

Abstract
Patients with the heritable cancer disease, Lynch syndrome, carry germline variants in the MLH1, MSH2, MSH6 and PMS2 genes, encoding the central components of the DNA mismatch repair system. Loss-of-function variants disrupt the DNA mismatch repair system and give rise to a detrimental increase in the cellular mutational burden and cancer development. The treatment prospects for Lynch syndrome rely heavily on early diagnosis; however, accurate diagnosis is inextricably linked to correct clinical interpretation of individual variants. Protein variant classification traditionally relies on cumulative information from occurrence in patients, as well as experimental testing of the individual variants. The complexity of variant classification is due to (1) that variants of unknown significance are rare in the population and phenotypic information on the specific variants is missing, and (2) that individual variant testing is challenging, costly and slow. Here, we summarise recent developments in high-throughput technologies and computational prediction tools for the assessment of variants of unknown significance in Lynch syndrome. These approaches may vastly increase the number of interpretable variants and could also provide important mechanistic insights into the disease. These insights may in turn pave the road towards developing personalised treatment approaches for Lynch syndrome.
Subject terms: Protein function predictions, Colon cancer, DNA mismatch repair
Introduction
Lynch syndrome (LS) is a hereditary cancer predisposition disease, caused by germline variants that impair the DNA mismatch repair (MMR) system and lead to the accumulation of spontaneously acquired somatic mutations. Thus, patients with LS suffer from a high cumulative lifetime cancer risk compared to the general population. As a result, LS underlies 3% of all colorectal cancer (CRC) cases, and 8% of cases in young CRC patients (<50 years) [1, 2]. This makes LS the most common cause of genetically predisposed CRC, giving rise to its previous term hereditary non-polyposis colorectal cancer (HNPCC). Accordingly, LS was the first familial cancer disorder to be described and was later found to cause 2% of all endometrial cancers and predispose patients to a range of other cancers, including stomach, brain and ovarian cancer [3–5].
The main therapeutic approach for LS-derived CRC is the partial or complete surgical removal of the colon or colon-rectum [6, 7], while prophylactic surgery has been suggested as an approach to treat LS-derived gynaecological cancers [8–10]. Although chemotherapeutic treatments are given as supplementary treatment in the later stages of LS-derived CRC, its effectiveness remains unclear [6], and probably new treatments such as immunotherapy will in time be implemented in combination with personalised medicine [11]. Thus, the strongest clinical tool at hand is an early diagnosis, together with frequent surveillance and surgical removal of early adenomas [5]. Importantly, early diagnosis relies on a thorough understanding of the underlying germline variant as well as the disease mechanism that relates to the specific variant, which is the focus of this review.
DNA mismatch repair
The MMR system repairs spontaneously arising somatic mutations. During replication, the DNA polymerase may incorporate mismatched nucleotides. The polymerase is a highly “faithful” enzyme and performs proofreading, which reduces the error rate [12]. However, the proofreading works in strong cooperation with the MMR system, which is recruited to sites of replication to correct mismatched base pairs [13] and is necessary to bring down the overall mutation rate [12, 14]. Thus, loss of MMR function is detrimental to genome integrity and sets the stage for cancer development.
LS patients carry germline loss of function (LoF) variants in one of the four key genes involved in MMR: MutL homolog 1 (MLH1), MutS homolog 2 (MSH2), postmeiotic segregation increased 2 (PMS2) and MutS homolog 6 (MSH6). In more rare cases, LS is caused by constitutional epimutations [15] that lead to the silencing of the MLH1 gene [16, 17] or the MSH2 gene [18]. Moreover, germline variants of MutS homolog 3 (MSH3) have been implicated in LS when occurring together with other low-risk alleles [19].
The MMR proteins form heterodimers, MutSα (MSH2-MSH6) and MutLα (MLH1-PMS2) (Fig. 1a), which play key and distinct roles in the MMR pathway (Fig. 1b) [20]. Through the MSH6 protein, MutSα binds the DNA at the site of the mismatch, thus detecting base-base mismatches and small insertion/deletion loops (IDLs) [21]. An alternative complex (MutSβ) consisting of MSH2 and MSH3 corrects larger IDLs and exists in a 1:10 ratio to MutSα. Although the binding mechanism of MSH3 to DNA is unlike that of MSH6, redundancy between MutSα and MutSβ has been suggested [22].
Fig. 1. The human DNA mismatch repair (MMR) system.
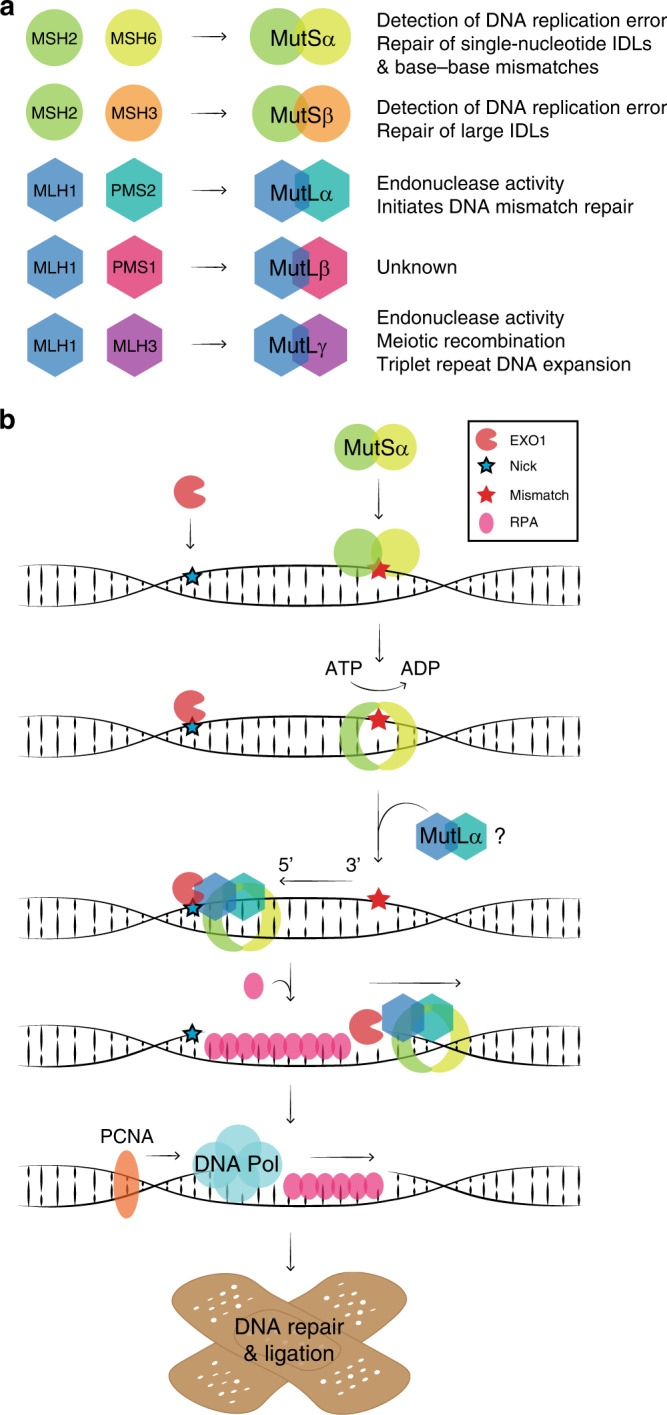
a The MSH2 protein forms dimers with MSH6 (MutSα) and MSH3 (MutSβ). The MLH1 protein forms dimers with PMS2 (MutLα), PMS1 (MutLβ) and MLH3 (MutLγ). Heterodimer functions are listed. IDL, insertion/deletion loop. b A schematic illustration of the 5’ to 3’ MMR pathway. EXO1 binds a nick in the newly synthesised DNA strand 5’ to the mismatch. MutSα recognises the mismatch and undergoes an ATP-dependent conformational change, which locks the complex around the DNA to form a sliding clamp. MutSα moves along the DNA strand and interacts with MutLα, which further binds the DNA. MutSα/MutLα binds EXO1 and moves in the 5’ to 3’ direction allowing for the excision of the mismatch by EXO1. RPA protects the unpaired strand until the DNA polymerase bound to PCNA repairs the strand, after which the DNA ligase seals off any remaining nicks (not shown).
