

Abstract
HMGB1 is a dual-function protein that acts as a chromatin-binding protein and as a danger-associated molecular pattern (DAMP) when released from activated immune cells or injured tissue. In much of the HMGB1 literature, immunomodulatory effects of extracellular HMGB1 are proposed to depend on its oxidation state. However, many of the foundational studies for this model have been retracted or flagged with expressions of concern. The literature on HMGB1 oxidation reveals a diversity of redox proteoforms of HMGB1 that are inconsistent with current models of redox modulation regulating HMGB1 secretion. A recent study of acetaminophen toxicity has identified previously unrecognized HMGB1 oxidized proteoforms. HMGB1 undergoes oxidative modifications that could serve as pathology-specific biomarkers and drug targets.
Keywords: A-box. B-box, DAMP, DNA-binding, HMGB1, immune response, plasma biomarker, post-translational modification, RAGE, TLR
Tweetable abstract
HMGB1 undergoes oxidative modifications that could serve as pathology-specific biomarkers and drug targets.
Graphical abstract
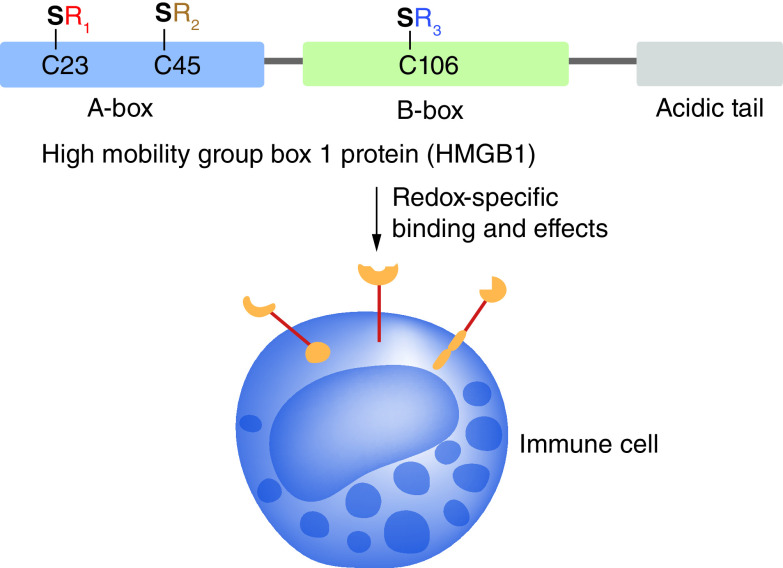
HMGB1, a non-histone 30 kDa chromosomal protein, was first isolated as a heparin binding protein (p30) with neurite outgrowth activity [1] and was originally called amphoterin [2]. The primary structure of HMGB1 (also known as HMG1) was deduced from its nucleotide sequence as a 215 amino acid single-chain polypeptide [3]. It contains A-box (9–79) and B-box (88–162) DNA-binding domains, an acidic C-terminal tail (186–215), and several redox-sensitive amino acids, including three highly conserved cysteine (Cys)-residues. (Figure 1). The N-terminal methionine-1 (M1) is lost during translation leaving glycine-2 (G2) at the amino terminus, although some reports refer to it as G1 and shift the labeling on all the other amino acids by -1.
Figure 1. . Amino acid sequence of HMGB1 with select redox-sensitive amino acids bolded and underlined (M, Y, C).

A-box = amino acids 9–79, B-box = amino acids 88–162, acidic tail at C-terminus = amino acids 186–215 [4].
HMGB1 is a multi-functional protein that acts as a DNA chaperone as well as a damage-associated molecular pattern (DAMP) with immune-stimulating effects. HMGB1 localizes to the nucleus where it binds DNA, facilitates ATP-utilizing chromatin assembly factor/chromatin accessibility complex (ACF/CHRAC)‐dependent nucleosome sliding [5] and allows assembly of transcription factors [6,7]. The regulatory effects of HMGB1 on transcription are such that it is highly conserved across species and produced in all nucleated eukaryotic cells [8]. Extracellular HMGB1 can induce a range of immunological effects including inflammation [9], migration of maturing dendritic cells [10], and release of cytokines such as tumor necrosis factor α (TNFα) [11]. Neutralizing anti-HMGB1 antibodies significantly decrease the lethality of injuries from acetaminophen-overdose, demonstrating HMGB1's contribution to pathological inflammation [12].
Although the presence of HMGB1 in the nucleus is supported by its binding to DNA and chromatin protein partners, it has also been suggested that the intracellular localization of HMGB1 is regulated by post-translational modifications (PTMs). For example, it was reported that post-translational methylation of HMGB1 at lysine-43 (named as lysine-42 with N-terminal glycine-1) is responsible for its cytoplasmic localization in neutrophils [13]. HMGB1 was shown to be secreted from monocytes via a non‐classical, vesicle‐mediated secretory pathway [14]. Bonaldi et al. suggested that hyperacetylation of HMGB1 was required to redirect it toward secretion from monocytes [15]. In contrast, Youn and Shin found that phosphorylation of six serine residues in purported nuclear localization sequence (NLS) regions was required in order to control the nucleocytoplasmic shuttling of HMGB1 and redirect it toward secretion [16]. PTMs of HMGB1 may additionally modulate receptor binding of HMGB1 released extracellularly and therefore alter its extracellular function.
HMGB1 can be released in multiple pathological contexts and bind to a variety of extracellular receptors. Release of HMGB1 can occur passively from necrotic cells [9] or during regulated apoptosis of certain cell types [17]. Since nuclear HMGB1 is bound tightly to chromatin during apoptosis and secondary necrosis, the majority of HMGB1 released during apoptosis is likely to come from the cytoplasmic pool of HMGB1, which could vary in amount by cell type [17]. Active secretion of HMGB1 from activated cells is also possible, at least in part through an innate immune response mediated by both the cluster of differentiation (CD)14‐ and TNF [18]. The immunological effects exerted by extracellular HMGB1 (also known as amphoterin) are mediated through multiple receptors, including the receptor for advanced glycation end products (RAGE) (Figure 2) [19]. Secreted HMGB1 directly activates toll-like receptor (TLR) 2 and TLR4 [20]; whereas TLR9-dependent activation by DNA-containing immune complexes is mediated by HMGB1 and RAGE [21]. Therefore, HMGB1 binds to and activates multiple TLRs [22]. In addition, HMGB1 can recruit inflammatory cells to damaged tissues by forming a complex with chemokine ligand 12 (CXCL12) and binding to chemokine receptor 4 (CXCR4) (Figure 2) [23].
Figure 2. . HMGB1 induces cytokine and chemokine effects through multiple binding partners RAGE, TLR2 and TLR4, via a complex with CXCL12/CXCR4, and TLR9 via a complex with RNA/DNA.

Binding to the individual receptors might be redox sensitive [19–22].
Most studies of HMGB1 oxidation states and their effects on function either only broadly characterize HMGB1 as reduced and oxidized or allow for a limited number of oxidized Cys-residue proteoforms. Many studies additionally characterize HMGB1 oxidation state via gel mobility shifts or targeted mutation techniques that may not account for all proteoforms present. A priori limitation of the possible HMGB1 oxidized proteoforms simplifies experimental models but may not accurately reflect the complexity of HMGB1 oxidation and its effects on immune function in vivo.
