

Abstract
The implications of nutrient starvation due to aging on the degeneration of the retinal pigment epithelium (RPE) is yet to be fully explored. We examined the involvement of AMPK activation in mitochondrial homeostasis and its relationship with the maintenance of a healthy mitochondrial population and epithelial characteristics of RPE cells under nutrient starvation. Nutrient starvation induced mitochondrial senescence, which led to the accumulation of reactive oxygen species (ROS) in RPE cells. As nutrient starvation persisted, RPE cells underwent pathological epithelial-mesenchymal transition (EMT) via the upregulation of TWIST1, a transcription regulator which is activated by ROS-induced NF-κB signaling. Enhanced activation of AMPK with metformin decelerated mitochondrial senescence and EMT progression through mitochondrial biogenesis, primed by activation of PGC1-α. Thus, by facilitating mitochondrial biogenesis, AMPK protects RPE cells from the loss of epithelial integrity due to the accumulation of ROS in senescent mitochondria under nutrient starvation.
Keywords: AMP-activated protein kinase, Epithelial-mesenchymal transition, Mitochondrial biogenesis, Nutrient starvation, Reactive oxygen species
INTRODUCTION
The retinal pigment epithelium (RPE) is located between the Bruch’s membrane (BrM) and the retina, and consists of a monolayer of postmitotic epithelial cells which do not undergo further differentiation (1). As aging progresses, the RPE becomes dysfunctional, with a decrease in its cell numbers, contributing to the disruption of retinal homeostasis (2). Aging also diminishes the circulatory function of the choriocapillaris (CC), the blood vessels behind the RPE and BrM which supply nutrients and oxygen to the retina. Since nutritional support for the RPE is provided by the CC, deficiencies in CC function with age may cause nutrient starvation in the RPE (3). Although the RPE is known to protect the retina against tissue stress and/or retinal damage, the detailed molecular mechanisms occurring during aging are not fully understood.
Under conditions of nutrient starvation, such as glucose withdrawal, RPE cells predominantly rely on mitochondria for ATP production, increasing mitochondrial oxidative phosphorylation (OXPHOS) (4). Reactive oxygen species (ROS), such as hydroxyl radicals, superoxide, and hydrogen peroxide (H2O2) are often produced by the mitochondrial electron transport chain (mETC), which produces ATP for cell survival under nutrient starvation (5). However, as glucose withdrawal persists without the supply of other nutrients, mitochondria in the RPE display depolarization accumulation with a decrease in oxygen consumption rate (OCR) (6). It has been suggested that the ability of RPE to tolerate oxidative stress can be increased by well-controlled autophagy, which removes senescent and dysfunctional mitochondria (7).
The general RPE stress responses are characterized by epithelial-mesenchymal transition (EMT), including RPE trans-differentiation, leading to the loss of RPE characteristics (8). Because cell-cell junctions are essential for maintaining RPE integrity, progression of EMT is likely to cause the deconstruction of epithelia-formed permeability barriers and RPE degeneration (9). It has been suggested that ROS plays a critical role during the EMT process in cancerous epithelial cells by the loss of cell-cell junctions with degradation of junction proteins such as CDH1/E-cadherin (10). The expression of junction proteins, during EMT, is repressed by transcription factors, including Snail, Slug, Twist and ZEB, and activation of NF-κB signaling is involved in the EMT process by stimulating the expression of these transcription factors (11-13). Notably, increased ROS levels have been observed to activate NF-κB signaling, leading to EMT-related morphological changes (14).
AMP-activated protein kinase (AMPK) is a major player in the modulation of the metabolic system to cope with nutrient deficiency in cells (15). AMPK has been known to be responsible for maintaining mitochondrial homeostasis through mitophagy and mitochondrial biogenesis, by activation of the upstream kinase in mitophagy (ULK1) and peroxisome proliferator-activated receptor-γ (PPARγ) co-activator 1α (PGC1-α), respectively (16). However, little is known about mitochondrial homeostasis in nutrient-starved RPE in terms of AMPK activation and the subsequent maintenance of RPE integrity.