The MMR pathway is bidirectional. The 5’ to 3’ repair proceeds as follows (Fig. 1b): first, a 5’ nick in the newly synthesised strand, which occurs randomly during replication and serves as strand discrimination to the MMR, allows exonuclease 1 (EXO1) to bind [23]. ATP exchange converts the mismatch-bound MutSα into a sliding clamp locked around the DNA [24]. The conformational change releases MutSα from the site of the mismatch and allows it to move along the DNA strand. Moreover, the change permits interaction with MutLα, which subsequently binds the DNA [24]. The MutSα/MutLα complex binds EXO1, after which 5’ to 3’ movement of the MutSα/MutLα/EXO1 complex allows for excision by EXO1 assisted by the single-stranded DNA binding protein complex replication protein A (RPA), which protects the exposed single-stranded DNA. Then, the DNA polymerase in complex with the proliferating cell nuclear antigen (PCNA) sliding clamp as well as the DNA ligase finish the repair process. The 3’ and 5’ directed repair processes seem to proceed differently, although some details remain to be resolved. For example, an in vitro study of the yeast MMR system suggests that 3’ to 5’ repair merely requires action from MutSα, EXO1 and RPA, while 5’ to 3’ repair requires additional action from MutLα and PCNA [25, 26]. Because EXO1 only excises DNA in a 5’ to 3’ direction, the main role of MutLα might be to travel with PCNA in the 5’ direction and nick the newly synthesised strand 5’ to the mismatch. This action is stimulated by PCNA and performed by PMS2 in an MLH1-dependent manner [26–28]. Moreover, whether EXO1 is essential for MMR also remains unclear [20, 29].
MLH1 forms alternative dimers with MLH3 and PMS1 (Fig. 1a). The MLH1-MLH3 dimer (MutLγ) plays a role in meiotic recombination [30] and triplet repeat DNA expansion [31], while the function of the MLH1-PMS1 dimer (MutLβ) is unknown, but may play a minor role in MMR [32]. Lastly, besides DNA repair, the MMR pathway promotes the DNA damage response, including cell cycle arrest and cell death, which further explains the tumorigenic load in MMR-deficient cells [33] and why LS tumours are often associated with resistance to a range of chemotherapies, including temozolomide, 5-fluorouracil and cisplatin [34].
Diagnosis of Lynch syndrome
LS-derived tumours are recognised by loss of one of the four key MMR proteins visualised by immunohistochemical staining of the tumour cells, and by the acquisition of a microsatellite instability (MSI) phenotype. Microsatellites are spans of short tandem repeats within the DNA, which are especially prone to acquiring frameshift mutations, due to DNA polymerase slippage. Loss of MMR results in a hypermutagenic MSI phenotype with altered microsatellite patterns, which is considered a hallmark of LS [35–37]. However, MSI is not exclusive to LS, and is seen in as much as 15% of CRCs, most of which are caused by spontaneous hypermethylation of the MLH1 promoter, leading to MMR loss [37, 38]. Thus, next to the molecular analysis of the tumour, the diagnosis must also rely on the family cancer history, individual cancer history and age of cancer onset. A variety of risk assessment tools have been used over the years: the Amsterdam criteria II [39], the revised Bethesda guidelines [40], simple CRC risk assessment tools [41], and more recently, computational prediction models [42]. Probably, these will be gradually complemented by sequence-based gene panel analyses of the germline. This development is likely to be accelerated by the need for individualised treatment depending on in which gene a pathogenic variant is detected.
Moreover, the diagnosis should involve evaluation of the specific germline variant, and accordingly, the disease course differs dramatically. It is estimated that there is a high general risk in the population (1:279) of carrying a LS-linked MMR variant [43]. Most LS variants are detected in the PMS2 (1:714) and MSH6 (1:758) genes, whereas MLH1 and MSH2 variants are less common (1:1946 and 1:2148, respectively). However, there is a difference in disease penetrance, which seems to be correlated inversely with the population frequencies, since most LS-linked cancers arise from variants in the MLH1 or MSH2 genes [6, 44, 45]. The difference in penetrance has been recognised in several studies [3, 5, 6, 46], but is not well understood. Importantly, penetrance is affected by environmental factors and the individual genetic makeup of patients, like co-segregating germline variants and modifier genes that may cause stronger or milder clinical effects. Thus, diverging penetrance patterns between individual patients is a major caveat for the classification of disease-linked protein variants. Lastly, while LS is dominantly inherited, disease development follows Knudson’s two-hit hypothesis [47], and thus relies on a “second hit” of the wild-type allele to ablate the MMR function and significantly increase the risk for further mutations [48], meaning the expressivity of the disease ultimately also relies on the timing of the second hit.
Notably, the MMR proteins are not equally dependent on each other, which may partially explain this difference in penetrance. For instance, the MSH2 protein is stabilised by MSH6, causing MSH2 levels to drop upon loss of MSH6 [49]. Moreover, interaction with MLH1 rescues PMS2 from degradation [50, 51], thus leaving PMS2 protein levels to depend on the presence of MLH1 [52], while MLH1 levels do not rely on PMS2. This means that the cell effectively can lose both MLH1 and PMS2 if MLH1 is destabilised [51], which is why the loss of both MLH1 and PMS2 visualised by immunohistochemical staining may indicate a germline MLH1 variant. Indeed, this depends on the specific variant, as some MLH1 variants induce solitary loss of PMS2 [53]. Likely, these variants alter the PMS2-interaction interface of the MLH1 protein, thus preventing the stabilisation of PMS2.
The specific variants also define how the disease is expressed. For example, some studies find MSH6-linked cancers to induce a high risk of gynaecological cancers [3, 5, 54, 55], which indicates that the genes play different roles in tumorigenesis. Conclusively, a thorough description of individual disease-causative LS variants is crucial for correct diagnosis and treatment of patients.
Variants and disease mechanism
To improve diagnosis, known LS-linked MMR variants have been collected in the InSiGHT database (http://insight-database.org/) and assigned to one of five classes of pathogenicity based on the data published on each variant [56]. The types of germline variants distribute differently between the classifications. For example, synonymous substitutions or intron variants would likely have no effect on the given MMR protein structure or function and predominate in class 1 (benign) and 2 (likely benign) [56]. In contrast, large IDLs, non-sense (truncating) and splice site variants would, in most cases, cause significant alterations to the protein sequence, expression level and structure. These will obviously be disruptive to the protein function and are highly represented in class 4 (likely pathogenic) and class 5 (pathogenic) [56]. However, missense variants are represented in all five different InSiGHT categories and make up the majority of variants in class 3 (variants of uncertain significance, VUS) and also constitute a significant part of the classified pathogenic MLH1 (40%), MSH2 (30%), MSH6 (50%) and PMS2 (60%) variants [45].
Missense variants are abundant in the VUS pool because their effects on a protein may range from undetectable to detrimental and greatly depend on the type and location of the amino acid substitution. Even a conservative amino acid substitution within or near an active site or binding interface may affect protein function, however, many non-conservative substitutions occur outside the active site, and thus mainly affect the stability of the protein.
Most protein structures are only marginally stable [57], and exist in an equilibrium between a folded and unfolded state. However, the folded and functional state of the protein—the native structure—is under normal conditions more stable than the unfolded state, which drives the equilibrium towards this native state. A germline variant that changes the protein sequence may destabilise the protein structure and shift the equilibrium towards the unfolded state. In the unfolded state, the protein may expose degradation signals (degrons) that are buried in the native structure and therefore becomes vulnerable to degradation. The exposed degrons recruit the cellular protein quality control (PQC) system, which directs the protein for proteasomal degradation (Fig. 2a). The PQC and degradation of misfolded proteins have recently been reviewed [58–62]. Briefly, most misfolded proteins are degraded through the ubiquitin–proteasome system (UPS) and thus rely on ubiquitin conjugation achieved by the sequential actions of three enzymes: a ubiquitin-activating enzyme (E1), a ubiquitin-conjugating enzyme (E2) and a ubiquitin-protein ligase (E3) (Fig. 2a). Following activation by an E1 and transfer to an E2, the substrate-specific E3 enzyme covalently links the ubiquitin molecule to the target protein. This mediates the binding of the substrate to the 26S proteasome, which in turn catalyzes the degradation of the target protein.
