

Abstract
BACKGROUND:
Neuropeptides are contained in nearly every neuron in the central nervous system and can be released not only from nerve terminals but also from somatodendritic sites. Cholecystokinin (CCK), among the most abundant neuropeptides in the brain, is expressed in the majority of midbrain dopamine neurons. Despite this high expression, CCK function within the ventral tegmental area (VTA) is not well understood.
METHODS:
We confirmed CCK expression in VTA dopamine neurons through immunohistochemistry and in situ hybridization and detected optogenetically induced CCK release using an enzyme-linked immunosorbent assay. To investigate whether CCK modulates VTA circuit activity, we used whole-cell patch clamp recordings in mouse brain slices. We infused CCK locally in vivo and tested food intake and locomotion in fasted mice. We also used in vivo fiber photometry to measure Ca2+ transients in dopamine neurons during feeding.
RESULTS:
Here we report that VTA dopamine neurons release CCK from somatodendritic regions, where it triggers long-term potentiation of GABAergic (gamma-aminobutyric acidergic) synapses. The somatodendritic release occurs during trains of optogenetic stimuli or prolonged but modest depolarization and is dependent on synaptotagmin-7 and T-type Ca2+ channels. Depolarization-induced long-term potentiation is blocked by a CCK2 receptor antagonist and mimicked by exogenous CCK. Local infusion of CCK in vivo inhibits food consumption and decreases distance traveled in an open field test. Furthermore, intra-VTA–infused CCK reduced dopamine cell Ca2+ signals during food consumption after an overnight fast and was correlated with reduced food intake.
CONCLUSIONS:
Our experiments introduce somatodendritic neuropeptide release as a previously unknown feedback regulator of VTA dopamine cell excitability and dopamine-related behaviors.
Nearly every neuron in the brain contains neuropeptides (1). Acting at G protein–coupled receptors, they can regulate neuronal excitability by modulating voltage-gated ion channels and modify synaptic transmission mediated by classical neurotransmitters (1,2). Neuropeptides are synthesized in somata and dendrites and are then cleaved and stored in large dense core vesicles, which are axonally transported and released from nerve terminals. However, neuropeptides are also released at somatodendritic sites (2–10). The best-studied examples of somatodendritic peptide release are oxytocin and vasopressin, which serve autocrine and paracrine roles in regulating hypothalamic neuron excitability (7,11). Large dense core vesicles that contain peptides are found in similar numbers in axons and dendrites (12–14), indicating that somatodendritic peptide release may serve a functional role as important as neuropeptide release from nerve terminals.
The first example of somatodendritic neurotransmitter release was dopamine release from ventral tegmental area (VTA) and substantia nigra neurons (15). Although dopamine cells express several neuropeptides, somatodendritic neuropeptide release from these neurons has not been described. Originally identified in the gut as a modulator of digestive function, cholecystokinin (CCK) is one of the most highly expressed neuropeptides in the brain (16,17). CCK is coex-pressed with dopamine in most VTA neurons in rats, monkeys, mice, and humans (18–24).
Controlling the excitability of midbrain dopamine neurons is critical because changes in dopamine release shape multiple physiological phenomena ranging from motor function to motivation and learning (25–31). Both local and extrinsic GABAergic (gamma-aminobutyric acidergic) afferents control the firing of dopamine neurons, and thus plasticity of inhibitory synapses has a major influence on these circuits (32–35). We previously reported that inhibitory GABAergic synapses in the VTA undergo long-term potentiation (LTP) following afferent stimulation paired with depolarization (36,37). These studies indicated an NMDA receptor–independent mechanism for LTP that requires depolarization of the postsynaptic dopamine cell and is blocked by postsynaptic chelation of Ca2+, which are features consistent with somatodendritic release of a neuro-modulator mediating LTP induction. Here we report that somatodendritic release of CCK underlies LTP of specific GABAergic synapses onto VTA dopamine neurons. We also demonstrate that CCK depresses VTA dopamine cell activity, feeding, and locomotion. Our work suggests that CCK released somatodendritically from dopamine neurons exerts potent behavioral effects.
METHODS AND MATERIALS
All procedures were carried out in accordance with the guidelines of the National Institutes of Health for animal care and use and were approved by the Administrative Panel on Laboratory Animal Care from Stanford University. Pitx3-GFP, DAT-IRES-Cre, Ai32, syt7 knockout (KO), Ai14, and C57BL/6 male and female mice were bred in-house.
Electrophysiology
VTA-containing horizontal slices were cut in a choline-based solution at a thickness of 220 μm and submerged in artificial cerebrospinal fluid until being transferred to the recording chamber. Slices were perfused at 28 °C to 32 °C in artificial cerebrospinal fluid containing DNQX, APV (2-amino-5-phos-phonopentanoic acid), and strychnine. Whole-cell patch clamp recordings were made from VTA neurons.
Enzyme-Linked Immunosorbent Assay
DAT-IRES-Cre mice were injected with AAV-DJ-EF1a-DIO-hChR2(H134R)-EYFP into the VTA. One week later, horizontal slices from 5 mice were cut and a supernatant sample was collected; 2 seconds of 20-Hz blue light pulses were delivered (6 minutes) and the supernatant was again collected. CCK concentrations were measured using an enzyme-linked immunosorbent assay kit.
Behavioral Tests
Mice had intra-VTA saline or CCK (1000 pmol/0.5 μL) administered in a counterbalanced, crossover design, with at least 5 days between infusions. Mice were fasted overnight and then received local bilateral intra-VTA infusions. Immediately after drug infusions, mice were given access to a preweighed food pellet and food intake was assessed at 1 hour. For open field locomotion, after intra-VTA infusions, distance traveled and velocity were measured using video-tracking software.
Fiber Photometry
Fiber photometry was performed as previously described (38,39). DAT-IRES-Cre mice were unilaterally injected with AAV9-hSyn-FLEX-jGCaMP8m into the VTA and implanted with dual optical fiber/multiple fluid injection cannulas that allow microinjections into the recording site. After 3 weeks, mice were habituated to palatable fruit loops in the home cage for 1 week and to microinjection and photometry procedures. On recording days, immediately after microinjections, mice were connected to patch cables. After a 5-minute baseline, fruit loops were placed in the cage and mice were allowed to consume freely for 30 minutes.
Statistics
Sample sizes were determined from our previous experiments and from related literature for electrophysiology experiments (40,41), behavior (42–52), and fiber photometry (38,39,51,53–58). All data are presented as mean ± standard error. Differences were deemed significant with p < .05. Data were first tested using the Shapiro-Wilk test for assessing normality. Data that passed this test were tested using a parametric test, while data that did not display a normal distribution were tested using a nonparametric test for significance (Table S1). We did not observe sex-specific trends in any of our experiments with CCK, although brain punches from the VTA of male rats have been reported to have more CCK than that of females (59); no differences were noted across the estrus cycle (not measured in our experiments) (60). In brain punches, differences in CCK concentrations may result from nerve terminals or cell bodies other than dopamine neurons.
RESULTS
Depolarization of a Postsynaptic Dopamine Neuron Potentiates GABAA Inhibitory Postsynaptic Currents
We previously found that low-frequency stimulation (LFS) paired with sustained modest depolarization induces LTP at GABAA synapses in the VTA, but the mechanisms were unclear (36) (Figure 1A–C). We hypothesized that even in the absence of LFS, depolarization could release a signaling molecule from the recorded dopamine neuron that in turn elicits LTP. To test whether depolarization alone could potentiate GABAergic synapses, we recorded GABAA receptor inhibitory postsynaptic currents (IPSCs) evoked using a stimulating electrode placed caudal to the VTA. Depolarization (−40 mV for 6 minutes) caused LTP of a magnitude similar to that triggered by LFS plus depolarization (Figure 1D–F), with a relatively slow onset. Thus, LFS is not required, but instead depolarization alone is sufficient to initiate LTP. We measured paired-pulse ratios and coefficient of variance (1/CV2) before and after depolarization but observed no differences either in paired-pulse ratios (0.93 ± 0.06 vs. 0.86 ± 0.06; p = .34) or 1/CV2 (19.6 ± 7.3 vs. 26.5 ± 9.9; p > .9999). To further test whether depolarization of dopamine cells is sufficient to induce LTP, we expressed ChR2 (channelrhodopsin-2) in the VTA of DAT-IRES-Cre mice and stimulated dopamine cells with blue light. Optogenetic stimulation trains also triggered LTP of evoked GABAergic IPSCs (Figure 1G–I).
Figure 1.