In this study, we investigated the role of AMPK in mitochondrial biogenesis under nutrient starvation, in which ROS levels increase owing to the accumulation of senescent mitochondria. We further examined whether AMPK activation could decelerate EMT progression in RPE cells under prolonged starvation conditions. The results of our study indicate that AMPK-induced mitochondrial biogenesis compensates for the loss of nascent and functional mitochondria, contributing to the deceleration of RPE cell degeneration under nutrient starvation.
RESULTS AND DISCUSSION
Nutrient starvation induces mitochondrial senescence and ROS accumulation
Since the accumulation of ROS is a known feature of mitochondrial senescence, we first examined the generation of ROS in RPE cells exposed to nutrient-depleted conditions by monitoring DCF-DA fluorescence (Fig. 1A). We observed that fluorescence increased in a starvation and time-dependent manner up to 9 h and then reduced at 12 h (red arrow in Fig. 1A). Interestingly, cellular ROS levels further increased after 12 h of continued nutrient starvation. To explain this fluctuation in ROS levels in RPE cells under prolonged nutrient starvation, we hypothesized that starvation-induced ROS generation could be regulated by AMPK, as a main sensor of cellular nutrient status (17).
Fig. 1.

AMPK activation decreases the level of ROS produced by senescent mitochondria under nutrient starvation. ARPE-19 cells were starved with HBSS medium for 0-36 h. (A) Fluorescence microscopy images of DCF-DA-stained ARPE-19 cells under nutrient starvation for 0-36 h. Scale bar: 275 μm. Bar graph indicates the fluorescence intensity of DCF, which was normalized to starvation 0 h. The data are presented as the mean ± SEM, n = 3. ***P < 0.005. (B) Western blot analysis of AMPK and p-AMPK under nutrient starvation for 0-36 h. The graph indicates the ratio of p-AMPK to total AMPK. (C) Confocal fluorescence microscopy images of MitoTimer-expressing cells under nutrient starvation for 0-36 h. Scale bar: 10 μm. Bar graph represents the red-to-green fluorescence ratio of MitoTimer. Data are presented as the mean ± SEM, n = 3. *P < 0.05. (D) Confocal fluorescence microscopy images of MitoTracker Red-stained cells under nutrient starvation for 0-36 h. Nuclei were stained with DAPI (blue). Scale bar: 10 μm. (E) TEM images showing mitochondrial internal structures of ARPE-19 cells under nutrient starvation for 0 or 36 h. Green- and yellow-colored arrows indicate healthy and damaged mitochondria, respectively. Scale bar: 500 nm.
To test this hypothesis, we first monitored AMPK phosphorylation in RPE cells under nutrient starvation conditions (Fig. 1B). Depletion of nutrients caused an increase in the levels of phosphorylated AMPK up to 12 h, but the extent of AMPK phosphorylation decreased after 12 h. We next investigated mitochondrial turnover in cells under nutrient starvation conditions by tracing the fluorescence of MitoTimer protein (Fig. 1C). Green and red fluorescence of MitoTimer indicate nascent and senescent mitochondria, respectively (18). During the initial 12 h of nutrient starvation, RPE cells displayed yellow fluorescence in the merged image, indicating that nascent and senescent mitochondria were present together. However, red fluorescence was dominant in cells under prolonged starvation conditions after 12 h, suggesting the accumulation of senescent mitochondria.
We further investigated the shape of mitochondria to infer mitochondrial senescence in RPE cells under nutrient starvation conditions (Fig. 1D). Mitochondria maintained their general morphology by showing elongated tubular forms for up to 12 h of nutrient starvation. However, consistent with the previous results (Fig. 1C), mitochondria under prolonged nutrient starvation displayed enlarged spherical forms. To further confirm changes in mitochondrial morphology and senescence, RPE cells under prolonged nutrient starvation were monitored by transmission electron microscopy (TEM) (Fig. 1E). RPE cells under nutrient starvation for up to 36 h exhibited extensive disruption of structural integrity with deteriorated mitochondrial cristae, when compared to non-starved RPE cells harboring healthy normal-appearing mitochondria with clear inner membranes and dense matrix space. Taken together, our results show that prolonged nutrient starvation causes mitochondrial senescence in RPE cells, and that AMPK phosphorylation decreases with ROS accumulation.