Fig. 2. Proteasomal degradation of misfolded proteins.
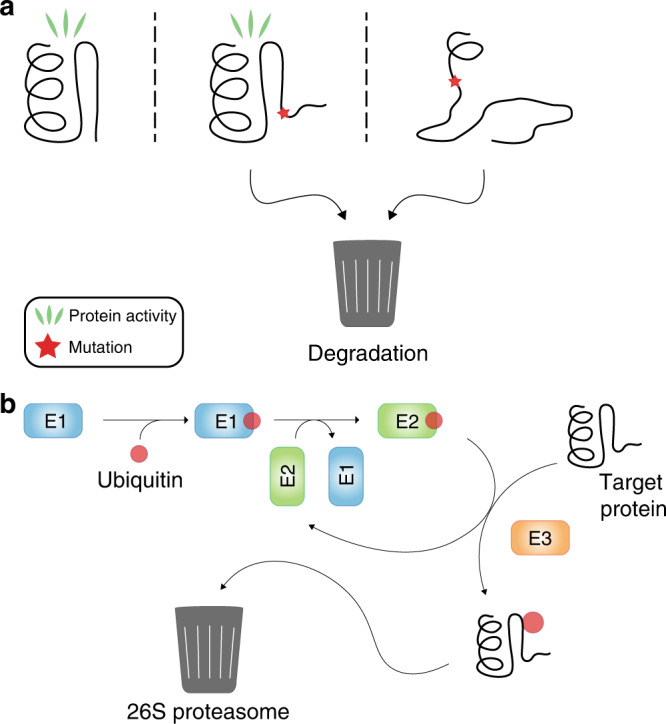
a Overview of the ubiquitin–proteasome system (UPS). A ubiquitin moiety is activated by an E1 enzyme and transferred to an E2 enzyme. From here, the ubiquitin is transferred to the target protein by the means of an E3 ubiquitin-protein ligase. Ubiquitination promotes binding at the proteasome and subsequent degradation of the target protein. b A wild-type protein (left) mainly exists in the functional native structure that is not degraded. Mutations affect the protein structure to mild (centre) or more severe (right) degrees and may obstruct the protein function. Both mildly and severely misfolded proteins risk undergoing proteasomal degradation.
In theory, even mild alterations to or destabilizations of the protein structure can cause protein degradation (Fig. 2b). Accordingly, the UPS also regulates the cellular abundance of wild-type proteins, including the MSH2 protein [49, 63–65]. However, the degradation of misfolded protein variants occurs more rapidly and is a well-established disease mechanism that appears to be the underlying cause for many cases of LS, causing low cellular levels of MSH2 and MLH1 variants [66–69], which is diagnosed by immunohistochemical staining of LS tumours. In some cases of LS, the unstable variant may still be functional. In the case of MSH2, inhibition of the UPS was shown to restore MMR function in cells with structurally unstable but functional MSH2 variants [70]. In these specific cases, inhibiting degradation could serve as a prophylactic treatment for patients carrying these variants [71]. Ideally, the rescue of a functional variant from degradation would restore the MMR function sufficiently and relieve the patient from the increased cancer risk—a strong incitement for obtaining a mechanistic understanding of individual LS variant effects at the molecular level [72].
The effects of variants of uncertain significance
Interpretation of genetic variants is inherently difficult. Close to 90% of LS-linked missense variants are classified as VUS [66], and correct classification is a large and difficult task, which needs to be solved before clinicians can accurately diagnose and treat many suspected LS patients. The majority of variants identified by sequencing have never been observed in the human population before, hence the simple lack of preceding events in the population causes most variants to be classified as VUS [73]. Of course, the vast number of VUS is not specific for MMR proteins, and the majority (99%) of missense variants, including LS-linked VUSs, are rare [74] and have only been observed once or a few times in patients, which makes it difficult to determine potential disease causality. In contrast, variants that occur frequently in the population, such as founder mutations [75], are well-described, e.g., the highly penetrant MSH2 A636P variant, which underlies a third of all LS cancers in the Ashkenazi Jewish population [76]. A variant can also be categorised as a VUS if there exists conflicting evidence for its pathogenicity. Reasons for this could include co-segregation with a pathogenic variant in another MMR gene, inconclusive IHC or MSI analysis, or patients with a low prevalence of LS cancers among relatives. In such cases, functional assays can help elucidate the variant effect on MMR and thus reclassify the variant as likely pathogenic/benign [77–79].
Ideally, clinicians should have access to a complete map of the effects of every potential missense variant within a given protein. Once they detect a rare variant, a complete map of variant effects could be an excellent resource for classifying such rare variants. Figure 3 presents an overview of the methods used for variant interpretation. Traditionally, low-throughput studies of function and abundance are applied retrospectively to analyse individual variants and ensure correct classification (Fig. 3a). Functional testing of individual variants in the MMR genes will possibly be made more readily available in the clinic with flow cytometry-based MMR assays that can be performed directly in blood cells from individuals that meet the Amsterdam II criteria [80]. However, individual verification is a slow, costly and labour-intensive process and applying such analysis to all possible missense variants in a protein is a daunting task.
Fig. 3. Overview of tools used for testing individual variant effects.

a Low-throughput lab-based experimental tools, i.e., individual variant testing. b Computational prediction tools, i.e., FoldX, GEMME, etc. c High-throughput lab-based experimental tools, i.e., MAVEs. Created with BioRender.com.
Computational methods for variant interpretation
The implementation of computational prediction tools for missense variant effects has been an important step towards shedding light on the VUS pool. These tools were developed using both evolutionary conservation and structural data as input, and most are trained on clinically labelled data of benign and pathogenic variants (Fig. 3b) [73]. As evolution selects for protein function, residues that are critical for catalysis, protein or ligand binding, or structural stability can all be identified in this manner. However, methods based exclusively on sequence conservation will therefore on their own not provide any mechanistic information as to why a particular variant might be damaging. Conversely, structure-based methods may pinpoint variants that are likely to cause protein destabilization or loss of key interactions, but these methods will be blind to variants that may destroy enzymatic function without affecting the global protein stability. Combining evolution-based methods with structural stability calculations can provide an understanding of the various loss of function mechanisms inflicted by protein variants [81].
A detailed discussion of the many different approaches to predict variant effects and pathogenicity is beyond the scope of this review. These include, however, specific models for MMR genes and LS [82, 83]. Many of the models for variant effect prediction, such as PolyPhen-2 [84], PROVEAN [85], SIFT [86], EVMutation [87] and GEMME [88], generally use multiple sequence alignments of homologous proteins as input to construct a statistical model that estimates the likelihood of a given variant, and outputs an evolutionary sequence score. The algorithms consider both the conservation at individual positions as well as the co-evolution of amino acid pairs, the co-evolution term being the major difference between the algorithms. Evolutionary sequence analysis is also the basis of the recently published mutational effect predictor DeepSequence [89] and pathogenicity predictor EVE [90], and these types of prediction methods have been shown to work well for both MSH2 [67, 68, 90] and MLH1 [50].
Computational modelling of amino acid substitutions directly on the 3D structure of the given protein allows the prediction of the change in folding energy between the protein variant and the wild-type. The tools Rosetta [91] and FoldX [92] predict the change in stability with an accuracy of about 1 kcal mol−1. The FoldX algorithm was developed to predict the structural destabilization afflicted by a missense substitution within a protein [93]. In simplified terms, these algorithms utilise information about the protein structure to calculate the difference in stability of a missense protein variant and the wild-type protein. Thus, one can predict the individual effects of all possible variants at any position within a structure. Several previous studies of the MMR proteins have found a correlation between LS and destabilised missense MMR variants [70, 94, 95]. Thus, in previous research, the FoldX and Rosetta algorithms were used to produce stability predictions of all possible missense variants of MSH2 [68], MLH1 [50] and a range of other proteins [96–98]. Here, it was shown that the effects of the majority of previously described missense variants with known clinical consequences could be accurately predicted, and that the predictions matched with cellular measurements of the individual variant abundances and protein function. Thus, an advantage of structure-based disease predictors is that they help provide the molecular mechanism underlying the damaging effect of a variant.
