

Abstract
Biochemical studies of integral membrane proteins are often hampered by low purification yields and technical limitations such as aggregation causing in vitro manipulations to be challenging. The ability of controlling proteins in live cells bypasses these limitations while broadening the scope of accessible questions owing to the proteins being in their native environment. Here we take advantage of the intein biorthogonality to mammalian systems, site specificity, fast kinetics, and auto processing nature as an attractive option for modifying surface proteins. Using EGFR as a model, we demonstrate that the split-intein pair AvaN/ NpuC can be used to efficiently and specifically modify target membrane proteins with a synthetic adduct for downstream live cell application.
Keywords: split intein, protein modification, EGFR
Graphical Abstract
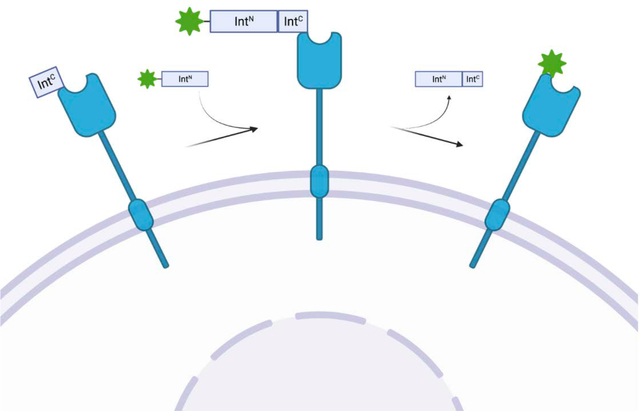
Split-inteins represent a unique avenue for the modification of proteins in cells as they carryout protein splicing in a traceless, auto processing, site specific manner that is biorthogonal to mammalian systems. Here we utilize split-inteins to label extracellular portion of EGFR with a synthetic fluorophore for live cell imaging.
Introduction
A detailed understanding of a protein’s function and regulation requires approaches that wield control of its composition at a residue-specific level. Classic in vitro biochemical methods to investigate proteins have provided a wealth of knowledge about their structure and function through the intricate and selective modifications. However, complex cellular milieus combined with the lack of biocompatible chemical strategies for introducing synthetic moieties has restricted most studies of transmembrane proteins to an in vitro set up that does not represent their native membrane context. Membrane proteins are a diverse and heterogenous group that make up over 30 % of the protein coding-genome and play key roles in a variety of cellular functions including signaling, mobility, and communication.[1] Additionally, over 60 % of all druggable targets are found on the cell surface and are important players in pathological states.[2] For example, the epidermal growth factor receptor (EGFR) has been shown to regulate multiple processes, including proliferation and survival, thus its mutant forms often drive disease states such as cancer.[3] However, EGFR, like other membrane proteins, is generally a poor substrate for in vitro investigation due to low solubility, requirement of a lipid membrane for correct folding, specific co- or post-translational modifications that determine their function, and effects of the membrane microcompartment composition.[4–5] These constraints in working with membrane proteins limit the capacity to adequately interrogate their functions. In addition, because the plasma membrane is a dynamic environment whose contents are constantly changing in response to various stimuli, studying these proteins in isolation is proven challenging.
Development of techniques to manipulate and study proteins in vivo remains an active and sought-after area. The most common and widely used methods like ligand labeling, tag-probe systems, or proximity-based labeling have allowed for the interrogation of target proteins in cells in real time.[6–7] In fact, synthetically labeled epidermal growth factor (EGF) is commonly used to visualize EGFR when bound,[8] and both SNAP-Tag and HaloTag have been successfully employed to EGFR in live cells enabling the visualization of ligand binding and internalization.[9–10]. However, these techniques depend upon strong ligand interactions and the incorporation of large appendages (20–30 kDa) in the target protein that can reduce localization precision.[11] In addition, these methods can lack temporal control and are limited to one modification at a time.[6–7] A complementary approach that overcomes these limitations through near-traceless engineering with access to a larger range of modifications allows for the study of membrane proteins in even greater detail.
Inteins are a family of proteins that catalyze their own excision from polypeptide sequences they are embedded in through a process called splicing. Mechanistically, intein splicing occurs through a series of N- to S- acyl shifts that result in the ligation of the flanking (“extein”) sequences with a native peptide bond.[12] A small subset of inteins are expressed in a “split” form and carryout protein splicing in trans, where they ligate two separate peptide or protein fragments.[13] Due to this unique traceless ligation inteins represent an exciting avenue for in vivo applications for synthetic protein manipulation. The utility of split-intein technology for engineering proteins in vitro and in vivo has been demonstrated through a range of applications allowing the incorporation of a diverse set of modifications into proteins.[13–16] Recent work in this area has extended to the modification of the extracellular domain of membrane proteins. For example, the interferon receptor subunit IFNAR1 was appended with a short peptide fluorophore and imaged in live cells through the use of an artificially split intein combined with a high affinity interaction pair to help induce splicing between the split inteins.[17] The introduction of a peptide with different lysine analogs to the extracellular domain of P2X2R was facilitated with tandem protein trans-splicing with the Cfa and Ssp intein pairs.[18] While both instances represent exciting uses of inteins for protein modifications in cells, the former requires extra factors to drive efficient protein splicing, and the latter involves complete protein reassembly rather than site-specific derivatization and suffers from exceedingly low yields.
Here, we develop a general platform utilizing ultra-fast split inteins for the modification of the extracellular domain of surface proteins. We selected EGFR as our model membrane protein as the signaling and trafficking dynamics of EGFR and its pathological variants remain important aspects in understanding both its role in the development of diseases such as cancer and responses to targeted therapy. We reasoned that a split-intein approach would allow for direct manipulation of the extracellular domain EGFR. We aimed to develop this method to be solely intein driven and attain appreciable splicing yields to expand the toolbox of intein-mediated protein engineering in cells. Using a widely characterized ultra-fast intein pair, we were able to modify EGFR with a synthetic fluorophore in live cells and demonstrate the generality of this method in investigating other membrane proteins as well.
