

Abstract
Objective:
The paucity of longitudinal natural history studies in MPZ-neuropathy remains a barrier to clinical trials. We have completed a longitudinal natural history study in patients with MPZ-neuropathies across 13 sites of the Inherited Neuropathy Consortium.
Methods:
Change in Charcot Marie Tooth Examination scores (CMTES) and Rasch modified CMTES (CMTES-R) scores were evaluated using longitudinal regression over a 5-year period in subjects with MPZ-neuropathy. Data from 139 patients with MPZ-neuropathy were examined.
Results:
The average baseline CMTES and CMTES-R scores were 10.84 (SD 6.0, range 0 – 28) and 14.60 (SD= 7.56, range 0 – 32), respectively. A mixed regression model showed significant change in CMTES at years 2–5 [mean change from baseline of 0.87 points at 2 years (p=0.008)]. Subgroup analysis revealed greater change in CMTES at 2 years in subjects with axonal as compared to demyelinating neuropathy [mean change of 1.30 points, (p=0.016) versus 0.06 points, (p=0.889)]. Patients with a moderate baseline neuropathy severity also showed more notable change, by estimate, than those with mild or severe neuropathy [mean 2 year change of 1.14 for baseline CMTES 8–14 (p=0.025), versus −0.03 for baseline CMTES 0–7 (p=0.958) and 0.25 for baseline CMTES ≥15 (p=0.6897)]. The progression in patients harboring specific MPZ mutations was highly variable.
Interpretation:
CMTES scores are sensitive to change over time in adult patients with axonal but not demyelinating forms of MPZ-neuropathy. Change in CMTES was greatest in patients with moderate baseline disease severity. These findings will inform future clinical trials of MPZ-neuropathies.
Keywords: Charcot-Marie-Tooth Disease, CMT1B, Hereditary Neuropathy, Myelin protein zero, natural history
Introduction:
Dominant mutations in the Myelin Protein Zero (MPZ) gene account for 5% of all of forms of genetically confirmed cases of Charcot Marie Tooth disease (CMT), and 10% of all genetically confirmed demyelinating forms of CMT1–3. MPZ-neuropathies are genetically heterogeneous, with over 200 different disease-causing mutations identified to date4. Genotype-phenotype correlation studies have identified 3 distinct types of MPZ-neuropathy, including an infantile form (clinical presentation prior to 3 years of age with severely slowed motor conduction velocities (NCV; <15 m/s), a childhood-onset (first or second decade) demyelinating neuropathy (CMT1B), and an adult-onset axonal neuropathy (CMT2I)4–10. The clinical presentation of MPZ-neuropathies is similar to other forms of CMT, with foot deformities, distal muscle weakness and atrophy, and length-dependent sensory loss. Some mutations are associated with additional clinical features such as tonic pupils, dysphagia, hearing loss, and neuropathic pain8.
MPZ encodes the MPZ/P0 protein, which is expressed almost exclusively by myelinating Schwann cells, and is the major protein in peripheral nerve myelin11. MPZ is a member of the immunoglobulin (Ig) supergene family and is a homophilic adhesion molecule that is required for the normal compaction of myelin12. The proposed biological mechanisms by which mutations in MPZ result in neuropathy are diverse and include gain-of-function (including dominant negative effects) and loss-of-function mechanisms13, 14. Some mutations result in retention of the mutant MPZ protein in the ER, whereas other MPZ mutants have abnormal interactions with wild type MPZ in the myelin sheath15.
The striking biological and phenotypic heterogeneity of MPZ-neuropathies are a challenge in developing candidate therapies, gathering accurate natural history data, and designing effective clinical trials. In this study, we have evaluated the natural history of MPZ-neuropathies within the multi-center Inherited Neuropathies Consortium over a 5-year period. Disease progression was assessed using the CMT Examination Score (CMTES) as well as the Rasch analysis-based weighted CMT Examination Score (CMTES-R), which are commonly employed clinical outcome assessments in CMT that have previously demonstrated responsiveness to changes in patients with CMT1A16.
Methods:
Standard protocol approvals, registrations, and patient consents
All sites participating in the Inherited Neuropathy Consortium (INC) natural history study (protocol 6601) received Institutional Review Board (IRB)/Ethics Board approval for the study. All patients or their guardian signed consent forms. This trial was registered at www.clinicaltrials.gov (ID number NCT01193075).
Patients:
Patients with MPZ mutations were recruited from the INC, which is a member of the NIH Rare Diseases Clinical Research Network (RDCRN; rarediseasesnetwork.org/). Data were collected as part of the INC natural history study (protocol 6601) between February 2009 and July 2020 from a total of 13 sites within the INC (TABLE 1). Patients were examined by clinical investigators who had received training and were certified in the proper use of the CMTES, a validated 7-item, 28-point composite score based on patients’ symptoms (3 items), and examination findings (4 items)17, 18. CMTES scores were then converted into their Rasch-modified scores (CMTES-R), with total scores of 3218. As previously defined in a cohort of patients with CMT1A, CMTES subgroups were defined as follows: mild 0–7, moderate 8–14, and severe 15+16. Patients were evaluated at yearly intervals, and the CMTES scores were recorded at each visit.
TABLE 1 –
Number of patients per Inherited Neuropathies Consortium (INC) site.
| INC Site | Number of Subjects Contributed |
|---|---|
| C. Besta Neurological Institute | 42 |
| National Hospital for Neurology and Neurosurgery | 33 |
| University of Iowa | 28 |
| University of Pennsylvania | 10 |
| University of Rochester | 7 |
| Wayne State University | 7 |
| John Hopkins University | 3 |
| Stanford University | 3 |
| University of Michigan | 2 |
| Children’s Hospital of Philadelphia | 1 |
| Harvard/Massachusetts General Hospital | 1 |
| Nemours Children’s Hospital | 1 |
| Vanderbilt University Medical Center | 1 |
Patients were diagnosed with MPZ-neuropathies based on clinical evidence of sensory and/or motor peripheral neuropathy (including length-dependent sensory loss, weakness, and atrophy of the distal musculature and decreased deep tendon reflexes), nerve conduction studies (NCS), and confirmatory genetic testing for a mutation in the MPZ gene. A Clinical Laboratory Improvement Amendments-certified laboratory in the United States or an equivalent certified testing facility outside of the United States performed all genetic testing.
Statistical Methods:
Only patients with mutations in the MPZ gene were included. The CMTES and CMTES-R scores over time were analyzed with longitudinal regression. Unstructured residual correlation matrices were used to maximize flexibility. The sample was determined on the bases of available cases and the ability to fit the longitudinal regression model. The covariance matrices of longitudinal models account for missing response data, so long as the missingness is not “not at random”. Site to site variance was accounted for with random intercepts. The fixed effect of time was considered both as a categorical variable and as a continuous variable with a linear effect on the response. The categorical and linear time models were compared to each other using model fit statistics and plots. Gender, age, axonal vs demyelinating, and baseline CMTES scores were examined for interaction effects with categorical time for baseline and the first 2 years of follow up. Change from baseline was used as the model outcome when baseline CMTES score was an explanatory variable. T and F tests were performed on linear combinations of the model parameters. The performance of the CMTES-R vs CMTES was evaluated by comparing mean change from baseline to 2 years to the SD of the change, the standardized response mean (SRM = mean change/SD change), for the first 2-year change. SRM values of 0.20 to 0.49, 0.50 to 0.79, and ≥0.80 reflect low, moderate, and high responsiveness, respectively19. Estimated annual mean score and changes and their SDs were obtained from the model parameters. The main outcomes were progression of CMTES and CMTES-R from baseline to follow up year 2.