HMGB1 release has been associated with oxidative stress in a wide range of pathological conditions. Reactive oxygen species (ROS) and reactive nitrogen species (RNS) can act both as redox signaling messengers and agents of oxidative damage depending on their concentration and a cell's antioxidant capacity. Many pathological conditions associated with extracellular HMGB1 release such as drug-induced hepatic necrosis, myocardial ischemia/reperfusion injury, and sepsis are also associated with excessive generation of ROS/RNS [24–29]. Originating from the mitochondria or other sources, ROS/RNS generated during pathological events may include hydrogen peroxide, superoxide and peroxynitrite as well as hydroxyl, nitrite, and carbonate radicals among others, each of which has its own reactivity and specificity [30]. Radical scavengers and antioxidants that would normally prevent the accumulation of ROS/RNS in cells are often depleted in conditions of pathological oxidative stress, increasing the likelihood of interaction between oxidants and intracellular proteins like HGMB1. Extracellular HMGB1 can therefore reasonably be expected to have exposure to a range of ROS/RNS species and concentrations depending on the context of release.
A range of unexplored oxidative modifications on Cys-, Met-, and other residues are possible and may account for the inconsistency in functional consequences of oxidation. There are five possible oxidation states of the three Cys-residues (Cys-disulfide [31], sulfenic acid (Cys-SOH) [32], sulfinic acid (Cys-SO2H) [33], Cys-SO3H [32,34], S-nitroso-Cys [35,36]) meaning that there are thousands of possible proteoforms without considering the additional complexity that could arise from oxidation of the Met-residues [37,38] or oxidation and nitration of the seven tyrosine residues (Figure 3) [39,40].
Figure 3. . A great number (>1E3) of oxidized modification combinations are possible depending on the ROS/RNS species present and their respective concentrations.

ROS and RNS like hydrogen peroxide (H2O2), peroxynitrite (ONOO-), superoxide (O2•-) and other radical oxidants can induce formation of reversible and irreversible Cys-residue modifications [31–36].
Oxidative modifications have distinct stability, reversibility and cellular lifetime, which can all inform on the biological context in which each would be expected to occur. Exposure of a Cys-residue to ROS results first in formation of a Cys-SOH, and although protein Cys-SOHs have been detected in cells [41], they are readily further oxidized to Cys-SO2s [42] or spontaneously form a disulfide bond with a free sulfhydryl group [43]. Like disulfides, Cys-SOHs are readily converted to thiols by reducing agents such as dithiothreitol (DTT), which must be considered during sample preparation and analysis of oxidized proteins. In contrast, Cys-SO2Hs are resistant to non-enzymatic reduction [44] but are readily further oxidized to Cys-SO3H. Cys-SNO is formed when nitric oxide (NO), peroxynitrite (ONOO-) or other S-nitrosylating agent reacts with a free sulfhydryl to form this readily reversible nitrosylated residue [45,46]. Met-residues are also readily oxidized upon ROS exposure to give Met-sulfoxide (Met-SO). Similar to Cys-SOH, Met-SO can be enzymatically reduced as well as further oxidized to Met-sulfone (Met-SO2) [47]. Variation in the concentration of ROS/RNS and duration of intracellular exposure in different pathological conditions creates potential for a multitude of different HMGB1 proteoforms with a mix of Cys-, Met- and other amino acid oxidation states.
Oxidative modifications, in a similar manner to all PTMs, may have unique structural and functional consequences that affect extracellular binding relationships and inform the immunological response to secreted HMGB1. Cys-oxidation under non-pathological conditions can allow Cys-residues to act as regulatory redox switches, responding to the local redox environment and modifying protein function accordingly. The NF-E2-related factor 2 (Nrf2)/ Kelch-like ECH-associated protein 1 (Keap1) system, for instance, allows release of Nrf2 into the nucleus in response to oxidative stress via oxidation of three Cys-residues in Keap1, which under reducing conditions confines Nrf2 to the cytosol ready for ubiquitination and degradation [48]. Under both pathological and non-pathological conditions, Cys-residue oxidation can have steric and hydrophobicity effects that alter protein structure and therefore receptor binding. Protein oxidation and the subsequent structural changes are often associated with loss of function, as was proposed in the incumbent HMGB1 oxidation state function paradigm. However, protein oxidation can sometimes induce a gain-of-function. For instance, oxidation of the plasma protein α2-macroglobulin increases its binding affinity for secreted cytokines, encouraging resolution of inflammation following release of oxidants from neutrophils [49]. Terminal oxidation of Cys-residues to Cys-SO3Hs has traditionally been viewed as exclusively inactivating in literature reports, but there is a possibility for future discovery of immune-activating effects, especially considering its association with pathological oxidation. Met-oxidation similarly has potential to alter protein structure and often occurs in parallel to Cys-oxidation; oxidation of α2-macroglobulin, for example, involves not only Cys-oxidation but also oxidation of Met-residues to Met-SO [49]. Met-SO has a “stiffer” and more polar side chain than an unmodified Met-residue with a similar hydrophobicity to lysine, a positively charged amino acid [37]. Oxidation of Met-residues has traditionally been associated with protein inactivation much like Cys-SO3H, but evidence is growing for alternative functions [37]. For instance, oxidation of Met-45 of inhibitor of kappa B-alpha (IkBα) increases its resistance to protein-degradation, thereby enhancing its ability to inhibit transcription factor nuclear factor kappa B NFkB) [50]. Therefore, additional studies are required to determine precisely how terminal Cys-oxidative modifications and Met-oxidations alter HMGB1 structure and to assess the subsequent changes to extracellular receptor binding affinity.
Unreliable reports of HMGB1 & oxidized HMGB1 as biomarkers
Findings in many previous studies have encouraged interest in HMGB1's potential utility as a biomarker for a range of pathological conditions. Unfortunately, many of these studies were conducted using serum rather than plasma [51]. It was shown, using matched plasma and serum samples from twenty healthy control subjects, that HMGB1 is secreted when blood is allowed to clot to form serum [51]. Clotting resulted in a 30-fold increase in HMGB1 concentrations from 0.2 ng/ml in plasma to 6 ng/ml in serum. This would make it almost impossible to detect an increase in the release of HMGB1 from tissues into the plasma. Therefore, measurements in serum only reveal the capacity of blood cells to produce HMGB1 and do not reflect the circulating levels of HMGB1. Despite this fundamental problem, serum is still often used in biomarker studies, to quantify the HMGB1 that is secreted from tissues in different pathological conditions. Furthermore, much of the literature on HMGB1 and its oxidized forms as biomarkers is confused by retractions and temporary removal of papers [52–60] or expressions of concern about the data reported [31,61–70]. Studies that have monitored HMGB1 as a biomarker that must now be re-evaluated because of these issues include those on epilepsy [60], drug-induced liver injury [68], acetaminophen hepatotoxicity [61,65], asbestos exposure [67], and autophagy [69]. Studies that have specifically examined HMGB1 oxidation that require re-evaluation include, dietary regulation of acetaminophen toxicity [61], cytokine activity [63], macrophage activation syndrome [66], inflammasome activation [56], and neutrophil-mediated injury [65].
