

Abstract
Pulmonary fibrosis refers to the development of diffuse parenchymal abnormalities in the lung that cause dyspnea, cough, hypoxemia, and impair gas exchange, ultimately leading to respiratory failure. Though pulmonary fibrosis can be caused by a variety of underlying etiologies, ranging from genetic defects to autoimmune diseases to environmental exposures, once fibrosis develops it is irreversible and most often progressive, such that fibrosis of the lung is one of the leading indications for lung transplantation. This review aims to provide a concise summary of the recent advances in our understanding of the genetics and genomics of pulmonary fibrosis, Idiopathic Pulmonary Fibrosis in particular, and how these recent discoveries may be changing the clinical approach to diagnosing and treating patients with fibrotic interstitial lung disease.
Introduction
Pulmonary fibrosis refers to irreversible scarring of the lung parenchyma that can be the end-result of many types of interstitial lung disease (ILD). ILDs are a heterogeneous set of lung diseases characterized by inflammation and/or collagen deposition in the interstitium, the space between the alveoli. Because efficient gas exchange depends on the transit of oxygen and carbon dioxide molecules across the alveolar epithelium, such inflammation and fibrosis leads to impaired gas exchange, restrictive physiology (as measured by spirometry and plethysmography with reduced lung volumes), hypoxemia, dyspnea, cough, and, when progressive, respiratory failure.
This manuscript aims to present a concise clinical summary of the most common forms of pulmonary fibrosis and to review the most recent advances in translational study in this area. This review will focus on Idiopathic Pulmonary Fibrosis (IPF), one of the most common ILDs, due to the breadth of literature focusing on this diagnosis, but also because of the implications findings in IPF have for understanding of other fibrotic lung diseases.
Clinical Presentation of Pulmonary Fibrosis
It is common for patients to come to clinical attention when they experience shortness of breath, decreased exercise tolerance, or cough that cannot be explained by more common conditions, such as infections, cardiac abnormalities, or airways diseases such as chronic obstructive pulmonary disease (COPD) and asthma. Many pulmonary fibrosis patients have been seen by numerous physicians and have been treated empirically with antibiotics, anti-allergy, and/or anti-inflammatory medications before they are accurately diagnosed.
Clinically, pulmonologists and ILD specialists classify their patients’ diseases based on careful synthesis of the patient’s family history, occupational and environmental history, symptomatology, radiologic patterns on high resolution CT scans (HRCTs) of the chest (Figure 1), and, when obtained, histopathology (Figure 2). Multi-disciplinary conferences, during which pathologists, radiologists, and pulmonologists confer are recommended at this time as the most effective way of diagnosing patients in whom there is any diagnostic uncertainty and determining the best courses of treatment (1).
Figure 1. Radiologic Features of Selected Forms of Pulmonary Fibrosis.
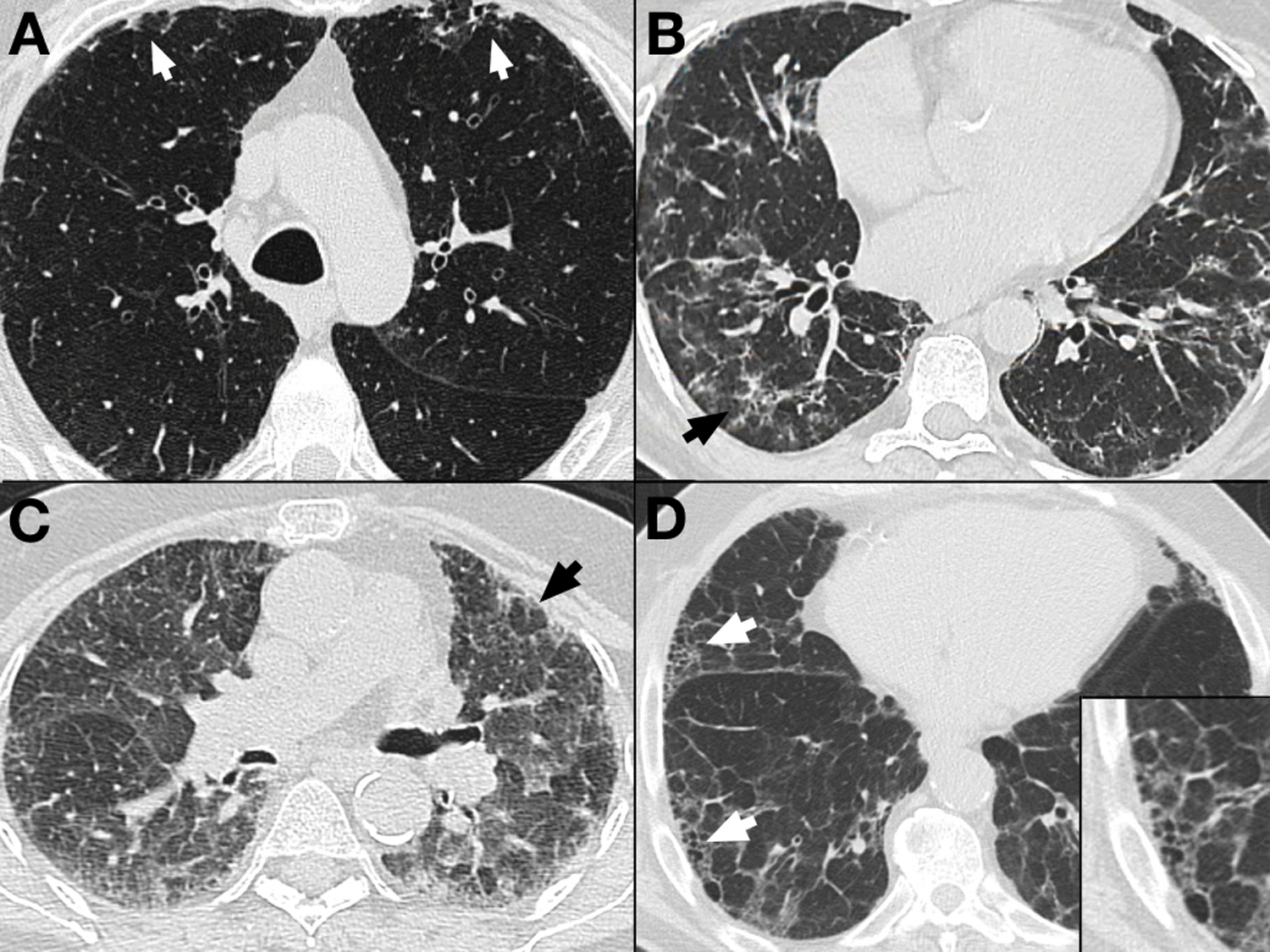
A. Axial image from a high resolution computed tomography (HRCT) scan of a patient with mild fibrotic changes in the lung, including bilateral peripheral reticular abnormality (arrows).
B. HRCT image from a patient with Sjogren’s disease (an autoimmune disease) and associated fibrotic interstitial lung disease (ILD). This image illustrates peripheral reticular abnormality more advanced than in (A), but it also shows areas of ground glass opacity (arrow) commonly seen in connective tissue disease related ILDs.
C. Expiratory HRCT image of a patient with significant fibrotic hypersensitivity pneumonitis (HP). This image illustrates ground glass opacities, significant reticular abnormality, as well as areas of lobular air trapping (arrow) indicating involvement of the small airways. This finding is common in HP and other ILDs that affect small airways, thereby leading to air trapping during expiration.
D. HRCT image from a patient with Idiopathic Pulmonary Fibrosis (IPF). The patient has extensive reticular abnormality, a relatively paucity of ground glass abnormality, as well as honeycombing (arrows), the term for peripheral stacked cystic abnormality that is characteristic of Usual Interstitial Pneumonia (UIP), the radiologic and pathologic pattern associated with IPF. The inset image shows a magnification of one of the honeycombed areas.
Images courtesy of Dr. Christopher J. G. Sigakis, Columbus Radiology/Radiology Partners.
Figure 2. Pathologic Findings in Selected Forms of Pulmonary Fibrosis.

A. Hematoxylin and eosin (H&E) staining of lung tissue from patient with asbestos-related lung fibrosis. Areas of fibrotic lung (arrowhead) are noted beside areas of relatively normal alveolar tissue (white arrow). Inset: Higher-power view of lung tissue from the same patient where asbestos fibers can be seen embedded in the fibrotic lung tissue (black arrow).
B. H&E staining of lung tissue from patient with fibrotic hypersensitivity pneumonitis. Lung tissue shows extensive fibrosis. Inset: the lung tissue here shows evidence of cellular infiltrate and poorly formed granulomas (black arrow), a hallmark of this disease.
C. Low power view of fibrotic non-specific interstitial pneumonia (NSIP) pattern of interstitial lung disease characteristic of patients with autoimmune diseases. This H&E stained tissue shows areas of septal thickening (white arrow). Inset: Higher-power view of lung tissue from the same patient shows areas of cellular infiltrate characteristic of NSIP (black arrow).
D. Usual Interstitial Pneumonia (UIP) pattern of fibrosis in lung tissue from a patient with Idiopathic Pulmonary Fibrosis (IPF), revealing severe fibrosis with sharp transition to non-fibrotic alveolar lung tissue, often referred to as “temporal heterogeneity.” Inset: Higher-power view of lung tissue from the same patient illustrating a fibroblastic focus (black arrow) at the leading edge of the fibrotic area, also characteristic of the UIP pattern.
Images courtesy of Dr. Haiying Zhang, Baylor University Medical Center at Dallas.
Causes of Pulmonary Fibrosis
In case of ILD and pulmonary fibrosis, underlying systemic diseases or exposures (in the environment or due to occupation) can be linked to the disease. However, in a subset of ILDs, no cause can be identified, in which case they are deemed Idiopathic Interstitial Pneumonias (IIPs, Table 1) (2), the most common of which is IPF (Figure 1D). The incidence and mortality of IPF is increasing (3,4). In the last few decades, careful study of large cohorts from numerous centers has revealed that in many cases, a significant proportion of disease risk in IIPs and subsets of pulmonary fibrosis is driven by both rare and common genetic variants (5,6). These discoveries have led to the identification of potential therapeutic targets and sub-phenotyping of disease.
