

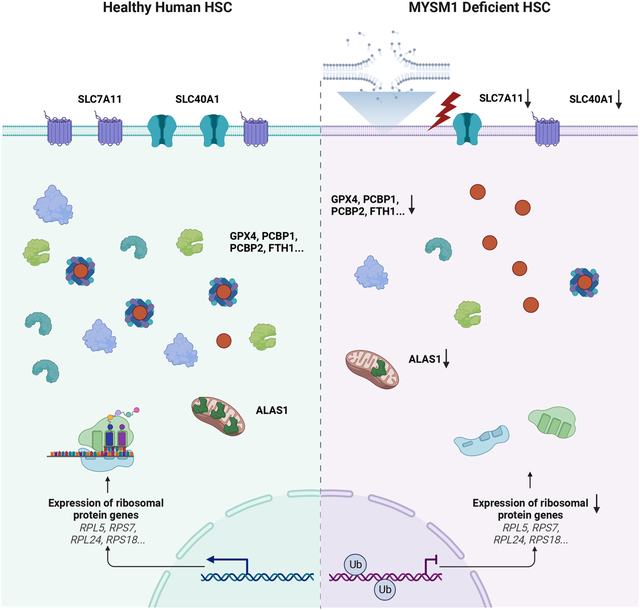
Summary
Hematopoietic stem cells (HSCs) have a number of unique physiologic adaptations that enable lifelong maintenance of blood cell production, including a highly regulated rate of protein synthesis. Yet the precise vulnerabilities that arise from such adaptations have not been fully characterized. Here, inspired by a bone marrow failure disorder due to loss of the histone deubiquitinase MYSM1, characterized by selectively disadvantaged HSCs, we show how reduced protein synthesis in HSCs results in increased ferroptosis. HSC maintenance can be fully rescued by blocking ferroptosis, despite no alteration in protein synthesis rates. Importantly, this selective vulnerability to ferroptosis not only underlies HSC loss in MYSM1 deficiency, but also characterizes a broader liability of human HSCs. Increasing protein synthesis rates via MYSM1 overexpression makes HSCs less susceptible to ferroptosis, more broadly illuminating the selective vulnerabilities that arise in somatic stem cell populations as a result of physiologic adaptations.
Graphical Abstract
Introduction
Lifelong production of blood and immune cells, or hematopoiesis, requires the continuous activity of rare hematopoietic stem cells (HSCs) that reside in the bone marrow1,2. These HSCs primarily remain in a quiescent state and can be activated to maintain hematopoiesis in homeostatic conditions or under the context of stressors, such as infections3. Alterations to HSC function can cause bone marrow failure or promote progression to pre-malignant states and blood cancers. As a result of the sensitivity of HSCs to genotoxic or other stress-induced insults that can impair their effectiveness over the lifespan, these cells have acquired a variety of adaptations that can help to sustain their function. HSCs have a number of metabolic adaptations that include reliance on glycolysis and autophagy to enable sufficient energy generation without excess oxidant stress4–7. In addition, HSCs are characterized by low and highly-regulated rates of protein synthesis, which needs to be balanced with the rate of protein degradation, to maintain effective blood cell production8–11. While considerable insights have been generated from studies of HSCs conducted through targeting specific genes or pathways, the full spectrum of adaptations found in HSCs and the vulnerabilities arising as a result remain incompletely understood.
Studies of human disorders characterized by HSC loss or dysfunction provide a unique opportunity to gain new insights into the selective vulnerabilities found in these cells2. For example, studies of Fanconi anemia, which is characterized by a variety of mutations in the homologous recombination repair pathway, have revealed a role for these proteins in preventing HSC genotoxicity resulting from endogenous aldehydes that promote DNA crosslinking12,13. In an analogous manner, we reasoned that studies of a bone marrow failure disorder attributable to biallelic loss-of-function of the histone deubiquitinase MYSM1 could reveal additional and previously unappreciated pathways that are important for human HSC maintenance. The phenotype of these patients is distinct from other bone marrow failure syndromes and therefore this disease provides an opportunity to identify new regulatory aspects found in HSCs. In this disorder, there is progressive HSC loss with a reduced level of mature blood and immune cell production. Importantly, HSC transplantation and somatic reversion can rescue the phenotype observed in these patients14–18. Here, we set out to faithfully model this disorder with genome editing in human HSCs and conduct in depth mechanistic studies to decipher how this loss of MYSM1 can compromise HSC function. Our studies reveal a selective vulnerability of all human HSCs to ferroptosis and uncover a unique physiologic role for this distinct form of cell death in human hematopoiesis.
Results
Loss of MYSM1 compromises human HSC function
Bone marrow failure syndrome 4 (OMIM #618116) is a rare autosomal recessive genetic disorder characterized by mutations in histone deubiquitinase MYSM1. Over a dozen patients with this form of bone marrow failure due to a number of distinct mutations have been reported to date14–20. The majority of these mutations cause complete loss-of-function of MYSM1 (Figure 1A). Importantly, this form of bone marrow failure is characterized by reduced production of all hematopoietic lineages and reduced cellularity of the bone marrow, suggesting a general defect impacting HSCs. Consistent with this, at least one patient has had somatic reversion in a subpopulation of HSCs enabling complete correction of their hematologic and immune phenotypes and other patients have been cured with HSC transplantation16,21.
Figure 1. Loss of MYSM1 in CD34+ HSPCs recapitulates bone marrow failure phenotype observed in patients.
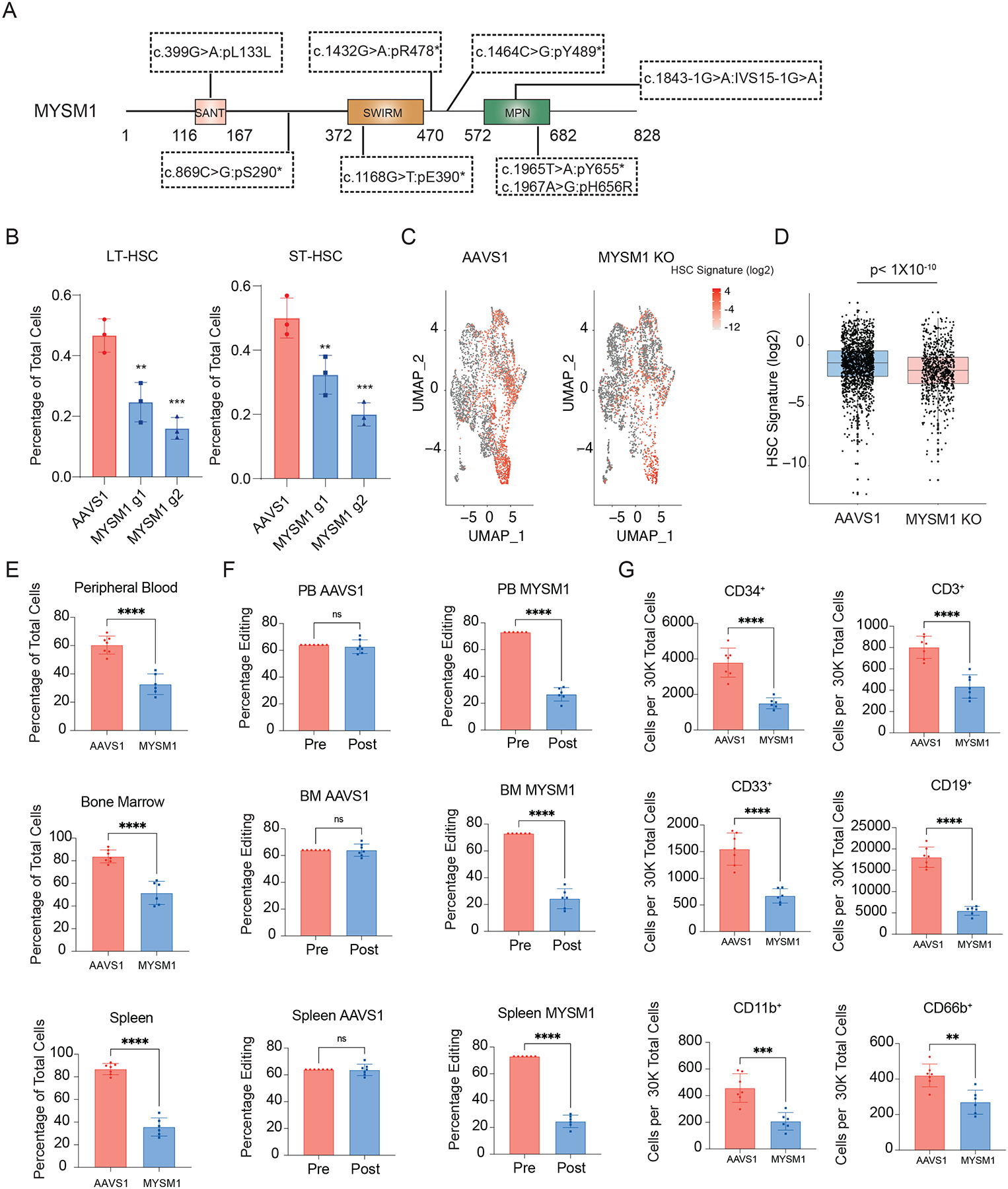
(A) Comprehensive schematic diagram of MYSM1 mutants identified in patients. Asterisks represent stop codons. (B) Quantification of LT- and ST-HSC populations in AAVS1 and MYSM1 edited CD34+ HSPCs. (C) Uniform manifold approximation and projection (UMAP) plots of 4,293 (AAVS1) and 3,871 (MYSM1 KO) CD34+CD45RA−CD90+ cells, highlighted according to average z-score normalized HSC gene signature. (D) Box plot of z-score normalized HSC signature expression of all cells of AAVS1 and MYSM1 KO groups. (E) Percentage of total human CD45+ cells after RBC depletion from indicated sites of NBSGW mice xenotransplanted with AAVS1 and MYSM1 edited cord blood CD34+ HSPCs. (F) Percentage of edits of cells before and after xenotransplantation from indicated sites of NBSGW mice xenotransplanted with AAVS1 and MYSM1 edited cord blood CD34+ HSPCs. (G) Cell numbers per 30,000 RBC depleted bone marrow cells of progenitor cells (CD34+), T cells (CD3+), myeloid (CD33+, CD11b+), B cells (CD19+) and granulocytes (CD66b+).
While mouse knockouts of Mysm1 do display some hematologic phenotypes22–26, there are notable differences in comparison to what is observed in human patients. For example, the mouse knockouts of Mysm1 lack progression to the pancytopenia observed in humans and have distinct hematologic phenotypes, such an increase, rather than a decrease in platelet counts. To more fully define the human HSC defects arising with MYSM1 mutations, we sought to faithfully model the loss-of-function mutations observed in patients using CRISPR-Cas9 genome editing27,28. Two days after Cas9 ribonucleoprotein (RNP) delivery, we observed an overall editing efficiency of greater than 90% with two independent MYSM1 gRNAs (Figure S1A). Among the edits, the majority were predicted to result in nonsense mutations and a significant reduction of protein level was observed by three days post-editing (Figure S1B–D). Concomitantly, we observed a reduction of both long term (LT-) and short term phenotypic HSCs (ST-HSCs) (Figure 1B, S1E). Given the imprecision of surface markers to confidently delineate molecularly-defined HSCs2,29, we performed single cell RNA sequencing (scRNA-seq) on AAVS1 control and MYSM1 edited CD34+CD45RA−CD90+ cells and observed a significant reduction of cells harboring a molecular signature known to enrich for HSCs with MYSM1 editing30 (Figure 1C–D).
To fully characterize the role of MYSM1 in human hematopoiesis, we transplanted AAVS1 control and MYSM1 edited cord blood CD34+ HSPCs into the NOD.Cg-KitW41JTyr+PrkdcscidIl2rgtm1Wjl/ThomJ (NBSGW) strain of immunodeficient and Kit mutant mice29,31,32. Consistent with the in vitro HSC depletion phenotype, there was significantly lower human CD45+ cell engraftment in the peripheral blood, bone marrow, and spleen at 4 months post-transplantation (Figure 1E, S1E), when hematopoiesis will predominantly be driven by transplanted HSCs. Importantly, the overall editing efficiency of cells was also dramatically reduced compared to CD34+ HSPCs before transplantation (Figure 1F), suggesting a strong selection against MYSM1 deficient cells. Based upon both the reduced bone marrow engraftment (~2.5-fold reduction) and depletion of MYSM1 edited alleles (~3-fold reduction), we estimate an ~8-fold loss of HSCs. Analysis of all major lineages confirmed that the repopulation defect broadly impacted all lineages (Figure 1G). Collectively, this data confirmed that normal MYSM1 expression is essential for human HSC maintenance both in vitro and in vivo, and genome editing of MYSM1 in HSPCs can faithfully recapitulate the bone marrow failure phenotypes observed in patients with biallelic mutations. Importantly, this model enabled us to ask critical questions about the molecular basis for HSC loss in this disorder, which remains enigmatic despite a number of phenotypic studies in mouse models.
Reduced protein synthesis rates in HSCs with MYSM1 loss
To begin to define how MYSM1 loss can compromise human HSC function, we examined the scRNA-seq data for those cells that had the molecular HSC signature and found that there was a significant downregulation of gene sets and genes implicated in ribosomal biogenesis and translation regulation (Figure 2A–C). Given that this downregulation appeared to be subtle, we confirmed that the expression of various ribosomal proteins and eukaryotic initiation factor (EIF) encoding mRNAs was significantly reduced in CD34+CD45RA−CD90+ cells (Figure S2A). Moreover, gene set enrichment analysis (GSEA) reinforced the finding that genes involved in translation initiation and ribosome formation were significantly depleted in MYSM1 edited HSCs (Figure 2D, S2B, Table S1–2). Importantly, the protein levels of many ribosomal proteins were also significantly reduced (Figure 2E). These findings are consistent with a recent report using a Mysm1 knockout mouse, where Mysm1 was shown to be important for transcription of ribosomal protein genes through occupancy at the proximal promoters and transcriptional activation18.
Figure 2. Loss of MYSM1 results in reduced cellular translation.
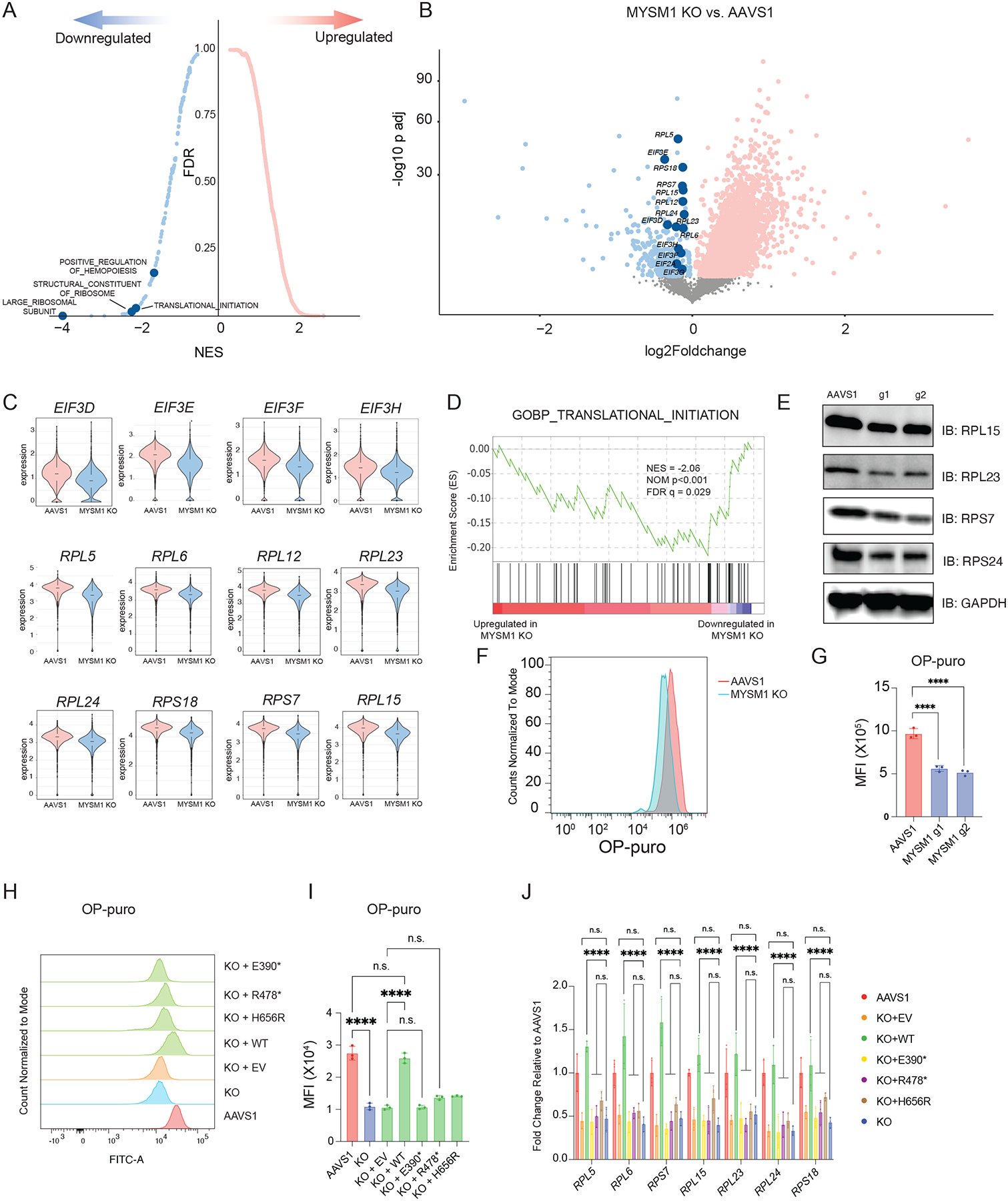
(A) Top enriched signatures in upregulated and downregulated genes in MYSM1 KO cells with average z-score normalized HSC gene signature > 0.2, highlighting pathways directly related to translation regulation. (B) Volcano plot of MYSM1 KO vs. AAVS1 gene expression in HSCs, highlighting genes that are involved in ribosomal biogenesis and translational initiation. (C) Representative violin plots of ribosomal protein gene expression in AAVS1 and MYSM1 KO HSCs. (D) Gene set enrichment analysis (GSEA) showing translation initiation is significantly downregulated in MYSM1 KO cells. (E) Western blot analysis of representative ribosomal protein levels of CD34+CD45RA−CD90+ sorted cells with AAVS1 or MYSM1 editing. (F) Representative flow cytometric histogram of O-Propargyl-puromycin based translation rate analysis on CD34+CD45RA−CD90+ sorted cells with AAVS1 or MYSM1 editing. (G) Quantification of mean fluorescence intensity (MFI) of O-Propargyl-puromycin based translation rate analysis on CD34+CD45RA−CD90+ sorted cells with AAVS1 or MYSM1 editing. (H) Representative flow cytometric histogram of O-Propargyl-puromycin based translation rate analysis on CD34+CD45RA−CD90+ sorted cells with AAVS1 or MYSM1 editing, and MYSM1 edited cells transduced with indicated overexpression constructs. (I) Quantification of mean fluorescence intensity (MFI) of O-Propargyl-puromycin based translation rate analysis on CD34+CD45RA−CD90+ sorted cells with AAVS1 or MYSM1 editing, and MYSM1 edited cells transduced with indicated overexpression constructs. (J) Real time PCR analysis of representative ribosomal protein expression of AAVS1 or MYSM1 edited cells, and MYSM1 edited cells transduced with indicated overexpression constructs.
