTs in combination with suppression of the offending protein’s production could lead to meaningful therapeutic outcomes where monotherapy might fail. In LSDs, a combination of gene therapy or enzyme-replacement therapy with MTs can be similarly advantageous, addressing simultaneously both the primary genetic defect and the harm caused by secondary storage of aggregated proteins or sphingolipids.
A possible medical use of MTs as antivirals is currently limited to topical applications because the antiviral activity of CLR01 is rapidly lost upon incubation in serum, most likely due to preferential binding to abundant serum proteins, such as albumin (Röcker et al., 2018b). This finding seems to be in conflict with the proven therapeutic effects of CLR01 in animal models of amyloid diseases described earlier. However, as discussed above, the concentrations of CLR01 required for preventing amyloid formation in vitro and especially for facilitating degradation of toxic oligomers and aggregates in vivo are orders of magnitude lower than those needed for blocking viral infection (Röcker et al., 2018b). Thus, it is plausible that due to binding to serum proteins, the effective concentration of CLR01 is not sufficient for systemic antiviral activity but does achieve anti-amyloid activity. This interpretation is supported by findings that CLR01 retains antiviral activity in semen, urine, saliva, and cerebrospinal fluid—body fluids containing substantially lower albumin and total protein concentrations than serum (Röcker et al., 2018b). Whether advanced MTs similarly lose antiviral activity in serum or whether MTs can be modified in ways that preclude serum protein binding will be addressed in ongoing studies. Until these questions are answered, the proposed administration of MTs for anti-viral applications must be topical, either formulated as a microbicide to protect from acquiring sexually transmitted pathogens such as HIV-1 or HSV-2 (Lump et al., 2015; Röcker et al., 2018b) or applied as nasal or oral sprays to protect or treat respiratory viral pathogens. Such sprays may be particularly important against emerging enveloped viruses, such as SARS-CoV-2.
VII. Conclusion
MTs are broad-spectrum inhibitors of protein aggregation and facilitators of the clearance of abnormal protein assemblies. A lead compound, CLR01, inhibits aggregation of approximately 20 amyloidogenic proteins and many of their disease-associated variants involved in neurodegenerative diseases and other proteinopathies. The broad-spectrum activity of MTs is achieved thanks to their unique mechanism of action, binding to exposed Lys and Arg residues, primarily in disordered proteins, rather than to a specific protein. At the same time, the moderate affinity and high on-off rate of the binding allows MTs to disrupt the process of abnormal protein self-assembly without affecting the structure or function of normal proteins.
An important feature of MTs is their ability to dissociate pre-formed fibrils of different amyloidogenic proteins. This ability allows them to be used not just for prevention of disease in the prodromal stage but also for treatment at later stages, when protein aggregation and deposition already has taken place. Dissociation of pre-formed fibrils of disease-associated protein in vitro is a slow process as the labile binding of the MT is the only force preventing monomers from re-associating with the fibril (Fig. 9), whereas, in vivo, the MTs join the cellular clearance mechanisms, facilitating substantially more efficient dissociation of existing aggregates, as has been demonstrated for Aβ, α-syn, and SOD1. A distinct case is functional amyloids, such as those in bacterial biofilm or semen-associated amyloids that enhance HIV infection. These amyloids are held together by weaker forces than those that cause proteinopathy and can be destroyed efficiently by MTs themselves.
The anti-aggregation and clearance facilitation activities of MTs translate into protection of cells, tissues and organisms from the cytotoxicity of amyloidogenic proteins. In support of the process-specific mechanism of these compounds, CLR01 has been shown to protect multiple cell types, including cell lines and primary cultured cells from the toxicity caused by many different proteins at concentrations well below those that cause cytotoxicity of the compound itself. Recent experiments have shown that MTs concentrate in the same compartments where the cells direct misfolded and aggregated proteins, primarily lysosomes, in neurons, astrocytes, kidney cells, and likely many other cell types. The concentration of MTs in these compartments, and their action facilitating clearance of amyloidogenic proteins, not only help cells mitigate the toxicity of protein oligomers and aggregates but also prevents prion-like cell-to-cell spreading of the offending proteins, as has been demonstrated in biosensor cell lines and animal models.
The demonstration of CLR01’s therapeutic efficacy in multiple cellular and animal models and the high safety profile of the compound support development of MTs as therapeutic agents against multiple proteinopathies and related conditions. The high efficacy has been demonstrated despite suboptimal pharmacokinetics, thanks to the atypical pharmacodynamics of CLR01 and its slow clearance from the brain, which allow it to act catalytically and achieve strong therapeutic effects even when administered at low doses (e.g., 0.14 mg/Kg) at intervals substantially less frequent (twice weekly) that would be required based simply on its plasma half-life (2–3 hours). At the same time, recent advances in chemistry needed for functionalization of the bridgehead groups suggest that derivatives and/or pro-drugs with improved pharmacokinetics can be obtained in the near future, which would offer even more attractive drug candidates for formal development and initiation of clinical trials.
Acknowledgments
GB gratefully acknowledges support from the Jim Easton Consortium for Alzheimer’s Drug Discovery and Biomarker Development at UCLA, NIH/NIEHS [Grant P01-ES016732], NIH/NINDS [Grant P50-NS38367], NIH/NIA [Grant P50-AG016570], NIH/NCRR [Grant UL1-TR000124], NIH/NIA [Grant R01-AG050721], NIH/NIA [Grant RF1-AG054000], Michael J. Fox Foundation 10220, Team Parkinson/Parkinson Alliance, RJG Foundation 20095024, Cure Alzheimer’s Fund 20152631, Judith & Jean Pape Adams Charitable Foundation, RGK Foundation 20143057, CurePSP Foundation 600-6-15, MSA Coalition 20170367, Cure Sanfilippo Foundation and Sanfilippo Children’s Foundation. MII acknowledges support from UM Protein Folding Disease Initiative. ESG was funded by the Deutsche Forschungsgemeinschaft (DFG; German Research Foundation) under Germany’s Excellence Strategy–RESOLV-EXC-2033–Project number 390677874 and the Collaborative Research Center CRC 1093 Supramolecular Chemistry on Proteins, subproject A8. TS gratefully acknowledges financial support from the German Research Foundation DFG (CRC 1093: Supramolecular Chemistry on Proteins) and from the European Union [Horizon 2020 Grant: Fightn Cov].
Abbreviations
- AA
reactive amyloidosis
- Aβ
amyloid β-protein
- AD
Alzheimer’s disease
- AL
amyloidosis light chain
- ALP
autophagy-lysosome pathway
- ALS
amyotrophic lateral sclerosis
- APP
amyloid β-protein precursor
- AS-PLA
α-Synuclein proximity ligation assay
- BBB
blood–brain barrier
- CBD
corticobasal degeneration
- CFP
cyan fluorescent protein
- CNS
central nervous system
- CTE
chronic traumatic encephalopathy
- DLB
dementia with Lewy bodies
- DOPC
2-dioleoyl-sn-glycero-3-phosphocholine
- DRM
desmin-related myopathy
- ECD-MS
electron-capture dissociation coupled with mass-spectrometry
- ERK2
extracellular signal-regulated kinase 2
- FAM
6-fluorescein amidite
- FRET
fluorescence-resonance energy transfer
- FTD
frontotemporal dementia
- FUS
fused in sarcoma
- GCI
glial cytoplasmic inclusions
- GFAP
glial fibrillary acidic protein
- GFP
green-fluorescent protein
- GSN
gelsolin
- GUVs
giant unilamellar vesicles
- HCMV
human cytomegalovirus
- HD
Huntington’s diseases
- HMW
high-molecular-weight
- hpf
hours post-fertilization
- HSF1
heat shock transcription factor 1
- i.c.v.
intracerebroventricular
- IHC
immunohistochemistry
- IM-MS
ion mobility spectroscopy–mass spectrometry
- iPSC
induced pluripotent stem-cell
- ITC
isothermal calorimetry
- LBs
Lewy bodies
- LC
light chain
- LSDs
lysosomal storage diseases
- MPS III
mucopolysaccharidosis type III
- MSA
multiple system atrophy
- MTBD
microtubule binding domain
- MTs
molecular tweezers
- NPC
Niemann-Pick disease type-C
- p-tau
hyperphosphorylated tau
- PAP
prostatic acid phosphatase
- PD
Parkinson’s disease
- PFFs
preformed fibrils
- PLP
proteolipid protein
- polyQ
polyglutamine
- PS1
presenilin 1
- PSM
phenol-soluble modulin
- PSP
progressive supranuclear palsy
- QM/MM
quantum mechanics/molecular mechanics
- RAN
repeat-associated non-AUG
- RBE
rat brain extract
- REMD
replica exchange molecular dynamics
- SCI
spinal-cord injury
- SEM
semenogelin
- SN
substantia nigra
- SNc
substantia nigra pars compacta
- SOD1
superoxide dismutase 1
- SUMO
small ubiquitin-like modifier
- α-syn
α-synuclein;
- TAMRA
5-carboxytetramethylrhodamine
- TDP-43
transactive response DNA binding protein 43 kDa
- TEM
transmission electron microscopy
- TH
tyrosine hydroxylase
- ThS
thioflavin-S
- ThT
thioflavin-T
- ULP1
ubiquitin-like protease 1
- UPS
ubiquitin-proteasome system
- VMAT2
vesicular monoamine transporter 2
- YFP
yellow fluorescent protein
- ZF
zebrafish
Authorship Contributions
Participated in the research design: Shahpasand-Kroner, Siddique, Malik, Linares, Ivanova, Ichida, Weil, Münch, Sanchez-Garcia, Klärner, Schrader, Bitan.