Axonal vs demyelinating neuropathy status was determined according to the recorded ulnar motor nerve conduction velocity (NCV) at their first visit, with demyelinating neuropathy defined by velocities less than or equal to 38 m/s, and axonal neuropathy defined as an ulnar motor NCV greater than 38 m/s20. If ulnar NCV was not available, axonal versus demyelinating status was determined based on the examiners’ specification. Demographics and baseline characteristics were compared between the axonal and demyelinating groups with chi-square/Fisher exact association tests for categorical variables, and with T-tests for continuous and scale variables. Associations between baseline CMTES and demographics and baseline characteristics were tested with T-tests and ANOVA type models for categorical predictors, and with linear regression for continuous and scale predictors.
Specific mutations to either amino acids or nucleotides were studied descriptively. For each participant, both the first available 2-year change in CMTES was calculated, and a regression line for all CMTES observations on a participant was fit. Spaghetti plots of CMTES were generated.
Thresholds for mild, moderate, and severe CMTES were established by applying a discriminant analysis for established CMTES categories. The same methods and data were previously used as in Fridman et al.16
Univariate alpha was set to 0.05. Statistical analysis was performed with SAS 9.4.
Results:
The INC evaluated patients with MPZ-neuropathies between February 2009 and July 2020. Data from 139 participants with at least 1 CMTES/CMTES-R observation done either at their initial visit or during a subsequent visit in the first 5 years of follow up were included (TABLE 2). Baseline CMTES and CMTES-R scores were available for 136 patients, and complete CMTNS scores were available for 65 participants (TABLE 2). 74 (53%) of the participants were female and 65 (47%) were male. The mean age of the participants was 46 years (SD 18, range 4 – 76); with 1 participant had missing age data). 68 participants (49%) were 50 years of age or older, and 18 (13%) were under 21 years of age. 122 (88%) identified as white, 2 (1.5%) identified as Hispanic and ethnicity data was missing for 9 participants. 36 participants (31.3%) reported a delay in ambulation as defined by walking later than 15 months of age; data was missing for 24 patients. Most patients [115 (84.6%)] remained ambulatory, with 42 participants (31%) requiring assistive devices for walking (including ankle foot orthotics and canes/walking sticks), and 21 (15%) requiring a wheelchair (data for walking assistance and wheelchair use were missing for 2 and 3 participants, respectively). Foot deformities were present in 93 patients (69%), and hearing loss in 30 patients (25%). (Foot deformity and hearing loss data were missing for 4 and 17 patients respectively). Data for pupillary abnormalities was only available for 28 patients (111 missing), of which 4 (14%) were reported to have abnormalities.
TABLE 2 –
Demographics and Baseline Statistics.
| # of Patients at Baseline | 139 |
|
| |
| Age, years (mean +/−SD) | 46.0 +/− 18.2 |
|
| |
| Gender (M/F) | 65 (46.8%) / 74 (53.2%) |
|
| |
| Race (white yes/no) | 122 (87.8%) / 17 (12.2%) |
|
| |
| Walked Before 15 Months (yes/no) | 79 (68.7%) / 36 (31.3%) |
|
| |
| Demyelinating / Axonal | 73 (54.5%) / 61 (45.5%) |
|
| |
| Wheelchair use (yes/no) | 21 (15.4%) / 115 (84.6%) |
|
| |
| Walking supports needed (yes/no) | 42 (30.7%) / 95 (69.3%) |
|
| |
| Ulnar MNCV Categories (m/s) | |
| <= 15 | 29 (28.7%) |
| 15 – 37 | 14 (13.9%) |
| 38 – 45 | 20 (19.8%) |
| > 45 | 38 (37.6%) |
|
| |
| Foot deformity (yes/no) | 93 (68.9%) / 42 (31.1%) |
|
| |
| Pupil abnormalities (yes/no) | 4 (14.3%) / 24 (85.7%) |
|
| |
| Hearing loss (yes/no) | 30 (24.6%) / 92 (75.4%) |
|
| |
| Hip dysplasia (yes/no) | 0 (0.0%) / 129 (100.0%) |
134 participants had available NCS data or a designation of having either demyelinating or axonal neuropathy. Of these participants, the majority [73 (54%)] had a demyelinating neuropathy, with 29 of 101 patients with available NCS data (29%) having an ulnar motor NCV of less than or equal to 15 m/s (table 2). 9 of the patients designated as having demyelinating neuropathy, had an ulnar CMAP of <2 mV, however their ulnar NCV were severely slowed (<15 m/s), suggesting that axonal loss was not the cause of NCV slowing in the majority of these participants. As compared to those with demyelinating neuropathy, patients with axonal neuropathy were older (mean of 55 vs 38 years, p value < 0.0001), (70% vs 32% >=50 years, p<0.0001), (3.3% vs 21.9% <21 years, p=0.018), more likely to be white (97% vs 79%, p = 0.003), less likely to have foot deformity (56% vs 80%, p = 0.003), were more likely to have walked by 15 months of age (96% vs 48%, p < 0.0001) and reported difficulty with walking at a later age (47 vs 13 months, p < 0.0001), were less likely to require a wheelchair (6.8% vs 20.6%, p=0.025), and had a lower baseline average CMTES scores (mean of 9.6 vs 11.8, p value = 0.036). There were no statistically significant differences between the axonal and demyelinating groups for any of the other demographic variables (TABLE 3).
TABLE 3 -.
Clinical Characteristics of Axonal Versus Demyelinating MPZ-Neuropathy.
| Axonal | Demyelinating | P-value | |
|---|---|---|---|
| # of Patients | 61 | 73 | |
| Age, years (mean +/-SD) | 55.16 (+/−13.13) | 38.26 (+/−18.54) | <0.0001 |
| Gender (M/F) | 33 (54.1%) / 28 (45.9%) | 29 (39.7%) / 44 (60.3%) | 0.0966 |
| Age difficulty walking, months (mean +/−SD) | 46.83 (+/−11.95) | 12.73 (+/−17.49) | <0.0001 |
| Walked before 15 months (yes/no) | 48 (96.0%) / 2 (4.0%) | 30 (48.4%) / 32 (51.6%) | <0.0001 |
| Walking supports needed (yes/no) | 18 (30.0%) / 42 (70.0%) | 23 (31.5%) / 50 (68.5%) | 0.8515 |
| Wheelchair use (yes/no) | 4 (6.8%) / 55 (93.2%) | 15 (20.5%) / 58 (79.5%) | 0. 0251 |
| Foot deformity (yes/no) | 34 (55.7%) / 27 (44.3%) | 56 (80.0%) / 14 (20.0%) | 0.0028 |
| Pupil abnormalities (yes/no) | 1 (10.0%) / 9 (90.0%) | 3 (16.7%) / 15 (83.3%) | 1.0000 |
| Hearing loss (yes/no) | 12 (22.2%) / 42 (77.8%) | 18 (28.6%) / 45 (71.4%) | 0.4330 |
| AFO(s) needed (yes/no) | 16 (26.2%) / 45 (73.8%) | 25 (40.3%) / 37 (59.7%) | 0.0974 |
| Hip dysplasia (yes/no) | 0 (0.0%) / 60 (100.0%) | 0 (0.0%) / 66 (100.0%) | - |
| Baseline CMTES-R (mean +/−SD) | 13.34 (+/−6.58) | 15.56 (+/−8.30) | 0.0915 |
| Baseline CMTES (mean +/−SD) | 9.62 (+/−4.73) | 11.79 (+/−6.87) | 0.0361 |
The average baseline CMTES score was 10.8 (SD = 6.0, range 0 – 28) and the average CMTES-R score was 14.6 (SD = 7.6, range 0 – 32), indicating a moderate range of severity. Baseline CMTES scores were associated with older age (p=0.001), non-white race (p=0.018), designation of demyelinating as opposed to axonal neuropathy (p value = 0.036) and lower MNCV (p=0.012), not walking before 15 months (p=0.036) and younger age of difficulty walking (p=0.018), likelihood of requiring walking support (p<0.0001), or a wheelchair (p<0.0001), and having hearing loss (p=0.013).