Results and Discussion
The split-intein based method we envisioned relied on an expressed portion involving the target protein (using EGFR as a model) and the synthetic deliverable portion (involving the desired chemical modification) (Scheme 1). In this case, the IntC is cloned upstream of the N-terminus of EGFR after a membrane signal peptide and this construct is expressed on the plasma membrane. The synthetic portion is generated by expressed protein ligation (EPL) of a purified recombinant IntN fragment ligated to a short peptide fragment (generated by solid phase peptide synthesis, SPPS) carrying the desired synthetic modification. The semi-synthetic fragment is then added to the media of the cells expressing the IntC-EGFR allowing splicing to occur on the membrane and the generation of the modified surface protein in minutes.
Scheme 1. A Protein trans-splicing method to generate semi-synthetically modified transmembrane proteins in live cells.
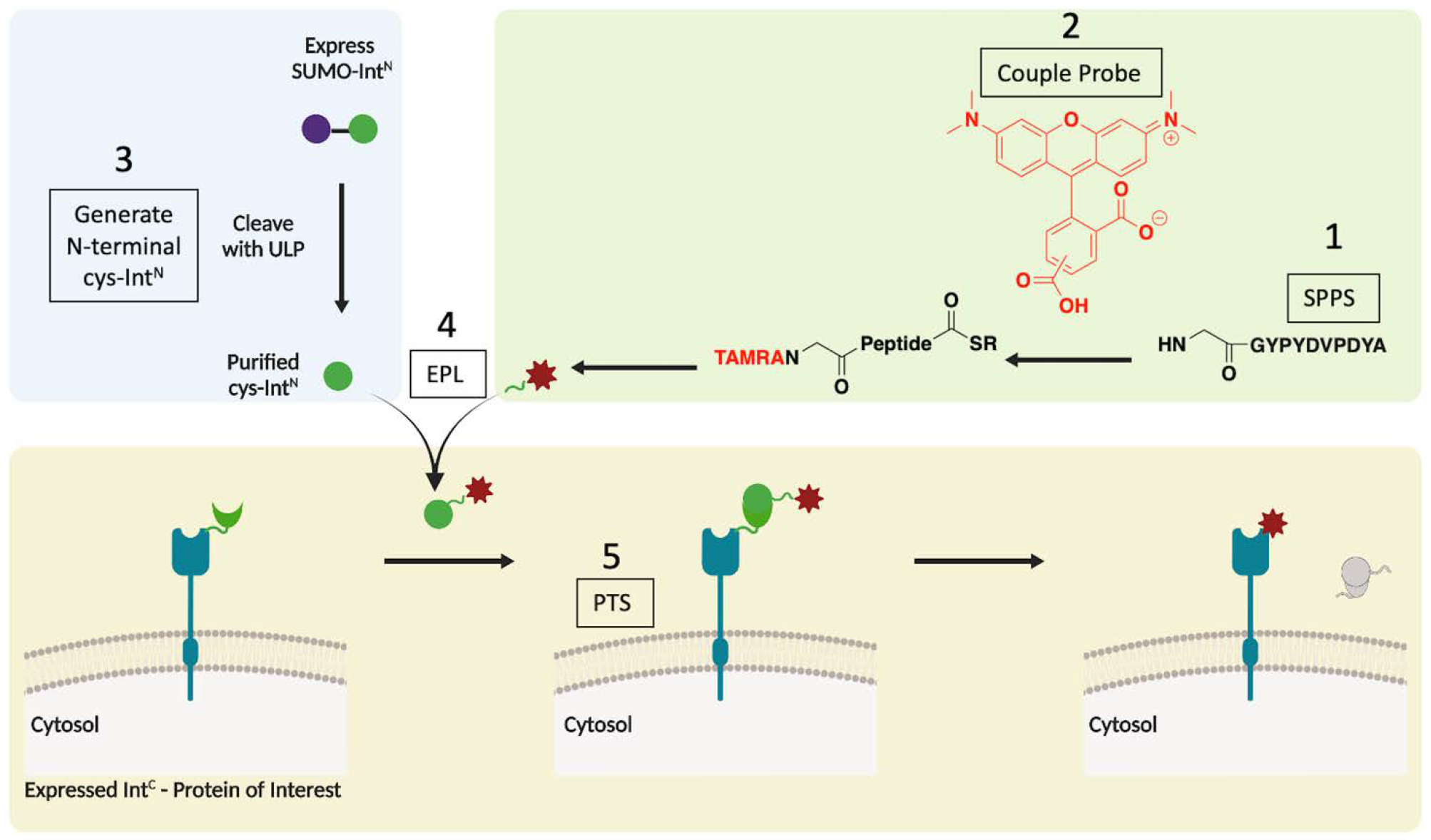
Green box: synthesis of modified peptide (SPPS- solid phase peptide synthesis, TAMRA – tetramethylrhodamine). Blue Box: purification of intein with N-terminal cysteine (ULP – ubiquitin-like-specific protease, EPL – expressed protein ligation). Yellow box: expressed intein construct and protein trans splicing (PTS) on cells. Briefly, the target peptide is generated by SPPS (1) and coupled to a synthetic probe (2). The recombinantly purified IntN (3) is ligated to the peptide by EPL (4). The product is added to the media of cells expressing IntC-fused protein of interest (POI), and PTS occurs in minutes on the cellular surface (5).