Current paradigm for oxidized HMGB1 as a mediator & a biomarker
It has been widely proposed that the oxidation state of HMGB1 and its extracellular function are linked in a trinary relationship through its three Cys-residues - Cys-23, Cys-45, and Cys-106 (Figure 4). This model suggests that HMGB1 Cys- residues could be fully reduced, oxidized with a disulfide between Cys-23 and Cys-45 with Cys-106 reduced, or terminally oxidized at all three Cys-residues [71]. The three oxidation states were proposed to correspond to chemokine functionality, cytokine functionality, and inactivity, respectively. Sequential release of each oxidized HMGB1 proteoform from injured cells as pathological oxidative stress progressed would therefore correspond to each stage of inflammation - leukocyte recruitment, leukocyte activation, and eventual resolution of inflammation (reviewed in Lu et al.) [72].
Figure 4. . Currently accepted paradigm for the effect of Cys-oxidation on the activity of HMGB1.

(A) Reduced HMGB1acts a chemokine through CXCR4. (B) HMGB1 with a thiol Cys-106 and a disulfide between Cys-23 and Cys-45 acts as a cytokine through RAGE, TRL's and TIM3. (C) Terminally oxidized HMGB1 is functionally inactive [71,72].
Apart from retracted or flagged publications, the accepted paradigm for HMGB1 oxidation-function was supported by the finding that under mild oxidizing conditions, disulfide bond formation occurs between Cys-23 and Cys-45, but Cys-106 remains in the reduced form, and that this bond formation regulates the function of HMGB1 in response to oxidation [73]. Additional support for the accepted paradigm was derived from the finding that caspase-mediated oxidation of HMGB1 at Cys-106 induced immunological tolerance in apoptotic cells, whereas formation of the Cys-23/Cys-45 disulfide did not affect the activity of HMGB1 in this setting [74]. In contrast, binding of histone H1 to DNA was modulated by formation of the Cys-23/Cys-45 disulfide [75].
However, oxidation of all three Cys-residues was found to improve TLR4 binding, cytokine release and immune responses [76]. In addition, oxidative stress elicited platelet/leukocyte inflammatory interactions through oxidative modifications to the three Cys-residues in HMGB1 [77]. Furthermore, LPS-induced kidney sepsis resulted in HMGB1 oxidation to the Cys-23/Cys-45 disulfide, which correlated with the ability of HMGB1 to induce inflammation [78]. Terminal oxidation of Cys-106 in HMGB1 to a cysteic acid (Cys-SO3H) was found to promote apoptosis and increase cell death upon exposure to chemotherapeutic agents [79]. It is noteworthy that mutation of Cys-106 to a serine residue inhibited its nuclear localization, ability to bind to TLR4, and inhibited cytokine release [80]. Yang et al. suggested that this provided evidence that Cys-oxidation to Cys-SO3H would have a similar effect [63]. However, there is an expression of concern related to this latter suggestion and no additional evidence has been obtained to date. Consequently, additional studies will be required to definitively establish how HMGB1 activity is modulated by such site-specific oxidations. Finally, analysis of HMGB1 proteoforms released in a cell-based model of APAP-overdose contradicted the currently accepted paradigm both in the types of oxidative modifications and their respective contexts of release [81]. Redox-injured cells released oxidized HMGB1 as well as fully reduced HMGB1, which was previously associated with release from activated immune cells (Figure 5A & B). Furthermore, two HMGB1 proteoforms not described in the previously accepted paradigm were detected: one with a mix of disulfide and terminally oxidized-Cys, the other with an intermolecular disulfide linkage at Cys-106 to an unidentified intracellular binding partner(s) (Figure 5C & D) [81]. Even without considering additional HMGB1 oxidized proteoforms, the proposed model of oxidation states and their relationship to function are clearly not as straightforward as suggested in numerous previous reports.
Figure 5. . Oxidized HMGB1 proteoforms detected by liquid chromatography-tandem mass spectrometry (LC–MS/MS) following treatment of hepatocarcinoma cells with high concentrations of acetaminophen typically found in overdose patients.

Oxidized HMGB1 proteoforms as context-specific biomarkers
Regardless of the abundance of oxidative modifications and their functional consequences, certain modifications might have the potential to serve as pathology-specific biomarkers. Extracellular HMGB1 can originate from activated immune cell secretions in addition to other pathological sources. The presence and quantity of extracellular HMGB1 are therefore more indicative of general inflammation than specific pathological states. Oxidized proteoforms of HMGB1, however, may only arise in certain pathological conditions, thereby allowing for the potential identification of pathology-specific biomarkers. They have previously been proposed as potential biomarkers and drug targets by Venereau et al. [82] and others. However, future studies will need to avoid building on ideas emanating from the substantial number publications that have been retracted or temporarily removed [52–60] or statements of concern issued [31,61–70].
There are many pathological conditions associated with oxidative stress that result in the release of extracellular HMGB1 and which may be suitable for developing an oxidized HMGB1-based biomarker. Myocardial ischemia and reperfusion injury, particularly as a function of myocardial infarction, is associated with oxidative stress in the form of excessive hydrogen peroxide, superoxide and peroxynitrite generation [25]. Interestingly, peroxynitrite induces HMGB1 release by cardiac cells in vitro and HMGB1 upregulation in the infarcted myocardium in vivo [83]. In addition, oxidized HMGB1 was found to play a role in doxorubicin-induced peroxynitrite-dependent myocardial apoptosis [84]. Drug-induced liver injury (DILI) is the most common cause of fatal liver failure [85] and is often a consequence of reactive quinone metabolites that lead to excessive generation of ROS and RNS, as is the case in acetaminophen overdose, the most common cause of DILI [85–87]. Oxidized HMGB1 is secreted during acetaminophen-induced oxidant stress and cell injury in cultured mouse hepatocytes [88]. Similarly, oxidized HMGB1 mediates hepatic injury after murine liver ischemia-reperfusion [89]. HMGB1 release induced by liver ischemia involves TLR4-dependent ROS production and calcium-mediated signaling [90]. Finally, sterile inflammation is thought to be mediated by HMGB1 proteoforms that are released from acetaminophen-injured hepatocytes and this activation can be prevented by heparin analogs that bind to HMGB1 [12]. In most cases, the precise oxidation state of HMGB1 derived from pathological conditions has not been unambiguously characterized, opening up the possibility that unique proteoforms are among the oxidized fractions. Future work characterizing the oxidation state of HMGB1's Cys- and Met-residues as well as other residues of interest such as tyrosine, might yield specific, predictive biomarkers.
The lack of unambiguous HMGB1 oxidized proteoform characterization in different pathological conditions can, at least in part, be attributed to the challenging analytical requirements. Specific antibodies for each Cys-oxidation state are not available and although anti-Met-SO antibodies have been produced and sold commercially, subsequent studies have shown that these antibodies are not specific [91]. Reactive, reversible modifications sensitive to the redox environment pose a challenge for sample preparation, requiring care that endogenous modifications are not lost, while ensuring that artifactual modifications are not created. Despite these challenges, technologies sufficient for robust characterization of oxidized proteoforms exist and may aid in the development of specific biomarkers (reviewed in [92]). Derivatization and subsequent analysis by western blotting, LC–MS/MS or other methods can provide oxidation state information. Readily reversible Cys-oxidation states such as disulfide linkages can be differentiated from free thiols with derivatization, followed by reduction and additional derivatization, which can then be analyzed by quantitative LC–MS/MS [81]. In addition, Cys-SOH, a reversible oxidative modification, can be detected using dimedone-based chemical probes [93]. Dimedone reacts selectively with the sulfur atom in Cys-SOH to form a stable thioether linkage that can be attached to biotin reporter tags and detected directly by LC–MS/MS [94]. Cys-SNO can similarly be detected using derivatization approaches such as the biotin switch technique, wherein free thiols are first blocked by a thiol-specific methanethiolation reagent such as methyl methanesulfonate, followed by ascorbate-mediated conversion of Cys-SNO groups back to Cys-residues and S-biotinylation of the Cys-free thiols [95]. Purification of the biotinylated proteins with streptavidin columns followed by western blotting with specific antibodies facilitates characterization of the proteins that originally had Cys-SNO residues [96]. Specific sites of Cys-CNO cysteine residues can be characterized by replacing the biotinylation step with derivatization using an isotope coded affinity tag (ICAT), followed by trypsin digestion, and LC–MS/MS analysis of the resulting tryptic peptides [97]. Modifications resistant to non-enzymatic reversal such as Cys-SO2H, Cys-SO3H, Met-SO, and Met-SO2 can be detected directly by LC–MS/MS [81].