AMPK maintains a healthy mitochondrial population through mitochondrial biogenesis
Based on the finding that RPE cells display an increase in ROS and senescent mitochondria under nutrient starvation after 12 h (Fig. 1), we examined whether AMPK activation led to a decrease in ROS accumulation in RPE cells under nutrient starvation conditions (Fig. 2A). RPE cells were treated with either compound C (Comp C), a known inhibitor of AMPK activation (Supplementary Fig. 1), or chloramphenicol (CAP), an inhibitor of mitochondrial protein translation. Consistent with the results shown in Fig. 1A, ROS fluorescence intensity was rarely observed in control cells under nutrient starvation for 12 h. However, in both Comp C- and CAP-treated cells, ROS fluorescence was significantly increased in cells under nutrient starvation for 12 h. This result suggests that starvation-induced ROS accumulation can be reduced by either AMPK activation or mitochondrial biogenesis.
Fig. 2.

Inhibition of AMPK-dependent mitochondrial biogenesis increases ROS and mitochondrial senescence during nutrient starvation. ARPE-19 cells were starved with HBSS medium in the presence or absence of Comp C (5 μM) and CAP (50 μg/ml) for 12 h. (A) Fluorescence microscopy images of DCF-DA-stained ARPE-19 cells. Scale bar: 275 μm. Bar graph indicates the fluorescence intensity of DCF, which was normalized to starvation 0 h in the presence or absence of Comp and CAP. Data are presented as the mean ± SEM, n = 3. **P < 0.01. (B) MitoTracker Red-stained cells under nutrient starvation for 12 h in the presence or absence of Comp C and CAP. Nuclei were stained with DAPI (blue). Scale bar: 10 μm. (C) Images in MitoTimer-expressing cells under nutrient starvation for 12 h in the presence or absence of Comp C and CAP. Scale bar: 10 μm. Bar graph represents the red-to-green fluorescence ratio of MitoTimer. Data are presented as the mean ± SEM, n = 3. **P < 0.01 and ***P < 0.005. (D) Flow cytometric assay for percentage population of cells with dysfunctional mitochondria in the presence or absence of Comp C and CAP. Bar graphs indicate the percentage of cells with dysfunctional mitochondria. Data are presented as the mean ± SEM, n = 3. *P < 0.05. (E) PGC1-α-immunostained (green) ARPE-19 cells under nutrient starvation for 12 h in the presence or absence of Comp C. Nuclei were stained with DAPI (blue). Scale bar: 10 μm. (F) Quantitative real-time PCR of ARPE-19 cells under nutrient starvation for 12 h in the presence or absence of Comp C. Bar graph indicates the relative mRNA level of TFAM which was normalized to starvation 0 h in the presence or absence of Comp C. Data are presented as the mean ± SEM, n = 3. ***P < 0.005.
Next, we tested whether the inhibition of AMPK activation or mitochondrial biogenesis affects mitochondrial morphology in RPE cells under nutrient starvation. When cells were nutrient-starved for 12 h in the presence of Comp C or CAP, round shape with perinuclear clustering of mitochondria was observed, which reflects abnormal mitochondrial morphology (Fig. 2B). In accordance with this morphological disturbance in mitochondria, the red-green fluorescence ratio of MitoTimer was approximately 6-fold higher than that of cells without inhibitor treatments (Fig. 2C). These results indicate that the population of nascent mitochondria diminished with AMPK inactivation and/or inhibition of mitochondrial biogenesis in RPE cells under nutrient starvation.
