

Abstract
Amyotrophic lateral sclerosis (ALS) is a progressive neurodegenerative disease affecting both upper and lower motor neurons (MNs) with large unmet medical needs. Multiple pathological mechanisms are considered to contribute to the progression of ALS, including neuronal oxidative stress and mitochondrial dysfunction. Honokiol (HNK) has been reported to exert therapeutic effects in several neurologic disease models including ischemia stroke, Alzheimer's disease and Parkinson's disease. Here we found that honokiol also exhibited protective effects in ALS disease models both in vitro and in vivo. Honokiol improved the viability of NSC-34 motor neuron-like cells that expressed the mutant G93A SOD1 proteins (SOD1-G93A cells for short). Mechanistical studies revealed that honokiol alleviated cellular oxidative stress by enhancing glutathione (GSH) synthesis and activating the nuclear factor erythroid 2-related factor 2 (NRF2)-antioxidant response element (ARE) pathway. Also, honokiol improved both mitochondrial function and morphology via fine-tuning mitochondrial dynamics in SOD1-G93A cells. Importantly, honokiol extended the lifespan of the SOD1-G93A transgenic mice and improved the motor function. The improvement of antioxidant capacity and mitochondrial function was further confirmed in the spinal cord and gastrocnemius muscle in mice. Overall, honokiol showed promising preclinical potential as a multiple target drug for ALS treatment.
KEY WORDS: Amyotrophic lateral sclerosis, Glutathione, Honokiol, Mitochondrial biogenesis, Mitochondrial dynamics, NRF2, Oxidative stress, SOD1-G93A
Graphical abstract
Honokiol decreased the oxidative damage by activating NRF2–GSH pathway. Meanwhile, honokiol improved mitochondrial dysfunction via regulating the mitochondrial biogenesis and mitochondrial fusion. Taken together, honokiol exerted neuroprotection in ALS models.
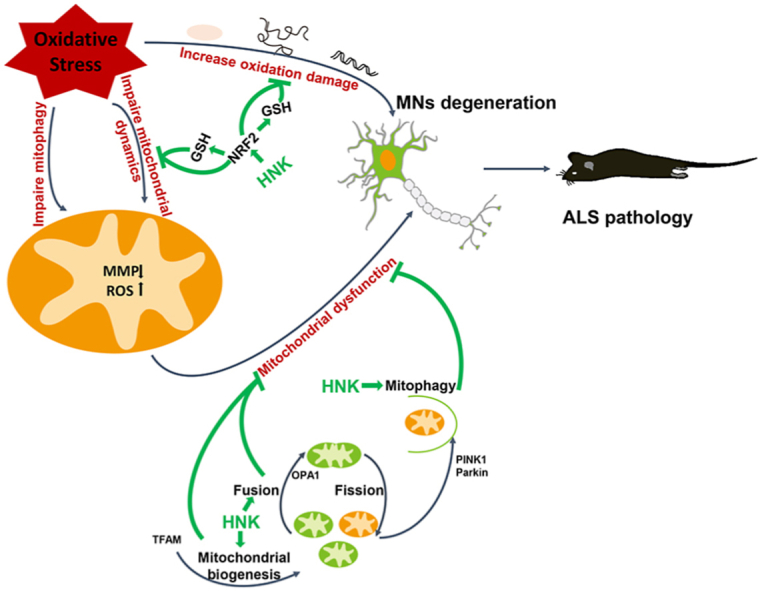
1. Introduction
Amyotrophic lateral sclerosis (ALS) is a progressive neurodegenerative disease that causes degeneration and death of the lower and upper motor neurons (MNs) which involves the cortex, brainstem, and spinal cord. The gradual muscle denervation leads to muscle weakness, atrophy, and paralysis, eventually respiratory failure and death. The incidence of ALS was estimated to be between 0.6 and 3.8 per 100,000 person-years1. ALS patients have a hidden onset however rapid progress and mean survival of 3–5 years after the disease onset. Among the ALS patients, nearly 90%–95% belong to sporadic ALS (sALS) most with unknown etiology2, while approximately 5%–10% is familial ALS (fALS)3 associated with known gene mutations. Cu/Zn superoxide dismutase 1 (SOD1) is the leading cause for ALS in China, and account for 25.3% of Chinese fALS cases4,5. Although a number of candidates have been shown to delay disease progression in preclinical trials, riluzole and edaravone are only two drugs approved by US Food and Drug Administration for ALS treatment. However, their effects are limited and are only able to extend the survival by a few months6.
The pathogenic mechanisms of ALS remain poorly understood. Numerous molecular mechanisms involved in these pathologies have been reported, such as glutamate excitotoxicity, mitochondrial dysfunction, alterations in axonal transport, oxidative stress, accumulation of misfolded proteins, and neuroinflammation7, 8, 9, 10. Oxidative stress has been demonstrated to play a key role in pathogenesis of ALS. The high levels of oxidative stress markers, such as products of lipid peroxidation (and altered lipid metabolism), protein oxidation, DNA oxidation, stress proteins, oxidatively modified proteins, and reactive oxygen species (ROS) have been revealed in ALS patients and SOD1-G93A transgenic mice11. Nuclear factor erythroid 2-related factor 2 (NRF2) is a redox-sensitive transcription factor and a master regulator of numerous antioxidant enzymes which protects cells from oxidative stress12,13. NRF2 has also been reported to regulate the synthesis of glutathione (GSH). GSH is the most important small-molecule antioxidant in mammalian cells and play essential roles in ROS elimination and repair of oxidized proteins14.
The accumulation of oxidative damage due to increased dysfunction in GSH homeostasis within cells has been reported to contribute to ALS progression15. Besides, oxidative stress status may trigger other pathological processes in ALS. Increased ROS level leads to more glutamate accumulating in the synapse and more glutamate receptors stimulated, which further causes an increase in calcium influx into the MNs and mitochondria, leading to the mitochondrial dysfunction16,17. Mitochondria is the central site of production of ROS. Under stressful conditions, mitochondrial quality control, a group of adaptive responses that regulate mitochondrial protein turnover, mitochondrial fusion, mitochondrial fission and mitophagy, is activated to preserve mitochondrial structure and function18. Alterations in the mitochondrial structure and disruption of the mitochondrial function have been postulated as the core in the pathophysiology of ALS, including dysregulation of mitochondrial proteins19, increased production of mitochondrial ROS, decreased production of ATP, and compromised mitochondrial quality control20, 21, 22. The fragmentation of mitochondria and abnormal mitochondrial morphology have been described in ALS and have pronounced effects on normal mitochondrial function23. The decrease of mitochondrial biogenesis and disruption of mitochondrial fusion-fission balance are common signatures in ALS patients24. In addition, some evidence suggested that the mitochondrial-dependent apoptotic pathways might involve in neurodegeneration in ALS25, 26, 27
Honokiol (HNK), one of the main bioactive components isolated from the traditional Chinese herb Magnolia Officinalis, has shown multiple pharmacological functions, such as antioxidant, anti-inflammation, anti-bacterial, and anti-tumor28, 29, 30. Besides, honokiol is a small polyphenol molecule, due to its strong lipophilic property, honokiol can readily permeate the blood–brain barrier and blood–cerebrospinal fluid barrier. At present, it has been reported to exert potent neuroprotective effects in multiple animal models of central nerve system diseases, including cerebrovascular injury, spinal cord injury, anxiety, epilepsy, and cognitive disorders diseases probably due to inhibiting oxidative stress and neuronal excitotoxicity, alleviating neuroinflammation, and regulating mitochondrial function28,29,31. Honokiol was reported to display therapeutic activity on the motor deficits and neuronal degeneration in Parkinson's disease by decreasing oxidative stress and inflammation32. In addition, honokiol was shown a beneficial effect on the cognitive impairment in APP/PS1 via ameliorating the mitochondrial dysfunction33. Honokiol alleviated brain edema and neurobehavioral deficits after subarachnoid hemorrhage through increasing mitochondrial fusion thus maintaining mitochondrial morphology, protecting mitochondrial function, and promoting neural survival34. Furthermore, honokiol was applied for patent (200310121303.0) for ischemic stroke treatment, and the clinical trials would be started soon in China. However, to our knowledge, no study was performed to assess the effects of honokiol on ALS disease.
In this study, we evaluated the neuroprotective effect of honokiol on the ALS disease by using stably-overexpressed SOD1-G93A mouse Motor Neuron NSC-34 cells, and SOD1-G93A transgenic mice. Moreover, the mechanisms underlying the efficacy of honokiol were investigated, such as mitochondrial dysfunction and oxidative stress.
2. Materials and methods
2.1. Free radical scavenging tests
The detection of DPPH free radical scavenging, pyrogallol autoxidation, Fenton's reaction and oxygen radical absorbance capacity determination (ORAC) were described in Supporting Information.
2.2. Measurement of oxidative stress damage and related protein expression in yeast library
A library of 44 in-frame GFP fusion proteins involved in oxidative stress damage relative pathways of Saccharomyces cerevisiae (No. 95702, ATCC 201388, Invitrogen, Waltham, MA, USA) was used to measure the influence of honokiol on oxidative stress induced by H2O2 in the yeast system (Supporting Information Table S1). A housekeeping gene PGK1 was selected as an internal control for plate normalization. The details of the yeast construction, marker selection, yeast culture and the data analysis were as described previously35. Briefly, the yeast library was constructed by oligonucleotide-directed homologous recombination to tag each open reading frame (ORF) with Aequorea victoria GFP (S65T) in the chromosomal location at the 3′ end. The yeast strains selected were seeded in clear bottom black 384-well plates and grown with minimal synthetic defined (SD) medium for 4–6 h at 30 °C until the cultures reached early exponential growth (OD600 about 0.2–0.4). Three concentrations of honokiol (final concentrations of 0.111, 0.333 and 1 μmol/L) dissolved in the SD medium were added into the wells in the honokiol-treated groups and the same volume of SD medium were added in the model group. H2O2 was added in the model group and honokiol-treated groups at the final concentration of 10 mg/L as an injurant to induce oxidative stress. The plates were then placed into a microplate reader (Synergy H1 Multi-Mode, BioTek, Winooski, VT, USA) for GFP signal (filters with 485 nm excitation and 535 nm emission for protein expression) measurements. In the meantime, the absorbance of OD600 for cell growth was detected as the correction of the cell number. The signals were measured every 5 min after double orbital fast shake for 1 min and lasted for 2 h. The tests were repeated in triplicate. The data processing and analysis methods were described in Supporting Information in detail.
