


Abstract
Cell cycle regulation is of paramount importance for all forms of life. Here, we report that a conserved and essential cell cycle-specific transcription factor (designated as aCcr1) and its viral homologs control cell division in Sulfolobales. We show that the transcription level of accr1 reaches peak during active cell division (D-phase) subsequent to the expression of CdvA, an archaea-specific cell division protein. Cells over-expressing the 58-aa-long RHH (ribbon-helix-helix) family cellular transcription factor as well as the homologs encoded by large spindle-shaped viruses Acidianus two-tailed virus (ATV) and Sulfolobus monocaudavirus 3 (SMV3) display significant growth retardation and cell division failure, manifesting as enlarged cells with multiple chromosomes. aCcr1 over-expression results in downregulation of 17 genes (>4-fold), including cdvA. A conserved motif, aCcr1-box, located between the TATA-binding box and the translation initiation site of 13 out of the 17 highly repressed genes, is critical for aCcr1 binding. The aCcr1-box is present in the promoters and 5′ UTRs of cdvA genes across Sulfolobales, suggesting that aCcr1-mediated cdvA repression is an evolutionarily conserved mechanism by which archaeal cells dictate cytokinesis progression, whereas their viruses take advantage of this mechanism to manipulate the host cell cycle.
INTRODUCTION
Cell cycle regulation is of fundamental importance for all organisms. DNA replication, chromosome segregation and cell division are tightly coordinated during the bacterial cell cycle, which ensures that one round of replication occurs per division event and division does not jeopardize genomic integrity (1). By contrast, in eukaryotes, the cell cycle is tightly coordinated through three sets of factors: (i) an ensemble of cell cycle-regulated proteins, including cyclin-dependent kinases (Cdk)-cyclin complexes and related kinases (2,3), (ii) various metabolic enzymes and related metabolites and (iii) reactive-oxygen species (ROS) and cellular redox status (4). Cyclin-dependent kinases are the engine of sequential progression through the eukaryotic cell cycle. Cyclins bind substrates and target the Cdks to specific subcellular locations. The formation of cyclin-Cdk complex results in Cdk activation. The oscillations of the cyclins are brought about by the fluctuations in cyclin gene expression and degradation by the ubiquitin mediated proteasome pathway (5).
The mechanism underlying the cell cycle regulation in Archaea, the third domain of life, remains elusive. Two major cell division machineries are present in Archaea. Whereas euryarchaea depend on the FtsZ-based bacterial-like system, most members of the TACK superphylum, including order Sulfolobales, as well as Asgardarchaeota employ the ESCRT-III/Vps4-based cell division machinery (also called Cdv system) (6–9). Whereas euryarchaea, similar to bacteria, do not display features of the eukaryotic-like cell cycle, crenarchaea and Sulfolobales, in particular, display eukaryotic-like cell cycle (10). The latter progresses through a pre-replicative growth period called the G1 phase, followed by the chromosome replication stage (S phase), a second period of cellular growth (G2 phase), and rapid genome segregation and cell division periods, known as the M and D phases, respectively (11). No bona fide cyclin homolog has been identified in archaea. Although some proteins possess ‘cyclin box’ domains (e.g. transcription initiation factor B) (12), their functioning as genuine cyclins has not been demonstrated. Certain eukaryotic-like serine/threonine kinases, implicated in stress response in Sulfolobales species, such as Sulfolobus acidocaldarius and Saccharolobus islandicus (formally Sulfolobus islandicus) (13–17), exhibit cyclic transcription patterns (18), but their roles in cell cycle control remain to be investigated. Furthermore, it was recently reported that degradation of cell division protein ESCRT-III (CdvB) by the proteasome drives cell division progression in S. acidocaldarius (19). Collectively, these lines of evidence imply that Sulfolobales cells have a simplified eukaryotic-like cell cycle regulation system. Therefore, elucidation of the cell cycle regulation in archaea, especially in Sulfolobales, could provide insights into the origin and evolution of the eukaryotic cell cycle regulation mechanism.
The archaeal cell cycle is likely to be regulated, at least partly, on the transcriptional level. The archaeal transcription apparatus is a unique mixture of eukaryotic-like and bacterial-like components (20,21). Archaea possess three transcription initiation factors (TBP, TFB, TFEa/b), two elongation factors (Spt4/5, Elf1), and one termination factor aCPSF1 (or FttA) that are all homologous to the eukaryotic transcription factors, and have an RNA Pol II-like polymerase (20–22). Despite the eukaryotic-like makeup of the core transcription machinery, archaea also commonly encode bacterial-like transcription factors with ribbon-helix-helix (RHH) and helix-turn-helix motifs. Many archaeal transcription factors have been implicated in the regulation of metabolic processes and response to environmental stresses (20,23). Notably, archaea appear to encode fewer transcription factors compared to bacteria, and the regulation may also depend on more complex pathways through post-translational modifications, such as phosphorylation (20) and methylation (24,25). In halophilic euryarchaea, an RHH family transcription factor, CdrS, plays a central role in the cell division regulation (26). CdrS is a global transcriptional regulator, controlling the expression of ftsZ and genes linked to other metabolic and regulatory processes, likely allowing cells to properly coordinate growth, division and metabolic activity (26). In another halophile, Halobacterium salinarum, the gene coding for the CdrS is co-transcribed with the cell division gene ftsZ2, and the gene encoding CdrL, another RHH transcription factor which binds the promoter of cdrS-ftsZ (27). The cdrS-ftsZ2 locus is well conserved across the Euryarchaeota, especially within the Halobacteria (27), suggesting a general cell division regulation mechanism in euryarchaea.
In Crenarchaeota, transcriptional regulation of the cell cycle has not been elucidated. Interestingly, we recently found that large spindle-shaped viruses of Sulfolobales are able to induce cell enlargement, by manipulating the archaeal cell cycle for virus production (28). This raises an intriguing question about how the host cell cycle is regulated by these viruses at the transcriptional level. In this study, we identified a small RHH family transcription factor, named aCcr1 (for archaeal cell cycle regulator 1), that is essential for cell viability and is involved in the control of cell division in S. islandicus REY15A. We found that aCcr1 homologs are widespread in Sulfolobales and their viruses. Over-expression of the cellular and viral aCcr1 homologs leads to cell enlargement and growth retardation. Transcriptomic analysis revealed that a number of genes, notably cdvA, are strongly downregulated in cells overexpressing aCcr1. Consistently, the purified cellular and viral aCcr1 proteins bind to the promoter and 5′ UTR region of cdvA, specifically at a conserved 9-nt motif (aCcr1-box). Our results demonstrate that aCcr1 plays a key role in cell division regulation and imply that it is also involved in the cell cycle manipulation by viruses.
MATERIALS AND METHODS
Strains and growth conditions
Saccharolobus islandicus REY15A was grown aerobically at 75°C in STV medium containing mineral salt, 0.2% (w/v) sucrose (S), 0.2% (w/v) tryptone (T), and a mixed vitamin solution (V). Saccharolobus islandicus REY15A(E233S)(ΔpyrEFΔlacS), hereafter E233S, was grown in STVU (STV supplemented with 0.01% (w/v) uracil) medium. The medium was adjusted to pH 3.3 with sulfuric acid, as described previously (29). SCV medium containing 0.2% (w/v) casamino acid (C) was used for screening and cultivating uracil prototrophic transformants. ATV medium containing 0.2% (w/v) D-arabinose (A) was used for protein expression. Culture plates were prepared using gelrite (0.8% [w/v]) by mixing 2 × STV and an equal volume of 1.6% gelrite. The strains constructed and used in this study are listed in the Supplementary information (Supplementary Table S1).
Bright-field and immunofluorescence microscopy
For bright-field microscopy analysis, 5 μl of cell suspension at the indicated time points were examined under a NIKON TI-E inverted fluorescence microscope (Nikon, Japan) in differential interference contrast (DIC) mode. Immunofluorescence microscopy analysis was carried out as previously described (28). Briefly, S. islandicus REY15A cells were collected and pelleted down at 5000 g for 5 min, re-suspended in 300 μl PBS buffer (137 mM NaCl, 2.7 mM KCl, 10 mM Na2HPO4·12H2O, 2 mM KH2PO4, pH 7.4), and fixed by addition of 700 μl cold absolute ethanol and kept at 4°C for at least 2 h. The fixed cells were washed for 3 times with PBST (PBS plus 0.05% Tween-20) to remove ethanol. Primary antibodies against ESCRT-III (HuaAn Biotechnology Co., Hangzhou, Zhejiang, China) were added with a dilution of 1:1000 in PBST and incubated at 4°C overnight. The cells were washed 3 times and then incubated with the goat anti-rabbit secondary antibodies Alexa Fluor® 488 (1:1000, Thermo Fisher Scientific, USA) for ESCRT-III, and Concanavalin A Alexa Fluor 647 Conjugate (50 μg/ml, Invitrogen™, Thermo Fisher Scientific, USA) for S-layer, and kept at 4°C for 2–4 h. The localization of ESCRT-III was observed under a SP8 confocal microscope, and the data were analysed using Leica Application Suite X (LAS X) software (Leica).
