

Abstract
Pathogenic somatic MTOR variants in the cerebral cortex are a frequent cause of focal cortical dysplasia (FCD). We describe a child with drug and surgery‐resistant focal epilepsy due to FCD type II who developed progressive enlargement and T2 signal hyperintensity in the ipsilateral caudate and lentiform nuclei. Histopathology of caudate nucleus biopsies showed dysmorphic neurons, similar to those in resected cortex. Genetic analysis of frontal and temporal cortex and caudate nucleus identified a pathogenic somatic MTOR variant [NM_004958.4:c.4375G > C (p.Ala1459Pro)] that was not present in blood‐derived gDNA. The mean variant allele frequency ranged from 0.4% to 3.2% in cerebral cortex and up to 5.4% in the caudate nucleus. The basal ganglia abnormalities suggest more widespread, potentially hemispheric dysplasia in this patient, consistent with the pathogenic variant occurring in early cerebral development. This finding provides a potential explanation for persistent seizures in some patients with seemingly complete resection of FCD or disconnection of a dysplastic hemisphere.
Keywords: brain malformation, dysmorphic neuron, epilepsy, genetics, somatic mosaicism
1. INTRODUCTION
Recent studies have identified somatic mosaicism as a major cause of malformations of cortical development including focal cortical dysplasia (FCD) and hemimegalencephaly (HME). 1 , 2 Type II FCD (FCDII) and HME are associated with variants affecting genes in the mTOR signaling pathway, with somatic MTOR variants accounting for 20%–32% of reported cases. 1 , 2 Patients with FCD often undergo epilepsy surgery but >40% have ongoing seizures for various reasons, including inaccurate seizure localization or incomplete resection of dysplasia or the epileptogenic zone. 3 We present the clinical, neuroimaging, histopathological, and genetic findings of a child with an MTOR‐mediated FCD and ipsilateral basal ganglia dysplasia, the latter potentially contributing to her postoperative seizures.
2. MATERIALS AND METHODS
This study was approved by the Human Research Ethics Committee at the Royal Children's Hospital, Melbourne (ID 29077). Written informed consent was obtained from the participant's parents.
The patient had drug‐resistant focal epilepsy and underwent three epilepsy surgeries. Information was obtained from medical records, scalp and intracranial EEG recordings, PET and MRI scans, operative records and photographs, and histopathology.
Resected brain tissue was snap frozen or stored as formalin‐fixed paraffin‐embedded (FFPE) tissue at the time of surgery. Genomic DNA preparation, HaloPlexHS targeted panel sequencing, droplet digital PCR (ddPCR) and immunohistochemistry were performed as previously described. 4 The assay ID of the Custom Taqman SNP Genotyping Assay for MTOR NM_004958.4:c.4375G > C was AN2XHET (Thermo Fisher Scientific) and immunostaining utilized anti‐Phospho‐S6 Ribosomal Protein (Ser235/236) antibody (Cell‐Signalling, #2211, #4858).
3. RESULTS
3.1. Epilepsy history and surgeries
The child is a left‐handed 10‐year‐old girl with mild intellectual disability who presented at age 11 months with focal seizures characterized by left eye blinking, tachycardia, breath‐holding, and right arm dystonia with tremor. These rapidly escalated to multiple daily frequency, despite trials of several anti‐seizure medications (ASM). Video‐EEG showed focal slowing, interictal spike‐slow wave discharges, and ictal rhythms in the left anterior temporal region. MRI showed reduced volume, poor gray‐white differentiation, and unusual sulcation in the left anterior temporal lobe. 18F‐FDG‐PET demonstrated mild relative hypometabolism in the left temporal lobe, insula, and perisylvian cortex. A left anterior temporal neocortical resection was performed at age 2.1 years.
