

Abstract
The Zinc-Activated Channel (ZAC) is an atypical member of the Cys-loop receptor (CLR) superfamily of pentameric ligand-gated ion channels, with its very different endogenous agonists and signalling properties. In this study, a compound library screening at ZAC resulted in the identification of 2-(5-bromo-2-chlorobenzamido)-4-methylthiazole-5-methyl ester (1) as a novel ZAC antagonist. The structural determinants for ZAC activity in 1 were investigated by functional characterization of 61 analogs at ZAC expressed in Xenopus oocytes by two-electrode voltage clamp electrophysiology, and couple of analogs exerting more potent ZAC inhibition than 1 were identified (IC50 values: 1–3 μM). 1 and N-(4-(tert-butyl)thiazol-2-yl)-3-fluorobenzamide (5a, TTFB) were next applied in studies of the functional properties and the mode of action of this novel class of ZAC antagonists. TTFB was a roughly equipotent antagonist of Zn+- and H+ -evoked ZAC signaling and of spontaneous ZAC activity, and the slow on-set of its channel block suggested that its ZAC inhibition is state-dependent. TTFB was found to be a selective ZAC antagonist, exhibiting no significant agonist, antagonist or modulatory activity at 5-HT3A, α3β4 nicotinic acetylcholine, α1β2γ2s GABAA or α1 glycine receptors at 30 μM. 1 displayed largely noncompetitive antagonism of Zn2+-induced ZAC signalling, and TTFB was demonstrated to target the transmembrane and/or intracellular domains of the receptor, which collectively suggests that the N-(thiazol-2-yl)-benzamide analog acts a negative allosteric modulator of ZAC. We propose that this first class of selective ZAC antagonists could constitute useful pharmacological tools in future explorations of the presently poorly elucidated physiological functions governed by this CLR.
Keywords: Cys-loop receptor (CLR), Pentameric ligand-gated ion channel (pLGIC), Zinc-Activated Channel (ZAC), N-(thiazol-2-yl)-benzamide analogs, Negative allosteric modulator (NAM), State-dependent inhibition
1. Introduction
The pentameric ligand-gated ion channels in the Cys-loop receptor (CLR) superfamily are mediators of the fast signalling mediated by the classical neurotransmitters acetylcholine (ACh), serotonin (5-hydroxy-tryptamine, 5-HT), γ-aminobutyric acid (GABA) and glycine [1], The nicotinic ACh, GABAA, 5-HT3 and glycine receptors (nAChRs, GABAARs, 5-HT3Rs and GlyRs, respectively) exert and regulate a plethora of physiological functions in the central nervous system and in the periphery, and the receptors are targeted by drugs currently used to treat insomnia/sleep disorders, anxiety, epilepsy, nicotine addiction, muscle spasms and nausea/emesis and are being pursued as putative therapeutic targets for numerous other indications [2–8]. The membrane-embedded pentameric CLR complex is composed of an extracellular domain (ECD) made up by the N-termini of the five subunits, a transmembrane domain (TMD) consisting of the four membrane-spanning α-helices from each of the subunits, and an intracellular domain (ICD) made up by the second intracellular loops of the five subunits [9–13]. Signal transduction through the classical CLR is initiated by agonist binding to orthosteric sites in subunit interfaces in the ECD, which triggers opening of and ion flux through the ion channel in the transmembrane domain (TMD) until the receptor undergoes deactivation or desensitization [12–14]. These conformational transitions of the CLR are highly sensitive to ligand binding to allosteric sites in the pentameric complex, and positive and negative allosteric modulators (PAMs and NAMs, respectively) figure prominently as pharmacological tools and therapeutics for the classical CLRs [4,15–20].
The Zinc-Activated Channel (ZAC) is in many ways an atypical CLR [21,22]. ZAC constitutes the fifth branch of the phylogenetic tree over mammalian CLRs and exhibits low amino acid sequence homology with other CLR subunits [21]. When expressed in HEK293 and COS-7 cells, ZAC forms homomeric cation-selective channels activated by zinc (Zn2+), copper (Cu2+) and protons (H+), with the divalent cations Ca2+ and Mg2+ inhibiting channel signalling [21,22]. In addition to the vastly different sizes and physico-chemical properties of the identified ZAC agonists compared to the classical CLR neurotransmitters, the signaling properties of the recombinant ZAC are also very distinct from those displayed by the classical CLRs, with ZAC exhibiting considerable levels of spontaneous activity and very slow desensitization kinetics in patch-clamp recordings from HEK293 and COS-7 cells [21,22]. Albeit an atypical CLR, the existence of a ligand-gated ion channel acting as an in vivo sensor of fluctuations in endogenous zinc, copper and/or pH levels does not seem unlikely. However, in contrast to the detailed insight into in vivo functions of the classical CLRs obtained through decades of research, very little is known about the physiological roles governed by ZAC. Whereas the human ZACN gene is conserved in most other mammalian species, orthologous genes are absent from the rat and mouse genomes, which in part explains the few studies of the physiological functions of the receptor performed to date. ZAC mRNA has been detected in fetal and adult human brain and in pancreas, placenta, prostate, thyroid, trachea and stomach tissues [21,23]. Recently, ZAC has been proposed to be expressed in thymus and lymph organs and to be important for T-cell division [24], but presently data to support this or any other claims for physiological functions of the receptor are very sparse.
Explorations into the in vivo functions governed by ZAC and the therapeutic potential in the receptor would be facilitated by the availability of potent and selective pharmacological tools. The curare alkaloid tubocurarine (TC) has been shown to inhibit ZAC signalling with low-micromolar IC50 values [21,25], but the fact that TC is a promiscuous antagonist displaying comparable or even higher antagonist potencies at several other CLRs makes it unsuited as a pharmacological tool for ZAC [26,27]. A recent patent application has reported that a wide range of structurally diverse ligands (including ATP, dye compounds, heparin and tricyclic antidepressants) modulate ZAC signalling at mid-micromolar to millimolar concentration ranges [24]. While this suggests that ZAC analogously to other CLRs is susceptible to allosteric modulation, the moderately potent ZAC modulation and the potent off-target activities that characterize all of these compounds also render them inapplicable as pharmacological tools [24]. In the present work, we have attempted to remedy this lack of pharmacological tools by a search for novel modulators of ZAC based on a screening of a small compound library at the receptor. We report the discovery of the first class of selective ZAC antagonists and present the functional properties of and the mode of action for their receptor inhibition.
2. Materials and methods
2.1. Materials
ZnCl2, CuCl2, 5-HT, GABA, (S)-nicotine, glycine and all chemicals for buffers were purchased from Sigma-Aldrich (St. Louis, MO), and culture medium, serum, antibiotics and trypsin for cell culture were obtained from Invitrogen (Paisley, UK). The 1,680 compound-library (the same library as applied in a previous study [28]) was purchased from Chembridge Corporation (San Diego, CA), and analogs of the identified screening hit (compound 1) were obtained from Chembridge Corporation, Enamine (Kiev, Ukraine) and ChemDiv (San Diego, CA). The PolyFect transfection reagent and the FLIPR Membrane Potential Red (FMP) assay kit were obtained from Qiagen (Hilden, Germany) and Molecular Devices (Crawley, UK), respectively. Defolliculated stage V-VI oocytes harvested from female Xenopus laevis frogs were obtained from Lohmann Research Equipment (Castrop-Rauxel, Germany). The construction of cDNAs for wild-type (WT) human ZAC (ZACThl128 [25]) and other CLR subunits (ZAC-pCIneo, ZAC-pUNIV, m5-HT3A-pUNIV, hαl-GlyR-pUNIV, hαl -GABAAR-pCDNA3.1 hβ2-GABAAR-pCDNA3.1, hγ2S-GABAAR-pCDNA3.1, hα3-nAChR-pCDNA3.1 and hβ4-nAChR-pCDNA3.1) and for chimeric subunits (m5-HT3A/ZAC-pUNIV and ZAC/hα1-Gly-pUNIV) have been described previously (the prefixes “h”, “m” and “r” denote himian, mouse and rat, respectively) [25,29–31]. The HEK293, h5-HT3A-HEK293 and rα3β4-nAChR-HEK293 cell lines were generous gifts from Drs. Jakob L. Hansen, Jan Egebjerg and Ken J. Kellar, respectively, and the construction of the stable HEK293 cell line expressing the human excitatory amino acid transporter subtype 3 (hEAAT3) has been described previously [32].
2.2. Cell culture and construction of the ZAC-HEK293 cell line
HEK293 cells were propagated in culture medium [Dulbecco’s Modified Eagle Medium Glutamax™-I supplemented with penicillin (100 U/mL), streptomycin (100 μg/mL) and 5% dialyzed fetal bovine serum] in a humidified atmosphere at 37 °C and 5% CO2. The ZAC-HEK293, h5-HT3A-HEK293 and rα3β4-nAChR-HEK293, and hEAAT3-HEK293 cell lines were grown in the same culture medium supplemented with 1 mg/mL G-418. For the construction of the stable monoclonal ZAC-HEK293 cell line, 5 × 105 HEK293 cells were seeded in a 10 cm tissue culture dish, and 24 h later the cells were transfected with a total of 8 μg ZAC-pCIneo using PolyFect. 16 h after, the transfection medium were replaced with culture medium supplemented with 2 mg/mL G-418, and the cells were propagated in this medium for the following 3–4 weeks. Antibiotic-resistant individual cell clones were isolated and amplified in the continued presence of 1 mg/mL G-418. The individual clones were screened for functionality in the FMP assay (section 2.3).
2.3. FLIPR® Membrane Potential Red (FMP) assay
The screening of the 1,680 compounds in the compound library for antagonist activity at the ZAC-HEK293 cell line was performed in the FMP assay essentially as previously described [33]. The day before the assay, cells were split into poly-D-lysine-coated black 96-well plates with clear bottoms (BD Biosciences, Palo Alto, CA) (6 × 104 cells/well). The following day, the culture medium was aspirated and the wells were washed once with 100 μl assay buffer (140 mM NaCl, 4.7 mM KCl, 2.5 mM CaCl2, 1.2 mM MgCl2, 11 mM HEPES, 10 mM D-glucose, pH 7.4), after which 100 μl assay buffer supplemented with FMP dye (0.5 mg/ml) and the various test compounds were added to the wells. The plate was then incubated at 37 °C for 30 min and assayed in a NOVOStar plate reader (BMG Labtechnologies, Offenburg, Germany) measuring emission at 560 nm caused by excitation at 530 nm before and up to 1 min after the addition of 33 μl of agonist solution. In the screening, the 1,680 compounds in the compound library were tested at assay concentrations in the 10–30 μM range, using Cu2+ as agonist at an assay concentration of 200 μM.