Flow cytometry analysis
The procedure for the flow cytometry analysis followed the reported method (28,30). Briefly, approximately 3 × 107 cells were collected for flow cytometry analysis. Cells were harvested at the indicated time points and fixed with 70% cool ethanol overnight (>12 h). The fixed cells were then pelleted at 800 g for 20 min. The cells were re-suspended and washed with 1 ml of PBS buffer. Finally, the cells were pelleted again and resuspended in 100 μl of staining buffer containing 50 μg/ml propidium iodide (PI) or SuperGreen. After staining for 30 min, the DNA content was analysed using the ImageStreamX MarkII Quantitative imaging analysis for flow cytometry system (Merck Millipore, Germany), which was calibrated with non-labelled beads with a diameter of 2 μm. The data from at least 20 000 cells were collected for each sample and the data of the single cells were analysed with the IDEAS software.
Transcriptome analysis
Strains of Sis/pSeSD and Sis/pSeSD-aCcr1 were cultured in ATV medium under the conditions as described above. For transcriptomic analysis, culture was inoculated with an initial OD600 of 0.05. The cells were pelleted at 6000 g for 10 min after 12 h of cultivation when the OD600 reached approximately 0.2. The pellet was resuspended in 1 ml PBS buffer. The cells were pelleted again and stored at −80°C. Total RNA was extracted using the Trizol reagent (Ambion, Austin, TX, USA). Total amounts and the integrity of RNA were assessed using the RNA Nano 6000 Assay Kit of the Bioanalyzer 2100 system (Agilent Technologies, CA, USA). Transcriptomic analysis was performed by Novogene (Beijing, China). About 3 μg of high-quality RNA per sample was used for the construction of RNA-Seq libraries. Firstly, mRNA was purified from the total RNA by depleting the rRNA using the biotin-labelled probes against rRNA. First strand cDNA was synthesized using random hexamer primer and the M-MuLV reverse transcriptase. Then, RNaseH was used to degrade the template RNA. For the second strand of cDNA synthesis by DNA polymerase I, dUTP was used to replace the dTTP in the dNTP mixture of deoxyribonucleotides. The remaining overhangs were converted into blunt ends via the exonuclease/polymerase activities. After adenylation of 3′ ends of DNA fragments, adaptors with hairpin loop structures were ligated for hybridization. Then, the USER enzyme was used to degrade the second dU-containing strand of cDNA. In order to select cDNA fragments of preferentially 370–420 bp in length, the library fragments were purified with AMPure XP system (Beckman Coulter, Beverly, USA). After PCR amplification, the product was purified by AMPure XP beads to obtain the libraries, which were sequenced using the Illumina NovaSeq 6000. Clean reads were aligned to the reference genome sequence of S. islandicus REY15A (31). The resulting data were then analysed by Fragments Per Kilobase of transcript sequence per Million base pairs sequenced (FPKM) analysis to reveal expression levels of all genes in the S. islandicus genome. Differential genome expression analysis (over-expression of aCcr1 versus empty vector) was performed using the DEGSeq R package. The resulting P-values were adjusted using the Benjamini and Hochberg's approach for controlling the false discovery rate padj < 0.05 and |log2(foldchange)| > 1 were set as the threshold for significantly differential expression. The ranscriptome experiments were performed in three biological repeats.
Cell cycle synchronization
S. islandicus REY15A cells were synchronized as previously described (30,32) with slight modifications. Briefly, cells were first grown aerobically at 75°C with shaking (145 rpm) in 30 ml of STV medium. When the OD600 reached 0.6–0.8, the cells were transferred into 300 ml STV medium with an initial estimated OD600 of 0.05. When the OD600 reached 0.15–0.2, acetic acid was added at a final concentration of 6 mM and the cells were blocked at G2 phase of the cell cycle after 6 h treatment. Then, the cells were collected by centrifugation at 3000 g for 10 min at room temperature to remove the acetic acid and washed twice with 0.7% (w/v) sucrose. Finally, the cells were resuspended into 300 ml of pre-warmed STV medium and cultivated as above for subsequent analysis.
Quantitative reverse transcription PCR (RT-qPCR)
Quantitative reverse transcription PCR (RT-qPCR) was used to validate some of the RNA-Seq data. Samples from the control and the aCcr1-over-expression strains were collected at indicated time points (same as for the transcriptome analysis). Total RNA was extracted using SparkZol (SparkJade Co., Shandong, China). First-strand cDNAs were synthesized from the total RNA according to the protocol of the First Strand cDNA Synthesis Kit (Accurate Biotechnology Co., Hunan, China) for RT-qPCR. The resulting cDNA preparations were used to evaluate the mRNA levels of the target genes by qPCR using the SYBR Green Premix Pro Taq HS qPCR Kit (Accurate Biotechnology Co., Hunan, China) and the gene specific primers (Supplementary Table S2). PCR was performed in an CFX96™ (Bio-Rad) with the following steps: denaturing at 95°C for 30 s, followed by 40 cycles of 95°C for 5 s and 60°C for 30 s. Relative amounts of mRNAs were evaluated using the comparative Ct method with 16S rRNA as the reference. All qPCR primers used had an amplification efficiency between 90% and 110%.
Protein purification and chemical cross-linking
To purify the aCcr1 (SiRe_0197) and its mutant proteins from E. coli, cells harbouring plasmids pET22b-aCcr1-C-His (for the wild type cellular protein), pET22b-aCcr1-R2A-C-His, pET22b-aCcr1-K7A-C-His, pET22b-aCcr1-R27A-C-His (for the DNA binding site deficient cellular protein mutants) and pET22b-ATV_gp29-C-His and pET22b-SMV3_gp63-C-His (for virus-derived aCcr1 homologs) were grown in 2 L of LB medium at 37°C with shaking until the optical density OD600 reached 0.4∼0.6, when 1.0 mM IPTG was added into the cultures and the cells were then grown at 37°C for 4 h with shaking. The cells were harvested by centrifugation at 7000 g for 10 min and then resuspended in the lysis buffer A (50 mM Tris-HCl [pH 7.4], 200 mM NaCl and 5% glycerol). Then, the cells were crushed with an ultrasonic crusher at 40% power, working at 5 s intervals for 5 s until the cell lysate became clear and cell debris was removed by centrifugation at 12 000 g for 15 min. The supernatant was incubated at 70°C for 20 min, centrifuged at 12 000 g for 15 min again, and then filtered through a membrane filter (0.45 μm). The samples were loaded on to a Ni-NTA agarose column (Invitrogen) pre-equilibrated with buffer A. Finally, the target protein was eluted with buffer A containing 300 mM imidazole. The eluted sample was analysed using a 18% SDS-PAGE gel. The protein samples were concentrated by ultrafiltration using an Amicon Ultra-3KDa concentrator (Millipore). For further purification, size exclusion chromatography was performed using a Superdex 200 increase 10/300 column (GE Healthcare). The protein concentration was determined by the Bradford method using bovine serum albumin as the standard. To assay the oligomeric status, 20 μl of wild-type aCcr1 protein (1 mg/ml) were incubated with increasing concentrations of glutaraldehyde (0.01–0.16%) on ice at 4°C for 15 min. The reaction was then stopped by the addition of SDS-PAGE loading buffer, after which the samples were electrophoresed by 20% SDS–PAGE, and the gel was stained with Coomassie blue R-250.
Western blotting
Antibodies against TBP, CdvA and ESCRT-III were produced using synthetic specific peptides (amino acids 18–31, SIPNIEYDPDQFPG for TBP [SiRe_1138]; 13–25, GQKVKDIYGREFG for CdvA [SiRe_1173]; 194–208 IEQSSRVSQSRPAVR for ESCRT-III [SiRe_1174]). Antibody against SisCcr1 (Ccr1 from S. islandicus REY15A) was produced using purified recombinant proteins purified from E. coli. Antibodies against TBP, CdvA and aCcr1 were produced in rabbit, and CdvA in rat. All the antibodies were produced by HuaAn Biotechnology Co. (Hangzhou, Zhejiang, China). For standard western blotting analysis, 2 × 108 cells (with or without induction) at the indicated times were collected by centrifugation at 5000 g for 10 min and resuspended in 20 μl PBS buffer. After the addition of 5 μl 5 × loading buffer, the samples were treated at 100°C for 10 min and analysed by SDS-PAGE. The proteins in the PAGE gel were transferred onto a PVDF membrane at 30 mA for 16 h at 4°C. Membranes were blocked with 5% (w/v) skimmed milk for 2 h at room temperature. The membrane was washed and incubated with a primary antibody and then the secondary anti-rabbit HRP conjugate antibody (TransGen Biotech company, Beijing, China) following the manufacturer’s instructions. Finally, the membranes were imaged using an Amersham ImageQuant 800 biomolecular imager (Cytiva).
Sequence analysis of the promoters and the 5′ UTRs of genes highly repressed by aCcr1
Sequences (60 bp preceding the start codon) of the 16 genes down-regulated (>4-fold change) in the aCcr1 overexpression strain were retrieved from the genome sequence of S. islandicus REY15A. These sequences were then used for de novo motif discovery using MEME (Multiple EM for Motif Elicitation) with default settings (33).
Electrophoretic mobility shift assay (EMSA)
Substrates used in EMSA experiments were generated by annealing the complementary oligonucleotides with 5′ FAM-labelled oligonucleotides (Supplementary Table S2). The reaction mixture (20 μl) containing 2 nM of the FAM-labelled substrates and different concentrations of aCcr1 or the mutant proteins was incubated at 37°C for 30 min in binding buffer (25 mM Tris–HCl, pH 8.0, 25 mM NaCl, 5 mM MgCl2, 10% glycerol, 1 mM dithiothreitol). After the reaction, samples were loaded onto a 10% native PAGE gel buffered with 0.5 × Tris–borate–EDTA (TBE) solution. DNA–protein complexes were separated at 200 V for 60 min. The resulting fluorescence was visualized by an Amersham ImageQuant 800 biomolecular imager (Cytiva).