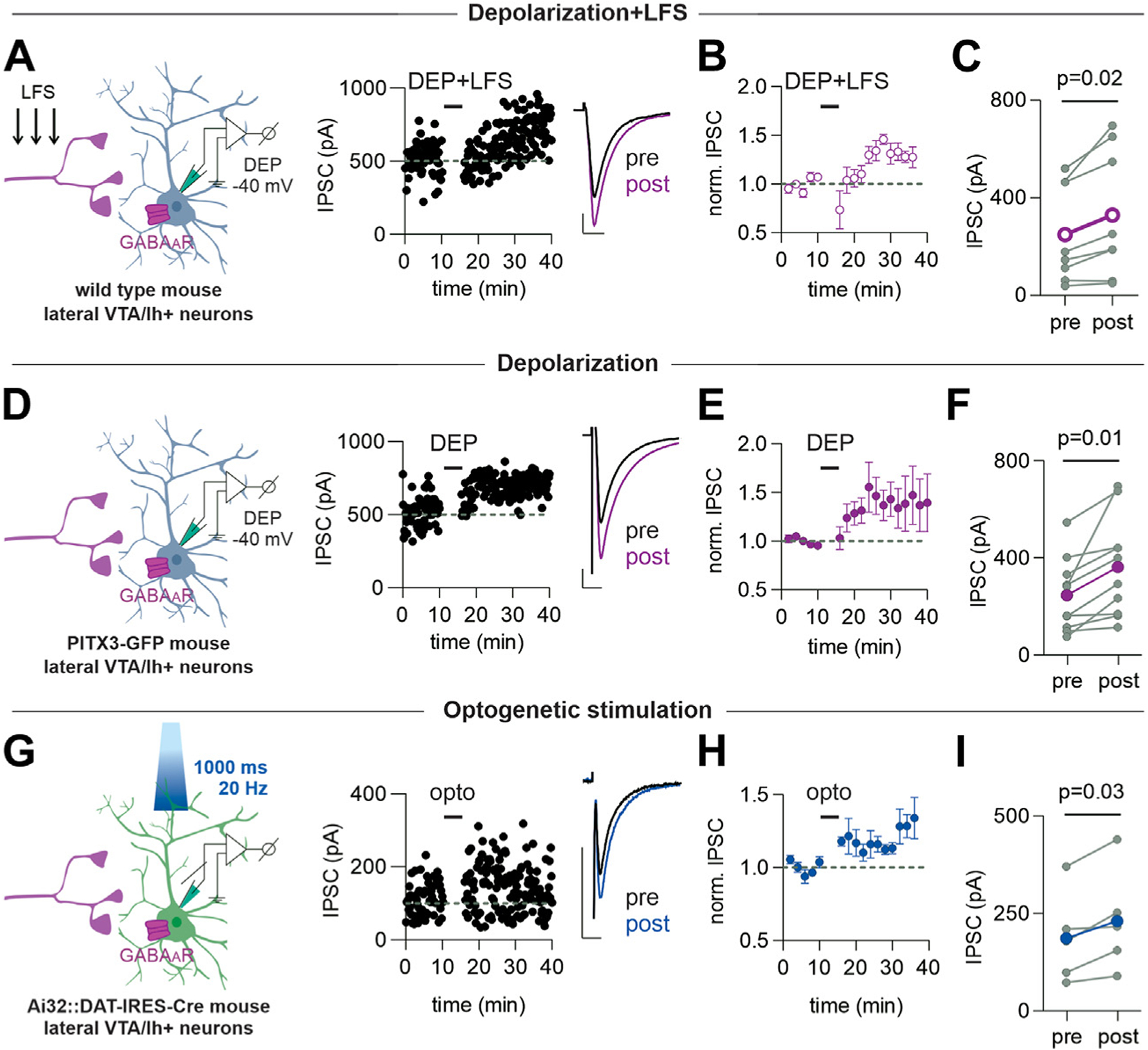
Depolarization alone potentiates IPSCs in the VTA dopamine cells. Left-hand diagrams in this and all figures illustrate the experimental design. (A) Representative time course and example IPSCs (inset) before and after a 6-minute depolarization of the recorded dopamine neuron from −70 to −40 mV with simultaneous afferent LFS (1 Hz) (DEP+LFS). (B) Averaged IPSC amplitudes before and after DEP+LFS (n = 8 cells/8 mice). (C) IPSC amplitudes before and after DEP+LFS (n = 8 cells/8 mice, 5 cells from male mice, 3 from female mice). Paired t test, p = .02, df = 7. For this and all figures, colored symbols/lines represent the mean. Error bars represent SEM. (D) Representative time course and example IPSCs before and after a 6-minute depolarization of the recorded neuron from −70 to −40 mV (DEP). Data for (A–C) are from wild-type mice. (E) Averaged IPSC amplitudes before and after DEP (n = 10 cells/8 mice). (F) IPSC amplitudes before and after DEP (n = 10 cells/8 mice; 7 cells from male mice, 3 from female mice). Paired t test, p = .01, df = 9. (G) Representative time course and example IPSCs from a VTA dopamine neuron expressing channelrhodopsin-2 (Ai32:DAT-IRES-Cre mouse) before and after optical stimulation using trains of light (20 Hz, 1000 ms) delivered for 6 minutes in current clamp. (H) Time course of averaged IPSC amplitudes before and after optical stimulation (n = 5 cells/3 mice). (I) IPSC amplitudes before and after optical stimulation (n = 5 cells/3 mice, 2 cells from male mice, 3 from female mice). Paired t test, p = .03, df = 4. Scale bars = 100 pA, 10 ms. DEP, depolarization; GFP, green fluorescent protein; IPSC, inhibitory postsynaptic current; LFS, low-frequency stimulation; norm., normalized; opto, optogenetic stimulation; VTA, ventral tegmental area.
Dopamine has long been known to be released somatodendritically from midbrain dopamine neurons and is therefore an obvious candidate to mediate LTP. Blocking both D1 and D2 receptor classes did not prevent depolarization-induced LTP, indicating that dopamine release is not necessary for this form of LTP (Figure 2A–C). Instead some other signaling molecule released upon depolarization may be required.
Figure 2.

Depolarization-induced long-term potentiation is not blocked by dopamine receptor antagonists. (A) Representative time course and example IPSCs before and after DEP+LFS in the presence of sulpiride (150 nM) and SCH 23390 (10 μM). (B) Time course of averaged IPSC amplitudes before and after DEP+LFS (n = 8 cells/4 mice). (C) IPSC amplitudes before and after DEP+LFS (n = 8 cells/4 mice, 8 cells from female mice). Paired t test, p = .02, df = 7. Scale bars = 100 pA, 10 ms. D1R, dopamine D1 receptor; D2R, dopamine D2 receptor; DEP, depolarization; GFP, green fluorescent protein; IPSC, inhibitory postsynaptic current; LFS, low-frequency stimulation; norm., normalized; VTA, ventral tegmental area.
Dopamine Neurons Contain CCK and Release It With Optogenetic Stimulation
GFP-positive cells from Pitx3-GFP mice are concordant with tyrosine hydroxylase–expressing neurons in the lateral VTA (40). Using these mice, we confirmed that CCK is highly expressed in VTA dopamine cells (Figure S1A–C) (75.3 ± 1.8% GFP-positive neurons were also CCK-positive), as shown previously. Consistent with this finding, we found a 77.5 ± 1.6% overlap of messenger RNA for DAT and CCK using fluorescence in situ hybridization (22) (Figure S1D–F).
It was previously found that in rat midbrain slices, high K+ depolarization evoked CCK release that was measured using radioimmunoassay. However, this protocol is expected to release CCK from both somatodendritic and axon terminal sites (41). In a more targeted approach, we used DAT-IRES-Cre mice selectively expressing ChR2 in dopamine cells and measured CCK through an enzyme-linked immunosorbent assay. After optogenetic stimulation, the CCK concentration in the supernatant increased (Figure 3C), demonstrating that CCK release observed was from dopamine neurons.
Figure 3.