We further examined mitochondrial functionality in nutrient-starved RPE cells under AMPK inactivation and/or inhibition of mitochondrial biogenesis by measuring the mitochondrial membrane potential (ΔΨM) (Fig. 2D). Mitochondrial dysfunction induces a decrease in ΔΨM, which can be identified by measuring MitoTracker double-stained cells by flow cytometry. MitoTracker Green accumulates in the mitochondrial matrix regardless of ΔΨM, representing the mass of mitochondria in cells, and MitoTracker Red stains polarized mitochondria with a negative membrane potential, indicating functional mitochondria (19). When RPE cells were subjected to nutrient starvation for 12 h, the cellular population with functional mitochondria did not change. In contrast, when nutrient-starved cells were treated with Comp C or CAP, the cellular population with dysfunctional mitochondria was significantly increased (Fig. 2D). Taken together, these results suggest that AMPK inactivation and the inhibition of mitochondrial biogenesis induce an increase in the senescent mitochondrial population with features of swollen shape, increased senescent mitochondrial protein (i.e., MitoTimer Red), and mitochondrial dysfunction in RPE cells under nutrient starvation.
It is known that PGC1-α, one of the main transcription factors for mitochondrial biogenesis, is phosphorylated by AMPK and translocated to the nucleus for the upregulation of the mitochondrial transcription factor A (TFAM) (16, 20). We examined the activation of PGC1-α by monitoring PGC1-α nuclear translocation under nutrient starvation conditions (Fig. 2E). PGC1-α was predominantly present in the cytoplasm prior to starvation and was translocated to the nucleus after continued nutrient withdraw. However, PGC1-α rarely translocated to the nucleus when the Comp C was used. Thus, the activation of AMPK is likely to induce the translocation of PGC1-α to the nucleus. In the same context, we observed that AMPK inhibition led to the failure of TFAM transcription under nutrient starvation conditions (Fig. 2F). Thus, our results indicate that activation of AMPK leads to the activation of PGC1-α and TFAM, which are responsible for mitochondrial biogenesis in RPE cells under nutrient starvation conditions.
Activated AMPK protects mitochondrial senescence and epithelial-mesenchymal transition
Since AMPK activation attenuates ROS through mitochondrial biogenesis under nutrient starvation conditions (Fig. 2), we investigated whether AMPK activation can also attenuate mitochondrial senescence and EMT progression in RPE cells under prolonged nutrient starvation. To enhance the activation of AMPK, metformin (Met), a drug used to treat diabetes mellitus patients, was employed (21). We first examined whether ROS accumulation was decreased by Met-assisted AMPK activation under prolonged starvation (Fig. 3A). When cells were consistently exposed to prolonged nutrient starvation for up to 36 h, ROS fluorescence increased substantially. In contrast, accumulation of ROS fluorescence was rarely observed after Met-treatment during prolonged nutrient starvation (36 h).
Fig. 3.

AMPK activation promotes mitochondrial quality control under nutrient starvation. ARPE-19 cells were starved with HBSS medium in the presence or absence of Met (1 mM; pre-treated for 2 h) for 36 h. (A) Fluorescence microscopy images of DCF-DA-stained ARPE-19 cells. Scale bar: 275 μm. Bar graph indicates the fluorescence intensity of DCF, which was normalized to starvation 0 h in the presence or absence of Met. Data are presented as the mean ± SEM, n = 3. **P < 0.01. (B) Images of PGC1-α-immunostained (green) ARPE-19 cells under nutrient starvation for 36 h in the presence or absence of Met. Nuclei were stained with DAPI (blue). Scale bar: 10 μm. (C) MitoTracker Red-stained cells under nutrient starvation for 36 h in the presence or absence of Met. Nuclei were stained with DAPI (blue). Scale bar: 10 μm. (D) TEM images showing mitochondrial internal structures of cells under nutrient starvation for 36 h in the presence or absence of Met. Green- and yellow-colored arrows indicate healthy and damaged mitochondria, respectively. Scale bar: 500 nm.