Conducted experiments: Shahpasand-Kroner, Siddique, Malik, Linares.
Performed data analysis: Shahpasand-Kroner, Siddique, Malik, Linares, Ichida, Weil, Münch, Sanchez-Garcia, Schrader, Bitan.
Wrote or contributed to the writing of the manuscript: Shahpasand-Kroner, Siddique, Malik, Weil, Münch, Sanchez-Garcia, Klärner, Schrader, Bitan.
Footnotes
This work was supported by National Institutes of Health National Institutes of Environmental Health Sciences [Grant P01-ES016732], National Institutes of Neurologic Disorders and Stroke [Grant P50-NS38367], National Institute of Aging [Grants P50-AG016570, R01-AG050721, and RF1-AG054000], and National Center of Research Resources [Grant UL1-TR000124]; Michael J. Fox Foundation [Grant 10220]; Team Parkinson/Parkinson Alliance, RJG Foundation [Grant 20095024]; Cure Alzheimer’s Fund [Grant 20152631]; RGK Foundation [Grant 20143057]; CurePSP Foundation [Grant 600-6-15], MSA Coalition [Grant 20170367]; University of Michigan Protein Folding Disease Initiative; German Research Foundation under Germany’s Excellence Strategy RESOLV-EXC-2033 [Grant 390677874]; the Collaborative Research Center CRC 1093 Supramolecular Chemistry on Proteins; and the European Union [Horizon 2020 Grant: Fightn Cov].
G.B., T.S., and F.-G.K. are coauthors and coinventors of 2009 International Patent No. PCT/US2010/026419, USA Patent No. 8,791,092, European Patent No. EP2403859 A2. G.B. is a coauthor and coinventor of 2013 US Patent No. 10,918,657 and 2018 International Patent Application No. PCT/US2019/029221, EU application No. 19850534.9, and International Patent Application No. PCT/US2019/029222, EU application No. 19841367.6. G.B., T.S., J.M., and R.M. are coauthors and coinventors of 2018 International Patent PCT/EP2010/000437, US Patent No. US 8481484 B2, European Patent No. EP 2493859 A1.
H.S.-K. and I.S. contributed equally to this work.
Current address: Department of Integrative Biology and Physiology, University of California, Los Angeles, California.
References
- Acharya S, Safaie BM, Wongkongkathep P, Ivanova MI, Attar A, Klärner FG, Schrader T, Loo JA, Bitan G, Lapidus LJ (2014) Molecular basis for preventing α-synuclein aggregation by a molecular tweezer. J Biol Chem 289:10727–10737. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Acosta SA, Tajiri N, de la Pena I, Bastawrous M, Sanberg PR, Kaneko Y, Borlongan CV (2015) α-synuclein as a pathological link between chronic traumatic brain injury and Parkinson’s disease. J Cell Physiol 230:1024–1032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Adegbuyiro A, Sedighi F, Pilkington AW 4th, Groover S, Legleiter J (2017) Proteins containing expanded polyglutamine tracts and neurodegenerative disease. Biochemistry 56:1199–1217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ahmad B, Chen Y, Lapidus LJ (2012) Aggregation of α-synuclein is kinetically controlled by intramolecular diffusion. Proc Natl Acad Sci USA 109:2336–2341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ahmad B, Lapidus LJ (2012) Curcumin prevents aggregation in α-synuclein by increasing reconfiguration rate. J Biol Chem 287:9193–9199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alder K, Pascher F, Vagt H (1942) The accumulation of malein acid-anhydride. Chemische Berichte 75:1501–1514. [Google Scholar]
- Ali FE, Barnham KJ, Barrow CJ, Separovic F (2004) Metal catalyzed oxidation of tyrosine residues by different oxidation systems of copper/hydrogen peroxide. J Inorg Biochem 98:173–184. [DOI] [PubMed] [Google Scholar]
- Andrade F, Aldámiz-Echevarría L, Llarena M, Couce ML (2015) Sanfilippo syndrome: overall review. Pediatr Int 57:331–338. [DOI] [PubMed] [Google Scholar]
- Antzutkin ON, Balbach JJ, Leapman RD, Rizzo NW, Reed J, Tycko R (2000) Multiple quantum solid-state NMR indicates a parallel, not antiparallel, organization of β-sheets in Alzheimer’s β-amyloid fibrils. Proc Natl Acad Sci USA 97:13045–13050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arndt JR, Brown RJ, Burke KA, Legleiter J, Valentine SJ (2015) Lysine residues in the N-terminal huntingtin amphipathic α-helix play a key role in peptide aggregation. J Mass Spectrom 50:117–126. [DOI] [PubMed] [Google Scholar]
- Arnold F, Schnell J, Zirafi O, Stürzel C, Meier C, Weil T, Ständker L, Forssmann WG, Roan NR, Greene WC, et al. (2012) Naturally occurring fragments from two distinct regions of the prostatic acid phosphatase form amyloidogenic enhancers of HIV infection. J Virol 86:1244–1249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arora RD, Khan YS(2021) Motor neuron disease, in StatPearls, Treasure Island, FL. [PubMed] [Google Scholar]
- Arriagada PV, Growdon JH, Hedley-Whyte ET, Hyman BT (1992) Neurofibrillary tangles but not senile plaques parallel duration and severity of Alzheimer’s disease. Neurology 42:631–639. [DOI] [PubMed] [Google Scholar]
- Atasoy B, Bayramoglu F, Hoekelek T (1994) A concise synthesis of the 2,3-dimethylene-1,4-methano-1,2,3,4-tetrahydronaphthalene and its reaction with singlet oxygen. Tetrahedron 50:5753–5764. [Google Scholar]
- Atlasi RS, Malik R, Corrales CI, Tzeplaeff L, Whitelegge JP, Cashman NR, Bitan G (2018) Investigation of anti-SOD1 antibodies yields new structural insight into SOD1 misfolding and surprising behavior of the antibodies themselves. ACS Chem Biol 13:2794–2807. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Attar A, Bitan G (2014) Disrupting self-assembly and toxicity of amyloidogenic protein oligomers by “molecular tweezers”—from the test tube to animal models. Curr Pharm Des 20:2469–2483. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Attar A, Chan WT, Klärner FG, Schrader T, Bitan G (2014) Safety and pharmacological characterization of the molecular tweezer CLR01—a broad-spectrum inhibitor of amyloid proteins’ toxicity. BMC Pharmacol Toxicol 15:23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Attar A, Ripoli C, Riccardi E, Maiti P, Li Puma DD, Liu T, Hayes J, Jones MR, Lichti-Kaiser K, Yang F, et al. (2012) Protection of primary neurons and mouse brain from Alzheimer’s pathology by molecular tweezers. Brain 135:3735–3748. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bartels T, Choi JG, Selkoe DJ (2011) α-Synuclein occurs physiologically as a helically folded tetramer that resists aggregation. Nature 477:107–110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beck M (2018) Treatment strategies for lysosomal storage disorders. Dev Med Child Neurol 60:13–18. [DOI] [PubMed] [Google Scholar]
- Bengoa-Vergniory N, Faggiani E, Ramos-Gonzalez P, Kirkiz E, Connor-Robson N, Brown LV, Siddique I, Li Z, Vingill S, Cioroch M, et al. (2020) CLR01 protects dopaminergic neurons in vitro and in mouse models of Parkinson’s disease. Nat Commun 11:4885. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Benkhoff J, Boese R, Klärner FG (1997) Synthesis of sterically rigid macrocycles by the use of pressure-induced repetitive Diels-Alder reactions. Liebigs Annalen 3:501–516. [Google Scholar]
- Bernal-Conde LD, Ramos-Acevedo R, Reyes-Hernández MA, Balbuena-Olvera AJ, Morales-Moreno ID, Argüero-Sánchez R, Schüle B, Guerra-Crespo M (2020) Alpha-synuclein physiology and pathology: a perspective on cellular structures and organelles. Front Neurosci 13:1399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bhaumik M, Muller VJ, Rozaklis T, Johnson L, Dobrenis K, Bhattacharyya R, Wurzelmann S, Finamore P, Hopwood JJ, Walkley SU, et al. (1999) A mouse model for mucopolysaccharidosis type III A (Sanfilippo syndrome). Glycobiology 9:1389–1396. [DOI] [PubMed] [Google Scholar]
- Bi M, Gladbach A, van Eersel J, Ittner A, Przybyla M, van Hummel A, Chua SW, van der Hoven J, Lee WS, Müller J, et al. (2017) Tau exacerbates excitotoxic brain damage in an animal model of stroke. Nat Commun 8:473. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bier D, Mittal S, Bravo-Rodriguez K, Sowislok A, Guillory X, Briels J, Heid C, Bartel M, Wettig B, Brunsveld L, et al. (2017) The molecular tweezer cLR01 stabilizes a disordered protein-protein interface. J Am Chem Soc 139:16256–16263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bier D, Rose R, Bravo-Rodriguez K, Bartel M, Ramirez-Anguita JM, Dutt S, Wilch C, Klärner FG, Sanchez-Garcia E, Schrader T, et al. (2013) Molecular tweezers modulate 14-3-3 protein-protein interactions. Nat Chem 5:234–239. [DOI] [PubMed] [Google Scholar]
- Bitan G (2019) Disease-modifying therapy for proteinopathies: can the exception become the rule? Prog Mol Biol Transl Sci 168:277–287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bitan G, Fraldi A, Auricchio A, Monaco A (2018) Inhibition of lipofuscin aggregation by molecular tweezers. International Application No. PCT/US2019/029221