Longitudinal CMTES and CMTES-R data was available for 67, 44, 38, 34 and 31 participants at years 1 through 5, respectively. The overall time effect was strongly statistically significant (p value < 0.0001), indicating that the CMTES progresses over time. From baseline to the year 2 follow up, the average CMTES increased by an estimated 0.87 points (95% CI: (0.23, 1.50), p = 0.008), and a significant change as compared to baseline was also evident at years 3, 4, and 5 (FIGURES 1 and 2). The linear model estimated an average CMTES increase of 0.73 units per 2 years (95% CI: (0.46, 1.00), p value < 0.0001). However, a linear approximation of the time trend was questionable (likelihood ratio test p value = 0.03; AIC increase of 2.4). CMTES-R scores increased from baseline to 2 years by an estimate of 0.96 points (95% CI: 0.10–1.82, p = 0.030) and a significant change as compared to baseline was present at years 3, 4, and 5 (FIGURE 3). As compared to the CMTES-R, CMTES had a higher SRM for change from baseline to 2 years (0.37 versus 0.30). Progression of the CMTNS was not examined due to the limited sample size.
FIGURE 1 – CMTES scores over 5 years Legend:
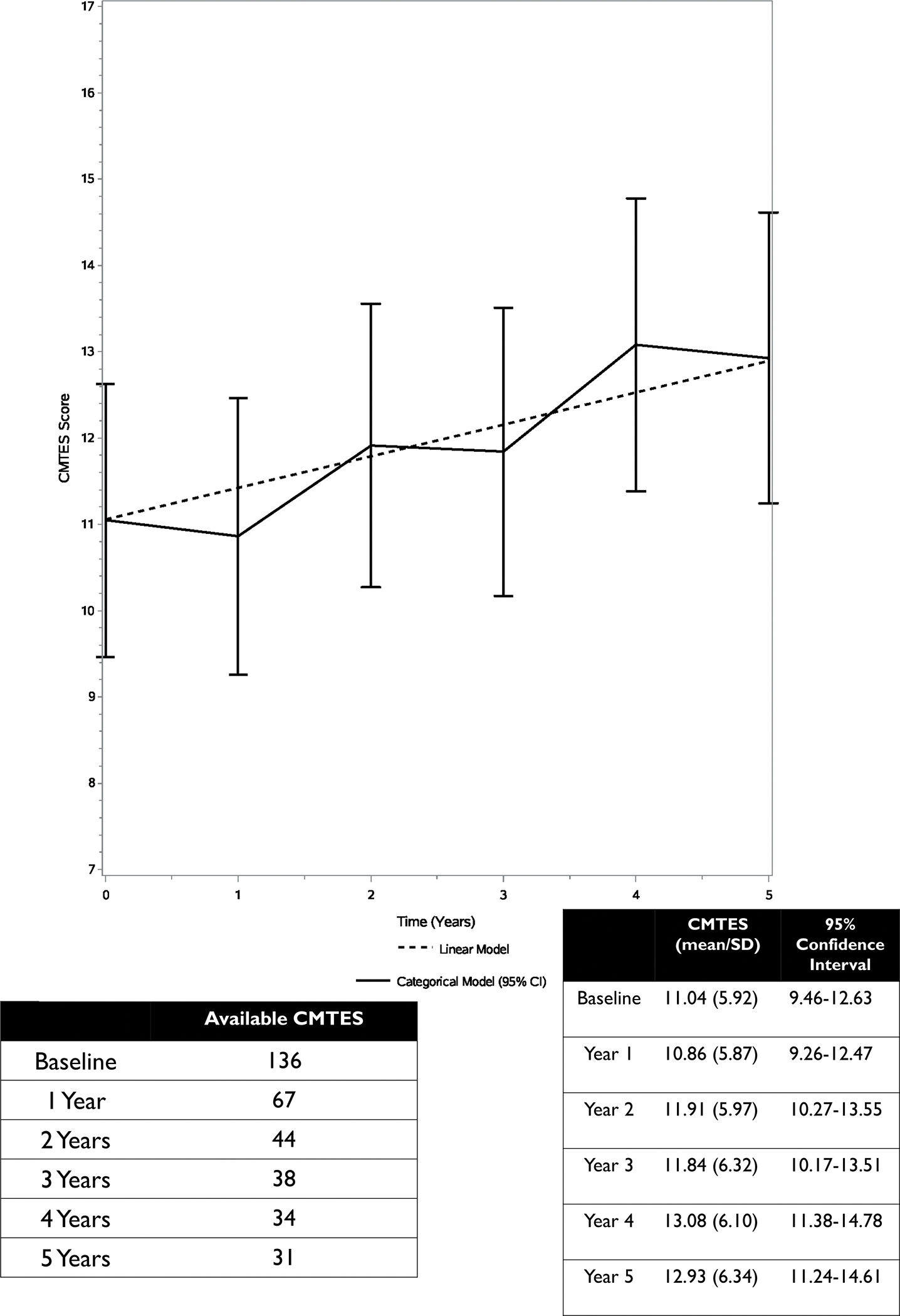
CMTES scores over a 5-year period. CMTES scores increase gradually over subsequent years with relatively linear progression. Bars are 95% confidence intervals.
FIGURE 2 – CMTES change from baseline over 5 years Legend:
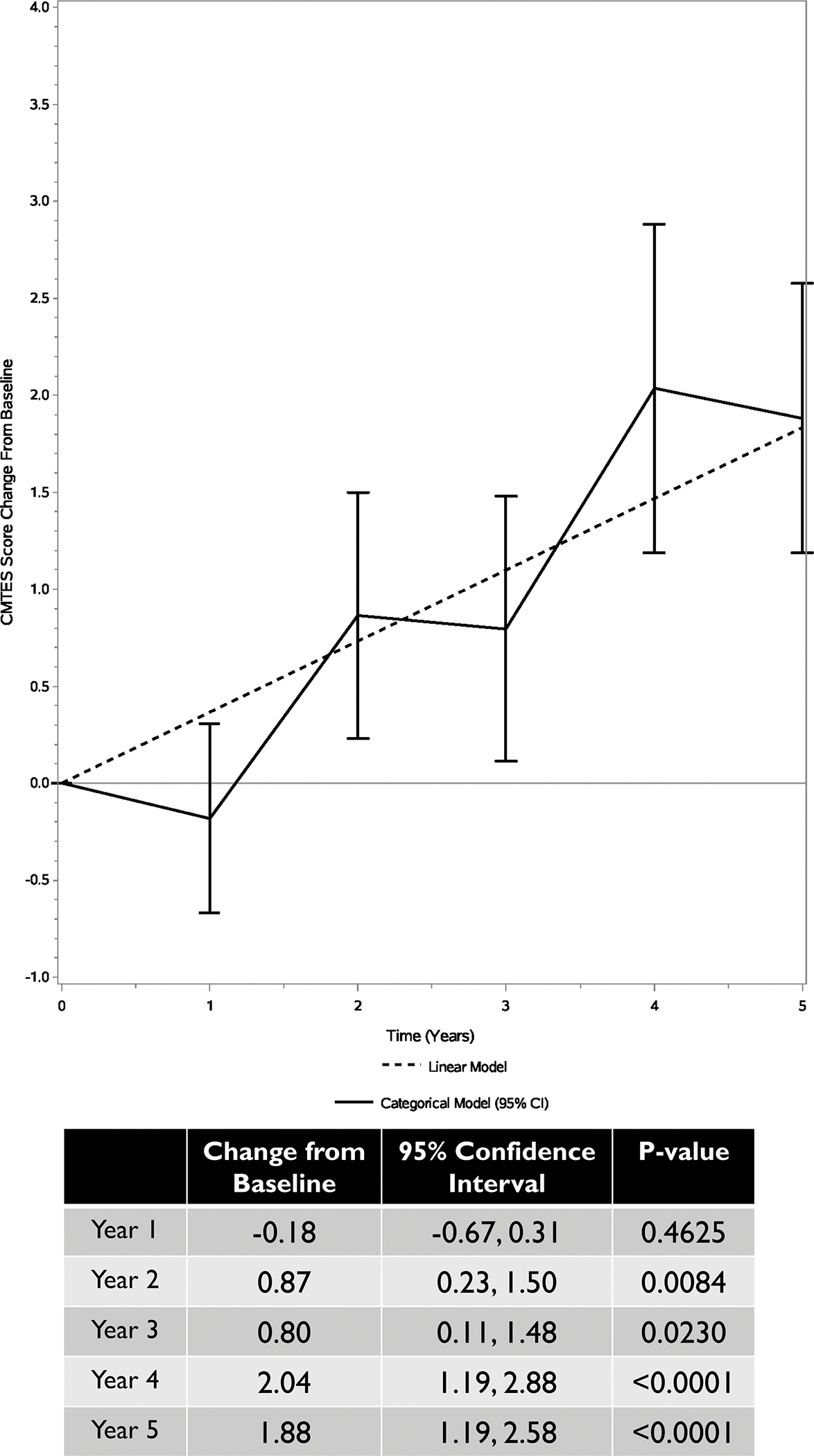
Change in CMTES scores from baseline over a 5-year period. Bars are the 95% confidence intervals.