While our data suggested that the global transcriptional program of ribosome biogenesis was downregulated, we sought to directly measure the impact on protein translation rates in HSCs. We therefore labeled CD34+CD45RA−CD90+ cells with O-Propargyl-puromycin (OP-puro) to assess translation rates in this population using flow cytometry10,33. The rate of protein translation rate was reduced to ~50% in MYSM1 edited HSCs compared to AAVS1 control edited cells (Figure 2F–G), directly demonstrating altered mRNA translation due to the loss of MYSM1 that causes reduced transcription of ribosomal proteins and other genes18. Importantly, we could confirm that this loss of protein synthesis was specifically due to MYSM1 activity by lentiviral rescue of edited cells, which could restore OP-puro incorporation and ribosomal protein gene expression to the levels observed in healthy HSCs (Figure S2C, Figure 2H–J). Importantly, several mutations found in patients were unable to elicit a similar rescue, validating the loss-of-function due to these alleles (Figure 2H–J).
Compromised HSC function in MYSM1 deficiency due to ferroptosis
While we and others have observed reduced ribosomal protein gene expression upon MYSM1 loss with accompanying reduced protein synthesis, how this would compromise HSC function remained unclear. HSCs are known to have a highly regulated and generally low rate of protein synthesis10, but the specific liabilities that arise from these adaptations have not been fully defined. To identify additional causes for the HSC loss, we examined the gene expression changes identified through scRNA-seq. Interestingly, we observed a large number of upregulated genes in MYSM1 edited HSCs, suggesting that some of these genes may provide insights into altered pathways underlying the observed loss of HSCs. Among the upregulated pathways, we found that there was a significant enrichment in oxidant detoxification, antioxidant, and iron binding genes (Figure 3A, S3B, Table S1–2). Genes involved in iron metabolism and antioxidant activity were significantly upregulated, whereas genes involved in iron absorption, such as TFRC, were significantly reduced (Figure 3B–C, S3A). These data suggested that the cells might undergo significant oxidative stress and this may be accompanied by dysregulated iron handling.
Figure 3. Loss of MYSM1 disrupts iron handling and results in increased ferroptosis in HSCs.
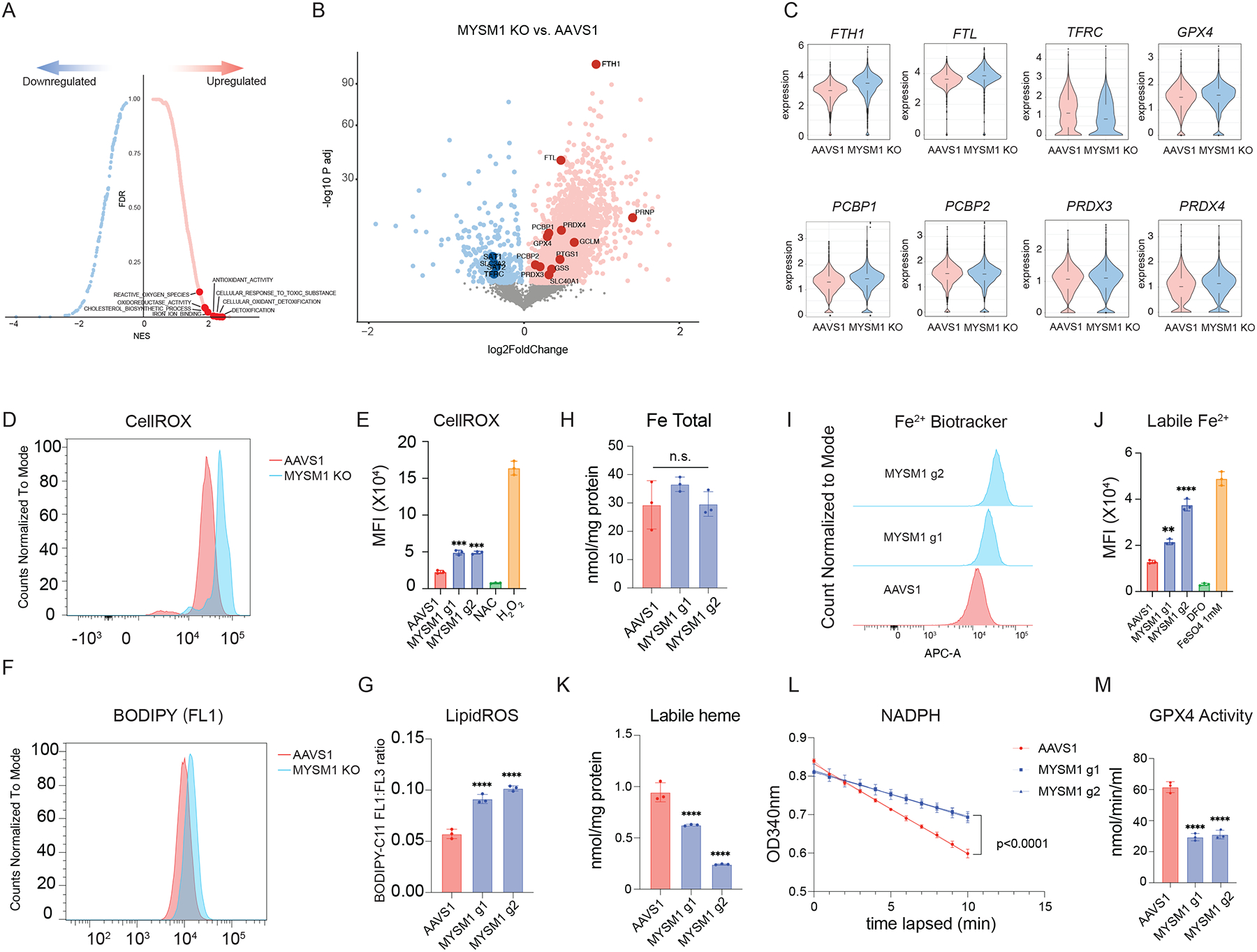
(A) Top enriched signatures in upregulated and downregulated genes in MYSM1 KO cells with average z-score normalized HSC gene signature > 0.2, highlighting pathways relevant to iron metabolism and ferroptosis. (B) Volcano plot of MYSM1 KO vs. AAVS1 gene expression in HSCs, highlighting genes that are involved in iron transport, storage and metabolism. (C) Representative violin plots of iron transport, storage and metabolism gene expression in AAVS1 and MYSM1 KO HSCs. (D) Representative flow cytometric histogram of cellular ROS level of sorted CD34+CD45RA−CD90+ cells measured by CellROX dye. (E) Quantification of mean fluorescence intensity (MFI) of cellular ROS level of sorted CD34+CD45RA−CD90+ cells measured by CellROX dye. (F) Representative flow cytometric histogram of oxidized BODIPY dye of sorted CD34+CD45RA−CD90+ cells. (G) Quantification of cellular lipid peroxidation level of sorted CD34+CD45RA−CD90+ cells measured by ratio of oxidized and non-oxidized BODIPY dye. (H) Quantification of total intracellular iron level of AAVS1 and MYSM1 edited cells. (I) Representative flow cytometric histogram of ferrous iron level of sorted CD34+CD45RA−CD90+ cells as measured by Fe2+ biotracker dye. (J) Quantification of mean fluorescence intensity (MFI) of labile ferrous iron (Fe2+) level of sorted CD34+CD45RA−CD90+ cells as measured by Fe2+ biotracker dye. (K) Quantification of total intracellular labile hemin level of AAVS1 and MYSM1 edited cells. (L) Time lapsed reduction of NADPH availability in GPX4 assay reaction measure by OD340. (M) Calculated intracellular GPX4 activity of sorted CD34+CD45RA−CD90+ cells of AAVS1 and MYSM1 edited cells.
To directly assess how these gene expression changes were impacting the cells, we measured overall oxidative stress in CD34+CD45RA−CD90+ cells using the dye CellROX, which can measure all reactive oxygen species (ROS), and found that MYSM1 edited cells displayed globally increased ROS levels (Figure 3D–E). Moreover, we found that lipid peroxidation levels, as measured by staining with the lipid dye BODIPY C11, were significantly increased in MYSM1 edited HSCs (Figure 3F–G). Additionally, we observed elevated lipid peroxidation levels in the human CD34+CD45RA−CD90+ HSC-enriched cell population from the bone marrow of NBSGW mice after 16 weeks of transplantation with MYSM1 edited cord blood HSPCs (Figure S4A). While we saw gene expression changes in iron regulating genes that would suggest protection from a cell death process such as ferroptosis34–36, the elevated lipid peroxidation suggested increased ferroptosis in this setting. Indeed, while we observed no change of total intracellular iron levels between AAVS1 control and MYSM1 edited HSCs, the intracellular concentration of redox active ferrous iron (Fe2+) was significantly higher in MYSM1 edited HSCs than controls (Figure 3H–J). Interestingly, one of the key iron handling processes37, heme synthesis, appeared to be inhibited and intracellular labile heme levels were also reduced in MYSM1 edited cells (Figure 3K). Additionally, although we observed a transcriptional upregulation of GPX4 mRNA in the HSC scRNA-seq data, the GPX4 activity in MYSM1 edited HSCs was significantly reduced (Figure 3L–M). Together, our data suggested that MYSM1 edited cells suffer from a significant upregulation of ROS that may underlie the HSC dysfunction and which appeared to arise from impaired iron handling. These features collectively suggested that the intracellular iron-dependent form of cell death, ferroptosis, may be activated in this setting and underlie the loss of HSCs. Notably this form of cell death is distinct from apoptosis, necroptosis, and other cell death pathways that have been reported.
We wanted to further confirm that the HSC loss we observed with MYSM1 deficiency was due to ferroptosis and not another form of cell death. Annexin V and PI staining are commonly used to assess apoptosis, but also are detected in the setting of ferroptosis38–40. Consistent with this, we indeed detected increased annexin V and PI staining in CD34+CD45RA−CD90+ MYSM1-deficient cells (Figure S4B). To more specifically assess for ferroptosis with morphologic features that characterize this process36, we examined the AAVS1 or MYSM1-edited CD34+CD45RA−CD90+ cells with electron microscopy, which revealed characteristic morphologic features of ferroptosis, including shrunken mitochondria and reduced mitochondrial cristae at levels observed in a well-characterized cell line treated with ferroptosis inducer FIN56 (Figure 4A–E). Additionally, MYSM1 deficient CD34+CD45RA−CD90+ cells also lacked expression of cleaved PARP or caspases, as would be found in apoptotic cells, and they also lacked phosphorylated MLKL, RIP, and RIPK3, as would be found in the setting of necroptosis (Figure S4C). Together, our data strongly suggested that MYSM1-deficient HSCs are lost by ferroptosis and not by another form of cell death.
Figure 4. MYSM1-deficient HSCs harbor signature ferroptosis features.
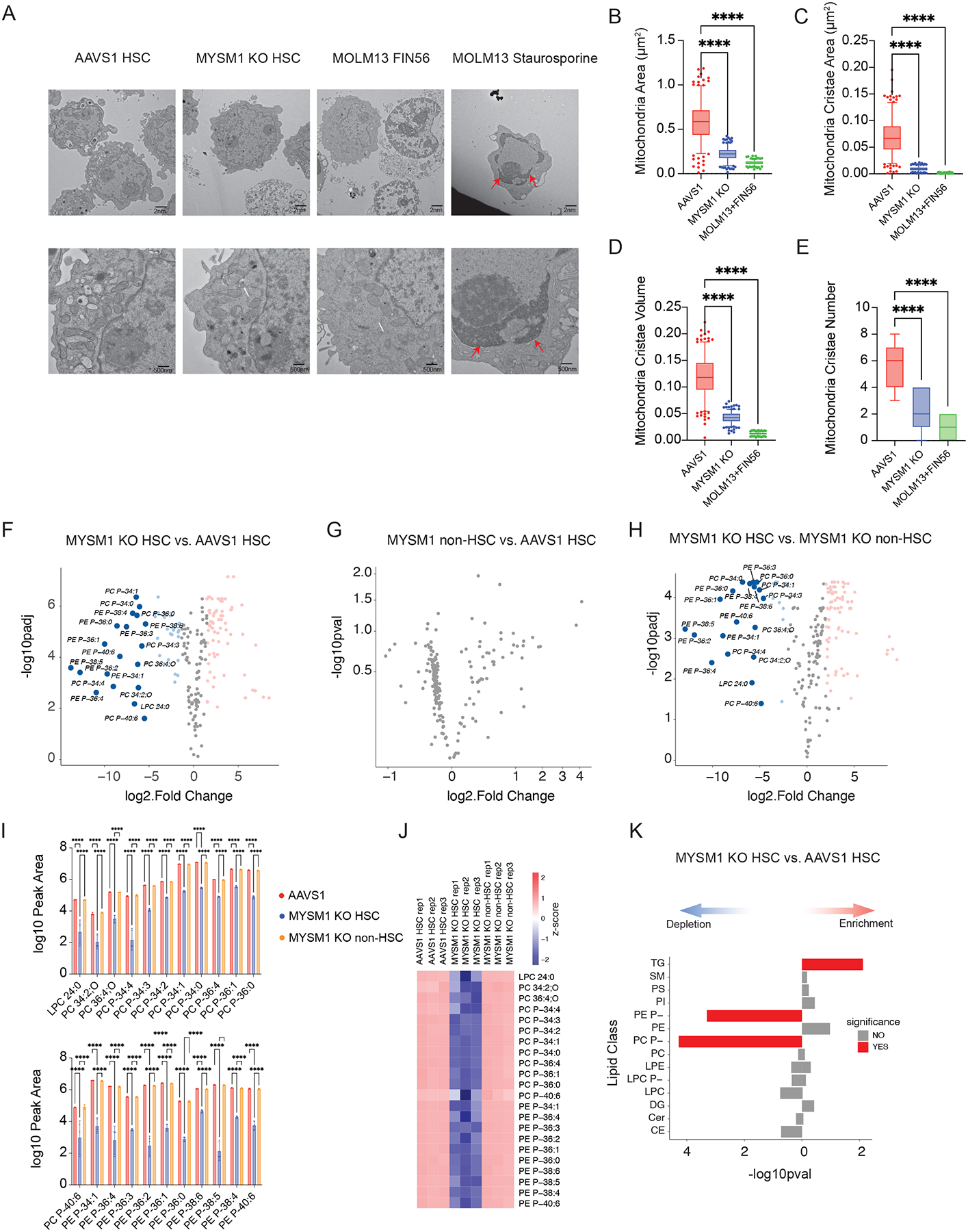
(A) Representative electron microscopic images of sorted CD34+CD45RA−CD90+ cells nucleofected with AAVS1 and MYSM1 gRNA. MOLM13 cell line treated with 5μM FIN56 for 10 hours was used as positive control for ferroptosis, and MOLM13 cell line treated with 1μM staurosporine for 5 hours was used as control for apoptosis. Top row scale bar = 2nm. Bottom row scale bar = 500nm. White arrows: shrunken mitochondria; Red arrows: chromatin condensation. (B) Quantification of mitochondria area of 10 mitochondria per cell for a total of 25 cells each sample (total of 250 mitochondria). Individual points out of 5 to 95 percentile were plotted and shown. (C) Quantification of mitochondria cristae area of 10 mitochondria per cell for a total of 25 cells each sample (total of 250 mitochondria). Individual crista areas were summed up for the total cristae surface area of each mitochondrion. Individual points out of 5 to 95 percentile were plotted and shown. (D) Quantification of mitochondria cristae volume, defined as the ratio of total cristae surface area of each mitochondrion to mitochondrial area, of 10 mitochondria per cell for a total of 25 cells each sample (total of 250 mitochondria). Individual points out of 5 to 95 percentile were plotted and shown. (E) Quantification of the cristae number of 10 mitochondria per cell for a total of 25 cells each sample (total of 250 mitochondria). (F) Volcano plots of differentially expressed lipids of sorted CD90+CD133+ MYSM1 KO cells compared to AAVS1 control. (G) Volcano plots of differentially expressed lipids of sorted CD90− MYSM1 KO cells compared to AAVS1 control. (H) Volcano plots of differentially expressed lipids of sorted CD90+CD133+ MYSM1 KO cells compared to CD90− MYSM1 KO cells. (I) Representative bar plots of lipids depleted in CD90+CD133+ MYSM1 KO cells. (J) Heatmap of lipids depleted in CD90+CD133+ MYSM1 KO cells. (K) Lipid species enrichment of CD90+CD133+ MYSM1 KO cells compared to AAVS1 controls by over-representation analysis. TG: triacylglycerol; SM: sphingomyelin; PS: phosphatidylserine; PI: phosphotidylinositol; PE P-: 1-O-alkenyl-glycerophosphoethanolamine; PE: diacyl glycerophosphoethanolamine; PC P-: 1-O-alkenyl-glycerophosphocholine; PC: diacyl glycerophosphocholine; LPE: lyso-diacyl glycerophosphoethanolamine; LPC P-: lyso-1-O-alkenyl-glycerophosphocholine; DG: diacylglycerol; Cer: ceramide; CE: cholesterol ester.