Phylogenetic analysis
aCcr1 homologs were collected by PSI-BLAST (2 iterations against the RefSeq database at NCBI; E = 1e-05) (34). The collected sequences were then clustered using MMseq2 (35) to 90% identity over 80% of the protein length. Sequences were aligned using MAFFT v7 (36) and the resultant alignment trimmed using trimal (37), with the gap threshold of 0.2. Maximum likelihood phylogenetic analysis was performed using IQ-Tree (38), with the best selected amino acid substitution model being LG + I + G4. The branch support was assessed using SH-aLRT (39).
Chromatin immunoprecipitation (ChIP-seq)
Chromatin immunoprecipitation (ChIP-Seq) was performed according to Takemata et al. (40) with slight modifications. Briefly, the cells were collected 3 h after synchronization, cross-linked by adding 1% formaldehyde for 15 min and quenched with a final concentration of 125 mM glycine. The cells were pelleted by centrifugation at 5000 g for 10 min and washed with PBS. The cells were then resuspended in TBS-TT buffer (20 mM Tris, 150 mM NaCl, 0.1% Tween-20, 0.1% Triton X-100, pH 7.5) and fragmented by sonication until the DNA fragments were of 200–500 bp. After centrifugation (10 000 g for 15 min), a 100 μl aliquot of the DNA-containing supernatant was kept apart to be uses as an input control and the remaining sample was divided into two aliquots. One aliquot was incubated with anti-aCcr1 antibody-coated protein A beads (Cytiva) and the other was incubated with pre-immune serum-coated protein A beads, which served as a nonspecific binding control (Mock control). The immuno-complexes were collected by centrifugation and washed by five consecutive 5 min incubations with 1 ml TBS-TT with vigorous shaking at room temperature. Beads were then washed once with TBS-TT containing 500 mM NaCl and once with TBS-TT containing 0.5% Tween-20 and 0.5% Triton X-100. Finally, the immuno-complexes were disrupted by resuspending the beads in a buffer containing 20 mM Tris (pH 7.8), 10 mM EDTA and 0.5% SDS, and heating the sample for 30 min at 65°C. Beads were removed, and the DNA was recovered by treating samples with 10 g/ml proteinase K for 6 h at 65°C, then for 10 h at 37°C. After incubation at 60°C for 10 h, the samples were collected and the captured DNA was purified by using the DNA Cycle-Pure Kit (Omega), according to the manufacturer’s instructions. The input samples were treated as above without the addition of antiserum and beads. The purified DNA was used for ChIP-Seq library preparation. The library was constructed by Novogene Corporation (Beijing, China). Subsequently, pair-end sequencing of sample was performed on Illumina platform (Illumina, CA, USA). Library quality was assessed on the Agilent Bioanalyzer 2100 system. Clean data were obtained by removing low quality reads as well as reads containing adapters, poly-N, from the raw dataset. All the downstream analyses were based on the clean, high quality reads. Reference genome and gene model annotation files were downloaded from GenBank. The reads were then aligned to the REY15A genome (31) and the entire genome was scanned with a specific window size using MACS2 (version 2.1.0) (41) to calculate the read enrichment level and IP enrichment regions for peak calling. A q-value threshold of 0.05 was used for all datasets. After peak calling, the distribution along the chromosome, peak width, fold enrichment, significance level, and peak summit number per peak were visualized. Peaks located at -100 to 0 of the genes were extracted and calculated. Finally, the MEME-ChIP tool (33) was applied for discovering the motifs in the peak regions.
RESULTS
The cyclically transcribed gene aCcr1 is essential for cell viability
Seven transcription factors displayed cyclic expression patterns in Sulfolobus acidocaldarius from the microarray-based genome-wide transcriptomic analysis (18), including one of the three eukaryotic transcription initiation factor IIB homologs, Tfb2 (Saci_1341), an RHH domain protein (CopG family, Saci_0942), a DtxR family protein (Saci_1012), a Tet family protein (Saci_1107), an Lrp/AsnC family protein (Saci_2136), and two HTH domain-containing proteins (Saci_0102 and Saci_0800). Except for the Tet and the Lrp/AsnC family proteins, these transcription factors are conserved in S. islandicus REY15A, suggesting that they play important roles in cell cycle regulation. To test this hypothesis, we focused on SiRe_0197, an RHH domain protein of S. islandicus REY15A. We named it aCcr1 (for archaeal Cell cycle regulator 1) based on the results described below. aCcr1 is a 58-amino acid protein (Supplementary Figure S1A) with an isoelectric point of 9.45 and a predicted molecular mass of 6.9 kDa. Using structural modelling, we predicted the structure of aCcr1 which was further modelled as a dimer (Supplementary Figure S1B), similar to other RHH proteins (42). Indeed, glutaraldehyde cross-linking experiment confirmed that in solution the dominant form of aCcr1 is a dimer (Supplementary Figure S2A and B). Based on the available RHH protein-DNA structures, the positively charged amino acid residues R2, K7, and R27 were predicted to interact with the major groove of the dsDNA via the two-stranded β-sheet (Supplementary Figure S1B).
To investigate the archaeal cell cycle regulation mechanism, we performed transcriptomic analysis using synchronized S. islandicus REY15A cells (30,32) (Figure 1A). Addition of acetic acid to the medium presumably results in starvation responses due to respiration uncoupling, leading to arrest of cells in the G2 phase of the cell cycle. We analysed the changes in the transcription levels of aCcr1 and cell division genes cdvA, escrt-III and vps4. As expected, all of them exhibited cyclic patterns (Figure 1B). Importantly, the transcription levels of cdvA peaked at about 60 min following the removal of acetic acid, while the levels of aCcr1, escrt-III and vps4 reached their maxima at approximately 120 min after the release of the cell cycle arrest (Figure 1B). This result confirms that aCcr1 (SiRe_0197) is likely a cell division specific transcription factor in S. islandicus REY15A.
Figure 1.

The gene accr1 is cyclically transcribed. (A) Flow cytometry profiles of samples of a synchronized S. islandicus REY15A (E233S) culture in one cell cycle. The cells were synchronized at G2 phase after treated with 6 mM acetic acid for 6 h before released by removing the acetic acid. The cultures were collected at different time points (0, 1.0, 1.5, 2.0, 2.5, 3.0, 3.5, 4.0, 5.0 and 6.0 h) and subjected to flow cytometry analysis. Cells started to divide at 2.0 h as shown by the appearance of cells with one copy chromosome (1C) and the ratio of dividing cells reached the highest level at about 3–3.5 h. As the cell cycle proceeded, cells with two copy chromosomes (2C) became dominant at 5 h. (B) Changes of transcription levels of accr1 and the cell division genes based on the transcriptomic data of the synchronized cell culture. The cells were collected at 0, 1, 2, 3, 4 and 5 h after cell arrest release. The transcriptome experiments were performed in three biological repeats.
To understand the importance of aCcr1 for the cell, we attempted to knock out aCcr1 using an endogenous CRISPR-based genome editing system in S. islandicus REY15A (43) (Supplementary Figure S3). However, all attempts (at least five times) failed to yield any viable knockout clones, implying that aCcr1 is probably an essential gene, consistent with the results of the previously reported genome-wide mutagenesis in another S. islandicus strain (44). This result suggests that the putative regulatory role of aCcr1 is indispensable for the cell survival.
Over-expression of aCcr1 results in cell enlargement and the DNA binding activity of aCcr1 is critical for its cell division regulation
To probe the in vivo function of aCcr1, we attempted to obtain strains in which the levels of aCcr1 are down- or up-regulated. Unfortunately, the knockdown analysis could not be pursued because the ccr1 gene in S. islandicus REY15A lacks a suitable protospacer necessary for the endogenous CRISPR-based silencing method (43,45). However, a series of aCcr1 over-expression strains, including those over-expressing the wild-type aCcr1 as well as putative DNA-binding deficient mutants aCcr1(R2A), aCcr1(K7A) and aCcr1(R27A) (Supplementary Figure S1B), were obtained (Supplementary Table S1). Compared with the control, cells over-expressing aCcr1 showed an obvious growth retardation (Figure 2A) and exhibited greatly enlarged cell sizes (Figure 2B and 2C) and increased amounts of DNA (Figure 2D). The average diameter of the cells reached a maximum of 4.58 μm at 24 h after induction (Figure 2C). These phenotypes are indicative of cell division defects in cells over-expressing aCcr1. To verify whether the observed cell division defect is dependent on the DNA binding activity of aCcr1, we compared the growth and cell sizes of the strains over-expressing the wild-type aCcr1 and DNA-binding deficient mutants R2A, K7A, and R27A. As shown in Supplementary Figure S4A–C, all the cells over-expressing the mutant proteins displayed normal growth and cell morphology. The expression of the wild-type and mutant aCcr1 proteins was confirmed by Western blotting analysis (Supplementary Figure S4D). These results indicate that the positively charged residues play a critical role in the function of aCcr1 and suggest that the DNA binding activity is essential for cell division regulation.
Figure 2.