Depolarization of dopamine neurons releases CCK and triggers long-term potentiation acting on CCK2R receptors. (A) Diagram of experimental design for ELISA. (B) Representative calibration curve from 1 experiment. (C) CCK concentration before and after optogenetic stimulation of the VTA dopamine neurons (6 experiments, 5 mice/experiment, 19 male and 11 female mice). Wilcoxon paired test, p = .03. (D) Representative time course and example IPSCs before and after a 6-minute depolarization of the recorded neuron from −70 to −40 mV with simultaneous afferent LFS (1 Hz) (DEP+LFS) in the presence of LY225910 (1 μM). (E) Time course of averaged IPSC amplitudes before and after DEP+LFS (n = 7 cells/4 mice). Colored symbols/lines represent the mean response across all cells; error bars represent SEM. (F) IPSC amplitudes before and after DEP+LFS in the presence of LY225910 (n = 7 cells/4 mice, 6 cells from male mice, 1 cell from a female mouse). Wilcoxon paired test, p = .58. (G) Representative time course and example IPSCs before and after a 6-minute depolarization of the recorded dopamine neuron from −70 to −40 mV (DEP) in the presence of LY225910 (1 μM). (H) Averaged IPSC amplitudes before and after DEP (n = 6 cells/3 mice). (I) IPSC amplitudes before and after DEP in the presence of LY225910 (n = 6 cells/3 mice, 4 cells from male mice, 2 from female mice). Paired t test, p = .51, df = 5. Scale bars = 100 pA, 10 ms. (J) Representative confocal images of fluorescent in situ hybridization on a coronal slice through the VTA. Dat (green), Cck (red), Cck2r (magenta), and merge: overlay of all 3 signals. Scale bar at high magnification = 20 μm. CCK, cholecystokinin; CCK2R, cholecystokinin 2 receptor; DEP, depolarization; ELISA, enzyme-linked immunosorbent assay; GFP, green fluorescent protein; IPSC, inhibitory postsynaptic current; LFS, low-frequency stimulation; norm., normalized; opto, optogenetic; VTA, ventral tegmental area.
LTP Is Blocked by a CCK2 Receptor Antagonist
Might CCK release occurring during depolarization cause LTP? CCK acts via 2 G protein–coupled receptors, CCK1R and CCK2R (61,62); CCK2R is predominant in the brain (63–66). We therefore bath applied the CCK2R antagonist, LY225910, and tested whether depolarization was still capable of inducing LTP. Neither depolarization paired with LFS (Figure 3D–F) nor depolarization alone (Figure 3G–I) potentiated IPSCs in the presence of the CCK2R antagonist. Using fluorescence in situ hybridization, we did not detect expression of CCK1R (data not shown) or CCK2R messenger RNA within the VTA (Figure 3J), indicating that these receptors are not likely to be found in somata or glial cells within the VTA. These results suggest that CCK acting via the CCK2R is necessary for LTP and that CCK2Rs are likely to be localized on afferent nerve terminals rather than in any VTA cell population.
CCK Occludes LTP of IPSCs and Acts in a Synapse-Specific Manner
If depolarization-induced CCK release from a single recorded dopamine neuron is sufficient to trigger LTP (Figure 1D–F), then application of exogenous CCK should mimic this. As expected, bath application of CCK elicited LTP with a time course and of a magnitude similar to LTP evoked with depolarization (Figure 4A–C). If CCK release is part of the mechanism underlying depolarization-induced LTP, bath application of CCK would be expected to prevent further potentiation by depolarization. After CCK application, depolarization did not elicit further LTP, arguing for a shared mechanism (Figure 4D–F).
Figure 4.

CCK is necessary and sufficient to potentiate caudally evoked but not rostrally evoked IPSCs. (A) Representative time course and example IPSCs evoked in a dopamine neuron with a stimulating electrode placed caudal to the VTA (diagram) before and during bath application of CCK (0.1 μM). (B) Averaged caudally evoked IPSC amplitudes before and during the presence of CCK (n = 8 cells/7 mice). (C) Caudally evoked IPSC amplitudes before and after application of CCK (n = 8 cells/7 mice, 3 cells from male mice, 5 from female mice). Paired t test, p = .04, df = 7. (D) Representative time course and example IPSCs before and after DEP+LFS in the presence of CCK. CCK (1 μM) was present for at least 20 minutes before DEP+LFS. (E) Time course of averaged IPSC amplitudes before and after DEP+LFS (n = 7 cells/5 mice). (F) IPSC amplitudes before and after DEP+LFS (n = 7 cells/5 mice, 2 cells from male mice, 5 cells from female mice). Wilcoxon paired test, p > .999. (G) Representative time course and example IPSCs evoked in a dopamine neuron with a stimulating electrode placed rostrally within the VTA (diagram) before and after bath application of CCK (0.1 μM). (H) Averaged rostrally evoked IPSC amplitudes before and after CCK application (n = 7 cells/5 mice). (I) Rostrally evoked IPSC amplitudes before and after CCK application (n = 7 cells/5 mice, 5 cells from male mice, 2 cells from female mice). Paired t test, p = .86, df = 6. Scale bars = 100 pA, 10 ms. CCK, cholecystokinin; DEP, depolarization; GFP, green fluorescent protein; IPSC, inhibitory postsynaptic current; LFS, low-frequency stimulation; LTP, long-term potentiation; norm., normalized; VTA, ventral tegmental area.
In the experiments above, we used a stimulating electrode positioned caudal to the VTA. We next asked whether CCK-induced potentiation exhibits synapse specificity. In contrast to CCK-induced LTP of caudally evoked IPSCs, IPSCs evoked with a rostrally placed stimulating electrode were unaffected by CCK (Figure 4G–I), consistent with previous results (36,67). All our recordings to this point were localized to the lateral VTA region in Ih+ neurons. We also tested for CCK-induced LTP in GFP+, Ih− dopamine neurons in the medial VTA and found that CCK does not potentiate synapses on these cells (Figure S2). CCK also failed to potentiate caudally evoked IPSCs from identified VTA GABAergic neurons. Together our data demonstrate that CCK is necessary and sufficient for selective potentiation of caudally evoked IPSCs onto specific VTA cell populations.
CCK Does Not Modulate D2-IPSCs
Coexpression of CCK and dopamine and their shared ability to undergo somatodendritic release from dopamine neurons prompted us to investigate their interplay. Train-evoked postsynaptic currents mediated by release of dopamine acting at D2 autoreceptors are a convenient readout of somatodendritic dopamine release (D2-IPSCs) (68). To test the possibility that CCK modifies somatodendritic dopamine release, we simultaneously evoked D2-IPSCs and GABAA IPSCs in the same cells and bath-applied CCK (Figure 5A, B). While CCK potentiated GABAergic IPSCs as expected, it had no effect on D2-IPSCs (Figure 5C, D). Thus, CCK does not appear to influence somatodendritic release of dopamine.
Figure 5.

CCK potentiates GABAA synapses but does not affect D2-IPSCs. (A) Representative examples of (a) composite IPSCs (scale bar = 100 pA, 1s); (b) D2-IPSCs (scale bar = 10 pA, 10 ms); and (c) GABAA IPSCs (scale bar = 100 pA, 10 ms) recorded simultaneously from a single dopamine neuron before and after CCK (0.1 μM) bath application. (B) Representative example time course of simultaneously recorded D2-IPSCs (green) and GABAA IPSCs (purple). (C) Averaged IPSC amplitudes before and after CCK bath application (n = 10 cells/9 mice; 6 cells from male mice, 4 from female mice). (D) D2-IPSC (left) and GABAA (right) IPSC amplitudes before and after the application of CCK (n = 10 cells/9 mice). Wilcoxon paired test: D2-IPSCs, p > .99; GABAA IPSCs, p = .04. CCK, cholecystokinin; GABA, gamma-aminobutyric acid; IPSC, inhibitory postsynaptic current; norm., normalized.
CCK Release Requires T-Type Calcium Channels and Synaptotagmin-7
A rise in intracellular calcium is generally required for somatodendritic release of peptides, but the calcium sources can be diverse (38). Consistent with a critical role for intracellular Ca2+, inclusion of the calcium chelator BAPTA in the recording pipette prevented depolarization-induced LTP (Figure 6A–C). Our setup precluded contributions from NMDA receptors and/or AMPA receptors, while the prolonged depolarization protocol (6 minutes) limited the possible contributions of most voltage-gated calcium channels (VGCCs) because they are rapidly inactivated. However, tonic influx of calcium through T-type VGCCs can occur at membrane potentials close to rest (39,42–44). Therefore, we next tested NiCl2 at a concentration (50 μM) that primarily blocks T-type VGCCs (39,45,46). Depolarization did not potentiate IPSCs, indicating that T-type channels may be required for CCK release. In contrast, D2-IPSCs could still be evoked (4/4 cells) (Figure 6D–F).
Figure 6.