- Blake CC, Geisow MJ, Oatley SJ, Rérat B, Rérat C (1978) Structure of prealbumin: secondary, tertiary and quaternary interactions determined by Fourier refinement at 1.8 A. J Mol Biol 121:339–356. [DOI] [PubMed] [Google Scholar]
- Blanco LP, Evans ML, Smith DR, Badtke MP, Chapman MR (2012) Diversity, biogenesis and function of microbial amyloids. Trends Microbiol 20:66–73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bradford JW, Li S, Li XJ (2010) Polyglutamine toxicity in non-neuronal cells. Cell Res 20:400–407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Breiden B, Sandhoff K (2019) Lysosomal glycosphingolipid storage diseases. Annu Rev Biochem 88:461–485. [DOI] [PubMed] [Google Scholar]
- Brenner S, Braun B, Read C, Weil T, Walther P, Schrader T, Münch J, von Einem J (2021) The molecular tweezer CLR01 inhibits antibody-resistant cell-to-cell spread of human cytomegalovirus. Viruses 13:13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bridier A, Briandet R, Thomas V, Dubois-Brissonnet F (2011) Resistance of bacterial biofilms to disinfectants: a review. Biofouling 27:1017–1032. [DOI] [PubMed] [Google Scholar]
- Brooks SP, Dunnett SB (2009) Tests to assess motor phenotype in mice: a user’s guide. Nat Rev Neurosci 10:519–529. [DOI] [PubMed] [Google Scholar]
- Brown DG, Shorter J, Wobst HJ (2020) Emerging small-molecule therapeutic approaches for amyotrophic lateral sclerosis and frontotemporal dementia. Bioorg Med Chem Lett 30:126942. [DOI] [PubMed] [Google Scholar]
- Bugg CW, Isas JM, Fischer T, Patterson PH, Langen R (2012) Structural features and domain organization of huntingtin fibrils. J Biol Chem 287:31739–31746. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bullock AN, Henckel J, Fersht AR (2000) Quantitative analysis of residual folding and DNA binding in mutant p53 core domain: definition of mutant states for rescue in cancer therapy. Oncogene 19:1245–1256. [DOI] [PubMed] [Google Scholar]
- Busch DJ, Morgan JR (2012) Synuclein accumulation is associated with cell-specific neuronal death after spinal cord injury. J Comp Neurol 520:1751–1771. [DOI] [PubMed] [Google Scholar]
- Bush AI (2003) The metallobiology of Alzheimer’s disease. Trends Neurosci 207–214.26: 207–214. [DOI] [PubMed] [Google Scholar]
- Butler DN, Snow RA (1975) Chemistry of proximal-bond systems. Part III. Synthesis of substituted benzopolycyclic hydrocarbons. Can J Chem 53:256–262. [Google Scholar]
- Buxbaum J (1996) The amyloidoses. Mt Sinai J Med 63:16–23. [PubMed] [Google Scholar]
- Çakar A, Durmuş-Tekçe H, Parman Y (2019) Familial amyloid polyneuropathy. Noro Psikiyatri Arsivi 56:150–156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cardoso I, Almeida MR, Ferreira N, Arsequell G, Valencia G, Saraiva MJ (2007) Comparative in vitro and ex vivo activities of selected inhibitors of transthyretin aggregation: relevance in drug design. Biochem J 408:131–138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chang HY, Sang TK, Chiang AS (2018) Untangling the tauopathy for Alzheimer’s disease and parkinsonism. J Biomed Sci 25:54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chesselet MF, Richter F, Zhu C, Magen I, Watson MB, Subramaniam SR (2012) A progressive mouse model of Parkinson’s disease: the Thy1-aSyn (“Line 61”) mice. Neurotherapeutics 9:297–314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheung GY, Joo HS, Chatterjee SS, Otto M (2014) Phenol-soluble modulins—critical determinants of staphylococcal virulence. FEMS Microbiol Rev 38:698–719. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chiò A, Logroscino G, Traynor BJ, Collins J, Simeone JC, Goldstein LA, White LA (2013) Global epidemiology of amyotrophic lateral sclerosis: a systematic review of the published literature. Neuroepidemiology 41:118–130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clavaguera F, Hench J, Goedert M, Tolnay M (2015) Invited review: prion-like transmission and spreading of tau pathology. Neuropathol Appl Neurobiol 41:47–58. [DOI] [PubMed] [Google Scholar]
- Cohen AS (1967) Amyloidosis. N Engl J Med 277:574–583. [DOI] [PubMed] [Google Scholar]
- Conway KA, Lee SJ, Rochet JC, Ding TT, Harper JD, Williamson RE, Lansbury PT Jr (2000) Accelerated oligomerization by Parkinson’s disease linked α-synuclein mutants. Ann N Y Acad Sci 920:42–45. [DOI] [PubMed] [Google Scholar]
- Conway KA, Rochet JC, Bieganski RM, Lansbury PT Jr (2001) Kinetic stabilization of the α-synuclein protofibril by a dopamine-α-synuclein adduct. Science 294:1346–1349. [DOI] [PubMed] [Google Scholar]
- Daoutsali E, Hailu TT, Buijsen RAM, Pepers BA, van der Graaf LM, Verbeek MM, Curtis D, de Vlaam T, van Roon-Mom WMC (2021) Antisense oligonucleotide-induced amyloid precursor protein splicing modulation as a therapeutic approach for Dutch-type cerebral amyloid angiopathy. Nucleic Acid Ther 31:351–363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Delenclos M, Burgess JD, Lamprokostopoulou A, Outeiro TF, Vekrellis K, McLean PJ (2019) Cellular models of α-synuclein toxicity and aggregation. J Neurochem 150:566–576. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deng HX, Hentati A, Tainer JA, Iqbal Z, Cayabyab A, Hung WY, Getzoff ED, Hu P, Herzfeldt B, Roos RP, et al. (1993) Amyotrophic lateral sclerosis and structural defects in Cu,Zn superoxide dismutase. Science 261:1047–1051. [DOI] [PubMed] [Google Scholar]
- Desai HV, Aronow WS, Peterson SJ, Frishman WH (2010) Cardiac amyloidosis: approaches to diagnosis and management. Cardiol Rev 18:1–11. [DOI] [PubMed] [Google Scholar]
- Desplats P, Lee HJ, Bae EJ, Patrick C, Rockenstein E, Crews L, Spencer B, Masliah E, Lee SJ (2009) Inclusion formation and neuronal cell death through neuron-to-neuron transmission of α-synuclein. Proc Natl Acad Sci USA 106:13010–13015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Despres C, Di J, Cantrelle FX, Li Z, Huvent I, Chambraud B, Zhao J, Chen J, Chen S, Lippens G, et al. (2019) Major differences between the self-assembly and seeding behavior of heparin-induced and in vitro phosphorylated tau and their modulation by potential inhibitors. ACS Chem Biol 14:1363–1379. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Di J, Siddique I, Li Z, Malki G, Hornung S, Dutta S, Hurst I, Ishaaya E, Wang A, Tu S, et al. (2021) The molecular tweezer CLR01 improves behavioral deficits and reduces tau pathology in P301S-tau transgenic mice. Alzheimers Res Ther 13:6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dickson DW (2001) α-Synuclein and the Lewy body disorders. Curr Opin Neurol 14:423–432. [DOI] [PubMed] [Google Scholar]
- Dunham NW, Miya TS (1957) A note on a simple apparatus for detecting neurological deficit in rats and mice. J Am Pharm Assoc 46:208–209. [DOI] [PubMed] [Google Scholar]
- Dutt S, Wilch C, Gersthagen T, Talbiersky P, Bravo-Rodriguez K, Hanni M, Sánchez-García E, Ochsenfeld C, Klärner FG, Schrader T (2013a) Molecular tweezers with varying anions: a comparative study. J Org Chem 78:6721–6734. [DOI] [PubMed] [Google Scholar]
- Dutt S, Wilch C, Gersthagen T, Wölper C, Sowislok AA, Klärner F-G, Schrader T (2013b) Linker effects on amino acid and peptide recognition by molecular tweezers. Eur J Org Chem 2013:7705–7714. [Google Scholar]
- Easterhoff D, Ontiveros F, Brooks LR, Kim Y, Ross B, Silva JN, Olsen JS, Feng C, Hardy DJ, Dunman PM, et al. (2013) Semen-derived enhancer of viral infection (SEVI) binds bacteria, enhances bacterial phagocytosis by macrophages, and can protect against vaginal infection by a sexually transmitted bacterial pathogen. Antimicrob Agents Chemother 57:2443–2450. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Evans WR, Hendriksz CJ (2017) Niemann-Pick type C disease–the tip of the iceberg? A review of neuropsychiatric presentation, diagnosis and treatment. BJPsych Bull 41:109–114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fauvet B, Mbefo MK, Fares MB, Desobry C, Michael S, Ardah MT, Tsika E, Coune P, Prudent M, Lion N, et al. (2012) α-Synuclein in the central nervous system and from erythrocytes, mammalian cells and E. coli exists predominantly as a disordered monomer. J Biol Chem 287:15345–15364. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ferreira N, Pereira-Henriques A, Attar A, Klärner FG, Schrader T, Bitan G, Gales L, Saraiva MJ, Almeida MR (2014) Molecular tweezers targeting transthyretin amyloidosis. Neurotherapeutics 11:450–461. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Filocamo M, Morrone A (2011) Lysosomal storage disorders: molecular basis and laboratory testing. Hum Genomics 5:156–169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fitzmaurice AG, Rhodes SL, Cockburn M, Ritz B, Bronstein JM (2014) Aldehyde dehydrogenase variation enhances effect of pesticides associated with Parkinson disease. Neurology 82:419–426. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fitzmaurice AG, Rhodes SL, Lulla A, Murphy NP, Lam HA, O’Donnell KC, Barnhill L, Casida JE, Cockburn M, Sagasti A, et al. (2013) Aldehyde dehydrogenase inhibition as a pathogenic mechanism in Parkinson disease. Proc Natl Acad Sci USA 110:636–641. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Flemming HC, Wingender J, Szewzyk U, Steinberg P, Rice SA, Kjelleberg S (2016) Biofilms: an emergent form of bacterial life. Nat Rev Microbiol 14:563–575. [DOI] [PubMed] [Google Scholar]