Table 1.
American Thoracic Society/European Respiratory Society Classification of Idiopathic Interstitial Pneumonias Based on Multidisciplinary Diagnoses
|
Major idiopathic interstitial pneumonia
Idiopathic pulmonary fibrosis (IPF) Idiopathic nonspecific interstitial pneumonia (NSIP) Respiratory bronchiolitis-interstitial lung disease (RB-ILD) Desquamative interstitial pneumonia (DIP) Cryptogenic organizing pneumonia (COP) Acute interstitial pneumonia (AIP) |
|
Rare idiopathic interstitial pneumonias
Idiopathic lymphoid interstitial pneumonia (LIP) Idiopathic pleuroparenchymal fibroelastosis |
| Unclassifiable idiopathic interstitial pneumonia |
Table adapted from Travis, W.D., Costabel, U., Hansell, D.M., King, T.E., Lynch, D.A., Nicholson, A.G., Ryerson, C.J., Ryu, J.H., Selman, M., Wells, A.U., Behr, J., Bouros, D., Brown, K.K., Colby, T. V., Collard, H.R., Cordeiro, C.R., Cottin, V., Crestani, B., Drent, M., Dudden, R.F., Egan, J., Flaherty, K., Hogaboam, C., Inoue, Y., Johkoh, T., Kim, D.S., Kitaichi, M., Loyd, J., Martinez, F.J., Myers, J., Protzko, S., Raghu, G., Richeldi, L., Sverzellati, N., Swigris, J., Valeyre, D., 2013. An official American Thoracic Society/European Respiratory Society statement: Update of the international multidisciplinary classification of the idiopathic interstitial pneumonias. Am. J. Respir. Crit. Care Med. 188, 733–748.
Environmental and Occupational Causes of Pulmonary Fibrosis
Individuals exposed to metal particles, dust, and fumes are at higher risk of pulmonary fibrosis, presumably due to interference with the regular pulmonary inflammatory response to injury. However, the pathogenesis of this is poorly understood (7). In this section, some of the common causes of pulmonary fibrosis related to environmental exposures will be reviewed.
One of the most well-recognized occupational causes of pulmonary fibrosis is asbestosis. Asbestos refers to naturally occurring fibers used commercially in construction, shipbuilding, and the automotive industries due to its strength, flexibility, and resistance to degradation (8). Asbestos exposure can lead to myriad lung abnormalities, including lung cancer, mesothelioma, and pleural disease; when it causes ILD, the condition is known as “asbestos,” and can phenotypically be indistinguishable from other forms of ILD such as IPF, though the presence of pleural plaques is pathognomonic for asbestos exposures (8,9). Demonstration of asbestos fibers in the lung tissue also confirms suspected diagnoses ((8); Figure 2A).
A rare alkaline earth metal beryllium (Be) is used in high technology industries (aerospace, ceramics, electronics, nuclear defense) can cause ILD in susceptible individuals – this condition is known as chronic beryllium disease (CBD). Dependent on the individual patient’s genetic susceptibility as well as the type and duration of exposure, CBD develops in 1–16% of those with Be exposure (10). CBD is defined by non-caseating granulomatous inflammation in the lungs which can be indistinguishable from sarcoidosis – therefore, careful occupational/exposure history, as well as testing for sensitization, is required to make the diagnosis. A third of patients with CBD who go untreated will experience progressive respiratory failure (10–12). While Be exposure is a requirement for CBD development, extensive translational studies have shown that genetic susceptibility is central to disease pathogenesis. HLA-DPB1 alleles with a glutamic acid at position 69 on the beta-chain (BetaGlu69) are strongly linked to the disease, with the most common allele being HLA-DPB1*02:01 (10,13). Those with the BetaGlu69 polymorphism have a 10-fold increased risk of developing disease, and those CBD patients without this polymorphism have other alleles that lead to functionally important amino acid substitutions (10).
Hypersensitivity pneumonitis (HP), also known as extrinsic allergic alveolitis, refers to parenchymal lung disease characterized by inflammation of the lung parenchyma due to organic antigen inhalation. Numerous antigens have been identified and associated with HP, though the most common are avian and microbial agents (14). Acute forms of HP occur, but chronic HP develops in the setting of persistent exposure to the offending antigen. This leads to parenchymal abnormalities (frequently including pulmonary fibrosis; Figures 1C and 2C), progressive respiratory failure and, if no intervention is made, death. The radiologic and pathologic findings of HP can vary, but poorly formed granulomas on histopathology are a hallmark of this disease (Figure 2B) (14,15). In its chronic stages, HP can appear similar to other ILDs, making an exposure history particularly critical to accurate diagnosis.
Autoimmune Diseases Commonly Associated with Pulmonary Fibrosis
Many auto-immune diseases are associated with ILD and pulmonary fibrosis—for some patients, pulmonary fibrosis can be the presenting symptoms of their systemic autoimmune disease, and for many it is a source of significant morbidity. Systemic sclerosis (SSc) and rheumatoid arthritis (RA) are two such autoimmune diseases that frequently affect the lungs, though many others (such as systemic lupus erythematous, Sjogren’s Disease, and polymyositis, to name a few) can also cause pulmonary fibrosis (Figure 1B).
SSc, often called scleroderma, is an autoimmune disease in which pulmonary fibrosis is a frequent cause of mortality (16). In SSc, excess connective tissue (collagen) is deposited in numerous tissues, often most prominently in the skin, but also in the heart, microvasculature, and the lung. It is diagnosed through the detection of antinuclear antibodies in the serum (anti-RNA polymerase, anti-centromere, anti-topoisomerase [ATA]), each of which correspond to clinical subsets of disease. ATA, for instance, is also frequently called “anti-Scl-70”: patients with this phenotype are prone to lung fibrosis (17). Different pathologic findings are observed in SSc-ILD: most commonly nonspecific interstitial pneumonia (NSIP, 77.5% in one observational study), though Usual Interstitial Pneumonia (UIP), a specific pattern of fibrosis characteristic of IPF and associated with poor prognosis, is not uncommonly observed (15%) (18). Patients are treated with immunosuppressive medications, which sometimes leads to improvement or stabilization, but the clinical course is variable, and there are many patients with progressive and fatal lung disease.
Pulmonary involvement of some kind occurs in approximately 60–80% of patients with rheumatoid arthritis (RA). In terms of RA-ILD, the precise prevalence is unknown, and can range from 1–58% depending on the methodology utilized to detect it, and in a subset of cases RA affects the lungs before any joint manifestations are present (19). Unlike other autoimmune diseases, RA-ILD’s most common histopathologic pattern is UIP. Those with the UIP pattern tend to be older and have poorer prognoses (19–22). This prevalence of UIP pattern and other similarities between the diseases has led to comparisons between the two diseases in terms of genetic risk.
Idiopathic Pulmonary Fibrosis (IPF)
IPF is the most common and most severe of the IIPs, and therefore it has received the most attention in terms of pulmonary fibrosis research. Since the year 2000, a meta-analysis of epidemiology on IPF conservatively estimates an incidence range of 3–9 cases per 100,000 per year in North America and Europe, with a lower incidence in East Asia and South America (4). More recently, estimates of IPF prevalence in the United States has been estimated to be 18 per 100,000 persons (2008–2010) (23). In IPF, HRCT reveals interstitial fibrosis with a UIP pattern (Figure 1D) characterized by sub-pleural and basilar predominant reticular abnormality, honeycombing, and traction bronchiectasis with a relative paucity of ground glass (24,25). When HRCT pattern is not diagnostic, patients are referred to surgical lung biopsy—in IPF, histopathology will also reveal a UIP pattern, defined by honeycomb change, fibroblastic foci, and minimal inflammatory cells (Figure 2D).
IPF is a relentlessly progressive disease with most patients dying within 3–5 years of diagnosis (26,27). Though two drugs have been recently approved by the Federal Drug Administration (FDA) to treat IPF and have been shown to slow disease progression, they do not reverse pulmonary fibrosis (28,29), and lung transplantation remains the only “curative” therapy. It is more common in men than in women, is associated with smoking, and most patients present to clinical care between the ages of 50 and 75. The incidence of this disease had risen in recent years, but has plateaued between 2005–2010 (3,4,23).
As its name implies, the etiology of IPF remains an area of active research. Various hypotheses have been interrogated in terms of determinants of disease onset and progression, including recurrent aspiration (30) and abnormal or exuberant cell metabolism (31,32).However, over the past three decades, advances in genetic approaches, technology, and study design also have revealed that a significant proportion of disease risk is inherited. Early studies in Europe hinted at a genetic basis of disease, where they found that familial forms of disease accounted for 2–4% of cases (33–37). These familial forms of disease were often indistinguishable from the “sporadic” versions of IPF in terms of presenting radiology, clinical exam, and histopathology, other than the fact that familial patients often presented at earlier ages (35,38). One of the strongest individual risk factors for IPF is a family history of the disease (39,40).
Recent Advances in Genetics and Pulmonary Fibrosis
As described above, early epidemiologic studies had identified family history of disease as a risk factor for IPF. However, as with other forms of pulmonary fibrosis, inheritance patterns in this disease rarely follow Mendelian patterns of inheritance, suggesting a relationship between genetic and environmental factors in disease pathogenesis. Numerous genetic variants and mutations have been, over the past few decades, associated with IPF. In this review, we will focus on variants in surfactant-related proteins, telomere-pathway genes, and common variants more recently identified utilizing linkage and genome-wide association studies.