Seizures continued with identical semiology and left‐sided scalp EEG abnormalities. Repeat MRI suggested possible dysplasia in the left superior temporal gyrus anteriorly and the uncus. Repeat 18F‐FDG‐PET showed widespread hypometabolism affecting the left temporal, parieto‐occipital, and insula regions. She then underwent two further epilepsy surgeries, each with prior subdural and depth EEG monitoring from her left frontal, temporal, central, and insula cortex; completion of a left anterior temporal lobectomy at age 4.8 years, and resection of left orbitofrontal, opercular, and anterior insular cortex at age 8.3 years. Again, seizures with identical semiology and left‐sided scalp EEG abnormalities continued following both surgeries, with no benefit from trials of further ASM, pulse steroids, intravenous gammaglobulin, ketogenic diet, and vagus nerve stimulation. Additionally, sirolimus was trialed for 6 weeks, reaching therapeutic dose, without reduction in seizures.
3.2. Basal ganglia abnormality and biopsies
MRI at age 2.1 years demonstrated T2 hyperintensity in the left caudate nucleus, lentiform nucleus, and anterior limb of internal capsule, not present on MRI at age 1.8 years. Serial MRI over the next year showed progressive enlargement and T2 hyperintensity of the left caudate nucleus, without contrast enhancement (Figure 1A–D ). Biopsies of the left caudate nucleus at age 2.6 years and 3.9 years revealed no evidence of tumor or chronic encephalitis. CSF analysis was normal. From age 3.9 to 10 years, serial MRIs showed the basal ganglia abnormality remained stable. FDG‐PET at age 3.1 years demonstrated normal metabolic activity in the left basal ganglia and thalamus but at age 6.3 years showed marked hypometabolism in the left basal ganglia and thalamus (Figure 1E,G ). During the third surgery, a depth electrode was passed stereotactically into the left lentiform nucleus near the caudate. Brief intraoperative electrocorticography (1–2 min) under anesthetic demonstrated no epileptiform activity. Notably, this does not exclude the possible involvement of basal ganglia in seizure generation, as epileptogenic lesions do not always exhibit epileptiform activity on electrocorticography, for example, hypothalamic harmatomas and dysembryoplastic neuroepithelial tumors.
FIGURE 1.
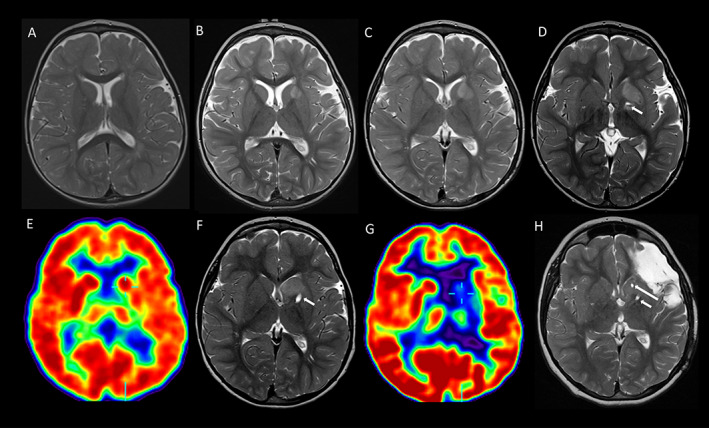
Serial T2‐weighted axial MRI and FDG‐PET images (radiological view) demonstrating the left basal ganglia abnormalities. MRI at age 1.6 (A), 2.1 (B), 2.4 (C), 2.6 (D), 3.9 (F), and 10 (H) years demonstrate initial progression then stability of the left basal ganglia abnormality, maximal in the caudate head. Tracts from the first (short arrow) and second (long arrow) biopsies are evident in (D, F, and H). FDG‐PET at age 3.1 years (E) shows normal metabolism in both basal ganglia and thalami, and relative hypometabolism in the left hemisphere. FDG‐PET at age 6.3 years (G) shows marked hypometabolism in the left basal ganglia and thalamus.
3.3. Histopathology and genetic testing
Histopathology of temporal, frontal, and insula cortical specimens (n = 12) from all surgeries showed dysmorphic neurons. Specimens from the anterior insula at the third surgery also showed balloon cells, therefore meeting the criteria for FCDIIb pathology according to the ILAE guidelines. 5 The dysmorphic neurons and balloon cells stained positive for phosphorylated S6 protein, confirming the upregulation of mTOR pathway (Figure 2A–D ). Histopathology of basal ganglia biopsies was reviewed by three pathologists from two independent institutions. The consensus was of likely dysmorphic neurons with increased size, irregular cytoplasmic outlines, and prominent nucleoli all atypical for normal basal ganglia neurons (Figure 2E,F). Unfortunately, immunostaining to confirm mTOR pathway upregulation could not be performed due to insufficient tissue.