2.3. Cell culture and transfection
Mouse Motor Neuron-Like Hybrid Cell Line (NSC-34, Shanghai Hongshun Biotechnology Company Limited, Shanghai, China) were cultured in Dulbecco's modified Eagle medium (Invitrogen) supplemented with 10% FBS and 1% penicillin (100 units/mL)/streptomycin (100 μg/mL) in a humidified atmosphere of 5% CO2 at 37 °C.
cDNAs of G93A mutant of human SOD1 was cloned into pcDNA3.1-EGFP vector. Both plasmids were constructed and purchased from Changsha Youbao Biotechnology Company Limited (Hunan, China). Transfections of cDNA constructs were carried out using Lipofectamine 3000 (Invitrogen) following the manufacturer's instructions. 800 μg/mL G418 was used to screen resistant clones.
2.4. MTT assay and Hoechst dyeing
NSC-34 cells transfected with SOD1-G93A (further referred as SOD1-G93A cells) or pcDNA3.1-EGFP vector (further referred as pEGFP cells) were seeded into 96 wells flat-bottom plates. SOD1-G93A cells were treated with different concentrations of honokiol range from 1 to 10 μmol/L or culture medium for 24 h. For MTT assay, MTT solution (5 mg/mL in PBS) was added. After 4 h of incubation, DMSO was added to dissolve formazan crystals. Absorbance at 570 nm was measured using the microplate reader and relative cell viability was calculated compared with pEGFP cells. For Hoechst assay, a black bottom transparent enzyme label plate was used for seeding cells. Drugs treatments were the same as the procedures mentioned above followed by Hoechst dyeing for 10 min. Cells were washed for three times with PBS. The fluorescence with 350 nm excitation and 461 nm emission signal was measured using the fluorescence microplate reader and relative cell viability was calculated compared with pEGFP cells.
2.5. Cell apoptosis assay
Annexin V-Alexa Fluor 647/PI Cell Apoptosis Detection Kit (Yeasen, Shanghai, China) was used to detect the apoptosis level of SOD1-G93A cells after incubation with honokiol for 24 h. Cells were digested by 0.25% trypsin without EDTA and resuspend by 1× binding buffer. 5 μL Annexin V-Alexa Fluor 647 and 10 μL PI were incubated with cells for 15 min at room temperature. Fluorescence intensity of Annexin V-Alexa Fluor 647 (APC channel) and PI (PE channel) were detected by BD FACSVerse flow cytometer (BD Biosciences, San Jose, Canada). Quantification was achieved with FlowJo software.
2.6. RNA sequencing
Three groups of cells were prepared for RNA sequencing, including pEGFP control cells, SOD1-G93A model cells and SOD1-G93A cells treated with honokiol for 12 h. Total RNA was extracted and the constructed cDNA library was sequenced using BGIseq 500 (BGI-Shenzhen, China). The reads were mapped to mouse assembly (GRCm38.p6) reference genome using HISAT2 (v2.0.4). Differential gene expression was examined with DESeq2 (v1.4.5) where the Q value is less than or equal to 0.05.
2.7. Malondialdehyde (MDA) assay, 8-hydroxy-2′-deoxyguanosine (8-OHdG) and protein carbonyl (PCO) ELISA assay
Cells treated with honokiol or culture medium for 24 h were collected in PBS. Cells were lysed by sonication and following centrifuged at 4 °C for 10 min at 20,000 × g. The supernatant was collected for MDA assay using a cell MDA assay kit (A003-4, Nanjing Jiancheng Bioengineering Institute, Nanjing, China) following the manufacturer's instructions. Cells were lysed by freeze-thawed for 3 times and following centrifuged at 4 °C for 10 min at 20,000 × g. The supernatant was collected for 8-OHdG assay using a mouse 8-OHdG ELISA kit (CSB-E10527m, CUSBIO, Wuhan, China) and PCO assay using a mouse PCO ELISA kit (JL46483, Jianglai Biotechnology, Shanghai, China). The results were normalized to protein concentration.
2.8. DNA damage alkaline comet assay
The alkaline comet assay was performed as previously described35. All the procedures were performed in the dark with triplicates. The slides were stained with YeaRed Nucleic Acid Gel Stain (10202ES76, Yeasen). Images were captured by a fluorescence microscope (37XB, Shanghai Optical Instrument Factory, Shanghai, China). At least twenty-five cells of each treatment were measured by software CASP randomly.
2.9. Total antioxidant capacity assay (T-AOC), GSH and GSSG content assay, and NADPH assay
Cells treated with honokiol or culture medium for 24 h were collected as indicated. T-AOC assay was performed according to the manufacturer's instructions of a T-AOC Assay Kit with FRAP method (S0116, Beyotime, Shanghai, China). GSH and GSSG content assay was performed according to the manufacturer's instructions of a GSH and GSSG Assay Kit (S0053, Beyotime). NADPH content assay was performed according to the manufacturer's instructions of a NADPH Assay Kit with WST-8 (S0179, Beyotime).
2.10. Glutathione reductase (GSR) assay and catalase (CAT) assay
Cells treated with honokiol or culture medium for 24 h were collected in Cell lysis buffer for Western and IP (P0013, Beyotime). GSR assay was performed according to the manufacturer's instructions of GSR Assay Kit with DTNB (S0055, Beyotime). CAT assay was performed according to the manufacturer's instructions of CAT Assay Kit (S0051, Beyotime).
2.11. Mitochondrial membrane potential (MMP) determination, ATP content assay, and mitochondrial respiratory chain complex function assay
For MMP analysis by flow cytometry, cells treated with honokiol or culture medium for 24 h were incubated with tetramethylrhodamine ethyl ester (TMRE) working solution at a concentration of 100 nmol/L (87917, Sigma–Aldrich, St. Louis, MO, USA) at 37 °C for 0.5 h. TMRE fluorescence was collected using PE channel detected by flow cytometry. Quantification was achieved with FlowJo software. ATP content assay was performed according to the manufacturer's instructions of Enhanced ATP Assay Kit (S0027, Beyotime). The vitality measurement of the mitochondrial respiratory chain complex II and IV are measured by the Mitochondrial Respiratory Chain Complex Vitality Determination Kit (BC3235 and BC0945, Solarbio, Beijing, China) according to the manufacturer's instructions.
2.12. Mitochondrial stress test
Seahorse XF Cell Mito Stress Test kit (103015-100, Agilent Tec-hnologies, Palo Alto, CA, USA) was used to assess the metabolic function of mitochondrial by following parameters: basal respiration, ATP production-coupled respiration, maximal respiration, spare capacity, and non-mitochondrial respiration. During the test, oligomycin, FCCP, and a mixture of rotenone and antimycin A working solutions were added to each well sequentially to determine the parameters. The oxygen consumption rate is measured by the Seahorse XFe96 Analyzer (Agilent Techno-logies).
2.13. Mitochondrial morphology observation
Cells treated with honokiol or culture medium for 24 h was incubated with MitoTracker™ Deep Red FM (M22426, Invitrogen) working solution at 37 °C for 15 min according to the manufacturer's instructions. The Mitochondrial morphology of each group were observed by a Leica TCS SP8 (Leica Microsystems, Buffalo Grove, IL, USA) and analyzed by MiNA (Mitochondrial Network Analysis) of Image J.