Overexpression of aCcr1 leads to remarkable cell enlargement. (A) Growth curves of cells over-expressing C-terminal His-tagged aCcr1. The cells were inoculated into 30 ml induction medium ATV to a final estimated OD600 of 0.03. The growth was monitored at OD600. Each value was based on data from three independent measurements. Cells harboring the empty plasmid pSeSD were used as a control. (B) Bright-field microscopy (DIC) of cells over-expressing aCcr1. Cells cultured in the induction medium were taken at different time points and observed under a NIKON-E microscope; Scale bars: 2 μm. (C) Cell size statistics of the data in (B). Cell cultures were sampled at the indicated time points and observed under the microscope. The diameters of ∼100 cells were measured using ImageJ software for each culture repeat. (D) Flow cytometry analysis of the DNA content of Sis/pSeSD and Sis/pSeSD-aCcr1 cells cultured in MATV media.
The cell division gene cdvA is strongly downregulated in cells over-expressing aCcr1
To identify which genes are transcriptionally regulated by aCcr1 and to gain insight into how aCcr1 over-expression influences the cell division, we conducted comparative transcriptomic analysis of the aCcr1 over-expressing strain and the control carrying an empty vector pSeSD. Samples were taken at 12 h after arabinose induction and subjected to transcriptomic analysis. In total, 76 and 124 genes were up- and down-regulated by more than two folds, respectively (Supplementary Tables S3,S4; Supplementary data S1; Figure 3). If 4-fold was taken as a threshold, 4 and 17 genes were up- and down-regulated, respectively (Figure 3 and Table 1). Intriguingly, cdvA (sire_1173), the archaea-specific cell division gene (8,9,46), was among the most highly down-regulated genes (Figure 3 and Table 1). During cell division, CdvA binds to the chromosome and membrane, forming a ring-like structure, then recruits ESCRT-III to the mid-cell for cell division (47). Over-expression of aCcr1 leads to increase in cell diameter and DNA content, indicative of failure in cell division. A very similar phenotype was obtained when cdvA transcription was downregulated using the CRISPR knockdown technology (30).
Figure 3.

Volcano plot of differentially expressed genes in strain Sis/pSeSD-aCcr1 compared to the control Sis/pSeSD. X-axis, fold change in gene expression. Y-axis, significance of fold change. Genes exhibiting > 2-fold (i.e. –1 > log2 > +1) up-and down-regulated with significance are highlighted in red and green, respectively, whereas those that showed a <2-fold change in differential gene expression or with no significance are shown in blue.
Table 1.
Summary of genes with > 4 fold down-regulated mRNA levels in the aCcr1 overexpression strain based on comparative transcriptomic analysis*
| Gene identity | Protein function | Sis/pSeSD-aCcr1 vs Sis/pSeSD log2 Fold Change |
|---|---|---|
| SiRe_0624 | Blue (type 1) copper domain-containing protein | -3.458533126 |
| SiRe_0623 | Uncharacterized membrane protein | -3.261849277 |
| SiRe_0670 | Zinc finger SWIM domain-containing protein | -3.153753639 |
| SiRe_2056 | Serine/threonine protein kinase | -3.077324891 |
| SiRe_0332 | Cellulase (endo 1,4 beta glucanase), putative (CelB) | -2.944384326 |
| SiRe_0764 | CRISPR-associated, Csa3a | -2.922021684 |
| SiRe_2100 | SedA, Membrane protein involved in DNA uptake | -2.918474234 |
| SiRe_0019 | Queuine/archaeosine tRNA-ribosyltransferase | -2.906286786 |
| SiRe_0086 | ATPase, predicted component of phage defense system | -2.817790738 |
| SiRe_0018 | Queuine tRNA-ribosyltransferase related protein | -2.789165379 |
| SiRe_1173 | Cell division protein CdvA | -2.669806305 |
| SiRe_2057 | Uncharacterized membrane protein | -2.249728764 |
| SiRe_1917 | AIR synthase related protein | -2.226944702 |
| SiRe_0691 | Thermopsin-like protease | -2.224399679 |
| SiRe_1464 | Uncharacterized protein | -2.069938647 |
| SiRe_0269 | Uncharacterized protein | -2.045724686 |
| SiRe_1239 | Zn-dependent alcohol dehydrogenase | -2.030511419 |
*The cell division protein CdvA is shown in bold.
aCcr1 binds to the promoter and 5′ UTR of cdvA at a conserved motif, aCcr1-box
To test whether aCcr1 binds to the promoter of cdvA, we performed the EMSA analysis using a DNA substrate including the sequence located upstream of the translation start site and containing the PcdvA promoter and the 5′UTR (5′ untranslated region). Purified aCcr1 protein (0, 0.1, 0.2, 0.4 and 0.8 μM) was incubated with fluorescein (FAM)-labelled DNA substrates (Figure 4A). When an oligonucleotide with the sequence corresponding to the distal region (-100 to -51) of the PcdvA promoter was used as a substrate, no retardation in electrophoretic mobility was observed (Figure 4A and 4C). In contrast, when the substrate contained the PcdvA sequence proximal to the first cdvA codon (-50 to -1), electrophoretic mobility was retarded in a protein concentration-dependent manner (Figure 4A and 4C). Therefore, the aCcr1 binding site was localized within the -50 to -1 region, covering the BRE (TFIIB recognition element), TATA-box, and the 5′ UTR. To further characterize the DNA binding activity of aCcr1, we expressed in E. coli and purified the site-directed mutants of aCcr1, aCcr1(R2A), aCcr1(K7A) and aCcr1(R27A) (Supplementary Table S1, Figure S1B and S5A). All the mutants lost the ability to bind to the cdvA promoter (Supplementary Figure S5B). Thus, we confirmed that R2, K7 and R27 are critical for DNA binding.
Figure 4.

aCcr1 binds to the promoter and 5′ UTR of cdvA at the aCcr1-box motif. (A) Schematic organization of Cdv genes and cdvA promoter in the genome of S. islandicus REY15A. The sequences of the upper region (-100 - -51), lower region (-50 - -1) and its variant mutants used in the EMSA analysis are listed. The aCcr1-box motif is highlighted in yellow. Dots and the underlined indicate truncated and substitutive sequences, respectively. The oligonucleotides are labelled with FAM fluorescence at the 5′-ends for EMSA. (B) A conserved motif (designated as aCcr1-box) identified in the promoter and 5′ UTR regions of the highly repressed genes due to aCcr1 over-expression. A total of 15 promoter sequences (-60 - -1) of the down-regulated (>4-fold) genes plus promoter of cdvA from Sulfolobus acidocaldarious were used as the input for De Novo motif discovery by MEME server with the default setting. The height of the letter indicates the relative similarity to that of consensus one. (C) EMSA of aCcr1 binding to different regions of the promoter of cdvA and its mutants. The 5′FAM-labelled and corresponding complementary nucleotide sequences are listed in Supplementary Table S2. The labelled oligonucleotides were annealed with the respective complementary strands as described in the Materials and Methods for the EMSA assay. The reaction was performed at 37°C for 30 min and analysed on a 10% native PAGE (see ‘Materials and Methods’). Each reaction contained 2 nM of the FAM-labelled probe and 0, 0.1, 0.2, 0.4 or 0.8 μM aCcr1 protein. (D) Quantification of the results in (C). The values were obtained from three independent experiments. Error bars indicate standard deviation.
To understand the substrate binding specificity of aCcr1, we analysed the nucleotide sequences corresponding to the putative promoter regions and 5′ UTRs of all 17 genes repressed in the aCcr1 over-expression strain (Tables 1 and 2; Figure 5). For convenience, we use the term ‘promoter’ to denote regions encompassing both the promoter and the 5′ UTR, if not specified otherwise. The 5′ UTRs in Sulfolobales are generally short in most genes. Notably, sire_0018 and sire_0019 are within the same operon, and so are sire_0623 and sire_0624, whereas a bidirectional promoter sequence is apparently shared by sire_2056 and sire_2057 (Figure 5). All 15 promoter sequences of aCcr1-repressed S. islandicus REY15A genes were retrieved and subjected to de novo motif discovery using the MEME server. We found that 10 promoters (of 12 genes) contain one or two copies of a 9 bp motif A(T/G)G(A)TA(G)A(T/G)TACN, which we name the aCcr1-box (Figures 4B and 5; Table 2). Therefore, 12 out of 17 genes significantly repressed by aCcr1 possess the aCcr1 box, whereas the remaining 5 genes lack this motif. We confirmed the importance of the motif for aCcr1 binding to the promoters of cdvA by EMSA. As shown in Figure 4C and 4D, deletion of either of the aCcr1-boxes reduced the binding affinity, while deletion or replacement of both motifs greatly impaired the binding of aCcr1. Because most of the sites are located between the TATA-box and the translation start site, binding to the promotor by aCcr1 would prevent the formation of transcriptional pre-initiation complex, leading to transcriptional repression. While the repression of cdvA by aCcr1 is probably the main mechanism for cell division failure in the aCcr1 over-expression strain, the physiological functions of repression of other genes by aCcr1 need further investigation.
Table 2.