- Fogerson SM, van Brummen AJ, Busch DJ, Allen SR, Roychaudhuri R, Banks SM, Klärner FG, Schrader T, Bitan G, Morgan JR (2016) Reducing synuclein accumulation improves neuronal survival after spinal cord injury. Exp Neurol 278:105–115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fokkens M, Schrader T, Klärner FG (2005) A molecular tweezer for lysine and arginine. J Am Chem Soc 127:14415–14421. [DOI] [PubMed] [Google Scholar]
- Friedler A, Veprintsev DB, Hansson LO, Fersht AR (2003) Kinetic instability of p53 core domain mutants: implications for rescue by small molecules. J Biol Chem 278:24108–24112. [DOI] [PubMed] [Google Scholar]
- Gallyas F (1971) Silver staining of Alzheimer’s neurofibrillary changes by means of physical development. Acta Morphol Acad Sci Hung 19:1–8. [PubMed] [Google Scholar]
- Gessel MM, Wu C, Li H, Bitan G, Shea JE, Bowers MT (2012) Aβ(39-42) modulates Aβ oligomerization but not fibril formation. Biochemistry 51:108–117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ghiglieri V, Calabrese V, Calabresi P (2018) α-Synuclein: from early synaptic dysfunction to neurodegeneration. Front Neurol 9:295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goedert M (2015) Neurodegeneration. Alzheimer’s and Parkinson’s diseases: the prion concept in relation to assembled Aβ, tau, and α-synuclein. Science 349:1255555. [DOI] [PubMed] [Google Scholar]
- Goedert M, Jakes R, Spillantini MG, Hasegawa M, Smith MJ, Crowther RA (1996) Assembly of microtubule-associated protein tau into Alzheimer-like filaments induced by sulphated glycosaminoglycans. Nature 383:550–553. [DOI] [PubMed] [Google Scholar]
- Golde TE, Borchelt DR, Giasson BI, Lewis J (2013) Thinking laterally about neurodegenerative proteinopathies. J Clin Invest 123:1847–1855. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grad LI, Yerbury JJ, Turner BJ, Guest WC, Pokrishevsky E, O’Neill MA, Yanai A, Silverman JM, Zeineddine R, Corcoran L, et al. (2014) Intercellular propagated misfolding of wild-type Cu/Zn superoxide dismutase occurs via exosome-dependent and -independent mechanisms. Proc Natl Acad Sci USA 111:3620–3625. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gupta R, Lan M, Mojsilovic-Petrovic J, Choi WH, Safren N, Barmada S, Lee MJ, Kalb R (2017) The proline/arginine dipeptide from hexanucleotide repeat expanded C9ORF72 inhibits the proteasome. eNeuro 4:ENEURO.0249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gurney ME, Pu H, Chiu AY, Dal Canto MC, Polchow CY, Alexander DD, Caliendo J, Hentati A, Kwon YW, Deng HX, et al. (1994) Motor neuron degeneration in mice that express a human Cu,Zn superoxide dismutase mutation. Science 264:1772–1775. [DOI] [PubMed] [Google Scholar]
- Hadrovic I, Rebmann P, Klärner FG, Bitan G, Schrader T (2019) Molecular lysine tweezers counteract aberrant protein aggregation. Front Chem 7:657. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hardy J, Selkoe DJ (2002) The amyloid hypothesis of Alzheimer’s disease: progress and problems on the road to therapeutics. Science 297:353–356. [DOI] [PubMed] [Google Scholar]
- Hardy JA, Higgins GA (1992) Alzheimer’s disease: the amyloid cascade hypothesis. Science 256:184–185. [DOI] [PubMed] [Google Scholar]
- Hayden EY, Putman J, Nunez S, Shin WS, Oberoi M, Charreton M, Dutta S, Li Z, Komuro Y, Joy MT, et al. (2019) Ischemic axonal injury up-regulates MARK4 in cortical neurons and primes tau phosphorylation and aggregation. Acta Neuropathol Commun 7:135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heid C, Sowislok A, Schaller T, Niemeyer F, Klärner FG, Schrader T (2018) Molecular tweezers with additional recognition sites. Chemistry 24:11332–11343. [DOI] [PubMed] [Google Scholar]
- Herrera-Vaquero M, Bouquio D, Kallab M, Biggs K, Nair G, Ochoa J, Heras-Garvin A, Heid C, Hadrovic I, Poewe W, et al. (2019) The molecular tweezer CLR01 reduces aggregated, pathologic, and seeding-competent α-synuclein in experimental multiple system atrophy. Biochim Biophys Acta Mol Basis Dis 1865:165513. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Herzog G, Shmueli MD, Levy L, Engel L, Gazit E, Klärner FG, Schrader T, Bitan G, Segal D (2015) The lys-specific molecular tweezer, clr01, modulates aggregation of the mutant p53 DNA binding domain and inhibits its toxicity. Biochemistry 54:3729–3738. [DOI] [PubMed] [Google Scholar]
- Hijaz BA, Volpicelli-Daley LA (2020) Initiation and propagation of α-synuclein aggregation in the nervous system. Mol Neurodegener 15:19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hogeweg A, Sowislok A, Schrader T, Beuck C (2017) An NMR method to pinpoint supramolecular ligand binding to basic residues on proteins. Angew Chem Int Ed Engl 56:14758–14762. [DOI] [PubMed] [Google Scholar]
- Holmes BB, Furman JL, Mahan TE, Yamasaki TR, Mirbaha H, Eades WC, Belaygorod L, Cairns NJ, Holtzman DM, Diamond MI (2014) Proteopathic tau seeding predicts tauopathy in vivo. Proc Natl Acad Sci USA 111:E4376–E4385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoop CL, Lin HK, Kar K, Hou Z, Poirier MA, Wetzel R, van der Wel PC (2014) Polyglutamine amyloid core boundaries and flanking domain dynamics in huntingtin fragment fibrils determined by solid-state nuclear magnetic resonance. Biochemistry 53:6653–6666. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hourahine B, Aradi B, Blum V, Bonafé F, Buccheri A, Camacho C, Cevallos C, Deshaye MY, Dumitrică T, Dominguez A, et al. (2020) DFTB+, a software package for efficient approximate density functional theory based atomistic simulations. J Chem Phys 152:124101. [DOI] [PubMed] [Google Scholar]
- Iqbal K, Liu F, Gong CX, Grundke-Iqbal I (2010) Tau in Alzheimer disease and related tauopathies. Curr Alzheimer Res 7:656–664. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jabbari E, Duff KE (2021) Tau-targeting antibody therapies: too late, wrong epitope or wrong target? Nat Med 27:1341–1342. [DOI] [PubMed] [Google Scholar]
- Jackson JW, Sparer T (2018) There is always another way! Cytomegalovirus’ multifaceted dissemination schemes. Viruses 10:383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Janezic S, Threlfell S, Dodson PD, Dowie MJ, Taylor TN, Potgieter D, Parkkinen L, Senior SL, Anwar S, Ryan B, et al. (2013) Deficits in dopaminergic transmission precede neuron loss and dysfunction in a new Parkinson model. Proc Natl Acad Sci USA 110:E4016–E4025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jayaraman M, Kodali R, Sahoo B, Thakur AK, Mayasundari A, Mishra R, Peterson CB, Wetzel R (2012) Slow amyloid nucleation via α-helix-rich oligomeric intermediates in short polyglutamine-containing huntingtin fragments. J Mol Biol 415:881–899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jin LW, Shie FS, Maezawa I, Vincent I, Bird T (2004) Intracellular accumulation of amyloidogenic fragments of amyloid-beta precursor protein in neurons with Niemann-Pick type C defects is associated with endosomal abnormalities. Am J Pathol 164:975–985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Joerger AC, Fersht AR (2007) Structure-function-rescue: the diverse nature of common p53 cancer mutants. Oncogene 26:2226–2242. [DOI] [PubMed] [Google Scholar]
- Jucker M, Walker LC (2013) Self-propagation of pathogenic protein aggregates in neurodegenerative diseases. Nature 501:45–51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Karpinar DP, Balija MB, Kügler S, Opazo F, Rezaei-Ghaleh N, Wender N, Kim HY, Taschenberger G, Falkenburger BH, Heise H, et al. (2009) Pre-fibrillar α-synuclein variants with impaired β-structure increase neurotoxicity in Parkinson’s disease models. EMBO J 28:3256–3268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Katsuno M, Tanaka F, Sobue G (2012) Perspectives on molecular targeted therapies and clinical trials for neurodegenerative diseases. J Neurol Neurosurg Psychiatry 83:329–335. [DOI] [PubMed] [Google Scholar]