2.14. Western blot
Whole cell lysates were prepared using RIPA lysis buffer (P0013B, Beyotime), supplemented with phosphatase inhibitors (04906845001, Roche, Indianapolis, IN, USA) and complete EDTA-free protease inhibitor mixtures (4693116001, Roche). The spinal cord tissue and gastrocnemius muscle tissue were prepared using RIPA lysis buffer supplemented with complete EDTA-free protease inhibitor mixtures and phosphatase inhibitors. The tissues were sonicated in the ice water bath until uniform lysates were prepared. The cell lysates and tissue lysates were then centrifuged at 4 °C for 0.5 h. The supernatant was collected for further protein quantification. For tissue lysates, one more centrifuge was needed. The mitochondrial protein was isolated by using a Cell Mitochondria Isolation Kit (C3601, Beyotime). Briefly, the cell lysates were collected in the mitochondrial isolation buffer from the kit and homogenized 20–30 times. The homogenate was then centrifuged at 4 °C for 10 min at 600 × g. The supernatant was collected and the centrifuged at 4 °C for 10 min at 11,000 × g. The mitochondrial precipitation was dissolved in the mitochondrial lysis from the kit. The protein quantification was performed by the Pierce™ BCA protein assay kit (23227, ThermoFisher Scientific, Carlsbad, CA, USA) following the manufacturer's instructions. As previously described36, protein samples were then diluted to the same concentration and boiled in loading buffer at 100 °C for 5 min. For Western blot, the protein samples were separated on SDS-polyacrylamide gels during the electrophoresis process. Separated proteins were then transferred electrophoretically onto PVDF membranes. Membranes were blocked by the 5% skimmed milk solution for 2 h at room temperature and then incubated at 4 °C overnight with antibodies against BCL2 (26539-1-AP, 1:1000), BAX (50599-2-Ig, 1:2000), cytochrome c (10993-1-AP, 1:2000), ATM (27156-1-AP, 1:1000), NRF2 (16396-1-AP, 1:1000), GCLC (12601-1-AP, 1:2000), GCLM (14241-1-AP, 1:2000), OPA1 (27733-1-AP, 1:1000), MFN2 (12186-1-AP, 1:2000), FIS1 (10956-1-AP, 1:1000), p62 (18420-1-AP, 1:1000), GFAP (26825-1-AP, 1:2000), HSP90 (13171-1-AP, 1:2000), GAPDH (60004-1-Ig, 1:5000) from Proteintech (Rosemont, IL, USA); GSR (sc-133245, 1:500), GSS (sc-166882, 1:500), VDAC1 (sc-390996, 1:500), PGC-1α (sc-518025, 1:500), NRF1 (sc-515360, 1:500), TFAM (sc-166965, 1:500) from Santa Cruz Biotechnology (Dallas, TX, USA); PINK1 (BC100-494S, 1:500) from Novus biological (Littleton, CO, USA); 4-HNE (ab46545, 1:1000), HO-1 (ab68477, 1:2000), NQO1 (ab80588, 1:1000) from Abcam (Cambridge, UK). Phospho-Histone H2AX (Ser139) (2577S, 1:1000), Lamin A/C (4777S, 1:1000) from Cell Signaling Technology (Danvers, MA, USA). IBA1 (016-20001, 1:1000) from Wako (Osaka, Japan). β-Actin (TA-09, 1:2000) from ZSGB-BIO (Beijing, China). Following anti-mouse or anti-rabbit IgG-HRP (Thermo Fisher Scientific) were used as secondary antibodies for 1.5 h at room temperature, signals were detected using ImageQuant LAS 4000 (GE Healthcare).
2.15. Antioxidant response element (ARE)-luciferase reporter assay
The HEK293-ARE reporter cells were seeded into 96-well plates and treated as indicated. The luciferase activity was detected using the Steady-Glo Luciferase Assay Reagent (Promega, Madison, WI, USA) according to the manufacturer's instructions. The luminescence intensity was measured by the Synergy H1 Hybrid Multi-Mode Microplate Reader (BioTek, Winooski, VT, USA).
2.16. RNA extraction, reverse transcription and quantitative RT-PCR analysis
Trizol reagent (15596026, Invitrogen) was used to isolate RNA samples from SOD1-G93A cells. cDNA was synthesized using Hifair® III 1st Strand cDNA Synthesis SuperMix for qPCR (gDNA digester plus) kit (11123ES60, Yeasen). Hieff UNICON® Power qPCR SYBR Green Master Mix (11197ES08, Yeasen) were used for RT-qPCR analysis under the ABI 7900HT Real-Time PCR System (Applied Biosystems, Carlsbad, CA, USA). The expression of mRNAs was normalized to Actb. The ΔΔCt method was used to calculate the relative fold change. Primers used for Hmox1 amplification: Forward 5′-CAGAAGAGGCTAAGACCGCC-3′; Reverse 5′-GCAGTATCTTGCACCAGGCTA-3′; Primers used for Nqo1 amplification: Forward 5′-CCATGTACGACAACGGTCCT-3′; Rev-erse 5′-GCAGGATGCCACTCTGAATC-3′; Primers used for Gclc amplification: Forward 5′-TGCTGTCCCAAGGCTCGC-3′; Reve-rse 5′-ACTCCACCTCGTCACCCCA-3′; Primers used for Gclm amplification: Forward 5′-ACAATGACCCGAAGAACTGC-3′; Reverse 5′-GAGCTGGAGTTAAGAGCCCC-3′; Primers used for Gss amplification: Forward 5′-GACAACGAGCGAGTTGGGA-3′; Reverse 5′-TGAATGGGGCATACGTCACC-3′; Primers used for Gsr amplification: Forward 5′-GACTGCCTTTACCCCGATGT-3′; Reverse 5′-TGAATGCCAACCACCTTTTC-C-3′; Primers used for Ppargc1a amplification: Forward 5′-CTGC-GGGATGATGGAGACAG-3′; Reverse 5′-TCGTTCGACCTGCGTAAAGT-3′; Primers used for Opa1 amplification: Forward 5′ATACTGGGATCTG-CTGTTGG-3′; Reverse 5′-AAGTCAGGCCAATCACTT-3′; Prim-ers used for Mfn2 amplification: Forward 5′-AAGCTTGGACAGG-TGGAGTC-3′; Reverse 5′-GAGC-AGGGACATCTCGTTTC-3′; Primers used for Fis1 amplification: Forward 5′-GCTGGTTCTG-TGTCCAAGAGCA-3′; Reverse 5′-GAC-ATAGTCCCGCTGT-TCCTCT-3′; Primers used for Actb amp-lification: Forward 5′-CGCAGCCACTGTCGAGTC-3′; Reverse 5′-GTCATCCATGGCGAACTGGT-3′.
2.17. Animals
Male hSOD1-G93A [B6SJL-Tg (SOD1∗G93A)/J] and cohort wild type mice were introduced from the Jackson Laboratories (Bar Harbor, ME, USA). Animals were fed in single cage, in controlled conditions of temperature (21–25 °C), humidity (45%–50%) and 12 h light/dark cycles, with ad libitum access to water and a standard rodent diet. All animal care and experimental procedures complied with the Animal Management Rule of the Ministry of Health, People's Republic of China (document No. 55, 2001) and were approved by the Laboratory Animal Ethics Committee of Chinese Academy of Medical Science, Beijing, China.
2.18. Drug administration
Honokiol injections were provided by School of Pharmaceutical Sciences, Peking University. Edaravone were purchased from Simcere Pharmaceutical Group (Nanjing, China). The experiment was divided into two batches. In the behavioral test and survival record, mice were randomly divided into six groups: (1) control group: wild type mice treated with vehicle; (2) model group: SOD1-G93A transgenic mice treated with vehicle; (3) positive drug group: SOD1-G93A transgenic mice treated with 15 mg/kg edaravone; (4) 10 μg/kg honokiol group: SOD1-G93A transgenic mice treated with 10 μg/kg honokiol; (5) 30 μg/kg honokiol group: SOD1-G93A transgenic mice treated with 30 μg/kg honokiol; (6) 90 μg/kg honokiol group-SOD1-G93A transgenic mice treated with 90 μg/kg honokiol. Mice were administrated drugs or vehicle control by intraperitoneal injection every day from the disease onset to end-stage of disease. The second batch mice were used for biochemical and histological studies. Mice were administrated drugs or vehicle control by intraperitoneal injection every day starting from aged 11 to 18 weeks, and then terminally anesthetized.
2.19. Rotarod test
The rotarod test was used to assess the motor function of the animals. Mice were trained on the rotarod daily for three days before recording the data. During the test, each mouse was placed on a 3.5 cm rotating rod (XR1514, Xinruan, Shanghai, China) with the same direction at the speed of 16 rpm within 180 s. The fall latency was recorded for all trials when the mouse fell from the rotarod. Once the latency was longer than 180 s, the score was recorded as 180 s. Each experiment was repeated three times, and the best score is recorded as the final score.
2.20. Hanging wire test
The Hanging wire test was used to assess the motor function of the animals. During the test, each mouse was placed on a cage cover, and gently knock the cover to impel the mice griping the cover. Flip the cover to make the mice upside down. The fall latency was recorded for all trials, once the latency is longer than 90 s, 90 s is record as the final score. Each experiment was repeated three times, and the best score is recorded as the final score.
2.21. Gripping test
The Gripping test was used to assess the motor function of the animals. During the test, each mouse was placed on the grip strength meter (XR-YLS-13A, Xinruan). Pull the mice tails gently to impel the mice griping the board with their forelimbs or hindlimbs, and then pull the mice tail backward rapidly with a uniform speed until mice release their grip. The gripping strength was recorded.
2.22. Histology, immunohistochemistry and immunofluorescence
At 18 weeks of age, SOD1-G93A transgenic and wild-type mice were sacrificed. Spinal cord and gastrocnemius muscle tissues were freshly isolated and fixed in 4% PFA. Immunofluorescence was performed on paraffin embedded sections (5 μm). Three gastrocnemius muscle sections of individual mice were stained with hematoxylin & eosin (H&E). Three spinal cord sections of individual mice were stained with Nissl (C0117, Beyotime) for evaluation of MNs. The levels of glial fibrillary acidic protein (GFAP) and ionized calcium binding adapter molecule 1 (IBA1) expression in the spinal cord were characterized by immunofluorescence. The spinal cord tissue sections were deparaffinized, rehydrated, antigen repaired and blocked by 10% goat serum working solution for 1 h at room temperature. The sections were incubated at 4 °C overnight with antibodies against GFAP (M0761, Dako, Copenhagen, Denmark) and IBA1 (019-19741, Wako). Following goat anti-rabbit IgG (H + L) highly cross-adsorbed secondary antibody, Alexa Fluor Plus 488 (A32731, Thermo Fisher Scientific) was used as secondary antibodies for 1 h at room temperature and examined by a fluorescence microscope. The level of choline acetyltransferase (ChAT) expression in the spinal cord was characterized by immunohistochemistry. The spinal cord tissue sections were deparaffinized, rehydrated, antigen repaired and then treated with 3% (v/v) H2O2 in PBS for 10 min, followed by blocking 10% goat serum working solution for 1 h at room temperature. The sections were incubated at 4 °C overnight with antibodies against ChAT (ab178850, Abcam). GTVision III Detection System/Mo&Rb (Including DAB) Kit (Gene Tech, Shanghai, China) was used for visualization and following examined by a light microscope.