FIGURE 2.
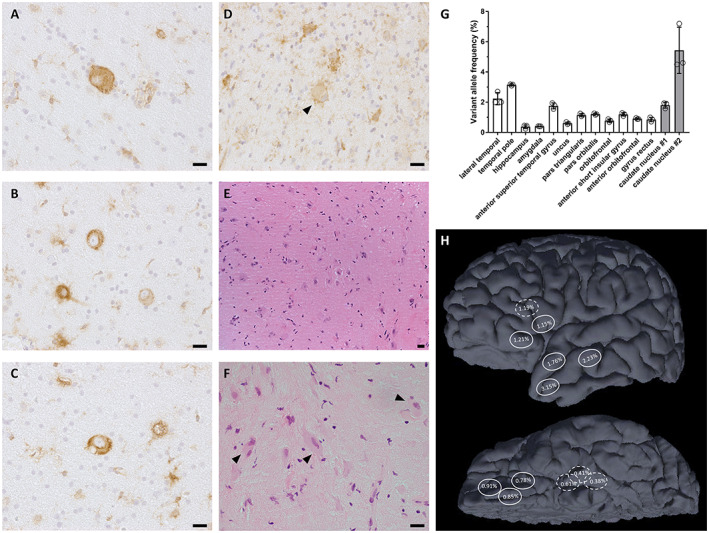
Somatic MTOR variant was detected in dysplastic cortex and basal ganglia. Immunostaining of phosphorylated S6 protein (Ser235/236) revealed (A–C) dysmorphic neurons and (D) balloon cells with mTOR pathway hyperactivation in cortical specimens. (E, F) Hematoxylin and eosin staining of basal ganglia biopsies showed dysmorphic neuron morphology with cytomegaly, irregular cytoplasmic outlines, and prominent nucleoli. (G) Droplet digital PCR confirmed the MTOR NM_004958.4:c.4375G > C (p.Ala1459Pro) variant in 12 frozen cortical specimens from three epilepsy surgeries (white bars), and two FFPE basal ganglia specimens from biopsy at age 2.6 years (gray bars), with mean VAF between 0.4% and 5.4%. Error bars represent standard deviation, n = 3 independent experiments. (H) Approximate locations of cortical specimens in which VAF was determined, taken from operative photographs and notes. Hatched line indicates location deep to the cortical surface. (A–F): ×400 magnification, (E): ×200 magnification, scale bars = 20 μm
Targeted panel sequencing identified a somatic MTOR NM_004958.4:c.4375G > C (p.Ala1459Pro) variant in the temporal pole specimen (13/749 reads, 1.7% VAF) from the first surgery. This variant was validated by ddPCR with 3.2% mean VAF. Subsequent ddPCR analysis of 11 additional frozen specimens of cortex from all three epilepsy surgeries (Figure 2G ) revealed the somatic variant in all samples, with the mean VAF between 0.4% and 3.2%. The somatic variant was also identified in two FFPE basal ganglia biopsy specimens (caudate nucleus #1 and #2) taken at age 2.6 years, with mean VAF 1.8% and 5.4% (Figure 2H ).