To further characterize the observed alterations that accompany the induction of ferroptosis in HSCs, we performed lipidomic analyses on the CD34+CD45RA−CD90+CD133+ HSC-enriched fraction of AAVS1 control and MYSM1 edited cells. We also compared the MYSM1 edited HSC enriched fraction to the non-HSC fraction of bulk progenitors that are CD34+CD90−. Consistent with prior analyses in the setting of ferroptosis41,42, there was a significant depletion of a wide spectrum of polyunsaturated phospholipid species, which are known to be most profoundly impacted with the lipid peroxidation that characterizes ferroptosis (Figure 4F–H, S4D–E, Tables S3–5). Critically, these changes were only observed within the HSC enriched compartment upon MYSM1 editing and were absent from the more differentiated progenitor compartment (CD34+CD90−). Importantly, while previous studies have focused on ester linked phospholipids, our data shows that the vast majority of phospholipids depleted are ether linked phospholipids, specifically the 1-O-alkenyl-glycerophospholipids in human HSCs, whose functional importance in ferroptosis has been highlighted in recent studies43 (Figure 4I–J, S4F–G, Table S3–5). Indeed, our data shows that while a few ester linked phospholipids are depleted, the majority of depleted lipids are ether linked phospholipids (Figure 4K). These data reveal that ether linked, but not ester linked, phospholipids are more susceptible to lipid peroxidation and ferroptosis due to loss of MYSM1 in HSCs. Accompanying these changes, we also noted an upregulation of triglyceride species, which may be due to changes following lipid peroxidation in the setting of ferroptosis (Figure 4K).
We next sought to directly test whether ferroptosis plays a role in the HSC depletion observed due to MYSM1 loss. We treated cells harboring loss of MYSM1 with a number of well established ferroptosis inhibitors including deferoxamine (DFO), ferrostatin-1 (Fer-1), and vitamin E, as well as the mitochondrially targeted antioxidant mitoTempo that will reduce ROS in the mitochondria, but will not alter ferroptosis44. Critically, all ferroptosis inhibitors could fully rescue the loss of LT- and ST-HSCs due to MYSM1 deficiency without any alteration in the parental CD34+CD45RA− progenitor population (Figure 5A). In contrast and consistent with the specificity of this rescue, the mitochondrially targeted antioxidant mitoTempo did not rescue the LT- or ST-HSC populations (Figure 5A). Moreover, all ferroptosis inhibitors were able to fully rescue and reduce the elevation of pan-ROS levels and lipid peroxidation in MYSM1 edited cells without impacting the compromised protein synthesis rates (Figure 5B–D, S5). This finding shows how ferroptosis underlies the loss of HSCs in MYSM1 deficiency and lies downstream of the reduced protein synthesis that occurs in this disorder.
Figure 5. Inhibition of ferroptosis rescues HSC depletion due to MYSM1 loss.
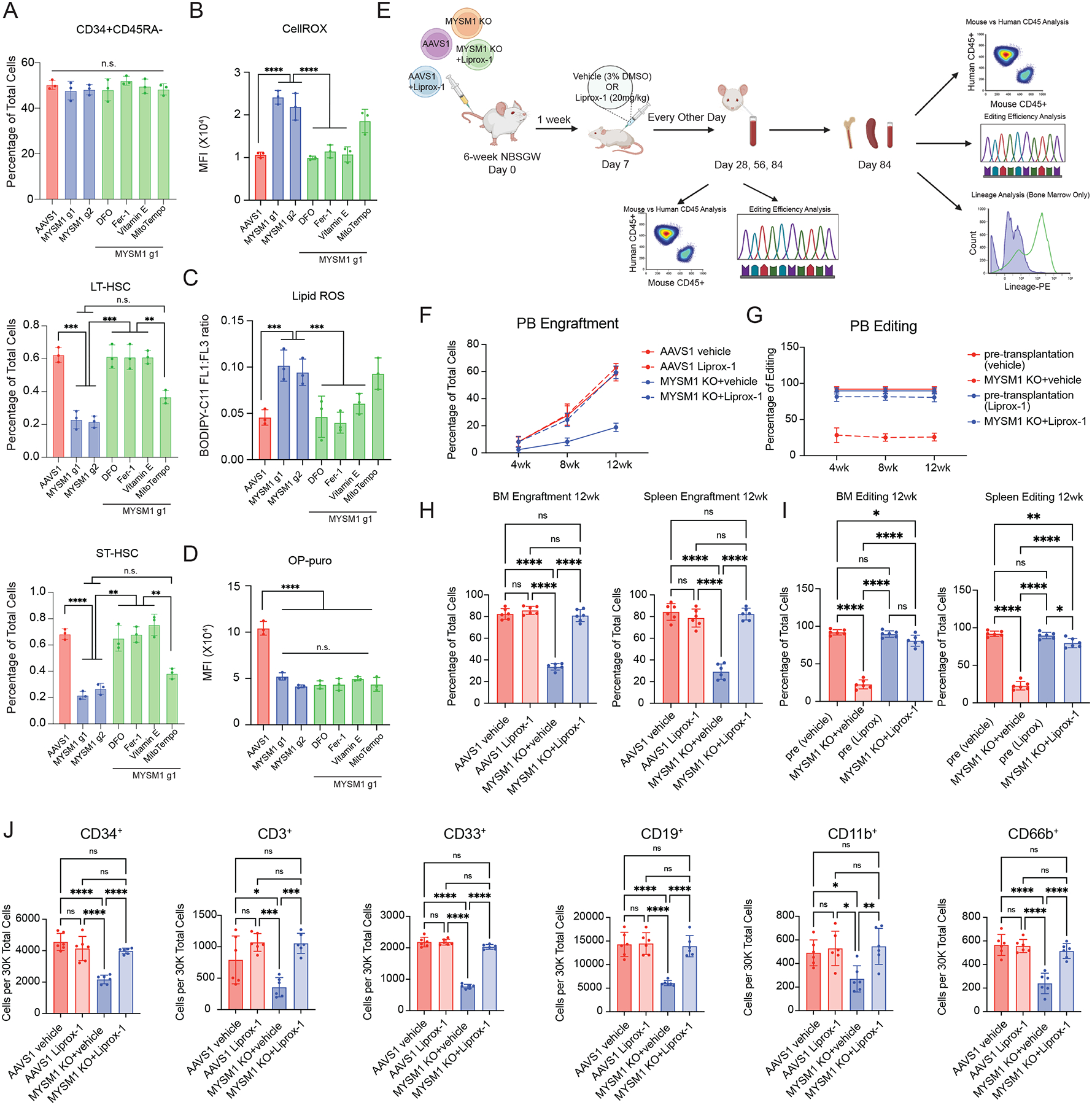
(A) Quantification of CD34+CD45RA−, ST-HSC, and LT-HSC populations in AAVS1 or MYSM1 edited cells, and MYSM1 edited cells treated with indicated chemicals. (B) Quantification of mean fluorescence intensity (MFI) of cellular ROS level of sorted CD34+CD45RA−CD90+ cells with AAVS1 or MYSM1 editing, and MYSM1 edited cells with indicated chemical treatments measured by CellROX dye. (C) Quantification of cellular lipid peroxidation level of sorted CD34+CD45RA−CD90+ cells with AAVS1 or MYSM1 editing, and MYSM1 edited cells with indicated chemical treatments measured by ratio of oxidized and non-oxidized BODIPY dye. (D) Quantification of mean fluorescence intensity (MFI) of O-Propargyl-puromycin based translation rate analysis on CD34+CD45RA−CD90+ sorted cells with AAVS1 or MYSM1 editing, and MYSM1 edited cells with indicated chemical treatments. (E) Schematic of in vivo rescue xenotransplantation experiment. (F) Percentage of total human CD45+ cells after RBC depletion from peripheral blood of NBSGW mice xenotransplanted with AAVS1 and MYSM1 edited cord blood CD34+ HSPCs treated with either vehicle (3%DMSO) or 20mg/kg Liproxstatin-1. Peripheral blood was harvested through retro-orbital plexus and analyzed at 4, 8 and 12 weeks post-transplantation. (G) Percentage of edits of MYSM1 edited cells before and after xenotransplantation from peripheral blood of NBSGW mice xenotransplanted with AAVS1 and MYSM1 edited cord blood CD34+ HSPCs treated with vehicle (3%DMSO) or 20mg/kg Liproxstatin-1. (H) Percentage of total human CD45+ cells after RBC depletion from bone marrow (left) or spleen (right) of NBSGW mice xenotransplanted with AAVS1 and MYSM1 edited cord blood CD34+ HSPCs treated with either vehicle (3%DMSO) or 20mg/kg Liproxstatin-1. (I) Percentage of edits of MYSM1 edited cells before and after xenotransplantation from bone marrow (left) or spleen (right) of NBSGW mice xenotransplanted with AAVS1 and MYSM1 edited cord blood CD34+ HSPCs treated with vehicle (3%DMSO) or 20mg/kg Liproxstatin-1. (J) Cell numbers per 30,000 RBC depleted bone marrow cells of progenitor cells (CD34+), T cells (CD3+), myeloid (CD33+, CD11b+), B cells (CD19+) and granulocytes (CD66b+) of NBSGW mice xenotransplanted with AAVS1 and MYSM1 edited cord blood CD34+ HSPCs treated with either vehicle (3%DMSO) or 20mg/kg Liproxstatin-1.
To extend our findings to an in vivo setting, we transplanted AAVS1 and MYSM1 edited cord blood CD34+ HSPCs into NBSGW mice and treated the mice with vehicle controls (3% DMSO) or ferroptosis inhibitor liproxstatin-1 (Figure 5E). Mice treated with liproxstatin-1 showed significantly improved engraftment of MYSM1-deficient human HSPCs across all time points surveyed, including once long-term engraftment had been achieved at 12 weeks (Figure 5F). Additionally, the MYSM1 edits were preserved in this rescue, in contrast to the depletion generally observed, further supporting improved engraftment of MYSM1 deficient HSCs (Figure 5G). A near complete rescue was observed in the bone marrow, spleen, and across all lineages (Figure 5H–J). Collectively, our data show that in vivo inhibition of ferroptosis can fully rescue the human HSC defects accompanying MYSM1 loss.
Induction of ferroptosis in MYSM1 deficient HSCs due to translational downregulation of protective factors
Having identified increased ferroptosis as a vulnerability underlying dysfunction of HSCs with MYSM1 loss, we wanted to better delineate the underlying mechanisms. Though iron transport, storage, and metabolism as well as antioxidant genes were transcriptionally upregulated or remained unchanged, the levels of key proteins including GPX4, SLC7A11, and FTH1 were all significantly reduced in HSPCs and most profoundly reduced in the primitive HSC-enriched CD34+CD45RA−CD90+ compartment, which could be rescued by re-expressing WT MYSM1, but not the disease-associated loss-of-function mutants (Figure 6A–B, S6A–B). Given the observed reduction in global protein synthesis, we wondered if this downregulation could specifically impact certain pathways more selectively, including the factors that would ordinarily constrain ferroptosis, analogous to our prior findings in human hematopoietic lineage commitment45. To directly assess this and define specific alterations in mRNA translation in an unbiased manner, we performed ribosome profiling with massively parallel sequencing (ribo-seq) to analyze specific changes in mRNA translation upon loss of MYSM146. Quality control using triplet periodicity and other features confirmed the production of high-quality ribo-seq data from these rare primary cell populations (Figure S6C). Importantly, we observed MYSM1 as one of the most significantly downregulated genes (Figure 6C), which is expected given the loss-of-function edits that occur in the gene and serves to validate the assay. Strikingly, we found that genes involved in protection from ferroptosis were significantly downregulated in terms of translational efficiency (TE) in MYSM1 edited cells, in contrast to the observed transcriptional upregulation of the mRNAs encoding these genes. Pathways that were transcriptionally enriched such as ferroptosis, cholesterol and lipid synthesis, and antioxidant activity were all ranked among the most depleted pathways (Figure 6D–E, S6D, Table S6–7). Moreover, analysis of the TEs for specific genes confirmed a significant reduction. In contrast, pathways that were transcriptionally depleted were highly enriched pathways from the TE analysis (Figure 6D, S6E), suggesting a reciprocal compensatory mechanism between transcription and translation across many pathways in MYSM1 edited HSCs. A motif analysis on 5’UTRs of translationally downregulated mRNAs identified 5’UTR motifs that tend to be translated less efficiently due to loss of MYSM1 and demonstrated that these motifs were enriched on factors that ordinarily serve to constrain ferroptosis, providing a molecular basis for the observed selective downregulation of these factors (Figure 6G–H). To test whether the 5’UTR motifs determine the translation efficiency in the setting of MYSM1 deficiency, we engineered the 5’ UTRs of translationally downregulated (FTH1, SLC40A1 and ALAS1) or upregulated (HSPB1 and RELB) genes by introducing repetitive advantageous motifs or disadvantageous motifs, respectively, and delivered in vitro transcribed modified mRNAs into MYSM1 deficient CD34+ HSPCs. Notably, when the 5’UTRs of FTH1, SLC40A1 and ALAS1 were engineered with advantageous motifs, the translation of those genes were partially restored (Figure 6I). Similarly, when HSPB1 and RELB were modified to harbor disadvantageous 5’UTR motifs, their translation was similarly impaired (Figure 6I). Together, our data show that reduced translation rates due to loss of MYSM1 result in HSC depletion through less efficient translation of ferroptosis protective genes.
Figure 6. Reduced cellular translation due to loss of MYSM1 preferentially decreases translation efficiency of iron transport, storage and metabolism genes, resulting in overall decrease of protein level.