2.23. Statistics analysis
All data are presented as the mean ± standard error of mean (SEM). Student's t-test was used for the comparison between two groups. One-way ANOVA or repeated measures ANOVA, followed by post hoc LSD test were used for the comparisons among three or more groups (SPSS 16.0). Kaplan–Meier analysis with log rank statistic test was used for the comparisons of the survival curves. Differences at P < 0.05 were considered statistically significant.
3. Results
3.1. Honokiol showed strong antioxidant capacity in vitro and protected the yeast against H2O2 induced oxidative damage
The chemical structure of honokiol was shown in Fig. 1A. The radical (DPPH, •OH, and ) scavenging activities of honokiol were evaluated in comparison with vitamin c (Vc) and edaravone. According to oxygen radical absorbance activity, both honokiol and edaravone showed a stronger antioxidant capacity than Vc (Fig. 1B). The ORAC of honokiol at 10 μmol/L was 8706.6 μmol Trolox/g, over three times larger than Vc. The ability of honokiol to scavenge DPPH were 15.3%, 20.0%, 26.8%, and 40.6%, respectively, at different concentrations ranging from 5 to 40 μmol/L (Fig. 1C). Honokiol exerted a moderate DPPH scavenging activity which was weaker than Vc and edaravone. The hydroxyl radicals scavenging activity of honokiol was much stronger than that of Vc and edaravone. The hydroxyl radicals scavenging activity of honokiol at concentrations from 100 to 800 μmol/L were 14.6%, 33.7%, 73.8%, and 100%, respectively, which exhibited a concentration-dependent scavenging activity (Fig. 1D). Fig. 1E shows a moderate superoxide radical scavenging activity of both honokiol and edaravone compared with Vc. The highest superoxide radicals scavenging activity reached 20.3% at 800 μmol/L of honokiol. Taken together, honokiol shows strong radicals scavenging capacity and antioxidant capacity in vitro.
Figure 1.

Honokiol showed strong radicals scavenging capacity and antioxidant capacity in vitro. (A) Chemical structure of honokiol; (B) oxygen radical absorbance capacity (n = 3); (C) scavenging capacity of DPPH radicals (n = 3); (D) scavenging capacity of hydroxyl radicals (n = 3); (E) scavenging capacity of superoxide radicals of Vc, edaravone, and honokiol (n = 4); (F) Temporal protein expression profiles of biomarkers indicative of different oxidative stress-related pathways upon exposure to H2O2 in the yeast (n = 3). Data are presented as the mean ± SEM.
To gain further insights into the antioxidant capacity of honokiol, the magnitude of expression changes of 44 proteins in the classical oxidative stress pathway were quantified at three concentrations in the yeast (Fig. 1F). The oxidant H2O2 upregulated a multiple protein expressions associated to oxidative stress damage in this yeast library, including sensor/regulator related proteins (YBP1, SLN1, MSN2, HAP2, HAP3, CUP2, MAC1, AFT1), the glutathione/glutaredoxin defense system (GSH1, GSH2, GRX1, GRX2, GPX1, HYR1, GPX2, GLR1, GTT1, GTT2, YCF1), the thioredoxin defense system (TRX1, TRX2, TSA1, TSA2, AHP1, PRX1, DOT5, SRX1, TRR1, TRR2), the enzymatic system (SOD1, SOD2, CCS1, CTT1, CCP1, GND2, ZWF1, PGM2) and the metal ion homeostasis defense system (CUP1-1). Interestingly, honokiol treatment (1 μmol/L) reduced the expressions of most oxidative stress-related proteins which were upregulated by H2O2, including MSN2, HAP3, CUP2, AFT1, GRX1, GPX1, TRX2, TSA1, TSA2, PRX1, SRX1, TRR1, TRR2, SOD1, SOD2, CCS1, CTT1, CCP1, GND2, ZWF1, and PGM2. The results indicate that honokiol exhibited a protective effect against H2O2-induced oxidative stress in the yeast.
3.2. Honokiol improved the cell viability of SOD1-G93A cells and protected SOD1-G93A cells from apoptosis
Given the radicals scavenging capacity in vitro and protective function in the yeast of honokiol, we determined whether honokiol had a protective role in the SOD1-G93A cell model. Compared with the pEGFP control group, the cell viability was significantly decreased to 86.3% after transfecting G93A SOD1, which showed the cytotoxic effects of the mutant form of SOD1. However, honokiol (3 and 10 μmol/L) significantly reversed the cell viability to 95.2% and 96.4%, respectively, compared with the SOD1-G93A model group model (Fig. 2A). The Hoechst dyeing assay was used to confirm the effects of honokiol on the cell viability, the Hoechst fluorescence intensity was significantly increased in 10 μmol/L honokiol-treated cells showing that honokiol improved the cell viability of SOD1-G93A cells (Supporting Information Fig. S1).
Figure 2.

Honokiol promoted the cell viability and reduced apoptosis in SOD1-G93A cells. (A) Cell viability analyzed by MTT assays (n = 3); (B) Apoptosis assay analyzed by flow cytometry, and early apoptosis (C) and late apoptosis (D) were analyzed by FlowJo (n = 4); (E) Western blot analysis and quantification of BCL2 and BAX in total cell lysates (n = 4); (F) Western blot analysis and quantification of cytochrome c in mitochondrial and cytoplasm cell lysates (n = 5). Cells were incubated with honokiol for 24 h in the experiments. One-way ANOVA followed by post-hoc LSD test was used for the comparisons among three or more groups (SPSS 16.0). Data are presented as the mean ± SEM. #P < 0.05, ##P < 0.01, ###P < 0.001 versus pEGFP control group; ∗P < 0.05, ∗∗P < 0.01, ∗∗∗P < 0.001 versus SOD1-G93A model group.
Annexin V-PI method was used to further verify the protective roles of honokiol in apoptosis. SOD1-G93A transfection significantly increased early (Annexin V positive and PI negative cells) and late (Annexin V and PI double positive cells) apoptosis to 127.5% and 138.5%, respectively, relative to the pEGFP control group. After treatment with honokiol, the early apoptosis rate obviously decreased to 91.8% and 79.8% (3 and 10 μmol/L) (Fig. 2B and C), and the late apoptosis rate also decreased to 90.0% and 65.2% (3 and 10 μmol/L, Fig. 2B and D). Then, the expressions of apoptosis-related proteins B-cell lymphoma 2 (BCL2), BCL2-associated X (BAX), and cytochrome c were detected. BCL2 expression was significantly increased (Fig. 2E) and cytoplasmic release of cytochrome c was markedly decreased (Fig. 2F) after the treatment of honokiol for 24 h compared with the SOD1-G93A model group. No alterations were observed in BAX protein level (Fig. 2E) nor mitochondrial protein level of cytochrome c (Fig. 2F). Together, these results suggest that honokiol treatment could alleviate SOD1-G93A induced neurotoxicity.
Moreover, we examined the protein expression of SOD1-G93A by Western blot and flow cytometry. The results show that honokiol had no direct influence on the clearance of the mutant SOD1 (Supporting Information Fig. S2).
3.3. Honokiol regulated redox homeostasis and mitochondrial function
In order to elucidate the main mechanism of honokiol in neuroprotection, we exploited RNA sequencing to analyze the differentially expressed genes after honokiol treatment. Totally 590 differentially genes were identified, including 187 upregulated genes (Supporting Information Table S2) and 403 downregulated genes (Supporting Information Table S3) after 12 h honokiol treatment at the concentration of 10 μmol/L (Fig. 3A and B). Kyoto Encyclopedia of Genes and Genomes (KEGG) enrichment analysis, Gene Set Enrichment Analysis (GSEA), and Gene Ontology (GO) enrichment analysis were used to clarify the potential mechanism. According to the KEGG enrichment analysis, we found GSH metabolism pathway was enriched in the honokiol treatment group (Fig. 3C). The transcription levels of six genes related to GSH metabolism were significantly changed after honokiol treatment including four up-regulated genes (Gsr, Gclm, Odc1, and Srm1) and two down-regulated genes (Idh1 and Ggt7). GSEA further showed that honokiol might affect several biological processes including mitochondrial translation, protein importing into mitochondrial matrix and positive regulation of ATPase activity (Fig. 3D). According to these results, we speculated that honokiol might improve the cellular redox homeostasis via decreasing oxidative stress and improving the mitochondrial function. Given several biological processes related to the mitochondrial function, we further used GO enrichment to analyze the differential expressed genes in connection with mitochondrial. The results show that honokiol affected a series of mitochondrial biological processes, including mitochondrial organization and establishment of protein localization to mitochondrion (Supporting Information Fig. S3). Honokiol increased the transcription levels of Gfer, Thg1l, Iba57 and Bcat1 which involved in the process of mitochondrial biogenesis and protein localization to mitochondrion. Honokiol also decreased the transcription levels of several genes including Tfrc, Mgarp, Stat2, and Lpin1 which related to mitochondrial fission, mitochondrial fusion inhibition, and collapse of the mitochondrial network. The results indicate that honokiol might regulate the mitochondrial biogenesis and the mitochondrial fusion–fission balance. According to the RNA sequencing results, we deduced that honokiol might exert neuroprotection due to maintaining redox homeostasis and regulating mitochondrial function in ALS cellular model.
Figure 3.

Honokiol regulated the antioxidant capacity and the mitochondrial function in SOD1-G93A cells. (A) Heatmap of the differential expressed genes; (B) Cluster analysis of the differential expressed genes; (C) KEGG enrichment analysis of the differential expressed genes; (D) GSEA of the differential expressed genes between SOD1-G93A model group and honokiol-treated group (n = 3).