Translational Studies in Surfactant Proteins and IPF
The first IPF-associated variants identified were discovered in familial patients (41–44). Coding and noncoding mutations in the gene encoding surfactant protein C (SFTPC) as well as coding mutations in surfactant protein A were segregated with disease and were not found in controls (42,43). SPC, one of the main surfactant proteins expressed in human alveoli, functions by altering surface tension thereby preventing alveolar collapse. The protein is expressed throughout the epithelium during development, but in adult lung tissue it localizes to type II alveolar epithelial cells (AECs). SFTPC mutations have been linked to disease in adult and pediatric cases and was first reported in a family in which autosomal dominant inheritance was observed, leading to a lack of mature SPC in the lungs of parent and child (43). Other studies have illustrated that impairment or lack of SPC in the lungs of humans can itself cause fibrosis, potentially through impairing normal lung repair processes (45,46). Murine experiments also suggest that SPC deficiencies can lead to fibrosis in various animal models (42,47,48).
In subsequent studies, SFTPC variants have been commonly observed in pediatric cases of interstitial lung disease (49) as well as adult familial pulmonary fibrosis cohorts (50–55). Some authors have suggested that in specific cohorts, up to 25% of IPF patients with familial disease may have SFTPC mutations (55). However, in studies of sporadic IPF patients, SFTPC mutations are far less commonly found. Other surfactant proteins also associated with disease include surfactant protein A (SFTPA) where mutations in the carbohydrate recognition domain were found in numerous families with disease (44). As with surfactant protein C, these SFTPA mutations led to retention of precursor protein in the ER, leading to ER stress/UPR in AECs. Other surfactant-associated proteins, such as the ATP-binding cassette transporter (ABCA3) is expressed in AEC lamellar bodies, and have also been associated with surfactant deficiencies as well as interstitial lung disease in children (56,57) and teenagers (58), but these have not been found in adult pulmonary fibrosis mutations.
The mechanism through which SFTPC mutations and SPC abnormalities lead to fibrosis is not clear, but processing of the precursor protein (pro-SPC) and associated AEC dysfunction may drive pathogenesis (46,59). The pro-SPC molecule requires endoplasmic reticulum (ER) and secretory pathway processing before it can be delivered to the alveolar space. Many disease-associated mutations lead to the creation of pro-SPC molecules that cannot be appropriately processed, thereby creating protein accumulation in the ER and activation of the unfolded protein response (UPR) (55,60). This UPR has been associated with AEC injury (61–63).
A recent series of experiments in mice has provide insight into the role that SFTPC mutations and type II cells may be playing in human lung fibrosis. Investigators from the group that initially described a mutation in SFTPC leading to an isoleucine-to-threonine substitution at position 73 of the pro-protein created a mouse with a corresponding knock-in mutation (64). Mice expressing this abnormal SPC in their alveolar type II cells showed evidence of markedly abnormal post-translational processing, alveolar type II cell hyperplasia, evidence of spontaneous lung injury and remodeling including fibroblastic foci, hypoxemia, and abnormal macroautophagy (64). In addition, the mice had evidence of elevated levels of cytokines in their bronchoalveolar lavage fluid that have been associated with IPF in humans, suggesting that the pathways triggered by this mutant SPC in mice may reflect the biology observed in human disease (64).
Telomere Pathway Abnormalities and IPF
Telomeres are noncoding repetitive nucleotide (TTAGGG) sequences at the ends of chromosomes that protect them from degradation during cell replication. Telomerase is the complex of proteins and RNA that catalyzes the addition of these repeats to the ends of chromosomes. The telomerase complex includes the telomerase reverse transcriptase (TERT) and the telomerase RNA component (TERC), both of which are essential for normal function and integrity. Shortening of the telomere and associated genetic sequence variation, has been associated with myriad diseases (65), including pulmonary fibrosis. Numerous studies of familial and sporadic IPF cases and their kindred have identified disease-associated mutations in various telomere pathway genes (TERT, TERC, RTEL1, PARN, NAF1, DKC1, TINF2), and some studies suggest that TERT and TERC mutations can be observed in up to one-sixth of pulmonary fibrosis families (66,67).
The story of telomere-pathway genes and fibrosis begins with dyskeratosis congenital (DKC), a syndrome of abnormal skin pigmentation, nail dystrophy and oral leukoplakia that can also affect bone marrow and cause pulmonary fibrosis in 20% of cases (68). Though there are mutations in the gene DKC1 that are causative of this disease, other forms of disease were linked to TERT and TERC sequence variants (69–71), and in 2005 Armanios and colleagues discovered a TERT mutation that appears to be causative in a DKC family whose phenotype was predominated by pulmonary fibrosis (69). Subsequent study of familial IPF led to the discovery of TERT and TERC mutations associated with affected family members; not only did disease-associated variants segregate with disease, but also these identified mutations demonstrated decreased telomerase activity and telomere shortening in peripheral blood lymphocytes (66).
Other groups reported similar findings in separate familial IPF cohorts. Utilizing linkage analysis in highly informative families, Tsakiri and colleagues were able to zero-in on TERT as a candidate gene, thereby discovering two separate coding mutations in adult FIP families (67). Just as the other investigators had found, Tsakiri’s study also identified that these mutations were associated with decreased enzymatic activity and telomere length; they subsequently also identified a TERT mutation in a sporadic case of ILD, but were unable to find it in ethnically matched control subjects (67).
Based on these findings, investigators then examined the relationship between telomere length itself and pulmonary fibrosis, independent of genetic mutation (72,73). In both familial and sporadic forms of disease, about a quarter of subjects showed evidence of telomere shortening, with peripheral blood leukocyte telomere length below the 10th percentile for age-matched controls. Additionally, lung tissue examination also revealed AECs with shortened telomeres (72). Therefore, even without known coding mutations in the relevant genes, telomeres in pulmonary fibrosis patients appeared to be shorter than age-matched controls. Furthermore, extrapulmonary manifestations of so-called “telomeropathy” were noted, including aplastic anemia and liver cirrhosis in a subset of patients (72,74,75). More recent work has utilized exome-sequencing techniques to identify rare variants in other genes within the telomerase pathway. Variants in RTEL1 and PARN have been associated with familial IPF (34,76,77), and as with the TERT and TERC mutation carriers, affected subjects with these sequence variants showed evidence of shortened telomeres in peripheral blood leukocytes.
Due to the many clinical commonalities noted between RA-ILD and IPF, telomere pathway genes have been examined in familial pulmonary fibrosis and RA-ILD. A French study recently discovered that subjects with RA-ILD carried mutations in coding regions of TERT, RTEL1, PARN, and SFTPC, genes that had been identified in studies of families enriched for pulmonary fibrosis not associated with RA (78). Not only were these coding mutations prevalent in their RA-ILD cohort, but the telomeres for these patients were also shortened, suggesting that the mutations were causing functional abnormalities in telomerase (78).
Though the genetic epidemiology of these variants suggest a strong link between telomerase and pulmonary fibrosis, the precise mechanism of this connection remains unknown. However, in vivo studies suggest that perhaps these variants lead to impaired injury response in the pulmonary epithelium (79).
Common Variants and IPF: MUC5B and Others
More recent studies of the genetic basis of IPF have leveraged larger cohorts of subject with disease to find more common disease-associated variants. “Common variants” are defined by the frequency of the less common (minor) allele—usually, common variants are defined in any individual study as those with a minor allele frequency (MAF) in the relevant population > 0.05.
In 2008, the first genome-wide association study (GWAS) in IPF was performed in Japan, which identified a common variant (rs2736100) in intron 2 of TERT associated with IPF risk (80). Utilizing a much larger cohort of affected subjects in 2011, Seibold and colleagues performed a linkage analysis followed by targeted sequencing and genotyping to identify a single nucleotide polymorphism (SNP) on chromosome 11 in the promoter region of the MUC5B gene (rs35705950) associated with both familial IPF and sporadic IPF. MUC5B codes for mucin 5B, a major component of mucin found in the lung as well as cervix and saliva in humans. Using case-control analyses of non-Hispanic whites (NHWs), the authors determined that those with the T allele had significantly increased odds ratios (ORs) for disease (81). Because this study utilized both sporadic and familial cases, they were able to note that the MAF for sporadic disease (0.375) was similar to that for familial disease (0.338). Lung tissue from IPF subjects and controls showed that subjects with IPF had a 14-fold increase in MUC5B gene expression. Unaffected subjects with the T-allele also had a 37-fold increase in gene expression. Also, the mucin was located on immunopathology to the honeycomb cyst, a pathologic hallmark of the disease (81,82).
The rs35705950 disease-association has been replicated by numerous groups (6,83–89), making it the strongest and most well-replicated single genetic risk factor for disease. However, it should be noted that in the NHW population, a T allele at this position is common and found in about 19% of individuals (MAF=0.09) without disease. Since IPF does not occur in 19% of NHWs, the presence of the variant alone is insufficient to cause disease – and since about half of subjects with IPF do not have the variant either, it is also not necessary for disease. Therefore, there is likely synergy between other factors (inherited or environmental) and rs35705950 driving disease pathogenesis—this remains an area of active research. Furthermore, studies of various ethnic groups indicate that the role of rs35705950 in disease risk may be group-specific. It is a risk factor for IPF in Mexican cohorts, but is rare in Korean cohorts (89). Overall, the MAF for rs35705950 appears to correlate with the prevalence of IPF in any given population – NHWs are at higher risk in general for developing disease compared to Hispanics and Asians, and the disease is thought to be rare in African populations (90) in which the MAF is vanishingly low (91). Therefore, the role of rs35705950 or any other genetic variant in disease risk is likely to be specific to different ethnic groups.
Initially, the relationship of the MUC5B promoter polymorphism to disease risk appeared to be quite specific to IPF and not to other processes that can result in similar pulmonary fibrosis. Studies of systemic sclerosis-ILD (84,86), sarcoidosis (84), chronic obstructive pulmonary disease, and asthma have not shown significant associations between the MUC5B promoter polymorphism and disease (92). However, a more recent study of two separate cohorts of chronic hypersensitivity pneumonitis (HP) showed that the minor allele at rs35705950 was associated with disease and that the MAFs observed in the cohorts were similar to those observed in other IPF cohorts (93), thereby linking the genetics of chronic HP and IPF. Subsequently, rheumatoid arthritis ILD (RA-ILD) cohorts have been examined—and the rs35705950 minor allele is more common in RA patients that develop ILD, in particular ILD with the UIP fibrosis pattern characteristic of IPF (94). Therefore, the association between the MUC5B promoter polymorphism and disease appears to be specific to select forms of pulmonary fibrosis (IPF, RA-ILD, and chronic HP), all of which are most frequently characterized by the UIP pattern of fibrosis on HRCT imaging or histopathology. Given the link between UIP and MUC5B, it may be the case that the rs35705950 predisposes individuals to this specific UIP pattern of fibrotic response to recurrent lung injury.