- Kayed R, Dettmer U, Lesné SE (2020) Soluble endogenous oligomeric α-synuclein species in neurodegenerative diseases: Expression, spreading, and cross-talk. J Parkinsons Dis 10:791–818. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kirkitadze MD, Bitan G, Teplow DB (2002) Paradigm shifts in Alzheimer’s disease and other neurodegenerative disorders: the emerging role of oligomeric assemblies. J Neurosci Res 69:567–577. [DOI] [PubMed] [Google Scholar]
- Kirkitadze MD, Condron MM, Teplow DB (2001) Identification and characterization of key kinetic intermediates in amyloid β-protein fibrillogenesis. J Mol Biol 312:1103–1119. [DOI] [PubMed] [Google Scholar]
- Klärner F-G, Burkert U, Kamieth M, Boese R, Benet-Buchholz J (1999) Molecular tweezers as synthetic receptors: molecular recognition of electron-deficient aromatic and aliphatic substrates. Chemistry 5:1700–1707. [Google Scholar]
- Klärner F-G, Polkowska J, Panitzky J, Seelbach UP, Burkert U, Kamieth M, Baumann M, Wigger AE, Boese R, Bläser D (2004) Effect of substituents on the complexation of aromatic and quinoid substrates with molecular tweezers and clips. Eur J Org Chem 7:1405–1423. [Google Scholar]
- Klärner FG, Benkhoff J, Boese R, Burkert U, Kamieth M, Naatz U (1996) Molecular tweezers as synthetic receptors in host-guest chemistry: Inclusion of cyclohexane and self-assembly of aliphatic side chains. Angew Chem Int Ed Engl 35:1130–1133. [Google Scholar]
- Klärner FG, Burkert U, Kamieth M, Boese R (2000) Molecular tweezers as synthetic receptors: molecular recognition of neutral and cationic aromatic substrates. A comparison between the supramolecular structures in crystal and in solution. J Phys Org Chem 13:604–611. [Google Scholar]
- Klärner FG, Kahlert B (2003) Molecular tweezers and clips as synthetic receptors. Molecular recognition and dynamics in receptor-substrate complexes. Acc Chem Res 36:919–932. [DOI] [PubMed] [Google Scholar]
- Klärner FG, Kahlert B, Nellesen A, Zienau J, Ochsenfeld C, Schrader T (2006) Molecular tweezer and clip in aqueous solution: unexpected self-assembly, powerful host-guest complex formation, quantum chemical 1H NMR shift calculation. J Am Chem Soc 128:4831–4841. [DOI] [PubMed] [Google Scholar]
- Klärner FG, Schrader T (2013) Aromatic interactions by molecular tweezers and clips in chemical and biological systems. Acc Chem Res 46:967–978. [DOI] [PubMed] [Google Scholar]
- Kluve-Beckerman B, Hardwick, Du L, Benson MD (2011) AA amyloidosis: potential therapy with antisense oligonucleotides. Amyloid 18(Suppl 1):200–202. [DOI] [PubMed] [Google Scholar]
- Knopman DS, Jones DT, Greicius MD (2021) Failure to demonstrate efficacy of aducanumab: An analysis of the EMERGE and ENGAGE trials as reported by Biogen, December 2019. Alzheimers Dement 17:696–701. [DOI] [PubMed] [Google Scholar]
- Knowles TP, Vendruscolo M, Dobson CM (2014) The amyloid state and its association with protein misfolding diseases. Nat Rev Mol Cell Biol 15:384–396. [DOI] [PubMed] [Google Scholar]
- Lacor PN, Buniel MC, Furlow PW, Clemente AS, Velasco PT, Wood M, Viola KL, Klein WL (2007) Abeta oligomer-induced aberrations in synapse composition, shape, and density provide a molecular basis for loss of connectivity in Alzheimer’s disease. J Neurosci 27:796–807. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Larsson M, Pettersson T, Carlström A (1985) Thyroid hormone binding in serum of 15 vertebrate species: isolation of thyroxine-binding globulin and prealbumin analogs. Gen Comp Endocrinol 58:360–375. [DOI] [PubMed] [Google Scholar]
- Lasagna-Reeves CA, Castillo-Carranza DL, Sengupta U, Clos AL, Jackson GR, Kayed R (2011) Tau oligomers impair memory and induce synaptic and mitochondrial dysfunction in wild-type mice. Mol Neurodegener 6:39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lashuel HA, Lai Z, Kelly JW (1998) Characterization of the transthyretin acid denaturation pathways by analytical ultracentrifugation: implications for wild-type, V30M, and L55P amyloid fibril formation. Biochemistry 37:17851–17864. [DOI] [PubMed] [Google Scholar]
- Lazo ND, Grant MA, Condron MC, Rigby AC, Teplow DB (2005) On the nucleation of amyloid β-protein monomer folding. Protein Sci 14:1581–1596. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Le MH, Taghuo K ES, Schrader T (2022) Molecular tweezers—a new class of potent broad-spectrum antivirals against enveloped viruses. Chem Commun (Camb) 58:2954–2966. [DOI] [PubMed] [Google Scholar]
- Lee HH, Choi TS, Lee SJ, Lee JW, Park J, Ko YH, Kim WJ, Kim K, Kim HI (2014) Supramolecular inhibition of amyloid fibrillation by cucurbit[7]uril. Angew Chem Int Ed Engl 53:7461–7465. [DOI] [PubMed] [Google Scholar]
- Lee SJ, Desplats P, Sigurdson C, Tsigelny I, Masliah E (2010) Cell-to-cell transmission of non-prion protein aggregates. Nat Rev Neurol 6:702–706. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li Z, Siddique I, Hadrović I, Kirupakaran A, Li J, Zhang Y, Klärner FG, Schrader T, Bitan G (2021) Lysine-selective molecular tweezers are cell penetrant and concentrate in lysosomes. Commun Biol 4:1076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Limoli DH, Jones CJ, Wozniak DJ (2015) Bacterial extracellular polysaccharides in biofilm formation and function. Microbiol Spectr 3:10.1128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin HK, Boatz JC, Krabbendam IE, Kodali R, Hou Z, Wetzel R, Dolga AM, Poirier MA, van der Wel PCA (2017) Fibril polymorphism affects immobilized non-amyloid flanking domains of huntingtin exon1 rather than its polyglutamine core. Nat Commun 8:15462. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lindau M, Almkvist O, Kushi J, Boone K, Johansson SE, Wahlund LO, Cummings JL, Miller BL (2000) First symptoms—frontotemporal dementia versus Alzheimer’s disease. Dement Geriatr Cogn Disord 11:286–293. [DOI] [PubMed] [Google Scholar]
- Lipinski CA, Lombardo F, Dominy BW, Feeney PJ (2001) Experimental and computational approaches to estimate solubility and permeability in drug discovery and development settings. Adv Drug Deliv Rev 46:3–26. [DOI] [PubMed] [Google Scholar]
- Liu T, Bitan G (2012) Modulating self-assembly of amyloidogenic proteins as a therapeutic approach for neurodegenerative diseases: strategies and mechanisms. ChemMedChem 7:359–374. [DOI] [PubMed] [Google Scholar]
- Lopes DH, Attar A, Nair G, Hayden EY, Du Z, McDaniel K, Dutt S, Bravo-Rodriguez K, Mittal S, Klärner FG, et al. (2015) Molecular tweezers inhibit islet amyloid polypeptide assembly and toxicity by a new mechanism. ACS Chem Biol 10:1555–1569. [DOI] [PubMed] [Google Scholar]
- Lorizate M, Sachsenheimer T, Glass B, Habermann A, Gerl MJ, Kräusslich HG, Brügger B (2013) Comparative lipidomics analysis of HIV-1 particles and their producer cell membrane in different cell lines. Cell Microbiol 15:292–304. [DOI] [PubMed] [Google Scholar]
- Lovell MA, Robertson JD, Teesdale WJ, Campbell JL, Markesbery WR (1998) Copper, iron and zinc in Alzheimer’s disease senile plaques. J Neurol Sci 158:47–52. [DOI] [PubMed] [Google Scholar]
- Luk KC, Kehm V, Carroll J, Zhang B, O’Brien P, Trojanowski JQ, Lee VM (2012) Pathological α-synuclein transmission initiates Parkinson-like neurodegeneration in nontransgenic mice. Science 338:949–953. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lulla A, Barnhill L, Bitan G, Ivanova MI, Nguyen B, O’Donnell K, Stahl MC, Yamashiro C, Klärner FG, Schrader T, et al. (2016) Neurotoxicity of the Parkinson disease-associated pesticide ziram is synuclein-dependent in zebrafish embryos. Environ Health Perspect 124:1766–1775. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lump E, Castellano LM, Meier C, Seeliger J, Erwin N, Sperlich B, Stürzel CM, Usmani S, Hammond RM, von Einem J, et al. (2015) A molecular tweezer antagonizes seminal amyloids and HIV infection. eLife 4:e05397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maina KN, Smet-Nocca C, Bitan G (2022) Using FRET-based biosensor cells to study the seeding activity of tau and α- synuclein, in Protein Aggregation: Methods and Protocols (Cieplak A ed) pp. 125–145, Springer, New York. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Malik R, Di J, Nair G, Attar A, Taylor K, Teng E, Klärner FG, Schrader T, Bitan G (2018) Using molecular tweezers to remodel abnormal protein self-assembly and inhibit the toxicity of amyloidogenic proteins. Methods Mol Biol 1777:369–386. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Malik R, Meng H, Wongkongkathep P, Corrales CI, Sepanj N, Atlasi RS, Klärner FG, Schrader T, Spencer MJ, Loo JA, et al. (2019) The molecular tweezer CLR01 inhibits aberrant superoxide dismutase 1 (SOD1) self-assembly in vitro and in the G93A-SOD1 mouse model of ALS. J Biol Chem 294:3501–3513. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Malishev R, Salinas N, Gibson J, Eden AB, Mieres-Perez J, Ruiz-Blanco YB, Malka O, Kolusheva S, Klärner FG, Schrader T, et al. (2021) Inhibition of Staphylococcus aureus biofilm-forming functional amyloid by molecular tweezers. Cell Chem Biol 28:1310–1320.e5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mallon M, Dutt S, Schrader T, Crowley PB (2016) Protein camouflage: supramolecular anion recognition by ubiquitin. ChemBioChem 17:774–783. [DOI] [PubMed] [Google Scholar]