3.4. Honokiol alleviated oxidative stress damage by activating NRF2/GSH signaling pathway in SOD1-G93A cells
Several oxidative stress damage markers were detected to further confirm the protective role of honokiol. ROS moiety was visualized with the fluorescent probe DHE to assess the effects of honokiol on ROS production. The result shows that SOD1-G93A cells had a much higher level of DHE intensity relative to the pEGFP control group, however, treatment with honokiol for 24 h significantly decreased the DHE intensity (Fig. 4A). MDA and 4-hydroxynonenal (4-HNE) content were detected to assess the effects of honokiol on lipid peroxidation damage. Compared with the pEGFP control group, the MDA content of SOD1-G93A cells was robustly increased, while the 4-HNE protein levels were increased but did not reach a significant difference. However, honokiol strongly decreased both the MDA and 4-HNE protein levels at the concentration of 10 μmol/L compared with SOD1-G93A model cells for 24 h treatment (Fig. 4B and C). The Western blot of 4-HNE protein showed only several bands might because that the antibody had high specificity or affinity for certain 4-HNE modified proteins that weigh 55–70 kDa. The PCO level was used to assess the effects of honokiol on protein oxidative damage. Compared with the pEGFP control group, the PCO content of SOD1-G93A cells was significantly increased; however, honokiol of 1 μmol/L decreased PCO content by 22.3% (P < 0.05, Fig. 4D). 8-OHdG content is indicator of DNA oxidative damage and the comet assay were usually used to assess DNA damage. Honokiol significantly decreased the level of 8-OHdG and the tail DNA rate induced by the mutant SOD1 expressing at 3 and 10 μmol/L for 24 h incubation, suggesting that honokiol might play DNA protective role in SOD1-G93A cells (Fig. 4E–G). We further detected the protein expressions of γH2AX and ataxia telangiectasia-mutated (ATM) which are related to DNA damage. The results showed that both γH2AX (P = 0.08) and ATM (P = 0.06) expressions had a decreasing tendency after 24 h honokiol incubation at the concentration of 10 μmol/L (Supporting Information Fig. S4).
Figure 4.

Honokiol alleviated the oxidative stress damage in SOD1-G93A cells. Effect of honokiol on (A) DHE intensity analyzed by flow cytometry (n = 3); (B) MDA content (n = 3); (C) 4-HNE protein level (n = 4); (D) PCO level (n = 3); (E) 8-OHdG level (n = 4); (F) and (G) DNA damage analyzed by comet assay (n = 3). Scale bars, 100 μm; (H) T-AOC capacity (n = 3); (I) NADPH content (n = 4); (J) GSH content (n = 5); (K) CAT activity (n = 3); (L) GSR activity (n = 4). Cells were incubated with honokiol for 24 h in the experiments. One-way ANOVA followed by post hoc LSD test was used for the comparison among three or more groups (SPSS 16.0). Data are presented as the mean ± SEM. ##P < 0.01, ###P < 0.001 versus pEGFP control group; ∗P < 0.05, ∗∗P < 0.01, ∗∗∗P < 0.001 versus SOD1-G93A model group.
In addition, the levels of antioxidant capacity, including T-AOC, NAPDH content, GSH content, CAT activity and GSR activity were measured after 24 h honokiol treatment. The T-AOC was concentration-dependently elevated by 31% (P < 0.05) and 65% (P < 0.001) at 3 and 10 μmol/L of honokiol, respectively, compared with the SOD1-G93A model group (Fig. 4H). The NADPH content was significantly increased by 52% (P < 0.05) and 58% (P < 0.05, Fig. 4I) and the GSH/GSSG ratio was significantly raised by 66% (P < 0.05) and 91% (P < 0.05) at 3 and 10 μmol/L of honokiol, respectively, compared with the SOD1-G93A model group (Fig. 4J). Apart from increasing the antioxidants, honokiol also significantly increased CAT activity and GSR activity at the concentration of 10 μmol/L (Fig. 4K and L) relative to the SOD1-G93A model group.
NRF2 is well known as a key transcription factor of the antioxidant response by regulating cytoprotective genes, including the antioxidant GSH pathway. ARE-luciferase reporter assay confirmed that honokiol had an NRF2 activation effect (Fig. 5A). Honokiol also stimulated the nuclear translocalization of NRF2 (Fig. 5B and C). The Western blot result showed honokiol significantly increased the nuclear NRF2 expression at 3 and 10 μmol/L for 24 h treatment (Fig. 5C). The Western blot method was used to detect the NRF2 protein level in the nucleus to determine NRF2 activation. The band of NRF2 is expected to run within 95–110 kDa as double bands in SDS-PAGE37 and was marked in the nuclear section. The band appeared in the cytoplasm part in our result should be unspecific bands. In addition, transcription levels of NRF2-target genes including Hmox1, Nqo1, Gclm, Gss and Gsr were significantly upregulated after 12 h honokiol treatment at 10 μmol/L (Fig. 5D). We further observed that honokiol obviously lifted the protein expressions of these NRF2-target genes including heme oxygenase-1 (HO-1), NAD(P)H: quinone oxidoreductase 1 (NQO1), glutamate–cysteine ligase catalytic subunit (GCLC), glutamate–cysteine ligase modifier subunit (GCLM), glutathione synthetase (GSS) and GSR at the concentration of 10 μmol/L (Fig. 5E). The results indicate that NRF2/GSH signaling pathway was activated by honokiol treatment and might involve in the neuroprotection of honokiol. To confirm the involvement of NRF2–GSH pathway, NRF2 inhibitor ML38538,39 (2.5 μmol/L) was used to incubate together with honokiol for 24 h in the MTT assay. Compared with the model group, 10 μmol/L honokiol significantly raised viability of SOD1-G93A cells; however, when incubating with ML385 together, honokiol didn't show improvement in the cell viability compared with the cells simply incubated with ML385 (Supporting Information Fig. S5). The result indicates that ML385 nullified the improvement of honokiol in the cell viability which confirmed that NRF2–GSH pathway might play an important role in the neuroprotection of honokiol.
Figure 5.

Honokiol activated the NRF2 anti-oxidant pathway in the SOD1-G93A cells. (A) Luminescence intensity of HEK293-ARE reporter cells after treatment with indicated concentrations of honokiol for 6, 12 and 24 h (n = 3); (B) Representative images of NRF2 nuclear translocalization after 24 h honokiol treatment. Scale bars, 8 μm; (C) Western blot analysis and quantification of NRF2 in nuclear and cytoplasmic cell lysates after 24 h honokiol treatment (n = 4); (D) RT-qPCR analysis of transcription levels of Hmox1, Nqo1, Gclm, Gclc, Gss and Gsr after 12 h honokiol treatment (n = 3–7); (E) Western blot analysis and quantification of HO-1, NQO1, GCLC, GCLM, GSS and GSR in total cell lysates after 24 h honokiol treatment (n = 3–6). One-way ANOVA followed by post hoc LSD test was used for the comparison among three or more groups (SPSS 16.0). Data are presented as the mean ± SEM. #P < 0.05, ###P < 0.001 versus pEGFP control group; ∗P < 0.05, ∗∗P < 0.01, ∗∗∗P < 0.001 versus SOD1-G93A model group.
3.5. Honokiol restored the mitochondrial function and morphology by ameliorating mitochondrial biogenesis and fusion–fission balance
Besides oxidative stress, mitochondrial dysfunction is a common pathology in ALS. Mutant G93A SOD1 disrupted MMP, whereas honokiol-treated cells exhibited higher MMP indicating less mitochondrial membrane depolarization (Fig. 6A). Besides, honokiol significantly boosted the ATP level compared with SOD1-G93A model cells (Fig. 6B). Mitochondrial oxidative respiration was measured by the Seahorse XF Cell Mito Stress Test kit. As shown in Supporting Information Fig. S6, honokiol of 10 μmol/L significantly enhanced the spare respiratory capacity of SOD1-G93A cells (P < 0.05) for 24 h treatment. In addition, the activity of mitochondrial complex II and complex IV were also increased after 24 h treatment of honokiol at the concentration of 10 μmol/L compared with SOD1-G93A model group (Fig. 6C and D). The mitochondrial morphology analysis showed the mean network size and footprint of the mitochondrial of the SOD1-G93A cells were significantly decreased relative to the pEGFP group; however, honokiol of 10 μmol/L increased the mean network size (P = 0.07) and the footprint (P < 0.001) for 24 h treatment (Fig. 6E, H and J) compared with the SOD1-G93A cells. No significant alterations were observed in individuals, networks, and mean branch length (Fig. 6F, G, and I).
Figure 6.

Honokiol preserved the mitochondrial function and mitochondrial morphology in the SOD1-G93A cells. (A) The MMP assessment analyzed by flow cytometry with the fluorescent probe TMRE (n = 3–4); (B) ATP content analysis (n = 3); (C) Mitochondrial complex II activity determination (n = 5); (D) Mitochondrial complex IV activity determination (n = 6); (E) Representative confocal microscopy images of mitochondrial morphology stained by mitotracker. Scale bars, 8 μm; (F–J) Mitochondrial morphology assessment including individuals, networks, mean network size, mean branch length, and footprint analyzed by MiNA. Cells were incubated with honokiol for 24 h in the experiments (n = 3). One-way ANOVA followed by post hoc LSD test was used for the comparisons among three or more groups (SPSS 16.0). Data are presented as the mean ± SEM. ###P < 0.001 versus pEGFP control group; ∗P < 0.05, ∗∗∗P < 0.001 versus SOD1-G93A model group.