The intein pair we chose to employ consists of the IntC fragment evolved from the Nostoc punctiforme split intein (NpuC), and the IntN fragment from Annabaena variabilis (AvaN). The NpuC intein has rapid splicing kinetics (half-life of one minute in vitro) at 37 °C, is tolerant to most extein flanking sequences, and is smaller in size (39 amino acids).[16, 19] The AvaN belongs to the same family of split inteins and has been shown to be a strong splicing partner of NpuC.[20] In the initial experiments, the native extein sequences of NpuC (CFN) and AvaN (AEY) were used for optimal splicing, however, these sequences can be shortened or removed to reduce the trace left by the intein splicing and maintain a more native protein sequence.
To track the splicing events in a facile manner, we utilized an exchange of high affinity tags. Specifically, the NpuC contained an N-terminal Flag tag and AvaN an N-terminal HA tag. As illustrated in the scheme in Figure 1A, following intein splicing, the EGFR product carries an N-terminal HA tag that has replaced the Flag-NpuC. In this way we can detect the splicing through the formation of the HA-EGFR product. To begin, we conducted splicing with the recombinant HA-AvaN carrying no modification. This allowed us to characterize the splicing reaction in detail before adding the complexity of a synthetic modification. We first transfected HEK293T cells with the Flag-IntC-EGFR construct. We confirmed the exclusive membrane localization of the exogenous construct with only a slight reduction in expression levels compared to the non-intein control (Supplemental Figure 1). Next, purified HA-IntN was added to the media at a final concentration of 20 μM. The cells were left to incubate at 37 °C and collected at different time points over the course of 90 minutes. The membrane fractions were then isolated, separated by SDS-PAGE, and analyzed by western blot with anti-Flag and anti-HA, with Na/K ATPase as a membrane fraction loading control (Figure 1B–C). In tracking the formation of the HA-EGFR product, we detected it as early as 5 minutes after the addition of the IntN, and it appears to plateau between 30 and 60 minutes. To determine the optimal concentration of IntN required for efficient reaction we examined a range of concentrations and noted that concentrations as low as 5 μM still produces a similar amount of product, although slightly lower compared to 10 or 20 μM (Supplemental Figure 2). Given that some synthetic cargos could be difficult to produce, it is important to note that lower concentrations are feasible with this system. Finally, as a measure to confirm the subcellular localization of the product, we repeated the experiment and analyzed the cells by immunofluorescence using antibodies against Flag or HA and imaged by confocal microscopy. Gratifyingly, we not only observe clear plasma membrane expression of the IntC-EGFR construct, but also the appearance of the HA-EGFR product only when both halves of the split intein are present in the system (Figure 1D, Supplemental Figure 3).
Figure 1. EGFR is site-specifically modified using a split intein system in live cells.
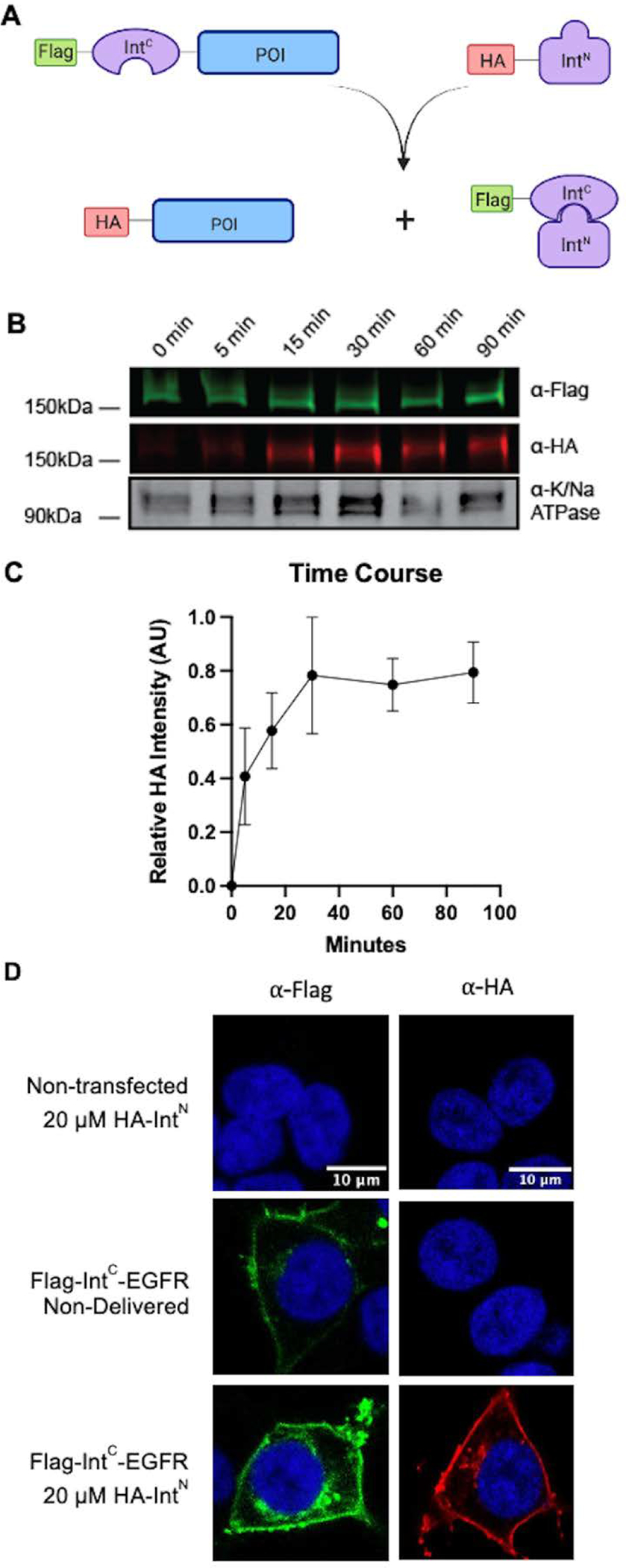
A) Scheme illustrating epitope tag exchange with split-intein splicing. B) Time course of splicing following the formation of HA-EGFR product. HEK293T cells were transfected with Flag-NpuC-EGFR and then treated with 20 μM of HA-AvaN over the course of 90 minutes. The membrane fraction was then extracted and analyzed by western blot (anti-Flag – green, anti-HA – red, anti-Na/K ATPase – gray scale). C. Quantification of B (n=3, error bars represent SEM) D) Immunofluorescence HEK293T cells transfected with Flag-NpuC-EGFR and incubated with HA-AvaN. Cells were fixed with paraformaldehyde and then stained with either anti-Flag (green) or anti-HA (red) and DAPI (blue) followed by imaging.