Multiplexed assays of variant effects
In another attempt to truly overcome the issues of variant interpretation, new laboratory-based high-throughput approaches have been developed, known as deep mutational scanning or multiplexed assays of variant effect (MAVE) technologies (Fig. 3c) [99]. These assays score the effects of all possible missense variants of a protein, and have been reported to outperform several computational prediction tools [66, 74, 100, 101]. However, in some cases, computational predictions have also been observed to outperform MAVEs in variant classification [90, 102].
MAVE technologies can be used to measure function, abundance, or interaction of libraries consisting of all possible single amino acid variants in a protein of interest. For example, the Variant Abundance by Massively Parallel sequencing (VAMP-seq) method measures abundance of fluorescently tagged protein variants, and thus scores their individual difference in pathogenicity in a high-throughput manner, when low abundance is the cause of loss of function [103]. Moreover, one can use the MAVE methods to test for variant function, by applying a selective pressure to the variant library, which was recently done for MSH2.
Multiplexed assays of MSH2 function
Recently, the function of missense MSH2 variants has been assayed in a high-throughput manner by Ollodart et al. [69] and Jia et al. [66]. In yeast, Ollodart et al. selected for LoF MSH2 variants by measuring the cellular mutation rate in a library of 185 different MSH2 variants. They selected cells with high mutation rates by measuring their resistance to canavanine, which is only tolerated by cells with a mutated CAN1 gene. With this method, they successfully distinguished pathogenic and benign MSH2 variants. At an even larger scale, Jia et al. utilise a human MSH2 knockout cell line in which they introduce a saturated library of MSH2 variants, including nearly all possible single amino acid missense variants. Treatment with 6-thioguanine (6TG) is toxic to MMR-proficient cells and allowed the authors to select for MMR-deficient MSH2 variants and identify these by sequencing. From this study, they estimate that 10–11% of missense variants are deleterious to MSH2 function [66] and that 7-8% of MSH2 VUS show a pathogenic phenotype, meaning the vast majority of MSH2 VUS are likely benign. This fits with predictions based on analyses of protein stability and sequence conservation that show that MSH2 is relatively tolerant to missense variants, and that most variants are predicted to have only minor effects [67, 68].
Current challenges in high-throughput variant classification
However, despite the promising progress, both functional MAVE analyses and computational prediction tools still wrongly classify some variants. In case of MAVE studies, misclassifications can occur if the employed assay is not sufficiently sensitive or does not capture all functional aspects of the protein. Further, it is important to establish guidelines for how to use MAVE functional data in clinical variant classification, as in addition to the guidelines that already exist for low-throughput functional assays [104]. Including functional data has been demonstrated to significantly aid in the reclassification of VUS [77].
Regarding computational pathogenicity predictions, the precision of the current prediction tools is promising, and it could certainly be a strong supportive tool in the clinic. Although some tools are challenged by reports of low sensitivity and a high number of false positives [105, 106], the advantages of computational prediction tools might in some situations outweigh their weaknesses, as the applicability, cost and efficiency is remarkable compared to individual variant testing. Indeed, new tools are being developed and perfected to overcome some of the inherent problems with the prediction algorithms [107, 108].
MAVE data can serve as useful benchmarks for computational variant effect predictors [73, 81, 87, 90, 102, 109], and may also be used to train variant effect predictors. This will likely lead to refined predictors in the near future.
Potential for restabilization of disease-causing MSH2 variants
MAVE technologies may also help to uncover particular disease mechanisms on the molecular level, for instance, by helping to determine which gene variants disrupt catalytic activity, interactions, or cause destabilization. For those variants that are degraded, the methods may shed light on the specificity of the PQC system, i.e., how does the PQC engage with proteins and what characterises the PQC-bound elements within the target proteins. For example, a recent study in E. coli shows how expression of PQC proteins, specifically the Lon protease, alters the mutational landscape of a model protein, and thus constrains which variants are allowed [110]. In a similar manner, the cellular PQC apparatus will also ultimately decide the threshold for which destabilised missense variants in MMR components will be pathogenic. Combining MAVE experiments with perturbations of the PQC apparatus will potentially allow the identification of variants that will regain function upon increased protein levels (Fig. 2b). This pool of variants is interesting, as they would be targetable from a clinical perspective. In principle, stabilisation of the protein would prevent disease development in the patient, and so far, this approach is used for the rescue of the cystic fibrosis CFTR F508Δ variant [111–113]. Proof-of-concept studies in yeast have demonstrated that restabilization of some pathogenic MSH2 variants can restore MMR function [70]. Thus, it may be possible to prevent cancer development for LS patients that carry “rescuable” MSH2 variants through the development of pharmaceuticals that act to increase the cellular amount of natively folded MSH2.
Concluding remarks
MAVE-based experimental approaches combined with computational biology have already brought us key insights for VUS in Lynch Syndrome. Importantly, these tools have provided us with new approaches to study basic scientific questions focused on how individual variants operate through effects on catalysis, interactions or protein stability, and this knowledge can in turn improve the diagnosis and potential treatment of not only Lynch syndrome but also other genetic diseases.
At the moment, use of computational evidence is restricted to “supporting evidence only” according to clinical guidelines such as those provided by the ACMG-AMP [114]. These guidelines specify that multiple computational predictions may not be considered independent due to concerns over overlap in the underlying algorithms and the data that they are trained on. While it is true that some programmes use similar approaches and are thus not independent, others can be considered complementary. Moreover, rapid growth of sequence databases can lead to improved variant consequence predictions, such that the guidelines with respect to computational results should perhaps be revised.
The value of including MAVE data in clinical variant classification was demonstrated in a recent study on the TP53, PTEN and BRCA1 proteins [115], where MAVE data was integrated with available clinical data to correctly classify VUS. The same approach may be applied to MSH2, for which both MAVE and clinical data exist. Similar prospective studies for other MMR proteins could help provide broader knowledge on MMR variant effects and enable personalised treatment for patients suffering from Lynch Syndrome. Indeed, the importance of Lynch Syndrome and the promise of patient benefits from personalised approaches is also recognised in its listing among genes to be prioritised for systematic assessment of variant consequences [116, 117].
Author contributions
ABA and SVN prepared the figures. ABA, SVN, IB, AS, KLL and RHP wrote the manuscript.
Funding
Our research in this area is funded by the Novo Nordisk Foundation Challenge programme PRISM (NNFOC180033950; to AS, KLL and RHP), the Lundbeck Foundation (R272-2017-4528 to AS), and the Danish Council for Independent Research, Natural Sciences (7014-00039B to RHP).
Competing interests
The authors declare no competing interests.
Ethics approval and consent to participate
Not applicable.
Consent to publish
Not applicable.
Footnotes
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Contributor Information
Sofie V. Nielsen, Email: svnielsen@bio.ku.dk
Kresten Lindorff-Larsen, Email: lindorff@bio.ku.dk.
Rasmus Hartmann-Petersen, Email: rhpetersen@bio.ku.dk.