Distribution of aCcr1 box in the promoter and 5′ UTRs of the highly down-regulated genes in the aCcr1 overexpressing strain and CdvA homologs from other archaeal species*
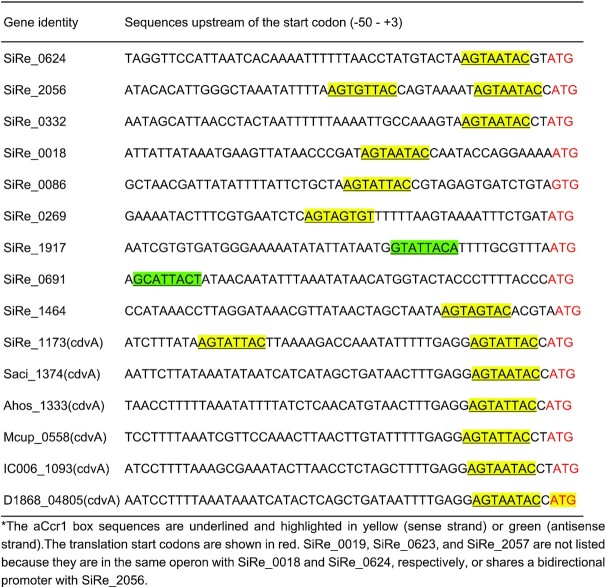
|
Figure 5.

Distribution of aCcr1-box in the highly repressed gene promoters and 5′ UTRs. ‘+’ and ‘–’ represent coding and non-coding strands, respectively. The red box represents the aCcr1-box.
Interestingly, aCcr1-boxes are present in the promoters of cdvA homologs from other Sulfolobales species, e.g. S. acidocaldarius DSM639 (saci_1374), Acidianus hospitalis W1 (ahos_1333), Metallosphaera cuprina Ar-4 (mcup_0558), Sulfuracidifex tepidarius (ic006_1093) and Stygiolobus azoricus (d1868_04805) (Table 2), suggesting that aCcr1 binding to cdvA promoter is conserved across Sulfolobales. To test if the aCcr1 homologs from other crenarchaeal species are functional in vivo, we over-expressed in S. islandicus REY15A SacCcr1 (Ccr1 from S. acidocaldarius), which differs from SisCcr1 by four residues (Y23T, M38L, L43T, and R47T) (Supplementary Figure S1A). As expected, the SacCcr1 over-expression strain showed phenotypes similar to those observed in cells over-expressing SisCcr1 (Supplementary Figure S7). Given that aCcr1-box motifs are present in the promoters of cdvA genes across Sulfolobales (Table 2), the mechanism of aCcr1-mediated control of cell division through repression of CdvA is likely to be conserved as well, at least, in members of the Sulfolobales.
ChIP-seq analysis reveals multiple aCcr1 binding sites
As aCcr1 is predicted to be a transcription factor, we performed ChIP-Seq analysis to identify the binding sites in vivo using a rabbit-derived antibody specifically recognizing the aCcr1 purified from E. coli. Cells were collected 3 h after synchronization when the transcription level of accr1 reached the highest level. A total of 307 loci at promoter regions were enriched, with 298 being positioned between -50 and 0 (Supplementary Tables S3 and S4, Figure 6A). Interestingly, promoters of 38 of the 124 down-regulated (>2-folds) genes, among which 17 were highly down-regulated (>4-fold), in the aCcr1-overexpression strain were enriched (Figure 3 and Supplementary Table S3) and the predicted binding site motif obtained from ChIP-Seq analysis matches well with the aCcr1-box sequence (Figures 4B, 6B and 6C). In contrast, promoters of only 7 of the 76 up-regulated (>2-fold) genes, among which one is highly upregulated (>4-fold), were enriched (Supplementary Data 2, Figure 3). Since most of the genes identified by ChIP-Seq did not show any significant transcriptional change in the transcriptome of the aCcr1-overexpressing strain, we speculate that some of these genes themselves are expressed at low levels in the wild-type strain and their transcription is already repressed by the background level of aCcr1 under normal conditions. In addition, 242 and 61 enriched sites are localized within genes and at intergenic regions (but outside of the promoter regions), respectively. It is unclear what is the functional significance of the aCcr1 binding to these sites. Overall, our ChIP-Seq results support the conclusion that aCcr1 functions mostly as a repressor of a number of genes including cdvA, although the full extent of the aCcr1 functionality needs further investigation.
Figure 6.

Identification of the aCcr1 DNA binding sites in vivo by Chip-seq. (A) Overview of the genomic binding profile of aCcr1 as monitored by Chip-seq. The sample before immunoprecipitation was used as input. Immunoprecipitation performed with the pre-immune antiserum was used as a mock control. The red shows the genome binding profile as monitored by ChIP-seq by the aCcr1 antibody. The boxed (in green) indicates transcription at cdvA loci. (B) The promoter regions of cdvA was a highly enriched site by Chip-seq. The schematic representation of the genomic organization and the binding sequences of the cdvA was shown, with the aCcr1-boxes in green and the translation start codon (ATG) being underlined. (C) Sequence logo of the aCcr1 binding representing MEME predictions of ChIP-seq enriched sequences. The arrows indicate the complementary nucleotides in the palindromic motif.
Over-expression of aCcr1 affects cell cycle progression due to specific downregulation of cdvA and other cell division genes
The growth retardation and cell enlargement phenotypes of the aCcr1 over-expression strain suggested that the cell division genes could be downregulated. Cell division in Sulfolobales is dependent on the eukaryotic-like ESCRT machinery, which comprises the archaea-specific protein CdvA, four ESCRT-III homologs (ESCRT-III [CdvB], ESCRT-III-1 [CdvB1], ESCRT-III-2 [CdvB2], ESCRT-III-3 [CdvB3]), and an AAA + ATPase Vps4 (also known as CdvC) (8,9,46). CdvA binds to DNA and membrane (47,48) and then recruits ESCRT-III to the mid-cell, where it forms a ring-like structure and drives cell division (47). Vps4 binds to ESCRT-III and other ESCRT-III homologs (46) and, upon ATP hydrolysis, drives the disassembly of the ESCRT-III rings, thereby promoting the cell division process (9,19). In a previous study, we reported that infection of S. islandicus REY15A with STSV2 led to transcriptional downregulation of the cell division genes, including cdvA, escrt-III, escrt-III-1, escrt-III-2, escrt-III-3 and vps4, which resulted in dramatic increase in the size and DNA content of infected cells (28). To confirm the regulatory role of aCcr1 in cell cycle progression, we analysed the aCcr1 over-expression strain by flow cytometry using synchronized cells. We added D-arabinose 3 h after the addition of acetic acid (Figure 7A). The expression of aCcr1 was induced 1.0 h before the acetic acid was removed. S. islandicus REY15A (E233S) containing the empty plasmid was used as a control. As shown in Figure 7B, subpopulation of cells containing a single chromosome copy (1C) could be observed at 0–4 h and up to the end of the normal cell cycle in the control, but not in the aCcr1 over-expression strain. Thus, over-expression of aCcr1 inhibited cell division. Western blotting results showed that in the control cells, CdvA exhibited a cyclic pattern with higher expression at 0–1 h (Figure 7C). In contrast, the CdvA levels remained low in the aCcr1 over-expression cells, consistent with the RT-qPCR results and transcriptome data. Thus, we established that binding of aCcr1 to the promoter of cdvA represses the gene expression and reduces CdvA level in the cell, leading to cell division failure.
Figure 7.

Over-expression of aCcr1 stalls cell division and impairs the ESCRT-III contractile ring formation. (A) Schematic showing the cell synchronization and induction of aCcr1 over-expression with arabinose (0.2%). Time for acetic acid treatment and arabinose induction are indicated. E233S containing the empty plasmid (Sis/pSeSD) was used as a control. (B) Flow cytometry profiles of DNA content distribution of cells 0–4 h after acetic acid removal. (C) Western blotting using anti-CdvA antibody in strain Sis/pSeSD-aCcr1 and the control at 0–5 h after acetic acid removal. TBP (TATA-box binding protein) was used as a loading control. The conditions for the western blotting were as described in the Materials and Methods. Two gels and blots were used for each antibody with the same cell sample preparations and detection, except for CdvA, where 4 min was taken for exposure instead of 40 s. (D) Immuno-fluorescence microscopy showing the formation of contractile rings using the primary antibody against ESCRT-III (CdvB) and goat anti-rabbit secondary antibody Alexa Fluor® 488. The S-layer was stained with Concanavalin A, Alexa Fluor 647 Conjugate. Shown are representative images. M, molecular size marker.
aCcr1 over-expression jeopardizes the formation of the ESCRT-III ring
To further characterize how the cell division is affected by the over-expression of aCcr1, we performed fluorescence microscopy analysis on the synchronized cells over-expressing aCcr1 using anti-ESCRT-III antibodies. Cells cultured at 3–4 h after removal of acetic acid were sampled and analysed. In the control cells carrying the empty vector, ESCRT-III displayed mid-cell localization and formed ring-like structures in 7.2% of the cells (Figure 7D). In the aCcr1 over-expressing strain, ESCRT-III ring was barely visible (0.8%). Presumably, in the absence of CdvA, ESCRT-III cannot be recruited to the membrane (Figure 7D). This result further reinforces the hypothesis that aCcr1 regulates cell division via cdvA repression.
aCcr1 homologs are widespread in sulfolobales and their viruses
To explore the diversity and distribution of aCcr1 in the domain Archaea, we performed PSI-BLAST search using aCcr1 from S. islandicus REY15A as a query. Given that RHH domain proteins are widespread in archaea and orthology of short divergent proteins is not straightforward to assess, we restricted our focus to proteins displaying relatively high sequence similarity to aCcr1 and which could be retrieved after two iterations of PSI-BLAST. The search yielded multiple hits in hyperthermophilic crenarchaea of the orders Sulfolobales and Desulfurococcales (Figure 8). We note, however, that further PSI-BLAST iterations revealed additional homologs in a broader diversity of archaea, including those from other phyla. In Sulfolobales genomes, aCcr1 genes are encoded within orthologous loci and have conserved gene synteny (Supplementary Figure S6). Interestingly, all cdvA promoter and 5′ UTR sequences of the Sulfolobales species contain at least one aCcr1-box motif at a conserved position (Table 2).