2.4. Xenopus oocytes and two-electrode voltage clamp (TEVC) recordings
The cDNAs for the various WT and chimeric CLR subunits were linearized and subsequently transcribed and capped mMessage mMachine T7 RNA transcription kit (Ambion, Waltham, MA). Different amounts of cRNA mixtures were injected for ZAC (9.2–36.8 nL in a concentration of 0.05 μg/μL), 1ια3β4 nAChR (9.2–36.8 nL in a concentration of 0.1 μg/μL of each subunit in a ratio of 1:1), hα1 GlyR (9.2–36.8 nL in a concentration of 0.05 μg/μL), hα1β2γ2s GABAAR (9.2–36.8 nL in a concentration of 0.05 μg/μL of each subunit in a ratio of 1:1:3), m5-HT3AR (9.2–36.8 nL in a concentration of 0.05 μg/μL), m5-HT3A/ZAC (9.2–36.8 nL in a concentration of 0.207 μg/μL), and ZAC/hai-Gly (9.2–36.8 nL in a concentration of 0.206 μg/μL). Oocytes were incubated in a sterile modified Barth’s solution [88 mM NaCl, 1 mM KCl, 15 mM HEPES (pH 7.5), 2.4 mM NaHCO3, 0.41 mM CaCl2, 0.82 mM MgSO4, 0.3 mM Ca(NO3)2, 100 U/ml penicillin and 100 μg/ml strep-tomycin] at 18 °C for 2 days after injection.
On the day of experiment, all compound dilutions were prepared in a saline solution [115 mM NaCl, 2.5 mM KCl, 10 mM MOPS (pH 7.5), 1.8 mM CaCl2, 0.1 mM MgCl2] and pH adjusted to 7.5 (if needed). Oocytes were placed in a recording chamber continuously perfused with this saline solution, and the compounds were applied in the perfusate. Both voltage and current electrodes were agar-plugged with 3 M KCl with a resistance of 0.2–2.0 MΩ. Oocytes were voltage-clamped at −50 mV (unless otherwise stated) by a Gene Clamp 500B amplifier (Axon Instruments, Union City, CA), while current signals were digitized by a Digidata 1320. Currents were recorded using pCLAMP 10 (Molecular Devices, Sunnyvale, CA). The recordings were performed at room temperature.
In all recordings, the compounds or compound combinations were applied in the bath until the peak current decayed to a steady state (up to 30 sec at the most). At the beginning or end (wherever appropriate) of all recordings determining concentration-response relationships at ZAC, two consecutive applications of a Zn2+ concentration giving rise to a maximal Zn2+ current (Zn2+ Imax) were applied to the perfusate, and it was verified that these consecutive applications elicited responses of comparable current amplitudes (±20%). The functional properties of the modulators at the CLRs were determined by pre-application of the modulator to the perfusate 30 sec and followed by co-application of the modulator and the agonist to the perfusate. In all recordings, washes of 1–5 min were executed between the applications to prevent receptor desensitization, the duration of these washes depending on the kinetics of the respective receptors and the “stickyness” of the respective compounds. For determination of the concentration-inhibtion relationships exhibited by the modulators at ZAC, agonist concentrations around EC50 (1 mM Zn2+ and 1 μM H+/pH 6.0) were used. For the selectivity and mode-of-action studies, the agonist concentrations used for different CLRs or chimeric CLRs were in the EC20–EC40 range (verified on the day of experiment), as this enabled us to investigate both for putative positive and negative receptor modulation exerted by TTFB (hα3β4 nAChR, 3 μM (S)-nicotine; m5-HT3AR, 2 μM 5-HT; hα1 GlyR, 100 μM glycine; hα1β2γ2S GABAAR, 30 μM GABA; m5-HT3A/ZAC, 0.3 μM 5-HT; ZAC/hɑ1-Gly, 2 μM Zn2+).
2.5. Data analysis
Data from the FMP assay was analysed using KaleidaGraph 3.08 (Synergy Software, Reading, PA). Concentration-response curves for Cu2+ and concentration-inhibition curves for compound 1 were constructed based on the difference in the relative fluorescence units (RFU) between the maximal fluorescence recorded before and after application of Cu2+. The data were fitted to sigmoidal curves with variable slopes using nonlinear regression, and EC50 (Cu2+) and IC50 (compound 1) values were derived from these equations.
Data analysis of the results from the TEVC recordings were performed using Clampfit software version 10.5 (Molecular Devices, Crawley, UK) and GraphPad Prism version 7.0c (GraphPad Software, Inc. La Jolla, CA). Data for the test compounds were normalized to either the maximal agonist-evoked current or to an EC20–EC50-mediated response elicited by the agonist on each oocyte (specified in each case). Unless otherwise stated, data for agonist-induced responses in the presence of the antagonists were extracted from the plateau (or steady-state) of the agonist-evoked current. Concentration-response and concentration-inhibition curves were fitted in GraphPad Prism by nonlinear regression using the equation for sigmoidal dose-response with variable slope. I-V relationships were analyzed by fitting the curves with a third order polynomial model. Each data point represents the mean ± S.E.M. value of recordings performed on at least five oocytes from at least two different batches.
3. Results
3.1. Discovery of a novel ZAC antagonist
In search for novel ZAC modulators we developed a stable ZAC-HEK293 cell line to enable screening of a compound library at the receptor. ZAC-HEK293 clones were screened for functionality in the FMP assay by testing the ability of Cu2+ (200 μM) to elicit a significant increase in fluorescence levels (arising from the depolarization of the cells caused by ZAC activation). In the clone eventually used for the screening, Cu2+ consistently mediated concentration-dependent responses and displayed an average EC50 value of 26 μM (PEC50 ± S.E.M.: 4.59 ± 0.17, n = 4) in the FMP assay (Fig. 1A). Thus, Cu2+ exhibited a ~6-fold lower agonist potency in this assay than that determined for the metal ion at ZAC-expressing COS-7 cells in patch-clamp recordings [22]. Importantly, however, application of Cu2+ at concentrations up to 300 μM did not evoke any responses in HEK293 or h5-HT3A-HEK293 cells in the FMP assay (data not shown), indicating that the Cu2+-induced response in the ZAC-HEK293 cells was indeed mediated through the receptor. On this basis we decided to perform the compound library screening.
Fig. 1. Identification of a novel ZAC antagonist.

A. Concentration-response curve for Cu2+ at ZAC-HEK293 cells in the FMP assay. Data are from a single representative experiment performed in duplicate (of a total of 4) and are given as ΔRFU (mean ± S.D.) values. B. Data obtained for one 96-well plate (out of of a total of 21 96-well plates) tested in the screening at the ZAC-HEK293 cell line in the FMP assay using Cu2+ (200 μM) as agonist, more spefically the 96-well plate containing compound 1. The data for the 16 wells in the 96-well plate containing controls [where the cells in the absence of any test compound were challenged with either pure buffer (red) or buffer supplemented with Cu2+ (200 μM) (blue)] and for the 80 wells in the plate containing various test compounds that either displayed negligible effects on the Cu2+ (200 μM)-induced response (black), were fluorescent or mediated non-specific effects (brown), mediated non-specific or toxic effects (purple), exhibited significant inhibition that not be reproduced in subsequent experiments (grey), and compound 1 (green) are indicated. C. Chemical structure of compound 1. D. Concentration-inhibition curve for compound 1 at ZAC-HEK293 cells in the FMP assay using Cu2+ (200 μM) as agonist. Data are from a single representative experiment performed in duplicate (of a total of 3) and are given as ΔRFU (mean ± S.D.) values.
The 1,680 compounds in the compound library were screened for antagonist activity (at assay concentrations in the 10–30 μM range) at the ZAC-HEK293 cells in the FMP assay using Cu2+ (200 μM) as agonist (~8-fold higher concentration than Cu2+ EC50, Fig. 1A). Several compounds from the library were found to mediate substantial reductions in the Cu2+-evoked fluorescence response in the ZAC-HEK293 cells in the assay (Fig. 1B). However, the majority of these hits were ruled out as ZAC antagonists in subsequent experiments, where the apparent ZAC inhibition exhibited by some of the compounds in the screening could not be reproduced, while other screening hits were found to also reduce the glutamate-induced response in hEAAT3-HEK293 cells in the FMP assay, suggesting that their effects were either non-specific or due to cell toxicity (data not shown). Nevertheless, the screening resulted in the identification of one verifiable ZAC antagonist: 2-(5-bromo-2-chlor-obenzamido)-4-methylthiazole-5-methyl ester (compound 1, Fig. 1B and 1C). Compound 1 was found to mediate concentration-dependent inhibition of the Cu2+ (200 μM)-evoked response in the ZAC-HEK293 cells in the FMP assay, exhibiting an IC50 value of 6.1 μM (pIC5o ± S. E.M.: 5.21 ± 0.13, n = 3) (Fig. 1D). Moreover, compound 1 did not affect the glutamate-induced response in hEAAT3-HEK293 cells, the 5-HT-induced response in h5-HT3A-HEK293 cells or the (S)-nicotine-induced response in rα3β4-nAChR-HEK293 cells in the FMP assay significantly at concentrations up to 100 μM (data not shown). Thus, 1 seemed to exert its effects on the Cu2+-evoked response in ZAC-HEK293 cells in the FMP assay through the receptor.
3.2. Functional properties of compound 1 at ZAC in Xenopus oocytes
The antagonism displayed by 1 at the ZAC-HEK293 cells in the FMP assay was subsequently verified at ZAC expressed in Xenopus oocytes in TEVC recordings. The signalling properties exhibited by ZAC in this assay has been characterized in detail in a previous study [25], and insights gained from that study guided the design of these experiments. 1 was found to inhibit the currents evoked by 1 mM Zn2+ (~EC50) through ZAC-expressing oocytes in a concentration-dependent manner. Interestingly, 1 appeared to mediate partial inhibition of the receptor, since the fitted concentration-inhibition curve for the compound plateaued at a response level constituting ~31% of that evoked by Zn2+ (1 mM) on its own, with 1 displaying an IC50 of 4.1 μM (pIC50 ± S.E.M: 5.39 ±0.17, n = 6) within this −69% inhibition range (Fig. 2A). Even though 1 thus seemingly displayed partial antagonist activity at ZAC, the comparable inhibition exerted by 30 μM and 100 μM 1 and the resulting plateau of the concentration-inhibition curve could also be a reflection of limited solubility of the compound at high concentrations, and it should be noted that several close structural analogs to 1 all displayed full antagonism at ZAC (see section 3.3.4). In view of this, we find it unlikely that 1 is a true partial antagonist of the receptor.