Following the validation of our design in cells, we sought to further determine its utility for downstream experimentation and as a more broadly applicable platform. First, we assessed what, if any, impact the addition of the inteins has on EGFR’s function. As seen in Figure 2A, both pre- and post-spliced EGFR are stimulated by the native epidermal growth factor (EGF) ligand and induce the activation of downstream signaling cascades as demonstrated by the detection of phosphorylated ERK.[21] To determine the stability of the spliced EGFR product, for the benefit of further experimentations post splicing, we reacted Flag-IntC-EGFR transfected cells with HA-IntN as described previously and followed the presence of the product over 24 hours. For every time point, the membrane fraction was isolated, separated by SDS-PAGE, and analyzed by western blot with anti-Flag and anti-HA. We found that the spliced HA-EGFR product exists in the plasma membrane at least 24 hours after splicing, although the quantity lowers over time, implying a wide window in which downstream experimentation with the post-spliced construct can be performed (Figure 2B, 2C). Finally, while we used EGFR as our model substrate for the proof-of-concept experiments, we envisioned this methodology being widely applicable and thus tested the same strategy on additional membrane proteins. We chose an additional member of the ErbB family, ErbB2, which shares the same topology as EGFR, as well as two disease mutants of EGFR with missing portions of the extracellular domain, EGFR vII and EGFR vIII, that have been found in numerous cancers including glioblastomas and lung cancer[22–23]. Interestingly, while all the proteins successfully splice, the variance of the yield of splicing was high, with ErbB2 having the lowest overall splicing (Figure 2C). In addition, we carried out splicing with three more monotopic membrane proteins of different sizes, CD40, CTLA4, and PD1, demonstrating successful splicing of membrane proteins of different topologies, although too at varying yields (Figure 2D). The differences in splicing efficiency across different target proteins suggest that while split-inteins can be applied more broadly, there could be sensitivity to the protein structure and local environment that may require additional optimization and fine tuning of reaction conditions.
Figure 2. Intein splicing is nondisruptive, long lasting, and generalizable.
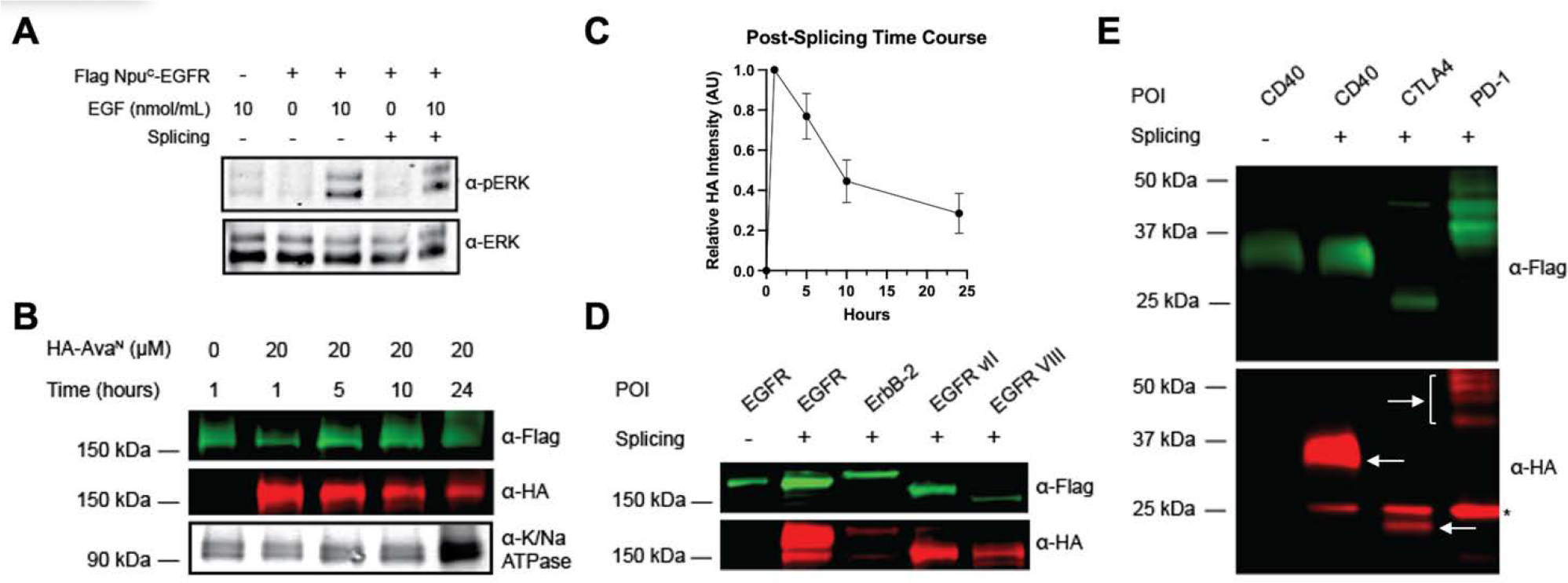
A) HEK293T cells were transfected with Flag-NpuC-EGFR and then treated with 20 μM of HA-AvaN. Cells were then starved overnight in serum free media before being treated with 10 ng/mol of EGF for 30 minutes. Next, the cells were harvested, lysate was separated on SDS-PAGE and phosphorylation of ERK was analyzed by western blot. B) HEK293T cells were transfected with Flag-NpuC-EGFR and then treated with 20 μM of HA-AvaN for 1 hr. Cells were then harvested at the indicated time points post splicing and the membrane fraction was extracted and analyzed by western blot (anti-Flag – green, anti-HA – red, anti-Na/K ATPase – gray scale, refer to Supplemental