- Marciniuk K, Taschuk R, Napper S (2013) Evidence for prion-like mechanisms in several neurodegenerative diseases: potential implications for immunotherapy. Clin Dev Immunol 2013:473706. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martin D, Thompson MA, Nadler JV (1993) The neuroprotective agent riluzole inhibits release of glutamate and aspartate from slices of hippocampal area CA1. Eur J Pharmacol 250:473–476. [DOI] [PubMed] [Google Scholar]
- Martin ZS, Neugebauer V, Dineley KT, Kayed R, Zhang W, Reese LC, Taglialatela G (2012) α-Synuclein oligomers oppose long-term potentiation and impair memory through a calcineurin-dependent mechanism: relevance to human synucleopathic diseases. J Neurochem 120:440–452. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martínez-González L, Rodríguez-Cueto C, Cabezudo D, Bartolomé F, Andrés-Benito P, Ferrer I, Gil C, Martín-Requero Á, Fernández-Ruiz J, Martínez A, et al. (2020) Motor neuron preservation and decrease of in vivo TDP-43 phosphorylation by protein CK-1δ kinase inhibitor treatment. Sci Rep 10:4449. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mason AJ, Hurst I, Malik R, Siddique I, Solomonov I, Sagi I, Klärner FG, Schrader T, Bitan G (2020) Different inhibitors of Aβ42-induced toxicity have distinct metal-ion dependency. ACS Chem Neurosci 11:2243–2255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McCutchen SL, Lai Z, Miroy GJ, Kelly JW, Colón W (1995) Comparison of lethal and nonlethal transthyretin variants and their relationship to amyloid disease. Biochemistry 34:13527–13536. [DOI] [PubMed] [Google Scholar]
- Melov S (2002) “…and C is for clioquinol” —the Aβ Cs of Alzheimer's disease. Response. Trends in Neurosciences 25:123–124. [DOI] [PubMed] [Google Scholar]
- Mercken M, Vandermeeren M, Lübke U, Six J, Boons J, Van de Voorde A, Martin JJ, Gheuens J (1992) Monoclonal antibodies with selective specificity for Alzheimer tau are directed against phosphatase-sensitive epitopes. Acta Neuropathol 84:265–272. [DOI] [PubMed] [Google Scholar]
- Mittal S, Bravo-Rodriguez K, Sanchez-Garcia E (2018) Mechanism of inhibition of β amyloid toxicity by supramolecular tweezers. J Phys Chem B 122:4196–4205. [DOI] [PubMed] [Google Scholar]
- Monaco A, Fraldi A (2020) Protein aggregation and dysfunction of autophagy-lysosomal pathway: a vicious cycle in lysosomal storage diseases. Front Mol Neurosci 13:37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Monaco A, Maffia V, Sorrentino NC, Sambri I, Ezhova Y, Giuliano T, Cacace V, Nusco E, De Risi M, De Leonibus E, et al. (2020) The amyloid inhibitor CLR01 relieves autophagy and ameliorates neuropathology in a severe lysosomal storage disease. Mol Ther 28:1167–1176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moretti R, Torre P, Antonello RM, Cazzato G (2001) Fronto-temporal dementia versus Alzheimer disease. Arch Gerontol Geriatr Suppl 7:273–278. [DOI] [PubMed] [Google Scholar]
- Mullane K, Williams M (2020) Alzheimer’s disease beyond amyloid: can the repetitive failures of amyloid-targeted therapeutics inform future approaches to dementia drug discovery? Biochem Pharmacol 177:113945. [DOI] [PubMed] [Google Scholar]
- Mullard A (2021) FDA approval for Biogen’s aducanumab sparks Alzheimer disease firestorm. Nat Rev Drug Discov 20:496. [DOI] [PubMed] [Google Scholar]
- Münch J, Rücker E, Ständker L, Adermann K, Goffinet C, Schindler M, Wildum S, Chinnadurai R, Rajan D, Specht A, et al. (2007) Semen-derived amyloid fibrils drastically enhance HIV infection. Cell 131:1059–1071. [DOI] [PubMed] [Google Scholar]
- Nimmrich V, Ebert U (2009) Is Alzheimer’s disease a result of presynaptic failure? Synaptic dysfunctions induced by oligomeric β-amyloid. Rev Neurosci 20:1–12. [DOI] [PubMed] [Google Scholar]
- Nshanian M, Lantz C, Wongkongkathep P, Schrader T, Klärner F-G, Blümke A, Despres C, Ehrmann M, Smet-Nocca C, Bitan G, et al. (2019) Native top-down mass spectrometry and ion mobility spectrometry of the interaction of tau protein with a molecular tweezer assembly modulator. J Am Soc Mass Spectrom 30:16–23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Obach RS, Walsky RL, Venkatakrishnan K, Gaman EA, Houston JB, Tremaine LM (2006) The utility of in vitro cytochrome P450 inhibition data in the prediction of drug-drug interactions. J Pharmacol Exp Ther 316:336–348. [DOI] [PubMed] [Google Scholar]
- Oddo S, Caccamo A, Shepherd JD, Murphy MP, Golde TE, Kayed R, Metherate R, Mattson MP, Akbari Y, LaFerla FM (2003) Triple-transgenic model of Alzheimer’s disease with plaques and tangles: intracellular Abeta and synaptic dysfunction. Neuron 39:409–421. [DOI] [PubMed] [Google Scholar]
- Onufriev AV, Case DA (2019) Generalized born implicit solvent models for biomolecules. Annu Rev Biophys 48:275–296. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Outeiro TF, Putcha P, Tetzlaff JE, Spoelgen R, Koker M, Carvalho F, Hyman BT, McLean PJ (2008) Formation of toxic oligomeric α-synuclein species in living cells. PLoS One 3:e1867. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pattison JS, Robbins J (2008) Protein misfolding and cardiac disease: establishing cause and effect. Autophagy 4:821–823. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pham E, Crews L, Ubhi K, Hansen L, Adame A, Cartier A, Salmon D, Galasko D, Michael S, Savas JN, et al. (2010) Progressive accumulation of amyloid-β oligomers in Alzheimer’s disease and in amyloid precursor protein transgenic mice is accompanied by selective alterations in synaptic scaffold proteins. FEBS J 277:3051–3067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Platt FM (2014) Sphingolipid lysosomal storage disorders. Nature 510:68–75. [DOI] [PubMed] [Google Scholar]
- Prabhudesai S, Sinha S, Attar A, Kotagiri A, Fitzmaurice AG, Lakshmanan R, Ivanova MI, Loo JA, Klärner FG, Schrader T, et al. (2012) A novel “molecular tweezer” inhibitor of α-synuclein neurotoxicity in vitro and in vivo. Neurotherapeutics 9:464–476. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Przybyla M, Stevens CH, van der Hoven J, Harasta A, Bi M, Ittner A, van Hummel A, Hodges JR, Piguet O, Karl T, et al. (2016) Disinhibition-like behavior in a P301S mutant tau transgenic mouse model of frontotemporal dementia. Neurosci Lett 631:24–29. [DOI] [PubMed] [Google Scholar]
- Rahimi F, Li H, Sinha S, Bitan G (2016) Modulators of amyloid β-protein (Aβ) self-assembly, in Developing Therapeutics for Alzheimer’s Disease (Wolfe MS, ed) pp. 97–191, Academic Press, Boston. [Google Scholar]
- Redondo C, Damas AM, Olofsson A, Lundgren E, Saraiva MJM (2000) Search for intermediate structures in transthyretin fibrillogenesis: soluble tetrameric Tyr78Phe TTR expresses a specific epitope present only in amyloid fibrils. J Mol Biol 304:461–470. [DOI] [PubMed] [Google Scholar]
- Řezáč J (2017) Empirical self-consistent correction for the description of hydrogen bonds in DFTB3. J Chem Theory Comput 13:4804–4817. [DOI] [PubMed] [Google Scholar]