Fig. 2. Antagonist properties displayed by compound 1 at ZAC expressed in Xenopus oocytes in TEVC recordings.

A. Compound 1 inhibits Zn2+ -evoked currents through ZAC in oocytes. Representative traces of Zn2+ (1 mM) evoked currents in ZAC-expressing oocytes in the absence and in the presence of increasing concentrations of 1 (left), and averaged concentration-inhibition relationships for 1 and TC at Zn2+ (1 mM)-induced currents in the oocytes (right). Data for 1 are given as mean ± S.E.M. values (n = 6), and the tubocurarine (TC) data given for comparison are from a recent study [25], B. Compound 1 inhibits the spontaneous ZAC activity in oocytes. Representative traces of the effects of increasing concentrations of 1 on the leak current in ZAC-expressing oocytes (left), and averaged concentration-inhibition relationships for 1 and TC at tire spontaneous currents of ZAC in the oocytes (right). Data for 1 are given as mean ± S.E.M. values (n = 6), and the tubocurarine (TC) data given here for comparison are from a recent study [25]. C. Delineation of the mode of antagonism exerted by compound 1 at Zn2+ -evoked ZAC currents in oocytes. Left: Averaged concentration–response relationships displayed by Zn2+ at ZAC in tire absence and in the presence of various concentrations of 1. Data are given as mean ± S.E.M. values in % of the current evoked by 10 mM Zn2+ in the absence of 1 (I10 mM Zn2+) in the specific oocyte (n = 5–8). Right: The relative degrees of inhibition mediated by different concentration of 1 of the responses evoked by 10 mM, 3 mM, 1 mM and 0.3 mM Zn2+ through ZAC. Data (extracted from the data in Fig. 2C, left) are given as mean values in % of the current evoked by the specific Zn2+ concentration in the absence of 1 (IZn2+).
Analogously to the robust spontaneous activity exhibited by ZAC in mammalian cells [21,22], the channel also displays this characteristic when expressed in oocytes, albeit to a smaller degree [25], As can be seen from the traces in Fig. 2A, the 30 s preincubation of the ZAC-expressing oocyte with compound 1 prior to the co-application of 1 and Zn2+ onto the oocyte produced small but significant outward currents, reflecting a block of this spontaneous activity (Fig. 2A). When the direct effects of compound 1 at this spontaneous activity in ZAC-expressing oocytes subsequently were investigated in more detail, the antagonist was found to exhibit an IC50 value of 7.4 μM (PIC50 ± S.E.M: 5.13 ± 0.07, n = 8) at the receptor (Fig. 2B). The antagonist properties exhibited by TC at ZAC-expressing oocytes in parallel recordings are given in Fig. 2A and 2B for comparison (data are from a recent study [25]). In contrast to 1, TC mediated complete inhibition of the Zn2+ (1 mM)-induced response through ZAC (a fitted ~94% inhibition range), but with an IC50 of 3.2 μM (pIC50 ± S.E.M: 5.49 ± 0.04, n = 8) TC was equipotent with 1 as a ZAC antagonist (Fig. 2A) [25]. TC was also equipotent to 1 in its block of the spontaneous activity in ZAC-expressing oocytes with an IC50 of 3.4 μM (PIC50 ± S.E.M: 5.47 ± 0.04, n = 4) (Fig. 2B).
The antagonist properties of 1 at ZAC were next characterized in further detail by determining the concentration-response relationships for Zn2+ at the receptor in the absence and presence of various concentrations of the compound (Fig. 2C). Since several of the fitted concentration-response curves for Zn2+ obtained in presence of 1 did not reach convincingly saturation within the concentration ranges tested, it was not possible to extract reliable agonist EC50 values from the fitted curves, but visual inspection of the curves suggests that the presence of 1 in the assay reduces the agonist potency of Zn2+ at ZAC somewhat (Fig. 2C, left). However, much more pronounced than this effect on Zn2+ potency is the concentration-dependent 1-mediated suppression of the maximal current evoked by Zn2+ through ZAC, which is indicative of a non-competitive antagonism mode (Fig. 2C, left). In further support of this, extraction and depiction of the data (from Fig. 2C, left) in a different manner revealed that 1 displays very similar concentration-inhibition relationships at the currents evoked by 0.3 mM, 1 mM and 3 mM Zn2+ through ZAC, whereas the antagonist appears less potent at the Zn2+ (10 mM)-evoked current through the receptor (Fig. 2C, right). All in all, the antagonism exerted by 1 at ZAC thus appeared to be largely non-competitive in its nature.
3.3. Stnicture-activity relationship (SAR) of the N-(thiazol-2-yl)-benzamide analog at ZAC
To elucidate structural determinants for ZAC activity in compound 1 and to search for analogs possessing higher antagonist potency at ZAC compared to this lead, we performed a SAR study in which the functional properties of 61 commercially available N-(thiazol-2-yl)-benzamide analogs were characterized at ZAC expressed in oocytes in TEVC recordings. In the following, the SAR results will be presented in the form of five series of analogs (2a-i, 3a-k, 4a-o, 5a-i, 6a-q), which all were tested at ZAC at three concentrations (0.3-3-30 μM or 1-10-100 μM, depending on their respective solubility properties) (Figs. 3–5).
Fig. 3. N-(thiazol-2-yl)-benzamide analogs with modifications to the thiazole ring.

A. Chemical structures of Series 2 analogs (2a-i) and the functional properties exhibited by them as antagonists at ZAC in oocytes using 1 mM Zn2+ as agonist. Data ate given as mean ± S.E.M. values (n = 4–8). B. Chemical structures of Series 3 analogs (3a-k) and the functional properties exhibited by them as antagonists at ZAC in oocytes using 1 mM Zn2+ as agonist. Data are given as mean ± S.E.M. values (n = 3–8).
Fig. 5. Miscelleneous N-(thiazol-2-yl)-benzamide analogs.

Chemical structures of Series 6 analogs (6a-q) and the functional properties exhibited by them as antagonists at ZAC in oocytes using 1 mM Zn2+ as agonist. Data are given as mean ± S.E.M. values (n = 5–8).
3.3.1. Analogs with modifications to the thiazole ring
With Series 2 and 3, we probed the importance of the thiazole ring and different substitutions in the 4- and 5-positions of it for the ZAC activity in the N-(thiazol-2-yl)-benzamide analog (Fig. 3). Since the nine analogs in Series 2 all comprise a 5-bromo-2-chlorobenzamido moiety, the functional properties arising from different modifications to or substitutions of the thiazole ring in these analogs can be compared directly to those of 1. In contrast to the inactivity displayed by the 5-methyl analog 2a at ZAC, the 4-fert-butyl (2b) and 4-ethylacetyl (2c) analogs both displayed slightly more potent and efficacious ZAC inhibition than 1, whereas ZAC activity was reduced substantially by the introduction of a 4-(p-toIyl) substituent (2d) (Fig. 3A). Substitution of the thiazol-2-yl ring for a benzo[(i]thiazol-2-yl (2e), a 5-cyclohexyl-1,3,4-thiadiazole (2f) or other bulky ring systems (2g-i) also resulted in substantial loss of activity (Fig. 3A).
The 11 analogs in Series 3 all comprise a 3-bromobenzamido moiety, and thus differences in the functional properties exhibited by the analogs within this series are also directly attributable to their respective thiazole ring substituents. Since we were unable to obtain 2-(3-bromobenzamido)-4-methylthiazole-5-methyl ester from commercial sources, the functional consequences of elimination of the o-chloro substituent on the phenyl ring could not be assessed in a direct 1:1 comparison with 1. However, the fact that 2b and 3d, the corresponding 4-(tert-butyl)-thiazol-2-yl analogs from Series 2 and 3, exhibited comparable antagonist potencies suggested that the o-chloro substituent in the phenyl ring is not a key determinant of ZAC activity (Fig. 3A and 3B). Interestingly, very small changes to the 4-methyl and 5-methyl ester substituents on the thiazole ring of 1 yielded the inactive analogs 3a (4-ethyl, 5-methyl ester) and 3b (4-methyl, 5-ethyl ester) (Fig. 3B). The 4-methyl analog 3c also displayed weak ZAC activity, whereas introduction of a tert-butyl group in this position yielded a fairly potent analog (3d). Introduction of the bulky 5-bromothiophen-2-yl ring in this position eliminated ZAC activity completely (3e). The 5-nitro analog (3f) displayed an IC50 ~10 μM, whereas the 4,5-dimethyl analog 3g was a considerably weaker antagonist. Introduction of bulky aromatic/heteroaromatic substituents in the 4- and/or the 5-position of the thiazole ring essentially eliminated ZAC activity in the analogs (3h-j), and so did the substitution of thiazole for a 5-(tert-butyl)-l,3,4-thiadiazole ring (3k) (Fig. 3B).
3.3.2. Analogs with modifications to the phenyl ring
With the analogs in Series 4 and 5, we investigated the importance of the phenyl ring and the substitution pattern on it for the ZAC activity of the N-(thiazol-2-yl)-benzamide analog (Fig. 4). Because of the limited commercial availability of analogs in which the rest of the compound 1 structure was completely conserved, Series 4 contained both analogs comprising 5-methyl ester (4a-h) and 5-ethyl ester (4i-o) groups on the thiazole ring (Fig. 4A), and this difference should be kept in mind for SAR comparisons across this analog series. As for the 5-methyl ester analogs (4a-h), the o-tolyl (4a) and p-tolyl (4b) analogs were essentially inactive, whereas substitution of the 5-bromo-2-chlorophenyl ring in 1 with 3-iodophenyl (4c) or 3,5-dimethylphenyl (4d) rings did not alter its antagonist activity substantially (Fig. 4A). In contrast to 4d, the 3,5-dini-trophenyl analog (4e) was completely devoid of antagonist activity at ZAC, and the fact that substitution of the 3-bromo substituent in 1 for a nitro group yielded an inactive analog (4f) further substantiated that a m-nitro group is unfavourable for ZAC activity. Finally, the introduction of a p-ethoxy group or substitution of the phenyl ring for a 4-pyridine resulted in inactive analogs (4g,h). The 5-ethyl ester analogs (4i-o) were considerably less informative than the 5-methyl ester analogs, which in light of the inactivity of analog 3b was not particular surprising. Thus, the inactivity of six of these seven analogs could potentially be rooted in this 5-ethyl ester substituent on the thiazole ring. The o-chlorophenyl (4i) and 3,5-dichlorophenyl (4j) analogs and analog 4k comprising 5-chloro and 2-methoxy groups in the same positions of the phenyl ring as the 5-bromo and 2-chloro substituents in 1, respectively, were all inactive. The 3,4,5-trimethoxyphenyl analog (4l) displayed weak antagonist activity. Not surprisingly, the analog comprising a big N, N-diallylsulfamoyl group in the para-position of the phenyl ring (4m) was inactive, as were the 3-pyridin and 2-furan analogs (4n,o) (Fig. 4A).