References
- 1.Pearlman R, Frankel WL, Swanson B, Zhao W, Yilmaz A, Miller K, et al. Prevalence and spectrum of germline cancer susceptibility gene mutations among patients with early-onset colorectal cancer. JAMA Oncol. 2017;3:464–71. doi: 10.1001/jamaoncol.2016.5194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Yurgelun MB, Kulke MH, Fuchs CS, Allen BA, Uno H, Hornick JL, et al. Cancer susceptibility gene mutations in individuals with colorectal cancer. J Clin Oncol. 2017;35:1086–95. doi: 10.1200/JCO.2016.71.0012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Dominguez-Valentin M, Sampson JR, Seppälä TT, ten Broeke SW, Plazzer J-P, Nakken S, et al. Cancer risks by gene, age, and gender in 6350 carriers of pathogenic mismatch repair variants: findings from the Prospective Lynch Syndrome Database. Genet Med. 2020;22:15–25. doi: 10.1038/s41436-019-0596-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Hampel H, Frankel WL, Martin E, Arnold M, Khanduja K, Kuebler P, et al. Screening for the Lynch syndrome (hereditary nonpolyposis colorectal cancer) N Engl J Med. 2005;352:1851–60. doi: 10.1056/NEJMoa043146. [DOI] [PubMed] [Google Scholar]
- 5.Møller P, Seppälä T, Bernstein I, Holinski-Feder E, Sala P, Evans DG, et al. Cancer incidence and survival in Lynch syndrome patients receiving colonoscopic and gynaecological surveillance: first report from the prospective Lynch syndrome database. Gut. 2017;66:464–72. doi: 10.1136/gutjnl-2015-309675. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Boland PM, Yurgelun MB, Boland CR. Recent progress in lynch syndrome and other familial colorectal cancer syndromes. CA Cancer J Clin. 2018;68:217–31. doi: 10.3322/caac.21448. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Seppälä TT, Latchford A, Negoi I, Sampaio Soares A, Jimenez-Rodriguez R, Sánchez-Guillén L, et al. European guidelines from the EHTG and ESCP for Lynch syndrome: an updated third edition of the Mallorca guidelines based on gene and gender. Br J Surg. 2021;108:484–98. doi: 10.1002/bjs.11902. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Crosbie EJ, Ryan NAJ, Arends MJ, Bosse T, Burn J, Cornes JM, et al. The Manchester International Consensus Group recommendations for the management of gynecological cancers in Lynch syndrome. Genet Med J Am Coll Med Genet. 2019;21:2390–400. doi: 10.1038/s41436-019-0489-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Grindedal EM, Renkonen-Sinisalo L, Vasen H, Evans G, Sala P, Blanco I, et al. Survival in women with MMR mutations and ovarian cancer: a multicentre study in Lynch syndrome kindreds. J Med Genet. 2010;47:99–102. doi: 10.1136/jmg.2009.068130. [DOI] [PubMed] [Google Scholar]
- 10.Schmeler KM, Lu KH. Gynecologic cancers associated with Lynch syndrome/HNPCC. Clin Transl Oncol. 2008;10:313–7. doi: 10.1007/s12094-008-0206-9. [DOI] [PubMed] [Google Scholar]
- 11.Therkildsen C, Jensen LH, Rasmussen M, Bernstein I. An update on immune checkpoint therapy for the treatment of Lynch syndrome. Clin Exp Gastroenterol. 2021;14:181–97. doi: 10.2147/CEG.S278054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.StCharles JA, Liberti SE, Williams JS, Lujan SA, Kunkel TA. Quantifying the contributions of base selectivity, proofreading and mismatch repair to nuclear DNA replication in Saccharomyces cerevisiae. DNA Repair. 2015;31:41–51. doi: 10.1016/j.dnarep.2015.04.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Huang Y, Li G-M. DNA mismatch repair preferentially safeguards actively transcribed genes. DNA Repair. 2018;71:82–6. doi: 10.1016/j.dnarep.2018.08.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Preston BD, Albertson TM, Herr AJ. DNA replication fidelity and cancer. Semin Cancer Biol. 2010;20:281–93. doi: 10.1016/j.semcancer.2010.10.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Hitchins MP. Inheritance of epigenetic aberrations (constitutional epimutations) in cancer susceptibility. Adv Genet. 2010;70:201–43. doi: 10.1016/B978-0-12-380866-0.60008-3. [DOI] [PubMed] [Google Scholar]
- 16.Hitchins MP, Ward RL. Constitutional (germline) MLH1 epimutation as an aetiological mechanism for hereditary non-polyposis colorectal cancer. J Med Genet. 2009;46:793–802. doi: 10.1136/jmg.2009.068122. [DOI] [PubMed] [Google Scholar]
- 17.Ward RL, Dobbins T, Lindor NM, Rapkins RW, Hitchins MP. Identification of constitutional MLH1 epimutations and promoter variants in colorectal cancer patients from the Colon Cancer Family Registry. Genet Med. 2013;15:25–35. doi: 10.1038/gim.2012.91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Ligtenberg MJL, Kuiper RP, Chan TL, Goossens M, Hebeda KM, Voorendt M, et al. Heritable somatic methylation and inactivation of MSH2 in families with Lynch syndrome due to deletion of the 3′ exons of TACSTD1. Nat Genet. 2009;41:112–7. doi: 10.1038/ng.283. [DOI] [PubMed] [Google Scholar]
- 19.Duraturo F, Liccardo R, Cavallo A, De Rosa M, Grosso M, Izzo P. Association of low-risk MSH3 and MSH2 variant alleles with Lynch syndrome: probability of synergistic effects. Int J Cancer. 2011;129:1643–50. doi: 10.1002/ijc.25824. [DOI] [PubMed] [Google Scholar]
- 20.Jiricny J. The multifaceted mismatch-repair system. Nat Rev Mol Cell Biol. 2006;7:335–46. doi: 10.1038/nrm1907. [DOI] [PubMed] [Google Scholar]
- 21.Dufner P, Marra G, Räschle M, Jiricny J. Mismatch recognition and DNA-dependent stimulation of the ATPase activity of hMutSα is abolished by a single mutation in the hMSH6 subunit *. J Biol Chem. 2000;275:36550–5. doi: 10.1074/jbc.M005987200. [DOI] [PubMed] [Google Scholar]
- 22.Gupta S, Gellert M, Yang W. Mechanism of mismatch recognition revealed by human MutSβ bound to unpaired DNA loops. Nat Struct Mol Biol. 2012;19:72–8. doi: 10.1038/nsmb.2175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Reyes GX, Schmidt TT, Kolodner RD, Hombauer H. New insights into the mechanism of DNA mismatch repair. Chromosoma. 2015;124:443–62. doi: 10.1007/s00412-015-0514-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Groothuizen FS, Winkler I, Cristóvão M, Fish A, Winterwerp HH, Reumer A, et al. MutS/MutL crystal structure reveals that the MutS sliding clamp loads MutL onto DNA. eLife. 2015;4:e06744. doi: 10.7554/eLife.06744. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Bowen N, Smith CE, Srivatsan A, Willcox S, Griffith JD, Kolodner RD. Reconstitution of long and short patch mismatch repair reactions using Saccharomyces cerevisiae proteins. Proc Natl Acad Sci USA. 2013;110:18472–7. doi: 10.1073/pnas.1318971110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Jiricny J. Postreplicative mismatch repair. Cold Spring Harb Perspect Biol. 2013;5:a012633. doi: 10.1101/cshperspect.a012633. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Groothuizen FS, Sixma TK. The conserved molecular machinery in DNA mismatch repair enzyme structures. DNA Repair. 2016;38:14–23. doi: 10.1016/j.dnarep.2015.11.012. [DOI] [PubMed] [Google Scholar]
- 28.Gueneau E, Dherin C, Legrand P, Tellier-Lebegue C, Gilquin B, Bonnesoeur P, et al. Structure of the MutLα C-terminal domain reveals how Mlh1 contributes to Pms1 endonuclease site. Nat Struct Mol Biol. 2013;20:461–8. doi: 10.1038/nsmb.2511. [DOI] [PubMed] [Google Scholar]
- 29.Goellner EM, Putnama CD, Kolodnera RD. Exonuclease 1-dependent and independent mismatch repair. DNA Repair. 2015;32:24–32. doi: 10.1016/j.dnarep.2015.04.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Cannavo E, Sanchez A, Anand R, Ranjha L, Hugener J, Adam C, et al. Regulation of the MLH1–MLH3 endonuclease in meiosis. Nature. 2020;586:618–22. doi: 10.1038/s41586-020-2592-2. [DOI] [PubMed] [Google Scholar]