Conclusion
The paradigm for the extracellular function and oxidation state relationship of HMGB1 lacks sufficient supporting data in light of numerous retractions, temporary removal of papers, and expressions of concern [31,52–70]. The precise oxidized HMGB1 proteoforms present in a variety of pathological and non-pathological conditions is unclear as many studies have failed to account for all of the potential proteoforms or limited their analysis to a binary “reduced” and “oxidized”. The previously accepted paradigm for oxidative structural changes in HMGB1 (Figure 4) may be far too simple and overlook many previously unexplored proteoforms (Figure 3)
Future perspective
It is now possible to characterize oxidized HMGB1 proteoforms with high specificity and sensitivity by the use of immunoaffinity purification coupled with efficient derivatization and stable isotope dilution LC–MS/MS [81]. This should permit the quantification of HMGB1 and novel oxidized proteoforms in plasma form different pathological conditions. Plasma should be used instead of serum because the preparation of serum causes a 30-fold increase in HMGB1 levels that would prevent the detection of any HMGB1 release from tissues into the plasma [51]. Potential pathological conditions where HMGB1 could be involved include: alcoholic liver disease [98], asbestos exposure and mesothelioma [67], pleural mesothelioma [99], acetaminophen hepatotoxicity [62], senescence [76], systemic lupus erythematosus [100], sepsis [101], type 1 and Type 2 diabetes mellitus [102], breast cancer [103], pancreatic cancer [104], squamous cell carcinoma [105], non-small-cell lung cancer [106], Alzheimer's disease [107], epilepsy [60], Parkinson's disease [108], atrial fibrillation [109], coronary artery disease [110], heart failure [111], and coronary artery stenosis [112]. Therefore, tools are now available to probe the full range of potential oxidized HMGB1 proteoforms, which should ultimately lead to the validation of useful specific biomarkers for different diseases as well as potential therapeutic targets [82] in the relevant diseases.
Executive summary.
HMGB1 is a dual-function protein that acts as a DNA-binding protein in the nucleus as well as a DAMP when released from activated immune cells or injured tissue.
Unreliable reports of HMGB1 & oxidized HMGB1 as biomarkers
The paradigm for the extracellular function and oxidation state relationship of HMGB1 lacks sufficient supporting data in light of numerous retractions, temporary removal of papers, and expressions of concern.
HMGB1 is secreted when blood is allowed to clot to make serum. Many studies have quantified HMGB1 in serum rather than plasma, so they do not reflect circulating levels but just the capacity for blood cells to secrete HMGB1.
The precise oxidative HMGB1 proteoforms present in a variety of pathological and non-pathological conditions is unclear.
Current paradigm for oxidized HMGB1 as a mediator & a biomarker
Many studies have failed to account for all the potential proteoforms or limited their analysis to a binary “reduced” and “oxidized” proteoform analysis.
In view of the large number of potential oxidative HMGB1 proteoforms, there is ample opportunity for discovery of many specific oxidized proteoforms that might serve as biomarkers for specific pathological states.
Unique oxidative modifications may additionally have implications for the structure and function of HMGB1 in the extracellular space as individual oxidized proteoforms could have different activities on receptors such as TLRs that mediate cytokine release.
Oxidized HMGB1 proteoforms as context-specific biomarkers
Activation of sterile inflammation, which is thought to be mediated by HMGB1 released from acetaminophen-injured hepatocytes, can be prevented by heparin analogs that bind to HMGB1 and by anti-HMGB1 antibodies.
A recent study of acetaminophen-mediated HMGB1 release from hepatocytes has identified previously unrecognized oxidized HMGB1 proteoforms.
Conclusion
There is ample opportunity for the discovery of numerous context-specific proteoforms that might serve as biomarkers for specific pathological states.
Unique oxidative modifications have implications for the structure and function of HMGB1 in the extracellular space.
The previously accepted paradigm for oxidative structural changes and HMGB1 may be far too simple and overlook many previously unexplored proteoforms.
Future directions
The development of highly specific and sensitive methodology based on the use of immunoaffinity purification coupled with efficient derivatization and stable isotope dilution LC–MS/MS should permit the characterization and quantification of novel oxidized HMGB1 proteoforms in plasma (not serum) obtained from different pathological conditions.
Tools are now available to probe the full range of potential oxidized HMGB1 proteoforms, which should ultimately lead to the validation of useful specific biomarkers for different diseases as well as potential therapeutic targets in those diseases.
Footnotes
Author contributions
All authors (R Pirnie, KP Gillespie, C Mesaros, IA Blair) conceived of the review article and contributed to the final text.
Financial & competing interests disclosure
This publication was made possible by funding from P30ES013508 (IA Blair and C Mesaros) and T32ES019851 (R Pirnie) from the National Institute of Environmental Health Sciences (NIEHS), NIH, DHHS. The authors have no other relevant affiliations or financial involvement with any organization or entity with a financial interest in or financial conflict with the subject matter or materials discussed in the manuscript apart from those disclosed
No writing assistance was utilized in the production of this manuscript.
Open access
This work is licensed under the Creative Commons Attribution 4.0 License. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/
References
Papers of special note have been highlighted as: • of interest; •• of considerable interest
- 1.Rauvala H, Pihlaskari R. Isolation and some characteristics of an adhesive factor of brain that enhances neurite outgrowth in central neurons. J. Biol. Chem. 262(34), 16625–16635 (1987). [PubMed] [Google Scholar]; • This was the first report of HMGB1 (originally called amphoterin – see Reference 2) as a 30 kDa protein in young rat brain with activity as a neurite growth factor. Since this seminal finding, numerous other activites have been identified including its ability to act as a DNA chaperone, regulate autophogy, and induce cytokine release through binding to numerous different extracellular receptors.