Fig. 4. N-(thiazol-2-yl)-benzamide analogs with modifications to the phenyl ring.

A. Chemical structures of Series 4 analogs (4a-o) and the functional properties exhibited by them as antagonists at ZAC in oocytes using 1 mM Zn+ as agonist. Data are given as mean ± S.E.M. values (n = 4–8). B. Chemical structures of Series 5 analogs (5a-i) and the functional properties exhibited by them as antagonists at ZAC in oocytes using 1 mM Zn2+ as agonist. Data are given as mean ± S.E.M. values (n = 4–8).
The more potent and notable higher degree of ZAC inhibition mediated by 2b compared to 1 suggested that introduction of a 4-tert-butyl group on the thiazole ring was beneficial for the antagonist activity (Fig. 3A). With Series 5 we continued to probe the impact of the substituent pattern on the phenyl ring in nine analogs that all comprised this 4-(tert-butyl)thiazol-2-yl moiety (Fig. 4B). The 3-fluorophenyl analog 5a (N-(4-(tert-butyl)thiazol-2-yl)-3-flurobenzamide, TTFB) was roughly equipotent with 2b and mediated almost complete inhibition of ZAC at 10 μM, whereas the 2,3,4,5,6-pentafluoropentyl analog 5b was a considerably weaker ZAC antagonist (Fig. 4B). ZAC activity was also substantially decreased by the introduction of methyl (5c), ethoxy (5d) or acetyl (5e) groups in the 3-position of the phenyl ring, whereas the 3-dimethylamino analog (5f) was roughly equipotent with 2b (Fig. 4B). The 5-chloro-2-methoxyphenyl analog 5g displayed somewhat potent but apparent partial inhibition of ZAC. The 2-chloro-4,5-difluorophenyl analog 5h displayed lower potency as ZAC antagonist than 2b, which was somewhat surprising considering that some of the more potent analogs in this SAR study comprise m-fluoro (5a) and o-chloro (2b, 2c) substituents on the phenyl group. Finally, the fact that analogs 5i and 4l both were essentially inactive suggests that the 3,4,5-trimethoxy substitution pattern on the phenyl ring is detrimental to ZAC activity.
3.3.3. Miscellaneous analogs with multiple modifications
The 17 analogs in Series 6 all comprised more than one modification to the N-(thiazol-2-yl)-benzamide scaffold compared to 1, and most of these analogs were completely inactive at ZAC (Fig. 5). The inactivity of analog 6b was more or less expected, since the 2-chloro-4,5-difluoro-phenyl ring from the moderately potent analog 5h here is combined with the 5-methyl-thiazol-2-yl moiety known from 2a to be detrimental for ZAC activity. Moreover, judging from the inactivity of two other 2-chloro-4,5-difluorophenyl analogs, unsubstituted thiazol-2-yl and 5-acyl-4-methylthiazol-2-yl rings are also not beneficial for ZAC activity in the N-(thiazol-2-yl)-benzamide (6a and 6c). In light of the inactivity of analogs 2d and 3e the inactivity of analogs 6d-f that also comprise bulky substituents in the 4-position of the thiazole ring was also not particularly surprising. Since 6g comprises the same 4-ethylacetyl-substituted thiazol-2-yl ring as the potent 2c analog, the inactivity of this analog must be ascribed to its 3,5-dichlorophenyl moiety. The inactivity of analogs 6h-j could both arise from the 3-chloro or 2,4,6-tri-methyl substitutions at the phenyl ring or from the 4-acetate and the 5-carboxanride thiazole ring substients. Substitution of the 4-ferf-butyl group at the thiazole in 5f for a cyclopropyl ring (6l) resulted in decreased antagonist potency at ZAC, and introduction of a more bulky 3-substituent in the phenylring further reduced ZAC activity (6m) (Fig. 5). Finally, given the weak antagonist activity displayed by other analogs comprising 5-ethyl ester, 4-methyl/5-methyl, 4-methyl or aromatic 4-substituents substituents at the thiazole ring (2d, 3b, 3c, 3g), the inactivity of analogs 6n-p was not surprising.
3.3.4. Detailed functional characterization of selected analogs
Following the SAR study, the functional properties of four of the N-(thiazol-2-yl)-benzamide analogs were characterized in further detail at ZAC (Fig. 6, Table 1). 2b, 4c and 5a (TTFB) were equipotent as ZAC antagonists (IC50 values of 1–3 μM), with 1 and 3f being weaker antagonists. Also notably, and in agreement with observations in the SAR study, 2b, 4c and 5a (TTFB) displayed substantially more efficacious ZAC antagonism than 1, inhibiting the Zn2+-induced responses through the receptor completely (Fig. 6, Table 1).
Fig. 6. Antagonist properties exhibited by five N-(thiazol-2-yl)-benzamide analogs at ZAC expressed in Xenopus oocytes in TEVC recordings.

Averaged concentration-inhibition curves for analogs 1, 2b, 3f, 4c and TTFB (5a) at Zn2+ (1 mM)-induced currents in ZAC-expressing oocytes. Data are given as mean ± S.E.M. values (n = 5–8). Averaged IC50, pIC50, nH, range of inhibition and n values for the five analogs are given in Table 1.
Table 1.
Functional properties exhibited by five N-(thiazol-2-yl)-benzamide analogs at ZAC expressed in oocytes using Zn2+ (1 mM) as agonist determined by TEVC electro-physiology. IC50 (in μM), pIC50 ± S.E.M., Hill coefficient (nH ± S.E.M.) and the fitted inhibition range (in %) are given, and the number of experiments for the data are indicated (n). The averaged concentration-inhibition relationships for the five analogs at ZAC are presented in Fig. 6.
| IC50 (μM) | pIC50 ± S.E.M. | nH ± S.E.M. | Inhibition (%) | n | |
|---|---|---|---|---|---|
| 1 | 4.1 | 5.39 ± 0.17 | −1.3 ± 0.13 | 69 ± 3 | 6 |
| 2b | 1.3 | 5.88 ± 0.05 | −1.2 ± 0.12 | 88 ± 3 | 7 |
| 3f a | ~20 | ~4.7 | n.d. | n.d. | 4 |
| 4c | 1.0 | 5.99 ± 0.08 | −0.89 ± 0.06 | 78 ± 3 | 5–7 |
| 5a (TTFB) | 3.0 | 5.52 ± 0.04 | −1.5 ± 0.08 | 91 ± 3 | 6 |
n.d., not determinable.
The concentration-inhibition curve for this analog was not completed within the tested concentration range.
The IC50 and pIC50 values for the analog are estimated under the assumption that it would mediate complete inhibition.
3.4. Functional properties and mode of action of TTFB (5a) as a ZAC antagonist
The functional properties and mode of action of this novel class of ZAC antagonists were next characterized in further detail using TTFB (5a) as representative.
3.4.1. TTFB (5a) mediates equipotent and voltage-independent inhibition of agonist-evoked and spontaneous ZAC signaling
The antagonist properties exhibited by TTFB at ZAC using Zn2+ and H+ as agonists and at the spontaneous ZAC activity in oocytes are given in Fig. 7. TTFB inhibited the ZAC responses induced by both Zn2+ and H+ in a concentration-dependent manner (Fig. 7A–B). Interestingly, TTFB-mediated ZAC inhibition was characterized by a slow on-set, as the peak current initially produced by the agonist in the presence of TTFB was reduced to a lower plateau (the steady-state current) later during the 30 s of the agonist/TTFB co-application (Fig. 7A, left and 7B, left). Although this slow on-set of inhibition was observed both when using Zn2+ and H+ as agonists, the degree of it was particularly pronounced in the case of H+, which was reflected in the differences observed between the TTFB concentrations-inhibition relationships extracted from recorded peak and steady-state current amplitudes for the two agonists (Fig. 7A, right and 7B, right). Extraction of IC50 (PIC50 ± S.E.M.) values (from the steady-state currents) showed that TTFB displayed slightly higher antagonist potency at Zn2+-evoked than at H+-evoked ZAC signalling (3.0 μM, 5.52 ± 0.04, n = 7 and 8.5 μM, 5.07 ± 0.10, n = 6, respectively) (Fig. 7A and 7B). The antagonist potency exhibited by TTFB at the spontaneous ZAC activity in the oocytes was comparable to that at the Zn2+-evoked response (4.7 μM, 5.32 ± 0.04, n = 7) (Fig. 7C).
Fig. 7. Antagonist properties displayed by TTFB (5a) at ZAC expressed in Xenopus oocytes in TEVC recordings.