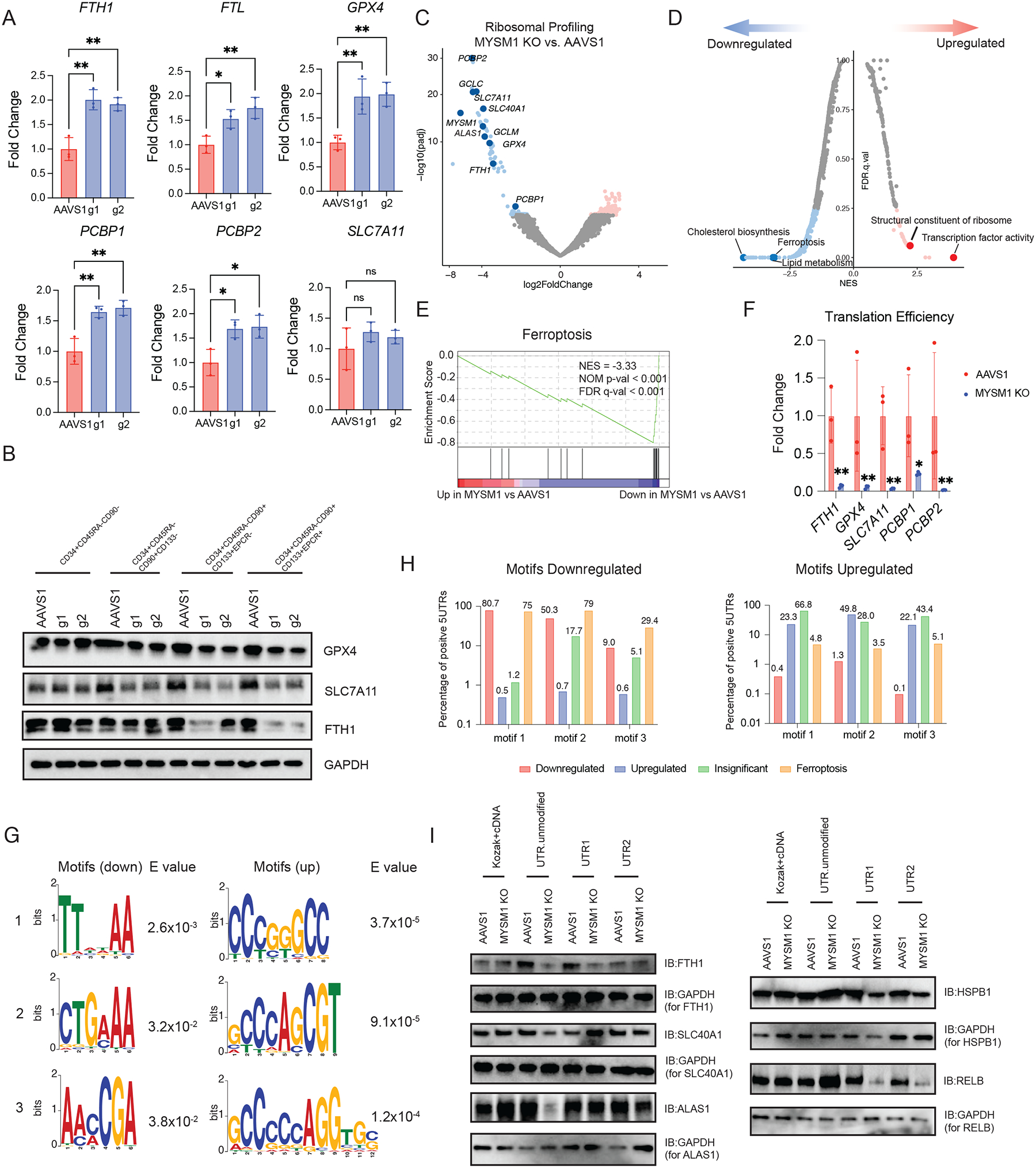
(A) Real time PCR analysis of iron transport, storage and metabolism gene expression of sorted CD34+CD45RA−CD90+CD133+ cells. (B) Western blot analysis of key iron handling and ferroptosis genes in indicated fractions of culture CD34+ HSPCs edited by AAVS1 control or MYSM1. (C) Volcano plot of MYSM1 KO vs. AAVS1 differentially translated genes, highlighting genes directly relevant to cellular iron transport, storage and metabolism, and ferroptosis. (D) Top enriched signatures in differentially translated genes in MYSM1 KO cells. (E) Gene set enrichment analysis (GSEA) showing ferroptosis protective gene translation is significantly impaired in MYSM1 KO cells. (F) Representative translation efficiency change of ferroptosis genes in MYSM1 KO cells. (G) Motif discovery analysis identified genes with specific 5’ UTR motif pattern tend to be less or more efficiently translated due to MYSM1 loss. (H) Percentage of 5’UTRs of genes of different groups in downregulated or upregulated motifs discovered in (G). (I) Western blot analysis of the translation of motif modified mRNA of MYSM1 deficient CD34+CD45RA−CD90+ cells. FTH1, SLC40A1 and ALAS 5’UTRs were engineered by placing three repeats of first two motifs identified in up side shown in (G) and HSPB1 and RELB were engineered by placing three repeats of first two motifs identified in down side shown in (G).
Healthy HSCs are selectively vulnerable to ferroptosis due to a low level of protein synthesis
We have thus far focused on the compromised HSC function due to MYSM1 deficiency causing reduced protein synthesis and increased ferroptosis, as a result. Given that low and highly-regulated protein synthesis is a critical adaptation in healthy HSCs8,10, we wondered if this vulnerability to ferroptosis may be more broadly present in HSCs. We employed several well-characterized small molecule inducers of ferroptosis including erastin, FIN56, FINO2, and RSL3, as well as genome editing of GPX4 to perturb human HSPCs. Remarkably, in contrast to the bulk population of human HSPCs, healthy HSCs were selectively vulnerable to ferroptosis via all of these distinct and complementary approaches (Figure 7A–B, S7A). All of these perturbations that resulted in ferroptosis in HSCs, increased global ROS levels and lipid peroxidation in the primitive HSC-enriched CD34+CD45RA−CD90+ compartment (Figure 7C–D), reinforcing the notion that HSC loss in the setting of these perturbations is accompanied by the expected alterations that characterize ferroptosis.
Figure 7. Normal hematopoietic stem cells (HSCs) are susceptible to ferroptosis induction and overexpression of MYSM1 can be protective due to increased translation.
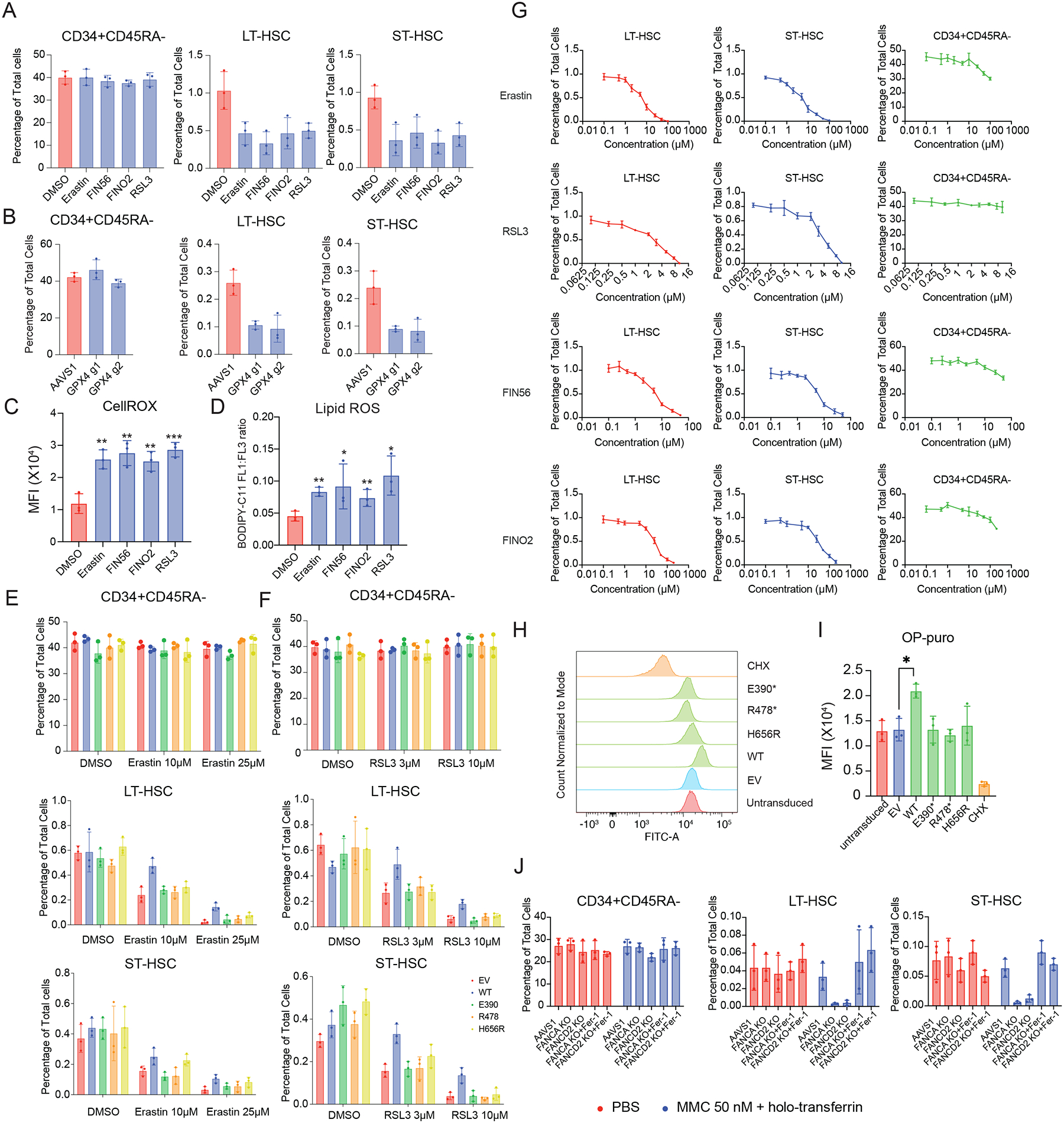
(A) Quantification of CD34+CD45RA−, ST-HSC, and LT-HSC populations in DMSO and indicated ferroptosis inducer-treated CD34+ HSPCs. (B) Quantification of CD34+CD45RA−, ST-HSC, and LT-HSC populations in AAVS1 or GPX4 edited cells. (C) Quantification of mean fluorescence intensity (MFI) of cellular ROS level of sorted CD34+CD45RA−CD90+ cells with DMSO or indicated ferroptosis inducer treatment measured by CellROX dye. (D) Quantification of cellular lipid peroxidation level of sorted CD34+CD45RA−CD90+ cells with DMSO or indicated ferroptosis inducer treatment measured by ratio of oxidized and non-oxidized BODIPY dye. (E) Quantification of CD34+CD45RA−, ST-HSC, and LT-HSC populations in DMSO and Erastin treated cells, and cells treated with Erastin and transduced with indicated constructs. (F) Quantification of CD34+CD45RA−, ST-HSC, and LT-HSC populations in DMSO and RSL3 treated cells, and cells treated with RSL3 and transduced with indicated constructs. (G) Viability curve of CD34+CD45RA−, ST-HSC, and LT-HSC populations, quantified by percentage of cells after five days culture with indicated concentration of ferroptosis inducing agent. (H) Representative flow cytometric histogram of O-Propargyl-puromycin based translation rate analysis on CD34+CD45RA−CD90+ sorted cells transduced with indicated constructs. (I) Quantification of mean fluorescence intensity (MFI) of O-Propargyl-puromycin based translation rate analysis on CD34+CD45RA−CD90+ sorted cells transduced with indicated constructs. (J) Quantification of LT- and ST-HSC populations in AAVS1, FANCA and FANCD2 edited CD34+ HSPCs induced with either PBS or low concentration of mitomycin c and holotransferrin treated with either DMSO or ferroptosis inhibitor Ferrostatin-1.
Given that prior studies have shown that HSCs have a characteristically low level of protein synthesis10 and we have demonstrated how reduced protein synthesis due to MYSM1 loss can induce ferroptosis, we wondered if overexpression of MYSM1 could augment protein synthesis in healthy HSCs and thereby protect the cells from ferroptosis. Induction of ferroptosis in the HSC compartment by either erastin or RSL3 at two different concentrations could be prevented by increasing the expression of normal MYSM1 (Figure 7E–F, S7B). We next performed a dose response curve analysis on CD34+ HSPCs with different ferroptosis inducing chemicals. Interestingly, we found that only the HSC populations exhibit high sensitivity to ferroptosis induction, but not the more differentiated CD34+CD45RA− progenitors (Figure 7G), suggesting that only HSCs are vulnerable to the induction of ferroptosis, while other hematopoietic cells, which would have higher rates of protein synthesis than HSCs, are not. Several disease-causal mutations in MYSM1 were unable to rescue this phenotype, reinforcing the need for full MYSM1 function to enable this rescue. Importantly, we observed an increase in protein synthesis rates in the HSC compartment with the normal but not disease-associated mutant forms of MYSM1 (Figure 7H–I).
To further extend these findings, we sought to test whether one of the most common genetic causes of HSC loss in humans, attributable to impairment of the DNA damage repair coordinating Fanconi anemia pathway47, would also trigger ferroptosis. We recreated two common genetic lesions that cause Fanconi anemia, loss of FANCA or FANCD2, by genome editing in human CD34+ HSPCs. We then treated FANCA or FANCD2 deficient cells with very low concentrations of mitomycin C to induce DNA cross-linking as seen in Fanconi anemia patients from endogenous aldehydes48,49. The resultant loss of LT- and ST-HSCs can be rescued by inhibiting ferroptosis with ferrostatin-1 (Fig 7J, S7C). This finding shows that under this model of Fanconi anemia, DNA cross-linking can trigger HSC loss, as occurs in humans in vivo with Fanconi anemia (over a longer duration that generally requires years), and this can be ameliorated by ferroptosis blockade. Together, we show that HSCs with genetic lesions found in Fanconi anemia are also sensitive to loss by ferroptosis, which demonstrates a broader relevance of our findings to other human diseases characterized by HSC loss. Collectively, we demonstrate how the low levels of protein synthesis that characterize HSCs confer a vulnerability to ferroptosis, which can be activated through a number of distinct pathways compromising HSC function and which can be mitigated by augmenting protein synthesis in HSCs.
Discussion
A milestone in our understanding of how the metabolic state of cells can be linked to cell survival and induction of cell death occurred ten years ago with the first description of ferroptosis35,36,50. Since that time, a considerable number of studies have characterized the molecular regulation of this process50–53, as well as how specific pathologic contexts can enable liabilities to cell death via ferroptosis, as exemplified by the sensitivity of cancer cells to ferroptosis43,54–58. However, despite the considerable advances made, the precise vulnerabilities of cell populations in physiologic settings to death via ferroptosis remains undefined. Here, through studies inspired by a rare inborn error impacting human HSCs due to deficiency of the histone deubiquitinase MYSM1, we have identified a distinct vulnerability of HSCs to ferroptosis. This sensitivity arises, at least in part, due to the low and highly-regulated rate of protein synthesis in these cells, which is thought to be critical for enabling homeostatic protein levels in these cells10,11. Through our in depth mechanistic studies, we are able to identify a key and previously undescribed connection that couples mRNA translation to the induction of ferroptosis and show how downregulation of mRNA translation impacts the translation of factors critical for ferroptosis, which harbor unique motifs in their 5’ UTRs. This adds to the emerging connections between the regulation of translation of specific factors, as is seen with selenoproteins, and sensitivity to ferroptosis59,60.
While a considerable body of work has focused upon the metabolic adaptations that are found in HSCs and which enable unique adaptations, including the use of autophagy by these cells to regulate metabolism6,7,61, the requirement for specific metabolites62,63, the fine-tuned regulation of iron homeostasis64, and the need for proteostasis10,11,65, the precise vulnerabilities arising from these adaptations remain poorly characterized. Human diseases that impact specific cell populations, as exemplified by MYSM1 deficiency that we study here, provide an opportunity to uncover the sensitivities of specific cells to activation of distinct pathways. In this context, we uncover a sensitivity of human HSCs to ferroptosis, which has broader implications in regulating these cells beyond the context of the rare bone marrow failure disorder that inspired this set of studies. Indeed, another more common form of bone marrow failure, Fanconi anemia, also appears to trigger HSC loss by ferroptosis.
Beyond the insights gained into fundamental pathways critical in the regulation of human HSCs, our findings also have important translational and clinical implications. First, agents that can chelate intracellular iron may enable expansion of HSCs, as suggested through studies of eltrombopag for the treatment of aplastic anemia64,66–68, a condition characterized by loss of HSCs in the bone marrow. While some of the observed activities may be attributable to stimulation of the thrombopoietin signaling axis by eltrombopag, experimental studies do suggest a role for limiting intracellular iron in promoting improved maintenance and expansion of HSCs64. Our findings emphasize the opportunities that may exist for helping preserve and augment HSC function in a number of bone marrow failure syndromes by restricting ferroptosis, including rare conditions such as MYSM1 deficiency, but also more common conditions characterized by HSC loss such as aplastic anemia and the myelodysplastic syndromes. Second, the observation that supraphysiologic oxygen can cause stress and impair HSC function may be attributable to the induction of lipid peroxidation and ferroptosis69. Blockage of ferroptosis may represent an ideal strategy for helping preserve HSC function in the setting of cell therapies requiring ex vivo manipulation. Third, ferroptosis is being actively pursued as a promising avenue for anti-cancer therapies, given the sensitivity of cancer cells to the induction of ferroptosis54,56. However, it is important to be aware of potential side effects of such agents and our findings suggest that HSC loss and resultant impaired hematopoiesis will be an issue that will require close monitoring as such agents are developed and tested. By further defining the underlying metabolic regulation that confers the observed vulnerability of HSCs to ferroptosis, a variety of clinical opportunities will likely arise to enable better treatments for therapy-resistant cancers and for intractable blood diseases.
Limitation of the study
Although we identified a relationship between mRNA translation and ferroptosis protection in human HSCs through the study of a rare bone marrow failure disorder, it is likely that the mechanisms we identified are highly cell type and context dependent. mRNA translation likely plays pivotal roles in ferroptosis regulation in other cell types, but this might occur through distinct mechanisms, which warrants further investigation. Moreover, further studies are needed to examine how HSCs in other diseases might be lost due to ferroptosis. For example, recent studies have found that accumulated intracellular iron can be problematic to HSCs in aged mouse models70, suggesting that our findings might have broader implications.
STAR Methods
Resource Availability
Lead contact
Further information and reagent requests should be directed to and will be fulfilled by the Lead Contact Vijay G. Sankaran (sankaran@broadinstitute.org)
Materials availability
All unique materials will be available upon request to the lead contact.