GWAS studies have identified risk loci for IPF other than the MUC5B locus on chromosome 11, though the rs35705950 variant remains the most well-replicated and strongest single genetic association with disease. In 2013, a case-control GWAS was performed in 1,616 subjects with fibrotic IIP and 4,683 control subjects without lung disease. This study confirmed previously identified risk loci (MUC5B, TERT, TERC), but also identified new loci and genes associated with disease: FAM13A at 4q22, DSP at 6p24, OBFC1 at 10q24, ATP11A at 13q4, DPP9 at 19q13, and loci without specific gene associations at 7q22 and 15q14–15 (5). It is estimated that variation at these loci may account for up to one third of disease risk (5,95). This GWAS was significant as it implicated genes associated with varied biological processes (host defense, DNA repair, cell-cell adhesion (5,96,97), thereby suggesting that the pathogenesis of disease may be multifaceted or that the disease may have numerous endotypes.
A separate GWAS confirmed the MUC5B promoter polymorphism association with IPF but also identified other risk-variants in TOLLIP and SPPL2C (98). In addition to identifying new risk variants for IPF, the study also illustrated differential mortality and potentially also differential therapeutic responses associated with variants in TOLLIP (98,99).
How do rare and common variants interact in terms of disease risk?
The studies described thus far have generally examined the role of individual variants and IPF risk. But the ways in which individual variants interact in order to influence disease risk remains poorly understood. Do common and rare variants have additive effects in terms of disease risk? How do different variants interact or modify risk? What is the link between specific variants and the biology of IPF? These remain areas of active research. Of note, one recent study examined rare protein-alerting variants in telomere-related genes (TERT, PARN, TERC, RTEL1) and the common MUC5B promoter polymorphism in IPF patients and found that IPF patients without the rs35705950 variant had a higher frequency of telomere-related variants (3% versus 7%, p=0.0004) (100). These findings suggest that telomere-variant related disease may be distinct in most patients compared to MUC5B related disease—though the significance of having both the MUC5B risk allele and a TERT variant remains unclear.
The significant and growing body of data on the genetic epidemiology of pulmonary fibrosis suggests that there are specific sequence variants that play a role in disease development, but that in many cases, these sequence variants themselves are not sufficient to cause disease. Given that common variants have repeatedly shown to be associated with a relatively rare disease, specific genetic variants may prime the pulmonary parenchyma to be prone to a fibrotic response to environmental insult or to systemic disease and inflammation.
The precise mechanism by which the common promoter polymorphism in MUC5B leads to increased risk of pulmonary fibrosis is an active area of research. In vivo and in vitro experiments indicate that overexpression of Muc5b in murine lung cells leads to impaired ciliary function (101). Transgenic overexpressing mice illustrated increased fibrotic response to intratracheal injury than their littermates, but did not develop spontaneous fibrosis; intriguingly, this sensitivity to injury was attenuated by the administration of a novel mucolytic compound (101). These data suggest a potential mechanism through which MUC5B polymorphisms might increase individual risk of fibrosis, but also identify a novel avenue for therapeutic investigation in this disease, mucolysis.
Sub-phenotyping Disease in IPF
While the link between many of the genetic risk variants described above and disease pathogenesis, large observational studies suggest that IPF, once thought to be a monolithic diagnosis, has numerous endotypes. In particular, telomere length, MUC5B genotype and ciliary gene expression are some of the potential genetic and genomic markers that show promise in helping to subcharacterize patients.
Disease Severity
An analyses of clinical trial data showed that IPF subjects with the minor allele (T) at the MUC5B promoter polymorphism rs35705950 had better survival compared to those with the GG (wild-type) genotype even when controlling for relevant covariates such as age, sex, presenting lung function, and treatment group (102). This is somewhat unexpected in that the MUC5B variant increases risk of disease, as the prior case-control studies had shown (5,6), and yet the disease phenotype for these individuals appears to be less severe than non-MUC5B variant forms of disease (102). A similar analysis was performed for a different common IPF risk variant in TOLLIP by Noth and colleagues (85) who found that the rs5743890 is associated with differential survival. The major allele (G) at rs5743890 is associated with decreased disease severity within diseased subjects, while minor allele carriers with disease had increased mortality (85). However, the relationship between the MUC5B risk variant and improved survival may be accounted for inclusion of subjects with disease that are biased due to the commonality of the genetic risk (103)—therefore, future prospective studies examining survival and its relationship to specific genotypes are needed to understand how common risk variants could be utilized in prognostication.
Telomere mutations and length are also associated with distinct but varied disease courses when compared to generalized cohorts of patients with IPF. In subjects with known telomerase pathway mutations, the prevalence of ILD increases with age – so much so that in TERT mutation carriers over the age of 60 had a prevalence of 60% (104,105). An observational study of pulmonary fibrosis patients with telomerase pathway mutations (in genes TERC, TERT, RTEL1, and PARN) found that those with TERC mutations were diagnosed at an earlier age relative to the other study subjects (106). This study also showed that the decline in lung function observed in large clinical trials of IPF was slower than those with telomerase pathway mutations regardless of the sub-phenotyping of their pulmonary fibrosis (106). This was a particularly important finding given that many of the patients with pulmonary fibrosis and telomerase-related mutations had clinical diagnoses other than IPF—only 46% had a clinical diagnosis of IPF, and others were labeled as hypersensitivity pneumonitis, pleuroparenchymal fibroelastosis, undifferentiated fibrosis, and ILD related to an underlying connective tissue disease (106). Despite this clinical heterogeneity observed in the pulmonary fibrosis phenotypes of these telomerase mutation carriers, the authors found that the disease was uniformly progressive and lethal (106).
Telomere mutations are associated with telomere shortening—but shortened telomeres themselves seem to associate with disease, implicating them in disease pathogenesis. Adjusting for age, sex, and severity of illness, telomere length has been found to be a predictor of transplant-free survival in IPF patients (107). Interestingly, this effect was found in IPF subjects but not in those with non-IPF ILD; in addition, the frequency of rare TERT mutations associated with IPF was low in the cohort (2–6%), suggesting that genetic variants outside of TERT, epigenetic changes, or other environmental factors could be responsible for the short telomeres in these patients (107).
Other studies sub-phenotyping IPF have utilized genomic technologies and focused on gene expression studies. In one study of lung tissue gene expression, differences in the expression of cilium related genes identified two sub-phenotypes of disease. This with high cilium gene expression showed more microscopic honeycombing and elevated tissue expression of MUC5B and MMP7 (108). A separate group has utilized peripheral blood mononuclear cell (PBMC) gene expression profiles, identifying a 52-gene signature that could predict outcomes in terms of transplant free survival (109,110).
Response to Medical Therapies and Lung Transplantation
In addition, a follow-up study examining a handful of specific IPF risk-variants in cohorts from the PANTHER-IPF trial (111) indicated that a different TOLLIP risk-variant (rs3750920) was associated with a differential response to pharmacotherapy, specifically to N-acetylcysteine (99), a medication that for all-comers with IPF is not associated with improved outcomes (112). In this study, those with the TT genotype at rs3750920 had decreased risk of the original trial’s composite endpoint (death, transplantation, hospitalization, and greater than 10% decrease in FVC), while those with the CC genotype at the same location had increased risk of the same (99). Other medications currently approved for the treatment of IPF (nintedanib (28) and pirfenidone (29)) have not been examined by genotype.
For candidates with the appropriate age and comorbidities, lung transplantation is the remaining therapeutic option for patients with advanced disease. However, given that telomeropathies are associated with numerous extra-pulmonary manifestations (cytopenias, liver cirrhosis, early graying of the hair) and that post-transplant management can include many non-transplant toxicities, investigators have examined telomerase pathway mutation carriers for differences in post-transplant outcomes. Indeed, retrospective analyses of telomerase mutation carriers in pulmonary fibrosis cohorts has revealed a greater incidence of post-transplant complications when these are compared to historical controls (113,114). It is thought that these observed differences in post-transplant complications may be driven by the hematologic abnormalities that could be secondary to bone marrow dysfunction described in telomeropathies (115). Larger more recent study of telomere length in post-transplant pulmonary fibrosis patients suggests that having telomere lengths < 10th percentile is associated with poor survival and faster onset of chronic allograft dysfunction—though no differences in acute rejection, cytopenias, infection, or renal dysfunction were noted (116).
These differences in clinical outcomes and response to available therapies certainly suggest that genetic and genomic techniques could be integrated into the clinical care of patients. Many of these relationships between genotype and genetics require validation in prospective studies, and the interpretation of clinical trials should require an assessment of phenotypic and genotypic variation within cohorts that clinicians generally lump together as “IPF.” Given the potential prognostic implications of some of these findings, their validation is important so that they can be integrated into shared decision-making with patients navigating clinical choices and multi-faceted lives. At this time, genotyping and sequencing are not generally part of routine care for IPF or for other forms of pulmonary fibrosis.
Genetic Variants in Non-IPF Forms of Pulmonary Fibrosis
The bulk of the research on genetics and pulmonary fibrosis has focused on IPF disease risk. However, as described with respect to the common MUC5B promoter polymorphism, there is also data suggesting an important relationship between inherited factors and non-IPF pulmonary fibrosis. Earlier studies revealed associations between the major histocompatibility complex (MHC) loci and HP in populations with different genetic backgrounds (117–120); additionally, polymorphisms in TIMP-3, TNFα, TAP genes, and PSMB8 have been associated with disease (118,121–125). Similarly, SSc, which frequently manifests as pulmonary fibrosis has also been associated with polymorphisms in genes involved in the immune system, such as CD247, TBX21, IRF5, STAT4, the MHC, and portions of the T-cell receptor (126–131). In the case of IRF5, genotype at the rs2004640 variant was associated with SSc-ILD (128).