Next, we investigated whether Met-treatment leads to continued activation of PGC1-α, which is involved in mitochondrial biogenesis (Fig. 3B). We observed translocation of PGC1-α to the nucleus in cells treated with Met under prolonged starvation. This result suggests that activated AMPK is likely to induce PGC1-α for mitochondrial biogenesis under prolonged nutrient starvation. Using the MitoTimer fluorescence assay, we observed that continued activation of AMPK with Met or AICAR (direct AMPK agonist) resulted in delayed mitochondrial senescence (Supplementary Fig. 2A, B). When cells were treated with Met for further activation of AMPK under prolonged starvation, there were more nascent mitochondria than the cells without Met-treatment. This result was further supported by the mitochondrial functionality assay performed under the same conditions (Supplementary Fig. 2C). Prolonged starvation significantly induced mitochondrial damage in cells, which was reduced by treatment with the AMPK activator Met. Thus, Met-treated RPE cells maintained mitochondrial quality control through AMPK activation under prolonged starvation conditions.
We next addressed whether Met-assisted AMPK activation can maintain intact mitochondrial morphology in RPE cells under prolonged nutrient starvation (Fig. 3C). Mitochondria with swollen shape and perinuclear clustering were distinctively observed in cells under prolonged starvation, while mitochondria displayed a normal reticulated shape in starved cells treated with Met. Furthermore, TEM image of prolonged starved RPE cells exhibited extensive structural damage in the mitochondria, such as enlarged shape and disrupted mitochondrial cristae, compared with starved-cells treated with the AMPK activator Met (Fig. 3D). Based on these results on mitochondrial morphology and functionality, we suggest that mitochondrial senescence is prevented by continued activation of AMPK through mitochondrial biogenesis, despite prolonged starvation in RPE cells.
Next, we examined whether prolonged nutrient starvation induces EMT in RPE cells and whether AMPK activation can delay EMT progression. To monitor EMT progression in RPE cells, the presence of CDH1/E-cadherin and vimentin (VIM), markers of epithelial and mesenchymal cells, respectively, was assessed by fluorescent monitoring. The epithelial marker CDH1 was present in the cell-cell junctions of RPE cells under nutrient starvation for up to 12 h, whereas the mesenchymal marker VIM was mainly observed in cells with persistent starvation for up to 24 h. Interestingly, this EMT progression was significantly reduced with Met-treatment in cells under prolonged starvation for up to 24 h, in which the fluorescence ratio of VIM-to-CDH1 was approximately 14-fold lower than that of cells without Met-treatment (Fig. 4A). The delay of EMT progression with AMPK activation was also demonstrated with the AMPK agonist (AICAR) treatment (Supplementary Fig. 3A). These results suggest that AMPK activation decelerates EMT progression in cells under prolonged starvation. To further confirm the effect of AMPK activation on the maintenance of epithelial characteristics, we tested whether AMPK inhibition by Comp C could accelerate EMT progression in RPE cells under nutrient starvation (Supplementary Fig. 3B). Significant progression of EMT was not observed in RPE cells under nutrient starvation for up to 12 h, but cells treated with Comp C showed a faster onset of EMT than cells without Comp C. Hence, we suggest that AMPK activation not only contributes to reduce EMT progression but also delays senescent changes in mitochondria in RPE cells under nutrient starvation conditions.
Fig. 4.