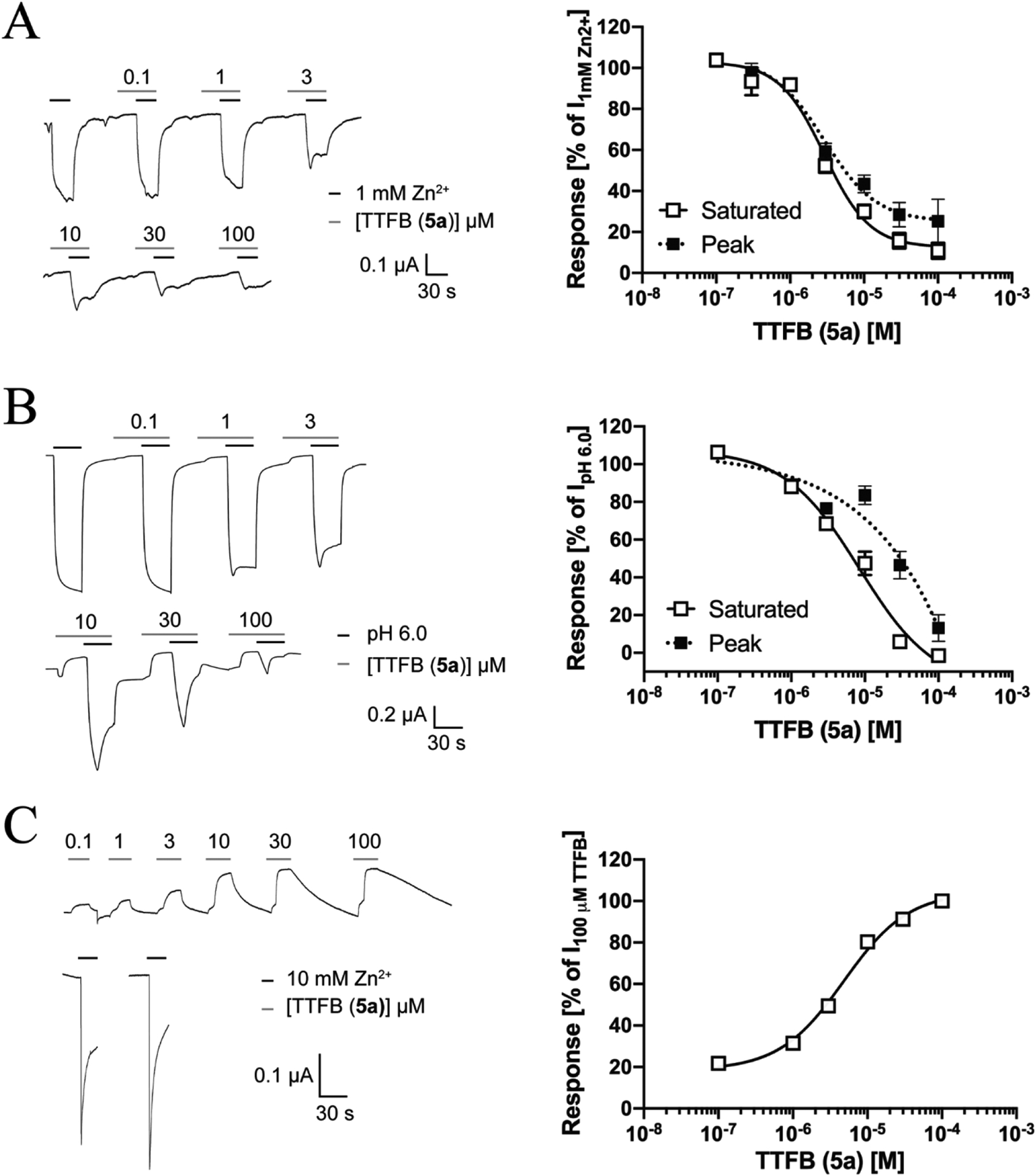
A. TTFB inhibits Zn2+ -evoked currents through ZAC in oocytes. Representative traces of Zn2+ (1 mM)-evoked currents in ZAC-expressing oocytes in the absence and in the presence of increasing concentrations of TTFB (left), and averaged concentration-inhibition relationship for TTFB (extracted from recorded saturated and peak responses) at the Zn2+ (1 mM)-induced currents in the oocytes (right). Data are given as mean ± S.E.M. values (n = 4–7). B. TTFB inhibits H+ -evoked currents through ZAC in oocytes. Representative traces of H+ (pH 6.0)-evoked currents in ZAC-expressing oocytes in the absence and in the presence of increasing concentrations of TTFB (left), and averaged concentration-inhibition relationship for TTFB (extracted from recorded saturated and peak responses) at the H+ (pH 6.0)-induced currents in the oocytes (right). Data are given as mean ± S. E.M. values (n = 4–5). C. TTFB inhibits the constitutive activity exhibited by ZAC in oocytes. Representative traces of the effects of increasing concentrations of TTFB on the leak current in ZAC-expressing oocytes (left), and averaged concentration-inhibition relationship for TTFB at the spontaneous currents of ZAC in the oocytes (right). Data are given as mean ± S.E.M. values (n = 7).
We also investigated the current–voltage (I-V) relationship of TTFB-mediated inhibition of spontaneous ZAC activity and assessed whether its inhibition of agonist-induced ZAC currents was voltage-dependent (Fig. 8). Analogously to the previously reported I-V-relationship for TC-mediated inhibition of spontaneous ZAC activity [21,25] the TTFB (100 μM)-induced currents recorded from ZAC-oocytes at holding potentials ranging from −60 mV to + 60 mV were characterized by considerably higher amplitudes at positive than at corresponding negative potentials, demonstrating that the spontaneous ZAC currents are outwardly rectifying, and TTFB exhibited an equilibrium potential of −11.5 mV (Fig. 8A). The ZAC inhibition mediated by TTFB was found to be voltage-independent, as the antagonist (10 μM) exerted comparable degrees of inhibition of H+-evoked currents in ZAC-oocytes voltage-clamped at −60 mV and + 60 mV (Fig. 8B). Notably, the slow on-set of TTFB-mediated ZAC inhibition was observed at both potentials (Fig. 8B, left).
Fig. 8. Current-voltage relationship of TTFB (5a)-induced currents and its voltage-independent inhibition of H+-evoked ZAC currents in Xenopus oocytes in TEVC recordings.

A. Current-voltage (I-V) relationship of TTFB-mediated inhibition of the spontaneous ZAC currents. Representative traces for currents evoked by applications of TTFB (100 μM) at different holding potentials (left) and the averaged IV-relationship displayed by TTFB (100 μM) at ZAC (right). Data given as leak-subtracted average current amplitudes normalized to the amplitude of currents recorded at −60 mV (mean ± S.E.M., n = 7). The IV curve was fitted with a third order polynomial model. B. Voltage-dependency of the ZAC inhibition mediated by TTFB. Representative traces of currents evoked by H+ (pH 6.5) in the absence and presence of TTFB (10 μM) under voltage-clamp of −60 mV and + 60 mV in the same ZAC-expressing oocyte (left) and averaged data for the TTFB (10 μM)-mediated inhibition of H+ (pH 6.5)-evoked currents in ZAC-expressing oocytes (mean ± S.E.M., n = 4) (right).
3.4.2. TTFB (5a) is a selective ZAC antagonist
The selectivity profile of TTFB as ZAC antagonist was assessed by investigating its functional properties at representatives from each of four classical CLR subfamilies: the m5-HT3AR, the hα3β4 nAChR, the hα1β2γ2s GABAAR and the hα1 GlyR (Fig. 9A). In these recordings, 30 μM TTFB was preapplied onto the oocyte for 30 s before TTFB (30 μM) was co-applied with an EC20–EC40 concentration of the agonist for the specific receptor, thus testing simultaneously for putative activity of TTFB as an agonist, as an antagonist or as a PAM at the receptor. TTFB (30 μM) did not display significant activity at any of these four representative classical CLRs (Fig. 9A).
Fig. 9. Selectivity profile and mode of action of TTFB (5a) as a ZAC antagonist.

A. TTFB is a selective ZAC antagonist. The modulation exerted by TTFB (30 μM) at agonist-induced signalling through ZAC, m5-HT3AR, hα3β4 nAChR, hα1β2γ2s GABAAR, and hα1 GlyR expressed in oocytes in TEVC recordings. EC20–EC40 concentrations of the agonists for the respective receptors (1 mM Zn2+, 2 μM 5-HT, 3 μM (S)-nicotine, 30 μM GABA and 100 μM glycine, respectively) were used for the recordings. Representative traces of agonist-evoked currents in oocytes expressing the respective receptors in the absence and in the presence of 30 μM TTFB (left), and averaged data for the currents measured in oocytes expressing the respective receptors in the presence of TTFB (30 μM), normalized to the current evoked by the agonist in the absence of TTFB (Iagonist) (right). The averaged data are given as mean ± S.E.M. values (n = 5–8). B. TTFB acts through the transmembrane and/or intracellular domains of ZAC. Left: Illustration of the topologies of WT ZAC, WT m5-HT3A, WT hα1 GlyR, m5-HT3A/ZAC and ZAC/hα1-Gly subunits and the pentameric complexes assembled from them. Middle and right: The modulation exerted by TTFB (30 μM) at the agonist-induced responses through the m5-HT3A/ZAC or ZAC/hα1-Gly receptors. EC20–EC40 agonist concentrations for m5-HT3A/ZAC or ZAC/hα1-Gly (0.3 μM 5-HT and 3 μM Zn2+, respectively) were used for the recordings. Representative traces of agonist-evoked currents in oocytes expressing chimeric m5-HT3A/ZAC or ZAC/hα1-Gly receptors in the absence and in the presence of 30 μM TTFB (middle), and averaged data for the Currents measured in oocytes expressing ZAC, m5-HT3AR, hα1-Gly, m5-HT3A/ZAC and ZAC/hα1-Gly in the presence of TTFB (30 μM), normalized to the current recorded in the absence of TTFB (Iagonist) (right). The averaged data for m5-HT3A/ZAC and ZAC/hα1-Gly are given as mean ± S.E.M. values (n = 6–7), and the averaged data for ZAC, m5-HT3AR and hα1 GlyR from Fig. 9A are presented for comparison.
3.4.3. TTFB (5a) acts through the TMD/ICD of ZAC
Functional receptors formed from chimeric subunits fusing the ECD of one CLR with the TMD and ICD of another have been used extensively to study the molecular basis for CLR signaling and to identify receptor domains involved in modulator binding [34–38]. In a companion paper [31] we report that chimeric m5-HT3A/ZAC and ZAC/hα1-Gly subunits both assemble into functional homomeric receptors when expressed in Xenopus oocytes (Fig. 9B, left). In this work, we took advantage of the fact that these two chimeric receptors comprise the ECD or the TMD-ICD of ZAC fused with the complementary domain from another CLR not modulated by TTFB (Fig. 9A) to delineate the ZAC domain targeted by the antagonist. Interestingly, TTFB (30 μM) mediated complete inhibition of the 5-HT-induced response through m5-HT3A/ZAC, whereas it displayed negligible effect on the Zn2+-induced response through ZAC/hα1-Gly (Fig. 9B, middle and right). These black-and-white modulatory properties of TTFB (30 μM) exhibited at the two chimeric receptors strongly indicates that the binding site of the modulator resides within the TMD and/or ICD of ZAC.
4. Discussion
In the present work we have discovered the first class of selective ZAC antagonists and delineated the functional properties and mode of action of these at the receptor.
4.1. Structure-activity relationship (SAR) of the N-(thiazol-2-yl)-benzamide analog as a ZAC antagonist
When developing modulators based on a hit from a compound library screening, chances of optimizing the pharmacological properties of the hit as well as the ability to elucidate the SAR in a systematic manner are very dependent on the analogs accessible. These limitations are also evident in the present study, where the SAR was based exclusively on commercially available analogs. Not all 61 analogs of 1 included in the study were informative with regard to the structural determinants for ZAC activity in the N-(thiazol-2-yl)-benzamide analog, and often the consequences of a modification in a certain position of this scaffold could not be assessed unambiguously due to differences in other parts of two analogs as well. Nevertheless, we propose that several interesting SAR observations about the N-(thiazol-2-yl)-benzamide analog as ZAC antagonist can be extracted from the study.