- 31.Kadyrova LY, Gujar V, Burdett V, Modrich PL, Kadyrov FA. Human MutLγ, the MLH1–MLH3 heterodimer, is an endonuclease that promotes DNA expansion. Proc Natl Acad Sci USA. 2020;117:3535–42. doi: 10.1073/pnas.1914718117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Campbell CS, Hombauer H, Srivatsan A, Bowen N, Gries K, Desai A, et al. Mlh2 is an accessory factor for DNA mismatch repair in Saccharomyces cerevisiae. PLOS Genet. 2014;10:e1004327. doi: 10.1371/journal.pgen.1004327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Gupta D, Heinen CD. The mismatch repair-dependent DNA damage response: mechanisms and implications. DNA Repair. 2019;78:60–9. doi: 10.1016/j.dnarep.2019.03.009. [DOI] [PubMed] [Google Scholar]
- 34.Lynch HT, Snyder CL, Shaw TG, Heinen CD, Hitchins MP. Milestones of Lynch syndrome: 1895–2015. Nat Rev Cancer. 2015;15:181–94. doi: 10.1038/nrc3878. [DOI] [PubMed] [Google Scholar]
- 35.De’ Angelis GL, Bottarelli L, Azzoni C, De’ Angelis N, Leandro G, Di Mario F, et al. Microsatellite instability in colorectal cancer. Acta Bio-Med Atenei Parm. 2018;89:97–101. doi: 10.23750/abm.v89i9-S.7960. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Imai K, Yamamoto H. Carcinogenesis and microsatellite instability: the interrelationship between genetics and epigenetics. Carcinogenesis. 2008;29:673–80. doi: 10.1093/carcin/bgm228. [DOI] [PubMed] [Google Scholar]
- 37.Yamamoto H, Imai K. Microsatellite instability: an update. Arch Toxicol. 2015;89:899–921. doi: 10.1007/s00204-015-1474-0. [DOI] [PubMed] [Google Scholar]
- 38.Herman JG, Umar A, Polyak K, Graff JR, Ahuja N, Issa JP, et al. Incidence and functional consequences of hMLH1 promoter hypermethylation in colorectal carcinoma. Proc Natl Acad Sci USA. 1998;95:6870–5. doi: 10.1073/pnas.95.12.6870. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Vasen HF, Watson P, Mecklin JP, Lynch HT. New clinical criteria for hereditary nonpolyposis colorectal cancer (HNPCC, Lynch syndrome) proposed by the International Collaborative group on HNPCC. Gastroenterology. 1999;116:1453–6. doi: 10.1016/S0016-5085(99)70510-X. [DOI] [PubMed] [Google Scholar]
- 40.Umar A, Boland CR, Terdiman JP, Syngal S, Chapelle A, de la, Rüschoff J, et al. Revised Bethesda Guidelines for hereditary nonpolyposis colorectal cancer (Lynch syndrome) and microsatellite instability. JNCI J Natl Cancer Inst. 2004;96:261–8. doi: 10.1093/jnci/djh034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Kastrinos F, Allen JI, Stockwell DH, Stoffel EM, Cook EF, Mutinga ML, et al. Development and validation of a colon cancer risk assessment tool for patients undergoing colonoscopy. Am J Gastroenterol. 2009;104:1508–18. doi: 10.1038/ajg.2009.135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Giardiello FM, Allen JI, Axilbund JE, Boland RC, Burke CA, Burt RW, et al. Guidelines on genetic evaluation and management of Lynch syndrome: a consensus statement by the US Multi-Society Task Force on colorectal cancer. J Am Coll Gastroenterol ACG. 2014;109:1159–79. doi: 10.1038/ajg.2014.186. [DOI] [PubMed] [Google Scholar]
- 43.Win AK, Jenkins MA, Dowty JG, Antoniou AC, Lee A, Giles GG, et al. Prevalence and penetrance of major genes and polygenes for colorectal cancer. Cancer Epidemiol Biomark Prev Publ Am Assoc Cancer Res Cosponsored Am Soc Prev Oncol. 2017;26:404–12. doi: 10.1158/1055-9965.EPI-16-0693. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Moreira L, Balaguer F, Lindor N, de la Chapelle A, Hampel H, Aaltonen LA, et al. Identification of Lynch syndrome among patients with colorectal cancer. J Am Med Assoc. 2012;308:1555–65. doi: 10.1001/jama.2012.13088. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Peltomäki P. Update on Lynch syndrome genomics. Fam Cancer. 2016;15:385–93. doi: 10.1007/s10689-016-9882-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Dominguez-Valentin M, Plazzer J-P, Sampson JR, Engel C, Aretz S, Jenkins MA, et al. No difference in penetrance between truncating and missense/aberrant splicing pathogenic variants in MLH1 and MSH2: a prospective Lynch Syndrome Database Study. J Clin Med. 2021;10:2856. doi: 10.3390/jcm10132856. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Knudson AG. Mutation and cancer: statistical study of retinoblastoma. Proc Natl Acad Sci USA. 1971;68:820–3. doi: 10.1073/pnas.68.4.820. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Hemminki A, Peltomäki P, Mecklin J-P, Järvinen H, Salovaara R, Nyström-Lahti M, et al. Loss of the wild type MLH1 gene is a feature of hereditary nonpolyposis colorectal cancer. Nat Genet. 1994;8:405–10. doi: 10.1038/ng1294-405. [DOI] [PubMed] [Google Scholar]
- 49.Zhang M, Xiang S, Joo H-Y, Wang L, Williams KA, Liu W, et al. HDAC6 deacetylates and ubiquitinates MSH2 to maintain proper levels of MutSα. Mol Cell. 2014;55:31–46. doi: 10.1016/j.molcel.2014.04.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Abildgaard AB, Stein A, Nielsen SV, Schultz-Knudsen K, Papaleo E, Shrikhande A, et al. Computational and cellular studies reveal structural destabilization and degradation of MLH1 variants in Lynch syndrome. eLife. 2019;8:e49138. doi: 10.7554/eLife.49138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Hinrichsen I, Weßbecher IM, Huhn M, Passmann S, Zeuzem S, Plotz G, et al. Phosphorylation-dependent signaling controls degradation of DNA mismatch repair protein PMS2. Mol Carcinog. 2017;56:2663–8. doi: 10.1002/mc.22709. [DOI] [PubMed] [Google Scholar]
- 52.Mohd AB, Palama B, Nelson SE, Tomer G, Nguyen M, Huo X, et al. Truncation of the C-terminus of human MLH1 blocks intracellular stabilization of PMS2 and disrupts DNA mismatch repair. DNA Repair. 2006;5:347–61. doi: 10.1016/j.dnarep.2005.11.001. [DOI] [PubMed] [Google Scholar]
- 53.Rosty C, Clendenning M, Walsh MD, Eriksen SV, Southey MC, Winship IM, et al. Germline mutations in PMS2 and MLH1 in individuals with solitary loss of PMS2 expression in colorectal carcinomas from the Colon Cancer Family Registry Cohort. BMJ Open. 2016;6:e010293. doi: 10.1136/bmjopen-2015-010293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.ten Broeke SW, van der Klift HM, Tops CMJ, Aretz S, Bernstein I, Buchanan DD, et al. Cancer risks for PMS2-associated Lynch syndrome. J Clin Oncol. 2018;36:2961–8. doi: 10.1200/JCO.2018.78.4777. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Wang C, Wang Y, Hughes KS, Parmigiani G, Braun D. Penetrance of colorectal cancer among mismatch repair gene mutation carriers: a meta-analysis. JNCI Cancer Spectr. 2020;4:pkaa027. [DOI] [PMC free article] [PubMed]
- 56.Thompson BA, Spurdle AB, Plazzer J-P, Greenblatt MS, Akagi K, Al-Mulla F, et al. Application of a five-tiered scheme for standardized classification of 2,360 unique mismatch repair gene variants lodged on the InSiGHT locus-specific database. Nat Genet. 2014;46:107–15. doi: 10.1038/ng.2854. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Williams PD, Pollock DD, Goldstein RA. Functionality and the evolution of marginal stability in proteins: inferences from lattice simulations. Evol Bioinforma Online. 2007;2:91–101. [PMC free article] [PubMed] [Google Scholar]
- 58.Abildgaard AB, Gersing SK, Larsen-Ledet S, Nielsen SV, Stein A, Lindorff-Larsen K, et al. Co-chaperones in targeting and delivery of misfolded proteins to the 26S proteasome. Biomolecules. 2020;10:1141. doi: 10.3390/biom10081141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Clausen L, Abildgaard AB, Gersing SK, Stein A, Lindorff-Larsen K, Hartmann-Petersen R. Chapter two - protein stability and degradation in health and disease. In: Advances in protein chemistry and structural biology. Donev R, editor. Vol. 114. London, United Kingdom: Academic Press; 2019. p. 61–83. [DOI] [PubMed]