- Richter F, Subramaniam SR, Magen I, Lee P, Hayes J, Attar A, Zhu C, Franich NR, Bove N, De La Rosa K, et al. (2017) A molecular tweezer ameliorates motor deficits in mice overexpressing α-synuclein. Neurotherapeutics 14:1107–1119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roan NR, Liu H, Usmani SM, Neidleman J, Müller JA, Avila-Herrera A, Gawanbacht A, Zirafi O, Chu S, Dong M, et al. (2014) Liquefaction of semen generates and later degrades a conserved semenogelin peptide that enhances HIV infection. J Virol 88:7221–7234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roan NR, Münch J, Arhel N, Mothes W, Neidleman J, Kobayashi A, Smith-McCune K, Kirchhoff F, Greene WC (2009) The cationic properties of SEVI underlie its ability to enhance human immunodeficiency virus infection. J Virol 83:73–80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roan NR, Sandi-Monroy N, Kohgadai N, Usmani SM, Hamil KG, Neidleman J, Montano M, Ständker L, Röcker A, Cavrois M, et al. (2017) Semen amyloids participate in spermatozoa selection and clearance. eLife 6:e24888. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roberts RF, Wade-Martins R, Alegre-Abarrategui J (2015) Direct visualization of α-synuclein oligomers reveals previously undetected pathology in Parkinson’s disease brain. Brain 138:1642–1657. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Röcker A, Roan NR, Yadav JK, Fändrich M, Münch J (2018a) Structure, function and antagonism of semen amyloids. Chem Commun (Camb) 54:7557–7569. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Röcker AE, Müller JA, Dietzel E, Harms M, Krüger F, Heid C, Sowislok A, Riber CF, Kupke A, Lippold S, et al. (2018b) The molecular tweezer CLR01 inhibits Ebola and Zika virus infection. Antiviral Res 152:26–35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rosen DR, Siddique T, Patterson D, Figlewicz DA, Sapp P, Hentati A, Donaldson D, Goto J, O’Regan JP, Deng HX, et al. (1993) Mutations in Cu/Zn superoxide dismutase gene are associated with familial amyotrophic lateral sclerosis. Nature 362:59–62. [DOI] [PubMed] [Google Scholar]
- Ruberg FL, Berk JL (2012) Transthyretin (TTR) cardiac amyloidosis. Circulation 126:1286–1300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Runwal G, Edwards RH (2021) The membrane interactions of synuclein: physiology and pathology. Annu Rev Pathol 16:465–485. [DOI] [PubMed] [Google Scholar]
- Salinas N, Colletier JP, Moshe A, Landau M (2018) Extreme amyloid polymorphism in Staphylococcus aureus virulent PSMα peptides. Nat Commun 9:3512. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sambri I, D’Alessio R, Ezhova Y, Giuliano T, Sorrentino NC, Cacace V, De Risi M, Cataldi M, Annunziato L, De Leonibus E, et al. (2017) Lysosomal dysfunction disrupts presynaptic maintenance and restoration of presynaptic function prevents neurodegeneration in lysosomal storage diseases. EMBO Mol Med 9:112–132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Santos SD, Fernandes R, Saraiva MJ (2010) The heat shock response modulates transthyretin deposition in the peripheral and autonomic nervous systems. Neurobiol Aging 31:280–289. [DOI] [PubMed] [Google Scholar]
- Saraiva MJ (1995) Transthyretin mutations in health and disease. Hum Mutat 5:191–196. [DOI] [PubMed] [Google Scholar]
- Saraiva MJ (2001) Transthyretin mutations in hyperthyroxinemia and amyloid diseases. Hum Mutat 17:493–503. [DOI] [PubMed] [Google Scholar]
- Sattentau Q (2008) Avoiding the void: cell-to-cell spread of human viruses. Nat Rev Microbiol 6:815–826. [DOI] [PubMed] [Google Scholar]
- Sawada H (2017) Clinical efficacy of edaravone for the treatment of amyotrophic lateral sclerosis. Expert Opin Pharmacother 18:735–738. [DOI] [PubMed] [Google Scholar]
- Schlecht LM, Peters BM, Krom BP, Freiberg JA, Hänsch GM, Filler SG, Jabra-Rizk MA, Shirtliff ME (2015) Systemic Staphylococcus aureus infection mediated by Candida albicans hyphal invasion of mucosal tissue. Microbiology (Reading) 161:168–181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schrader T, Bitan G, Klärner FG (2016) Molecular tweezers for lysine and arginine—powerful inhibitors of pathologic protein aggregation. Chem Commun (Camb) 52:11318–11334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schwartz K, Ganesan M, Payne DE, Solomon MJ, Boles BR (2016) Extracellular DNA facilitates the formation of functional amyloids in Staphylococcus aureus biofilms. Mol Microbiol 99:123–134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Selkoe DJ (2002) Alzheimer’s disease is a synaptic failure. Science 298:789–791. [DOI] [PubMed] [Google Scholar]
- Selkoe DJ (2008) Soluble oligomers of the amyloid β-protein impair synaptic plasticity and behavior. Behav Brain Res 192:106–113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shahim P, Politis A, van der Merwe A, Moore B, Ekanayake V, Lippa SM, Chou YY, Pham DL, Butman JA, Diaz-Arrastia R, et al. (2020) Time course and diagnostic utility of NfL, tau, GFAP, and UCH-L1 in subacute and chronic TBI. Neurology 95:e623–e636. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shankar GM, Bloodgood BL, Townsend M, Walsh DM, Selkoe DJ, Sabatini BL (2007) Natural oligomers of the Alzheimer amyloid-β protein induce reversible synapse loss by modulating an NMDA-type glutamate receptor-dependent signaling pathway. J Neurosci 27:2866–2875. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sharma PK, Rajpal N, Upadhyay S, Shaha D, Deo N (2021). Endocrinol Diabetes Nutr (Engl Ed) [published ahead of print]
- Shi KY, Mori E, Nizami ZF, Lin Y, Kato M, Xiang S, Wu LC, Ding M, Yu Y, Gall JG, et al. (2017) Toxic PRn poly-dipeptides encoded by the C9orf72 repeat expansion block nuclear import and export. Proc Natl Acad Sci USA 114:E1111–E1117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sigurdsson EM (2005) Histological staining of amyloid-β in mouse brains. Methods Mol Biol 299:299–308. [DOI] [PubMed] [Google Scholar]
- Silva AC, Lobo DD, Martins IM, Lopes SM, Henriques C, Duarte SP, Dodart JC, Nobre RJ, Pereira de Almeida L (2020) Antisense oligonucleotide therapeutics in neurodegenerative diseases: the case of polyglutamine disorders. Brain 143:407–429. [DOI] [PubMed] [Google Scholar]
- Sinha S, Du Z, Maiti P, Klärner FG, Schrader T, Wang C, Bitan G (2012a) Comparison of three amyloid assembly inhibitors: the sugar scyllo-inositol, the polyphenol epigallocatechin gallate, and the molecular tweezer CLR01. ACS Chem Neurosci 3:451–458. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sinha S, Lopes DH, Bitan G (2012b) A key role for lysine residues in amyloid β-protein folding, assembly, and toxicity. ACS Chem Neurosci 3:473–481. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sinha S, Lopes DH, Du Z, Pang ES, Shanmugam A, Lomakin A, Talbiersky P, Tennstaedt A, McDaniel K, Bakshi R, et al. (2011) Lysine-specific molecular tweezers are broad-spectrum inhibitors of assembly and toxicity of amyloid proteins. J Am Chem Soc 133:16958–16969. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sivanandam VN, Jayaraman M, Hoop CL, Kodali R, Wetzel R, van der Wel PC (2011) The aggregation-enhancing huntingtin N-terminus is helical in amyloid fibrils. J Am Chem Soc 133:4558–4566. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith BR, Santos MB, Marshall MS, Cantuti-Castelvetri L, Lopez-Rosas A, Li G, van Breemen R, Claycomb KI, Gallea JI, Celej MS, et al. (2014) Neuronal inclusions of α-synuclein contribute to the pathogenesis of Krabbe disease. J Pathol 232:509–521. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Solomonov I, Korkotian E, Born B, Feldman Y, Bitler A, Rahimi F, Li H, Bitan G, Sagi I (2012) Zn2+-Aβ40 complexes form metastable quasi-spherical oligomers that are cytotoxic to cultured hippocampal neurons. J Biol Chem 287:20555–20564. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sorrentino NC, Cacace V, De Risi M, Maffia V, Strollo S, Tedesco N, Nusco E, Romagnoli N, Ventrella D, Huang Y, et al. (2019) Enhancing the therapeutic potential of sulfamidase for the treatment of mucopolysaccharidosis IIIA. Mol Ther Methods Clin Dev 15:333–342. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spires-Jones TL, Attems J, Thal DR (2017) Interactions of pathological proteins in neurodegenerative diseases. Acta Neuropathol 134:187–205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sreedharan J, Brown RH Jr (2013) Amyotrophic lateral sclerosis: problems and prospects. Ann Neurol 74:309–316. [DOI] [PubMed] [Google Scholar]
- Stancu IC, Vasconcelos B, Terwel D, Dewachter I (2014) Models of β-amyloid induced tau-pathology: the long and “folded” road to understand the mechanism. Mol Neurodegener 9:51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stefanova N, Wenning GK (2015) Animal models of multiple system atrophy. Clin Auton Res 25:9–17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sulzer D, Edwards RH (2019) The physiological role of α-synuclein and its relationship to Parkinson’s disease. J Neurochem 150:475–486. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Talbiersky P, Bastkowski F, Klärner FG, Schrader T (2008) Molecular clip and tweezer introduce new mechanisms of enzyme inhibition. J Am Chem Soc 130:9824–9828. [DOI] [PubMed] [Google Scholar]
- Tanida I, Ueno T, Kominami E (2008) LC3 and autophagy. Methods Mol Biol 445:77–88. [DOI] [PubMed] [Google Scholar]
- Terazaki H, Ando Y, Fernandes R, Yamamura K, Maeda S, Saraiva MJ (2006) Immunization in familial amyloidotic polyneuropathy: counteracting deposition by immunization with a Y78F TTR mutant. Lab Invest 86:23–31. [DOI] [PubMed] [Google Scholar]