Data and code availability
The scRNA-seq and ribosome profiling datasets of AAVS1 or MYSM1 edited HSPCs have been deposited to NCBI Gene Expression Omnibus and are publicly available as of the date of publication. The GEO accession numbers of the datasets are included in the key resource table.
Key resources table
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| Anti-MYSM1 antibody | Abcam | ab-221725 |
| RPS24 Polyclonal antibody | ProteinTech | 14831-1-AP |
| RPS7 Polyclonal antibody | ProteinTech | 14491-1-AP |
| Ferroptosis Antibody Sampler Kit | Cell signaling Technology | 29650 |
| Brilliant Violet 421™ anti-human CD34 Antibody | BD Biosciences | 562577 |
| PE Mouse Anti-Human CD66b | BD biosciences | 561650 |
| PE Mouse Anti-Human CD33 | BD biosciences | 555450 |
| PE Mouse Anti-Human CD19 | BD biosciences | 555413 |
| RPL15 Polyclonal antibody | Proteintech | 16086-1-AP |
| RPL23 Polyclonal antibody | Proteintech | 10727-1-AP |
| Ferritin light chain Polyclonal antibody | Proteintech | 10727-1-AP |
| anti-CD90-PECy7 | BD biosciences | 561558 |
| APC-H7 Mouse Anti-Human CD45RA | BD biosciences | 560674 |
| Necroptosis Antibody Sampler Kit | Cell signaling Technology | 98110T |
| Apoptosis Antibody Sampler Kit | Cell signaling Technology | 9915T |
| hnRNP-E1 Polyclonal antibody | proteintech | 14523-1-AP |
| PCBP2 Polyclonal antibody | proteintech | 15070-1-AP |
| SLC40A1/FPN1 Polyclonal antibody | proteintech | 26601-1-AP |
| GCLM Polyclonal antibody | proteintech | 14241-1-AP |
| GCLC Polyclonal antibody | proteintech | 12601-1-AP |
| FITC Annexin V | Biolegend | 640906 |
| APC anti-human CD49c (integrin α3) Antibody | Biolegend | 343808 |
| ALAS1 Polyclonal antibody | proteintech | 16200-1-AP |
| HSP27 Polyclonal antibody | proteintech | 18284-1-AP |
| RELB Polyclonal antibody | proteintech | 25027-1-AP |
| Ferroportin/SLC40A1 Antibody - BSA Free | Novus Biologicals | NBP1-21502SS |
| BB700 Mouse Anti-Human CD133 | BD biosciences | 747638 |
| rat anti-human anti-EPCR-PE | Biolegend | 351904 |
| PE anti-human CD34 Antibody | Biolegend | 343506 |
| PE anti-human CD3 Antibody | Biolegend | 317308 |
| PE anti-human CD11b Antibody | Biolegend | 301306 |
| Monoclonal ANTI-FLAG® M2 antibody produced in mouse | Sigma Aldrich | F1804 |
| Chemicals, peptides, and recombinant proteins | ||
| Holo-transferrin Human | Sigma Aldrich | T0665 |
| Hygromycin B | Gibco | 10687010 |
| Erastin | Selleck | S7242 |
| FIN56 | Selleck | S8254 |
| FINO2 | Cayman Chemical | 25096 |
| Deferoxamine mesylate | Selleck | S5742 |
| Ferrostatin-1 | Selleck | S7243 |
| BODIPY™ 581/591 C11 | Invitrogen | D3861 |
| CellROX™ Green Reagent | Invitrogen | C10444 |
| MitoTEMPO (hydrate) | Cayman Chemical | 16621 |
| N-acetyl-L-Cysteine | Cayman Chemical | 20261 |
| (±)-α-Tocopherol Acetate | Cayman Chemical | 28399 |
| Polyethylenimine, Linear, MW 25000, Transfection Grade (PEI 25K™) | PolySciences | 23966-100 |
| Ambion™ RNase I, cloned, 100 U/μ | Invitrogen | AM2294 |
| GlycoBlue™ Coprecipitant (15 mg/mL) | Invitrogen | AM9515 |
| Cycloheximide | Sigma Aldrich | C7698 |
| Ultra Low Range DNA Ladder | Invitrogen | 10597012 |
| SYBR™ Gold Nucleic Acid Gel Stain (10,000X Concentrate in DMSO) | Invitrogen | S11494 |
| RSL3 | Selleck | S8155 |
| CC100 | StemCell Technologies | 2690 |
| BioTracker Far-red Labile Fe2+ Dye | Millipore Sigma | SCT037 |
| SM-164 1mg | Cell Signaling Technology | 56003S |
| Z-VAD-FMK | Selleck | S7023 |
| Recombinant Human TNF-α 10ug | Peprotech | 300-01A |
| Mitomycin C 10mg | Selleck | S8146 |
| Iron(II) sulfate heptahydrate | F7002-250G | Sigma Aldrich |
| Liproxstatin-1 | Cayman Chemical | 17730 |
| Staurosporine | Cayman chemical | 81590 |
| Methanol | Sigma Aldrich | 179337 |
| RPMI1640 medium | Life Technologies | 11875-119 |
| Opti-MEM | Gibco | 31985-062 |
| Ficoll-Paque Premium sterile solution | GE Healthcare | 17-5442-02 |
| 0.25% Trypsin (1X) | Gibco | 15050-057 |
| Blotting nonfat dry milk | BioRad | 1706404XTU |
| Cas9 protein, recombinant | IDT | 1081061 |
| Penicillin-Streptomycin 10,000u/ml | Life Technologies | 15140-122 |
| Recombinant Human TPO | Peprotech | 300-18-100UG |
| Formamide | Sigma Aldrich | F7503 |
| TBE Buffer (Tris-borate-EDTA) (10X) | Thermo Scientific | B52 |
| Annexin-V-Binding Buffer | Biolegend | 422201 |
| L-Glutamine 200mM for cell culture | GIBCO | 25-030-081 |
| SSC (20X), RNase-free | Invitrogen | AM9763 |
| DMEM High Glucose | Life Technologies | 11965-118 |
| Critical commercial assays | ||
| Glutathione Peroxidase Assay Kit | Cayman Chemical | 703102 |
| Protein Synthesis Assay Kit | Cayman Chemical | 601100 |
| Hemin Colorimetric Assay Kit | BioVision | K672 |
| Iron Colorimetric Assay Kit | BioVision | K390 |
| Kapa Library Quantification Kit - Complete Kit (Universal) | Roche | 7960140001 |
| mMESSAGE mMACHINE™ T7 Transcription Kit | Invitrogen | AM1344 |
| MEGAclear™ Transcription Clean-Up Kit | Invitrogen | AM1908 |
| Poly(A) Tailing Kit | Invitrogen | AM1350 |
| Deposited data | ||
| Single cell RNA seq | This paper | GSE221170 |
| Ribosome Profiling | This paper | GSE221170 |
| Experimental models: Cell lines | ||
| CD34+ HSPCs | Fred Hutch | |
| HEK293T | ATCC | CRL-3216 |
| Experimental models: Organisms/strains | ||
| NBSGW | JAX Laboratory | 026622 |
| Oligonucleotides | ||
| Alt-R® Cas9 Electroporation Enhancer | IDT | 1075916 |
| Universal miRNA Cloning Linker | New England Biolabs | S1315S |
| qPCR primers and genotyping/editing efficiency testing primers | Genewiz | Table S8 |
| Chemically synthesized sgRNA | Synthego | Table S9 |
| Recombinant DNA | ||
| MGC Fully Sequenced Human SLC40A1 cDNA | Horizon Discovery | MHS6278-202801506 |
| MGC Fully Sequenced Human FTH1 cDNA | Horizon Discovery | MHS6278-202755914 |
| MGC Fully Sequenced Human ALAS1 cDNA | Horizon Discovery | MHS6278-202831218 |
| MGC Human HSPB1 Sequence-Verified cDNA | Horizon Discovery | MHS6278-202756315 |
| MGC Human RELB Sequence-Verified cDNA | Horizon Discovery | MHS6278-202801567 |
| Software and algorithms | ||
| Graphpad Prism v.9.0.1 | Graphpad | RRID:SCR_002798 |
| Biorender | Biorender | RRID:SCR_018361 |
| Flowjo | Flowjo | RRID:SCR_008520 |
| ImageJ v1.52a | Wayne Rasband | RRID:SCR_003070 |
| R | The R Foundation | |
| STAR v2.7.10a | ||
| Bowtie2 v2.3.3 | ||
| RiboCode v1.2.15 | ||
| Cutadapt v4.1 | ||
| CellRanger v7.0.0 | 10xGenomics | |
| Seurat v4.0.2 | ||
| Other | ||
| In-Fusion® HD Cloning Plus | Takara/Clontech | 638909 |
| Q5 site directed mutagenesis kit | New England Biolabs | E0554S |
| iScript™ cDNA Synthesis Kit | BioRad | 1708891 |
| Monarch® DNA Gel Extraction Kit | New England Biolabs | T1020L |
| iQ™ SYBR® Green Supermix | BioRad | 1708882 |
| P3 Primary Cell 4D Nucelofector X Kit S | Lonza | V4XP-3032 |
| SuperScript™ III Reverse Transcriptase | Invitrogen | 18080044 |
| Chromium Next GEM Single Cell 3ʹ Kit v3.1, 16 rxns | 10XGenomics | PN-1000268 |
| Chromium Next GEM Chip G Single Cell Kit, 48 rxns | 10XGenomics | PN-1000120 |
| Dual Index Kit TT Set A, 96 rxns | 10XGenomics | PN-1000215 |
| CircLigase™ ssDNA Ligase | Lucigen | CL4111K |
| T4 RNA Ligase 2, truncated | New England Biolabs | M0242L |
| StemSpan™ SFEM II | StemCell Technologies | 09655 |
| Fetal Bovine Serum - Premium Select G21162 | Bio-Techne | S11550 |
| SUPERase•In™ RNase Inhibitor (20 U/μL) | Invitrogen | AM2694 |
| EasySep Human Cord Blood CD34 Positive Selection Kit II | StemCell Technologies | 17896 |
| Immobilon-P PVDF Membrane | Millipore Sigma | IPVH00010 |
| SPRIselect | Beckman Coulter | B23317 |
| Qubit™ 1X dsDNA HS Assay Kit | ThermoFisher | Q33231 |
| Zero Blunt™ TOPO™ PCR Cloning Kit for Sequencing, without competent cells 25rxn | Invitrogen | 450031 |
There is no original code generated in this study.
Any additional information or help in reanalyzing the datasets is available from the lead contact upon request.
Experimental model and subject details
CD34+HSPC culture
Human CD34+ HSPCs from mobilized peripheral blood of healthy adults were obtained from the Cooperative Center of Excellence in Hematology at the Fred Hutchinson Cancer Research Center. The cells were thawed and cultured in StemSpan SFEM II culture medium, supplemented with 1X CC100 (containing the cytokines FLT3L, SCF, IL3, and IL6), recombinant thrombopoietin (TPO) (100ng/ml), and UM171 (35nM), as we have described previously29
Xenotransplantation and animal models
All animal procedures were performed under the protocol approved by Boston Children’s Hospital Institutional Animal Care and Use Committee (IACUC). Cord blood was acquired from Dana-Farber Cancer Institute’s Pasquarello tissue core. CD34+ HSPCs were enriched by EasySep Human Cord Blood CD34+ positive selection kit (StemCell Technologies) according to the manufacturer’s instructions. Enriched cells were nucleofected by AAVS1 control and MYSM1 sgRNAs in a manner identical to what was done for adult CD34+ HSPCs, as described above. Three days post-nucleofection, a small fraction of cells will be harvested for ICE analysis of editing efficiency. The remaining cells will be intravenously injected into NBSGW immunodeficient and Kit mutant mice (JAX#026622) at 1×105 cells per mouse. Autoclaved sulfatrim antibiotic water was given and changed weekly to prevent infections. Peripheral blood, bone marrow, and spleen were harvested four months after transplantation for analysis of engraftment, MYSM1 editing efficiency, and lineage-specific markers. Specifically, peripheral blood was harvested through cardiac puncture right after the mice were sacrificed and collected in EDTA coated tubes. Red blood cells were depleted by adding 20X volume of ddH2O for 30 seconds, the osmolarity was balanced by adding 10X PBS solution. If the pellet appears to be red, a second round of RBC lysis with the same method was used to deplete remaining RBCs. For bone marrow cell harvest, femurs, and tibia were dissected and crushed by mortar and pestle in RPMI 1640 medium supplemented with 10% FBS. The cells were passed through 40 μm cell strainer. The cells were washed by ice-cold PBS once and washed cells were frozen down in 90% FBS + 10% DMSO. The majority of RBCs were automatically depleted by cryopreservation. Prior to analysis, the thawed bone marrow cells were further RBC depleted by the above method. For splenocyte isolation, spleens were dissected and minced. The minced spleen was digested by 1 mg/ml collagenase IV for 30 minutes at 37 degrees C. The digestion was stopped by addition of EDTA at a final concentration of 1 mM. Splenocytes were frozen in 90% FBS + 10% DMSO to deplete RBCs and stored in liquid nitrogen for subsequent analysis.
For liproxstatin-1 rescue experiments, 50 mg of liproxstatin-1 was initially dissolved in 0.5 ml of 100% DMSO. The dissolved liproxstatin-1, with any remaining residues from the vial, was transferred to 15 ml centrifuge tubes, which was further diluted and dissolved by about 14.5 ml of 100% PBS, resulting in a total 15ml liproxstatin-1 solution in about 3% DMSO (concentration at about 3.3 mg/ml). Prior to xenotransplantation, nucleofected cord blood CD34+ HSPCs (AAVS1 or MYSM1 edited) were treated with either 3% DMSO or 1μM of liproxstatin-1 prior to injection. Intravenously injected NBSGW mice were housed in presence of autoclave sulfatrim water 1 week before initiating regular intraperitoneal injections of vehicle (3% DMSO in PBS) or 20 mg/kg liproxstatin-1 every other day for a total of 12 weeks. The body weight of each animal was carefully monitored to observe any significant adverse effect of the treatment. The animals were anesthetized by isoflurane and retro-orbitally bled for peripheral blood at 4, 8, and 12 weeks post-transplant for assessments of engraftment. At the end of 12 weeks, the mice were sacrificed and harvested for bone marrow cells and splenocytes, as described above.
Method Details
CRISPR/Cas9 RNP nucleofection
The Cas9 ribonucleoprotein (RNP) complexes were assembled by mixing 50 pmol chemically-modified sgRNA and 75 pmol Cas9 protein and incubating at room temperature for 10–30 mins. The assembled RNP complex was then mixed with Lonza P3 primary cell nucleofection reagent in presence of 1μl of 100μM stock of IDT nucleofection enhancer. The mixture was delivered into CD34+ HSPCs by nucleofection using the Lonza 4D nucleofector system with the DZ100 program. The cells were harvested and genomic DNA was extracted 72 hours post-nucleofection. DNA fragments flanking the editing site at least 250 bp upstream and downstream were amplified and sent for Sanger sequencing to assess editing efficiencies. The .ab1 file was used for ICE analysis from Synthego website for prediction of editing efficiency and edit compositions (https://ice.synthego.com/#/).
MYSM1 overexpression cloning and lentivirus packaging
MYSM1 cDNA was amplified using CD34+ HSPC cDNA library and cloned into the pCRII vector using Zero Blunt TOPO cloning kit (Invitrogen). The sequence of amplified MYSM1 cDNA was verified by Sanger sequencing before subcloning into the pLVX overexpression vector using the In-Fusion cloning kit (Takara). Q5 site directed mutagenesis kit was used to make H656R mutant.
For lentiviral packaging HEK-293T cells were cultured in DMEM medium supplemented with 10% FBS and 1X penicillin/streptomycin. After one passage, the cells were plated into 10 cm dishes at 50% confluency. On the next day (within 24 hrs after plating), WT and mutant MYSM1 transfer plasmids were co-transfected with VSV-G and psPAX2 plasmids at 1:1:1 molar ratio using linear PEI at PEI:DNA=4:1 ratio. Viral supernatants were harvested twice at 48 and 72 hours post-transfection. Combined viral supernatant was centrifuged and filtered at 0.45 μm. Filtered viral supernatant was then concentrated by ultracentrifugation with a 2 ml 20% sucrose cushion at 25,000 rpm in the SW32 rotor of Beckman Coulter ultracentrifuge for 2hrs. After ultracentrifugation, the supernatant was decanted and viral pellets were resuspended in ice cold PBS.
Concentrated virus was added to CD34+ HSPCs at an MOI of 50 in presence of 8 μg/ml polybrene. The cells were then spin infected at 2,000 rpm for 90 mins at 37 C. After spin infection, the supernatant was removed and fresh culture medium was added. Hygromycin selection was started 36 hours post-transduction at a final concentration of 300 μg/ml.
Single cell RNA-seq analysis
Edited AAVS1 and MYSM1 KO cells were FACS sorted and ~50,000 CD34+CD45RA−CD90+ cells were collected. The sorted cells were spun down and resuspended at 1,000/μl. The scRNA-seq library was prepared using 10X Genomics scRNA-seq library prep kit according to manufacturer’s instructions. The sequencing results were pre-analyzed by the CellRanger pipeline to generate the matrix file, which was brought to downstream analysis. Normalization, scaling, and cell clustering was done in R by the Seurat v4.0.2 package. The top 1000 most highly variable genes were used to calculate the top 50 principal components. The HSC cluster was highlighted by averaging the z-score normalized expression of CD34, HLF, CRHBP for each cell, as previously described30. The HSC population, defined by the averaged z score of more than 0.5 for the HSC signature, was used for DEG calling by DESeq2. The DEG list was used to generate rnk file, which was used to run the GSEA analysis (https://www.gsea-msigdb.org/gsea/index.jsp)71.