- 60.Kohler V, Andréasson C. Hsp70-mediated quality control: should I stay or should I go? Biol Chem. 2020;401:1233–48. doi: 10.1515/hsz-2020-0187. [DOI] [PubMed] [Google Scholar]
- 61.Reinle K, Mogk A, Bukau B. The diverse functions of small heat shock proteins in the proteostasis network. J Mol Biol. 2022;434:167157. doi: 10.1016/j.jmb.2021.167157. [DOI] [PubMed] [Google Scholar]
- 62.Rosenzweig R, Nillegoda NB, Mayer MP, Bukau B. The Hsp70 chaperone network. Nat Rev Mol Cell Biol. 2019;20:665–80. doi: 10.1038/s41580-019-0133-3. [DOI] [PubMed] [Google Scholar]
- 63.Hernandez-Pigeon H, Laurent G, Humbert O, Salles B, Lautier D. Degadration of mismatch repair hMutSα heterodimer by the ubiquitin-proteasome pathway. FEBS Lett. 2004;562:40–4. doi: 10.1016/S0014-5793(04)00181-4. [DOI] [PubMed] [Google Scholar]
- 64.Wu Q, Huang Y, Gu L, Chang Z, Li G-M. OTUB1 stabilizes mismatch repair protein MSH2 by blocking ubiquitination. J Biol Chem. 2021;296:100466. doi: 10.1016/j.jbc.2021.100466. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Zhang M, Hu C, Tong D, Xiang S, Williams K, Bai W, et al. Ubiquitin-specific peptidase 10 (USP10) deubiquitinates and stabilizes MutS homolog 2 (MSH2) to regulate cellular sensitivity to DNA damage*. J Biol Chem. 2016;291:10783–91. doi: 10.1074/jbc.M115.700047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Jia X, Burugula BB, Chen V, Lemons RM, Jayakody S, Maksutova M, et al. Massively parallel functional testing of MSH2 missense variants conferring Lynch syndrome risk. Am J Hum Genet. 2021;108:163–75. doi: 10.1016/j.ajhg.2020.12.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Nielsen SV, Hartmann-Petersen R, Stein A, Lindorff-Larsen K. Multiplexed assays reveal effects of missense variants in MSH2 and cancer predisposition. PLOS Genet. 2021;17:e1009496. doi: 10.1371/journal.pgen.1009496. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Nielsen SV, Stein A, Dinitzen AB, Papaleo E, Tatham MH, Poulsen EG, et al. Predicting the impact of Lynch syndrome-causing missense mutations from structural calculations. PLoS Genet. 2017;13:e1006739. doi: 10.1371/journal.pgen.1006739. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Ollodart AR, Yeh C-LC, Miller AW, Shirts BH, Gordon AS, Dunham MJ. Multiplexing mutation rate assessment: determining pathogenicity of Msh2 variants in Saccharomyces cerevisiae. Genetics. 2021;218:iyab058. [DOI] [PMC free article] [PubMed]
- 70.Arlow T, Scott K, Wagenseller A, Gammie A. Proteasome inhibition rescues clinically significant unstable variants of the mismatch repair protein Msh2. Proc Natl Acad Sci USA. 2013;110:246–51. doi: 10.1073/pnas.1215510110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Kampmeyer C, Nielsen SV, Clausen L, Stein A, Gerdes A-M, Lindorff-Larsen K, et al. Blocking protein quality control to counter hereditary cancers. Genes Chromosomes Cancer. 2017;56:823–31. doi: 10.1002/gcc.22487. [DOI] [PubMed] [Google Scholar]
- 72.Stein A, Fowler DM, Hartmann-Petersen R, Lindorff-Larsen K. Biophysical and mechanistic models for disease-causing protein variants. Trends Biochem Sci. 2019;44:575–88. doi: 10.1016/j.tibs.2019.01.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Livesey BJ, Marsh JA. Using deep mutational scanning to benchmark variant effect predictors and identify disease mutations. Mol Syst Biol. 2020;16:e9380. doi: 10.15252/msb.20199380. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Starita LM, Ahituv N, Dunham MJ, Kitzman JO, Roth FP, Seelig G, et al. Variant interpretation: functional assays to the rescue. Am J Hum Genet. 2017;101:315–25. doi: 10.1016/j.ajhg.2017.07.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Ponti G, Castellsagué E, Ruini C, Percesepe A, Tomasi A. Mismatch repair genes founder mutations and cancer susceptibility in Lynch syndrome: mismatch repair genes founder mutations in Lynch syndrome. Clin Genet. 2015;87:507–16. doi: 10.1111/cge.12529. [DOI] [PubMed] [Google Scholar]
- 76.Foulkes WD, Thiffault I, Gruber SB, Horwitz M, Hamel N, Lee C, et al. The founder mutation MSH2*1906G->C is an important cause of hereditary nonpolyposis colorectal cancer in the Ashkenazi Jewish population. Am J Hum Genet. 2002;71:1395–412. doi: 10.1086/345075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Brnich SE, Rivera‐Muñoz EA, Berg JS. Quantifying the potential of functional evidence to reclassify variants of uncertain significance in the categorical and Bayesian interpretation frameworks. Hum Mutat. 2018;39:1531–41. doi: 10.1002/humu.23609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Houlleberghs H, Goverde A, Lusseveld J, Dekker M, Bruno MJ, Menko FH, et al. Suspected Lynch syndrome associated MSH6 variants: a functional assay to determine their pathogenicity. PLOS Genet. 2017;13:e1006765. doi: 10.1371/journal.pgen.1006765. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Houlleberghs H, Dekker M, Lantermans H, Kleinendorst R, Dubbink HJ, Hofstra RMW, et al. Oligonucleotide-directed mutagenesis screen to identify pathogenic Lynch syndrome-associated MSH2 DNA mismatch repair gene variants. Proc Natl Acad Sci USA. 2016;113:4128–33. doi: 10.1073/pnas.1520813113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Alim I, Loke J, Yam S, Templeton AS, Newcomb P, Lindor NM, et al. Cancer risk C (CR-C), a functional genomics test is a sensitive and rapid test for germline mismatch repair deficiency. Genet. Med. 2022;S1098360022007584. 10.1016/j.gim.2022.05.003. [DOI] [PMC free article] [PubMed]
- 81.Cagiada M, Johansson KE, Valanciute A, Nielsen SV, Hartmann-Petersen R, Yang JJ, et al. Understanding the origins of loss of protein function by analyzing the effects of thousands of variants on activity and abundance. Mol Biol Evol. 2021;38:3235–46. doi: 10.1093/molbev/msab095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Arora S, Huwe PJ, Sikder R, Shah M, Browne AJ, Lesh R, et al. Functional analysis of rare variants in mismatch repair proteins augments results from computation-based predictive methods. Cancer Biol Ther. 2017;18:519–33. doi: 10.1080/15384047.2017.1326439. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Niroula A, Vihinen M. Classification of amino acid substitutions in mismatch repair proteins using PON-MMR2. Hum Mutat. 2015;36:1128–34. doi: 10.1002/humu.22900. [DOI] [PubMed] [Google Scholar]
- 84.Adzhubei I, Jordan DM, Sunyaev SR. Predicting functional effect of human missense mutations using PolyPhen-2. Curr Protoc Hum Genet. 2013;Chapter 7:Unit7.20. [DOI] [PMC free article] [PubMed]
- 85.Choi Y, Chan AP. PROVEAN web server: a tool to predict the functional effect of amino acid substitutions and indels. Bioinformatics. 2015;31:2745–7. doi: 10.1093/bioinformatics/btv195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Kumar P, Henikoff S, Ng PC. Predicting the effects of coding non-synonymous variants on protein function using the SIFT algorithm. Nat Protoc. 2009;4:1073–81. doi: 10.1038/nprot.2009.86. [DOI] [PubMed] [Google Scholar]
- 87.Hopf TA, Ingraham JB, Poelwijk FJ, Schärfe CPI, Springer M, Sander C, et al. Mutation effects predicted from sequence co-variation. Nat Biotechnol. 2017;35:128–35. doi: 10.1038/nbt.3769. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Laine E, Karami Y, Carbone A. GEMME: a simple and fast global epistatic model predicting mutational effects. Mol Biol Evol. 2019;36:2604–19. doi: 10.1093/molbev/msz179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Riesselman AJ, Ingraham JB, Marks DS. Deep generative models of genetic variation capture the effects of mutations. Nat Methods. 2018;15:816–22. doi: 10.1038/s41592-018-0138-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Frazer J, Notin P, Dias M, Gomez A, Min JK, Brock K, et al. Disease variant prediction with deep generative models of evolutionary data. Nature. 2021;599:91–5. doi: 10.1038/s41586-021-04043-8. [DOI] [PubMed] [Google Scholar]