Mechanisms underlying somatodendritic depolarization–induced CCK release. (A) Representative time course and example IPSCs before and after a 6-minute DEP of the recorded neuron from −70 to −40 mV with BAPTA (30 mM) included in the patch pipette. To allow the BAPTA to diffuse into the cell, DEP was initiated at least 20 minutes after the start of whole-cell recording. (B) Time course of averaged IPSC amplitudes in BAPTA-loaded cells before and after DEP (n = 7 cells/4 mice). (C) IPSC amplitudes in BAPTA-loaded cells before and after DEP (n = 7 cells/4 mice, 2 cells from male mice, 5 cells from female mice). Paired t test, p = .54, df = 6. (D) Representative time course and example IPSCs before and after DEP in the presence of NiCl2 (50 μM). (E) Time course of averaged IPSC amplitudes before and after DEP (n = 6 cells/3 mice). (F) IPSC amplitudes before and after DEP in the presence of NiCl2 (n = 6 cells/3 mice, 5 cells from male mice, 1 cell from a female mouse). Paired t test, p = .29, df = 5. Pie chart indicates that in 4 of 4 neurons, D2-IPSCs could be evoked in the presence of NiCl2. (G) Representative time course and example IPSCs before and after DEP in a VTA dopamine neuron from a syt7 KO mouse. (H) Time course of averaged IPSC amplitudes before and after DEP in dopamine neurons from syt7 KO mice (n = 6 cells/3 mice). (I) IPSC amplitudes before and after DEP in cells from syt7 KO mice (n = 6 cells/3 mice, 4 cells from male mice, 2 cells from female mice). Pie chart indicates that in 6 of 7 syt7 KO neurons from syt7 KO mice, D2-IPSCs could not be evoked. Paired t test, p = .25, df = 5. Scale bars = 100 pA, 10 ms. D2, dopamine D2 receptor; DEP, depolarization; GFP, green fluorescent protein; IPSC, inhibitory postsynaptic current; KO, knockout; norm., normalized; VGCC, voltage-gated calcium channel; VTA, ventral tegmental area.
The release machinery required for somatodendritic release differs from that at nerve terminal active zones (38,47). Synaptotagmins-4 and −7 have been implicated in somatodendritic release of neuropeptides as well as dopamine (48–50). In syt7 KO mice, we found that depolarization-induced LTP was strongly attenuated (Figure 6G–I). D2-IPSCs in slices from these mice were also markedly reduced (6/7 cells). Our results suggest that both somatodendritic dopamine and CCK release require synaptotagmin-7.
Intra-VTA Delivery of CCK Reduces Food Intake, Locomotion, and Dopamine Neuron Firing Rate
What physiological processes might be controlled by the local release of CCK within the VTA? CCK-expressing VTA neurons project most strongly to the nucleus accumbens shell (24,51) and contribute to the regulation of reward behaviors (24). We therefore asked whether CCK infused into the VTA modulates consummatory behavior. After overnight food deprivation, wild-type mice received either CCK or saline microinjections locally into the VTA. Consumption of standard chow was measured over a 1-hour period. In a crossover design, intra-VTA delivery of CCK reduced food intake when compared with intra-VTA delivery of saline in the same animals (Figure 7B). Because midbrain dopamine neurons modulate motor behaviors, we also assessed the impact of intra-VTA CCK delivery on locomotor behavior in the open field and found that both total distance traveled and velocity were reduced after treatment with CCK (Figure 7C, D). Together, these behavioral data indicate that when released in the VTA, CCK inhibits both feeding and locomotor activity.
Figure 7.

Intra-VTA CCK infusion reduces food intake and dopamine cell firing rate in vitro. (A) Diagram of the experimental design for food intake or open field locomotion. (B) Food intake over the 1-hour period after bilateral intra-VTA infusion of either CCK or veh (n = 13, 11 male mice, 3 female mice). Cannula placement in the VTA was confirmed post hoc. Paired t test, p = .02, df = 13. (C) Total distance traveled in the open field apparatus over a 30-minute period after bilateral intra-VTA infusion of CCK or veh (n = 8 mice, 5 male mice, 3 female mice). Paired t test, p = .008, df = 7. (D) Velocity in the open field apparatus over a 30-minute period after bilateral intra-VTA infusion of CCK or veh (n = 8 mice, 5 male mice, 3 female mice). Wilcoxon paired test, p = .008. (E) Diagram of the experimental design for measuring cell attached firing rate. CCK or veh was infused bilaterally into the VTA of fasted mice, and at least 10 minutes later, slices were prepared and stored in aCSF without added CCK. (F) On-cell firing rate of GFP+ VTA neurons in slices from mice that received either saline or CCK in vivo (total n = 141 cells, 7 mice; veh/aCSF n = 77, 24 cells from male mice, 53 cells from female mice; CCK/aCSF n = 64, 14 cells from male mice, 50 cells from female mice). Mann-Whitney test, p = .02. Scale bar = 50 pA, 5 s. aCSF, artificial cerebrospinal fluid; CCK, cholecystokinin; GFP, green fluorescent protein; veh, vehicle; VTA, ventral tegmental area.
CCK behavioral effects might result from a decrease in dopamine cell activity. We tested whether in vivo intra-VTA CCK infusion modulates dopamine neuron firing rate in slices (Figure 7E, F; Figure S3). After infusion of CCK, GFP+ neurons from Pitx3-GFP mice displayed reduced firing rates compared with cells from vehicle-treated mice.
CCK may inhibit feeding behavior by decreasing dopamine cell excitability. We measured Ca2+ transients while mice engaged in home cage feeding after infusion of saline or CCK in a crossover design (Figure 8A). DAT-IRES-Cre mice were injected with AAV-FLEX-GCaMP8m and implanted with a cannula/optic fiber in the VTA. Fasted mice were microinjected with CCK or saline into the VTA and imaging was started. With saline infusion, dopamine cells displayed transient increases in activity time-locked to food consumption. However, after CCK, dopamine cell activity upon food consumption was significantly dampened (Figure 8C, D, F). The amount of food consumed was also significantly reduced with CCK infusion compared with saline infusion (Figure 8E). Moreover, the degree of CCK-induced dopamine cell inhibition and food intake reduction (compared with those measured after saline) were highly correlated (Figure 8G). Together, these data suggest that CCK signaling in the VTA reduces dopamine cell activity to suppress food intake and indicate a likely role for somatodendritically released CCK.
Figure 8.

Intra-VTA CCK infusion reduces dopamine cell calcium transients in vivo. (A) Schematic of fiber photometry configuration and experimental timeline. (B) Representative image of GCaMP8m in the VTA dopamine neurons, coronal slice. Dotted lines indicate dual cannula position. (C) Peristimulus histogram of time course of averaged GCaMP8m transient z scores event-locked to food consumption (n = 8 mice, 5 females, 3 males). (D) Quantification of peak z scores during food consumption after either saline or CCK infusion (n = 8 mice, 5 females, 3 males). Paired t test, p = .0006, df = 7. (E) Quantification of food intake during recording sessions over a 30-minute period (n = 8 mice, 5 females, 3 males). Paired t test, p = .004, df = 7. (F) Representative heat map of z score changes over all trials from individual mice; 0 time is the onset of food consumption. (G) CCK-induced dopamine cell inhibition and food intake are significantly correlated (n = 8 mice, 5 females, 3 males). Pearson’s r = 0.84. Paired t test, p = .009. Bold symbols/lines represent the mean response across all animals; error bars represent SEM. CCK, cholecystokinin; DA, dopamine; LED, light-emitting diode; veh, vehicle; VTA, ventral tegmental area.
DISCUSSION
The central nervous system signaling roles of neuropeptides are not well understood despite their broad distribution (1). For example, the expression of CCK in dopamine neurons was identified over 40 years ago (18), but even the deployment of modern tools has focused on CCK as a useful marker of cell groups rather than elucidating the functions of CCK itself (24,51). Here we report that CCK is released somatodendritically when dopamine cells are depolarized, and in turn it potentiates inhibitory synapses. Our results support the idea that locally released CCK is part of an inhibitory feedback mechanism, acting synergistically with somatodendritic dopamine release to inhibit dopamine neuron excitability and modulate dopamine-dependent behaviors. Given the widespread expression of CCK in other brain areas, this may be a general mechanism to regulate synaptic strength.
CCK Is Somatodendritically Released From Dopamine Cells
The CCK release we observe is likely predominantly of somatodendritic origin rather than from nerve terminals. First, only a small subset of dopaminergic nerve terminals in the VTA originate from VTA/substantia nigra pars compacta (52,53). Second, blocking T-type VGCCs with Ni2+ blocks the CCK-induced LTP, but is not expected to affect nerve terminal release of neurotransmitter (54). Finally, we evoked CCK release with subthreshold depolarization of a single dopamine cell, a stimulus unlikely to trigger nerve terminal release.
Optogenetic depolarization of dopamine neurons in the VTA triggers somatodendritic CCK release, which has not been accounted for in optogenetic studies to date. Moreover, other neuropeptides are expressed in dopamine neurons, and depolarization may trigger their release as well (55,56). Somatodendritic peptide release may be a common result of depolarizing stimuli and will need to be considered in studies using optogenetic activation of cell body and dendritic sites.