Figure 8.

aCcr1 homologs are widely distributed in Sulfolobales and Desulfurococcales as well as in the plasmids and viruses of Sulfolobales. aCcr1 homologs were collected by PSI-BLAST. The collected sequences were then clustered using MMseq2. The sequences were aligned using MAFFT v7 and the resultant alignment trimmed using trimal. Phylogenetic analysis was performed using IQ-Tree and the branch support was assessed using SH-aLRT. The details of the analysis were listed in the Materials and Methods. The aCcr1 homologs investigated in this study were indicated with the yellow stars.
Intriguingly, aCcr1 homologs are also conserved in several conjugative plasmids and genomes of Sulfolobales viruses from five different families, including Rudiviridae, Bicaudaviridae, Fuselloviridae, Ungulaviridae and Turriviridae (49) as well as in unclassified Sulfolobales viruses YNP1 and YNP2 (Figure 8). The viral, plasmid and cellular aCcr1 homologs display a complex evolutionary history with multiple horizontal gene transfers, even within the same family of viruses. The phylogeny splits into three major clades (I-III), with members of the Sulfolobales being distributed between clades I and III, and Desulfurococcales forming the clade II (Figure 8). Viruses are intermixed with the Sulfolobales within clades I and III, whereas plasmids are restricted to clade III. The horizontal gene transfer of aCcr1 homologs between viruses and cells suggests that viruses might have hijacked aCcr1 genes for manipulation of the cell cycle of their hosts.
Over-expression of viral aCcr1 homologs impairs cell cycle progression in S. Islandicus REY15A
In our previous study, we reported that upon infection with spindle-shaped viruses STSV2 and SMV1, the cell size of S. islandicus REY15 was enlarged (28). Unexpectedly, aCcr1 homologs could not be found in the genomes of the two viruses. However, aCcr1 homologs are encoded by other large spindle-shaped viruses. To investigate the functions of these viral aCcr1 homologs, we have chosen two viruses belonging to clade I, which also includes aCcr1 proteins of S. islandicus REY15A and S. acidocaldarius (Figure 8). We constructed strains over-expressing aCcr1 homologs from Acidianus two-tailed virus (ATV_gp29) and Sulfolobus monocaudavirus 3 (SMV3_gp63). Over-expression of the ATV_gp29 and SMV3_gp63 resulted in growth retardation and yielded enlarged cells with multiple chromosomes (Figure 9A–C), reminiscent of the cell phenotype induced by STSV2 and SMV1 infections (28). Thus, these results suggest a mechanism by which viruses can control the division of their host cells. Factors that induce cell enlargement in STSV2 and SMV1 need to be explored in future studies.
Figure 9.

Cells with over-expression of the aCcr1 homologs, ATV_gp29 from Acidianus Two-tailed Virus and SM3_gp63 from Sulfolobus monocaudavirus 3 have similar phenotype as those with over-expression of SisCcr1. (A) Growth curves of cells Sis/pSeSD-ATV_gp29 and Sis/pSeSD-SMV3_gp63 cultured in induction medium ATV. The cells were inoculated into 30 ml medium to a final estimated OD600 of 0.03 and the growth was monitored using spectrometer. Each value was based on data from three independent repeats. Cells harboring the empty plasmid pSeSD were used as a control. (B) Bright-field microscopy (DIC) and (C) flow cytometry of cells over-expressing ATV_gp29 and SMV3_gp63. Cells cultured in medium ATV were taken at different time and observed under an inverted fluorescence microscope for DNA content using ImageStreamX MarkII Quantitative imaging analysis flow cytometry (Merck Millipore, Germany); scale bars: 2 μm.
DISCUSSION
Studies on cell cycle progression and regulation in archaea could provide a key to understanding the eukaryogenesis, one of the most intriguing mysteries in biology. Recent research has shown that archaea of the Asgard superphylum, in particular, Heimdallarchaeota, are evolutionarily most closely related to eukaryotes (50–52). However, microorganisms from the Asgard superphylum are difficult to cultivate and no genetically tractable system has been established, which hinders the characterization of the putative archaeal ancestor of eukaryotes. Archaea of the TACK superphylum share many features with the Asgard superphylum and could serve as valuable models for understanding the evolution of eukaryotic-like features in archaea. In particular, genetically tractable members of the genera Sulfolobus and Saccharolobus represent one of such archaeal model systems with many eukaryotic signature proteins and a eukaryotic-like cell cycle (53,54). Mapping of the cell cycle regulatory mechanisms and identification of proteins involved in cell-cycle processes in these model archaea is crucial for understanding the basic biology of archaea (18) and the origin of Eukaryota. In this study, we identified a small RHH family transcription factor, named aCcr1, conserved in Sulfolobales and their viruses that can arrest the cell division of Saccharolobus islandicus. It binds to a conserved 9-bp palindromic motif, the aCcr1-box, within the promoter and 5′ UTR of cdvA, an archaea-specific component of the cell division machinery, thereby repressing the cdvA expression. Notably, sequence analysis showed that the aCcr1-box is present in all Sulfolobales cdvA promoters and 5′UTRs at equivalent positions. Thus, we hypothesize that aCcr1-mediated cell cycle control through repression of CdvA is conserved in this archaeal order.
In halophilic euryarchaea, which use the FtsZ-based bacterial-like system for the cell division (6,8,47), an RHH family transcription factor, CdrS, plays a central role in the regulation of the cell division. Interestingly, the cdrS-ftsZ2 locus shows conserved gene synteny across the Euryarchaeota, especially within the Halobacteria (Supplementary Figure S8) (27), suggesting a general cell division regulation mechanism in euryarchaea. Both aCcr1 and CdrS are small RHH proteins and although they are distantly related at the amino acid sequence level (only 15 amino acids are identical), the modelled 3D structures are very similar (Supplementary Figure S1C). Furthermore, the aCcr1-box is similar to the most conserved part of the putative CdrS binding motif (also a palindromic sequence) (26). On the other hand, aCcr1 seems to have multiple binding sites in the genome, whereas CdrS has a limited number of targets. While CdrS and aCcr1 play equivalent roles in the control of cell cycle progression, perhaps due to their ability to regulate the expression of the respective cell division genes, the difference in the numbers of targets between halophilic euryarchaea and Sulfolobales may be related to their respective cell cycle features. We hypothesize that the aCcr1/CdrS-mediated cell division regulation mechanism has originated before the divergence of archaeal lineages into those using the ESCRT-III-based and FtsZ-based cell division systems.
Apart from cdvA, 11 other genes were highly repressed (>4 folds) probably directly by aCcr1 in the aCcr1 over-expression strain. The proteins for which the corresponding promoters contain the aCcr1-box include ePK1, possible nutrient uptake related proteins (endoglucanase, SiRe_0332, and thermopsin-like protease, SiRe_0691), and a predicted component of anti-virus defence system ATPase (SiRe_0086) (Table 1). The ePK1 from S. islandicus and its homolog in S. acidocaldarius exhibited DNA damage agent-dependent changes at the transcriptional level or in phosphorylation status (14,23,55). Phosphorylation plays a crucial role in the cell cycle regulation in eukaryotic cells, and may also play a similar role in archaea. Further characterization of the regulation of these genes by aCcr1 and identification of the function of other multiple binding sites could help unravel the cell cycle regulation network in Archaea.
Many aCcr1 homologs are present in the genomes of archaeal viruses (Figure 8). Over-expression of the aCcr1 homologs from the Acidianus two-tailed virus (ATV_gp29) and Sulfolobus monocaudavirus 3 (SMV3_gp63) resulted in growth retardation and appearance of enlarged cells with multiple chromosomes (Figure 9). ATV and SMV3 are spindle-shaped viruses belonging to the family Bicaudaviridae (56), which also includes genetically distant but morphologically similar S. tengchongensis spindle-shaped virus 1 (STSV1), S. tengchongensis spindle-shaped virus 2 (STSV2), Acidianus tailed spindle virus (ATSV), and Sulfolobus monocaudavirus 1 (SMV1). We previously reported a virus-induced cell enlargement of S. islandicus REY15A by SMV1 and STSV2, illuminating the inherent plasticity of Sulfolobus cells, which might be relevant for eukaryogenesis (28). Although the regulators manipulating cell division in STSV2 and SMV1 are yet to be identified, the finding that ATV- and SMV3-encoded aCcr1 homologs induce cell enlargement suggests that a similar mechanism might be operating in STSV2- and SMV1-infected cells. By hijacking a key cell division regulator, viruses can manipulate the archaeal cell cycle, transforming the cell into a giant virion producing factory. A similar scenario might have also taken place in ancestral archaea, producing cells with sufficiently large volume, a prerequisite for eukaryogenesis.