- 91.Park H, DiMaio F, Baker D. CASP11 refinement experiments with ROSETTA. Proteins Struct Funct Bioinforma. 2016;84:314–22. doi: 10.1002/prot.24862. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Guerois R, Nielsen JE, Serrano L. Predicting changes in the stability of proteins and protein complexes: a study of more than 1000 mutations. J Mol Biol. 2002;320:369–87. doi: 10.1016/S0022-2836(02)00442-4. [DOI] [PubMed] [Google Scholar]
- 93.Schymkowitz J, Borg J, Stricher F, Nys R, Rousseau F, Serrano L. The FoldX web server: an online force field. Nucleic Acids Res. 2005;33:W382–8. doi: 10.1093/nar/gki387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Raevaara TE, Korhonen MK, Lohi H, Hampel H, Lynch E, Lönnqvist KE, et al. Functional significance and clinical phenotype of nontruncating mismatch repair variants of MLH1. Gastroenterology. 2005;129:537–49. doi: 10.1016/j.gastro.2005.06.005. [DOI] [PubMed] [Google Scholar]
- 95.Takahashi M, Shimodaira H, Andreutti-Zaugg C, Iggo R, Kolodner RD, Ishioka C. Functional analysis of human MLH1 variants using yeast and in vitro mismatch repair assays. Cancer Res. 2007;67:4595–604. doi: 10.1158/0008-5472.CAN-06-3509. [DOI] [PubMed] [Google Scholar]
- 96.De Baets G, Van Durme J, Reumers J, Maurer-Stroh S, Vanhee P, Dopazo J, et al. SNPeffect 4.0: on-line prediction of molecular and structural effects of protein-coding variants. Nucleic Acids Res. 2012;40:D935–9. doi: 10.1093/nar/gkr996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Gerasimavicius L, Liu X, Marsh JA. Identification of pathogenic missense mutations using protein stability predictors. Sci Rep. 2020;10:15387. doi: 10.1038/s41598-020-72404-w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Pey AL, Stricher F, Serrano L, Martinez A. Predicted effects of missense mutations on native-state stability account for phenotypic outcome in phenylketonuria, a paradigm of misfolding diseases. Am J Hum Genet. 2007;81:1006–24. doi: 10.1086/521879. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Fowler DM, Fields S. Deep mutational scanning: a new style of protein science. Nat Methods. 2014;11:801–7. doi: 10.1038/nmeth.3027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Raraigh KS, Han ST, Davis E, Evans TA, Pellicore MJ, McCague AF, et al. Functional assays are essential for interpretation of missense variants associated with variable expressivity. Am J Hum Genet. 2018;102:1062–77. doi: 10.1016/j.ajhg.2018.04.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Sun S, Yang F, Tan G, Costanzo M, Oughtred R, Hirschman J, et al. An extended set of yeast-based functional assays accurately identifies human disease mutations. Genome Res. 2016;26:670–80. doi: 10.1101/gr.192526.115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Jepsen MM, Fowler DM, Hartmann-Petersen R, Stein A, Lindorff-Larsen K. Chapter 5 - Classifying disease-associated variants using measures of protein activity and stability. In: Protein homeostasis diseases. Pey AL, editor. London, United Kingdom: Academic Press; 2020, p. 91–107.
- 103.Matreyek KA, Starita LM, Stephany JJ, Martin B, Chiasson MA, Gray VE, et al. Multiplex assessment of protein variant abundance by massively parallel sequencing. Nat Genet. 2018;50:874–82. doi: 10.1038/s41588-018-0122-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Brnich SE, On behalf of the Clinical Genome Resource Sequence Variant Interpretation Working Group. Abou Tayoun AN, Couch FJ, Cutting GR, Greenblatt MS, et al. Recommendations for application of the functional evidence PS3/BS3 criterion using the ACMG/AMP sequence variant interpretation framework. Genome Med. 2020;12:3. doi: 10.1186/s13073-019-0690-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Ernst C, Hahnen E, Engel C, Nothnagel M, Weber J, Schmutzler RK, et al. Performance of in silico prediction tools for the classification of rare BRCA1/2 missense variants in clinical diagnostics. BMC Med Genomics. 2018;11:35. doi: 10.1186/s12920-018-0353-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Miosge LA, Field MA, Sontani Y, Cho V, Johnson S, Palkova A, et al. Comparison of predicted and actual consequences of missense mutations. Proc Natl Acad Sci USA. 2015;112:E5189–98. doi: 10.1073/pnas.1511585112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Grimm DG, Azencott C-A, Aicheler F, Gieraths U, MacArthur DG, Samocha KE, et al. The evaluation of tools used to predict the impact of missense variants is hindered by two types of circularity. Hum Mutat. 2015;36:513–23. doi: 10.1002/humu.22768. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Wu Y, Li R, Sun S, Weile J, Roth FP. Improved pathogenicity prediction for rare human missense variants. Am J Hum Genet. 2021. 10.1016/j.ajhg.2021.08.012. [DOI] [PMC free article] [PubMed]
- 109.Høie MH, Cagiada M, Beck Frederiksen AH, Stein A, Lindorff-Larsen K. Predicting and interpreting large-scale mutagenesis data using analyses of protein stability and conservation. Cell Rep. 2022;38:110207. doi: 10.1016/j.celrep.2021.110207. [DOI] [PubMed] [Google Scholar]
- 110.Thompson S, Zhang Y, Ingle C, Reynolds KA, Kortemme T. Altered expression of a quality control protease in E. coli reshapes the in vivo mutational landscape of a model enzyme. eLife. 2020;9:e53476. doi: 10.7554/eLife.53476. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Brusa I, Sondo E, Falchi F, Pedemonte N, Roberti M, Cavalli A. Proteostasis regulators in cystic fibrosis: current development and future perspectives. J Med Chem. 2022. 10.1021/acs.jmedchem.1c01897. [DOI] [PMC free article] [PubMed]
- 112.Hutt DM, Herman D, Rodrigues APC, Noel S, Pilewski JM, Matteson J, et al. Reduced histone deacetylase 7 activity restores function to misfolded CFTR in cystic fibrosis. Nat Chem Biol. 2010;6:25–33. doi: 10.1038/nchembio.275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Okiyoneda T, Veit G, Dekkers JF, Bagdany M, Soya N, Xu H, et al. Mechanism-based corrector combination restores ΔF508-CFTR folding and function. Nat Chem Biol. 2013;9:444–54. doi: 10.1038/nchembio.1253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Richards S, Aziz N, Bale S, Bick D, Das S, Gastier-Foster J, et al. Standards and guidelines for the interpretation of sequence variants: a joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet Med. 2015;17:405–24. doi: 10.1038/gim.2015.30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Fayer S, Horton C, Dines JN, Rubin AF, Richardson ME, McGoldrick K, et al. Closing the gap: systematic integration of multiplexed functional data resolves variants of uncertain significance in BRCA1, TP53, and PTEN. Am J Hum Genet. 2021;S0002-9297(21)00411–0. 10.1016/j.ajhg.2021.11.001. [DOI] [PMC free article] [PubMed]
- 116.Kuang D, Truty R, Weile J, Johnson B, Nykamp K, Araya C, et al. Prioritizing genes for systematic variant effect mapping. Bioinformatics. 2020;36:5448–55. doi: 10.1093/bioinformatics/btaa1008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Manolio TA, Fowler DM, Starita LM, Haendel MA, MacArthur DG, Biesecker LG, et al. Bedside back to bench: building bridges between basic and clinical genomic research. Cell. 2017;169:6–12. doi: 10.1016/j.cell.2017.03.005. [DOI] [PMC free article] [PubMed] [Google Scholar]