Somatodendritic CCK Release Requires T-Type Ca2+ Channels and Synaptotagmin-7
CCK release is blocked by chelation of calcium in the dopamine cell, consistent with the reported requirement for extracellular Ca2+ in high K+-induced CCK release from midbrain slices (41). Among VGCCs, T-type channels are likely candidates to contribute to depolarization-induced CCK release based on their voltage dependence (42,44), and our data with NiCl2 support this hypothesis (57). It is somewhat surprising that a 6-minute depolarization to −40 mV releases CCK because dopamine neurons exhibit relatively depolarized membrane potentials, firing spontaneously in vivo and in vitro. Trains of optogenetic depolarizing pulses that may mimic more physiologically relevant stimuli also release CCK, triggering LTP (Figure 1G, H) and allowing biochemical detection of CCK release (Figure 3A–C).
Synaptotagmin-1 is likely to account entirely for fast dopamine release from striatal nerve terminals (48,58,69). However, even in dopamine axon fields in the striatum, in syt1 KO mice, high K+-dependent and action potential–dependent dopamine release still occur. This finding indicates that another Ca2+ sensor, hypothesized to be synaptotagmin-7, is responsible for an additional slower form of release (58,70). Synaptotagmin-7 is found in somatodendritic compartments (48), and knockdown of synaptotagmin-7 was also reported to decrease somatodendritic dopamine release from cultured neurons (48). In our experiments, both CCK release (using depolarization-induced LTP as a proxy for CCK release) and the D2-IPSC (a readout of somatodendritic dopamine release) were absent in the syt7 KO mice. Our data support a model in which Ca2+ entry through T-type Ca2+ channels, perhaps augmented by store-operated Ca2+ release (71,72), promotes the movement and fusion of CCK-containing vesicles to the plasma membrane through a synaptotagmin-7–dependent mechanism (73).
Somatodendritic Release of Dopamine and CCK May Occur Independently
Theoretically, vesicles that contain dopamine could also contain CCK, analogously with adrenal chromaffin cells that contain both catecholamines and peptides (74,75). CCK release clearly potentiates inhibitory synapses after a single dopamine neuron is voltage-clamped to −40 mV (without evoking action potentials) (Figure 1A–F), while D2-IPSCs are tetrodotoxin sensitive (68). The Ca2+ chelator, BAPTA (10 mM), in the intracellular solution does not block the D2-IPSC (32), but in our experiments 30 mM BAPTA does prevent depolarization-induced LTP (Figure 5A–C) (37). Furthermore, our data suggest that CCK release is blocked by 50 μM Ni2+, while somatodendritic dopamine release was relatively unaffected by T-type channel block (76,77). Finally, if the trains we used to evoke D2-IPSCs had caused significant release of CCK, GABAA IPSCs should have undergone LTP, preventing further potentiation upon CCK application (Figure 5C, D). Together, the evidence suggests that somatodendritic release of dopamine and CCK can occur independently. However, because we only tested a few release protocols, simultaneous release remains a possibility, and genetically encoded sensors may be useful tools to test this idea.
Somatodendritic Release of Dopamine and CCK Act Synergistically Over Different Timescales
Somatodendritically released dopamine hyperpolarizes dopamine cells and is thought to provide feedback inhibition after a burst of action potentials, on a timescale of milliseconds to seconds (70,78). In contrast, CCK initiates a gradual, far slower–onset LTP of GABAergic afferents over many minutes, and the potentiation lasts for at least an hour. Because neuropeptides can diffuse relatively long distances before being degraded, modulation by CCK may not be localized, unlike somatodendritically released dopamine, which undergoes rapid reuptake. The timescales of neuromodulation of dopamine neuron excitability by dopamine and CCK may be analogous to the separate roles of endocannabinoid modulation: local and brief in the case of depolarization-induced suppression of inhibition and excitation (79) and longer-lasting in the case of endocannabinoid LTD (80).
CCK Modulates Local Circuit Function
Rather than upregulating all inhibitory synapses, CCK acts on a specific afferent population or subcircuit. Exogenous CCK or depolarization that releases CCK selectively potentiates only caudally evoked but not rostrally evoked IPSCs (Figure 4) (67). CCK also has no effect on IPSCs recorded in VTA GABAergic neurons, nor on dopamine cells in the medial VTA, even though medial dopamine cells appear to contain CCK (Figures S1 and S2). Furthermore, depolarization potentiates synapses from periaqueductal gray (37) but not those from the rostromedial tegmental nucleus (81,82). The simplest model to explain these features is that CCK does not exert autocrine effects on dopamine cells but instead acts on specific susceptible GABAergic terminals that express the CCK2R. Our fluorescence in situ hybridization data support this idea, indicating very low levels of CCK2Rs on somata or astrocytes in the VTA (Figure 3J). However, we did not detect changes in paired-pulse ratio and 1/CV2 that often accompany presynaptically maintained LTP. Additional work is needed to further explore this novel neuropeptide-dependent LTP mechanism.
Other than the periaqueductal gray (37), known GABAergic populations that target the mouse VTA include the bed nucleus of the stria terminalis, VTA, dorsal raphe, ventral pallidum, lateral hypothalamus, and nucleus accumbens (83–88). Among these regions, CCK2R binding in rat was reported in the bed nucleus of the stria terminalis, nucleus accumbens, and lateral hypothalamus, suggesting that afferents from one or more of these populations may be potentiated by CCK.
CCK Infused Into the VTA Decreases Food Consumption and Dopamine Cell Activity
Early studies in rats reported that CCK can transiently excite VTA dopamine cells (89–91), but other studies found that CCK potentiates dopamine-induced inhibition of dopamine cell firing in vitro and in vivo (90,92). This inhibition is consistent with our recordings showing that CCK infusion in vivo reduces dopamine cell firing rate (Figure 7E, F). These effects were seen minutes to hours after CCK exposure, leaving open the possibility that CCK may induce transient excitation followed by persistent inhibition. Our Ca2+ imaging data also support a critical role for CCK in inhibiting dopamine cell activity in vivo, consistent with LTP of inhibitory synaptic transmission.
Dopamine neurons fire during consumption of food and water and are essential for motivation for food rewards (93,94), although they may not be necessary for feeding per se (26,95–97). We find that bilateral intra-VTA CCK infusion in mice inhibits feeding after an overnight fast, as previously reported in rats 2 hours postinfusion (98). In addition to blunting food-evoked Ca2+ activity in dopamine cells, CCK microinfusions concomitantly reduced food intake. Furthermore, the magnitudes of these reductions were significantly correlated, perhaps indicating a causal role for CCK-induced dopamine neuron inhibition in feeding suppression.
What Is the Role of CCK in the VTA?
VTA dopamine neurons may release CCK as a negative feedback modulator to tune down their own activity. This mechanism might be critical for the calibration of food intake, preventing animals from consuming more than necessary, independent of peripheral CCK (99). Previous work has shown that insulin, which is elevated in plasma after feeding, depresses excitatory synapses on dopamine neurons after feeding (100,101), and similarly, leptin reduces dopamine cell firing rates and suppresses food intake (102). If CCK is released upon feeding, it may act synergistically with insulin and leptin to inhibit dopamine cells and suppress the motivation to continue feeding.
We observed a modest decrease in locomotion after CCK microinfusions. Our intra-VTA CCK infusions may have reached the substantia nigra, which modulates motor behavior where dopamine neurons also colocalize CCK (18,103). VTA projections to substantia reticulata could also be influenced by local CCK release (104). We previously found that optogenetically driving periaqueductal gray–to-VTA afferents promoted an immobility phenotype in mice (37); because these synapses are potentiated by depolarization, they could contribute to CCK-mediated reduction in locomotion.
Dopaminergic projections from the VTA are required for motivated and natural reinforcing behaviors. Feeding and drugs of abuse increase dopamine release (105). Local CCK potentiates inhibitory synapses and depresses dopamine cell firing in the lateral VTA, suggesting that somatodendritic CCK release may exert an inhibitory influence on reinforcement.
Somatodendritic Release May Be a Common Feature of Neuropeptide Signaling
Our data show that a neuropeptide can be released somatodendritically with modest stimuli and exert persistent effects on synaptic strength. Beyond the well-established examples of vasopressin and oxytocin release from supraoptic nucleus (2), somatodendritic release of galanin is hypothesized to exert feedback inhibition on locus coeruleus noradrenergic cells (8,9,106), and neuropeptide Y may also provide autocrine modulation via dendritic release from reticular thalamic neurons (107). Intriguingly, CCK is also required for glutamatergic LTP in auditory neocortex (10). Although this could result from axon terminal release, somatodendritically released CCK may also have a role. Reminiscent of what we report in the VTA, CCK is released in the dorsomedial hypothalamus by repetitive depolarization of postsynaptic cells and acts on CCK2R to transiently potentiate GABAergic synapses (5,6). Peptide signaling adds complexity and richness to neural circuits.