FIGURE 3 – Influence of axonal vs. demyelinating disease type on change from baseline to 2 years Legend:
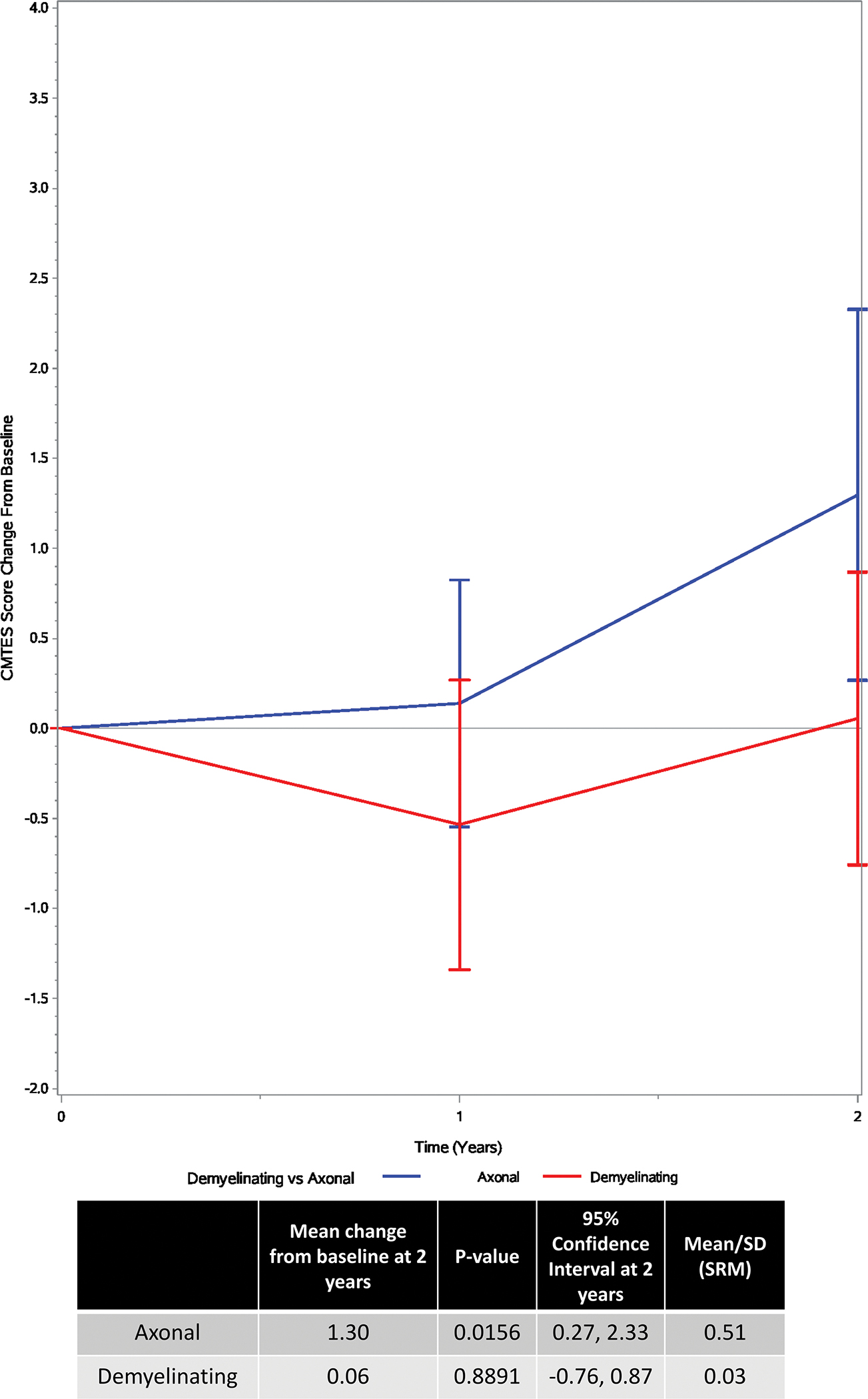
Categorical model showing change in CMTES based on axonal vs. demyelinating disease type over a 2-year period. Red = demyelinating model. Blue = axonal model. Bars are the 95% confidence intervals.
The type of neuropathy (demyelinating vs. axonal), gender, and age were examined as effect modifiers for the progression of CMTES over 2 years of follow up. Complete data was used for consistency; 131 patients were included in the analysis: 130 at baseline, 63 at follow up year 1, and 42 at follow up year 2. 62 (47%) were male and 69 (53%) were female. Mean age was 46.3 years (SD = 18.1, range: 3.9 to 76.4). 60 (46%) were classified as axonal and 71 (54%) as demyelinating. Mean baseline CMTES was 10.8 (SD=6.1, range = 0 to 28); 1 observation was missing. Neither gender nor age significantly modified CMTES progression form baseline to follow up year 2 (p values = 0.500 and 0.972 respectively). The test for type of neuropathy as a modifier of CMTES progression to follow up year 2 was also non-significant (p=0.057).
By estimate, CMTES progression decreased, as age increased, however, the overall test for progression at any age was non-significant (p value = 0.062). Female patients increased by an estimated 1.11 units (95% CI: (0.01, 2.22), p value = 0.049) from baseline to 2 year follow up, whereas the change for male patients was non-significant (estimate = 0.64, 95% CI: (−0.29, 1.57), p value = 0.170). Based on sample size, participants were divided into axonal and demyelinating neuropathy groups, as opposed to the 3 phenotypic groups previously described in MPZ-neuropathies8. Axonal patients increased on the CMTES by an estimated 1.30 points (95% CI: (0.27, 2.33), p value = 0.016, SRM = 0.51) from baseline to 2 year follow up, whereas the change for demyelinating patients was non-significant (estimate = 0.06, 95% CI: (−0.76, 0.87), p value = 0.889, SRM = 0.03) (FIGURE 3). To reiterate, the difference in progression between axonal and demyelinating participants was non-significant (p value = 0.057), possibly because the test for effect modification was underpowered.
Baseline CMTES, classified as mild (0–7), moderate (8–14), and severe (15+), was also considered as an effect modifier of CMTES progression. Change scores from baseline to follow up years 1 and 2 were fit to a longitudinal regression model (FIGURE 4). Data was available for 83 patients: 21 mild, 40 moderate, and 22 severe. There were 66 available observations at follow up year 1, and 44 at follow up year 2. Only the moderate group showed statistically significant progression from baseline to follow up year 2 (estimate = 1.14, 95% CI: (0.16, 2.12), p value = 0.025, SRM = 0.49). The mild group was estimated to decrease non-significantly by −0.03 (95% CI: (−1.36, 1.30), p value = 0.958, SRM = −0.02), while the severe group increased non-significantly by 0.25 (95% CI: (−1.09, 1.59), p value = 0.690, SRM = 0.11). The overall test for baseline CMTES category modifying the progression of CMTES from baseline to follow up year 2 was non-significant, however (p = 0.281). Furthermore, none of the pair-wise comparisons were statistically significant.