4. DISCUSSION
We present a child with refractory focal epilepsy due to a MTOR‐mediated FCDIIb who had an unusual basal ganglia abnormality. Despite the initial growth of this abnormality, there was no evidence of neoplasia or inflammation in the biopsies and further growth halted spontaneously. Notably, the basal ganglia imaging changes seemingly correlated with seizure burden at the time. There were histopathological features suggestive of dysplasia, consistent with previous reports of dysmorphic neurons in the basal ganglia of patients with FCD. 6 , 7 Deep sequencing revealed somatic MTOR mosaicism in both the basal ganglia and overlying cortex. The somatic MTOR c.4375G > C (p.Ala1459Pro) variant is recently reported in an individual affected by FCDIIb. 8 In addition, variants affecting the same amino acid residue including c.4375G > T (p.Ala1459Ser) and c.4376C > A (p.Ala1459Asp) are associated with FCD and HME. 1 , 2 , 9 , 10 , 11 The alanine 1459 residue is located within the FAT domain of the MTOR protein, and functional assays have shown that the alanine to proline substitution results in hyperactivation of the mTOR pathway. 12 , 13
Somatic MTOR variants are reported in FCD and HME, 1 , 2 but only in the cerebral cortex. In this study, we describe a somatic MTOR variant in basal ganglia and cerebral cortex, suggesting that the somatic mosaicism occurred early in hemispheric development and may be extensive in distribution throughout the left hemisphere. The basal ganglia are derived from the telencephalon (caudate nucleus, putamen, and globus pallidus), diencephalon (subthalamic nucleus), and mesencephalon (substantia nigra). 14 Cerebral cortex is also derived from the telencephalon, and so shares the same developmental lineage as the caudate nucleus. This suggests that a common progenitor cell likely acquired the somatic variant before the structural specification of the cerebral cortex and basal ganglia. Although the exact extent of somatic mosaicism is unknown, the early PET imaging abnormalities and high somatic MTOR VAF in the basal ganglia suggest that the entire hemisphere may harbor the somatic variant, resulting in a widespread, potentially hemispheric dysplasia. This contrasts to bottom‐of‐sulcus dysplasia, in which somatic MTOR variants have been shown to be highly localized to the bottom of a single sulcus, reflecting that the variant likely occurred later in cerebral cortex development. 4
The involvement of the basal ganglia in epileptogenesis of FCD is incompletely understood. While the basal ganglia and thalamus play critical roles in seizure propagation and epileptic networks, 15 there is less certainty as to whether they can generate epileptic activity. Given the mounting evidence for epileptogenicity of dysmorphic neurons in FCD and tuberous sclerosis complex, 16 , 17 it is conceivable that the dysmorphic neurons in our patient's dysplastic basal ganglia could play an active role in seizure generation. This notion is supported by previous reports of successful seizure control following the resection of basal ganglia structures in patients with FCD who had ongoing seizures despite the removal of cortical lesions. 6 , 7 Although the underlying genetic basis for these patients is not known, histopathological examination of the resected basal ganglia showed dysmorphic neurons, demonstrating that dysplasia of the basal ganglia can occur in patients with FCD and may contribute to seizure generation. 6 , 7
Somatic MTOR pathogenic variants causing basal ganglia dysplasia may explain why some patients with HME and FCD have postoperative seizures, noting that current approaches to hemispheric malformations such as functional hemispherotomy leave the ipsilateral basal ganglia intact. The dysplastic, ipsilateral basal ganglia that remain following a hemispherotomy might produce seizure manifestations referable to the connected contralateral hemisphere, via minor commissural pathways, potentially leading clinicians to conclude that the patient has a bilateral malformation. Although a somatic MTOR variant was reported in the contralateral hemisphere of a patient with bilateral asymmetric hemispheric dysplasia, 18 and somatic MTOR variants at low VAF in a normal appearing contralateral hemisphere are hypothesized as the reason for failed hemispheric surgery, 18 there is no EEG or clinical evidence to support this as the reason for our patient's ongoing seizures. In our patient, further epilepsy surgery has not been undertaken. Notably, sirolimus treatment did not result in reduction in epileptic seizures in our patient. Similarly, a clinical trial that investigated the efficacy of sirolimus in FCD showed that the reduction of focal seizures did not meet the predetermined level of statistical significance. 19
5. CONCLUSION
In summary, we report a somatic MTOR variant, histological features suggestive of dysplasia, and potential seizure‐related MRI and PET changes in the basal ganglia ipsilateral to FCDIIb in a child with drug‐ and surgery‐resistant focal epilepsy. Somatic mosaicism involving both deep and cortical gray matter suggests a hemispheric malformation in our patient, which may include the basal ganglia in the epileptogenic process. These findings are potentially relevant to other patients with FCD and persistent postoperative seizures, specifically those having undergone hemispherotomy with basal ganglia preservation.