Supplementary Material
ACKNOWLEDGMENTS AND DISCLOSURES
Funding support was from National Institutes of Health (Grant No. R01 DA011289 [to JAK]), National Institute on Drug Abuse (Grant No. 5T32DA035165-08 [to CEM]), National Institute for Neurological Disorders and Stroke (Grant Nos. 1F32NS123012-01 [to CEM], T32 DA035165 [to MPB]), and Stanford Dean’s award (to VMD).
We thank Dr. Elizabeth Steinberg for helpful discussions and Dr. Neir Eshel and the STAAR Lab at Stanford for support with fiber photometry experiments. We thank Dr. Susan Sesack and Dr. John Williams for helpful suggestions.
Footnotes
Partial advances of this project were published in abstract/poster forms in the following conferences: Society for Neuroscience Annual Meeting (2021), Society for Neuroscience Annual Meeting (2020), Argentinian Society for Neuroscience Annual Meeting (2020). A previous version of this article was published as a preprint on bioRxiv: https://doi.org/10.1101/2022.01.14.476405.
The authors report no biomedical financial interests or potential conflicts of interest.
Supplementary material cited in this article is available online at https://doi.org/10.1016/j.biopsych.2022.06.011.
REFERENCES
- 1.Smith SJ, Hawrylycz M, Rossier J, Sümbül U (2020): New light on cortical neuropeptides and synaptic network plasticity. Curr Opin Neurobiol 63:176–188. [DOI] [PubMed] [Google Scholar]
- 2.Ludwig M, Leng G (2006): Dendritic peptide release and peptide-dependent behaviours. Nat Rev Neurosci 7:126–136. [DOI] [PubMed] [Google Scholar]
- 3.Wagner JJ, Terman GW, Chavkin C (1993): Endogenous dynorphins inhibit excitatory neurotransmission and block LTP induction in the hippocampus. Nature 363:451–454. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Krawczyk M, Mason X, DeBacker J, Sharma R, Normandeau CP, Hawken ER, et al. (2013): D1 dopamine receptor-mediated LTP at GABA synapses encodes motivation to self-administer cocaine in rats. J Neurosci 33:11960–11971. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Crosby KM, Baimoukhametova DV, Bains JS, Pittman QJ (2015): Postsynaptic depolarization enhances GABA drive to dorsomedial hypothalamic neurons through somatodendritic cholecystokinin release. J Neurosci 35:13160–13170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Crosby KM, Murphy-Royal C, Wilson SA, Gordon GR, Bains JS, Pittman QJ (2018): Cholecystokinin switches the plasticity of GABA synapses in the dorsomedial hypothalamus via astrocytic ATP release. J Neurosci 38:8515–8525. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Brown CH, Ludwig M, Tasker JG, Stern JE (2020): Somatodendritic vasopressin and oxytocin secretion in endocrine and autonomic regulation. J Neuroendocrinol 32:e12856. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Hökfelt T, Barde S, Xu ZD, Kuteeva E, Rüegg J, Le Maitre E, et al. (2018): Neuropeptide and small transmitter coexistence: Fundamental studies and relevance to mental illness. Front Neural Circuits 12:106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Vila-Porcile E, Xu ZQ, Mailly P, Nagy F, Calas A, Hökfelt T, Landry M (2009): Dendritic synthesis and release of the neuropeptide galanin: Morphological evidence from studies on rat locus coeruleus neurons. J Comp Neurol 516:199–212. [DOI] [PubMed] [Google Scholar]
- 10.Chen X, Li X, Wong YT, Zheng X, Wang H, Peng Y, et al. (2019): Cholecystokinin release triggered by NMDA receptors produces LTP and sound-sound associative memory. Proc Natl Acad Sci U S A 116:6397–6406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Ludwig M, Pittman QJ (2003): Talking back: Dendritic neurotransmitter release. Trends Neurosci 26:255–261. [DOI] [PubMed] [Google Scholar]
- 12.Lipka J, Kapitein LC, Jaworski J, Hoogenraad CC (2016): Microtubule-binding protein doublecortin-like kinase 1 (DCLK1) guides kinesin-3-mediated cargo transport to dendrites. EMBO J 35:302–318. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Zahn TR, Angleson JK, MacMorris MA, Domke E, Hutton JF, Schwartz C, Hutton JC (2004): Dense core vesicle dynamics in Caenorhabditis elegans neurons and the role of kinesin UNC-104. Traffic 5:544–559. [DOI] [PubMed] [Google Scholar]
- 14.Persoon CM, Moro A, Nassal JP, Farina M, Broeke JH, Arora S, et al. (2018): Pool size estimations for dense-core vesicles in mammalian CNS neurons. EMBO J 37:e99672. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Cheramy A, Leviel V, Glowinski J (1981): Dendritic release of dopamine in the substantia nigra. Nature 289:537–542. [DOI] [PubMed] [Google Scholar]
- 16.Crawley JN (1985): Comparative distribution of cholecystokinin and other neuropeptides. Why is this peptide different from all other peptides? Ann N Y Acad Sci 448:1–8. [DOI] [PubMed] [Google Scholar]
- 17.Rehfeld JF (2017): Cholecystokinin-from local gut hormone to ubiquitous messenger. Front Endocrinol (Lausanne) 8:47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Hökfelt T, Skirboll L, Rehfeld JF, Goldstein M, Markey K, Dann O (1980): A subpopulation of mesencephalic dopamine neurons projecting to limbic areas contains a cholecystokinin-like peptide: Evidence from immunohistochemistry combined with retrograde tracing. Neuroscience 5:2093–2124. [DOI] [PubMed] [Google Scholar]
- 19.Hökfelt T, Holets VR, Staines W, Meister B, Melander T, Schalling M, et al. (1986): Coexistence of neuronal messengers—An overview. Prog Brain Res 68:33–70. [DOI] [PubMed] [Google Scholar]
- 20.Seroogy K, Ceccatelli S, Schalling M, Hökfelt T, Frey P, Walsh J, et al. (1988): A subpopulation of dopaminergic neurons in rat ventral mesencephalon contains both neurotensin and cholecystokinin. Brain Res 455:88–98. [DOI] [PubMed] [Google Scholar]
- 21.Jayaraman A, Nishimori T, Dobner P, Uhl GR (1990): Cholecystokinin and neurotensin mRNAs are differentially expressed in subnuclei of the ventral tegmental area. J Comp Neurol 296:291–302. [DOI] [PubMed] [Google Scholar]
- 22.Schalling M, Friberg K, Seroogy K, Riederer P, Bird E, Schiffmann SN, et al. (1990): Analysis of expression of cholecystokinin in dopamine cells in the ventral mesencephalon of several species and in humans with schizophrenia. Proc Natl Acad Sci U S A 87:8427–8431. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Crawley JN, Corwin RL (1994): Biological actions of cholecystokinin. Peptides 15:731–755. [DOI] [PubMed] [Google Scholar]
- 24.Heymann G, Jo YS, Reichard KL, McFarland N, Chavkin C, Palmiter RD, et al. (2020): Synergy of distinct dopamine projection populations in behavioral reinforcement. Neuron 105:909–920.e5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Berke JD (2018): What does dopamine mean? Nat Neurosci 21:787–793. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Salamone JD, Correa M (2012): The mysterious motivational functions of mesolimbic dopamine. Neuron 76:470–485. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Berridge KC (2007): The debate over dopamine’s role in reward: The case for incentive salience. Psychopharmacology (Berl) 191:391–431. [DOI] [PubMed] [Google Scholar]
- 28.Friedman A (2014): Neuroscience. Jump-starting natural resilience reverses stress susceptibility. Science 346:555. [DOI] [PubMed] [Google Scholar]
- 29.Popescu AT, Zhou MR, Poo MM (2016): Phasic dopamine release in the medial prefrontal cortex enhances stimulus discrimination. Proc Natl Acad Sci U S A 113:E3169–E3176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Howe MW, Dombeck DA (2016): Rapid signalling in distinct dopaminergic axons during locomotion and reward. Nature 535:505–510. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Cox J, Witten IB (2019): Striatal circuits for reward learning and decision-making. Nat Rev Neurosci 20:482–494. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Beckstead MJ, Williams JT (2007): Long-term depression of a dopamine IPSC. J Neurosci 27:2074–2080. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Polter AM, Kauer JA (2014): Stress and VTA synapses: Implications for addiction and depression. Eur J Neurosci 39:1179–1188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Xin W, Edwards N, Bonci A (2016): VTA dopamine neuron plasticity – The unusual suspects. Eur J Neurosci 44:2975–2983. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Simmons DV, Petko AK, Paladini CA (2017): Differential expression of long-term potentiation among identified inhibitory inputs to dopamine neurons. J Neurophysiol 118:1998–2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.St Laurent R, Kauer J (2019): Synaptic plasticity at inhibitory synapses in the ventral tegmental area depends upon stimulation site. eNeuro 6:ENEURO.0137–19.2019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.St Laurent R, Martinez Damonte V, Tsuda AC, Kauer JA (2020): Periaqueductal gray and rostromedial tegmental inhibitory afferents to VTA have distinct synaptic plasticity and opiate sensitivity. Neuron 106:624–636.e4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Ludwig M, Apps D, Menzies J, Patel JC, Rice ME (2016): Dendritic release of neurotransmitters. Compr Physiol 7:235–252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Perez-Reyes E (2003): Molecular physiology of low-voltage-activated t-type calcium channels. Physiol Rev 83:117–161. [DOI] [PubMed] [Google Scholar]