The thiazole ring system was found to be integral for the ZAC activity of the N-(thiazol-2-yl)-benzamide analog, as substitution of this ring for other ring systems (2f-i, 3k) or fusion of it with phenyl or cycloalkyl rings (2e, 3j) yielded inactive analogs. Introduction of small electron-withdrawing substituents (methoxy, nitro) in the 5-position of the thiazole ring yielded some of the more potent analogs in the series (1, 3f), whereas analogs with electron-donating groups (methyl, phenyl) in this position were inactive (2a, 3g,i) (Fig. 6B). The apparent importance of the electronic characteristics of the 5-substituent could arise from a direct impact on its ability to form interactions in the modulator binding site or from an overall effect on the electron distribution in the thiazole ring. In light of the relative potent activity displayed by several 5-methoxy analogs (1, 4c,d), the inactivity displayed by all 5-ethoxy (3b, 4i-o) and 5-(N,N-dimethyl)amide (6i,j) analogs was surprising (Figs. 3–5). This suggests that the 5-substituent projects towards and potentially forms interactions with residues lining the modulator binding site and, if sufficiently big, will introduce a steric clash with these. Analogs unsubstituted at the 5-position included inactive compounds (2d, 3e) as well as both weak (3c) and potent ZAC antagonists (2b,c, 3d), a range in potency primarily attributable to the identity of the 4-substituent in their respective thiazole rings. Introduction of bulky aliphatic and ester groups in the 4-position (2c,b, 3d) increased ZAC activity substantially compared to analogs with a 4-methyl (3c), whereas ethyl or aromatic/heteroaromatic groups in this position was much less favourable for ZAC activity (2d, 3a,e) (Fig. 3).
The benzamide moiety was retained in 59 of the 62 analogs in the SAR study, which thus mainly focused on the importance of the phenyl ring substitution pattern for the ZAC activity of the N-(thiazol-2-yl)-benzamide analog, primarily with the 4a-h and 5a-i analogs. Introduction of a single ortho- or para-methyl group in the phenyl ring yielded inactive analogs (4a,b). As evidenced by the potent ZAC antagonism exhibited by the 5-bromo-2-chlorobenzamido analogs 1 and 2b,c, an ortho-chloro substituent can be accommodated in the N-(thiazol-2-yl)-benzamide without loss of activity, albeit the similar antagonist potencies displayed by 2b and 3d also suggest that this substituent does not contribute significantly to binding affinity. The inactivity of 4g contrasts the potent ZAC activity displayed by 1 and suggests that para-substitution in the phenyl ring is unfavorable for modulator binding. Finally, analogs comprising a single methyl, fluoro, iodo or N,N-dimethylamide group in the meta-position possessed potent activity (4c, 5a,c,f), whereas the m-aceto and m-ethoxy analogs 5d,e were inactive (Fig. 6B). Since ZAC activity thus was retained in analogs with fairly big meta-substituents, the contrasting activities exhibited by 4d,e and 6g indicate that m-substituent bulk could be more critical for the analogs with 3,5-disubstituted phenyl rings and that residues in the modulator binding pocket may present steric clash with one of the two meta-substitutions in the 4e (3,5-dinitro) and 6g (3,5-dichloro) analogs. Moreover, the fact that most analogs comprising a disubstituted phenyl ring (including 4a, b,e-h, 5d,e,i, 6g) were inactive raises another point. While no analogs comprising an unsubstituted phenyl group was included in the study, the m-fluoro analog TTFB (5a) was one of the most potent ZAC antagonists in the series, indicating that substitutions to the phenyl ring in the N-(thiazol-2-yl)-benzamide analog in general may not be beneficial for ZAC modulation.
While we obviously could have hoped for analogs with even more increased antagonist potencies at ZAC to come out of the SAR study, the functional properties exhibited by analogs 2b, 4c and 5a at ZAC still constitute improvement compared to those of 1 (Fig. 6A). It is also important to stress that derivatization strategies not pursued in this study potentially could form the basis for development of more potent ZAC antagonists. For example, since 4-tert-butyl/4-ethylacetyl and 5-nitro/5-methoxy substitutions to the thiazole ring seem to have isolated beneficial effects on ZAC activity in the N-(thiazol-2-yl)-benzamide analog (1, 2b,c, 3c,f), it would be interesting to probe whether combining these substituents in the N-(thiazol-2-yl)-benzamide analog would have additive or synergistic effects on ZAC antagonist potency.
4.2. Mode of action of the N-(thiazol-2-yl)-benzamide analog as ZAC antagonist
Our investigations into the underlying mechanism for the ZAC inhibition exerted by the N-(thiazol-2-yl)-benzamide analogs unequivocally demonstrate that the compounds act as NAMs at ZAC. In light of its pronounced selectivity for ZAC over m5-HT3AR and hα1 GlyR (Fig. 9A), the robust TTFB-mediated inhibition of m5-HT3A/ZAC signalling and its negligible effect on ZAC/hα1-Gly signalling pinpoints the N-(thiazol-2-yl)-benzamide analog binding site to reside within the TMD-ICD of ZAC (Fig. 9B). Since both Zn2+ and H+ have been proposed to act through the ECD of ZAC [31], this also concords with the largely non-competitive mode of inhibition displayed by compound 1 on Zn2+-induced ZAC signalling (Fig. 2C). The classification of the N-(thiazol-2-yl)-benzamide analog as a ZAC NAM could actually be argued to be premature, since it still remains a question whether Zn2+, Cu2+ and/or H+ or another yet unidentified transmitter is the “true” endogenous agonist for the receptor. However, given that the presently identified ZAC agonists act through the ECD and that any other putative “true” ZAC transmitter would have to be the first endogenous CLR agonist not acting through the ECD in order for the N-(thiazol-2-yl)-benzamide analog not to be a NAM, we nevertheless consider this a reasonable claim.
While a possible involvement of the ICD in N-(thiazol-2-yl)-benzamide binding to ZAC cannot be completely ruled out based on the black-and-white modulation displayed by TTFB at the m5-HT3A/ZAC and ZAC/hα1-Gly receptors, it seems unlikely (Fig. 9B). Classical CLR signaling is known to be modulated by phosphorylation of ICD residues and by binding of intracellular proteins to this domain [39,40], but to our knowledge no small-molecule CLR modulators have been reported to act through this domain. Moreover, the fact that the second intracellular loop in ZAC is very short (~40 residues) compared to the corresponding loops in the classical CLRs makes a putative involvement of this domain in TTFB binding even more unlikely. In contrast, a considerable amount of experimental data from structural biology, biophysical and mutagenesis studies over the years have demonstrated that the CLR TMD comprises several allosteric sites, and a multitude of modulators have been proposed to act through intra-subunit or inter-subunit sites, through sites formed by TMD regions and their surrounding lipid bilayer, through ECD/TMD interface sites or as regular ion channel blockers [12,28,41–47]. The insights into the molecular architecture of these allosteric sites could guide the search for the N-(thiazol-2-yl)-benzamide binding site in the ZAC TMD in future studies.
The interesting slow on-set of the TTFB-mediated ZAC inhibition can not be ascribed to overall slow on-binding of the antagonist to the receptor. Whereas TTFB seems unable to establish binding equilibrium with ZAC during the 30 s-preincubation step, the subsequent co-application of it with agonist clearly facilitates its receptor binding (Fig. 7A, 7B and 8B), and analogously, TTFB-mediated inhibition of spontaneous ZAC activity occurs immediately after ligand application and reaches a steady-state well within 30 s (Fig. 7C). Thus, we propose that the observed slow on-set of ZAC inhibition could be a reflection of state-dependent antagonism, where TTFB preferentially targets the active conformation (be it the constitutively active or the agonist-bound/open channel) over the resting conformation of ZAC. State-dependency in ion channel antagonists is often associated with channel blockers acting through sites in the ion pore [48–50] but it can also arise from other mechanisms [51], and thus this observation does not further elucidate the location of the N-(thiazol-2-yl)-benzamide binding site in the ZAC TMD. However, the substantially larger difference between peak and steady-state currents observed for TTFB-mediated inhibition of H+-evoked currents than for its inhibition of Zn2+-evoked currents is interesting, as it could have both molecular and kinetic origins. In the molecular scenario, the state-dependency of the TTFB-mediated inhibition is agonist-specific, meaning that the structural composition of the TTFB binding site, and with that the ability of the antagonist to bind to it, differs in the Zn2+- and H+-stabilized active ZAC conformations. While TTFB thus may exhibit little preference for the Zn2+-bound active conformation over the resting ZAC conformation, resulting in a modest difference between peak and steady-state currents (Fig. 7A), TTFB binding affinity to the proton-activated ZAC could be vastly higher than its binding affinity to the resting conformation, reflected in the big difference between the two currents for this agonist (Fig. 7B and 8B). In the kinetic scenario, TTFB displays similar preference for the Zn2+- and H+-bound active ZAC conformation over the resting conformation, and thus the different peak/steady-state current ratios observed for the two agonists are rooted in their different kinetic properties. H+-evoked currents through ZAC have been shown both to activate and decay considerably faster than those evoked by Zn2+ [22], and given the negligible desensitization of ZAC and the presumed limited contribution of deactivation in the continuous presence of agonist during the recordings, peak current amplitudes evoked in the presence of TTFB will mainly be determined by the activation kinetics of the agonist and how these compare to the kinetic properties of TTFB binding to the active ZAC conformation. Thus, at equi-effective Zn2+ and H+ concentrations, the faster activator of the two agonists (H+) will induce a higher maximal number of open channels during the initial phase of the agonist/TTFB co-application than the slower agonist (Zn+), and the change in the current arising from the subsequent establishment of the kinetic equilibrium between the agonist and TTFB (steady-state) will inevitably be bigger for the agonist having the bigger kinetic edge as there will be a greater accumulation of agonist-bound, blocked channels.
4.3. The N-(thiazol-2-yl)-benzamide analog as a pharmacological tool for ZAC
While TTFB and the other active N-(thiazol-2-yl)-benzamide analogs in this series represent the first selective ZAC ligands published to date, some considerations should be given to their applicability as pharmacological tools.
Although we only have probed the putative off-target effects of TTFB by testing it at one representative from each of the four classical CLR subfamilies, we propose that the pronounced selectivity exhibited by the antagonist for ZAC over m5-HT3AR, hα3β4 nAChR, hα1β2γ2S GABAAR and hα1 GlyR is likely to extend to the other members of the CLR superfamily (Fig. 9A). Considering the very low homology (<20% amino acid sequence identity) between ZAC and these CLRs [21], it is perhaps not surprising that it is possible to identify selective ZAC modulators. However, whether TTFB and its analogs overall are selective for ZAC over other off-targets is an entirely different matter. The N-(thiazol-2-yl)-benzamide scaffold has been used extensively in medicinal chemistry development of ligands for a diverse range of targets (exemplified in Fig. 10) [52–59]. Judging from the ZAC SAR determined in this study, all of these previously published compounds comprise additional moieties and/or substitutions at the N-(thiazol-2-yl)-benzamide scaffold that most likely would render them inactive at ZAC. Conversely, it can not be ruled out that the ZAC antagonists identified in this study could hold activity at some of these other targets, and a ligand displaying a functional potency of ~1 μM at its target can not be automatically assumed not to possess off-target activities in relevant concentration ranges.