Mitochondrial biogenesis is an important process for regulating the number of mitochondria in the cell. Peroxisome proliferator-activated receptor gamma coactivator-1 alpha (PGC-1α), nuclear respiratory factor 1 (NRF1) and mitochondrial transcription factor A (TFAM) were reported to take part in mitochondrial biogenesis. The PGC-1α and NRF1 expressions were markedly reduced in SOD1-G93A cells. Honokiol treatment for 12 h showed an increasing tendency for Ppargc1a transcription level (P = 0.08, Supporting Information Fig. S7) at 10 μmol/L and significantly upregulated the PGC-1α, NRF1, and TFAM protein expressions relative to the SOD1-G93A model group at 10 μmol/L. Therefore, honokiol improved mitochondrial biogenesis in the SOD1-G93A cells (Fig. 7A).
Figure 7.

Honokiol improved the mitochondrial biogenesis, mitochondrial fusion–fission balance and mitophagy. (A) Western blot analysis and quantification of PGC-1α, NRF1, and TFAM in total cell lysates (n = 4–6); (B) Western blot analysis and quantification of OPA1, MFN2, and FIS1 in total cell lysates (n = 3–5); (C) Western blot analysis and quantification of PINK1 and p62 in mitochondrial lysates (n = 3–4); (D) Representative confocal microscopy images of COX IV and LAMP1 co-immunofluorescence staining and the quantification of yellow fusion dots (n = 3). Scale bars, 10 μm. Cells were incubated with honokiol for 24 h in the experiments. One-way ANOVA followed by post hoc LSD test was used for the comparisons among three or more groups (SPSS 16.0). Data are presented as the mean ± SEM. #P < 0.05, ##P < 0.01 versus pEGFP control group; ∗P < 0.05, ∗∗P < 0.01, ∗∗∗P < 0.001 versus SOD1-G93A model group.
Mitochondrial dynamics and turnover are crucial to regulating cellular bioenergetics. Our further investigation found that SOD1-G93A overexpression disrupted normal mitochondrial fission/fusion balance. The transcription levels of mitochondrial fusion protein Opa1 and Mfn2 showed no significant difference between the pEGFP cells and SOD1-G93A cells and honokiol increased the transcription levels of Opa1 (P < 0.05) and Mfn2 (P = 0.07) at the concentration of 10 μmol/L for 12 h treatment (Fig. S7). The protein level of optic atrophy 1 (OPA1) significantly decreased (P < 0.05) and mitofusin-2 (MFN2) had a tendency for a decrease (P = 0.06) in the SOD1-G93A model group, while honokiol restored the protein expressions of both OPA1 and MFN2 at the concentration of 3 and 10 μmol/L (Fig. 7B). The transcription level of mitochondrial fission protein Fis1 was significantly decreased in the SOD1-G93A cells compared with pEGFP cells while honokiol significantly increased the Fis1 transcription level (Fig. S7). However, the protein level of fission 1 (FIS1) was obviously increased in the SOD1-G93A cell model while there was no alteration after honokiol treatment (Fig. 7B). Overall, according to the results, the SOD1-G93A cells exerted mitochondrial fission/fusion unbalance compared with pEGFP cells which showed lower mitochondrial fusion protein expression levels while honokiol significantly mediated the relative protein expressions and improved the mitochondrial dynamics.
We next examined the expression of mitophagy-related protein PTEN induced putative kinase 1 (PINK1) in the isolated mitochondrial protein of the cell lysates. We found PINK1 expression was increased to some content while didn't reach to statistical significance in the SOD1-G93A model group, meanwhile, substantial p62 was accumulated in the mitochondrial in the SOD1-G93A cells compared with pEGFP cells. Interestingly, honokiol of 10 μmol/L elevated the expressions of mitochondrial PINK1 while decreased the accumulation of mitochondrial p62 which indicated a higher mitophagy level (Fig. 7C). We further detected the co-localized puncta of cytochrome c oxidase subunit IV (COX IV) and lysosomal-associated membrane protein 1 (LAMP1) which indicated the success mitophagy. Honokiol obviously enhanced the co-localized yellow puncta showing improved mitophagy flux (Fig. 7D).
Meanwhile, we respectively co-incubated the mitochondrial biogenesis inhibitor doxycycline (10 μmol/L)40, 41, 42, mitochondrial fusion protein OPA1 inhibitor MYLS22 (10 μmol/L)43,44 and autophagy inhibitor chloroquine (50 μmol/L, inhibiting lysosomal acidification therefore blocking autophagy) and honokiol for 24 h and then performed MTT assay in SOD1-G93A cells to examine whether the protection of honokiol were disrupted. The results demonstrated that honokiol had not shown benefits in the cell viability when co-incubating with doxycycline compared with the cells simply incubating doxycycline (Fig. S5). Besides, honokiol showed a slight increasing (P = 0.06) in the cell viability when co-incubating with MYLS22 compared with the cells simply incubating MYLS22, but the difference didn't reach to statistical significance (Fig. S5). However, chloroquine did not affect the honokiol-induced cell viability elevation (Fig. S5). Taken together, these results suggest that both doxycycline and MYLS22 discounted honokiol's protection which further confirmed the involvement of the mitochondrial biogenesis and mitochondrial dynamics in the honokiol-exerted neuroprotection.
3.6. Honokiol improved motor functions and extended the lifespan of transgenic SOD1-G93A mice
Given the cytoprotective effects of honokiol exhibited in the SOD1-G93A cell model, we used the SOD1-G93A transgenic mice as an animal model to further assess the effects of honokiol (Fig. 8A). The weight of the SOD1-G93A mice was significantly decreased compared with WT mice, and no alterations were observed between model mice and honokiol-treated mice (Supporting Information Fig. S8). However, honokiol administration significantly improved motor functions of the transgenic SOD1-G93A mice. SOD1-G93A mice treated with 30 μg/kg honokiol had a longer latency in rotarod test (Fig. 8C) and hanging wire test (Fig. 8D) than vehicle mice which reflected the hindlimb motor function. No alteration was observed in the gripping test between the model group and honokiol groups (Fig. S8). More importantly, honokiol-treated mice had a longer survival relative to the model group (Fig. 8B). The average survival of the SOD1-G93A transgenic model group was 150 days, while the average survival was extended to 158 days after honokiol treatment at the dose of 30 μg/kg.
Figure 8.

Honokiol improved the motor function, extended the lifespan, and alleviated the pathology of the SOD1-G93A transgenic mice. (A) The experimental process of in vivo studies; (B) The survival determination (n = 6–8); (C) The rotarod latency determination (n = 6–8); (D) The hanging wire test latency determination (n = 6–8); (E) Immunohistochemistry of ChAT positive MNs of the spinal cord (n = 3). Scale bars, 200 μm; (F) Western blot analysis and quantification of GFAP and IBA1 in the spinal cord tissue lysates (n = 4); (G) and (H) Representative images of GFAP and IBA1 immunofluorescence staining of the spinal cord. Scale bars, 100 μm; (I) Representative images of H&E staining of the gastrocnemius muscle. Scale bars, 100 μm. Kaplan–Meier analysis with log rank statistic test was used for the comparisons of the survival curves among the indicated groups (SPSS 16.0). Repeated measures ANOVA followed by post hoc LSD test was used for the comparisons of rotarod latency and hanging wire test latency determination among the indicated groups (SPSS 16.0). Student's t-test was used for the comparison of the ChAT positive MNs between SOD1-G93A mice and honokiol-treated mice (SPSS 16.0). One-way ANOVA followed by post hoc LSD test was used for the comparisons among the indicated groups in the Western blot quantification (SPSS 16.0). Data are presented as the mean ± SEM. ##P < 0.01, ###P < 0.001 versus WT control group; ∗P < 0.05, ∗∗P < 0.01, ∗∗∗P < 0.001 versus SOD1-G93A model group.
3.7. Honokiol alleviated the ALS pathology via activating NRF2/GSH pathway and improving mitochondrial function in SOD1-G93A mice
Degeneration of MNs and glial cell reactivity in the central nervous system are important neuropathological features of ALS. We found the number of ChAT positive MNs of the spinal cord had an increasing tendency (P = 0.07) after honokiol administration compared with SOD1-G93A vehicle group (Fig. 8E). Meanwhile, the levels of GFAP and IBA1, the markers of astrocytes and microglia activations were significantly raised in SOD1-G93A transgenic mice. Fortunately, honokiol significantly decreased the levels of both GFAP and IBA1 (Fig. 8F–H).
Muscle atrophy is one of the major pathological changes in the gastrocnemius muscle of the SOD1-G93A mice. H&E staining showed that the myocyte in the WT group had a uniform size while the SOD1-G93A myocyte appeared groups of small and atrophy myocytes which were called “group atrophy” as the arrows shown. However, honokiol seemed to reverse the pathological abnormal (Fig. 8I).
To investigate the antioxidant capacity of honokiol in the SOD1-G93A mice, the NRF2/GSH related genes were examined. We did not find significant changes in the expressions of HO-1, NQO1, GCLC and GSS in honokiol-treated group (Fig. 9A and D). However, the expressions of GSR and GCLM were significantly increased in the spinal cord after honokiol treatment (Fig. 9A and D). Similar results were also observed in the gastrocnemius muscle, that honokiol increased the protein expressions of GCLC and GSR (P < 0.05), and partially lifted the expressions of GCLM (P = 0.08) and NQO1 (P = 0.09), while had no effects on HO-1 level (Fig. 9G and J). The results suggest that honokiol might regulate the NRF2/GSH pathway to exert antioxidant capacity in the spinal cord and gastrocnemius muscle of the SOD1-G93A mice.
Figure 9.