- 40.Maxwell SL, Ho HY, Kuehner E, Zhao S, Li M (2005): Pitx3 regulates tyrosine hydroxylase expression in the substantia nigra and identifies a subgroup of mesencephalic dopaminergic progenitor neurons during mouse development. Dev Biol 282:467–479. [DOI] [PubMed] [Google Scholar]
- 41.Freeman AS, Chiodo LA, Lentz SI, Wade K, Bannon MJ (1991): Release of cholecystokinin from rat midbrain slices and modulatory effect of D2DA receptor stimulation. Brain Res 555:281–287. [DOI] [PubMed] [Google Scholar]
- 42.Evans RC, Zhu M, Khaliq ZM (2017): Dopamine inhibition differentially controls excitability of substantia nigra dopamine neuron subpopulations through T-type calcium channels. J Neurosci 37:3704–3720. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Cain SM, Snutch TP (2010): Contributions of T-type calcium channel isoforms to neuronal firing. Channels (Austin) 4:475–482. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Tracy ME, Tesic V, Stamenic TT, Joksimovic SM, Busquet N, Jevtovic-Todorovic V, Todorovic SM (2018): CaV3.1 isoform of T-type calcium channels supports excitability of rat and mouse ventral tegmental area neurons. Neuropharmacology 135:343–354. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Zamponi GW, Bourinet E, Snutch TP (1996): Nickel block of a family of neuronal calcium channels: Subtype- and subunit-dependent action at multiple sites. J Membr Biol 151:77–90. [DOI] [PubMed] [Google Scholar]
- 46.Lee JH, Gomora JC, Cribbs LL, Perez-Reyes E (1999): Nickel block of three cloned T-type calcium channels: Low concentrations selectively block alpha1H. Biophys J 77:3034–3042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Kennedy MJ, Ehlers MD (2011): Mechanisms and function of dendritic exocytosis. Neuron 69:856–875. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Mendez JA, Bourque MJ, Fasano C, Kortleven C, Trudeau LE (2011): Somatodendritic dopamine release requires synaptotagmin 4 and 7 and the participation of voltage-gated calcium channels. J Biol Chem 286:23928–23937. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Zhang Z, Bhalla A, Dean C, Chapman ER, Jackson MB (2009): Synaptotagmin IV: A multifunctional regulator of peptidergic nerve terminals. Nat Neurosci 12:163–171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Delignat-Lavaud B, Ducrot C, Kouwenhoven W, Feller N, Trudeau LÉ (2022): Implication of Synaptotagmins 4 and 7 in Activity-Dependent Somatodendritic Dopamine Release. Open Biol 12:210339. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Poulin JF, Caronia G, Hofer C, Cui Q, Helm B, Ramakrishnan C, et al. (2018): Mapping projections of molecularly defined dopamine neuron subtypes using intersectional genetic approaches. Nat Neurosci 21:1260–1271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Bayer VE, Pickel VM (1990): Ultrastructural localization of tyrosine hydroxylase in the rat ventral tegmental area: Relationship between immunolabeling density and neuronal associations. J Neurosci 10:2996–3013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Deutch AY, Goldstein M, Baldino F Jr, Roth RH (1988): Telencephalic projections of the A8 dopamine cell group. Ann N Y Acad Sci 537:27–50. [DOI] [PubMed] [Google Scholar]
- 54.Dolphin AC, Lee A (2020): Presynaptic calcium channels: Specialized control of synaptic neurotransmitter release. Nat Rev Neurosci 21:213–229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Dore R, Krotenko R, Reising JP, Murru L, Sundaram SM, Di Spiezio A, et al. (2020): Nesfatin-1 decreases the motivational and rewarding value of food. Neuropsychopharmacology 45:1645–1655. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Perez-Bonilla P, Santiago-Colon K, Matasovsky J, Ramirez-Virella J, Khan R, Garver H, et al. (2021): Activation of ventral tegmental area neurotensin Receptor-1 neurons promotes weight loss. Neuropharmacology 195:108639. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Wolfart J, Roeper J (2002): Selective coupling of T-type calcium channels to SK potassium channels prevents intrinsic bursting in dopaminergic midbrain neurons [published correction appears in J Neurosci 2002;22:5250]. J Neurosci 22:3404–3413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Banerjee A, Lee J, Nemcova P, Liu C, Kaeser PS (2020): Synaptotagmin-1 is the Ca2+ sensor for fast striatal dopamine release. Elife 9: e58359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Frankfurt M, Siegel RA, Sim I, Wuttke W (1985): Cholecystokinin and substance P concentrations in discrete areas of the rat brain: Sex differences. Brain Res 358:53–58. [DOI] [PubMed] [Google Scholar]
- 60.Frankfurt M, Siegel RA, Sim I, Wuttke W (1986): Estrous cycle variations in cholecystokinin and substance P concentrations in discrete areas of the rat brain. Neuroendocrinology 42:226–231. [DOI] [PubMed] [Google Scholar]
- 61.Hill DR, Woodruff GN (1990): Differentiation of central cholecystokinin receptor binding sites using the non-peptide antagonists MK-329 and L-365,260. Brain Res 526:276–283. [DOI] [PubMed] [Google Scholar]
- 62.Dufresne M, Seva C, Fourmy D (2006): Cholecystokinin and gastrin receptors. Physiol Rev 86:805–847. [DOI] [PubMed] [Google Scholar]
- 63.Gaudreau P, Quirion R, St-Pierre S, Pert CB (1983): Characterization and visualization of cholecystokinin receptors in rat brain using [3H] pentagastrin. Peptides 4:755–762. [DOI] [PubMed] [Google Scholar]
- 64.Pélaprat D, Broer Y, Studler JM, Peschanski M, Tassin JP, Glowinski J, et al. (1987): Autoradiography of CCK receptors in the rat brain using [(3)H]Boc[Nle(28)(31)]CCK(27)-(33) and [(125)I]bolton-hunter CCK(8). Functional significance of subregional distributions. Neurochem Int 10:495–508. [DOI] [PubMed] [Google Scholar]
- 65.Niehoff DL (1989): Quantitative autoradiographic localization of cholecystokinin receptors in rat and guinea pig brain using 125I-Bolton-Hunter-CCK8. Peptides 10:265–274. [DOI] [PubMed] [Google Scholar]
- 66.Honda T, Wada E, Battey JF, Wank SA (1993): Differential gene expression of CCK(A) and CCK(B) receptors in the rat brain. Mol Cell Neurosci 4:143–154. [DOI] [PubMed] [Google Scholar]
- 67.Dacher M, Nugent FS (2011): Morphine-induced modulation of LTD at GABAergic synapses in the ventral tegmental area. Neuropharmacology 61:1166–1171. [DOI] [PubMed] [Google Scholar]
- 68.Beckstead MJ, Grandy DK, Wickman K, Williams JT (2004): Vesicular dopamine release elicits an inhibitory postsynaptic current in midbrain dopamine neurons. Neuron 42:939–946. [DOI] [PubMed] [Google Scholar]
- 69.Delignat-Lavaud B, Kano J, Ducrot C, Massé I, Mukherjee S, Giguère N, et al. (2021): The Calcium Sensor synaptotagmin-1 Is Critical for Phasic Axonal Dopamine Release in the Striatum and Mesencephalon, but Is Dispensable for Basic Motor Behaviors in Mice. bioRxiv. 10.1101/2021.09.15.460511. [DOI] [Google Scholar]
- 70.Hikima T, Lee CR, Witkovsky P, Chesler J, Ichtchenko K, Rice ME (2021): Activity-dependent somatodendritic dopamine release in the substantia nigra autoinhibits the releasing neuron. Cell Rep 35: 108951. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Berridge MJ (1998): Neuronal calcium signaling. Neuron 21:13–26. [DOI] [PubMed] [Google Scholar]
- 72.Rose CR, Konnerth A (2001): Stores not just for storage. Intracellular calcium release and synaptic plasticity. Neuron 31:519–522. [DOI] [PubMed] [Google Scholar]