- 2.Merenmies J, Pihlaskari R, Laitinen J, Wartiovaara J, Rauvala H. 30-kDa heparin-binding protein of brain (amphoterin) involved in neurite outgrowth. Amino acid sequence and localization in the filopodia of the advancing plasma membrane. J. Biol. Chem. 266(25), 16722–16729 (1991). [PubMed] [Google Scholar]
- 3.Tsuda K, Kikuchi M, Mori K, Waga S, Yoshida M. Primary structure of non-histone protein HMG1 revealed by the nucleotide sequence. Biochemistry 27(16), 6159–6163 (1988). [DOI] [PubMed] [Google Scholar]
- 4.Lotze MT, Tracey KJ. High-mobility group box 1 protein (HMGB1): nuclear weapon in the immune arsenal. Nat Rev Immunol 5(4), 331–342 (2005). [DOI] [PubMed] [Google Scholar]
- 5.Bonaldi T, Langst G, Strohner R, Becker PB, Bianchi ME. The DNA chaperone HMGB1 facilitates ACF/CHRAC-dependent nucleosome sliding. EMBO J. 21(24), 6865–6873 (2002). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Jayaraman L, Moorthy NC, Murthy KG, Manley JL, Bustin M, Prives C. High mobility group protein-1 (HMG-1) is a unique activator of p53. Genes Dev. 12(4), 462–472 (1998). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Melvin VS, Edwards DP. Coregulatory proteins in steroid hormone receptor action: the role of chromatin high mobility group proteins HMG-1 and -2. Steroids 64(9), 576–586 (1999). [DOI] [PubMed] [Google Scholar]
- 8.Calogero S, Grassi F, Aguzzi A et al. The lack of chromosomal protein Hmg1 does not disrupt cell growth but causes lethal hypoglycaemia in newborn mice. Nat. Genet. 22(3), 276–280 (1999). [DOI] [PubMed] [Google Scholar]
- 9.Scaffidi P, Misteli T, Bianchi ME. Release of chromatin protein HMGB1 by necrotic cells triggers inflammation. Nature 418(6894), 191–195 (2002). [DOI] [PubMed] [Google Scholar]
- 10.Dumitriu IE, Bianchi ME, Bacci M, Manfredi AA, Rovere-Querini P. The secretion of HMGB1 is required for the migration of maturing dendritic cells. J. Leukoc. Biol. 81(1), 84–91 (2007). [DOI] [PubMed] [Google Scholar]
- 11.Bartholdi D, Schwab ME. Expression of pro-inflammatory cytokine and chemokine mRNA upon experimental spinal cord injury in mouse: an in situ hybridization study. Eur. J. Neurosci. 9(7), 1422–1438 (1997). [DOI] [PubMed] [Google Scholar]
- 12.Arnold K, Xu Y, Sparkenbaugh EM et al. Design of anti-inflammatory heparan sulfate to protect against acetaminophen-induced acute liver failure. Sci Transl Med 12(535), (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]; •• Provides compelling evidence for the contribution of HMGB1 to pathological inflammation is such that neutralizing anti-HMGB1 antibodies significantly decrease the lethality of injuries from acetaminophen-overdose. In addition, it introduced the concept of using heparin
- 13.Ito I, Fukazawa J, Yoshida M. Post-translational methylation of high mobility group box 1 (HMGB1) causes its cytoplasmic localization in neutrophils. J. Biol. Chem. 282(22), 16336–16344 (2007). [DOI] [PubMed] [Google Scholar]; • The study of Ito et al. indicates how a PTM could alter the intracellular localization of HMGB1. The study reported that methylation of lysine-43 caused the cytoplasmic localization of HMGB1 in neutrophils.
- 14.Gardella S, Andrei C, Ferrera D et al. The nuclear protein HMGB1 is secreted by monocytes via a non-classical, vesicle-mediated secretory pathway. EMBO Rep 3(10), 995–1001 (2002). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Bonaldi T, Talamo F, Scaffidi P et al. Monocytic cells hyperacetylate chromatin protein HMGB1 to redirect it towards secretion. EMBO J. 22(20), 5551–5560 (2003). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Youn JH, Shin JS. Nucleocytoplasmic shuttling of HMGB1 is regulated by phosphorylation that redirects it toward secretion. J. Immunol. 177(11), 7889–7897 (2006). [DOI] [PubMed] [Google Scholar]
- 17.Bell CW, Jiang W, Reich CF 3rd, Pisetsky DS. The extracellular release of HMGB1 during apoptotic cell death. Am J Physiol Cell Physiol 291(6), C1318–1325 (2006). [DOI] [PubMed] [Google Scholar]
- 18.Chen G, Li J, Ochani M et al. Bacterial endotoxin stimulates macrophages to release HMGB1 partly through CD14- and TNF-dependent mechanisms. J. Leukoc. Biol. 76(5), 994–1001 (2004). [DOI] [PubMed] [Google Scholar]
- 19.Hori O, Brett J, Slattery T et al. The receptor for advanced glycation end products (RAGE) is a cellular binding site for amphoterin. Mediation of neurite outgrowth and co-expression of rage and amphoterin in the developing nervous system. J. Biol. Chem. 270(43), 25752–25761 (1995). [DOI] [PubMed] [Google Scholar]
- 20.Park JS, Svetkauskaite D, He Q et al. Involvement of toll-like receptors 2 and 4 in cellular activation by high mobility group box 1 protein. J. Biol. Chem. 279(9), 7370–7377 (2004). [DOI] [PubMed] [Google Scholar]
- 21.Yu M, Wang H, Ding A et al. HMGB1 signals through toll-like receptor (TLR) 4 and TLR2. Shock 26(2), 174–179 (2006). [DOI] [PubMed] [Google Scholar]
- 22.Park JS, Gamboni-Robertson F, He Q et al. High mobility group box 1 protein interacts with multiple Toll-like receptors. Am J Physiol Cell Physiol 290(3), C917–924 (2006). [DOI] [PubMed] [Google Scholar]
- 23.Schiraldi M, Raucci A, Munoz LM et al. HMGB1 promotes recruitment of inflammatory cells to damaged tissues by forming a complex with CXCL12 and signaling via CXCR4. J. Exp. Med. 209(3), 551–563 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Hanawa N, Shinohara M, Saberi B, Gaarde WA, Han D, Kaplowitz N. Role of JNK translocation to mitochondria leading to inhibition of mitochondria bioenergetics in acetaminophen-induced liver injury. J. Biol. Chem. 283(20), 13565–13577 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Levraut J, Iwase H, Shao ZH, Vanden Hoek TL, Schumacker PT. Cell death during ischemia: relationship to mitochondrial depolarization and ROS generation. Am J Physiol Heart Circ Physiol 284(2), H549–558 (2003). [DOI] [PubMed] [Google Scholar]
- 26.Martins PS, Kallas EG, Neto MC, Dalboni MA, Blecher S, Salomao R. Upregulation of reactive oxygen species generation and phagocytosis, and increased apoptosis in human neutrophils during severe sepsis and septic shock. Shock 20(3), 208–212 (2003). [DOI] [PubMed] [Google Scholar]