Honokiol activated the NRF2–GSH pathway and improved mitochondrial function in the spinal cord and gastrocnemius muscle of SOD1-G93A mice. (A) and (D) Western blot analysis and quantification of HO-1, NQO1, GCLC, GCLM, GSS and GSR in the spinal cord (n = 3–4); (B) and (E) Western blot analysis and quantification of PGC-1α, NRF1 and TFAM in the spinal cord (n = 4); (C) and (F) Western blot analysis and quantification of OPA1, MFN2 and FIS1 in the spinal cord (n = 4); (G) and (J) Western blot analysis and quantification analysis of HO-1, NQO1, GCLC, GCLM, GSS and GSR in the gastrocnemius muscle (n = 4); (H) and (K) Western blot analysis and quantification of PGC-1α, NRF1 and TFAM in the gastrocnemius muscle (n = 4–6); (I) and (L) Western blot analysis and quantification of OPA1, MFN2 and FIS1 in the gastrocnemius muscle (n = 4). One-way ANOVA followed by post hoc LSD test was used for the comparisons among three or more groups (SPSS 16.0). Data are presented as the mean ± SEM (n = 3–6, repeated independent experiments). #P < 0.05, ##P < 0.01 versus WT control group; ∗P < 0.05, ∗∗P < 0.01 versus SOD1-G93A model group.
We further detected the protein expressions related to mitochondrial biogenesis, mitochondrial fusion and fission, and mitophagy in the spinal cord. PGC-1α expression had a decreasing tendency in SOD1-G93A mice (P = 0.09) compared with WT mice. No significant changes were observed in NRF1 and TFAM expressions between WT mice and SOD1-G93A mice. Honokiol treatment significantly increased the protein expressions of PGC-1α, NRF1 and TFAM (P < 0.05, Fig. 9B and E). About mitochondrial fusion and fission, the OPA1 expression was significantly decreased while the MFN2 expression was decreased but the difference did not reach to statistical significance in SOD1-G93A mice. No alterations were detected in FIS1 expression between WT mice and SOD1-G93A mice. Honokiol treatment elevated the expression of OPA1 (P < 0.05, Fig. 9C and F). The results of gastrocnemius muscle were basically same as that in spinal cord. Honokiol significantly raised the expressions of NRF1 and TFAM and had an increasing tendency for PGC-1α (P = 0.09, Fig. 9H and K). Meantime, we observed that long-form OPA1 (>100 kDa) was the main form in WT mice while short-form OPA1 (<100 kDa) was the main form in SOD1-G93A mice. Interestingly, honokiol increased the long-form and short-form ratio of OPA1 (Fig. 9I and L). Whether in spinal cord or gastrocnemius muscle, we did not find any changes in the mitophagy-related protein-PINK1 among the three groups (Supporting Information Fig. S9).
4. Discussion
ALS is a fatal neurodegenerative disease characterized by the loss of the upper and lower MNs in the brain and spinal cord with no current effective treatments, although riluzole and edaravone have been approved in clinic. Honokiol is a pleiotropic compound isolated from the plant of Magnolia grandiflora, and has been reported to display neuroprotective effects on several CNS diseases, such as ischemic stroke, Alzheimer's disease and Parkinson's disease, due to its potent antioxidation, anti-excitotoxicity, mitochondrial protection and anti-neuroinflammation28,33,45, 46, 47, 48, 49. However, whether honokiol has a beneficial function in ALS is unknown. As honokiol can readily permeate the blood-cerebrospinal fluid, here we try to reveal the efficacy of honokiol on ALS50. The present work provides important evidence that honokiol shows an obvious neuroprotection in SOD1-G93A cellular model and transgenic mice. ALS-related cell model by stably transfecting an EGFP-tagged G93A mutated form of SOD1 plasmid into NSC-34 cell line (a mouse neural hybrid cell line produced by a fusion of motor neuron-enriched embryonic mouse spinal cord cells with mouse neuroblastoma)51, has been proved to suffer impaired proliferation and cell differentiation, being more sensitive to oxidative stress, and having more fragmentation of Golgi apparatus compared with control cells which mimic several pathologies in ALS cells. These results demonstrate the reliability of NSC34-hSOD1-G93A cells to be used as a model of ALS MNs52,53. Progressive loss of MNs is the main pathology in ALS54, we identified that honokiol improved the survival rate and alleviated the apoptosis in SOD1-G93A cells. Most importantly, honokiol slowed the progression of motor system dysfunction and prolonged the survival time in SOD1-G93A ALS transgenic mice. Honokiol had no significant effects on the weight loss and there were similar findings that the survival of mice was extended without improvement in body weight after treatment55,56. In this study, we did not find the significant changes in the gripping test which mainly examined the strength of forelimbs, while the increased latency in rotarod test and hanging wire test suggested that honokiol improved motor function by enhancing the hindlimbs strength and alleviating gastrocnemius pathology which developed earlier in the disease progress and were often more serious. Furtherly, we verified that honokiol partially reversed spinal MNs loss, but the difference was not significant, possibly because of high variability and small sample size. Interestingly, honokiol markedly alleviated the glia cells activation which were the obvious pathological changes in the spinal cord of ALS. As reported, honokiol might directly inhibit the activations of astrocyte and microglia, and then reduced neuroinflammation in several models including Alzheimer's disease animal model and Experimental Autoimmune Encephalomyelitis model57,58. Besides, honokiol was demonstrated to alleviate oxidative stress and reactive astrocyte induction through decreasing NADPH-oxidase and GFAP expressions in Parkinson's disease animal model46. The effects of honokiol on alleviating glial cells activation might be a comprehensive result of the anti-inflammation capacity and anti-oxidant capacity. In addition, progressive degeneration of skeletal muscles is an early pathological feature of ALS which likely occurs prior to MNs in ALS patients and in the SOD1-G93A ALS mouse model6,59,60. Honokiol mitigated the group atrophy of the gastrocnemius muscle compared with the SOD1-G93A model mice. Taken together, honokiol might play a therapeutic role in ALS by ameliorating motor system decline and pathological damage.
Increasing evidence highlights the importance of redox imbalance in the pathogenesis of ALS. ALS patients have high levels of oxidative stress markers61, and about 61% of the ALS patient samples display gene expression signatures consistent with a robust response to oxidative stress62. Mutant SOD1 disrupts the redox homeostasis of ALS through the abnormal production of ROS, reactive nitrogen and the formation of misfolded protein aggregates63. Here, RNA sequencing analysis hinted that honokiol might involve in regulating oxidative stress and mitochondrial function in mutant SOD1-G93A MNs. Thus, we evaluated the oxidative stress damage in the SOD1-G93A cells and found honokiol decreased the lipid peroxidation damage, protein oxidation damage, and DNA oxidation damage. It has been demonstrated that the T-AOC was decreased in the ALS patient cerebrospinal fluid64 while honokiol significantly increased the T-AOC in a dose dependent manner. In addition, honokiol had considerable effects with edaravone on oxygen radical absorbance and superoxide anion radical scavenging, a lower effect on DPPH scavenging, but an obvious higher effect on hydroxyl radical scavenging compared with edaravone in the in vitro evaluation of antioxidant capacity. By using a yeast oxidative stress library in which the yeast was each transfected an oxidative stress related gene with GFP tag, a series of antioxidant genes expressions were downregulated after honokiol treatment compared with H2O2 model group, implying honokiol had a strong anti-oxidative potential in ALS models.
There are two main systems to deal with oxidative stress in vivo, enzymatic system such as SOD, CAT, glutathione peroxidase, and thioredoxin peroxidase and non-enzymatic antioxidant system such as GSH, thioredoxin (TRX), Vc, and others. GSH and TRX are the two antioxidant systems depending on the sulfhydryl group. GSH, the reduced form of glutathione, a well-known antioxidant sulfhydryl (–SH) tripeptide, is cellular essential antioxidant substance to non-enzymatically react with ROS and prevent oxidative damage65. It has been reported that GSH was lower in the motor cortex of ALS patients as compared with healthy volunteers66. A reduction in the GSH/GSSG ratio had also been observed in the cerebrospinal fluid of ALS patients15,67. GSH depletion promotes neurological deficits, mitochondrial dysfunction68, and MNs degeneration69 in SOD1-G93A mice. In the study, honokiol exhibited a strong elevation in GSH/GSSG ratio in SOD1-G93A cells. One of the most important regulators of the antioxidant response in cells is NRF2-kelch-like ECH-associated protein 1 signal. It is known that GSH production is mainly regulated by NRF2 pathway70. When the cell is insulted by oxidative stress, NRF2 dissociates from kelch-like ECH-associated protein 1, translocates into the nucleus and binds the AREs in promoter regions, and then regulates the transcription of approximately 250 genes to build a multifaceted network that integrates cellular activities including detoxification reactions, the maintenance of both redox and protein homeostasis, and energy metabolism13. Notably, the impairment of NRF2 antioxidant pathway has been linked to ALS71,72. HO-1 is regulated by NRF2 and mainly catalyzes the catabolism of heme to ferrous iron, carbon monoxide and biliverdin acting as an important antioxidant enzyme. The degradation of heme is beneficial to prevent its pro-oxidative effect73. Besides, biliverdin and bilirubin are potent antioxidants which help to resist the oxidative stress. As a NRF2-target gene, NQO1 exerted detoxification of quinones mainly by reduction of quinones to hydroquinones which further decreased the electrophilic quinones and avoided the semiquinone radical and ROS generation74. Glutamate-cysteine ligase (GCL), composed of catalytic (GCLC) and regulatory/modifier (GCLM) subunits, is the rate-limiting enzyme for cellular GSH biosynthesis and regulated by NRF275. GSS which also involves in the biosynthesis of GSH, similarly, is a NRF2-related gene76, 77, 78. GSR which reduces the oxidized GSSG to GSH accompanied with NADPH oxidation that involves in the regeneration of GSH, is also regulated by NRF276, 77, 78, 79. According to the existing publications, honokiol exerted protective effects and alleviated oxidative stress via upregulating GSH synthesis in several models including the acute kidney injury ischemia model80, the renal ischemia and reperfusion injury model81, liver damage model82, and N-methyl-d-aspartic acid injury model83. Honokiol is expected to be a multi-target compound and here we demonstrated a significant increase of NRF2 nuclear translocation and upregulation of the NRF2-target genes including HO-1, NQO1, GCLC, GCLM, GSS and GSR after honokiol treatment in SOD1-G93A cells. As mentioned in the RNA sequencing result, GSH metabolism pathway was enriched in the honokiol treatment group and was verified in the GSH content assay. Therefore, we speculated honokiol exerted anti-oxidant function through affecting the complex NRF2–GSH pathway and involved in multiple anti-oxidant processes. Besides, the NRF2 inhibitor ML385 significantly blocked the improvements of honokiol on the cell viability, showing the importance of the NRF2–GSH pathway acted on the protection of honokiol. At the meantime, honokiol promoted GSH synthesis and regeneration through increasing GCLM, GCLC and GSR expression in the MNs and gastrocnemius muscle of SOD1-G93A transgenic mice. In the current study, we mainly explored the possible anti-oxidative mechanism of honokiol depending on the RNA sequencing results, and more researches related to other anti-oxidant pathways such as TRX system would be performed in further research. The existing results suggest that NRF2/GSH signal pathway involves in the anti-oxidative damage function of honokiol in ALS therapy.