Fig. 10. Examples of previously published ligands comprising the N-(thiazol-2-yl)-benzamide moiety.

The chemical structures, main targets and pharmacological activities reported for eight different ligands comprising the N-(thiazol-2-yl)-benzamide moiety [52–59].
Another question is whether TTFB and the other N-(thiazol-2-yl)-benzamide analogs presented here potentially could be applied as a pharmacological tool for studies of ZAC in vivo. We have not investigated the pharmacokinetic properties of TTFB, but given its moderate target potency and its limited solubility it seems unlikely that systemic administration of the drug would produce sufficiently high concentrations of the free (unbound) compound to achieve significant ZAC inhibition in vivo. Moreover, the absence of an orthologous ZACN gene in rat and mouse genomes also rules out that such studies can be performed in the species most often used for in vivo investigations. Instead, TTFB and its analogs could be useful tools in focused electrophysiological or other functional in vitro/ex vivo studies of native ZAC signalling by use of tissue or cell lines derived from human or other mammalian species.
5. Conclusion
In conclusion, TTFB and the other active N-(thiazol-2-yl)-benzamide analogs constitute the first class of selective antagonists of ZAC, and we propose that the antagonists represent a valuable addition to the limited pharmacological tool box presently available for ZAC and could aid future investigations into this enigmatic member of the CLR superfamily. The N-(thiazol-2-yl)-benzamide analogs were found to act as NAMs through a binding site in its TMD and to mediate state-dependent inhibition of ZAC, which further substantiates the complexity of CLR signalling and of the ligand-mediated modulation of them through allosteric sites.
Acknowledgements
This study was supported financially by the Danish Council of Independent Research for Medical Sciences, the Carlsberg Foundation, NIH-National Institute of Mental Health (MH097446), and National Institute of Neurological Disorders and Stroke (NS108378, NS111064 and NS111338). M.Sc. Louise M. Rahr and M.Sc. Sarah B. Thygesen are thanked for excellent work in connection with the compound library screening.
Abbreviations:
- ACh
acetylcholine
- 5-HT
5-hydioxytiyptaniine
- 5-HT3R
5-HT3 receptor
- CLR
Cys-loop receptor
- EAAT3
excitatory amino acid transporter subtype 3
- ECD
extracellular domain
- FMP
FLIPR Membrane Potential Red
- GABAAR
GABAA receptor
- GlyR
glycine receptor
- ICD
intracellular domain
- nAChR
nicotinic acetylcholine receptor
- NAM
negative allosteric modulator
- PAM
positive allosteric modulator
- SAR
structure-activity relationship
- TC
tubocurarine
- TEVC
two-electrode voltage clamp
- TMD
transmembrane domain
- TTFB
N-(4-(tert-butyl)thiazol-2-yl)-3-fluorobenzamide
- WT
wild type
- ZAC
Zinc-Activated Channel
Footnotes
CRediT authorship contribution statement
Nawid Madjroh: Data curation, Formal analysis, Investigation, Visualization, Writing - review & editing. Eleni Mellou: Data curation, Formal analysis, Writing - review & editing. Paul A. Davies: Formal analysis, Investigation, Supervision, Writing - review & editing. Pella C. Söderhielm: Formal analysis, Investigation, Supervision, Writing - review & editing. Anders A. Jensen: Conceptualization, Data curation, Formal analysis, Funding acquisition, Investigation, Project administration, Supervision, Visualization, Writing - original draft, Writing - review & editing.
Declaration of Competing Interest
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
References
- [1].Alexander SPH, Mathie A, Peters JA, Veale EL, Striessnig J, Kelly E, et al. , THE CONCISE GUIDE TO PHARMACOLOGY 2019/20: Ion channels, Br J Pharmacol 176 (Suppl 1) (2019) S142–S228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [2].Walstab J, Rappold G, Niesler B, 5-HT3 receptors: role in disease and target of drugs, Pharmacol Ther 128 (1) (2010) 146–169. [DOI] [PubMed] [Google Scholar]
- [3].Fakhfouri G, Rahimian R, Dyhrfjeld-Johnsen J, Zirak MR, Beaulieu J-M, Witkin JM, 5-HT3 receptor antagonists in neurologic and neuropsychiatric disorders: The iceberg still lies beneath the surface, Pharmacol Rev 71 (3) (2019) 383–412. [DOI] [PubMed] [Google Scholar]
- [4].Lynch JW, Zhang Y, Talwar S, Estrada-Mondragon A, Glycine Receptor Drug Discovery, Adv Pharmacol 79 (2017) 225–253. [DOI] [PubMed] [Google Scholar]
- [5].Chua HC, Chebib M, GABAA Receptors and the Diversity in their Structure and Pharmacology, Adv Pharmacol 79 (2017) 1–34. [DOI] [PubMed] [Google Scholar]
- [6].Engin E, Benham RS, Rudolph U, An Emerging Circuit Pharmacology of GABAA Receptors, Trends Pharmacol Sci 39 (8) (2018) 710–732. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [7].Taly A, Corringer P-J, Guedin D, Lestage P, Changeux J-P, Nicotinic receptors: allosteric transitions and therapeutic targets in the nervous system, Nat Rev Drug Discov 8 (9) (2009) 733–750. [DOI] [PubMed] [Google Scholar]
- [8].Bouzat C, Lasala M, Nielsen BE, Corradi J, Esandi MDC, Molecular function of α7 nicotinic receptors as drug targets, J Physiol 596 (10) (2018) 1847–1861. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [9].Hassaine G, Deluz C, Grasso L, Wyss R, Tol MB, Hovius R, Graff A, Stahlberg H, Tomizaki T, Desmyter A, Moreau C, Li X-D, Poitevin F, Vogel H, Nury H, X-ray structure of the mouse serotonin 5-HT3 receptor, Nature 512 (7514) (2014) 276–281. [DOI] [PubMed] [Google Scholar]
- [10].Morales-Perez CL, Noviello CM, Hibbs RE, X-ray structure of the human α4β2 nicotinic receptor, Nature 538 (7625) (2016) 411–415. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [11].Laverty D, Desai R, Uchański T, Masiulis S, Stec WJ, Malinauskas T, Zivanov J, Pardon E, Steyaert J, Miller KW, Aricescu AR, Cryo-EM structure of the human α1β3γ2 GABAA receptor in a lipid bilayer, Nature 565 (7740) (2019) 516–520. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [12].Polovinkin L, Hassaine Ghérici, Perot J, Neumann E, Jensen AA, Lefebvre SN, Corringer P-J, Neyton J, Chipot C, Dehez F, Schoehn G, Nury H, Conformational transitions of the serotonin 5-HT3 receptor, Nature 563 (7730) (2018) 275–279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [13].Hibbs RE, Gouaux E, Principles of activation and permeation in an anion-selective Cys-loop receptor, Nature 474 (7349) (2011) 54–60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [14].Gielen M, Thomas P, Smart TG, The desensitization gate of inhibitory Cys-loop receptors, Nat Commun 6 (2015) 6829. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [15].Sieghart W, Allosteric modulation of GABAA receptors via multiple drug-binding sites, Adv Pharmacol 72 (2015) 53–96. [DOI] [PubMed] [Google Scholar]
- [16].Davies PA, Allosteric modulation of the 5-HT3 receptor, Curr Opin Pharmacol 11 (1) (2011) 75–80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [17].Yevenes GE, Zeilhofer HU, Allosteric modulation of glycine receptors, Br J Pharmacol 164 (2011) 224–236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [18].Grupe M, Grunnet M, Bastlund JF, Jensen AA, Targeting α4β2 nicotinic acetylcholine receptors in central nervous system disorders: perspectives on positive allosteric modulation as a therapeutic approach, Basic Clin Pharmacol Toxicol 116 (3) (2015) 187–200. [DOI] [PubMed] [Google Scholar]
- [19].Chatzidaki A, Millar NS, Allosteric modulation of nicotinic acetylcholine receptors, Biochem Pharmacol 97 (4) (2015) 408–417. [DOI] [PubMed] [Google Scholar]
- [20].Kim JJ, Hibbs RE, Direct Structural Insights into GABAA Receptor Pharmacology, Trends Biochem Sci 46 (6) (2021) 502–517. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [21].Davies PA, Wang W, Hales TG, Kirkness EF, A novel class of ligand-gated ion channel is activated by Zn2+, J Biol Chem 278 (2) (2003) 712–717. [DOI] [PubMed] [Google Scholar]
- [22].Trattnig SM, Gasiorek A, Deeb TZ, Ortiz EJC, Moss SJ, Jensen AA, Davies PA, Copper and protons directly activate the zinc-activated channel, Biochem Pharmacol 103 (2016) 109–117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [23].Houtani T, Munemoto Y, Kase M, Sakuma S, Tsutsumi T, Sugimoto T, Cloning and expression of ligand-gated ion-channel receptor L2 in central nervous system, Biochem Biophys Res Commun 335 (2) (2005) 277–285. [DOI] [PubMed] [Google Scholar]
- [24].Chang Y, Modulators of Zinc Activated Channel (US 2019/0022121 Al), Dignity Health, Phoeniz, AZ, United States, 2019. [Google Scholar]