Real time PCR analysis
The total RNA of sorted or unsorted cells were harvested using RNeasy mini RNA purification kit according to manufacturer’s instructions. 500 ng (sorted cells) to 1 μg (unsorted cells) of total RNA was used for reverse transcription using iScript cDNA synthesis kit (Biorad). The cDNA product was diluted at 1:20 and 2 μl of diluted cDNA was used for real time PCR analysis using iQ SYBR green supermix (Biorad). Data was normalized by loading control (ACTB) and presented as fold change compared to control samples.
Immunoblotting analysis
Total protein lysate of sorted or unsorted cells was extracted by RIPA buffer in presence of protease inhibitor cocktail and PMSF on ice for 10 minutes followed by a 5-minute incubation with MNase at 37 C. The total lysate was then linearized by 1X SDS loading buffer and heated at 55 degrees C for 10 minutes. The lysate was run on 5–20% gradient gel and then transferred onto a PVDF membrane. The membrane was then blocked by 5% nonfat dry milk in TBS/T. The membrane was then incubated with primary antibodies at 1:1000 dilution at 4C overnight. HRP conjugated secondary antibodies were then incubated for 1hr at room temperature. Membrane was developed using ECL and imaged in the Biorad gel imaging system.
Flow cytometric analysis
All antibody-based flow cytometric analysis was performed as follows: cells were centrifuged and washed by ice fold 1X PBS. The washed cell pellets were resuspended by staining solution: 100μl of FACS buffer (sterile PBS+3%FBS) with FACS antibody (1:60 dilution) per 0.1 million cells. The cells were stained for 15–20 mins under the protection of aluminum foil at 4C. For human HSC phenotyping, cultured CD34+ HSPCs were stained with CD34, CD45RA, CD90, CD133, EPCR and ITGA3 antibodies. For engraftment analysis, the RBC depleted cells were stained with human and mouse CD45 antibodies. PE-conjugated antibodies of each lineage were co-stained with human and mouse CD45 antibodies individually for bone marrow lineage analysis of xenotransplantation. The stained cells were then centrifuged and the staining solution was discarded. The stained cells were washed with ice cold PBS three times. Washed cells were then resuspended at a concentration of 0.1 million cells per 1 ml FACS buffer. All flow cytometry data analysis was performed in FlowJo software.
Intracellular ROS analysis
Nucleofected cells were sorted for CD34+CD45RA−CD90+ and stained with CellROX dye according to manufacturer’s instructions. Briefly, the cells were stained CD34+ expansion medium at a final concentration of 5μM for 30 mins at 37 degrees C. The cells were spun down and the culture medium was removed. The cells were then washed by ice cold PBS three times and resuspended in the FACS buffer (PBS + 3% FBS). Labeled cells analyzed by BD Accuri C6 table top flow cytometer.
Lipid peroxidation analysis
Sorted CD34+CD45RA−CD90+ cells were stained with BODIPY C11 lipid probe (Invitrogen) according to the manufacturer’s instructions. Briefly, the cells were stained by 5 μM of BODIPY 581/591 C11 reagent in PBS as 37 degrees C for 30 mins. Labeled cells were washed and analyzed on BD Accuri C6 table top flow cytometer. For lipid peroxidation analysis, the peroxidation state of each group was calculated by mean fluorescence intensity (MFI) of FL1 channel to that of FL3 channel.
Cellular translation rate analysis
The cellular translation rate of sorted cells was measured by O-Propargyl-puromycin based translation assay kit (Cayman chemicals) according to manufacturer’s instructions. Briefly, the cells were stained with O-Propargyl-puromycin at a 1:40 dilution from the stock solution in culture medium at 37 degrees C for 30 minutes. Labeled cells were then washed with FACS buffer. The cells were fixed by the fixative reagent of the kit for 5 minutes at room temperature. Fixed cells were then stained with 5-FAM azide solution for 30 minutes at room temperature, which was then analyzed by flow cytometry.
Total and ferrous iron measurement
Total cellular iron level was measured using iron colorimetric assay kit (BioVision) according to manufacturer’s instructions. Briefly, cells were harvested and homogenized in the iron assay buffer. Lysed cells were cleared by centrifugation at 15,000g for 5 minutes at 4 degrees C. The supernatant was collected and a small fraction was split for Qubit protein quantification according to manufacturer’s instructions. The rest of cell lysates were quantified for total iron after the reaction as a measurement of OD593. Iron concentrations were calculated by normalizing total protein concentration.
Intracellular ferrous iron was measured by labeling cells with BioTracker Far-red Labile Fe2+ Dye (Millipore Sigma) at 5 μM for 90 mins in PBS at 37 degrees C and analyzed by flow cytometry.
Labile hemin analysis
Intracellular hemin level was measured by hemin colorimetric assay kit (BioVision) according to manufacturer’s instructions. Briefly, cells were lysed in the hemin assay buffer. 50 μl of cell lysates was used for quantification in 96 well plates. After a 30 minutes reaction, the plates were read for OD570. The final hemin concentration was normalized by total protein concentration.
GPX4 activity analysis
The cellular GPX4 activity was measured indirectly by GPX4 inhibitor screening assay kit (Cayman Chemicals) according to manufacturer’s instructions. Briefly, collected cells were lysed in GPX4 sample buffer containing 50mM Tris-HCl, pH7.5, 5mM EDTA and 1mM DTT. 20 μl of sample was used for each reaction. Dynamic plate reading for OD340 was started immediately after all reaction components were added. Plates were read every 1 minute for a total 15 minutes. GPX4 activity was calculated according to the manufacturer’s user manual.
Ribo-seq library preparation and analysis
Ribosome profiling sequencing libraries were constructed as previously described72. Briefly, cells were treated with cycloheximide at final concentration of 100 μg/ml for 5 minutes at 37 degrees C. The cells were then harvested in presence of same concentration of cycloheximide to extract cytoplasmic RNA in ribosome lysis buffer (20 mM Tris-HCl pH 7.4, 150 mM NaCl, 5 mM MgCl2, 1 mM DTT, 1% Triton X-100, Turbo DNase I 25U/ml). Extracted RNA was then digested by RNase I at room temperature for 45 minutes on a nutator. Digestion was stopped by adding SuperRNaseIn RNase inhibitor. The digested RNA was then loaded on ribosome buffer (lysis buffer without Triton-100 and DNase) with 2 ml of 1 M sucrose cushion and centrifuged at maximum speed on SW-32 rotor of Beckman Coulter ultracentrifuge at 4 degrees C for 7 hours. The supernatant was then poured out and the translucent pellet was then resuspended in 1 ml of Trizol reagent. RNA was extracted according to the Trizol reagent manufacturer’s instructions. Purified RNA was run in 15% polyacrylamide TBE-urea gel with synthesized 24 nt and 36 nt RNA oligos as markers for excision. Fragments between 24–36 nt were excised and extracted by freezing in dry ice bath with RNA extraction buffer (300 mM NaOAc pH 5.5, 1.0 mM EDTA, 0.25% SDS) for 30 minutes, followed by an overnight incubation at room temperature on a nutator. Diffused RNA in the RNA extraction buffer was then precipitated and purified by isopropanol. Purified RNA was then ligated with universal miRNA adaptor (NEB). Excessive adaptors were removed by running the ligation product in 15% polyacrylamide TBE-urea gel. Ligated RNA was then reverse transcribed by SuperScript III cDNA synthesis kit (Invitrogen). This first strand cDNA was purified from excessive RT primers through polyacrylamide gel electrophoresis and circularized by CircLigase. Circularized cDNA was PCR amplified to construct sequencing libraries and purified by polyacrylamide gel electrophoresis. Library was then quantified by KAPA library quantification kit and sequenced in Novaseq 6000 platform.
For analysis, raw FASTQ files were firstly trimmed by cutadapt with default setting using the universal miRNA adaptor sequence73. Reads shorter than 20 bp after trimming were removed by the “-m” option. Trimmed FASTQ files were firstly aligned against human rRNA index using bowtie aligner74. Unmatched FASTQ files were further aligned against the GRCh38 index using STAR75. Aligned bam files were analyzed by RiboCode software for 3-nt periodicity identification and a new GTF file containing actively translated ORFs was generated76, which was then used for count table generation by HTSeq software77.
Lipidomics analysis
Cells were FACS sorted directly into 100% isopropanol for lipid extraction. Analyses of polar and non-polar plasma lipids were conducted using an LC-MS system comprised of a Shimadzu Nexera X2 U-HPLC (Shimadzu Corp.) coupled to an Exactive Plus orbitrap mass spectrometer (Thermo Fisher Scientific). After centrifugation, to remove cellular debris, 10uL cell extracts in isopropanol were injected onto a 100 × 2.1 mm, 1.7 μm ACQUITY BEH C8 column (Waters). The column was eluted isocratically with 80% mobile phase A (95:5:0.1 vol/vol/vol 10mM ammonium acetate/methanol/formic acid) for 1 minute followed by a linear gradient to 80% mobile-phase B (99.9:0.1 vol/vol methanol/formic acid) over 2 minutes, a linear gradient to 100% mobile phase B over 7 minutes, then 3 minutes at 100% mobile-phase B. MS analyses were carried out using electrospray ionization in the positive ion mode using full scan analysis over 220–1100 m/z at 70,000 resolution and 3 Hz data acquisition rate. Other MS settings were: sheath gas 50, in source CID 5 eV, sweep gas 5, spray voltage 3 kV, capillary temperature 300°C, S-lens RF 60, heater temperature 300°C, microscans 1, automatic gain control target 1e6, and maximum ion time 100 ms. Raw data were processed using TraceFinder software (Thermo Fisher Scientific) for targeted peak integration and manual review of a subset of identified lipids and using Progenesis QI (Nonlinear Dynamics) for peak detection and integration of both lipids of known identity and unknowns. Lipid identities were determined based on comparison to reference plasma extracts and are denoted by total number of carbons in the lipid acyl chain(s) and total number of double bonds in the lipid acyl chain(s). Downstream statistical analysis and lipid class enrichment analysis were done by LipidSig software78.
Electron microscopy
We used the following as a fixative solution: 2.5% Glutaraldehyde 1.25% Paraformaldehyde and 0.03% picric acid in 0.1 M sodium cacodylate buffer (pH 7.4). Cells were fixed for at least 2 hours at RT in the above fixative by adding 2X fixative solution 1:1 to cell culture medium directly, washed in 0.1 M cacodylate buffer and postfixed with 1% osmium tetroxide (OsO4)/ 1.5% potassium ferrocyanide (KFeCN6) for 1 hour, washed 2x in water, 1x Maleate buffer (MB) 1x and incubated in 1% uranyl acetate in MB for 1 hour followed by 2 washes in water and subsequent dehydration in grades of alcohol (10 minutes each; 50%, 70%, 90%, 2×10min 100%). After dehydration propylene oxide was added to the dish and the cells were lifted off using a transfer pipet, pelleted, and infiltrated ON in a 1:1 mixture of propylene oxide and TAAB Epon (TAAB Laboratories Equipment Ltd, https://taab.co.uk). The following day the samples were embedded in TAAB Epon and polymerized at 60 degrees C for 48 hrs.
Ultrathin sections (about 60 nm) were cut on a Reichert Ultracut-S microtome, picked up on to copper grids stained with lead citrate and examined in a JEOL 1200EX transmission electron microscope or a TecnaiG2 Spirit BioTWIN and images were recorded with an AMT 2k CCD camera.
Image analysis
Electron microscopic images were analyzed in ImageJ software described previously79,80. Specifically, tiff formatted images were loaded and read by ImageJ. Scale was first set by the “set scale” option from the “Analyze” tab (1 pixel = 1 nm for all images). Free hand tool was then used to trace and outline each mitochondria and all cristae structure. The measurement was stored in the ROI manager. After all mitochondria and the cristae structures were outlined, click “measure” in the ROI manager to get the area measurement.
In vitro transcription
The template of in vitro transcription was prepared by PCR amplifying the cDNA of interest, including 5’ and 3’ UTRs. T7 promoter sequence (5’-TAATACGACTCACTATAGGGAGA-3’) was added at the 5’ the PCR fragment. For cDNA expression mRNAs only, Kozak consensus sequence (GCCACC) was included directly downstream of T7 promoter. For motif modified templates, three repetitive corresponding 5’ motifs were added directly downstream of the T7 promoter sequence. PCR amplified templates were then gel purified by the NEB gel purification kit. Multiple PCR reactions were run for each template so that the purified PCR fragment reached a concentration of at least 500ng/μl for a total of 25μl. The in vitro transcription reaction was done by mMESSAGE mMACHINE™ T7 Transcription Kit from Invitrogen according to the manufacturer’s instructions. Briefly, 1μg of template was used (~2μl) for each reaction mixture. The reaction was incubated at 37C for 2 hours. After incubation, 1μl of TURBO DNase was added directly to the reaction mixture and incubated for 15 mins at 37C to eliminate all remaining template DNA. The resulting template digested mRNA products were polyA modified by the Poly(A) Tailing Kit from Invitrogen according to the manufacturer’s instructions. The polyA modified mRNA products were purified by the MEGAclear™ Transcription Clean-Up Kit. For delivery of mRNA products, MYSM1 and corresponding gene of interest sgRNA RNPs were delivered first into CD34+ HSPCs to eliminate endogenous expression of each gene of interest, which interferes with the analysis of translation of mRNA.. Three days after RNP delivery, mRNA was delivered to the cells using the same program code (DZ100) of Lonza 4D nucleofector. The CD34+CD45RA−CD90+ cells were sorted and harvested for Western blot analysis after two days.
Chemical treatments
Chemicals (ferroptosis inducing reagents, ferroptosis inhibitor, mitoTempo, and mitomycin C) were dissolved in DMSO to make 10 mM stock solutions. Ferroptosis inducing agents (Erastin, RSL3, FIN56, FINO2) were used at indicated concentrations with DMSO as control treatment. For ferroptosis inhibitors, DFO and vitamin E were used at 10 μM, Ferrostatin (Fer-1) was used at 1μM. MitoTempo was used at 1μM. For mitomycin C treatment, the stock solution was diluted in sterile PBS to make a 50 μM diluted solution. The diluted stock solution was used 1:1000 in CD34+ HSPCs with PBS as control treatment. To facilitate HSC specific depletion, extra iron was added in the form of holo-transferrin. Specifically, holo-transferrin was dissolved in IMDM medium to make a 10 mg/ml (about 130 μM) stock solution. The cells were treated at a final concentration of 1.5 μM of holo-transferrin at the same time mitomycin c was added.
Quantification and Statistical Analysis
In all experiments, data were presented as mean±standard deviation. When comparing two samples, Two-tailed Student’s t test was used for testing statistical significance. When comparing three or more samples, Levene’s test was firstly used to test equality of variance. If the variance across all samples were tested insignificantly differed, One way or two way ANOVA with Dunnett’s test (for multiple comparison where no reference group is defined) or Tukey’s test (for multiple comparison where reference group is defined) as post-hoc analysis was used. If the variance across samples were tested to be significantly different, Kruskal-Wallis test was used instead of ANOVA, with Dunn test as the post-hoc multiple comparison test. All statistical tests were performed in Graphpad software or in R, when statistical tests were not available through Graphpad.
Supplementary Material
Table S2 GSEA analysis of differentially expressed genes of MYSM1 KO vs AAVS1, Related to Figure 1–3.
Table S3 Lipidomics of MYSM1 KO HSC vs AAVS1 HSC, Related to Figure 4.
Table S4 Lipidomics of MYSM1 KO non-HSC vs AAVS1 HSC, Related to Figure 4.
Table S5 Lipidomics of MYSM1 KO HSC vs MYSM1 KO non-HSC, Related to Figure 4.
Table S6 Differentially translated genes of MYSM1 KO vs AAVS1, Related to Figure 6.
Table S7 GSEA of differentially translated genes of MYSM1 KO vs AAVS1, Related to Figure 6.
Figure S1 CRISPR editing analysis and flow cytometric analysis of HSC phenotyping, related to Figure 1. (A) ICE analysis of percentage of edited cells by two independent MYSM1 gRNAs. (B) Prediction of edited species by two MYSM1 gRNAs. Major edited species all result in stop codons at very early sites of the MYSM1 gene. (C) Western blot validation of successful knockout of MYSM1. (D) Quantification of MYSM1 protein in AAVS1 and two MYSM1 gRNA edited CD34+ HSPCs. (E) Representative flow cytometric analysis of HSC phenotyping of AAVS1 control or MYSM1 edited cells and gating strategy. (F) Representative flow cytometric analysis of human vs. mouse CD45+ cells after RBC depletion from indicated sites of NBSGW mice xenotransplanted with AAVS1 and MYSM1 edited cord blood CD34+ HSPCs.