FIGURE 4 -. Influence of baseline disease severity Legend:
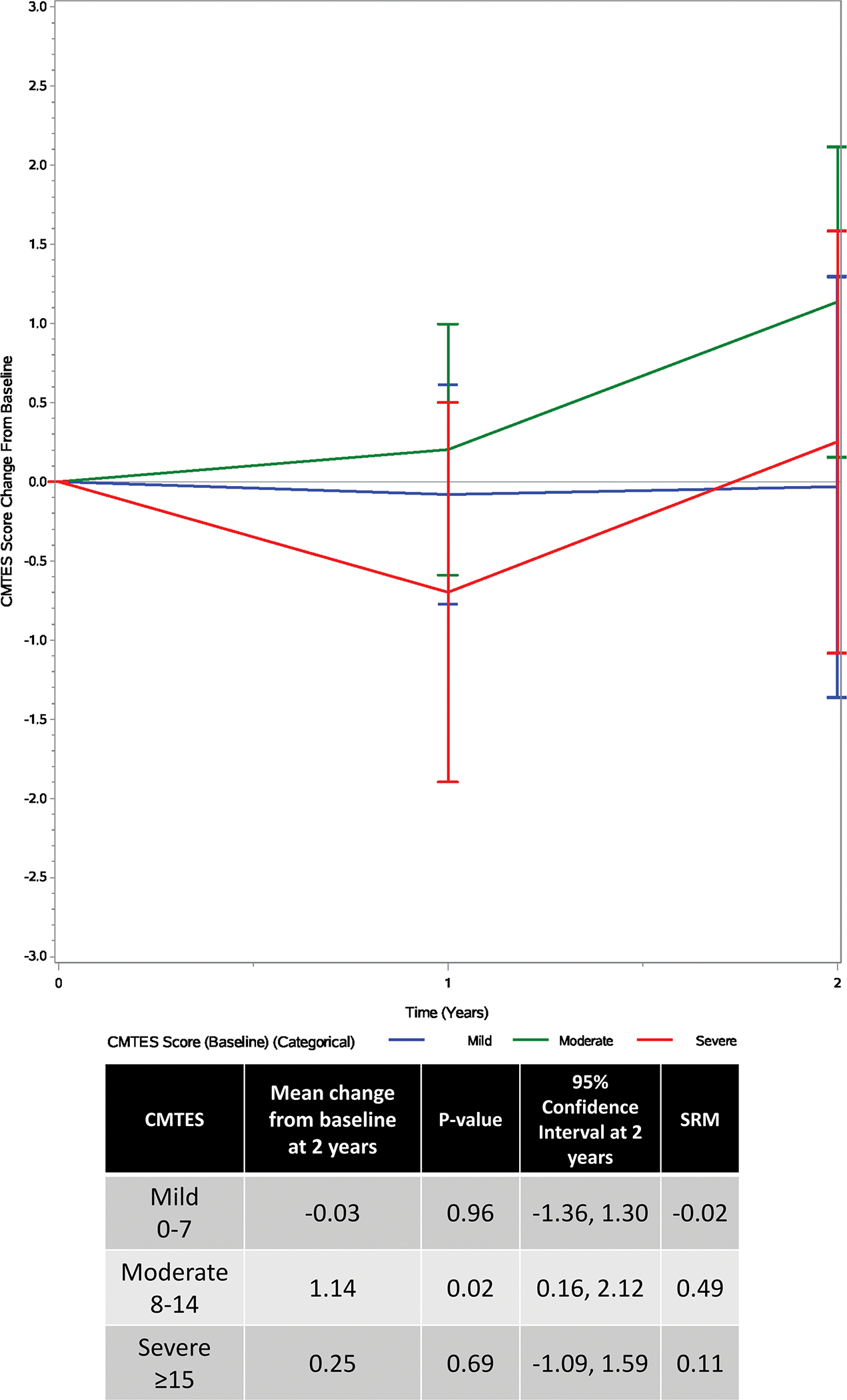
Categorical model showing change in CMTES based on disease severity at baseline over a 2-year period. Red=mild (CMTES 0–7), green=moderate (CMTES 8–14), blue=severe (CMTES ≥15). Bars are the 95% confidence intervals.
137 patients had available MPZ variant data. Variant details and clinical features are described in SUPPLEMENTAL TABLE 1. 32 patients had a participating family member making up 12 families total, however segregation data was not available for all patients. 44 unique variants were described, 1 of which appears to be novel and is classified as of uncertain significance. 39 variants are classified as pathogenic or likely pathogenic21. CMTES score data was available for 60 patients with 32 unique variants. The most common variant in our cohort was p.Pro70Ser, which was reported in 13 total participants, nine with CMTES scores available. All 13 patients had their initial visit in adulthood (42–76 years of age) and presented with an axonal neuropathy. Of the 10 with available data, all walked before 15 months. The highest baseline CMTES scores were reported in the p.Gly137Ser and p.Val102fs variants.
Progression on the CMTES varied amongst participants with different MPZ variants, as well as between patients with the same variant and patients from the same family (FIGURE 5). For example, in the 9 participants with the p.Pro70Ser variant, the 2-year change in CMTES showed an increase in 6 patients, remained stable in 1 patient, and decreased in 2 patients. Despite this variable progression, axonal or demyelinating phenotypes were largely consistent for individual mutations. Variants associated with the greatest 2-year change included p.Ser140Thr and p.Pro70Ser. In contrast, participants with the p.Arg98His and p.Lys236del variants showed a notable decrease in CMTES scores over 2 years.