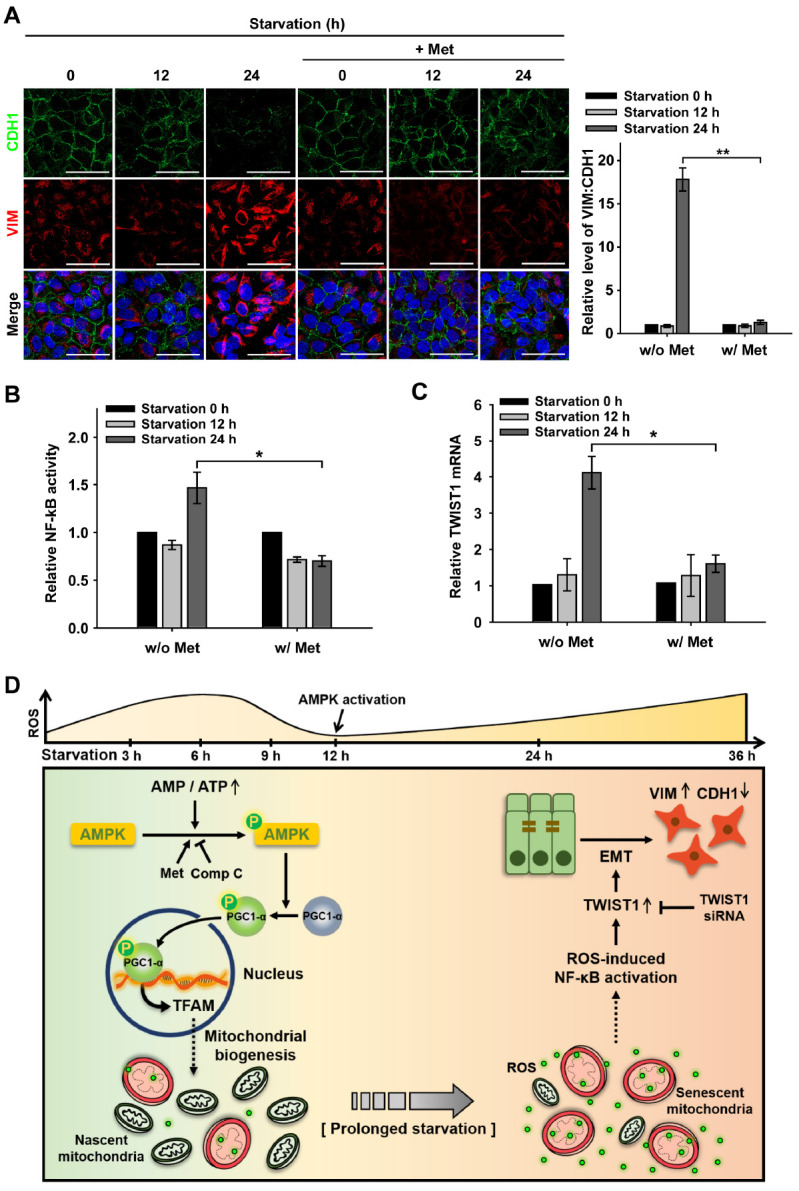
AMPK decelerates EMT by down-regulating TWIST1 under prolonged starvation. ARPE-19 cells were starved with HBSS medium in the presence or absence Met (1 mM; pre-treated for 2 h) for 0-24 h. (A) CDH1 and VIM-immunostained (green and red, respectively) ARPE-19 cells under nutrient starvation for 0-24 h in the presence or absence of Met. Nuclei were stained with DAPI (blue). Scale bar: 50 μm. Bar graph represents the VIM-to-CDH1 fluorescence ratio. Data are presented as the mean ± SEM, n = 3. **P < 0.01. (B) Dual-luciferase reporter assay for NF-κB activation in ARPE-19 cells under nutrient starvation for 0-24 h in the presence or absence of Met. Bar graph represents the activity of NF-κB luciferase which was normalized to Renilla luciferase. Data are presented as the mean ± SEM, n = 3. *P < 0.05. (C) Quantitative real-time PCR for the relative mRNA level of TWIST1 in ARPE-19 cells under nutrient starvation, which was normalized to starvation 0 h in the presence or absence of Met. Data are presented as the mean ± SEM, n = 3. *P < 0.05. (D) Proposed mechanism of the cellular fate of the RPE under nutrient starvation via AMPK activation and the epithelial-mesenchymal transition (EMT).