- 73.Tawfik B, Martins JS, Houy S, Imig C, Pinheiro PS, Wojcik SM, et al. (2021): Synaptotagmin-7 places dense-core vesicles at the cell membrane to promote Munc13–2- and Ca2+-dependent priming. Elife 10:e64527. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Crivellato E, Nico B, Ribatti D (2008): The chromaffin vesicle: Advances in understanding the composition of a versatile, multifunctional secretory organelle. Anat Rec (Hoboken) 291:1587–1602. [DOI] [PubMed] [Google Scholar]
- 75.Winkler H, Westhead E (1980): The molecular organization of adrenal chromaffin granules. Neuroscience 5:1803–1823. [DOI] [PubMed] [Google Scholar]
- 76.Chen BT, Moran KA, Avshalumov MV, Rice ME (2006): Limited regulation of somatodendritic dopamine release by voltage-sensitive Ca channels contrasted with strong regulation of axonal dopamine release. J Neurochem 96:645–655. [DOI] [PubMed] [Google Scholar]
- 77.Ford CP, Beckstead MJ, Williams JT (2007): Kappa opioid inhibition of somatodendritic dopamine inhibitory postsynaptic currents. J Neurophysiol 97:883–891. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Ford CP, Gantz SC, Phillips PE, Williams JT (2010): Control of extracellular dopamine at dendrite and axon terminals. J Neurosci 30:6975–6983. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Diana MA, Marty A (2004): Endocannabinoid-mediated short-term synaptic plasticity: Depolarization-induced suppression of inhibition (DSI) and depolarization-induced suppression of excitation (DSE). Br J Pharmacol 142:9–19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Heifets BD, Castillo PE (2009): Endocannabinoid signaling and long-term synaptic plasticity. Annu Rev Physiol 71:283–306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Jhou TC, Fields HL, Baxter MG, Saper CB, Holland PC (2009): The rostromedial tegmental nucleus (RMTg), a GABAergic afferent to midbrain dopamine neurons, encodes aversive stimuli and inhibits motor responses. Neuron 61:786–800. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Markovic T, Pedersen CE, Massaly N, Vachez YM, Ruyle B, Murphy CA, et al. (2021): Pain induces adaptations in ventral tegmental area dopamine neurons to drive anhedonia-like behavior. Nat Neurosci 24:1601–1613. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Soden ME, Chung AS, Cuevas B, Resnick JM, Awatramani R, Zweifel LS (2020): Anatomic resolution of neurotransmitter-specific projections to the VTA reveals diversity of GABAergic inputs. Nat Neurosci 23:968–980. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Faget L, Osakada F, Duan J, Ressler R, Johnson AB, Proudfoot JA, et al. (2016): Afferent inputs to neurotransmitter-defined cell types in the ventral tegmental area. Cell Rep 15:2796–2808. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Beier KT, Gao XJ, Xie S, DeLoach KE, Malenka RC, Luo L (2019): Topological organization of ventral tegmental area connectivity revealed by viral-genetic dissection of input–output relations. Cell Rep 26:159–167.e6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Beier KT, Steinberg EE, DeLoach KE, Xie S, Miyamichi K, Schwarz L, et al. (2015): Circuit architecture of VTA dopamine neurons revealed by systematic input–output mapping. Cell 162:622–634. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Zahm DS, Heimer L (1990): Two transpallidal pathways originating in the rat nucleus accumbens. J Comp Neurol 302:437–446. [DOI] [PubMed] [Google Scholar]
- 88.Kalivas PW, Churchill L, Klitenick MA (1993): GABA and enkephalin projection from the nucleus accumbens and ventral pallidum to the ventral tegmental area. Neuroscience 57:1047–1060. [DOI] [PubMed] [Google Scholar]
- 89.Skirboll LR, Grace AA, Hommer DW, Rehfeld J, Goldstein M, Hökfelt T, Bunney BS (1981): Peptide-monoamine coexistence: Studies of the actions of cholecystokinin-like peptide on the electrical activity of midbrain dopamine neurons. Neuroscience 6:2111–2124. [DOI] [PubMed] [Google Scholar]
- 90.Brodie MS, Dunwiddie TV (1987): Cholecystokinin potentiates dopamine inhibition of mesencephalic dopamine neurons in vitro. Brain Res 425:106–113. [DOI] [PubMed] [Google Scholar]
- 91.Stittsworth JD Jr, Mueller AL (1990): Cholecystokinin octapeptide potentiates the inhibitory response mediated by D2 dopamine receptors in slices of the ventral tegmental area of the brain in the rat. Neuropharmacology 29:119–127. [DOI] [PubMed] [Google Scholar]
- 92.Artaud F, Baruch P, Stutzmann JM, Saffroy M, Godeheu G, Barbeito L, et al. (1989): Cholecystokinin: Corelease with dopamine from nigrostriatal neurons in the cat. Eur J Neurosci 1:162–171. [DOI] [PubMed] [Google Scholar]
- 93.Zhou QY, Palmiter RD (1995): Dopamine-deficient mice are severely hypoactive, adipsic, and aphagic. Cell 83:1197–1209. [DOI] [PubMed] [Google Scholar]
- 94.van Zessen R, Phillips JL, Budygin EA, Stuber GD (2012): Activation of VTA GABA neurons disrupts reward consumption. Neuron 73:1184–1194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Mikhailova MA, Bass CE, Grinevich VP, Chappell AM, Deal AL, Bonin KD, et al. (2016): Optogenetically-induced tonic dopamine release from VTA-nucleus accumbens projections inhibits reward consummatory behaviors. Neuroscience 333:54–64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Boekhoudt L, Roelofs TJM, de Jong JW, de Leeuw AE, Luijendijk MCM, Wolterink-Donselaar IG, et al. (2017): Does activation of midbrain dopamine neurons promote or reduce feeding? Int J Obes (Lond) 41:1131–1140. [DOI] [PubMed] [Google Scholar]
- 97.Sandhu EC, Fernando ABP, Irvine EE, Tossell K, Kokkinou M, Glegola J, et al. (2018): Phasic stimulation of midbrain dopamine neuron activity reduces salt consumption. eNeuro 5:ENEURO.0064–18.2018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Blevins JE, Hamel FG, Fairbairn E, Stanley BG, Reidelberger RD (2000): Effects of paraventricular nucleus injection of CCK-8 on plasma CCK-8 levels in rats. Brain Res 860:11–20. [DOI] [PubMed] [Google Scholar]
- 99.May AA, Liu M, Woods SC, Begg DP (2016): CCK increases the transport of insulin into the brain. Physiol Behav 165:392–397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Labouèbe G, Liu S, Dias C, Zou H, Wong JC, Karunakaran S, et al. (2013): Insulin induces long-term depression of ventral tegmental area dopamine neurons via endocannabinoids. Nat Neurosci 16:300–308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Liu S, Globa AK, Mills F, Naef L, Qiao M, Bamji SX, Borgland SL (2016): Consumption of palatable food primes food approach behavior by rapidly increasing synaptic density in the VTA. Proc Natl Acad Sci U S A 113:2520–2525. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Hommel JD, Trinko R, Sears RM, Georgescu D, Liu ZW, Gao XB, et al. (2006): Leptin receptor signaling in midbrain dopamine neurons regulates feeding. Neuron 51:801–810. [DOI] [PubMed] [Google Scholar]
- 103.Savasta M, Ruberte E, Palacios JM, Mengod G (1989): The colocalization of cholecystokinin and tyrosine hydroxylase mRNAs in mesencephalic dopaminergic neurons in the rat brain examined by in situ hybridization. Neuroscience 29:363–369. [DOI] [PubMed] [Google Scholar]
- 104.He F, Zhang P, Zhang Q, Qi G, Cai H, Li T, et al. (2021): Dopaminergic projection from ventral tegmental area to substantia nigra pars reticulata mediates chronic social defeat stress-induced hypo-locomotion. Mol Neurobiol 58:5635–5648. [DOI] [PubMed] [Google Scholar]
- 105.Westerink BH, Teisman A, de Vries JB (1994): Increase in dopamine release from the nucleus accumbens in response to feeding: A model to study interactions between drugs and naturally activated dopaminergic neurons in the rat brain. Naunyn Schmiedebergs Arch Pharmacol 349:230–235. [DOI] [PubMed] [Google Scholar]
- 106.Pieribone VA, Xu ZQ, Zhang X, Grillner S, Bartfai T, Hökfelt T (1995): Galanin induces a hyperpolarization of norepinephrine-containing locus coeruleus neurons in the brainstem slice. Neuroscience 64:861–874. [DOI] [PubMed] [Google Scholar]
- 107.Sun QQ, Baraban SC, Prince DA, Huguenard JR (2003): Target-specific neuropeptide Y-ergic synaptic inhibition and its network consequences within the mammalian thalamus. J Neurosci 23:9639–9649. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.