Figure 4 for unedited version). C. Quantification of B (n=3, error bars represent SEM). D) HEK293T cells were transfected with the indicated Flag-NpuC-EGFR/ErbB2 variant and then treated with 20 μM of HA-AvaN for 1 hour. The membrane fraction was then extracted, separated on SDS-PAGE and analyzed by western blot (anti-Flag – green, anti-HA – red, anti-Na/K ATPase – gray scale). E) HEK293T cells were transfected with Flag-NpuC-CD40, Flag-NpuC-CTLA4, or Flag-NpuC-PD-1 and treated with 20 μM of HA-AVAN for 1 hour. The membrane fraction was extracted, separated on SDS-PAGE and analyzed by western blot (anti-Flag – green, anti-HA – red, arrows indicate expected size). *unspliced HA-AvaN
Having established and optimized the in cellulo splicing of EGFR, we next sought to introduce a synthetic modification onto the receptor. We designed the deliverable portion to be of a similar composition and carry a small organic fluorophore. In order to generate this construct, we used expressed protein ligation (EPL) to conjugate two halves. Briefly, the HA peptide coupled to an organic fluorophore (TAMRA) was synthesized on a CTC resin and the C-terminal was converted from a hydrazine to a thioester. This was then coupled the cys-AvaN, created by purifying with an N-terminal SUMO tag cleaved by ULP, to generate the final TAMRA-HA-AvaN (Supplemental Figure 5A, B). We confirmed the ligation of the fluorophore to the intein both by Coomassie/in gel Fluorescence (Figure 3A) and mass spectrometry (Supplemental Figure 5C). We repeated the splicing experiment on HEK293T cells expressing the IntC-EGFR to produce an N-terminally fluorophore tagged EGFR. As seen in Figure 3B splicing with the semi-synthetic cargo occurs with comparable efficiency to the recombinant cargo using the same concentrations and reaction time. In fact, we observed no appreciable difference in splicing kinetics or efficiency between the two. We next confirmed splicing by imaging the cells. HEK293T were transfected with Flag-IntC-EGFR, and then incubated with the synthetic TAMRA-HA-IntN. The cells were then imaged by live cell imaging to visualize the semi-synthetic EGFR, which were membrane localized only in the cells that received both intein fragments (Figure 3C). We verified the successful splicing in cells as described before, staining with antibodies against HA that detect the HA-EGFR product, and found appreciable overlap between the HA signal and the TAMRA signal (Supplemental Figure 6).
Figure 3. Generating a site-specific semi-synthetic EGFR in live cells.
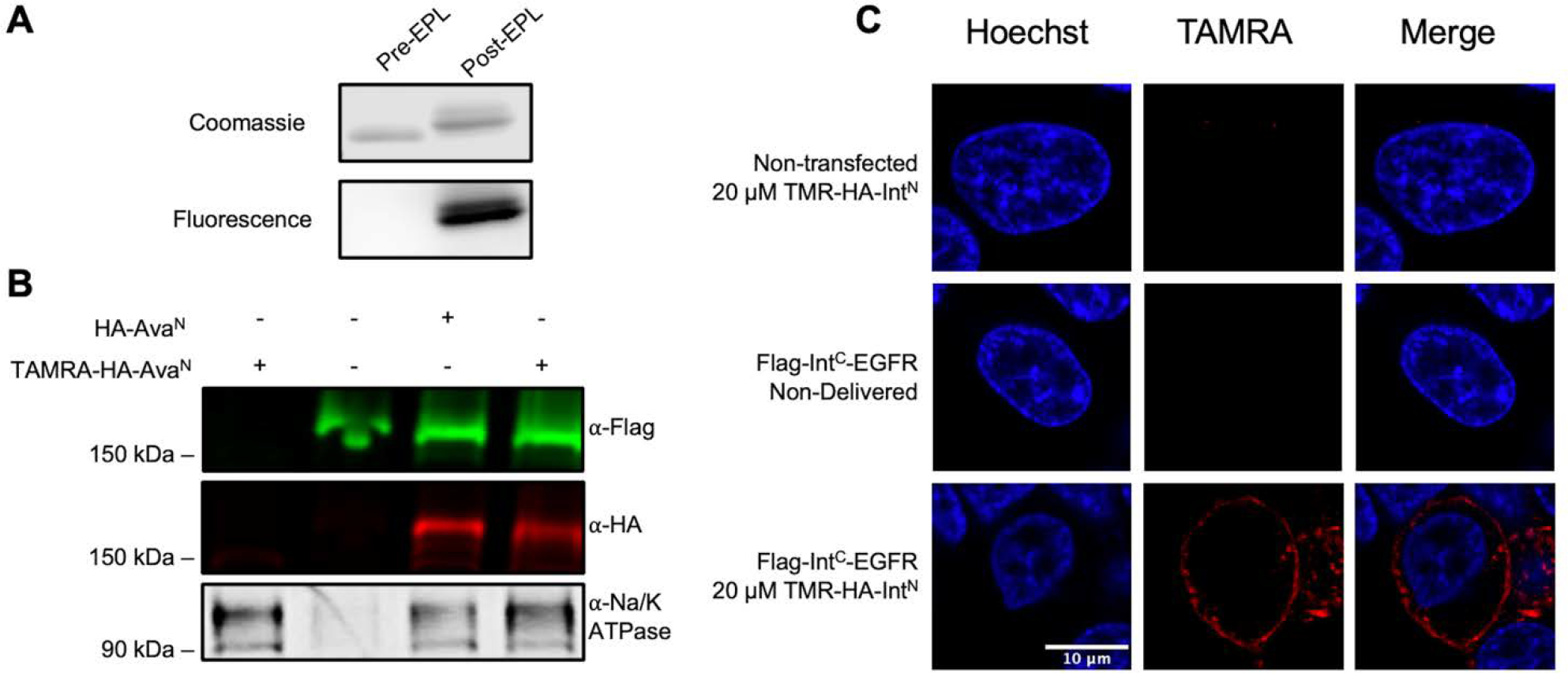
A) Coomassie and in gel fluorescence of AvaN −/+ the synthetic peptide. B) Comparison of splicing with recombinant vs synthetically modified AvaN. HEK293T cells were transfected with Flag-NpuC-EGFR and then treated with 20 μM of HA-AvaN or TAMRA-HA-AvaN for 60 minutes. The membrane fraction was then extracted, separated on SDS-PAGE and analyzed by western blot (anti-Flag – green, anti-HA – red, anti-Na/K ATPase – gray scale). C) Live cell images of HEK293T cells transfected with Flag-NpuC-EGFR and treated with TAMRA-HA-AvaN (red) and Hoechst (blue).