We found that transcriptional level of cdvA peaked at about 60 min following the removal of acetic acid, while the levels of aCcr1, escrt-III and vps4 reached their maxima at approximately 120 minutes after the release of the cell cycle arrest (Figure 1B). In S. acidocaldarius, the peak of aCcr1 (Saci_0942) transcription was about 30 min after that for CdvA, but coincided with those of ESCRT-III and Vps4 (18), exhibiting similar transcription pattern for accr1, escrt-III, and vps4. Exactly how aCcr1 regulates the expression of CdvA and other genes as well as how it is regulated at protein level remains unknown. Addressing these questions is partially hampered by failure to make a sensitive and sufficiently specific antibody against CdvA and detection of cyclic expression of accr1 in the wild-type cells in this study, which need to be solved in future investigations. Based on these results, we propose a scenario of cell division control by aCcr1 in S. islandicus REY15A. Following the initiation of cell division by CdvA, aCcr1 expression is activated by an unidentified factor, leading to a timely shut down of the CdvA expression. This repression is likely necessary to prevent further recruitment of ESCRT-III to the membrane at the mid-cell and to ensure that the cell division ring assembles only during the cytokinesis stage of the cell cycle (47). In summary, we have identified a key cell division regulator, a small RHH family protein aCcr1 that controls cell division in crenarchaea through repression of the factor which orchestrates the assembly of the cytokinesis machinery. This study opens doors for further dissection of the cell cycle regulation network in archaea.
DATA AVAILABILITY
All data supporting the findings of this study are available within the article and its Supplementary Information, or from the corresponding author upon reasonable request. The RNA-seq and ChIP-seq data for the overexpression strain were deposited under the accession number GSE218793, whereas the RNA-seq dada for the synchronization samples is available under GSE220819.
Supplementary Material
ACKNOWLEDGEMENTS
The work in the MK laboratory was supported by a grant from Ville de Paris (Emergence(s) project MEMREMA). We would like to thank all the lab members of the CRISPR and Archaea Biology Research Centre for helpful discussions.
Contributor Information
Yunfeng Yang, CRISPR and Archaea Biology Research Centre, Microbial Technology Institute and State Key Laboratory of Microbial Technology, Shandong University, Qingdao, 266237, P. R. China.
Junfeng Liu, CRISPR and Archaea Biology Research Centre, Microbial Technology Institute and State Key Laboratory of Microbial Technology, Shandong University, Qingdao, 266237, P. R. China; Institut Pasteur, Université Paris Cité, CNRS UMR6047, Archaeal Virology Unit, Paris, 75015, France.
Xiaofei Fu, CRISPR and Archaea Biology Research Centre, Microbial Technology Institute and State Key Laboratory of Microbial Technology, Shandong University, Qingdao, 266237, P. R. China.
Fan Zhou, CRISPR and Archaea Biology Research Centre, Microbial Technology Institute and State Key Laboratory of Microbial Technology, Shandong University, Qingdao, 266237, P. R. China.
Shuo Zhang, CRISPR and Archaea Biology Research Centre, Microbial Technology Institute and State Key Laboratory of Microbial Technology, Shandong University, Qingdao, 266237, P. R. China.
Xuemei Zhang, CRISPR and Archaea Biology Research Centre, Microbial Technology Institute and State Key Laboratory of Microbial Technology, Shandong University, Qingdao, 266237, P. R. China.
Qihong Huang, CRISPR and Archaea Biology Research Centre, Microbial Technology Institute and State Key Laboratory of Microbial Technology, Shandong University, Qingdao, 266237, P. R. China.
Mart Krupovic, Institut Pasteur, Université Paris Cité, CNRS UMR6047, Archaeal Virology Unit, Paris, 75015, France.
Qunxin She, CRISPR and Archaea Biology Research Centre, Microbial Technology Institute and State Key Laboratory of Microbial Technology, Shandong University, Qingdao, 266237, P. R. China.
Jinfeng Ni, CRISPR and Archaea Biology Research Centre, Microbial Technology Institute and State Key Laboratory of Microbial Technology, Shandong University, Qingdao, 266237, P. R. China.
Yulong Shen, CRISPR and Archaea Biology Research Centre, Microbial Technology Institute and State Key Laboratory of Microbial Technology, Shandong University, Qingdao, 266237, P. R. China.
SUPPLEMENTARY DATA
Supplementary Data are available at NAR Online.
FUNDING
National Key Research and Development Program of China [2020YFA0906800]; National Natural Science Foundation of China [31970546, 31670061 to Y.S.; 31970119 to J.N.; 31771380 to Q.S.]; State Key Laboratory of Microbial Technology.
Conflict of interest statement. None declared.
This paper is linked to: doi:10.1093/nar/gkad011.
REFERENCES
- 1. Dewachter L., Verstraeten N., Fauvart M., Michiels J.. An integrative view of cell cycle control in Escherichia coli. FEMS Microbiol. Rev. 2018; 42:116–136. [DOI] [PubMed] [Google Scholar]
- 2. Evans T., Rosenthal E.T., Youngblom J., Distel D., Hunt T.. Cyclin: a protein specified by maternal mRNA in sea urchin eggs that is destroyed at each cleavage division. Cell. 1983; 33:389–396. [DOI] [PubMed] [Google Scholar]
- 3. Galderisi U., Jori F.P., Giordano A.. Cell cycle regulation and neural differentiation. Oncogene. 2003; 22:5208–5219. [DOI] [PubMed] [Google Scholar]
- 4. Diaz-Moralli S., Tarrado-Castellarnau M., Miranda A., Cascante M.. Targeting cell cycle regulation in cancer therapy. Pharmacol. Ther. 2013; 138:255–271. [DOI] [PubMed] [Google Scholar]
- 5. Morgan D.O. Principles of CDK regulation. Nature. 1995; 374:131–134. [DOI] [PubMed] [Google Scholar]
- 6. Caspi Y., Dekker C.. Dividing the archaeal way: the ancient Cdv cell-division machinery. Front. Microbiol. 2018; 9:174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Liao Y., Ithurbide S., Evenhuis C., Lowe J., Duggin I.G.. Cell division in the archaeon haloferax volcanii relies on two FtsZ proteins with distinct functions in division ring assembly and constriction. Nat. Microbiol. 2021; 6:594–605. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Lindas A.C., Karlsson E.A., Lindgren M.T., Ettema T.J., Bernander R.. A unique cell division machinery in the Archaea. Proc. Natl. Acad. Sci. U.S.A. 2008; 105:18942–18946. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Samson R.Y., Obita T., Freund S.M., Williams R.L., Bell S.D.. A role for the ESCRT system in cell division in archaea. Science. 2008; 322:1710–1713. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Bernander R. Chromosome replication, nucleoid segregation and cell division in archaea. Trends Microbiol. 2000; 8:278–283. [DOI] [PubMed] [Google Scholar]
- 11. Lindas A.C., Bernander R.. The cell cycle of archaea. Nat. Rev. Microbiol. 2013; 11:627–638. [DOI] [PubMed] [Google Scholar]
- 12. Dexl S., Reichelt R., Kraatz K., Schulz S., Grohmann D., Bartlett M., Thomm M.. Displacement of the transcription factor B reader domain during transcription initiation. Nucleic Acids Res. 2018; 46:10066–10081. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Hoffmann L., Anders K., Bischof L.F., Ye X., Reimann J., Khadouma S., Pham T.K., van der Does C., Wright P.C., Essen L.O.et al.. Structure and interactions of the archaeal motility repression module ArnA-ArnB that modulates archaellum gene expression in sulfolobus acidocaldarius. J. Biol. Chem. 2019; 294:7460–7471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Huang Q., Lin Z., Wu P., Ni J., Shen Y.. Phosphoproteomic analysis reveals Rio1-related protein phosphorylation changes in response to UV irradiation in Sulfolobus islandicus REY15A. Front. Microbiol. 2020; 11:586025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Huang Q., Mayaka J.B., Zhong Q., Zhang C., Hou G., Ni J., Shen Y.. Phosphorylation of the archaeal Holliday junction resolvase hjc inhibits its catalytic activity and facilitates DNA repair in Sulfolobus islandicus REY15A. Front. Microbiol. 2019; 10:1214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Huang Q., Zhong Q., Mayaka J.B.A., Ni J., Shen Y.. Autophosphorylation and cross-phosphorylation of protein kinases from the Crenarchaeon Sulfolobus islandicus. Front. Microbiol. 2017; 8:2173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Reimann J., Lassak K., Khadouma S., Ettema T.J., Yang N., Driessen A.J., Klingl A., Albers S.V.. Regulation of archaella expression by the FHA and von Willebrand domain-containing proteins ArnA and ArnB in sulfolobus acidocaldarius. Mol. Microbiol. 2012; 86:24–36. [DOI] [PubMed] [Google Scholar]
- 18. Lundgren M., Bernander R.. Genome-wide transcription map of an archaeal cell cycle. Proc. Natl. Acad. Sci. U.S.A. 2007; 104:2939–2944. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Tarrason Risa G., Hurtig F., Bray S., Hafner A.E., Harker-Kirschneck L., Faull P., Davis C., Papatziamou D., Mutavchiev D.R., Fan C.et al.. The proteasome controls ESCRT-III-mediated cell division in an archaeon. Science. 2020; 369:eaaz2532. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Lemmens L., Maklad H.R., Bervoets I., Peeters E.. Transcription regulators in Archaea: homologies and differences with bacterial regulators. J. Mol. Biol. 2019; 431:4132–4146. [DOI] [PubMed] [Google Scholar]