- [25].Madjroh N, Davies PA, Smalley JL, Kristiansen U, Söderhielm PC, Jensen AA, Delineation of the functional properties exhibited by the Zinc-Activated Channel (ZAC) and its high-frequency Thr128Ala variant (rs2257020) in Xenopus oocytes, Pharmacol Res 169 (2021) 105653, 10.1016/j.phrs.2021.105653. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [26].Spirova EN, Ivanov IA, Kasheverov IE, Kudryavtsev DS, Shelukhina IV, Garifulina AI, Son LV, Lummis SCR, Malca-Garcia GR, Bussmann RW, Hennig L, Giannis A, Tsetlin VI, Silman I, Curare alkaloids from Matis Dart Poison: Comparison with d-tubocurarine in interactions with nicotinic, 5-HT3 serotonin and GABAA receptors, PLoS One 14 (1) (2019) e0210182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [27].Liu M, Dilger JP, Synergy between pairs of competitive antagonists at adult human muscle acetylcholine receptors, Anesth Analg 107 (2008) 525–533. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [28].Trattnig SM, Harpsøe K, Thygesen SB, Rahr LM, Ahring PK, Balle T, Jensen AA, Discovery of a novel allosteric modulator of 5-HT3 receptors: Inhibition and potentiation of Cys-loop receptor signaling through a conserved transmembrane intersubunit site, J Biol Chem 287 (30) (2012) 25241–25254. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [29].Jensen AA, Bergmann ML, Sander T, Balle T, Ginkgolide X is a potent antagonist of anionic Cys-loop receptors with a unique selectivity profile at glycine receptors, J Biol Chem 285 (13) (2010) 10141–10153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [30].Jensen AB, Hoestgaard-Jensen K, Jensen AA, Elucidation of molecular impediments in the α6 subunit for in vitro expression of functional α6β4* nicotinic acetylcholine receptors, J Biol Chem 288 (47) (2013) 33708–33721. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [31].Madjroh N, Mellou E, Æbelø L, Davies PA, Söderhielm PC, Jensen AA, Probing the molecular basis for signal transduction through the Zinc-Activated Channel (ZAC), Biochem Pharmacol 193 (2021), 114781, 10.1016/j.bcp.2021.114781. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [32].Jensen AA, Bräuner-Osborne H, Pharmacological characterization of human excitatory amino acid transporters EAAT1, EAAT2 and EAAT3 in a fluorescence-based membrane potential assay, Biochem Pharmacol 67 (11) (2004) 2115–2127. [DOI] [PubMed] [Google Scholar]
- [33].Jensen AA, Kristiansen U, Functional characterisation of the human αl glycine receptor in a fluorescence-based membrane-potential assay, Biochem Pharmacol 67 (9) (2004) 1789–1799. [DOI] [PubMed] [Google Scholar]
- [34].Eiselé J-L, Bertrand S, Galzi J-L, Devillers-Thiéry A, Changeux J-P, Bertrand D, Chimaeric nicotinic-serotonergic receptor combines distinct ligand binding and channel specificities, Nature 366 (6454) (1993) 479–483. [DOI] [PubMed] [Google Scholar]
- [35].Mihic SJ, Ye Q, Wick MJ, Koltchine VV, Krasowski MD, Finn SE, Mascia MP, Valenzuela CF, Hanson KK, Greenblatt EP, Harris RA, Harrison NL, Sites of alcohol and volatile anaesthetic action on GABAa and glycine receptors, Nature 389 (6649) (1997) 385–389. [DOI] [PubMed] [Google Scholar]
- [36].Gasiorek A, Trattnig SM, Ahring PK, Kristiansen U, Frølund B, Frederiksen K, Jensen AA, Delineation of the functional properties and the mechanism of action of TMPPAA, an allosteric agonist and positive allosteric modulator of 5-HT3 receptors, Biochem Pharmacol 110–111 (2016) 92–108. [DOI] [PubMed] [Google Scholar]
- [37].Price KL, Lummis SCR, Characterization of a 5-HT3-ELIC Chimera Revealing the Sites of Action of Modulators, ACS Chem Neurosci 9 (6) (2018) 1409–1415. [DOI] [PubMed] [Google Scholar]
- [38].Ghosh B, Tsao T-W, Czajkowski C, A chimeric prokaryotic-eukaryotic pentameric ligand gated ion channel reveals interactions between the extracellular and transmembrane domains shape neurosteroid modulation, Neuropharmacology 125 (2017) 343–352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [39].Langlhofer G, Villmann C, The intracellular loop of the glycine receptor: It’s not all about the size, Front Mol Neurosci 9 (2016) 41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [40].Coyle JE, Qamar S, Rajashankar KR, Nikolov DB, Structure of GABARAP in two conformations: implications for GABAA receptor localization and tubulin binding, Neuron 33 (1) (2002) 63–74. [DOI] [PubMed] [Google Scholar]
- [41].Limon A, Estrada-Mondragón A, Ruiz JMR, Miledi R, Dipicrylamine modulates GABAρ1 receptors through interactions with residues in the TM4 and Cys-loop domains, Mol Pharmacol 89 (4) (2016) 446–456. [DOI] [PubMed] [Google Scholar]
- [42].Hammer H, Bader BM, Ehnert C, Bundgaard C, Bunch L, Hoestgaard-Jensen K, et al. , A multifaceted GABAA receptor modulator: Functional properties and molecular mechanism of action of the sedative-hypnotic and recreational drug methaqualone (Quaalude), Mol Pharmacol 88 (2015) 401–420. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [43].Hosie AM, Wilkins ME, da Silva HMA, Smart TG, Endogenous neurosteroids regulate GABAA receptors through two discrete transmembrane sites, Nature 444 (7118) (2006) 486–489. [DOI] [PubMed] [Google Scholar]
- [44].Li G-D, Chiara DC, Sawyer GW, Husain SS, Olsen RW, Cohen JB, Identification of a GABAA receptor anesthetic binding site at subunit interfaces by photolabeling with an etomidate analog, J Neurosci 26 (45) (2006) 11599–11605. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [45].Paradiso K, Zhang J, Steinbach JH, The C terminus of the human nicotinic α4β2 receptor forms a binding site required for potentiation by an estrogenic steroid, J Neurosci 21 (17) (2001) 6561–6568. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [46].Nury H, Van Renterghem C, Weng Y, Tran A, Baaden M, Dufresne V, Changeux JP, Sonner JM, Delarue M, Corringer P-J, X-ray structures of general anaesthetics bound to a pentameric ligand-gated ion channel, Nature 469 (7330) (2011) 428–431. [DOI] [PubMed] [Google Scholar]
- [47].Chua HC, Christensen ETH, Hoestgaard-Jensen K, Hartiadi LY, Ramzan I, Jensen AA, Absalom NL, Chebib M, Barnes S, Kavain, the major constituent of the anxiolytic kava extract, potentiates GABAa Receptors: Functional characteristics and molecular mechanism, PLoS One 11 (6) (2016) e0157700. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [48].Halliwell RF, Peters JA, Lambert JJ. The mechanism of action and pharmacological specificity of the anticonvulsant NMDA antagonist MK-801: a voltage clamp study on neuronal cells in culture. Br J Pharmacol 1989;96:480–94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [49].Yoon KW, Covey DF, Rothman SM, Multiple mechanisms of picrotoxin block of GABA-induced currents in rat hippocampal neurons, J Physiol 464 (1993) 423–439. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [50].Winquist RJ, Pan JQ, Gribkoff VK, Use-dependent blockade of Cav2.2 voltage-gated calcium channels for neuropathic pain, Biochem Pharmacol 70 (4) (2005) 489–499. [DOI] [PubMed] [Google Scholar]
- [51].Hansen KB, Traynelis SF, Structural and mechanistic determinants of a novel site for noncompetitive inhibition of GluN2D-containing NMDA receptors, J Neurosci 31 (10) (2011) 3650–3661. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [52].Uto Y, Ogata T, Kiyotsuka Y, Miyazawa Y, Ueno Y, Kurata H, Deguchi T, Yamada M, Watanabe N, Takagi T, Wakimoto S, Okuyama R, Konishi M, Kurikawa N, Kono K, Osumi J, Novel and potent inhibitors of stearoyl-CoA desaturase-1. Part Π: Identification of 4-ethylamino-3-(2-hydroxyethoxy)-N-[5-(3-trifluoromethylbenzyl)thiazol-2-yl]benza mide and its biological evaluation, Bioorg Med Chem Lett 19 (15) (2009) 4159–166. [DOI] [PubMed] [Google Scholar]
- [53].Abbasi M, Raza H, Rehman A, Siddiqui S, Nazir M, Mumtaz A, Shah S, Seo S-Y, Hassan M, Synthesis, Antioxidant and In-Silico Studies of Potent Urease Inhibitors: N-(4-{[(4-Methoxyphenethyl)-(substituted)amino]sulfonyl}phenyl) acetamides, Drug Res (Stuttg) 69 (02) (2019) 111–120. [DOI] [PubMed] [Google Scholar]
- [54].lino T, Tsukahara D, Kamata K, Sasaki K, Ohyama S, Hosaka H, Hasegawa T, Chiba M, Nagata Y, Eiki J-I, Nishimura T, Discovery of potent and orally active 3-alkoxy-5-phenoxy-N-thiazolyl benzamides as novel allosteric glucokinase activators, Bioorg Med Chem 17 (7) (2009) 2733–2743. [DOI] [PubMed] [Google Scholar]
- [55].Sams AG, Mikkelsen GK, Larsen M, Langgård M, Howells ME, Schrøder TJ, Brennum LT, Torup L, Jorgensen EB, Bundgaard C, Kreilgård M, Bang-Andersen B, Discovery of phosphoric acid mono-{2-[(E/Z)-4-(3,3-dimethyl-butyryIamino)-3,5-difluoro-benzoylimino]-thiazol-3-ylmethyl} ester (Lu AA47070): a phosphonooxymethylene prodrug of a potent and selective hA(2A) receptor antagonist, J Med Chem 54 (3) (2011) 751–764. [DOI] [PubMed] [Google Scholar]
- [56].Satoh A, Nagatomi Y, Hirata Y, Ito S, Suzuki G, Kimura T, Maehara S, Hikichi H, Satow A, Hata M, Ohta H, Kawamoto H, Discovery and in vitro and in vivo profiles of 4-fluoro-N-[4-[6-(isopropylamino)pyrimidin-4-yl]-l,3-thiazol-2-yl]-N-methylbenzam ide as novel class of an orally active metabotropic glutamate receptor 1 (mGluRl) antagonist, Bioorg Med Chem Lett 19 (18) (2009) 5464–5468. [DOI] [PubMed] [Google Scholar]
- [57].Wu G, Qiu X-L, Zhou L, Zhu J, Chamberlin R, Lau J, Chen P-L, Lee W-H, Small molecule targeting the Hecl/Nek2 mitotic pathway suppresses tumor cell growth in culture and in animal, Cancer Res 68 (20) (2008) 8393–8399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [58].van Muijlwijk-Koezen JE, Timmerman H, Vollinga RC, Frijtag J, von Drabbe Kiinzel, de Groote M, Visser S, IJzerman AP, Thiazole and thiadiazole analogues as a novel class of adenosine receptor antagonists, J Med Chem 44 (5) (2001) 749–762. [DOI] [PubMed] [Google Scholar]
- [59].In E-J, Lee Y, Koppula S, Kim T-Y, Han J-H, Lee K-H, Kang T-B, Identification and Characterization of NTB451 as a Potential Inhibitor of Necroptosis, Molecules 23 (11) (2018) 2884, 10.3390/molecules23112884. [DOI] [PMC free article] [PubMed] [Google Scholar]