Conclusion
Although recombinant biochemical generation of proteins allows for their manipulation and more precise control of the target protein, it is intrinsically limited in the scope of information that can be gained. This is especially true of proteins that are less tolerant of purification and isolation, such as integral membrane proteins. Gaining synthetic access to proteins in their native environments can both contextualize information gained through in vitro experiments and enable “biochemical” dissection in a physiologically relevant environment. While a myriad of techniques have been developed that seek to achieve this, each has their own benefits and downsides. Because split-inteins are auto processing, bioorthogonal, and allow for traceless and site-specific modification, they represent a promising avenue for further development in the pursuit of live-cell protein engineering.
Here we describe and characterize a method to append synthetic molecules to the extracellular portion of membrane proteins using split-inteins. Using EGFR as a model protein, we demonstrate that with the split-intein pair NpuC/AvaN we can modify the N-terminal portion of EGFR embedded in a live cell membrane under concentrations as low as 2–5 μM in less than an hour. While slower than in vitro kinetics (most likely due to the more complex environment splicing takes place in), these times are appreciably fast for live cell experimentation. In addition, we show that the insertion of the inteins has little to no effect on protein expression and function making it relevant for studying things such as downstream signaling, and that this method can be applied to other monotropic proteins besides EGFR. Finally, using this method were able to insert a synthetic fluorophore onto EGFR and then image the modified receptor in live cells.
Inteins represent an exciting space in the quest to engineer proteins in live cells. A key aspect of intein chemistry allows for the site specificity in the introduction of the desired modification.[24] While in this method we utilized split-inteins to modify a target protein, the system enables labeling of a precise residue of the desired protein when designed correctly. As split inteins continue to be adapted to cellular systems, the next big advancement will be taking advantage of their site specificity, such as by incorporating a specific post-translational modification or photo-crosslinker to a protein domain. In fact, split-inteins were recently used to insert peptides carrying specific PTMs to the NaV1.5 channel, although yield of spliced product remained low and limited the suite of compatible downstream assays.[18] Inteins have also been applied towards the modification of intracellular targets, although this comes with the added hurdle of getting the intein-cargo delivered into the cell, which impacts the yield of spliced product.[16] Together, our work along with that of others demonstrates we have only scratched the surface of the potential of inteins to control and investigate proteins in live cells.
Experimental Section
General Materials and Methods.
All reagents were purchased form Fisher Scientific of Sigma-Aldrich unless otherwise stated. Size Exclusion chromatography was performed on an AKTA FPLC system from GE Healthcare equipped with a P-920 pump and UPC-900 monitor. Preparative purifications were conducted on an Agilent LC system on a C18 preparative column (15–20 um, 20×50 mm, flow rate 20 mL/ minute) or semi-preparative column (12 um, 10×250 mm, flow rate 2 mL/ minute) employing 0.1% TFA in water (Buffer A) and 90% acetonitrile, 0.1% TFA in water (Buffer B) as the mobile phases. HPLC Electrospray ionization MS was performed on an Agilent 6120 Quadrupole LC/MS spectrometer. Concentrations of DNA and proteins were measured spectroscopically with a NanoDrop 2000c (Thermo Scientific). Western blots were imaged on an Odyssey CLx Imaging System (Li-Cor).
Cloning of Mammalian Expressed Intein Constructs.
All constructs were generated using Gibson Assembly of IntC, protein of interest (POI), and back bone pFlag-EGFR (Velvet) from Bruce Beutler (Addgene plasmid 18788).[18] Cloning reagents (Gibson Master Mix, DNA polymerase, DPN1) were purchased from New England BioLabs. PCR amplifications were performed on a Bio-Rad T100™ Thermal Cycler.
Purification of HA-AvaN-GFP.