- 21. Lewis M., Stracker T.H.. Transcriptional regulation of multiciliated cell differentiation. Semin. Cell Dev. Biol. 2021; 110:51–60. [DOI] [PubMed] [Google Scholar]
- 22. Yue L., Li J., Zhang B., Qi L., Li Z., Zhao F., Li L., Zheng X., Dong X.. The conserved ribonuclease aCPSF1 triggers genome-wide transcription termination of archaea via a 3'-end cleavage mode. Nucleic Acids Res. 2020; 48:9589–9605. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Sun M., Feng X., Liu Z., Han W., Liang Y.X., She Q.. An Orc1/Cdc6 ortholog functions as a key regulator in the DNA damage response in Archaea. Nucleic Acids Res. 2018; 46:6697–6711. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Chu Y., Zhu Y., Chen Y., Li W., Zhang Z., Liu D., Wang T., Ma J., Deng H., Liu Z.J.et al.. aKMT catalyzes extensive protein lysine methylation in the hyperthermophilic Archaeon Sulfolobus islandicus but is dispensable for the growth of the organism. Mol. Cell. Proteomics. 2016; 15:2908–2923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Vorontsov E.A., Rensen E., Prangishvili D., Krupovic M., Chamot-Rooke J.. Abundant lysine methylation and N-terminal acetylation in sulfolobus islandicus revealed by bottom-up and top-down proteomics. Mol. Cell. Proteomics. 2016; 15:3388–3404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Liao Y., Vogel V., Hauber S., Bartel J., Alkhnbashi O.S., Maass S., Schwarz T.S., Backofen R., Becher D., Duggin I.G.et al.. CdrS is a global transcriptional regulator influencing cell division in Haloferax volcanii. Mbio. 2021; 12:e0141621. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Darnell C.L., Zheng J., Wilson S., Bertoli R.M., Bisson-Filho A.W., Garner E.C., Schmid A.K.. The ribbon-helix-helix domain protein CdrS regulates the tubulin homolog ftsZ2 to control cell division in Archaea. Mbio. 2020; 11:e01007–e01020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Liu J., Cvirkaite-Krupovic V., Baquero D.P., Yang Y., Zhang Q., Shen Y., Krupovic M.. Virus-induced cell gigantism and asymmetric cell division in archaea. Proc. Natl. Acad. Sci. U.S.A. 2021; 118:e2022578118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Deng L., Zhu H., Chen Z., Liang Y.X., She Q.. Unmarked gene deletion and host-vector system for the hyperthermophilic crenarchaeon Sulfolobus islandicus. Extremophiles. 2009; 13:735–746. [DOI] [PubMed] [Google Scholar]
- 30. Liu J., Cvirkaite-Krupovic V., Commere P.H., Yang Y., Zhou F., Forterre P., Shen Y., Krupovic M.. Archaeal extracellular vesicles are produced in an ESCRT-dependent manner and promote gene transfer and nutrient cycling in extreme environments. ISME J. 2021; 15:2892–2905. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Guo L., Brugger K., Liu C., Shah S.A., Zheng H., Zhu Y., Wang S., Lillestol R.K., Chen L., Frank J.et al.. Genome analyses of Icelandic strains of Sulfolobus islandicus, model organisms for genetic and virus-host interaction studies. J. Bacteriol. 2011; 193:1672–1680. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Lundgren M., Andersson A., Chen L., Nilsson P., Bernander R.. Three replication origins in Sulfolobus species: synchronous initiation of chromosome replication and asynchronous termination. Proc. Natl. Acad. Sci. U.S.A. 2004; 101:7046–7051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Bailey T.L., Boden M., Buske F.A., Frith M., Grant C.E., Clementi L., Ren J., Li W.W., Noble W.S.. MEME SUITE: tools for motif discovery and searching. Nucleic Acids Res. 2009; 37:W202–W208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Altschul S.F., Madden T.L., Schaffer A.A., Zhang J., Zhang Z., Miller W., Lipman D.J.. Gapped BLAST and PSI-BLAST: a new generation of protein database search programs. Nucleic Acids Res. 1997; 25:3389–3402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Gabler F., Nam S.Z., Till S., Mirdita M., Steinegger M., Soding J., Lupas A.N., Alva V.. Protein sequence analysis using the MPI bioinformatics toolkit. Curr. Protoc. Bioinform. 2020; 72:e108. [DOI] [PubMed] [Google Scholar]
- 36. Katoh K., Rozewicki J., Yamada K.D.. MAFFT online service: multiple sequence alignment, interactive sequence choice and visualization. Brief. Bioinform. 2019; 20:1160–1166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Capella-Gutierrez S., Silla-Martinez J.M., Gabaldon T.. trimAl: a tool for automated alignment trimming in large-scale phylogenetic analyses. Bioinformatics. 2009; 25:1972–1973. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Minh B.Q., Schmidt H.A., Chernomor O., Schrempf D., Woodhams M.D., Haeseler A., Lanfear R.. IQ-TREE 2: new models and efficient methods for phylogenetic inference in the genomic era. Mol. Biol. Evol. 2020; 37:1530–1534. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Guindon S., Dufayard J.F., Lefort V., Anisimova M., Hordijk W., Gascuel O.. New algorithms and methods to estimate maximum-likelihood phylogenies: assessing the performance of PhyML 3.0. Syst. Biol. 2010; 59:307–321. [DOI] [PubMed] [Google Scholar]
- 40. Takemata N., Samson R.Y., Bell S.D.. Physical and functional compartmentalization of archaeal chromosomes. Cell. 2019; 179:165–179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Zhang Y., Liu T., Meyer C.A., Eeckhoute J., Johnson D.S., Bernstein B.E., Nusbaum C., Myers R.M., Brown M., Li W.et al.. Model-based analysis of ChIP-Seq (MACS). Genome Biol. 2008; 9:R137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Schreiter E.R., Drennan C.L.. Ribbon-helix-helix transcription factors: variations on a theme. Nat. Rev. Microbiol. 2007; 5:710–720. [DOI] [PubMed] [Google Scholar]
- 43. Li Y., Pan S., Zhang Y., Ren M., Feng M., Peng N., Chen L., Liang Y.X., She Q.. Harnessing type I and type III CRISPR-cas systems for genome editing. Nucleic Acids Res. 2016; 44:e34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Zhang C., Phillips A.P.R., Wipfler R.L., Olsen G.J., Whitaker R.J.. The essential genome of the crenarchaeal model Sulfolobus islandicus. Nat. Commun. 2018; 9:4908. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Peng N., Han W., Li Y., Liang Y., She Q.. Genetic technologies for extremely thermophilic microorganisms of sulfolobus, the only genetically tractable genus of crenarchaea. Sci. China Life Sci. 2017; 60:370–385. [DOI] [PubMed] [Google Scholar]
- 46. Liu J., Gao R., Li C., Ni J., Yang Z., Zhang Q., Chen H., Shen Y.. Functional assignment of multiple ESCRT-III homologs in cell division and budding in Sulfolobus islandicus. Mol. Microbiol. 2017; 105:540–553. [DOI] [PubMed] [Google Scholar]
- 47. Samson R.Y., Obita T., Hodgson B., Shaw M.K., Chong P.L., Williams R.L., Bell S.D.. Molecular and structural basis of ESCRT-III recruitment to membranes during archaeal cell division. Mol. Cell. 2011; 41:186–196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Moriscot C., Gribaldo S., Jault J.M., Krupovic M., Arnaud J., Jamin M., Schoehn G., Forterre P., Weissenhorn W., Renesto P.. Crenarchaeal CdvA forms double-helical filaments containing DNA and interacts with ESCRT-III-like CdvB. PLoS One. 2011; 6:e21921. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Prangishvili D., Bamford D.H., Forterre P., Iranzo J., Koonin E.V., Krupovic M.. The enigmatic archaeal virosphere. Nat. Rev. Microbiol. 2017; 15:724–739. [DOI] [PubMed] [Google Scholar]
- 50. Imachi H., Nobu M.K., Nakahara N., Morono Y., Ogawara M., Takaki Y., Takano Y., Uematsu K., Ikuta T., Ito M.et al.. Isolation of an archaeon at the prokaryote-eukaryote interface. Nature. 2020; 577:519–525. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Liu Y., Makarova K.S., Huang W.C., Wolf Y.I., Nikolskaya A.N., Zhang X., Cai M., Zhang C.J., Xu W., Luo Z.et al.. Expanded diversity of Asgard archaea and their relationships with eukaryotes. Nature. 2021; 593:553–557. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Zaremba-Niedzwiedzka K., Caceres E.F., Saw J.H., Backstrom D., Juzokaite L., Vancaester E., Seitz K.W., Anantharaman K., Starnawski P., Kjeldsen K.U.et al.. Asgard archaea illuminate the origin of eukaryotic cellular complexity. Nature. 2017; 541:353–358. [DOI] [PubMed] [Google Scholar]
- 53. Bernander R., Poplawski A.. Cell cycle characteristics of thermophilic archaea. J. Bacteriol. 1997; 179:4963–4969. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Spang A., Caceres E.F., Ettema T.J.G.. Genomic exploration of the diversity, ecology, and evolution of the archaeal domain of life. Science. 2017; 357:eaaf3883. [DOI] [PubMed] [Google Scholar]
- 55. Gotz D., Paytubi S., Munro S., Lundgren M., Bernander R., White M.F.. Responses of hyperthermophilic crenarchaea to UV irradiation. Genome Biol. 2007; 8:R220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Prangishvili D., Krupovic M., Ictv Report C.. ICTV virus taxonomy profile: bicaudaviridae. J. Gen. Virol. 2018; 99:864–865. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
All data supporting the findings of this study are available within the article and its Supplementary Information, or from the corresponding author upon reasonable request. The RNA-seq and ChIP-seq data for the overexpression strain were deposited under the accession number GSE218793, whereas the RNA-seq dada for the synchronization samples is available under GSE220819.