- Tezel G, Luo C, Yang X (2007) Accelerated aging in glaucoma: immunohistochemical assessment of advanced glycation end products in the human retina and optic nerve head. Invest Ophthalmol Vis Sci 48:1201–1211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thakur AK, Jayaraman M, Mishra R, Thakur M, Chellgren VM, Byeon IJ, Anjum DH, Kodali R, Creamer TP, Conway JF, et al. (2009) Polyglutamine disruption of the huntingtin exon 1 N terminus triggers a complex aggregation mechanism. Nat Struct Mol Biol 16:380–389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tjernberg LO, Lilliehöök C, Callaway DJE, Näslund J, Hahne S, Thyberg J, Terenius L, Nordstedt C (1997) Controlling amyloid β-peptide fibril formation with protease-stable ligands. J Biol Chem 272:12601–12605. [DOI] [PubMed] [Google Scholar]
- Tõugu V, Karafin A, Zovo K, Chung RS, Howells C, West AK, Palumaa P (2009) Zn(II)- and Cu(II)-induced non-fibrillar aggregates of amyloid-β (1-42) peptide are transformed to amyloid fibrils, both spontaneously and under the influence of metal chelators. J Neurochem 110:1784–1795. [DOI] [PubMed] [Google Scholar]
- Townsend M, Shankar GM, Mehta T, Walsh DM, Selkoe DJ (2006) Effects of secreted oligomers of amyloid β-protein on hippocampal synaptic plasticity: a potent role for trimers. J Physiol 572:477–492. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trippier PC, Zhao KT, Fox SG, Schiefer IT, Benmohamed R, Moran J, Kirsch DR, Morimoto RI, Silverman RB (2014) Proteasome activation is a mechanism for pyrazolone small molecules displaying therapeutic potential in amyotrophic lateral sclerosis. ACS Chem Neurosci 5:823–829. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trusch F, Loebach L, Wawra S, Durward E, Wuensch A, Iberahim NA, de Bruijn I, MacKenzie K, Willems A, Toloczko A, et al. (2018) Cell entry of a host-targeting protein of oomycetes requires gp96. Nat Commun 9:2347. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsai WC, Hsu CC, Chung CY, Lin MS, Li SL, Pang JH (2007) The pentapeptide KTTKS promoting the expressions of type I collagen and transforming growth factor-β of tendon cells. J Orthop Res 25:1629–1634. [DOI] [PubMed] [Google Scholar]
- Verma M, Vats A, Taneja V (2015) Toxic species in amyloid disorders: oligomers or mature fibrils. Ann Indian Acad Neurol 18:138–145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vöpel T, Bravo-Rodriguez K, Mittal S, Vachharajani S, Gnutt D, Sharma A, Steinhof A, Fatoba O, Ellrichmann G, Nshanian M, et al. (2017) Inhibition of huntingtin exon-1 aggregation by the molecular tweezer CLR01. J Am Chem Soc 139:5640–5643. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vrinten DH, Hamers FF (2003) “CatWalk” automated quantitative gait analysis as a novel method to assess mechanical allodynia in the rat; a comparison with von Frey testing. Pain 102:203–209. [DOI] [PubMed] [Google Scholar]
- Wang J, Dickson DW, Trojanowski JQ, Lee VM (1999) The levels of soluble versus insoluble brain Abeta distinguish Alzheimer’s disease from normal and pathologic aging. Exp Neurol 158:328–337. [DOI] [PubMed] [Google Scholar]
- Wang W, Perovic I, Chittuluru J, Kaganovich A, Nguyen LT, Liao J, Auclair JR, Johnson D, Landeru A, Simorellis AK, et al. (2011) A soluble α-synuclein construct forms a dynamic tetramer. Proc Natl Acad Sci USA 108:17797–17802. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Y, Hall RA, Lee M, Kamgar-Parsi A, Bi X, Baudry M (2017) The tyrosine phosphatase PTPN13/FAP-1 links calpain-2, TBI and tau tyrosine phosphorylation. Sci Rep 7:11771. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Y, Tian D, Wei C, Cui V, Wang H, Zhu Y, Wu A, Yue Y (2020) Propofol attenuates α-synuclein aggregation and neuronal damage in a mouse model of ischemic stroke. Neurosci Bull 36:289–298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ward SM, Himmelstein DS, Lancia JK, Fu Y, Patterson KR, Binder LI (2013) TOC1: characterization of a selective oligomeric tau antibody. J Alzheimers Dis 37:593–602. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Watt NT, Whitehouse IJ, Hooper NM (2010) The role of zinc in Alzheimer’s disease. Int J Alzheimers Dis 2011:971021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weil T, Groß R, Röcker A, Bravo-Rodriguez K, Heid C, Sowislok A, Le MH, Erwin N, Dwivedi M, Bart SM, et al. (2020) Supramolecular mechanism of viral envelope disruption by molecular tweezers. J Am Chem Soc 142:17024–17038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wenger DA, Rafi MA, Luzi P (1997) Molecular genetics of Krabbe disease (globoid cell leukodystrophy): diagnostic and clinical implications. Hum Mutat 10:268–279. [DOI] [PubMed] [Google Scholar]
- Westermark P, Bergström J, Solomon A, Murphy C, Sletten K (2003) Transthyretin-derived senile systemic amyloidosis: clinicopathologic and structural considerations. Amyloid 10 (Suppl 1):48–54. [PubMed] [Google Scholar]
- Wilch C, Talbiersky P, Berchner-Pfannschmidt U, Schaller T, Kirsch M, Klärner FG, Schrader T (2017) Molecular tweezers inhibit PARP-1 by a new mechanism. Eur J Org Chem 2017:2223–2229. [Google Scholar]
- Williams DH, Bardsley B (1999) The vancomycin group of antibiotics and the fight against resistant bacteria. Angew Chem Int Ed Engl 38:1172–1193. [DOI] [PubMed] [Google Scholar]
- Wilson DM, Binder LI (1997) Free fatty acids stimulate the polymerization of tau and amyloid beta peptides. In vitro evidence for a common effector of pathogenesis in Alzheimer’s disease. Am J Pathol 150:2181–2195. [PMC free article] [PubMed] [Google Scholar]
- Winner B, Jappelli R, Maji SK, Desplats PA, Boyer L, Aigner S, Hetzer C, Loher T, Vilar M, Campioni S, et al. (2011) In vivo demonstration that α-synuclein oligomers are toxic. Proc Natl Acad Sci USA 108:4194–4199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wong PC, Pardo CA, Borchelt DR, Lee MK, Copeland NG, Jenkins NA, Sisodia SS, Cleveland DW, Price DL (1995) An adverse property of a familial ALS-linked SOD1 mutation causes motor neuron disease characterized by vacuolar degeneration of mitochondria. Neuron 14:1105–1116. [DOI] [PubMed] [Google Scholar]
- Xia G, Benmohamed R, Kim J, Arvanites AC, Morimoto RI, Ferrante RJ, Kirsch DR, Silverman RB (2011) Pyrimidine-2,4,6-trione derivatives and their inhibition of mutant SOD1-dependent protein aggregation. Toward a treatment for amyotrophic lateral sclerosis. J Med Chem 54:2409–2421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu N, Bitan G, Schrader T, Klärner FG, Osinska H, Robbins J (2017) Inhibition of mutant αB crystallin-induced protein aggregation by a molecular tweezer. J Am Heart Assoc 6:e006182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yadav K, Yadav A, Vashistha P, Pandey VP, Dwivedi UN (2019) Protein misfolding diseases and therapeutic approaches. Curr Protein Pept Sci 20:1226–1245. [DOI] [PubMed] [Google Scholar]
- Yamasaki TR, Holmes BB, Furman JL, Dhavale DD, Su BW, Song ES, Cairns NJ, Kotzbauer PT, Diamond MI (2019) Parkinson’s disease and multiple system atrophy have distinct α-synuclein seed characteristics. J Biol Chem 294:1045–1058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamazaki T, Chang TY, Haass C, Ihara Y (2001) Accumulation and aggregation of amyloid β-protein in late endosomes of Niemann-pick type C cells. J Biol Chem 276:4454–4460. [DOI] [PubMed] [Google Scholar]
- Yiannopoulou KG, Anastasiou AI, Zachariou V, Pelidou SH (2019) Reasons for failed trials of disease-modifying treatments for Alzheimer disease and their contribution in recent research. Biomedicines 7(4):97: PMC6966425. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yoshiyama Y, Higuchi M, Zhang B, Huang SM, Iwata N, Saido TC, Maeda J, Suhara T, Trojanowski JQ, Lee VM (2007) Synapse loss and microglial activation precede tangles in a P301S tauopathy mouse model. Neuron 53:337–351. [DOI] [PubMed] [Google Scholar]
- Yu H, Dee DR, Liu X, Brigley AM, Sosova I, Woodside MT (2015) Protein misfolding occurs by slow diffusion across multiple barriers in a rough energy landscape. Proc Natl Acad Sci USA 112:8308–8313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zaman M, Andreasen M (2020) Cross-talk between individual phenol-soluble modulins in Staphylococcus aureus biofilm enables rapid and efficient amyloid formation. eLife 9: PMC7732344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zheng X, Liu D, Klärner FG, Schrader T, Bitan G, Bowers MT (2015) Amyloid β-protein assembly: the effect of molecular tweezers CLR01 and CLR03. J Phys Chem B 119:4831–4841. [DOI] [PMC free article] [PubMed] [Google Scholar]