AUTHOR CONTRIBUTIONS
Wei Shern Lee: Conception and design of the study, acquisition and analysis of data, and drafting a significant portion of the manuscript or figures; Emma Macdonald‐Laurs: Conception and design of the study, acquisition and analysis of data, and drafting a significant portion of the manuscript or figures; Sarah EM Stephenson: Conception and design of the study and acquisition and analysis of data; Colleen D'Arcy: Conception and design of the study and acquisition and analysis of data; Duncan MacGregor: Conception and design of the study and acquisition and analysis of data; Richard J Leventer: Conception and design of the study, acquisition and analysis of data, supervision and funding procurement, and drafting a significant portion of the manuscript or figures; Wirginia Maixner: Conception and design of the study and acquisition and analysis of data; A Simon Harvey: Conception and design of the study, acquisition and analysis of data, and drafting a significant portion of the manuscript or figures; Paul J Lockhart: Conception and design of the study, acquisition and analysis of data, supervision and funding procurement, and drafting a significant portion of the manuscript or figures.
FUNDING INFORMATION
This study was funded by the National Health and Medical Research Council (GNT1128933 and GNT1161549) and the Brain Foundation. Additional funding was provided by the Independent Research Institute Infrastructure Support Scheme and the Victorian State Government Operational Infrastructure Program. RJL was supported by a Melbourne Children's Clinician Scientist Fellowship. PJL was supported by the Vincent Chiodo Foundation. EML is supported by the Clifford Family PhD scholarship & RTP scholarship. WSL was supported by the MCRI Early Career Research Fellowship.
CONFLICT OF INTEREST
None of the authors has any conflict of interest to disclose. We confirm that we have read the Journal's position on issues involved in ethical publication and affirm that this study is consistent with those guidelines.
IRB STATEMENT
We confirm that we have read the Journal's position on issues involved in ethical publication and affirm that this report is consistent with those guidelines. This study was not industry sponsored.
ACKNOWLEDGMENTS
We thank the family involved in this research and acknowledge the assistance of Kate Pope and Greta Gillies in manuscript preparation.
Lee WS, Macdonald‐Laurs E, Stephenson SEM, D’Arcy C, MacGregor D, Leventer RJ, et al. Basal ganglia dysplasia and mTORopathy: A potential cause of postoperative seizures in focal cortical dysplasia. Epilepsia Open. 2023;8:205–210. 10.1002/epi4.12678
Wei Shern Lee and Emma Macdonald‐Laurs contributed equally.
Richard J Leventer, Wirginia Maixner, A Simon Harvey and Paul J Lockhart contributed equally.
DATA AVAILABILITY STATEMENT
The data that support the findings of this study are available from the corresponding author upon reasonable request, subject to privacy and ethical restrictions as appropriate
REFERENCES
- 1. Baldassari S, Ribierre T, Marsan E, Adle‐Biassette H, Ferrand‐Sorbets S, Bulteau C, et al. Dissecting the genetic basis of focal cortical dysplasia: a large cohort study. Acta Neuropathol. 2019;138:885–900. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Sim NS, Ko A, Kim WK, Kim SH, Kim JS, Shim KW, et al. Precise detection of low‐level somatic mutation in resected epilepsy brain tissue. Acta Neuropathol. 2019;138:901–12. [DOI] [PubMed] [Google Scholar]
- 3. Blumcke I, Spreafico R, Haaker G, Coras R, Kobow K, Bien CG, et al. Histopathological findings in brain tissue obtained during epilepsy surgery. N Engl J Med. 2017;377:1648–56. [DOI] [PubMed] [Google Scholar]
- 4. Lee WS, Stephenson SEM, Pope K, Gillies G, Maixner W, Macdonald‐Laurs E, et al. Genetic characterization identifies bottom‐of‐sulcus dysplasia as an mTORopathy. Neurology. 2020;95:e2542–51. [DOI] [PubMed] [Google Scholar]