Figure S2 Transcriptional regulation of translation genes by MYSM1, related to Figure 2. (A) Additional gene set enrichment analysis (GSEA) showing ribosomal biogenesis and cellular translation are impaired with loss of MYSM1. (B) Real-time PCR validation of genes shown as downregulation due to loss of MYSM1 in scRNA-seq. (C) Western blot validation of expression of overexpression constructs of MYSM1 WT, E390*, R478* and H656R mutants.
Figure S3 Upregulation of ferroptosis protective genes and downregulation of ferroptosis promoting genes upon loss of MYSM1, related to Figure 3. (A) Additional violin plot of ferroptosis gene expression in AAVS1 and MYSM1 KO HSCs. (B) Additional gene set enrichment analysis (GSEA) showing that cellular oxidant detoxification and general detoxification processes were significantly stimulated in MYSM1 KO HSCs.
Figure S4 Increased oxidative stress and lipid peroxidation and depletion of membrane phospholipids upon loss of MYSM1, related to Figure 4. (A) Principal Component Analysis (PCA) of three replicates of the lipidomics of AAVS1 control, sorted CD90+CD133+ and CD90− MYSM1 KO cells. (B) Unsupervised hierarchical analysis of three replicates of AAVS1 control, sorted CD90+CD133+ and CD90− MYSM1 KO cells. (C) Bubble chart of selected enriched and depleted lipids of sorted CD90+CD133+ MYSM1 KO cells compared to AVVS1 control. (D) Bubble chart of selected enriched and depleted lipids of sorted CD90+CD133+ MYSM1 KO cells compared to CD90− MYSM1 KO cells. (E) Quantification of Annexin V−PI− (normal cells), Annexin V+PI− (early apoptosis cells), Annexin V+PI+ (late apoptosis cells) and Annexin V−PI+ (full dead cells) of AAVS1 or MYSM1 gRNA edited CD34+CD45RA−CD90+ cells. (F) Western blot analysis of apoptosis (left) and necroptosis (right) protein markers of AAVS1 or MYSM1 gRNA edited CD34+CD45RA−CD90+ cells. (G) Lipid ROS quantification of bone marrow CD34+CD45RA−CD90+ sorted cells after xenotransplantation of AAVS1 or MYSM1 edited cord blood CD34+ cells.
Figure S5 HSC rescue by ferroptosis inhibitor, related to Figure 5. Representative flow cytometric histogram of CellROX, oxidized BODIPY and O-Propargyl-puromycin signals of sorted CD34+CD45RA−CD90+ cells with AAVS1 or MYSM1 editing, and MYSM1 edited cells with indicated chemical treatments.
Figure S6 Quality control of ribo-seq experiment and GSEA of top genes whose translation is affected, related to Figure 6. (A) Quantification of protein levels of Fig 6B. (B) Western blot validation of rescue of ferroptosis protective gene expression by overexpressing MYSM1 WT, E390*, R478* and H656R mutants in CD34+CD45RA−CD90+ CD133+ MYSM1 deficient cells. (C) Identification of 3-nt periodicity of ribosome profiling sequencing (ribo-seq) data. (D) Additional gene set enrichment analysis (GSEA) showing that lipid synthesis related gene translation and mevalonate pathway translation were also significantly reduced in MYSM1 KO cells. (E) Additional gene set enrichment analysis (GSEA) showing that ribosome biogenesis and translation initiation gene translation was significantly increased.
Figure S7 Flow cytometric analysis of HSC phenotyping upon ferroptosis induction, related to Figure 7. (A) Prediction of edit species by two independent gRNAs of GPX4. (B) Representative flow cytometric analysis of HSC phenotyping of CD34+ HSPCs treated with Erastin at indicated concentration transduced with empty vector (EV) or wildtype MYSM1. (C) Schematic of Fanconi anemia HSC modeling experiment shown in Fig. 7J.
Highlights.
MYSM1 deficiency causes human HSCs loss by ferroptosis.
MYSM1-deficient HSCs have reduced translation of ferroptosis protective mRNAs.
Human HSCs, but not progenitors, are vulnerable to ferroptosis.
Human HSC vulnerability to ferroptosis arises due to low rates of protein synthesis.
Hematopoietic stem cells have a low rate of protein synthesis that makes them selectively vulnerable to ferroptosis.
Acknowledgements
We thank members of the Sankaran lab and a number of colleagues, including P. van Galen, S. Schreiber, and B. Will, for valuable comments and advice on this work. We thank the Harvard Medical School Electron Microscopy Facility (HMS-EMF) for electron microscopy imaging and consultation. This work was supported by the New York Stem Cell Foundation (NYSCF), a gift from the Lodish Family to Boston Children’s Hospital, the Edward P. Evans Foundation, and National Institute of Health (NIH) grants R01 DK103794, R01 CA265726, and R01 HL146500. V.G.S. is a NYSCF-Robertson Investigator.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Declaration of Interests
V.G.S. serves as an advisor to and/or has equity in Branch Biosciences, Ensoma, Novartis, Forma, Sana Biotechnology, and Cellarity. There are no other competing interests to declare.
Inclusion and Diversity
One or more of the authors of this paper self-identifies as an underrepresented ethnic minority in their field of research. One or more of the authors of this paper self-identifies as a gender minority in their field of research. One or more of the authors of this paper self-identifies as a member of the LGBTQIA+ community. We support inclusive, diverse, and equitable conduct of research.
References
- 1.Laurenti E & Göttgens B From haematopoietic stem cells to complex differentiation landscapes. Nature 553, 418–426 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Liggett LA & Sankaran VG Unraveling Hematopoiesis through the Lens of Genomics. Cell 182, 1384–1400 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Baldridge MT, King KY, Boles NC, Weksberg DC & Goodell MA Quiescent haematopoietic stem cells are activated by IFN-gamma in response to chronic infection. Nature 465, 793–797 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Tothova Z et al. FoxOs are critical mediators of hematopoietic stem cell resistance to physiologic oxidative stress. Cell 128, 325–339 (2007). [DOI] [PubMed] [Google Scholar]
- 5.Mortensen M et al. The autophagy protein Atg7 is essential for hematopoietic stem cell maintenance. J. Exp. Med 208, 455–467 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Ho TT et al. Autophagy maintains the metabolism and function of young and old stem cells. Nature 543, 205–210 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Dong S et al. Chaperone-mediated autophagy sustains haematopoietic stem-cell function. Nature 591, 117–123 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Magee JA & Signer RAJ Developmental Stage-Specific Changes in Protein Synthesis Differentially Sensitize Hematopoietic Stem Cells and Erythroid Progenitors to Impaired Ribosome Biogenesis. Stem Cell Reports 16, 20–28 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Signer RAJ et al. The rate of protein synthesis in hematopoietic stem cells is limited partly by 4E-BPs. Genes & Development vol. 30 1698–1703 Preprint at 10.1101/gad.282756.116 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Signer RAJ, Magee JA, Salic A & Morrison SJ Haematopoietic stem cells require a highly regulated protein synthesis rate. Nature 509, 49–54 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Kruta M et al. Hsf1 promotes hematopoietic stem cell fitness and proteostasis in response to ex vivo culture stress and aging. Cell Stem Cell 28, 1950–1965.e6 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Garaycoechea JI et al. Alcohol and endogenous aldehydes damage chromosomes and mutate stem cells. Nature 553, 171–177 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Garaycoechea JI et al. Genotoxic consequences of endogenous aldehydes on mouse haematopoietic stem cell function. Nature 489, 571–575 (2012). [DOI] [PubMed] [Google Scholar]
- 14.Alsultan A, Shamseldin HE, Osman ME, Aljabri M & Alkuraya FS MYSM1 is mutated in a family with transient transfusion-dependent anemia, mild thrombocytopenia, and low NK- and B-cell counts. Blood 122, 3844–3845 (2013). [DOI] [PubMed] [Google Scholar]
- 15.Li N et al. Further delineation of bone marrow failure syndrome caused by novel compound heterozygous variants of MYSM1. Gene 757, 144938 (2020). [DOI] [PubMed] [Google Scholar]
- 16.Le Guen T et al. An in vivo genetic reversion highlights the crucial role of Myb-Like, SWIRM, and MPN domains 1 (MYSM1) in human hematopoiesis and lymphocyte differentiation. J. Allergy Clin. Immunol 136, 1619–1626.e5 (2015). [DOI] [PubMed] [Google Scholar]
- 17.Zhan X et al. A novel compound heterozygous mutation of MYSM1 gene in a patient with bone marrow failure syndrome 4. Br. J. Biomed. Sci 78, 239–243 (2021). [DOI] [PubMed] [Google Scholar]
- 18.Belle JI et al. MYSM1 maintains ribosomal protein gene expression in hematopoietic stem cells to prevent hematopoietic dysfunction. JCI Insight 5, (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Bluteau O et al. A landscape of germ line mutations in a cohort of inherited bone marrow failure patients. Blood 131, 717–732 (2018). [DOI] [PubMed] [Google Scholar]
- 20.Ulirsch JC et al. The Genetic Landscape of Diamond-Blackfan Anemia. Am. J. Hum. Genet 103, 930–947 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Revy P, Kannengiesser C & Fischer A Somatic genetic rescue in Mendelian haematopoietic diseases. Nat. Rev. Genet 20, 582–598 (2019). [DOI] [PubMed] [Google Scholar]
- 22.Huo Y et al. MYSM1 is essential for maintaining hematopoietic stem cell (HSC) quiescence and survival. Med. Sci. Monit 24, 2541–2549 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Belle JI et al. p53 mediates loss of hematopoietic stem cell function and lymphopenia in Mysm1 deficiency. Blood 125, 2344–2348 (2015). [DOI] [PubMed] [Google Scholar]
- 24.Won H et al. Epigenetic control of dendritic cell development and fate determination of common myeloid progenitor by Mysm1. Blood 124, 2647–2656 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Huang XF et al. Mysm1 is required for interferon regulatory factor expression in maintaining HSC quiescence and thymocyte development. Cell Death Dis. 7, e2260 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Fiore A et al. Deubiquitinase MYSM1 in the Hematopoietic System and beyond: A Current Review. Int. J. Mol. Sci 21, (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Shen Y et al. A unified model of human hemoglobin switching through single-cell genome editing. Nat. Commun 12, 4991 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Wagenblast E et al. Functional profiling of single CRISPR/Cas9-edited human long-term hematopoietic stem cells. Nat. Commun 10, 4730 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Voit RA et al. A genetic disorder reveals a hematopoietic stem cell regulatory network co-opted in leukemia. Nat. Immunol 1–15 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Bao EL et al. Inherited myeloproliferative neoplasm risk affects haematopoietic stem cells. Nature 586, 769–775 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.McIntosh BE et al. Nonirradiated NOD,B6.SCID Il2rγ−/− KitW41/W41 (NBSGW) Mice Support Multilineage Engraftment of Human Hematopoietic Cells. Stem Cell Reports 4, 171–180 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Fiorini C et al. Developmentally-faithful and effective human erythropoiesis in immunodeficient and Kit mutant mice. Am. J. Hematol 92, E513–E519 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Hidalgo San Jose L et al. Modest Declines in Proteome Quality Impair Hematopoietic Stem Cell Self-Renewal. Cell Rep. 30, 69–80.e6 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Dixon SJ & Stockwell BR The role of iron and reactive oxygen species in cell death. Nat. Chem. Biol 10, 9–17 (2014). [DOI] [PubMed] [Google Scholar]
- 35.Stockwell BR et al. Ferroptosis: A Regulated Cell Death Nexus Linking Metabolism, Redox Biology, and Disease. Cell 171, 273–285 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Dixon SJ et al. Ferroptosis: an iron-dependent form of nonapoptotic cell death. Cell 149, 1060–1072 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Hentze MW, Muckenthaler MU, Galy B & Camaschella C Two to tango: regulation of Mammalian iron metabolism. Cell 142, 24–38 (2010). [DOI] [PubMed] [Google Scholar]
- 38.Nishizawa H et al. Lipid peroxidation and the subsequent cell death transmitting from ferroptotic cells to neighboring cells. Cell Death Dis. 12, 332 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Klöditz K & Fadeel B Three cell deaths and a funeral: macrophage clearance of cells undergoing distinct modes of cell death. Cell Death Discovery vol. 5 Preprint at 10.1038/s41420-019-0146-x (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Wu C et al. Induction of ferroptosis and mitochondrial dysfunction by oxidative stress in PC12 cells. Sci. Rep 8, 574 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Zhang Y et al. Imidazole Ketone Erastin Induces Ferroptosis and Slows Tumor Growth in a Mouse Lymphoma Model. Cell Chem Biol 26, 623–633.e9 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Yang WS et al. Peroxidation of polyunsaturated fatty acids by lipoxygenases drives ferroptosis. Proc. Natl. Acad. Sci. U. S. A 113, E4966–75 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Zou Y et al. Plasticity of ether lipids promotes ferroptosis susceptibility and evasion. Nature 585, 603–608 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Li J et al. Ferroptosis: past, present and future. Cell Death Dis. 11, 88 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Khajuria RK et al. Ribosome Levels Selectively Regulate Translation and Lineage Commitment in Human Hematopoiesis. Cell 173, 90–103.e19 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Ingolia NT, Hussmann JA & Weissman JS Ribosome Profiling: Global Views of Translation. Cold Spring Harb. Perspect. Biol 11, (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Niraj J, Färkkilä A & D’Andrea AD The Fanconi Anemia Pathway in Cancer. Annu Rev Cancer Biol 3, 457–478 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Rosado IV, Langevin F, Crossan GP, Takata M & Patel KJ Formaldehyde catabolism is essential in cells deficient for the Fanconi anemia DNA-repair pathway. Nat. Struct. Mol. Biol 18, 1432–1434 (2011). [DOI] [PubMed] [Google Scholar]
- 49.Langevin F, Crossan GP, Rosado IV, Arends MJ & Patel KJ Fancd2 counteracts the toxic effects of naturally produced aldehydes in mice. Nature 475, 53–58 (2011). [DOI] [PubMed] [Google Scholar]
- 50.Jiang X, Stockwell BR & Conrad M Ferroptosis: mechanisms, biology and role in disease. Nat. Rev. Mol. Cell Biol 22, 266–282 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Ingold I et al. Selenium Utilization by GPX4 Is Required to Prevent Hydroperoxide-Induced Ferroptosis. Cell 172, 409–422.e21 (2018). [DOI] [PubMed] [Google Scholar]
- 52.Yang WS et al. Regulation of ferroptotic cancer cell death by GPX4. Cell 156, 317–331 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Bersuker K et al. The CoQ oxidoreductase FSP1 acts parallel to GPX4 to inhibit ferroptosis. Nature 575, 688–692 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Hangauer MJ et al. Drug-tolerant persister cancer cells are vulnerable to GPX4 inhibition. Nature 551, 247–250 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Jiang L et al. Ferroptosis as a p53-mediated activity during tumour suppression. Nature 520, 57–62 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Viswanathan VS et al. Dependency of a therapy-resistant state of cancer cells on a lipid peroxidase pathway. Nature 547, 453–457 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Zou Y et al. A GPX4-dependent cancer cell state underlies the clear-cell morphology and confers sensitivity to ferroptosis. Nat. Commun 10, 1617 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Ubellacker JM et al. Lymph protects metastasizing melanoma cells from ferroptosis. Nature 585, 113–118 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Alborzinia H et al. Selenocysteine metabolism is a targetable vulnerability in MYCN-amplified cancers. bioRxiv 2022.05.17.492172 (2022) doi: 10.1101/2022.05.17.492172. [DOI] [Google Scholar]
- 60.Li Z et al. Ribosome stalling during selenoprotein translation exposes a ferroptosis vulnerability. Nat. Chem. Biol 1–11 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Filippi M-D & Ghaffari S Mitochondria in the maintenance of hematopoietic stem cells: new perspectives and opportunities. Blood 133, 1943–1952 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Cimmino L et al. Restoration of TET2 Function Blocks Aberrant Self-Renewal and Leukemia Progression. Cell 170, 1079–1095.e20 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Agathocleous M et al. Ascorbate regulates haematopoietic stem cell function and leukaemogenesis. Nature 549, 476–481 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Kao Y-R et al. Thrombopoietin receptor–independent stimulation of hematopoietic stem cells by eltrombopag. Sci. Transl. Med 10, eaas9563 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Mbong N et al. The Integrated Stress Response Activity Marks Stem Cells in Normal Hematopoiesis and Leukemia. Blood vol. 132 1276–1276 Preprint at 10.1182/blood-2018-99-110819 (2018). [DOI] [PubMed] [Google Scholar]
- 66.Desmond R et al. Eltrombopag restores trilineage hematopoiesis in refractory severe aplastic anemia that can be sustained on discontinuation of drug. Blood 123, 1818–1825 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Olnes MJ et al. Eltrombopag and improved hematopoiesis in refractory aplastic anemia. N. Engl. J. Med 367, 11–19 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Peffault de Latour R et al. Eltrombopag Added to Immunosuppression in Severe Aplastic Anemia. N. Engl. J. Med 386, 11–23 (2022). [DOI] [PubMed] [Google Scholar]
- 69.Mantel CR et al. Enhancing Hematopoietic Stem Cell Transplantation Efficacy by Mitigating Oxygen Shock. Cell 161, 1553–1565 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Kao Y-R et al. Cytoplasmic labile iron accumulates in aging stem cells perturbing a key rheostat for identity control. bioRxiv 2021.08.03.454947 (2021) doi: 10.1101/2021.08.03.454947. [DOI] [Google Scholar]
- 71.Subramanian A et al. Gene set enrichment analysis: a knowledge-based approach for interpreting genome-wide expression profiles. Proc. Natl. Acad. Sci. U. S. A 102, 15545–15550 (2005). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Ingolia NT, Brar GA, Rouskin S, McGeachy AM & Weissman JS The ribosome profiling strategy for monitoring translation in vivo by deep sequencing of ribosome-protected mRNA fragments. Nat. Protoc 7, 1534–1550 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Martin M Cutadapt removes adapter sequences from high-throughput sequencing reads. EMBnet. journal (2011). [Google Scholar]
- 74.Langmead B Aligning short sequencing reads with Bowtie. Curr. Protoc. Bioinformatics Chapter 11, Unit 11.7 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Dobin A et al. STAR: ultrafast universal RNA-seq aligner. Bioinformatics 29, 15–21 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Xiao Z et al. De novo annotation and characterization of the translatome with ribosome profiling data. Nucleic Acids Res. 46, e61 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Anders S, Pyl PT & Huber W HTSeq—a Python framework to work with high-throughput sequencing data. Bioinformatics 31, 166–169 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Lin W-J et al. LipidSig: a web-based tool for lipidomic data analysis. Nucleic Acids Res. 49, W336–W345 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Lam J et al. A Universal Approach to Analyzing Transmission Electron Microscopy with ImageJ. Cells 10, (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Schneider CA, Rasband WS & Eliceiri KW NIH Image to ImageJ: 25 years of image analysis. Nat. Methods 9, 671–675 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Table S2 GSEA analysis of differentially expressed genes of MYSM1 KO vs AAVS1, Related to Figure 1–3.