HA-AvaN-GFP-His was expressed in a pet3a vector and transformed into BL21 (DE3) cells. Cells were grown at 37 °C under ampicillin selection until reaching an OD of 0.6–0.8. The cultures were induced with 0.5 mM of ITPG at 16 °C for 16 hours. All purification steps were carried out at 4 °C. Cells were harvested by centrifugation (6000 ×g for 25 minutes) and resuspended in lysis buffer (200 mM NaCl, 50 mM Tris, 1 mM DTT, 1 mM PMSF) then sonicated for 2 minutes in 15 sec intervals at amplitude 25. After centrifugation at 30,000 ×g for 25 minutes the supernatant was loaded onto Ni-NTA beads equilibrated with lysis buffer and incubated with agitation for 1 hour. The flow-through was discarded, the beads were washed with lysis buffer plus 10mM of imidazole, and then the protein was eluted with elution buffer (lysis buffer with 500 mM of imidazole). The elution was injected onto a size-exclusion column (GE Life sciences preparative scale S 200 10/300). Fractions containing HA-AvaN-GFP were pooled, concentration was determined by nanodrop and used immediately.
Purification of cys-AVAN.
His-Sumo-cys-AVAN was expressed in a pet30 vector and transformed into BL21 (DE3) cells. Cells were grown at 37 °C under kanamycin selection until reaching an OD of 0.6–0.8. The cultures were induced with 0.5 mM of ITPG at 16 °C for 16 hours. Cells were harvested by centrifugation (6000 ×g for 25 minutes) and resuspended in extraction buffer (6M guanidinium chloride, 50 mM Tris, 1 mM DTT) then sonicated for 2 minutes in 15 sec intervals at amplitude 25 and agitated at room temperature for 1hr. After centrifugation at 30,000 ×g for 25 minutes the supernatant was loaded onto Ni-NTA beads equilibrated with extraction buffer and incubated with agitation at room tempera for 1 hr. The flow-through was discarded, the beads were washed with extraction buffer plus 10mM of imidazole, and then the protein was eluted with elution buffer (extraction buffer with 500 mM of imidazole). The elution was sequentially dialyzed to 4M then 1.5 M Urea, 50 mM Tris, 1 mM DTT, and then treated with ULP-1 (1:100 v/v of 4mg/mL enzyme) with agitation overnight at 4 °C. The elution was then purified on through preparative HPLC with a gradient of 30–70%. The purified cys-AVAN fractions were pooled and lyophilized, and the mass was verified by LC/MS.
Synthesis of TAMRA-HA Peptide.
The HA peptide (GYPYDVPDYA) was synthesized as a C-terminal hydrazine on a CTC resin by standard Fmoc-based solid phase peptide synthesis. 5(6)-TAMRA fluorophore (AnaSpec) was coupled under standard SPPS amino acid coupling conditions overnight. The peptide was cleaved with 95% TFA and purified by semipreparative HPLC (30–70% gradient). Fractions were pooled and lyophilized, and the mass confirmed by LC/MS.
Expressed Protein Ligation of TAMRA-HA-cys-AvaN.
The TAMRA-HA peptide hydrazide (3eq) was dissolved in pH 3 degassed ligation buffer (6M guanidinium chloride, 100 mM phosphate). 20 mM NaNO2 was added and this was left to incubate at −20 °C for 20 minutes. An equal volume of 200 mM of MESNA (Alfa Aesar) in ligation buffer was added and the pH was adjusted to 7.2 and thioester generation occurred at room temperature for 45 minutes. The thioester was then added to lyophilized cys-AVA (1eq) and the pH was adjusted to 7.5. The reaction was left to incubate at room temperature overnight. Completion of the reaction was confirmed by LC-MS, 100mM of TCEP was added to the reaction to reduce any disulfides and the product was purified by semi-preparative HPLC (40–90%).
Cell Culture.
WT HEK293T cells were cultured in DMEM supplemented with 10% Fetal Bovine Serum (FBS, Atlas Biologicals, Ft Collins, CO) and incubated at 37 °C under 5 % CO2. Transfections were done using Lipofectamine 2000 Transfection Reagent according to manufacturer’s protocol.
Splicing assay on cells.
HEK293T cells were transfected with Flag-NpuC-POI. Cells were then washed with fresh serum free media and incubated with 20 μM (or indicated concentration) of the AvaN construct in serum free media for 1 hour (or indicated time). Cells were then harvested, and the membrane fraction was extracted with Mem-PER™ Plus Membrane Protein Extraction Kit (Thermo Fischer). The membrane fractions were then analyzed by western blot with anti-HA (Thermo Fisher 26183) and anti-Flag antibodies (Sigma Aldrich F7425).
EGF activation assay.
HEK293T cells transfected with Flag-NpuC-EGFR were starved in serum free media overnight. EGFR was stimulated by adding 10 ng/mL of EGF (source) at 37 for 30 minutes. Cells were harvested with PBS and lysed with RIPA lysis buffer (1% NP-40, 0.5% w/v sodium deoxycholate, 0.1% SDS, cOmplete protease inhibitor (Roche) in PBS), and incubated for 30 minutes at 4 °C. The lysate was then analyzed by western blot with anti-AKT and anti-pAKT antibodies (CST 2920 and 4060).
Imaging of Fixed Cells.
HEK293T cells were grown in chamber covered glass and splicing was conducted as stated previously. Cells were washed with cold PBS and fixed with 1% paraformaldehyde for 15 minutes. Cells were then blocked with blocking buffer (10% goat serum, 1% BSA, 0.02% NaN3 in PBS) for 30 minutes at room temperature and then washed again with PBS. Anti-HA (Anti-HA High Affinity Roche) diluted 1:125 in 1% BSA in PBS was incubated at 4 °C overnight. Anti-Flag (Sigma F1804) diluted 1:500 in 1% BSA in PBS was incubated for 1 hour at room temperature. Secondary antibodies (Alexa Fluro 488 and 568) diluted 1:500 in 1% BSA in PBS were incubated for 1 hour at room temperature. Cells were stained with DAPI diluted 1:1000 in water for 10 minutes at room temperature. Cells were imaged on an inverted Leica TCS SP5 confocal microscope (Leica Microsystems, Germany) with a 40x/1.3NA oil-immersion objective.
Live Cell Imaging.