- 5. Najm I, Lal D, Alonso Vanegas M, Cendes F, Lopes‐Cendes I, Palmini A, et al. The ILAE consensus classification of focal cortical dysplasia: an update proposed by an ad hoc task force of the ILAE diagnostic methods commission. Epilepsia. 2022;63:1899–919. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Kaido T, Otsuki T, Kaneko Y, Takahashi A, Kakita A, Takahashi H, et al. Anterior striatum with dysmorphic neurons associated with the epileptogenesis of focal cortical dysplasia. Seizure. 2010;19:256–9. [DOI] [PubMed] [Google Scholar]
- 7. Kaido T, Otsuki T, Kakita A, Sugai K, Saito Y, Sakakibara T, et al. Novel pathological abnormalities of deep brain structures including dysplastic neurons in anterior striatum associated with focal cortical dysplasia in epilepsy. J Neurosurg Pediatr. 2012;10:217–25. [DOI] [PubMed] [Google Scholar]
- 8. Avansini SH, Puppo F, Adams JW, Vieira AS, Coan AC, Rogerio F, et al. Junctional instability in neuroepithelium and network hyperexcitability in a focal cortical dysplasia human model. Brain. 2022;145:1962–77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Nakashima M, Saitsu H, Takei N, Tohyama J, Kato M, Kitaura H, et al. Somatic mutations in the MTOR gene cause focal cortical dysplasia type IIb. Ann Neurol. 2015;78:375–86. [DOI] [PubMed] [Google Scholar]
- 10. Moller RS, Weckhuysen S, Chipaux M, et al. Germline and somatic mutations in the MTOR gene in focal cortical dysplasia and epilepsy. Neurol Genet. 2016;2:e118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Hanai S, Sukigara S, Dai H, Owa T, Horike SI, Otsuki T, et al. Pathologic active mTOR mutation in brain malformation with intractable epilepsy leads to cell‐autonomous migration Delay. Am J Pathol. 2017;187:1177–85. [DOI] [PubMed] [Google Scholar]
- 12. Grabiner BC, Nardi V, Birsoy K, Possemato R, Shen K, Sinha S, et al. A diverse array of cancer‐associated MTOR mutations are hyperactivating and can predict rapamycin sensitivity. Cancer Discov. 2014;4:554–63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Yang H, Jiang X, Li B, Yang HJ, Miller M, Yang A, et al. Mechanisms of mTORC1 activation by RHEB and inhibition by PRAS40. Nature. 2017;552:368–73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Heimer L, Ganglia B. The human brain and spinal cord: functional neuroanatomy and dissection guide. New York, NY: Springer US; 1983. p. 199–209. [Google Scholar]
- 15. Pizzo F, Roehri N, Giusiano B, Lagarde S, Carron R, Scavarda D, et al. The ictal signature of thalamus and basal Ganglia in focal epilepsy: a SEEG study. Neurology. 2021;96:e280–93. [DOI] [PubMed] [Google Scholar]
- 16. Cepeda C, Andre VM, Yamazaki I, et al. Comparative study of cellular and synaptic abnormalities in brain tissue samples from pediatric tuberous sclerosis complex and cortical dysplasia type II. Epilepsia. 2010;51(Suppl 3):160–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Stephenson SEM, Maixner WJ, Barton SM, D'Arcy C, Mandelstam SA, MacGregor D, et al. Resection of tuber centers only for seizure control in tuberous sclerosis complex. Epilepsy Res. 2021;171:106572. [DOI] [PubMed] [Google Scholar]
- 18. Guerrini R, Cavallin M, Pippucci T, Rosati A, Bisulli F, Dimartino P, et al. Is focal cortical dysplasia/epilepsy caused by somatic MTOR mutations always a unilateral disorder? Neurol Genet. 2021;7:e540. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Kato M, Kada A, Shiraishi H, Tohyama J, Nakagawa E, Takahashi Y, et al. Sirolimus for epileptic seizures associated with focal cortical dysplasia type II. Ann Clin Transl Neurol. 2022;9:181–92. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
The data that support the findings of this study are available from the corresponding author upon reasonable request, subject to privacy and ethical restrictions as appropriate