Table S3 Lipidomics of MYSM1 KO HSC vs AAVS1 HSC, Related to Figure 4.
Table S4 Lipidomics of MYSM1 KO non-HSC vs AAVS1 HSC, Related to Figure 4.
Table S5 Lipidomics of MYSM1 KO HSC vs MYSM1 KO non-HSC, Related to Figure 4.
Table S6 Differentially translated genes of MYSM1 KO vs AAVS1, Related to Figure 6.
Table S7 GSEA of differentially translated genes of MYSM1 KO vs AAVS1, Related to Figure 6.
Figure S1 CRISPR editing analysis and flow cytometric analysis of HSC phenotyping, related to Figure 1. (A) ICE analysis of percentage of edited cells by two independent MYSM1 gRNAs. (B) Prediction of edited species by two MYSM1 gRNAs. Major edited species all result in stop codons at very early sites of the MYSM1 gene. (C) Western blot validation of successful knockout of MYSM1. (D) Quantification of MYSM1 protein in AAVS1 and two MYSM1 gRNA edited CD34+ HSPCs. (E) Representative flow cytometric analysis of HSC phenotyping of AAVS1 control or MYSM1 edited cells and gating strategy. (F) Representative flow cytometric analysis of human vs. mouse CD45+ cells after RBC depletion from indicated sites of NBSGW mice xenotransplanted with AAVS1 and MYSM1 edited cord blood CD34+ HSPCs.
Figure S2 Transcriptional regulation of translation genes by MYSM1, related to Figure 2. (A) Additional gene set enrichment analysis (GSEA) showing ribosomal biogenesis and cellular translation are impaired with loss of MYSM1. (B) Real-time PCR validation of genes shown as downregulation due to loss of MYSM1 in scRNA-seq. (C) Western blot validation of expression of overexpression constructs of MYSM1 WT, E390*, R478* and H656R mutants.
Figure S3 Upregulation of ferroptosis protective genes and downregulation of ferroptosis promoting genes upon loss of MYSM1, related to Figure 3. (A) Additional violin plot of ferroptosis gene expression in AAVS1 and MYSM1 KO HSCs. (B) Additional gene set enrichment analysis (GSEA) showing that cellular oxidant detoxification and general detoxification processes were significantly stimulated in MYSM1 KO HSCs.
Figure S4 Increased oxidative stress and lipid peroxidation and depletion of membrane phospholipids upon loss of MYSM1, related to Figure 4. (A) Principal Component Analysis (PCA) of three replicates of the lipidomics of AAVS1 control, sorted CD90+CD133+ and CD90− MYSM1 KO cells. (B) Unsupervised hierarchical analysis of three replicates of AAVS1 control, sorted CD90+CD133+ and CD90− MYSM1 KO cells. (C) Bubble chart of selected enriched and depleted lipids of sorted CD90+CD133+ MYSM1 KO cells compared to AVVS1 control. (D) Bubble chart of selected enriched and depleted lipids of sorted CD90+CD133+ MYSM1 KO cells compared to CD90− MYSM1 KO cells. (E) Quantification of Annexin V−PI− (normal cells), Annexin V+PI− (early apoptosis cells), Annexin V+PI+ (late apoptosis cells) and Annexin V−PI+ (full dead cells) of AAVS1 or MYSM1 gRNA edited CD34+CD45RA−CD90+ cells. (F) Western blot analysis of apoptosis (left) and necroptosis (right) protein markers of AAVS1 or MYSM1 gRNA edited CD34+CD45RA−CD90+ cells. (G) Lipid ROS quantification of bone marrow CD34+CD45RA−CD90+ sorted cells after xenotransplantation of AAVS1 or MYSM1 edited cord blood CD34+ cells.
Figure S5 HSC rescue by ferroptosis inhibitor, related to Figure 5. Representative flow cytometric histogram of CellROX, oxidized BODIPY and O-Propargyl-puromycin signals of sorted CD34+CD45RA−CD90+ cells with AAVS1 or MYSM1 editing, and MYSM1 edited cells with indicated chemical treatments.
Figure S6 Quality control of ribo-seq experiment and GSEA of top genes whose translation is affected, related to Figure 6. (A) Quantification of protein levels of Fig 6B. (B) Western blot validation of rescue of ferroptosis protective gene expression by overexpressing MYSM1 WT, E390*, R478* and H656R mutants in CD34+CD45RA−CD90+ CD133+ MYSM1 deficient cells. (C) Identification of 3-nt periodicity of ribosome profiling sequencing (ribo-seq) data. (D) Additional gene set enrichment analysis (GSEA) showing that lipid synthesis related gene translation and mevalonate pathway translation were also significantly reduced in MYSM1 KO cells. (E) Additional gene set enrichment analysis (GSEA) showing that ribosome biogenesis and translation initiation gene translation was significantly increased.
Figure S7 Flow cytometric analysis of HSC phenotyping upon ferroptosis induction, related to Figure 7. (A) Prediction of edit species by two independent gRNAs of GPX4. (B) Representative flow cytometric analysis of HSC phenotyping of CD34+ HSPCs treated with Erastin at indicated concentration transduced with empty vector (EV) or wildtype MYSM1. (C) Schematic of Fanconi anemia HSC modeling experiment shown in Fig. 7J.
Data Availability Statement
The scRNA-seq and ribosome profiling datasets of AAVS1 or MYSM1 edited HSPCs have been deposited to NCBI Gene Expression Omnibus and are publicly available as of the date of publication. The GEO accession numbers of the datasets are included in the key resource table.
Key resources table
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| Anti-MYSM1 antibody | Abcam | ab-221725 |
| RPS24 Polyclonal antibody | ProteinTech | 14831-1-AP |
| RPS7 Polyclonal antibody | ProteinTech | 14491-1-AP |
| Ferroptosis Antibody Sampler Kit | Cell signaling Technology | 29650 |
| Brilliant Violet 421™ anti-human CD34 Antibody | BD Biosciences | 562577 |
| PE Mouse Anti-Human CD66b | BD biosciences | 561650 |
| PE Mouse Anti-Human CD33 | BD biosciences | 555450 |
| PE Mouse Anti-Human CD19 | BD biosciences | 555413 |
| RPL15 Polyclonal antibody | Proteintech | 16086-1-AP |
| RPL23 Polyclonal antibody | Proteintech | 10727-1-AP |
| Ferritin light chain Polyclonal antibody | Proteintech | 10727-1-AP |
| anti-CD90-PECy7 | BD biosciences | 561558 |
| APC-H7 Mouse Anti-Human CD45RA | BD biosciences | 560674 |
| Necroptosis Antibody Sampler Kit | Cell signaling Technology | 98110T |
| Apoptosis Antibody Sampler Kit | Cell signaling Technology | 9915T |
| hnRNP-E1 Polyclonal antibody | proteintech | 14523-1-AP |
| PCBP2 Polyclonal antibody | proteintech | 15070-1-AP |
| SLC40A1/FPN1 Polyclonal antibody | proteintech | 26601-1-AP |
| GCLM Polyclonal antibody | proteintech | 14241-1-AP |
| GCLC Polyclonal antibody | proteintech | 12601-1-AP |
| FITC Annexin V | Biolegend | 640906 |
| APC anti-human CD49c (integrin α3) Antibody | Biolegend | 343808 |
| ALAS1 Polyclonal antibody | proteintech | 16200-1-AP |
| HSP27 Polyclonal antibody | proteintech | 18284-1-AP |
| RELB Polyclonal antibody | proteintech | 25027-1-AP |
| Ferroportin/SLC40A1 Antibody - BSA Free | Novus Biologicals | NBP1-21502SS |
| BB700 Mouse Anti-Human CD133 | BD biosciences | 747638 |
| rat anti-human anti-EPCR-PE | Biolegend | 351904 |
| PE anti-human CD34 Antibody | Biolegend | 343506 |
| PE anti-human CD3 Antibody | Biolegend | 317308 |
| PE anti-human CD11b Antibody | Biolegend | 301306 |
| Monoclonal ANTI-FLAG® M2 antibody produced in mouse | Sigma Aldrich | F1804 |
| Chemicals, peptides, and recombinant proteins | ||
| Holo-transferrin Human | Sigma Aldrich | T0665 |
| Hygromycin B | Gibco | 10687010 |
| Erastin | Selleck | S7242 |
| FIN56 | Selleck | S8254 |
| FINO2 | Cayman Chemical | 25096 |
| Deferoxamine mesylate | Selleck | S5742 |
| Ferrostatin-1 | Selleck | S7243 |
| BODIPY™ 581/591 C11 | Invitrogen | D3861 |
| CellROX™ Green Reagent | Invitrogen | C10444 |
| MitoTEMPO (hydrate) | Cayman Chemical | 16621 |
| N-acetyl-L-Cysteine | Cayman Chemical | 20261 |
| (±)-α-Tocopherol Acetate | Cayman Chemical | 28399 |
| Polyethylenimine, Linear, MW 25000, Transfection Grade (PEI 25K™) | PolySciences | 23966-100 |
| Ambion™ RNase I, cloned, 100 U/μ | Invitrogen | AM2294 |
| GlycoBlue™ Coprecipitant (15 mg/mL) | Invitrogen | AM9515 |
| Cycloheximide | Sigma Aldrich | C7698 |
| Ultra Low Range DNA Ladder | Invitrogen | 10597012 |
| SYBR™ Gold Nucleic Acid Gel Stain (10,000X Concentrate in DMSO) | Invitrogen | S11494 |
| RSL3 | Selleck | S8155 |
| CC100 | StemCell Technologies | 2690 |
| BioTracker Far-red Labile Fe2+ Dye | Millipore Sigma | SCT037 |
| SM-164 1mg | Cell Signaling Technology | 56003S |
| Z-VAD-FMK | Selleck | S7023 |
| Recombinant Human TNF-α 10ug | Peprotech | 300-01A |
| Mitomycin C 10mg | Selleck | S8146 |
| Iron(II) sulfate heptahydrate | F7002-250G | Sigma Aldrich |
| Liproxstatin-1 | Cayman Chemical | 17730 |
| Staurosporine | Cayman chemical | 81590 |
| Methanol | Sigma Aldrich | 179337 |
| RPMI1640 medium | Life Technologies | 11875-119 |
| Opti-MEM | Gibco | 31985-062 |
| Ficoll-Paque Premium sterile solution | GE Healthcare | 17-5442-02 |
| 0.25% Trypsin (1X) | Gibco | 15050-057 |
| Blotting nonfat dry milk | BioRad | 1706404XTU |
| Cas9 protein, recombinant | IDT | 1081061 |
| Penicillin-Streptomycin 10,000u/ml | Life Technologies | 15140-122 |
| Recombinant Human TPO | Peprotech | 300-18-100UG |
| Formamide | Sigma Aldrich | F7503 |
| TBE Buffer (Tris-borate-EDTA) (10X) | Thermo Scientific | B52 |
| Annexin-V-Binding Buffer | Biolegend | 422201 |
| L-Glutamine 200mM for cell culture | GIBCO | 25-030-081 |
| SSC (20X), RNase-free | Invitrogen | AM9763 |
| DMEM High Glucose | Life Technologies | 11965-118 |
| Critical commercial assays | ||
| Glutathione Peroxidase Assay Kit | Cayman Chemical | 703102 |
| Protein Synthesis Assay Kit | Cayman Chemical | 601100 |
| Hemin Colorimetric Assay Kit | BioVision | K672 |
| Iron Colorimetric Assay Kit | BioVision | K390 |
| Kapa Library Quantification Kit - Complete Kit (Universal) | Roche | 7960140001 |
| mMESSAGE mMACHINE™ T7 Transcription Kit | Invitrogen | AM1344 |
| MEGAclear™ Transcription Clean-Up Kit | Invitrogen | AM1908 |
| Poly(A) Tailing Kit | Invitrogen | AM1350 |
| Deposited data | ||
| Single cell RNA seq | This paper | GSE221170 |
| Ribosome Profiling | This paper | GSE221170 |
| Experimental models: Cell lines | ||
| CD34+ HSPCs | Fred Hutch | |
| HEK293T | ATCC | CRL-3216 |
| Experimental models: Organisms/strains | ||
| NBSGW | JAX Laboratory | 026622 |
| Oligonucleotides | ||
| Alt-R® Cas9 Electroporation Enhancer | IDT | 1075916 |
| Universal miRNA Cloning Linker | New England Biolabs | S1315S |
| qPCR primers and genotyping/editing efficiency testing primers | Genewiz | Table S8 |
| Chemically synthesized sgRNA | Synthego | Table S9 |
| Recombinant DNA | ||
| MGC Fully Sequenced Human SLC40A1 cDNA | Horizon Discovery | MHS6278-202801506 |
| MGC Fully Sequenced Human FTH1 cDNA | Horizon Discovery | MHS6278-202755914 |
| MGC Fully Sequenced Human ALAS1 cDNA | Horizon Discovery | MHS6278-202831218 |
| MGC Human HSPB1 Sequence-Verified cDNA | Horizon Discovery | MHS6278-202756315 |
| MGC Human RELB Sequence-Verified cDNA | Horizon Discovery | MHS6278-202801567 |
| Software and algorithms | ||
| Graphpad Prism v.9.0.1 | Graphpad | RRID:SCR_002798 |
| Biorender | Biorender | RRID:SCR_018361 |
| Flowjo | Flowjo | RRID:SCR_008520 |
| ImageJ v1.52a | Wayne Rasband | RRID:SCR_003070 |
| R | The R Foundation | |
| STAR v2.7.10a | ||
| Bowtie2 v2.3.3 | ||
| RiboCode v1.2.15 | ||
| Cutadapt v4.1 | ||
| CellRanger v7.0.0 | 10xGenomics | |
| Seurat v4.0.2 | ||
| Other | ||
| In-Fusion® HD Cloning Plus | Takara/Clontech | 638909 |
| Q5 site directed mutagenesis kit | New England Biolabs | E0554S |
| iScript™ cDNA Synthesis Kit | BioRad | 1708891 |
| Monarch® DNA Gel Extraction Kit | New England Biolabs | T1020L |
| iQ™ SYBR® Green Supermix | BioRad | 1708882 |
| P3 Primary Cell 4D Nucelofector X Kit S | Lonza | V4XP-3032 |
| SuperScript™ III Reverse Transcriptase | Invitrogen | 18080044 |
| Chromium Next GEM Single Cell 3ʹ Kit v3.1, 16 rxns | 10XGenomics | PN-1000268 |
| Chromium Next GEM Chip G Single Cell Kit, 48 rxns | 10XGenomics | PN-1000120 |
| Dual Index Kit TT Set A, 96 rxns | 10XGenomics | PN-1000215 |
| CircLigase™ ssDNA Ligase | Lucigen | CL4111K |
| T4 RNA Ligase 2, truncated | New England Biolabs | M0242L |
| StemSpan™ SFEM II | StemCell Technologies | 09655 |
| Fetal Bovine Serum - Premium Select G21162 | Bio-Techne | S11550 |
| SUPERase•In™ RNase Inhibitor (20 U/μL) | Invitrogen | AM2694 |
| EasySep Human Cord Blood CD34 Positive Selection Kit II | StemCell Technologies | 17896 |
| Immobilon-P PVDF Membrane | Millipore Sigma | IPVH00010 |
| SPRIselect | Beckman Coulter | B23317 |
| Qubit™ 1X dsDNA HS Assay Kit | ThermoFisher | Q33231 |
| Zero Blunt™ TOPO™ PCR Cloning Kit for Sequencing, without competent cells 25rxn | Invitrogen | 450031 |
There is no original code generated in this study.
Any additional information or help in reanalyzing the datasets is available from the lead contact upon request.