- 27.Martin-Murphy BV, Holt MP, Ju C. The role of damage associated molecular pattern molecules in acetaminophen-induced liver injury in mice. Toxicol. Lett. 192(3), 387–394 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Xu H, Yao Y, Su Z et al. Endogenous HMGB1 contributes to ischemia-reperfusion-induced myocardial apoptosis by potentiating the effect of TNF-α/JNK. Am J Physiol Heart Circ Physiol 300(3), H913–921 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Wang H, Liao H, Ochani M et al. Cholinergic agonists inhibit HMGB1 release and improve survival in experimental sepsis. Nat. Med. 10(11), 1216–1221 (2004). [DOI] [PubMed] [Google Scholar]
- 30.Di Meo S, Reed TT, Venditti P, Victor VM. Role of ROS and RNS Sources in Physiological and Pathological Conditions. Oxid Med Cell Longev 2016, 1245049 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Lindahl M, Mata-Cabana A, Kieselbach T. The disulfide proteome and other reactive cysteine proteomes: analysis and functional significance. Antioxid Redox Signal 14(12), 2581–2642 (2011). [DOI] [PubMed] [Google Scholar]
- 32.Iwata H, Yamagami S, Baba A. Cysteine sulfinic acid in the central nervous system: specific binding of [35S]cysteic acid to cortical synaptic membranes–an investigation of possible binding sites for cysteine sulfinic acid. J. Neurochem. 38(5), 1275–1279 (1982). [DOI] [PubMed] [Google Scholar]
- 33.Chang YC, Huang CN, Lin CH, Chang HC, Wu CC. Mapping protein cysteine sulfonic acid modifications with specific enrichment and mass spectrometry: an integrated approach to explore the cysteine oxidation. Proteomics 10(16), 2961–2971 (2010). [DOI] [PubMed] [Google Scholar]
- 34.Manneberg M, Lahm HW, Fountoulakis M. Quantification of cysteine residues following oxidation to cysteic acid in the presence of sodium azide. Anal. Biochem. 231(2), 349–353 (1995). [DOI] [PubMed] [Google Scholar]
- 35.Marino SM, Gladyshev VN. Structural analysis of cysteine S-nitrosylation: a modified acid-based motif and the emerging role of trans-nitrosylation. J. Mol. Biol. 395(4), 844–859 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Gould N, Doulias PT, Tenopoulou M, Raju K, Ischiropoulos H. Regulation of protein function and signaling by reversible cysteine S-nitrosylation. J. Biol. Chem. 288(37), 26473–26479 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Hoshi T, Heinemann S. Regulation of cell function by methionine oxidation and reduction. J. Physiol. 531(Pt 1), 1–11 (2001). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Drazic A, Winter J. The physiological role of reversible methionine oxidation. Biochim. Biophys. Acta 1844(8), 1367–1382 (2014). [DOI] [PubMed] [Google Scholar]
- 39.Souza JM, Daikhin E, Yudkoff M, Raman CS, Ischiropoulos H. Factors determining the selectivity of protein tyrosine nitration. Arch. Biochem. Biophys. 371(2), 169–178 (1999). [DOI] [PubMed] [Google Scholar]
- 40.Ischiropoulos H. Biological selectivity and functional aspects of protein tyrosine nitration. Biochem. Biophys. Res. Commun. 305(3), 776–783 (2003). [DOI] [PubMed] [Google Scholar]
- 41.Seo YH, Carroll KS. Profiling protein thiol oxidation in tumor cells using sulfenic acid-specific antibodies. Proc Natl Acad Sci U S A 106(38), 16163–16168 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Woo HA, Chae HZ, Hwang SC et al. Reversing the inactivation of peroxiredoxins caused by cysteine sulfinic acid formation. Science 300(5619), 653–656 (2003). [DOI] [PubMed] [Google Scholar]
- 43.Rehder DS, Borges CR. Cysteine sulfenic acid as an intermediate in disulfide bond formation and nonenzymatic protein folding. Biochemistry 49(35), 7748–7755 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Broniowska KA, Hogg N. The chemical biology of S-nitrosothiols. Antioxid Redox Signal 17(7), 969–980 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Lowther WT, Brot N, Weissbach H, Matthews BW. Structure and mechanism of peptide methionine sulfoxide reductase, an “anti-oxidation” enzyme. Biochemistry 39(44), 13307–13312 (2000). [DOI] [PubMed] [Google Scholar]
- 46.Jaffrey SR, Erdjument-Bromage H, Ferris CD, Tempst P, Snyder SH. Protein S-nitrosylation: a physiological signal for neuronal nitric oxide. Nat Cell Biol 3(2), 193–197 (2001). [DOI] [PubMed] [Google Scholar]
- 47.Wu SM, Patel DD, Pizzo SV. Oxidized alpha2-macroglobulin (alpha2M) differentially regulates receptor binding by cytokines/growth factors: implications for tissue injury and repair mechanisms in inflammation. J. Immunol. 161(8), 4356–4365 (1998). [PubMed] [Google Scholar]
- 48.Saito R, Suzuki T, Hiramoto K et al. Characterizations of Three Major Cysteine Sensors of Keap1 in Stress Response. Mol. Cell. Biol. 36(2), 271–284 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Reddy VY, Desorchers PE, Pizzo SV et al. Oxidative dissociation of human alpha 2-macroglobulin tetramers into dysfunctional dimers. J. Biol. Chem. 269(6), 4683–4691 (1994). [PubMed] [Google Scholar]
- 50.Midwinter RG, Cheah FC, Moskovitz J, Vissers MC, Winterbourn CC. IkappaB is a sensitive target for oxidation by cell-permeable chloramines: inhibition of NF-kappaB activity by glycine chloramine through methionine oxidation. Biochem. J. 396(1), 71–78 (2006). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Weng L, Guo L, Vachani A, Mesaros C, Blair IA. Quantification of Serum High Mobility Group Box 1 by Liquid Chromatography/High-Resolution Mass Spectrometry: Implications for Its Role in Immunity, Inflammation, and Cancer. Anal. Chem. 90(12), 7552–7560 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]; •• Demonstrates that HMGB1 secreted from human blood samples during clotting was not acetylated, which was previously proposed as a modification required for secretion of HMGB1 into the extracellular environment. This finding called into question the relationship between different HMGB1 PTMs and their effects on protein localization and function and highlighted the need to use plasma rather than serum for HMGB1 determinations.