Earlier Identification of Pulmonary Fibrosis
The therapeutic options for patients with IPF remain limited—indeed, approved anti-fibrotics do not reverse disease, but instead slow down the rate of progression towards fatal respiratory failure, and lung transplantation is not available to many patients with this disease (28,29). Additionally, patients are often finally diagnosed with pulmonary fibrosis with advanced stage disease (27,90). Therefore, an active area of research in pulmonary fibrosis is the development of early detection and risk-stratification.
Early lung abnormalities can be identified on CT scans of the chest (Figure 1A). A study utilizing the Framingham Heart Study cohort found that the MUC5B promoter polymorphism rs35705950 is associated with radiographic evidence of interstitial lung abnormalities (ILAs), defined as radiologic evidence of some form of parenchymal lung abnormality (e.g., ground glass, centrilobular nodularity, cysts, honeycombing, traction bronchiectasis, reticular abnormality), in non-Hispanic white subjects. They were 2.8 times more common for each copy of the rs35705950 T allele (132). While ILAs are not equivalent to lung fibrosis, they are associated with increased mortality and pulmonary-related mortality (133,134). The association between the rs35705950 variant and ILAs is intriguing as it suggests a link between the genetic predisposition to ILA and the genetic predisposition to clinically meaningful fibrotic lung disease, IPF. The relationship between ILAs and clinically relevant pulmonary fibrosis continues to be studied, and further research will be needed to determine whether common genetic variants such as the MUC5B promoter polymorphism can assist in determining the significance of finding ILA in any given individual.
Conclusion
There is a growing body of evidence that pulmonary fibrosis develops due to an interplay between host factors and environmental and behavioral exposures. Further translational research in this field will be required in order to understand the biological mechanisms connecting genetic and genomic factors with disease pathophysiology. In addition, an understanding between biological host factors and environmental and behavioral exposures will be required in order to develop personalized approaches to this progressive and fatal disease.
Acknowledgements
The authors would like to thank Dr. Haiying Zhang (Department of Pathology, Baylor University Medical Center at Dallas) and Dr. Christopher J.G. Sigakis (Columbus Radiology/Radiology Partners) for their assistance with preparing the figures included in this manuscript. Dr. David Schwartz’s research is supported by the National Institutes of Health (NHLBI: P01 HL092870, R01 HL097163, R33 HL120770, UH2 HL123442) and the United States Department of Defense (W81XWH-17-1-0597). Dr. Schwartz is also an employee of Eleven P15, Inc., and has received personal fees from NuMedii, Inc., outside the submitted work.
Abbreviations:
- IPF
Idiopathic pulmonary fibrosis
- IPP
idiopathic interstitial pneumonia
- ILD
interstitial lung disease
Footnotes
All authors have read the journal’s authorship agreement and policy on disclosure of potential conflicts of interest.
References
- 1.Raghu G, Remy-Jardin M, Myers JL, Richeldi L, Ryerson CJ, Lederer DJ, et al. Diagnosis of idiopathic pulmonary fibrosis An Official ATS/ERS/JRS/ALAT Clinical practice guideline. Am J Respir Crit Care Med 2018;198(5):e44–68. [DOI] [PubMed] [Google Scholar]
- 2.Travis WD, Costabel U, Hansell DM, King TE, Lynch DA, Nicholson AG, et al. An official American Thoracic Society/European Respiratory Society statement: Update of the international multidisciplinary classification of the idiopathic interstitial pneumonias. Am J Respir Crit Care Med 2013;188(6):733–48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Hutchinson JP, McKeever TM, Fogarty AW, Navaratnam V, Hubbard RB. Increasing global mortality from idiopathic pulmonary fibrosis in the twenty-first century. Ann Am Thorac Soc 2014;11(8):1176–85. [DOI] [PubMed] [Google Scholar]
- 4.Hutchinson J, Fogarty A, Hubbard R, McKeever T. Global incidence and mortality of idiopathic pulmonary fibrosis: a systematic review. Eur Respir J 2015. Sep;46(3):795–806. [DOI] [PubMed] [Google Scholar]
- 5.Fingerlin TE, Murphy E, Zhang W, Peljto AL, Brown KK, Steele MP, et al. Genome-wide association study identifies multiple susceptibility loci for pulmonary fibrosis. Nat Genet. Nature Publishing Group; 2013. Jun;45(6):613–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Seibold MA, Wise A, Speer M, Steele M, Brown K, Lloyd JE, et al. A common MUC5B promoter polymorphism and pulmonary fibrosis. N Engl J Med 2011;364(16):1503–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Assad N, Sood A, Campen MJ, Zychowski KE. Metal-Induced Pulmonary Fibrosis. Curr Environ Heal Reports. Current Environmental Health Reports; 2018;(3):1–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Gulati M, Redlich CA. Asbestosis and environmental causes of usual interstitial pneumonia. Curr Opin Pulm Med 2015. Mar;21(2):193–200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Vehmas T, Oksa P. Chest HRCT signs predict deaths in long-term follow-up among asbestos exposed workers. Eur J Radiol 2014. Oct;83(10):1983–7. [DOI] [PubMed] [Google Scholar]
- 10.Fontenot AP, Falta MT, Kappler JW, Dai S, McKee AS. Beryllium-Induced Hypersensitivity: Genetic Susceptibility and Neoantigen Generation. J Immunol 2016;196(1):22–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Fontenot AP, Kotzin BL. Chronic beryllium disease: immune-mediated destruction with implications for organ-specific autoimmunity. Tissue Antigens 2003. Dec;62(6):449–58. [DOI] [PubMed] [Google Scholar]
- 12.Balmes JR, Abraham JL, Dweik RA, Fireman E, Fontenot AP, Maier LA, et al. An official American Thoracic Society statement: diagnosis and management of beryllium sensitivity and chronic beryllium disease. Am J Respir Crit Care Med 2014. Nov 15;190(10):e34–59. [DOI] [PubMed] [Google Scholar]
- 13.Richeldi L, Sorrentino R, Saltini C. HLA-DPB1 glutamate 69: a genetic marker of beryllium disease. Science 1993. Oct 8;262(5131):242–4. [DOI] [PubMed] [Google Scholar]
- 14.Spagnolo P, Rossi G, Cavazza A, Bonifazi M, Paladini I, Bonella F, et al. Hypersensitivity Pneumonitis: A Comprehensive Review. J Investig Allergol Clin Immunol 2015;25(4):237–50; quiz follow 250. [PubMed] [Google Scholar]
- 15.Silva CIS, Müller NL, Lynch DA, Curran-Everett D, Brown KK, Lee KS, et al. Chronic hypersensitivity pneumonitis: differentiation from idiopathic pulmonary fibrosis and nonspecific interstitial pneumonia by using thin-section CT. Radiology 2008. Jan;246(1):288–97. [DOI] [PubMed] [Google Scholar]
- 16.Tochimoto A, Kawaguchi Y, Yamanaka H. Genetic Susceptibility to Interstitial Lung Disease Associated with Systemic Sclerosis. Clin Med Insights Circ Respir Pulm Med 2015;9s1:135–40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Briggs DC, Vaughan RW, Welsh KI, Myers A, duBois RM, Black CM. Immunogenetic prediction of pulmonary fibrosis in systemic sclerosis. Lancet 1991;338(8768):661–2. [DOI] [PubMed] [Google Scholar]
- 18.Bouros D, Wells AU, Nicholson AG, Colby TV., Polychronopoulos V, Pantelidis P, et al. Histopathologic subsets of fibrosing alveolitis in patients with systemic sclerosis and their relationship to outcome. Am J Respir Crit Care Med 2002;165(12):1581–6. [DOI] [PubMed] [Google Scholar]
- 19.Spagnolo P, Lee JS, Sverzellati N, Rossi G, Cottin V. The Lung in Rheumatoid Arthritis: Focus on Interstitial Lung Disease. Arthritis Rheumatol 2018;70(10):1544–54. [DOI] [PubMed] [Google Scholar]
- 20.Kim EJ, Elicker BM, Maldonado F, Webb WR, Ryu JH, Van Uden JH, et al. Usual interstitial pneumonia in rheumatoid arthritis-associated interstitial lung disease. Eur Respir J 2010;35(6):1322–8. [DOI] [PubMed] [Google Scholar]
- 21.Chen ES, Wahlström J, Song Z, Willett MH, Wikén M, Yung RC, et al. T cell responses to mycobacterial catalase-peroxidase profile a pathogenic antigen in systemic sarcoidosis. J Immunol 2008. Dec 15;181(12):8784–96. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Yunt ZX, Chung JH, Hobbs S, Fernandez-Perez ER, Olson AL, Huie TJ, et al. High resolution computed tomography pattern of usual interstitial pneumonia in rheumatoid arthritis-associated interstitial lung disease: Relationship to survival. Respir Med 2017;126:100–4. [DOI] [PubMed] [Google Scholar]