FIGURE 5 –
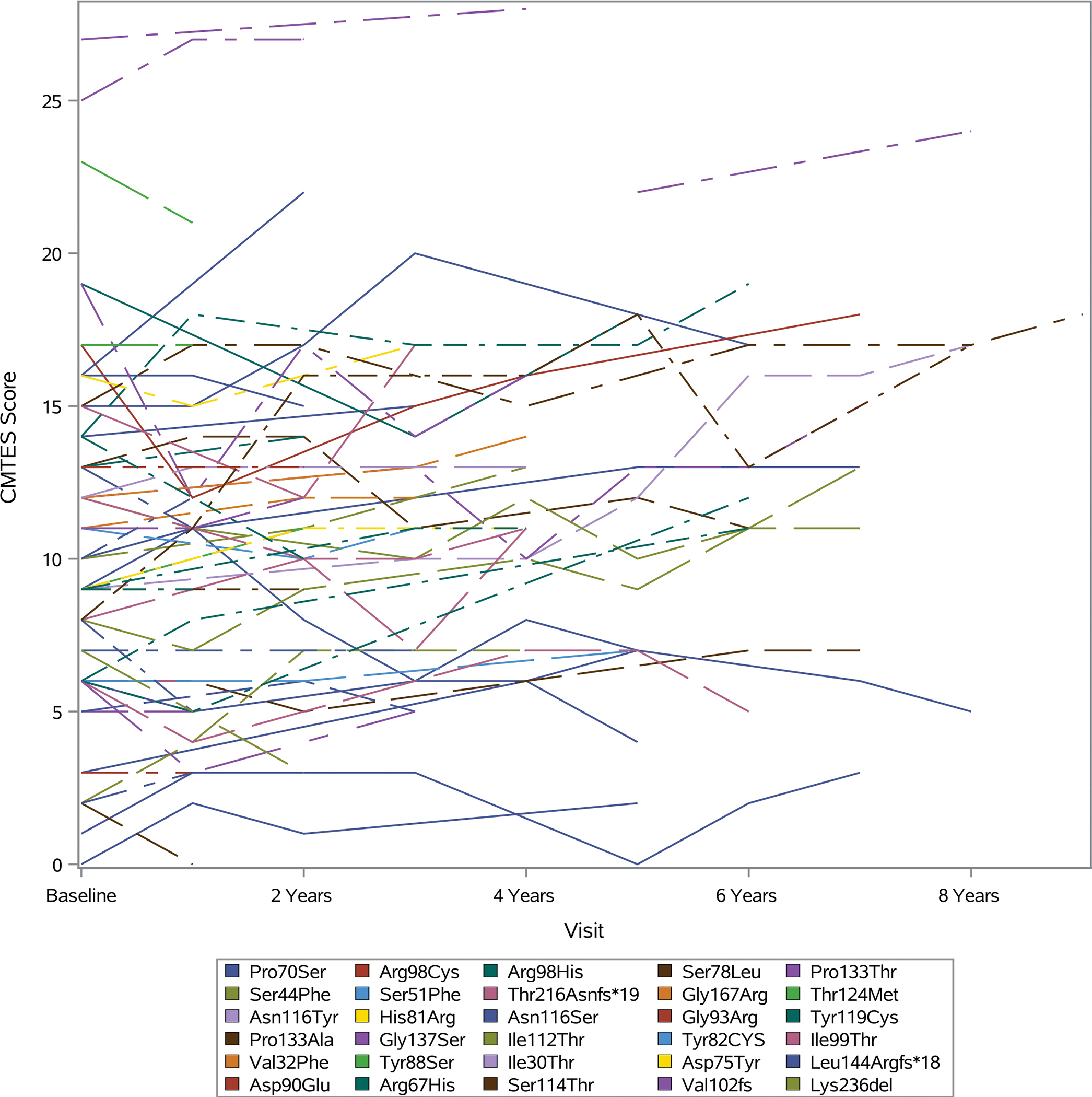
CMTES scores over 8 years for individual participants
Discussion:
To our knowledge, this is the largest longitudinal study of MPZ-neuropathies to date, and is the first study to evaluate mutation-specific natural history in this patient population. Our findings demonstrated progression in CMTES scores over time with an estimated change of 0.87 points (p = 0.008) from baseline to 2 years follow up, confirming that this clinical outcome assessment will be useful for future clinical trials. Importantly, by examining this large cohort, we were able to detect a qualitative difference in the 2-year change in the CMTES between participants with axonal versus demyelinating neuropathy [1.30 point increase for axonal (p=0.016), versus a non-significant increase of 0.06 points for demyelinating (p=0.889)]. The 2-year change in the axonal group was also associated with a higher SRM (0.51 versus 0.03), suggesting a higher level of responsiveness. It should be emphasized, however, that the difference between the 2 groups did not meet statistical significance (p=0.057), potentially owing to a lack of power. Additionally, CMTES was only sensitive to change in participants with moderate neuropathy (as defined by the baseline CMTES), and did not reliably capture progression in patients on the phenotypic extremes. Finally, the mutation-specific natural history in this cohort showed highly variable rates of progression among patients with different MPZ mutations, as well as among patients with shared genetic variants, underscoring the heterogeneity in the clinical progression of these disorders.
In keeping with prior studies, we observed an association between baseline CMTES scores and age (p=0.001), however, there was no evidence that the responsiveness of the CMTES differed according to age or gender16. Baseline scores were also associated with a younger age of walking difficulty (p=0.018), suggesting that earlier gait difficulty in MPZ-neuropathy may portend a more severe neuropathy in later life. The 2-year change in CMTES that we observed in MPZ-neuropathy is greater than that previously observed in CMT1A (0.4 points) and smaller than that seen in patients with CMT2A (0.97 points)16, 22. These change scores are consistent with the known differences in the clinical severity of these disorders, which together comprise the most common forms of AD hereditary neuropathy.
In contrast to our observations in CMT1A, we found that the SRM associated with the 2-year change in MPZ-neuropathy was slightly higher for unweighted CMTES scores than for the CMTES-R (0.37 versus 0.30, respectively), suggesting that the use of the CMTES-R introduces a greater variability in scores. This observation parallels prior findings in CMT2A (MFN2 mutations) reported by Pipis et al., which demonstrated a lower 2-year SRM for the CMTES-R than for the unweighted CMTES22. The CMTNS-R was developed based on a psychometric analysis using CMT1A data in an effort to allow for the detection of smaller differences in clinical severity and did result in an improved SRM in the CMT1A population16. Our current findings suggest that the CMTES-R could be further refined for use in more rapidly progressive neuropathies such as MPZ-neuropathy and CMT2A.
While there were fewer patients with axonal neuropathy in our cohort, they drove the overall change observed in CMTES scores. This finding is also consistent with the prior observations that demyelinating MPZ-neuropathy progresses less rapidly beyond adolescence, whereas axonal forms can progress rapidly in adulthood, with some patients losing ambulation within years of disease onset9, 23–25. The differences in natural history between axonal and demyelinating MPZ-neuropathy may be related to the varied pathophysiology of these conditions, with childhood-onset demyelinating forms more commonly resulting from developmental defects in myelin (i.e. dysmyelination), and axonal forms arising secondary to disruptions in MPZ-mediated signal transduction and Schwann cell-axonal interactions8, 26, 27. These data suggest that clinical trials in the adult population with MPZ-neuropathy employing the CMTES may be most informative in participants with axonal neuropathy. Additionally, our findings underscore the importance of ongoing natural history studies in children with demyelinating CMT1B using age appropriate scales such as the CMT Pediatric Scale (CMTPeds) and CMT Infant and Toddler Scale (CMTInfS), as progression in this patient population is likely to be greatest in childhood28, 29.
The finding that only participants with moderate neuropathy (CMTES scores of 8–14) showed progression in CMTES scores over 2 years also has important implications for future clinical trials. 2-year progression on the CMTES in the moderate group was 1.14, (p value = 0.025) with an SRM of 0.49. In contrast, the mild group actually showed a non-significant decrease in scores over 2 years, while the severe group showed a non-significant increase. The lack of responsiveness of the CMTES in severely affected patients mirrors our prior findings in CMT1A, underscoring that the CMTES is insensitive to the possible progression in those with severe neuropathy. In contrast, the lack of significant progression in patients with initial scores in the mild range of severity in this cohort differs from our observations in CMT1A, as mildly affected patients with CMT1A did have significant progression over 2 years on the CMTES-R16. Given that the severity categories employed in the current study were defined in a CMT1A population, further examining these categories in conjunction with other measures of neuropathy severity will be needed to determine their relevance in MPZ-neuropathy.
Genotype-phenotype correlations in our cohort were consistent with those defined by Sanmaneechai et al.8, although fewer patients in our cohort reported delayed milestones (TABLE 3). 1 new variant with an unknown classification was also identified: p.Pro105Ser, for which segregation analysis was not available. This variant has not been reported in the genome aggregation database (gnomAD), implying it is not present in the general population. The expected amino acid residues are highly conserved throughout species, and the pathogenic variant p.Pro105Thr affects the same codon21. Further characterization of this novel variant will be needed to determine their likelihood of pathogenicity.
The progression in CMTES among patients with differing MPZ mutations was highly variable, with the greatest progression over 2 years observed in a patient with the p.Ser140Thr variant and a patient with the p.Pro70Ser variant, both with axonal neuropathy. While the majority of individual MPZ variants did consistently manifest with either axonal or demyelinating phenotypes, as previously observed by Sanmaneechai et al., patients with the same MPZ variant demonstrated variable 2-year change on the CMTES, as specifically highlighted by the 13 participants with the p.Pro70Ser variant8. Our findings are purely descriptive and cannot be extrapolated to other patients with shared variants in MPZ, however it does appear that the natural history associated with individual MPZ mutations is complex and heterogeneous. These findings align with prior observations that specific cellular disruptions resulting from MPZ mutants do not correlate with specific clinical phenotypes8.