HEK293T cells were grown in chamber covered glass and splicing was conducted as stated previously. Nuclei were then stained with Hoechst (Thermo 33342) in a 1:10,000 dilution in PBS for 15 minutes at room temperature. Cells were imaged on an inverted Leica TCS SP5 confocal microscope (Leica Microsystems, Germany) with a 40x/1.3NA oil-immersion objective in a 37 °C chamber.
Supplementary Material
Acknowledgements
Work in the David lab is supported by the Josie Robertson Foundation, the Pershing Square Sohn Cancer Research Alliance, the NIH (CCSG core grant P30 CA008748, MSK SPORE P50 CA 192937 and R35 GM138386), the Parker Institute for Cancer Immunotherapy (PICI), the STARR Cancer Alliance award, and the Anna Fuller Trust. In addition, the David lab is supported by Mr. William H. Goodwin and Mrs. Alice Goodwin and the Common-wealth Foundation for Cancer Research and the Center for Experimental Therapeutics at MSKCC. DMR is supported by an MSTP grant from the NIH General Sciences (T32 GM007739) to the WCM/Rockefeller/Sloan Kettering Tri-Institutional MD-PhD Program. Figure schemes were created using BioRender. We thank E. Chan (MSK Molecular Cytology core facility) for advice on live cell imaging and analysis. We would also like to the thank members of the David lab for their support in refining the ideas in this paper.
Biography

Yael David received her B.S. in Biology from SUNY Stony Brook, as a Summa Cum Laude where she was awarded the Howard Hughes Medical Institute research fellowship to perform research in molecular neurobiology. She subsequently moved to the Weizmann Institute of Science in Israel, where she studied the mechanism and regulation of polyubiquitination. She then moved to the Chemistry Department at Princeton University where she combined her experience with Prof. Tom Muir’s expertise in peptide chemistry to develop novel tools towards the mechanistic investigation of histone post-translational modifications including their site-specific manipulation in live cells. In 2016, she brought her powerful research program to the Memorial Sloan Kettering Cancer Center. The David laboratory has performed highly innovative and interdisciplinary research driven by outstanding questions in Epigenetics.
Footnotes
Supplemental Information. Supplemental Figures 1–6 (PDF).
References
- [1].Scott DJ, Kummer L, Tremmel D, Plückthun A, Curr. Opin. Chem. Biol. 2013, 17, 427–435. [DOI] [PubMed] [Google Scholar]
- [2].Overington JP, Al-Lazikani B, Hopkins AL, Nat. Rev. Drug Discovery 2006, 5, 993–996. [DOI] [PubMed] [Google Scholar]
- [3].Herbst RS, Int. J. Radiat. Oncol. Biol. Phys. 2004, 59, S21–S26. [DOI] [PubMed] [Google Scholar]
- [4].Rawlings AE, Biochem. Soc. Trans. 2018, 46, 1541–1549. [DOI] [PubMed] [Google Scholar]
- [5].Krapf D, Curr. Opin. Cell Biol. 2018, 53, 15–21. [DOI] [PubMed] [Google Scholar]
- [6].Gingras AC, Abe KT, Raught B, Curr. Opin. Chem. Biol. 2019, 48, 44–54. [DOI] [PubMed] [Google Scholar]
- [7].Yano Y, Matsuzaki K, Biochim. Biophys. Acta, Biomembr. 2009, 1788, 2124–2131. [DOI] [PubMed] [Google Scholar]
- [8].McKay CS, Blake JA, Cheng J, Danielson DC, Pezacki JP, Chem. Commun. 2011, 47, 10040–10042. [DOI] [PubMed] [Google Scholar]
- [9].Sadaie W, Harada Y, Matsuda M, Aoki K, Mol. Cell. Bio. 2014, 34, 3272–3290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [10].Bosch PJ, Corrêa IR, Sonntag MH, Ibach J, Brunsveld L, Kanger JS, Subramaniam V, Biophys. J. 2014, 107, 803–814. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [11].Galbraith CG, Galbraith JA, Cell Sci J. 2011, 124, 1607–1611. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [12].Shah NH, Muir TW, Chem. Sci. 2014, 5, 446–461. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [13].Shah NH, Muir TW, Isr. J. Chem. 2011, 51, 854–861. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [14].Wood DW, Camarero JA, of Biol J. Chem. 2014, 289, 14512–14519. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [15].Topilina NI, Mills KV, Mobile DNA 2014, 5, 5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [16].David Y, Vila-Perelló M, Verma S, Muir TW, Nat. Chem. 2015, 7, 394–402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [17].Bhagawati M, Terhorst TME, Füsser F, Hoffmann S, Pasch T, Pietrokovski S, Mootz HD, Proc. Natl. Acad. Sci. U. S. A. 2019, 116, 22164–22172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [18].Khoo KK, Galleano I, Gasparri F, Wieneke R, Harms H, Poulsen MH, Chua HC, Wulf M, Tampé R, Pless SA, Nat. Commun. 2020, 11, 1–10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [19].Shah NH, Dann GP, Vila-Perelló M, Liu Z, Muir TW, J. Am. Chem. Soc. 2012, 134, 11338–11341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [20].Vila-Perelló M, Muir TW, Cell 2010, 143, 191–200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [21].Hackel PO, Zwick E, Prenzel N, Ullrich A, Curr. Opin. Cell Biol. 1999, 11, 184–189. [DOI] [PubMed] [Google Scholar]
- [22].Purba E, Saita E.-i., Maruyama I, Cells 2017, 6, 13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [23].Gan HK, Cvrljevic AN, Johns TG, FEBS J 2013, 280, 5350–5370. [DOI] [PubMed] [Google Scholar]
- [24].Maksimovic I, Ray D, Zheng Q, David Y, Methods Enzymol. 2019, 626, 203–222. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.