- 52.Hui CK, Cheung WW, Leung KW et al. Retracted: outcome and immune reconstitution of HBV-specific immunity in patients with reactivation of occult HBV infection after alemtuzumab-containing chemotherapy regimen. Hepatology 48(2), 1–10 (2008). [DOI] [PubMed] [Google Scholar]
- 53.Flores MBS, Rocha GZ, Damas-Souza DM et al. RETRACTED: obesity-induced increase in tumor necrosis factor-alpha leads to development of colon cancer in mice. Gastroenterology 143(3), 741–753 e744 (2012). [DOI] [PubMed] [Google Scholar]
- 54.Yang X, Wang H, Zhang M, Liu J, Lv B, Chen F. RETRACTED: HMGB1: a novel protein that induced platelets active and aggregation via Toll-like receptor-4, NF-kappaB and cGMP dependent mechanisms. Diagn Pathol 10, 134 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 55.Zhang J, Zhu JS, Zhou Z, Chen WX, Chen NW. RETRACTED: inhibitory effects of ethyl pyruvate administration on human gastric cancer growth via regulation of the HMGB1-RAGE and Akt pathways in vitro and in vivo. Oncol Rep 36(6), 3716 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Sun Q, Loughran P, Shapiro RA et al. RETRACTED: redox‐dependent regulation of hepatocyte absent in melanoma 2 inflammasome activation in sterile liver injury in mice. Hepatology 65(1), 253–268 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Su Z, Ni P, She P et al. RETRACTED: bio-HMGB1 from breast cancer contributes to M-MDSC differentiation from bone marrow progenitor cells and facilitates conversion of monocytes into MDSC-like cells. Cancer Immunol. Immunother. 66(3), 391–401 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 58.Francis B, Clarke JI, Walker LE et al. TEMPORARY REMOVAL: reference intervals for putative biomarkers of drug-induced liver injury and liver regeneration in healthy human volunteers. J. Hepatol. (2018). [DOI] [PubMed] [Google Scholar]
- 59.Palmblad K, Schierbeck H, Sundberg E et al. Retraction Note to: high systemic levels of the cytokine-inducing HMGB1 isoform secreted in severe macrophage activation syndrome. Mol. Med. 26(1), 131 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Walker LE, Frigerio F, Ravizza T et al. RETRACTED: molecular isoforms of high-mobility group box 1 are mechanistic biomarkers for epilepsy. J. Clinical Investigation 129(5), 2166–2166 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Antoine DJ, Williams DP, Kipar A, Laverty H, Park BK. RETRACTED: diet restriction inhibits apoptosis and HMGB1 oxidation and promotes inflammatory cell recruitment during acetaminophen hepatoxicity. Mol. Med. 16(11-12), 479–490 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 62.Antoine DJ, Jenkins RE, Dear JW et al. RETRACTED: molecular forms of HMGB1 and keratin-18 as mechanistic biomarkers for mode of cell death and prognosis during clinical acetaminophen hepatotoxicity. J. Hepatol. 56(5), 1070–1079 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 63.Yang H, Lundback P, Ottosson L et al. RETRACTION: redox modification of cysteine residues regulates the cytokine activity of high mobility group box-1 (HMGB1). Mol. Med. 18, 250–259 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 64.Andersson U, Antoine DJ, Tracey KJ. EXPRESSION OF CONCERN: the functions of HMGB1 depend on molecular localization and post-translational modifications. J. Intern. Med. 276(5), 420–424 (2014). [DOI] [PubMed] [Google Scholar]
- 65.Huebener P, Pradere JP, Hernandez C et al. EXPRESSION OF CONCERN: the HMGB1/RAGE axis triggers neutrophil-mediated injury amplification following necrosis. J. Clin. Invest. 125(2), 539–550 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Palmblad K, Schierbeck H, Sundberg E et al. EXPRESSION OF CONCERN: high systemic levels of the cytokine-inducing HMGB1 isoform secreted in severe macrophage activation syndrome. Mol. Med. 20, 538–547 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 67.Napolitano A, Antoine DJ, Pellegrini L et al. EXPRESSION OF CONCERN: HMGB1 and Its Hyperacetylated Isoform are Sensitive and Specific Serum Biomarkers to Detect Asbestos Exposure and to Identify Mesothelioma Patients. Clin Cancer Res 22(12), 3087–3096 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Hernandez C, Huebener P, Pradere JP, Antoine DJ, Friedman RA, Schwabe RF. EXPRESSION OF CONCERN; HMGB1 links chronic liver injury to progenitor responses and hepatocarcinogenesis. J. Clin. Invest. 128(6), 2436–2451 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Khambu B, Huda N, Chen X et al. EXPRESSION OF CONCERN: HMGB1 promotes ductular reaction and tumorigenesis in autophagy-deficient livers. J. Clin. Invest. 128(6), 2419–2435 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Yang H, Wang H, Wang Y et al. EXPRESSION OF CONCERN: the haptoglobin beta subunit sequesters HMGB1 toxicity in sterile and infectious inflammation. J. Intern. Med. 286(1), 115 (2019). [DOI] [PubMed] [Google Scholar]
- 71.Antoine DJ, Harris HE, Andersson U, Tracey KJ, Bianchi ME. A systematic nomenclature for the redox states of high mobility group box (HMGB) proteins. Mol. Med. 20, 135–137 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Lu B, Wang C, Wang M et al. Molecular mechanism and therapeutic modulation of high mobility group box 1 release and action: an updated review. Expert Rev Clin Immunol 10(6), 713–727 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Hoppe G, Talcott KE, Bhattacharya SK, Crabb JW, Sears JE. Molecular basis for the redox control of nuclear transport of the structural chromatin protein Hmgb1. Exp. Cell Res. 312(18), 3526–3538 (2006). [DOI] [PubMed] [Google Scholar]
- 74.Kazama H, Ricci JE, Herndon JM, Hoppe G, Green DR, Ferguson TA. Induction of immunological tolerance by apoptotic cells requires caspase-dependent oxidation of high-mobility group box-1 protein. Immunity 29(1), 21–32 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Polanska E, Pospisilova S, Stros M. Binding of histone H1 to DNA is differentially modulated by redox state of HMGB1. PLOS ONE 9(2), e89070 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Davalos AR, Kawahara M, Malhotra GK et al. p53-dependent release of Alarmin HMGB1 is a central mediator of senescent phenotypes. J. Cell Biol. 201(4), 613–629 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]; •• Davalos et. al. reported that oxidation of all three HMGB1 cysteine residues improved binding to TLR4, cytokine release and immune responses. These findings directly contradicted accepted models of HMGB1 oxidation-function.
- 77.Maugeri N, Rovere-Querini P, Baldini M et al. Oxidative stress elicits platelet/leukocyte inflammatory interactions via HMGB1: a candidate for microvessel injury in sytemic sclerosis. Antioxid Redox Signal 20(7), 1060–1074 (2014). [DOI] [PubMed] [Google Scholar]
- 78.Abdulmahdi W, Patel D, Rabadi MM et al. HMGB1 redox during sepsis. Redox Biol 13, 600–607 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Tang D, Kang R, Cheh CW et al. HMGB1 release and redox regulates autophagy and apoptosis in cancer cells. Oncogene 29(38), 5299–5310 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]; • This study showed that oxidation of HMGB1 cysteine residues to cysteic acids promotes apoptosis and enhances the effect of chemotherapeutic agents. These findings directly contradicted accepted models of HMGB1 oxidation-function that described HMGB1with cysteine residues oxidized to cysteic acids as functionally inert.
- 80.Yang H, Hreggvidsdottir HS, Palmblad K et al. A critical cysteine is required for HMGB1 binding to Toll-like receptor 4 and activation of macrophage cytokine release. Proc Natl Acad Sci U S A 107(26), 11942–11947 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]; • Reports the important finding that mutation of Cys-106 to a serine residue inhibited its nuclear localization, ability to bind to TLR4, and caused cytokine release. Unfortunately, it led to the led to the unlikely concept by other investigators that conversion of cysteine to a cysteic acid residue would have similar effect.
- 81.Pirnie R, Gillespie KP, Weng L, Mesaros C, Blair IA. Characterization and Quantification of Oxidized High Mobility Group Box 1 Proteoforms Secreted from Hepatocytes by Toxic Levels of Acetaminophen. Chem. Res. Toxicol. 35(10), 1893–1902 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]; •• Pirnie et al. described a pattern of HMGB1 oxidative modification in a cell-based model of oxidative pathology that directly contradicts the previously accepted paradigm of HMGB1 oxidation-function relationship. Novel HMGB1 proteoforms were detected while previously described proteoforms were not, calling into question the proposed specific immune-effector role of those previously described proteoforms.