- 23.Raghu G, Chen SY, Hou Q, Yeh WS, Collard HR. Incidence and prevalence of idiopathic pulmonary fibrosis in US adults 18–64 years old. Eur Respir J 2016;48(1):179–86. [DOI] [PubMed] [Google Scholar]
- 24.Lynch DA, Sverzellati N, Travis WD, Brown KK, Colby TV., Galvin JR, et al. Diagnostic criteria for idiopathic pulmonary fibrosis: A Fleischner Society White Paper. Lancet Respir Med 2017;2600(17):1–16. [DOI] [PubMed] [Google Scholar]
- 25.Raghu G, Collard HR, Egan JJ, Martinez FJ, Behr J, Brown KK, et al. An Official ATS/ERS/JRS/ALAT Statement: Idiopathic pulmonary fibrosis: Evidence-based guidelines for diagnosis and management. Am J Respir Crit Care Med 2011;183(6):788–824. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Bjoraker JA, Ryu JH, Edwin MK, Myers JL, Tazelaar HD, Schroeder DR, et al. Prognostic significance of histopathologic subsets in idiopathic pulmonary fibrosis. Am J Respir Crit Care Med 1998. Jan;157(1):199–203. [DOI] [PubMed] [Google Scholar]
- 27.Ley B, Collard HR, King TE. Clinical course and prediction of survival in idiopathic pulmonary fibrosis. Am J Respir Crit Care Med 2011. Feb 15;183(4):431–40. [DOI] [PubMed] [Google Scholar]
- 28.Richeldi L, du Bois RM, Raghu G, Azuma A, Brown KK, Costabel U, et al. Efficacy and safety of nintedanib in idiopathic pulmonary fibrosis. N Engl J Med 2014;370(22):2071–82. [DOI] [PubMed] [Google Scholar]
- 29.King TE, Bradford WZ, Castro-Bernardini S, Fagan EA, Glaspole I, Glassberg MK, et al. A phase 3 trial of pirfenidone in patients with idiopathic pulmonary fibrosis. N Engl J Med 2014;370(22):2083–92. [DOI] [PubMed] [Google Scholar]
- 30.Savarino E, Carbone R, Marabotto E, Furnari M, Sconfienza L, Ghio M, et al. Gastro-oesophageal reflux and gastric aspiration in idiopathic pulmonary fibrosis patients. Eur Respir J 2013;42(5):1322–31. [DOI] [PubMed] [Google Scholar]
- 31.Win T, Screaton NJ, Porter JC, Ganeshan B, Maher TM, Fraioli F, et al. Pulmonary 18F-FDG uptake helps refine current risk stratification in idiopathic pulmonary fibrosis (IPF). Eur J Nucl Med Mol Imaging. European Journal of Nuclear Medicine and Molecular Imaging; 2018;45(5):806–15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Justet A, Laurent-Bellue A, Thabut G, Dieudonné A, Debray MP, Borie R, et al. [18F]FDG PET/CT predicts progression-free survival in patients with idiopathic pulmonary fibrosis. Respir Res. Respiratory Research; 2017;18(1):1–10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Hodgson U, Laitinen T, Tukiainen P. Nationwide prevalence of sporadic and familial idiopathic pulmonary fibrosis: Evidence of founder effect among multiplex families in Finland. Thorax 2002;57(4):338–42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Cogan JD, Kropski JA, Zhao M, Mitchell DB, Rives L, Markin C, et al. Rare variants in RTEL1 are associated with familial interstitial pneumonia. Am J Respir Crit Care Med 2015. Mar 15;191(6):646–55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Marshall RP, Puddicombe a, Cookson WO, Laurent GJ Adult familial cryptogenic fibrosing alveolitis in the United Kingdom. Thorax 2000;55(2):143–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Lawson WE, Loyd JE. The genetic approach in pulmonary fibrosis: can it provide clues to this complex disease? Proc Am Thorac Soc 2006. Jun;3(4):345–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Loyd JE. Pulmonary fibrosis in families. Am J Respir Cell Mol Biol 2003. Sep;29(3 Suppl):S47–50. [PubMed] [Google Scholar]
- 38.Kropski JA, Mitchell DB, Markin C, Polosukhin VV., Choi L, Johnson JE, et al. A novel dyskerin ( DKC1 ) mutation is associated with familial interstitial pneumonia. Chest 2014;146(1). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.García-Sancho C, Buendía-Roldán I, Fernández-Plata MR, Navarro C, Pérez-Padilla R, Vargas MH, et al. Familial pulmonary fibrosis is the strongest risk factor for idiopathic pulmonary fibrosis. Respir Med 2011;105(12):1902–7. [DOI] [PubMed] [Google Scholar]
- 40.Bitterman PB, Rennard SI, Keogh BA, Wewers MD, Adelberg S, Crystal RG. Familial idiopathic pulmonary fibrosis. Evidence of lung inflammation in unaffected family members. N Engl J Med 1986;314(21):1343–7. [DOI] [PubMed] [Google Scholar]
- 41.Fernandez BA, Fox G, Bhatia R, Sala E, Noble B, Denic N, et al. A Newfoundland cohort of familial and sporadic idiopathic pulmonary fibrosis patients: clinical and genetic features. Respir Res. Respiratory Research; 2012;13(1):64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Lawson WE, Grant SW, Ambrosini V, Womble KE, Dawson EP, Lane KB, et al. Genetic mutations in surfactant protein C are a rare cause of sporadic cases of IPF. Thorax 2004;59(11):977–80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Nogee LM, Dunbar AE, Wert SE, Askin F, Hamvas A, Whitsett JA. A mutation in the surfactant protein C gene associated with familial interstitial lung disease. N Engl J Med 2001;344(8):573–9. [DOI] [PubMed] [Google Scholar]
- 44.Wang Y, Kuan PJ, Xing C, Cronkhite JT, Torres F, Rosenblatt RL, et al. Genetic Defects in Surfactant Protein A2 Are Associated with Pulmonary Fibrosis and Lung Cancer. Am J Hum Genet. The American Society of Human Genetics; 2009;84(1):52–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Amin RS, Wert SE, Baughman RP, Tomashefski JF, Nogee LM, Brody AS, et al. Surfactant protein deficiency in familial interstitial lung disease. J Pediatr 2001. Jul;139(1):85–92. [DOI] [PubMed] [Google Scholar]
- 46.Whitsett JA, Weaver TE. Hydrophobic surfactant proteins in lung function and disease. N Engl J Med 2002. Dec 26;347(26):2141–8. [DOI] [PubMed] [Google Scholar]
- 47.Glasser SW, Detmer EA, Ikegami M, Na C-L, Stahlman MT, Whitsett JA. Pneumonitis and emphysema in sp-C gene targeted mice. J Biol Chem 2003. Apr 18;278(16):14291–8. [DOI] [PubMed] [Google Scholar]
- 48.Glasser SW, Eszterhas SK, Detmer E a, Maxfield MD, Korfhagen TR. The murine SP-C promoter directs type II cell-specific expression in transgenic mice. Am J Physiol Lung Cell Mol Physiol 2005. Apr;288(4):L625–32. [DOI] [PubMed] [Google Scholar]
- 49.Kröner C, Reu S, Teusch V, Schams A, Grimmelt AC, Barker M, et al. Genotype alone does not predict the clinical course of SFTPC deficiency in paediatric patients. Eur Respir J 2015;46(1):197–206. [DOI] [PubMed] [Google Scholar]
- 50.Brasch F, Birzele J, Ochs M, Guttentag SH, Schoch OD, Boehler A, et al. Surfactant proteins in pulmonary alveolar proteinosis in adults. Eur Respir J 2004. Sep;24(3):426–35. [DOI] [PubMed] [Google Scholar]
- 51.Cameron HS, Somaschini M, Carrera P, Hamvas A, Whitsett JA, Wert SE, et al. A common mutation in the surfactant protein C gene associated with lung disease. J Pediatr 2005. Mar;146(3):370–5. [DOI] [PubMed] [Google Scholar]
- 52.Hamvas A, Nogee LM, White FV, Schuler P, Hackett BP, Huddleston CB, et al. Progressive lung disease and surfactant dysfunction with a deletion in surfactant protein C gene. Am J Respir Cell Mol Biol 2004. Jun;30(6):771–6. [DOI] [PubMed] [Google Scholar]
- 53.Nogee LM, Dunbar AE, Wert S, Askin F, Hamvas A, Whitsett JA. Mutations in the surfactant protein C gene associated with interstitial lung disease. Chest 2002. Mar;121(3 Suppl):20S–21S. [DOI] [PubMed] [Google Scholar]
- 54.Tredano M, Griese M, Brasch F, Schumacher S, de Blic J, Marque S, et al. Mutation of SFTPC in infantile pulmonary alveolar proteinosis with or without fibrosing lung disease. Am J Med Genet A 2004. Apr 1;126A(1):18–26. [DOI] [PubMed] [Google Scholar]
- 55.Van Moorsel CHM, Van Oosterhout MFM, Barlo NP, De Jong P a., Van Der Vis JJ, Ruven HJT, et al. Surfactant protein C mutations are the basis of a significant portion of adult familial pulmonary fibrosis in a dutch cohort. Am J Respir Crit Care Med 2010;182(11):1419–25. [DOI] [PubMed] [Google Scholar]
- 56.Bullard JE, Wert SE, Whitsett JA, Dean M, Nogee LM. ABCA3 mutations associated with pediatric interstitial lung disease. Am J Respir Crit Care Med 2005. Oct 15;172(8):1026–31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Bullard JE, Nogee LM. Heterozygosity for ABCA3 mutations modifies the severity of lung disease associated with a surfactant protein C gene (SFTPC) mutation. Pediatr Res 2007. Aug;62(2):176–9. [DOI] [PubMed] [Google Scholar]