Several missense variants in MPZ have specifically been reported to cause a late-onset, severe, axonal neuropathy with rapid progression. These variants tend to congregate around codon 70 in the extracellular and domain: p.Pro70Leu, p.Asp75Val, and p.Pro70Ser30, 31. In contrast, the 9 subjects in our cohort with the p.Pro70Ser variant demonstrated baseline CMTES scores in the mild to moderate range and showed variable progression on the CMTES that was not distinct from the other variants examined. The difference in the observed disease progression could be related to the possibility that genetic testing was selectively offered to patients with more severe clinical phenotypes in the past. The broader use of genetic testing more recently may have led to the identification of milder and more slowly progressing MPZ-neuropathies. The natural history of specific MPZ variants will therefore likely continue to clarify as more affected individuals are identified. Given the clinical heterogeneity of MPZ-neuropathy, as well as its varied gain of function mechanisms, future treatments will likely need to be personalized with focus on allele specific approaches32.
Our study is limited by sample size as well as by missing data, as not all patients returned every year and some had missing baseline CMTES/CMTES-R scores. We have addressed this by using longitudinal regression models to evaluate change in CMTES scores, allowing for missing years between visits. Our determination of axonal versus demyelinating neuropathy also relied on ulnar NCV, rather than more complete electrophysiological data. Sample size also limited our comparison of change in CMTES in the axonal and demyelinating groups beyond 2 years of follow up, precluding a comparison of previously described phenotypic groups in MPZ-neuropathy including early-onset, childhood (classic CMT1B)-onset, and adult-onset patients. Finally, our study focused on the CMTES as the measure of progression, and did not examine other established measures of neuropathy, such as the CMT-FOM, 10-meter walk test, or the Overall Neuropathy Limitations Scale (ONLS)19, 33, 34, many of which were not available when we began our study. For similar reasons we did not evaluate emerging objective biomarkers such as MRI of intramuscular fat accumulation or plasma neurofilament light chain concentrations, which will need to be evaluated in future studies35.
Taken together, our results suggest that the CMTES will serve as a valuable tool for clinical trials in axonal forms of MPZ-neuropathy. Based on our findings, a 2-year double-blinded, randomized, placebo-controlled trial in this population (powered to evaluate complete arrest in disease progression with 80% power and alpha of 0.05) would require 62 subjects in each arm. While this is still an ambitious number given the rarity of MPZ-neuropathy, the recent rise in gene testing has led to a rapid identification of new MPZ cases, with 76 new mutations reported between 2005 and 2018, the majority of which are axonal4. Additionally, the CMTES would not be used in isolation, but rather alongside other biomarkers, such as quantitative calf muscle fat accumulation on MRI (which shows significant 1 year change in CMT1A with an SRM of 0.83) allowing for further reductions in sample size35, 36. Importantly, we have shown that the CMTES is not an effective clinical outcome assessment for adult patients with early-onset, demyelinating forms of MPZ-neuropathy. These patients should therefore be evaluated in early life using pediatric clinical outcome assessments, such as the CMT pediatric and infant scores, and the recently developed pediatric CMT-specific quality of life outcome measure28, 29, 37.
Supplementary Material
Social Media If Published.
If you and/or a co-author has a Twitter handle that you would like to be tagged, please enter it here. N/A
-
What is the current knowledge on the topic?
Neuropathy related to mutations in the MPZ gene constitute the second most common forms of Charcot Marie Tooth disease type 1 (CMT1). Few studies have examined the natural history of MPZ-neuropathy, which remains a barrier to clinical trials.
-
What question did this study address?
This study aimed to assess whether the Charcot Marie Tooth Examination Score (CMTES) is an effective clinical measure of progression in MPZ-neuropathy and to define the rate of clinical progression over a 5-year time frame. The study also aimed to determine the optimal patient population for future treatment trials in MPZ-neuropathy.
-
What does this study add to our knowledge?
This study demonstrates that the CMTES is an effective clinical measure of MPZ-neuropathy and reflects the change in clinical severity of neuropathy over a 2-year time frame. Additionally, the findings suggest that adult participants with axonal neuropathy caused by mutations in the MPZ gene will serve as the optimal population in future clinical trials.
-
How might this potentially impact on the practice of neurology?
The findings of this study will allow clinicians to more effectively help their patients understand the prognosis of MPZ-neuropathy overall, and in regard to specific genetic variants in the gene. Additionally, these results will help guide patient selection for upcoming clinical trials.
Acknowledgements:
The authors would like to thank all the patients who participated in the INC and their families, without whom this study would not be possible. The authors would also like to thank the people working at INC sites who contributed to this study, especially Julian Blake, Betsy Burgos, Daniela Calabrese, Vinay Chaudhry, David Cornblath, Katy Eichinger, Tim Estilow, Claudia Gandioli, Audra Hamilton, Allan M Glanzman, Ahmet Hoke, Andrea Kelley, Livija Medne, Manoj Menezes, Joan Mountain, Sinead Murphy, Jillian Olsen, Alex Rossor, Oranee Sanmaneechai, Paola Saveri, Anna Sorey, Mariola Skorupinska, Janet Sowden and Andrea Swenson. This research was also supported by the National Institute for Health Research University College London Hospitals Biomedical Research Centre.
The Inherited Neuropathies Consortium (2U54NS065712–07) is a part of the Rare Diseases Clinical Research Network (RDCRN), an initiative of the Office of Rare Diseases Research (ORDR), NCATS. The INC is funded through a collaboration between NCATS and the NINDS. The INC also receives support from the MDA and CMTA. The funders had no role in the study design, data collection, data analysis, data interpretation, or writing of the manuscript.
VF reports funding support from NIDDK, 5K23DK118202–02. DNH reports grant support through U54 NS065712, NINDS, 5U01NS109403–03, CMT Association, 1R01DK115687–03, MDA, Friedreich’s Ataxia Research Alliance, Voyager Pharmaceuticals, and Acceleron Pharma. CJS reports funding support from Roche and Argenx. DP reports grant support from Telethon-UILDM, AFM-Telethon, and the Charcot-Marie-Tooth Association. SSS reports grant support from U54 NS0657, the Charcot-Marie-Tooth Association, and the Muscular Dystrophy Association. MMR reports grant support from U54 NS0657, the Muscular Dystrophy Association, Charcot-Marie-Tooth Association, the Medical Research Council (MRC) and the Wellcome trust. MES reports grant support from U54 NS0657, the Muscular Dystrophy Association, and the Charcot-Marie-Tooth Association.
Abbreviations:
- (CMT)
Charcot Marie Tooth disease
- (CMTES)
CMT Examination Score
- (CMTInfS)
CMT Infant and Toddler Scale
- (CMTPeds)
CMT Pediatric Scale
- (INC)
Inherited Neuropathy Consortium
- (MPZ)
Myelin Protein Zero
- (NCS)
nerve conduction study
- (NCV)
nerve conduction velocity
- (RDCRN)
Rare Diseases Clinical Research Network
- (CMTES-R)
Rasch modified CMT Examination Score
- (SRM)
standardized response mean
Footnotes
Potential Conflicts of Interest:
VF, SS, JB, KS, IM, EP, CP, GP, ML, FM, CB, SF, TG, LG, RS, JW, DNH, JL, CJS, TEL, JD, CEK, SWY, RS, RSF, SSS, DP, MMR, and MES have no potential conflicts of interest to report. SR is currently employed by a CRO (PRA Health Sciences) that works with pharmaceutical companies. Istituto Neurologico Carlo Besta receives donations for research from Pfizer, LAM Therapeutics, and Acceleron Pharma Inc.
Data Availability Statement:
Data not provided in the article because of space limitations as well as the study protocol and statistical analysis will be shared at the request of any qualified investigator for purposes of replicating procedures and results.