It has been known that EMT is induced by the transcription of EMT-related genes such as SNAI1 or TWIST1 following activation of NF-κB (10). To address the signaling pathway underlying EMT progression by AMPK activation, we examined the activation of NF-κB in RPE cells under prolonged starvation conditions using a dual-luciferase reporter assay (Fig. 4B). NF-κB activity was not elevated under nutrient starvation for up to 12 h but significantly increased after 24 h of starvation. However, NF-κB activation was significantly decreased in cells treated with the Met under nutrient starvation for 24 h. This result indicates that enhanced activation of AMPK attenuates starvation-induced NF-κB activation. We next investigated the transcription of EMT-related genes by monitoring TWIST1 and SNAI1 mRNA levels in cells under nutrient starvation (Supplementary Fig. 3C). TWIST 1 mRNA levels were significantly increased in cells undergoing continued starvation for up to 24 h (Supplementary Fig. 3C and Fig. 4C). The enhanced level of TWIST1 mRNA was reduced by Met treatment, implying that TWIST1, through NF-κB activation, is responsible for EMT progression in RPE cells under nutrient-depleted conditions. In addition, to confirm that TWIST1 is indeed involved in EMT during starvation, we examined whether EMT progression was attenuated in RPE cells transfected with TWIST1 siRNA (Supplementary Fig. 3D). Cells transfected with TWIST1 siRNA showed the presence of CDH1 in the cell-cell junctions, with little VIM fluorescence, even under nutrient starvation, while both non-transfected and control siRNA-transfected cells showed depleted CDH1 fluorescence under nutrient starvation. Collectively, our findings indicate that AMPK activation can decelerate TWIST1-mediated EMT progression in RPE cells, which leads to attenuation of RPE degeneration induced by nutrient starvation.
In conclusion, our studies demonstrate that activated AMPK downregulates the accumulation of ROS and mitochondrial senescence in RPE cells under nutrient starvation (Fig. 4D). As RPE cells were exposed to nutrient starvation, activation of AMPK maintained a healthy mitochondrial population by facilitating mitochondrial biogenesis through the activation of PGC1-α and subsequent TFAM transcription. Furthermore, stimulation of AMPK-induced mitochondrial biogenesis was beneficial for the efficient suppression of oxidative stress caused by nutrient starvation-induced senescent mitochondria. As starvation persisted, RPE cells underwent EMT, which induced RPE degeneration. EMT progression was facilitated by the activation of TWIST1 transcription, following the ROS-induced NF-κB pathway in RPE cells under prolonged starvation. We observed that enhanced activation of AMPK delayed the onset of EMT progression by reducing ROS accumulation under nutrient starvation. Our results provide an insight into the mechanisms of RPE degeneration by senescent mitochondria, suggesting that AMPK activation would be beneficial in impeding the progression of retinal degeneration.
MATERIALS AND METHODS
Detailed materials and methods are available in Supplementary Material.
Funding Statement
ACKNOWLEDGEMENTS This research was supported by a grant from the National Research Foundation of Korea (NRF) funded by the Korean government, Ministry of Science, and ICT (RS-2022-00165439).
Footnotes
CONFLICTS OF INTEREST
The authors have no conflicting interests.
REFERENCES
- 1.Chen M, Rajapakse D, Fraczek M, Luo C, Forrester JV, Xu HP. Retinal pigment epithelial cell multinucleation in the aging eye - a mechanism to repair damage and maintain homoeostasis. Aging Cell. 2016;15:436–445. doi: 10.1111/acel.12447. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Lidgerwood GE, Senabouth A, Smith-Anttila CJA, et al. Transcriptomic profiling of human pluripotent stem cell-derived retinal pigment epithelium over time. Genom Proteom Bioinform. 2021;19:223–242. doi: 10.1016/j.gpb.2020.08.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Lipecz A, Miller L, Kovacs I, et al. Microvascular contributions to age-related macular degeneration (AMD): from mechanisms of choriocapillaris aging to novel interventions. Geroscience. 2019;41:813–845. doi: 10.1007/s11357-019-00138-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Swerdlow RH. Mitochondrial medicine and the neurodegenerative mitochondriopathies. Pharmaceuticals (Basel) 2009;2:150–167. doi: 10.3390/ph2030150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Scherz-Shouval R, Elazar Z. Regulation of autophagy by ROS: physiology and pathology. Trends Biochem Sci. 2011;36:30–38. doi: 10.1016/j.tibs.2010.07.007. [DOI] [PubMed] [Google Scholar]