- 58.Young LR, Nogee LM, Barnett B, Panos RJ, Colby TV, Deutsch GH. Usual interstitial pneumonia in an adolescent with ABCA3 mutations. Chest 2008. Jul;134(1):192–5. [DOI] [PubMed] [Google Scholar]
- 59.Beers MF, Mulugeta S. Surfactant protein C biosynthesis and its emerging role in conformational lung disease. Annu Rev Physiol 2005;67:663–96. [DOI] [PubMed] [Google Scholar]
- 60.Lawson WE, Crossno PF, Polosukhin VV, Roldan J, Cheng D-S, Lane KB, et al. Endoplasmic reticulum stress in alveolar epithelial cells is prominent in IPF: association with altered surfactant protein processing and herpesvirus infection. Am J Physiol Lung Cell Mol Physiol 2008. Jun;294(6):L1119–26. [DOI] [PubMed] [Google Scholar]
- 61.Bridges JP, Wert SE, Nogee LM, Weaver TE. Expression of a human surfactant protein C mutation associated with interstitial lung disease disrupts lung development in transgenic mice. J Biol Chem 2003. Dec 26;278(52):52739–46. [DOI] [PubMed] [Google Scholar]
- 62.Mulugeta S, Maguire JA, Newitt JL, Russo SJ, Kotorashvili A, Beers MF. Misfolded BRICHOS SP-C mutant proteins induce apoptosis via caspase-4- and cytochrome c-related mechanisms. Am J Physiol Lung Cell Mol Physiol 2007. Sep;293(3):L720–9. [DOI] [PubMed] [Google Scholar]
- 63.Mulugeta S, Nguyen V, Russo SJ, Muniswamy M, Beers MF. A surfactant protein C precursor protein BRICHOS domain mutation causes endoplasmic reticulum stress, proteasome dysfunction, and caspase 3 activation. Am J Respir Cell Mol Biol 2005. Jun;32(6):521–30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Nureki S-I, Tomer Y, Venosa A, Katzen J, Russo SJ, Jamil S, et al. Expression of mutant Sftpc in murine alveolar epithelia drives spontaneous lung fibrosis. J Clin Invest 2018;128(9):4008–24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Armanios M, Blackburn EH. The telomere syndromes. Nat Rev Genet 2012;13(10):693–704. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Armanios MY, Chen JJ-L, Cogan JD, Alder JK, Ingersoll RG, Markin C, et al. Telomerase mutations in families with idiopathic pulmonary fibrosis. N Engl J Med 2007;356(13):1317–26. [DOI] [PubMed] [Google Scholar]
- 67.Tsakiri KD, Cronkhite JT, Kuan PJ, Xing C, Raghu G, Weissler JC, et al. Adult-onset pulmonary fibrosis caused by mutations in telomerase. Proc Natl Acad Sci U S A 2007;104(18):7552–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Vulliamy T, Dokal I. Dyskeratosis congenita. Semin Hematol 2006. Jul;43(3):157–66. [DOI] [PubMed] [Google Scholar]
- 69.Armanios M, Chen J-L, Chang Y-PC, Brodsky R a, Hawkins A, Griffin C a, et al. Haploinsufficiency of telomerase reverse transcriptase leads to anticipation in autosomal dominant dyskeratosis congenita. Proc Natl Acad Sci U S A 2005;102(44):15960–4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Vulliamy T, Marrone A, Szydlo R, Walne A, Mason PJ, Dokal I. Disease anticipation is associated with progressive telomere shortening in families with dyskeratosis congenita due to mutations in TERC. Nat Genet 2004. May;36(5):447–9. [DOI] [PubMed] [Google Scholar]
- 71.Vulliamy T, Marrone A, Goldman F, Dearlove A, Bessler M, Mason PJ, et al. The RNA component of telomerase is mutated in autosomal dominant dyskeratosis congenita. Nature 2001. Sep 27;413(6854):432–5. [DOI] [PubMed] [Google Scholar]
- 72.Alder JK, Chen JJ-L, Lancaster L, Danoff S, Su S, Cogan JD, et al. Short telomeres are a risk factor for idiopathic pulmonary fibrosis. Proc Natl Acad Sci U S A 2008;105(35):13051–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Cronkhite J, Xing C, Raghu G. Telomere shortening in familial and sporadic pulmonary fibrosis. Am J … 2008;178(7):729–37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Borie R, Tabèze L, Thabut G, Nunes H, Cottin V, Marchand-Adam S, et al. Prevalence and characteristics of TERT and TERC mutations in suspected genetic pulmonary fibrosis. Eur Respir J 2016;48(6):1721–31. [DOI] [PubMed] [Google Scholar]
- 75.Parry EM, Alder JK, Qi X, Chen JJ-L, Armanios M. Syndrome complex of bone marrow failure and pulmonary fibrosis predicts germline defects in telomerase. Blood 2011. May 26;117(21):5607–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Stuart BD, Choi J, Zaidi S, Xing C, Holohan B, Chen R, et al. Exome sequencing links mutations in PARN and RTEL1 with familial pulmonary fibrosis and telomere shortening. Nat Genet. Nature Publishing Group; 2015;47(5):512–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Kannengiesser C, Borie R, Menard C, Reocreux M, Nitschke P, Gazal S, et al. Heterozygous RTEL1 mutations are associated with familial pulmonary fibrosis. Eur Respir J 2015;46(249816):474–85. [DOI] [PubMed] [Google Scholar]
- 78.Juge P-A, Borie R, Kannengiesser C, Gazal S, Revy P, Wemeau-Stervinou L, et al. Shared genetic predisposition in rheumatoid arthritis-interstitial lung disease and familial pulmonary fibrosis. Eur Respir J 2017;49(5). [DOI] [PubMed] [Google Scholar]
- 79.Alder JK, Barkauskas CE, Limjunyawong N, Stanley SE, Kembou F, Tuder RM, et al. Telomere dysfunction causes alveolar stem cell failure. Proc Natl Acad Sci 2015;112(16):201504780. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Mushiroda T, Wattanapokayakit S, Takahashi a, Nukiwa T, Kudoh S, Ogura T, et al. A genome-wide association study identifies an association of a common variant in TERT with susceptibility to idiopathic pulmonary fibrosis. J Med Genet 2008;45(10):654–6. [DOI] [PubMed] [Google Scholar]
- 81.Seibold MA, Wise AL, Speer MC, Steel MP, Brown KK, Loyd JE, et al. A Common MUC5B Promoter Polymorphism and Pulmonary Fibrosis. N Engl J Med 2012;364(16):1503–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Seibold MA, Smith RW, Urbanek C, Groshong SD, Cosgrove GP, Brown KK, et al. The Idiopathic Pulmonary Fibrosis Honeycomb Cyst Contains A Mucocilary Pseudostratified Epithelium. PLoS One 2013;8(3). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Zhang Y, Noth I, Garcia JGN, Kaminski N. A Variant in the Promoter of MUC5B and Idiopathic Pulmonary Fibrosis NT5E Mutations and Arterial Calcifications. N Engl J Med 2011;364(16):1576–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Stock CJ, Sato H, Fonseca C, Banya W a S, Molyneaux PL, Adamali H, et al. Mucin 5B promoter polymorphism is associated with idiopathic pulmonary fibrosis but not with development of lung fibrosis in systemic sclerosis or sarcoidosis. Thorax 2013;68(5):436–41. [DOI] [PubMed] [Google Scholar]
- 85.Noth I, Zhang Y, Ma S-F, Flores C, Barber M, Huang Y, et al. Genetic variants associated with idiopathic pulmonary fibrosis susceptibility and mortality: a genome-wide association study. lancet Respir Med. Elsevier Ltd; 2013;1(4):309–17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Borie R, Crestani B, Dieude P, Nunes H, Allanore Y, Kannengiesser C, et al. The MUC5B variant is associated with idiopathic pulmonary fibrosis but not with systemic sclerosis interstitial lung disease in the European Caucasian population. PLoS One 2013. Jan;8(8):e70621. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Wei R, Li C, Zhang M, Jones-Hall YL, Myers JL, Noth I, et al. Association between MUC5B and TERT polymorphisms and different interstitial lung disease phenotypes. Transl Res. Mosby, Inc; 2014;163(5):494–502. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Horimasu Y, Ohshimo S, Bonella F, Tanaka S, Ishikawa N, Hattori N, et al. MUC5B promoter polymorphism in Japanese patients with idiopathic pulmonary fibrosis. Respirology 2015;20(3):439–44. [DOI] [PubMed] [Google Scholar]
- 89.Peljto AL, Selman M, Kim DS, Murphy E, Tucker L, Pardo A, et al. The MUC5B promoter polymorphism is associated with idiopathic pulmonary fibrosis in a mexican cohort but is rare among asian ancestries. Chest 2015. Feb 1;147(2):460–4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Ley B, Collard HR. Epidemiology of Idiopathic Pulmonary Fibrosis. Clin Epidemiol 2013;5:483–92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.(NCBI) NC for BI. dbSNP entry for rs35705950 p. http://www.ncbi.nlm.nih.gov/SNP/snp_ref.cgi?rs=357.