REFERENCES
- 1.Saporta AS, Sottile SL, Miller LJ, Feely SM, Siskind CE, Shy ME. Charcot-Marie-Tooth disease subtypes and genetic testing strategies. Ann Neurol 2011;69:22–33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Murphy SM, Laura M, Fawcett K, et al. Charcot-Marie-Tooth disease: frequency of genetic subtypes and guidelines for genetic testing. J Neurol Neurosurg Psychiatry 2012;83:706–710. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Nelis E, Van Broeckhoven C, De Jonghe P, et al. Estimation of the mutation frequencies in Charcot-Marie-Tooth disease type 1 and hereditary neuropathy with liability to pressure palsies: a European collaborative study. Eur J Hum Genet 1996;4:25–33. [DOI] [PubMed] [Google Scholar]
- 4.Callegari I, Gemelli C, Geroldi A, et al. Mutation update for myelin protein zero-related neuropathies and the increasing role of variants causing a late-onset phenotype. J Neurol 2019;266:2629–2645. [DOI] [PubMed] [Google Scholar]
- 5.Shy ME, Jani A, Krajewski K, et al. Phenotypic clustering in MPZ mutations. Brain 2004;127:371–384. [DOI] [PubMed] [Google Scholar]
- 6.Krajewski KM, Lewis RA, Fuerst DR, et al. Neurological dysfunction and axonal degeneration in Charcot-Marie-Tooth disease type 1 A. Brain 2000;123 ( Pt 7):1516–1527. [DOI] [PubMed] [Google Scholar]
- 7.Thomas PK, Marques W Jr., Davis MB, et al. The phenotypic manifestations of chromosome 17p11.2 duplication. Brain 1997;120 ( Pt 3):465–478. [DOI] [PubMed] [Google Scholar]
- 8.Sanmaneechai O, Feely S, Scherer SS, et al. Genotype-phenotype characteristics and baseline natural history of heritable neuropathies caused by mutations in the MPZ gene. Brain 2015;138:3180–3192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Bird TD, Kraft GH, Lipe HP, Kenney KL, Sumi SM. Clinical and pathological phenotype of the original family with Charcot-Marie-Tooth type 1B: a 20-year study. Ann Neurol 1997;41:463–469. [DOI] [PubMed] [Google Scholar]
- 10.Brennan KM, Bai Y, Shy ME. Demyelinating CMT--what’s known, what’s new and what’s in store? Neurosci Lett 2015;596:14–26. [DOI] [PubMed] [Google Scholar]
- 11.Hayasaka K, Himoro M, Sato W, et al. Charcot-Marie-Tooth neuropathy type 1B is associated with mutations of the myelin P0 gene. Nat Genet 1993;5:31–34. [DOI] [PubMed] [Google Scholar]
- 12.Volpi VG, Touvier T, D’Antonio M. Endoplasmic Reticulum Protein Quality Control Failure in Myelin Disorders. Front Mol Neurosci 2016;9:162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Grandis M, Vigo T, Passalacqua M, et al. Different cellular and molecular mechanisms for early and late-onset myelin protein zero mutations. Hum Mol Genet 2008;17:1877–1889. [DOI] [PubMed] [Google Scholar]
- 14.Xu W, Shy M, Kamholz J, et al. Mutations in the cytoplasmic domain of P0 reveal a role for PKC-mediated phosphorylation in adhesion and myelination. J Cell Biol 2001;155:439–446. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Wrabetz L, D’Antonio M, Pennuto M, et al. Different intracellular pathomechanisms produce diverse Myelin Protein Zero neuropathies in transgenic mice. J Neurosci 2006;26:2358–2368. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Fridman V, Sillau S, Acsadi G, et al. A longitudinal study of CMT1A using Rasch analysis based CMT neuropathy and examination scores. Neurology 2020;94:e884–e896. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Shy ME, Blake J, Krajewski K, et al. Reliability and validity of the CMT neuropathy score as a measure of disability. Neurology 2005;64:1209–1214. [DOI] [PubMed] [Google Scholar]
- 18.Sadjadi R, Reilly MM, Shy ME, et al. Psychometrics evaluation of Charcot-Marie-Tooth Neuropathy Score (CMTNSv2) second version, using Rasch analysis. J Peripher Nerv Syst 2014;19:192–196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Piscosquito G, Reilly MM, Schenone A, et al. Responsiveness of clinical outcome measures in Charcot-Marie-Tooth disease. Eur J Neurol 2015;22:1556–1563. [DOI] [PubMed] [Google Scholar]
- 20.Harding AE, Thomas PK. The clinical features of hereditary motor and sensory neuropathy types I and II. Brain 1980;103:259–280. [DOI] [PubMed] [Google Scholar]
- 21.Richards S, Aziz N, Bale S, et al. Standards and guidelines for the interpretation of sequence variants: a joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet Med 2015;17:405–424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Pipis M, Feely SME, Polke JM, et al. Natural history of Charcot-Marie-Tooth disease type 2A: a large international multicentre study. Brain 2020;143:3589–3602. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Bai Y, Ianokova E, Pu Q, et al. Effect of an R69C mutation in the myelin protein zero gene on myelination and ion channel subtypes. Arch Neurol 2006;63:1787–1794. [DOI] [PubMed] [Google Scholar]
- 24.Dacci P, Taroni F, Bella ED, et al. Myelin protein zero Arg36Gly mutation with very late onset and rapidly progressive painful neuropathy. J Peripher Nerv Syst 2012;17:422–425. [DOI] [PubMed] [Google Scholar]
- 25.Liu L, Li X, Zi X, et al. Two novel MPZ mutations in Chinese CMT patients. J Peripher Nerv Syst 2013;18:256–260. [DOI] [PubMed] [Google Scholar]
- 26.De Jonghe P, Timmerman V, Ceuterick C, et al. The Thr124Met mutation in the peripheral myelin protein zero (MPZ) gene is associated with a clinically distinct Charcot-Marie-Tooth phenotype. Brain 1999;122 ( Pt 2):281–290. [DOI] [PubMed] [Google Scholar]
- 27.Li J, Bai Y, Ianakova E, et al. Major myelin protein gene (P0) mutation causes a novel form of axonal degeneration. J Comp Neurol 2006;498:252–265. [DOI] [PubMed] [Google Scholar]
- 28.Mandarakas MR, Menezes MP, Rose KJ, et al. Development and validation of the Charcot-Marie-Tooth Disease Infant Scale. Brain 2018;141:3319–3330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Burns J, Ouvrier R, Estilow T, et al. Validation of the Charcot-Marie-Tooth disease pediatric scale as an outcome measure of disability. Ann Neurol 2012;71:642–652. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Laura M, Milani M, Morbin M, et al. Rapid progression of late onset axonal Charcot-Marie-Tooth disease associated with a novel MPZ mutation in the extracellular domain. J Neurol Neurosurg Psychiatry 2007;78:1263–1266. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Benedetti S, Previtali SC, Coviello S, et al. Analyzing histopathological features of rare charcot-marie-tooth neuropathies to unravel their pathogenesis. Arch Neurol 2010;67:1498–1505. [DOI] [PubMed] [Google Scholar]
- 32.Fratta P, Ornaghi F, Dati G, et al. A nonsense mutation in myelin protein zero causes congenital hypomyelination neuropathy through altered P0 membrane targeting and gain of abnormal function. Hum Mol Genet 2019;28:124–132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Eichinger K, Burns J, Cornett K, et al. The Charcot-Marie-Tooth Functional Outcome Measure (CMT-FOM). Neurology 2018;91:e1381–e1384. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Graham RC, Hughes RA. A modified peripheral neuropathy scale: the Overall Neuropathy Limitations Scale. J Neurol Neurosurg Psychiatry 2006;77:973–976. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Morrow JM, Evans MRB, Grider T, et al. Validation of MRC Centre MRI calf muscle fat fraction protocol as an outcome measure in CMT1A. Neurology 2018;91:e1125–e1129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Morrow JM, Sinclair CD, Fischmann A, et al. MRI biomarker assessment of neuromuscular disease progression: a prospective observational cohort study. Lancet Neurol 2016;15:65–77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Ramchandren S, Wu TT, Finkel RS, et al. Development and Validation of the Pediatric Charcot-Marie-Tooth Disease Quality of Life Outcome Measure. Ann Neurol 2021;89:369–379. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
Data not provided in the article because of space limitations as well as the study protocol and statistical analysis will be shared at the request of any qualified investigator for purposes of replicating procedures and results.