- 6.Song SB, Hwang ES. A rise in ATP, ROS, and mitochondrial content upon glucose withdrawal correlates with a dysregulated mitochondria turnover mediated by the activation of the protein deacetylase SIRT1. Cells. 2019;8:11. doi: 10.3390/cells8010011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Baek A, Son S, Baek YM, Kim DE. KRT8 (keratin 8) attenuates necrotic cell death by facilitating mitochondrial fission-mediated mitophagy through interaction with PLEC (plectin) Autophagy. 2021;17:3939–3956. doi: 10.1080/15548627.2021.1897962. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Zhao C, Yasumura D, Li X, et al. mTOR-mediated dedifferentiation of the retinal pigment epithelium initiates photoreceptor degeneration in mice. J Clin Invest. 2011;121:369–383. doi: 10.1172/JCI44303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Boles NC, Fernandes M, Swigut T, et al. Epigenomic and transcriptomic changes during human rpe emt in a stem cell model of epiretinal membrane pathogenesis and prevention by nicotinamide. Stem Cell Rep. 2020;14:631–647. doi: 10.1016/j.stemcr.2020.03.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Jiang JW, Wang K, Chen Y, Chen HN, Nice EC, Huang CH. Redox regulation in tumor cell epithelial-mesenchymal transition: molecular basis and therapeutic strategy. Sig Transduct Target Ther. 2017;2:17036. doi: 10.1038/sigtrans.2017.36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Chua HL, Bhat-Nakshatri P, Clare SE, Morimiya A, Badve S, Nakshatri H. NF-κB represses E-cadherin expression and enhances epithelial to mesenchymal transition of mammary epithelial cells: potential involvement of ZEB-1 and ZEB-2. Oncogene. 2007;26:711–724. doi: 10.1038/sj.onc.1209808. [DOI] [PubMed] [Google Scholar]
- 12.Julien S, Puig I, Caretti E, et al. Activation of NF-κB by Akt upregulates snail expression and induces epithelium mesenchyme transition. Oncogene. 2007;26:7445–7456. doi: 10.1038/sj.onc.1210546. [DOI] [PubMed] [Google Scholar]
- 13.Storci G, Sansone P, Mari S, et al. TNFalpha up-regulates SLUG via the NF-kappaB/HIF1alpha axis, which imparts breast cancer cells with a stem cell-like phenotype. J Cell Physiol. 2010;225:682–691. doi: 10.1002/jcp.22264. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Chatterjee R, Chatterjee J. ROS and oncogenesis with special reference to EMT and stemness. Eur J Cell Biol. 2020;99:151073. doi: 10.1016/j.ejcb.2020.151073. [DOI] [PubMed] [Google Scholar]
- 15.González A, Hall MN, Lin SC, Hardie DG. AMPK and TOR: the Yin and Yang of cellular nutrient sensing and growth control. Cell Metab. 2020;31:472–492. doi: 10.1016/j.cmet.2020.01.015. [DOI] [PubMed] [Google Scholar]
- 16.Picca A, Mankowski RT, Burman JL, et al. Mitochondrial quality control mechanisms as molecular targets in cardiac ageing. Nat Rev Cardiol. 2018;15:543–554. doi: 10.1038/s41569-018-0059-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Herzig S, Shaw RJ. AMPK: guardian of metabolism and mitochondrial homeostasis. Nat Rev Mol. 2018;19:121–135. doi: 10.1038/nrm.2017.95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Ferree AW, Trudeau K, Zik E, et al. MitoTimer probe reveals the impact of autophagy, fusion, and motility on subcellular distribution of young and old mitochondrial protein and on relative mitochondrial protein age. Autophagy. 2013;9:1887–1896. doi: 10.4161/auto.26503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Cottet-Rousselle C, Ronot X, Leverve X, Mayol JF. Cytometric assessment of mitochondria using fluorescent probes. Cytometry A. 2011;79:405–425. doi: 10.1002/cyto.a.21061. [DOI] [PubMed] [Google Scholar]
- 20.Marin TL, Gongol B, Zhang F, et al. AMPK promotes mitochondrial biogenesis and function by phosphorylating the epigenetic factors DNMT1, RBBP7, and HAT1. Sci Signal. 2017;10:eaaf7478. doi: 10.1126/scisignal.aaf7478. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Pernicova I, Korbonits M. Metformin-mode of action and clinical implications for diabetes and cancer. Nat Rev Endocrinol. 2014;10:143–156. doi: 10.1038/nrendo.2013.256. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.