- 92.Yang I V, Schwartz DA Epigenetics of idiopathic pulmonary fibrosis. Transl Res. Elsevier Inc; 2015;165(1):48–60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Ley B, Newton CA, Arnould I, Elicker BM, Henry TS, Vittinghoff E, et al. The MUC5B promoter polymorphism and telomere length in patients with chronic hypersensitivity pneumonitis: an observational cohort-control study. Lancet Respir Med. Elsevier Ltd; 2017;5(8):639–47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Juge P-A, Lee JS, Ebstein E, Furukawa H, Dobrinskikh E, Gazal S, et al. MUC5B Promoter Variant and Rheumatoid Arthritis with Interstitial Lung Disease. N Engl J Med 2018. Oct 20; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Mathai SK, Yang IV, Schwarz MI, Schwartz DA. Incorporating genetics into the identification and treatment of Idiopathic Pulmonary Fibrosis. BMC Med 2015;13(1):191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Yang IV, Fingerlin TE, Evans CM, Schwarz MI, Schwartz DA. MUC5B and Idiopathic Pulmonary Fibrosis. Ann Am Thorac Soc 2015. Nov;12 Suppl 2:S193–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Mathai SK, Schwartz DA, Warg LA. Genetic susceptibility and pulmonary fibrosis. Curr Opin Pulm Med 2014;20(5). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Noth I, Zhang Y, Ma SF, Flores C, Barber M, Huang Y, Broderick SM, Wade MS, Hysi P, Scuirba J, Richards TJ, Juan-Guardela BM, Vij R, Han MK, Martinez FJ, Kossen K, Seiwert SD, Christie JD, Nicolae D, Kaminski NGJ. Genetic variants associated with idiopathic pulmonary fibrosis susceptibility and mortality: a genome-wide association study. Lancet Respir Med 2013;1(4):309–17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Oldham JM, Ma SF, Martinez FJ, Anstrom KJ, Raghu G, Schwartz DA, et al. TOLLIP, MUC5B, and the response to N-acetylcysteine among individuals with idiopathic pulmonary fibrosis. Am J Respir Crit Care Med 2015;192(12):1475–82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Dressen A, Abbas AR, Cabanski C, Reeder J, Ramalingam TR, Neighbors M, et al. Analysis of protein-altering variants in telomerase genes and their association with MUC5B common variant status in patients with idiopathic pulmonary fibrosis: a candidate gene sequencing study. Lancet Respir Med. Elsevier Ltd; 2018. Jun 8;2600(18):1–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Hancock LA, Hennessy CE, Solomon GM, Dobrinskikh E, Estrella A, Hara N, et al. Muc5b overexpression causes mucociliary dysfunction and enhances lung fibrosis in mice. Nat Commun 2018;9(1):5363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Peljto AL, Zhang Y, Fingerlin TE, Ma S-F, Garcia JGN, Richards TJ, et al. Association between the MUC5B promoter polymorphism and survival in patients with idiopathic pulmonary fibrosis. JAMA 2013. Jun 5;309(21):2232–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Dudbridge F, Allen RJ, Sheehan NA, Schmidt AF, Lee JC, Jenkins RG, et al. Adjustment for index event bias in genome-wide association studies of subsequent events. bioRxiv 2018. Jan 1; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.de Leon AD, Cronkhite JT, Katzenstein AL a, Godwin JD, Raghu G, Glazer CS, et al. Telomere lengths, pulmonary fibrosis and telomerase (TERT) Mutations. PLoS One 2010;5(5):e10680. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Diaz De Leon A, Cronkhite JT, Yilmaz C, Brewington C, Wang R, Xing C, et al. Subclinical lung disease, macrocytosis, and premature graying in kindreds with telomerase (TERT) mutations. Chest 2011;140(3):753–63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Newton CA, Batra K, Torrealba J, Kozlitina J, Glazer CS, Aravena C, et al. Telomere-related lung fibrosis is diagnostically heterogeneous but uniformly progressive. Eur Respir J 2016;093096:1–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Stuart BD, Lee JS, Kozlitina J, Noth I, Devine MS, Glazer CS, et al. Effect of telomere length on survival in patients with idiopathic pulmonary fibrosis: An observational cohort study with independent validation. Lancet Respir Med. Elsevier Ltd; 2014;2(7):557–65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Yang IV, Coldren CD, Leach SM, Seibold M a, Murphy E, Lin J, et al. Expression of cilium-associated genes defines novel molecular subtypes of idiopathic pulmonary fibrosis. Thorax 2013;68:1114–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Herazo-Maya JD, Noth I, Duncan SR, Kim S, Ma S-F, Tseng GC, et al. Peripheral blood mononuclear cell gene expression profiles predict poor outcome in idiopathic pulmonary fibrosis. Sci Transl Med 2013. Oct 2;5(205):205ra136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Huang Y, Ma S-F, Vij R, Oldham JM, Herazo-Maya J, Broderick SM, et al. A functional genomic model for predicting prognosis in idiopathic pulmonary fibrosis. BMC Pulm Med. BMC Pulmonary Medicine; 2015;15(1):147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Raghu G, Anstrom KJ, King TE, Lasky JA, Martinez FJ. Prednisone, azathioprine, and N-acetylcysteine for pulmonary fibrosis. N Engl J Med 2012;366(21):1968–77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Idiopathic Pulmonary Fibrosis Clinical Research Network, Martinez FJ, de Andrade JA, Anstrom KJ, King TE, Raghu G Randomized trial of acetylcysteine in idiopathic pulmonary fibrosis. N Engl J Med 2014. May 29;370(22):2093–101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Tokman S, Singer JP, Devine MS, Westall GP, Aubert JD, Tamm M, et al. Clinical outcomes of lung transplant recipients with telomerase mutations. J Hear Lung Transplant. Elsevier; 2015;34(10):1318–24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Silhan LL, Shah PD, Chambers DC, Snyder LD, Riise GC, Wagner CL, et al. Lung transplantation in telomerase mutation carriers with pulmonary fibrosis. Eur Respir J 2014;44(1):178–87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Borie R, Kannengiesser C, Hirschi S, Le Pavec J, Mal H, Bergot E, et al. Severe hematologic complications after lung transplantation in patients with telomerase complex mutations. J Hear Lung Transplant. Elsevier; 2015;34(4):538–46. [DOI] [PubMed] [Google Scholar]
- 116.Newton CA, Kozlitina J, Lines JR, Kaza V, Torres F, Garcia CK. Telomere length in patients with pulmonary fibrosis associated with chronic lung allograft dysfunction and post–lung transplantation survival. J Hear Lung Transplant. Elsevier Inc; 2017;36(8):845–53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Ando M, Hirayama K, Soda K, Okubo R, Araki S, Sasazuki T. HLA-DQw3 in Japanese summer-type hypersensitivity pneumonitis induced by Trichosporon cutaneum. Am Rev Respir Dis 1989. Oct;140(4):948–50. [DOI] [PubMed] [Google Scholar]
- 118.Camarena A, Juarez A, Mejia M, Estrada A, Carrillo G, Falfan R, et al. Major histocompatibility complex and tumor necrosis factor-alpha polymorphisms in pigeon breeder’s disease. Am J Respir Crit Care Med 2001;163(7):1528–33. [DOI] [PubMed] [Google Scholar]
- 119.Selman M, Terán L, Mendoza A, Camarena A, Martínez-Cordero E, Lezama M, et al. Increase of HLA-DR7 in pigeon breeder’s lung in a Mexican population. Clin Immunol Immunopathol 1987. Jul;44(1):63–70. [DOI] [PubMed] [Google Scholar]
- 120.Selman M, Pardo A, King TE. Hypersensitivity pneumonitis: Insights in diagnosis and pathobiology. Am J Respir Crit Care Med 2012;186(4):314–24. [DOI] [PubMed] [Google Scholar]
- 121.Janssen R, Kruit A, Grutters JC, Ruven HJT, Van Moorsel CMH, Van Den Bosch JMM. TIMP-3 promoter gene polymorphisms in BFL [1]. Thorax 2005;60(11):974. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Hill MR, Briggs L, Montaño MM, Estrada A, Laurent GJ, Selman M, et al. Promoter variants in tissue inhibitor of metalloproteinase-3 (TIMP-3) protect against susceptibility in pigeon breeders’ disease. Thorax 2004;59(7):586–90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Camarena Á, Aquino-Galvez A, Falfán-Valencia R, Sánchez G, Montaño M, Ramos C, et al. PSMB8 (LMP7) but not PSMB9 (LMP2) gene polymorphisms are associated to pigeon breeder’s hypersensitivity pneumonitis. Respir Med 2010;104(6):889–94. [DOI] [PubMed] [Google Scholar]
- 124.Aquino-Galvez A, Camarena Á, Montaño M, Juarez A, Zamora AC, González-Avila G, et al. Transporter associated with antigen processing (TAP) 1 gene polymorphisms in patients with hypersensitivity pneumonitis. Exp Mol Pathol 2008;84(2):173–7. [DOI] [PubMed] [Google Scholar]
- 125.Schaaf BM, Seitzer U, Pravica V, Aries SP, Zabel P. Tumor necrosis factor-α −308 promoter gene polymorphism and increased tumor necrosis factor serum bioactivity in farmer’s lung patients. Am J Respir Crit Care Med 2001;163(2):379–82. [DOI] [PubMed] [Google Scholar]
- 126.Radstake TRDJ, Gorlova O, Rueda B, Martin J, Alizadeh BZ, Palomino-Morales R, et al. Genome-wide association study of systemic sclerosis identifies CD247 as a new susceptibility locus. Nat Genet 2010. May;42(5):426–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Gourh P, Agarwal SK, Divecha D, Assassi S, Paz G, Arora-Singh RK, et al. Polymorphisms in TBX21 and STAT4 increase the risk of systemic sclerosis: Evidence of possible gene-gene interaction and alterations in Th1/Th2 cytokines. Arthritis Rheum 2009;60(12):3794–806. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Dieudé P, Guedj M, Wipff J, Avouac J, Fajardy I, Diot E, et al. Association between the IRF5 rs2004640 functional polymorphism and systemic sclerosis: A new perspective for pulmonary fibrosis. Arthritis Rheum 2009;60(1):225–33. [DOI] [PubMed] [Google Scholar]
- 129.Rueda B, Broen J, Simeon C, Hesselstrand R, Diaz B, Suárez H, et al. The STAT4 gene influences the genetic predisposition to systemic sclerosis phenotype. Hum Mol Genet 2009;18(11):2071–7. [DOI] [PubMed] [Google Scholar]
- 130.Kawaguchi Y, Ota Y, Kawamoto M, Ito I, Tsuchiya N, Sugiura T, et al. Association study of a polymorphism of the CTGF gene and susceptibility to systemic sclerosis in the Japanese population. Ann Rheum Dis 2009. Dec;68(12):1921–4. [DOI] [PubMed] [Google Scholar]
- 131.Zhou X, Tan FK, Wang N, Xiong M, Maghidman S, Reveille JD, et al. Genome-wide association study for regions of systemic sclerosis susceptibility in a Choctaw Indian population with high disease prevalence. Arthritis Rheum 2003. Sep;48(9):2585–92. [DOI] [PubMed] [Google Scholar]
- 132.Hunninghake GM, Hatabu H, Okajima Y, Gao W, Dupuis J, Latourelle JC, et al. MUC5B promoter polymorphism and interstitial lung abnormalities. N Engl J Med 2013. Jun 6;368(23):2192–200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133.Putman RK, Hatabu H, Araki T, Gudmundsson G, Gao W, Nishino M, et al. Association Between Interstitial Lung Abnormalities and All-Cause Mortality. JAMA 2016;315(7):672. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134.Araki T, Putman RK, Hatabu H, Gao W, Dupuis J, Latourelle JC, et al. Development and Progression of Interstitial Lung Abnormalities in the Framingham Heart Study. Am J Respir Crit Care Med 2016;Epub ahead of print. [DOI] [PMC free article] [PubMed] [Google Scholar]