- 82.Venereau E, De Leo F, Mezzapelle R, Careccia G, Musco G, Bianchi ME. HMGB1 as biomarker and drug target. Pharmacol. Res. 111, 534–544 (2016). [DOI] [PubMed] [Google Scholar]
- 83.Loukili N, Rosenblatt-Velin N, Li J et al. Peroxynitrite induces HMGB1 release by cardiac cells in vitro and HMGB1 upregulation in the infarcted myocardium in vivo. Cardiovasc. Res. 89(3), 586–594 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Yao Y, Xu X, Zhang G, Zhang Y, Qian W, Rui T. Role of HMGB1 in doxorubicin-induced myocardial apoptosis and its regulation pathway. Basic Res. Cardiol. 107(3), 267 (2012). [DOI] [PubMed] [Google Scholar]
- 85.Ostapowicz G, Fontana RJ, Schiodt FV et al. Results of a prospective study of acute liver failure at 17 tertiary care centers in the United States. Ann. Intern. Med. 137(12), 947–954 (2002). [DOI] [PubMed] [Google Scholar]
- 86.Meyers LL, Beierschmitt WP, Khairallah EA, Cohen SD. Acetaminophen-induced inhibition of hepatic mitochondrial respiration in mice. Toxicol. Appl. Pharmacol. 93(3), 378–387 (1988). [DOI] [PubMed] [Google Scholar]
- 87.Bajt ML, Knight TR, Lemasters JJ, Jaeschke H. Acetaminophen-induced oxidant stress and cell injury in cultured mouse hepatocytes: protection by N-acetyl cysteine. Toxicol Sci 80(2), 343–349 (2004). [DOI] [PubMed] [Google Scholar]
- 88.Tsung A, Sahai R, Tanaka H et al. The nuclear factor HMGB1 mediates hepatic injury after murine liver ischemia-reperfusion. J. Exp. Med. 201(7), 1135–1143 (2005). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Tsung A, Klune JR, Zhang X et al. HMGB1 release induced by liver ischemia involves Toll-like receptor 4 dependent reactive oxygen species production and calcium-mediated signaling. J. Exp. Med. 204(12), 2913–2923 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Dragomir AC, Laskin JD, Laskin DL. Macrophage activation by factors released from acetaminophen-injured hepatocytes: potential role of HMGB1. Toxicol. Appl. Pharmacol. 253(3), 170–177 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Oien DB, Canello T, Gabizon R et al. Detection of oxidized methionine in selected proteins, cellular extracts and blood serums by novel anti-methionine sulfoxide antibodies. Arch. Biochem. Biophys. 485(1), 35–40 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.West JD. Experimental Approaches for Investigating Disulfide-Based Redox Relays in Cells. Chem. Res. Toxicol. 35(10), 1676–1689 (2022). [DOI] [PubMed] [Google Scholar]
- 93.Nelson KJ, Klomsiri C, Codreanu SG et al. Use of dimedone-based chemical probes for sulfenic acid detection methods to visualize and identify labeled proteins. Methods Enzymol. 473, 95–115 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Charles RL, Schroder E, May G et al. Protein sulfenation as a redox sensor: proteomics studies using a novel biotinylated dimedone analogue. Mol Cell Proteomics 6(9), 1473–1484 (2007). [DOI] [PubMed] [Google Scholar]
- 95.Jaffrey SR, Snyder SH. The biotin switch method for the detection of S-nitrosylated proteins. Science's STKE 2001(86), pl1–pl1 (2001). [DOI] [PubMed] [Google Scholar]
- 96.Forrester MT, Foster MW, Benhar M, Stamler JS. Detection of protein S-nitrosylation with the biotin-switch technique. Free Radic. Biol. Med. 46(2), 119–126 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Wu C, Liu T, Wang Y et al. Biotin Switch Processing and Mass Spectrometry Analysis of S-Nitrosated Thioredoxin and Its Transnitrosation Targets. Methods Mol. Biol. 1747, 253–266 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Ge X, Antoine DJ, Lu Y et al. High mobility group box-1 (HMGB1) participates in the pathogenesis of alcoholic liver disease (ALD). J. Biol. Chem. 289(33), 22672–22691 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Tabata C, Shibata E, Tabata R et al. Serum HMGB1 as a prognostic marker for malignant pleural mesothelioma. BMC Cancer 13, 205 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Abdulahad DA, Westra J, Bijzet J, Limburg PC, Kallenberg CG, Bijl M. High mobility group box 1 (HMGB1) and anti-HMGB1 antibodies and their relation to disease characteristics in systemic lupus erythematosus. Arthritis Res Ther 13(3), R71 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Karlsson S, Pettila V, Tenhunen J, Laru-Sompa R, Hynninen M, Ruokonen E. HMGB1 as a predictor of organ dysfunction and outcome in patients with severe sepsis. Intensive Care Med. 34(6), 1046–1053 (2008). [DOI] [PubMed] [Google Scholar]
- 102.Skrha J Jr, Kalousova M, Svarcova J et al. Relationship of soluble RAGE and RAGE ligands HMGB1 and EN-RAGE to endothelial dysfunction in type 1 and type 2 diabetes mellitus. Exp. Clin. Endocrinol. Diabetes 120(5), 277–281 (2012). [DOI] [PubMed] [Google Scholar]
- 103.Sun S, Zhang W, Cui Z et al. High mobility group box-1 and its clinical value in breast cancer. Onco Targets Ther 8, 413–419 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Wittwer C, Boeck S, Heinemann V et al. Circulating nucleosomes and immunogenic cell death markers HMGB1, sRAGE and DNAse in patients with advanced pancreatic cancer undergoing chemotherapy. Int. J. Cancer 133(11), 2619–2630 (2013). [DOI] [PubMed] [Google Scholar]
- 105.Qiu G, Li Y, Liu Z, Wang M, Ge J, Bai X. Clinical value of serum HMGB1 in diagnosis and prognosis of laryngeal squamous cell carcinoma. Med. Oncol. 31(12), 316 (2014). [DOI] [PubMed] [Google Scholar]
- 106.Niki M, Nakaya A, Kurata T et al. Immune checkpoint inhibitor re-challenge in patients with advanced non-small cell lung cancer. Oncotarget 9(64), 32298–32304 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Festoff BW, Sajja RK, van Dreden P, Cucullo L. HMGB1 and thrombin mediate the blood-brain barrier dysfunction acting as biomarkers of neuroinflammation and progression to neurodegeneration in Alzheimer's disease. J Neuroinflammation 13(1), 194 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Santoro M, Maetzler W, Stathakos P et al. In-vivo evidence that high mobility group box 1 exerts deleterious effects in the 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine model and Parkinson's disease which can be attenuated by glycyrrhizin. Neurobiol Dis 91, 59–68 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Wu Y, Zhang K, Zhao L, Guo J, Hu X, Chen Z. Increased serum HMGB1 is related to oxidative stress in patients with atrial fibrillation. J. Int. Med. Res. 41(6), 1796–1802 (2013). [DOI] [PubMed] [Google Scholar]
- 110.Yan XX, Lu L, Peng WH et al. Increased serum HMGB1 level is associated with coronary artery disease in nondiabetic and type 2 diabetic patients. Atherosclerosis 205(2), 544–548 (2009). [DOI] [PubMed] [Google Scholar]
- 111.Wang LJ, Lu L, Zhang FR, Chen QJ, De Caterina R, Shen WF. Increased serum high-mobility group box-1 and cleaved receptor for advanced glycation endproducts levels and decreased endogenous secretory receptor for advanced glycation endproducts levels in diabetic and non-diabetic patients with heart failure. Eur J Heart Fail 13(4), 440–449 (2011). [DOI] [PubMed] [Google Scholar]
- 112.Hu X, Jiang H, Bai Q et al. Increased serum HMGB1 is related to the severity of coronary artery stenosis. Clin. Chim. Acta 406(1-2), 139–142 (2009). [DOI] [PubMed] [Google Scholar]