Mitochondria–oxidative stress–neurodegenerative disease interaction is well evident in ALS as mentioned before10,84. The mitochondrial morphology abnormalities and mitochondrial function disruption is a common pathology appeared in ALS patients and ALS models. The impaired MMP, ATP production and mitochondrial respiratory chain complex were reported in ALS patients and ALS models85,86. The decrease of mitochondrial biogenesis and mitophagy, and elevation of mitochondrial fission level were observed in the peripheral blood mononuclear cells from the ALS patients24. In this study, the mean network size and the footprint of the mitochondrial morphology in SOD1-G93A cells were significantly decreased in SOD1-G93A cells as previous reported87,88, while honokiol showed an increasing tendency in the mean network size and evidently hoist the footprint of the mitochondrial, indicating alleviation of mitochondrial fragmentation. Meanwhile, honokiol treatment clearly enhanced the mitochondrial function, such as MMP, ATP content and the activity of mitochondrial respiratory chain complexes II and IV. To further investigate which pathway takes part in honokiol-regulated mitochondrial function, the key protein molecules of mitochondrial biogenesis (PGC-1α, NRF1, and TFAM), mitochondrial fusion and fission (OPA1, MFN2, and FIS1), and mitophagy pathway (PINK1 and p62) were ascertained. Similar to the known reports, the SOD1-G93A cells suffered impaired mitochondrial biogenesis, unbalanced mitochondrial fusion-fission with downregulated fusion proteins of OPA1 and MFN2 and upregulated protein level of FIS1, and impaired mitophagy with decreased mitochondrial-lysosome fusion. However, honokiol obviously boosted the expressions of the mitochondrial biogenesis related proteins PGC-1α, NRF1 and TFAM, and the mitochondrial fusion related proteins-OPA1 and MFN2. Besides, honokiol recruited more mitophagy related protein PINK1 to the mitochondrial and decreased the mitochondrial p62 accumulation, meanwhile increased mitochondrial–lysosome fusion dots, indicating that honokiol might improve the clearance of the damaged mitochondrial. The mitochondrial biogenesis inhibitor doxycycline and mitochondrial fusion protein OPA1 inhibitor MYLS22 discounted the effects of honokiol on the cell viability which showed the involvement of the mitochondrial dynamics in the protection of honokiol. Similar results were further confirmed in SOD1-G93A transgenic mice. Honokiol increased mitochondrial biogenesis through increasing the protein expression of PGC-1α, NRF1 and TFAM in ALS mice. Here we observed some different changes of OPA1 in ALS cells and tissues. Long OPA1 isoforms (L-OPA1) carry out inner membrane fusion when MMP is intact. However, L-OPA1 isoforms are cleaved to short (S-OPA1) isoforms upon loss of MMP, collapsing the mitochondrial network and promoting apoptosis89,90. In the SOD1-G93A cells and the spinal cord of the SOD1-G93A mice, the L-OPA1 was decreased obviously in ALS model group and honokiol clearly reversed the changes, while the S-OPA1 was unchanged in each group. However, in the gastrocnemius muscle, honokiol treatment significantly increased the L-OPA1 level. Meanwhile, the S-OPA1 significantly increased in model group and honokiol treatment reversed this change. Taken L-OPA1/S-OPA1 ratio as an index89, we found the ratio was significantly decreased in the gastrocnemius muscle of the SOD1-G93A mice while honokiol eliminated the reduction. No significant alterations were found in PINK1 protein levels SOD1-G93A mice, indicating mitophagy shows slight effect on honokiol-regulated mitochondrial function.
There are a few limitations in our study which need more research. First, the number of SOD1-G93A mice was not huge enough. Second, more experiments should be carried out around the oxidative stress and mitochondrial dysfunction in vivo. All in all, this is the first study to demonstrate the therapeutic effects of honokiol in the ALS through SOD1-G93A stably-expressing cells and SOD1-G93A transgenic mice in vitro and in vivo. Currently, honokiol has applied patent for ALS treatment in China (CN113057951A). The research provided a new strategy for ALS treatment.
5. Conclusions
Our study first demonstrated that honokiol protected the MNs from the toxicity of mutant SOD1-G93A in vitro and in vivo. In addition, we found that honokiol inhibited oxidative stress damage via NRF2/GSH signaling pathway and improved the mitochondrial dysfunction by increasing the mitochondrial biogenesis and maintaining the mitochondrial fusion–fission balance. Honokiol appears to be promising as a multitarget drugs for ALS treatment.
Acknowledgments
This project was supported by the grants from National Natural Science Foundation of China (Nos. 82073835 and 81872855), National Key R&D Program of China (No. 2019YFC1708901), CAMS Innovation Fund for Medical Sciences (No. 2021-I2M-1-028), Beijing Key Laboratory of New Drug Mechanisms and Pharmacological Evaluation Study (BZ0150, China), and Disciplines construction project (201920200802, China).
Footnotes
Peer review under responsibility of Chinese Pharmaceutical Association and Institute of Materia Medica, Chinese Academy of Medical Sciences.
Supporting data to this article can be found online at https://doi.org/10.1016/j.apsb.2022.07.019.
Contributor Information
Hongtao Jin, Email: jinhongtao@imm.ac.cn.
Shizhong Chen, Email: chenshizhong66@163.com.
Ying Peng, Email: ypeng@imm.ac.cn.
Author contributions
Ying Peng, Yujun Zhou and Jingshu Tang designed the experiments. Yujun Zhou, Xinhong Feng and Yong Zhang conducted biological experiments. Yujun Zhou and Jiaqi Lan conducted yeast experiments. Yujun Zhou and Yuying Kang conducted the in vivo experiments. Yujun Zhou, Hongyue Wang and Qiuyu Chen analyzed the data. Yujun Zhou and Yong Zhang drafted the manuscript. Ying Peng, Shizhong Chen and Hongtao Jin conceived and supervised the project. Yujun Zhou, Jingshu Tang, Yang Sun and Lei Wu participated in data analysis. Ying Peng revised and finalized the manuscript writing. All authors read and approved the final version of the manuscript.
Conflicts of interest
The authors have no conflicts of interest to declare.
Appendix A. Supporting information
The following is the Supplementary data to this article:
References
- 1.Longinetti E., Fang F. Epidemiology of amyotrophic lateral sclerosis: an update of recent literature. Curr Opin Neurol. 2019;32:771–776. doi: 10.1097/WCO.0000000000000730. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Pasinelli P., Brown R.H. Molecular biology of amyotrophic lateral sclerosis: insights from genetics. Nat Rev Neurosci. 2006;7:710–723. doi: 10.1038/nrn1971. [DOI] [PubMed] [Google Scholar]
- 3.Renton A.E., Chio A., Traynor B.J. State of play in amyotrophic lateral sclerosis genetics. Nat Neurosci. 2014;17:17–23. doi: 10.1038/nn.3584. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Chen L.X., Xu H.F., Wang P.S., Yang X.X., Wu Z.Y., Li H.F. SOD1 mutation spectrum and natural history of ALS patients in a 15-year cohort in southeastern China. Front Genet. 2021;12 doi: 10.3389/fgene.2021.746060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Liu X., He J., Gao F.B., Gitler A.D., Fan D. The epidemiology and genetics of amyotrophic lateral sclerosis in China. Brain Res. 2018;1693:121–126. doi: 10.1016/j.brainres.2018.02.035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Massopust R., Juros D., Shapiro D., Lopes M., Haldar S.M., Taetzsch T., et al. KLF15 overexpression in myocytes fails to ameliorate ALS-related pathology or extend the lifespan of SOD1G93A mice. Neurobiol Dis. 2021;162 doi: 10.1016/j.nbd.2021.105583. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.D'Ambrosi N., Cozzolino M., Carri M.T. Neuroinflammation in amyotrophic lateral sclerosis: role of redox (dys)regulation. Antioxid Redox Signal. 2018;29:15–36. doi: 10.1089/ars.2017.7271. [DOI] [PubMed] [Google Scholar]
- 8.Johann S., Heitzer M., Kanagaratnam M., Goswami A., Rizo T., Weis J., et al. NLRP3 inflammasome is expressed by astrocytes in the SOD1 mouse model of ALS and in human sporadic ALS patients. Glia. 2015;63:2260–2273. doi: 10.1002/glia.22891. [DOI] [PubMed] [Google Scholar]
- 9.Le Gall L., Anakor E., Connolly O., Vijayakumar U.G., Duddy W.J., Duguez S. Molecular and cellular mechanisms affected in ALS. J Pers Med. 2020;10:101. doi: 10.3390/jpm10030101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Obrador E., Salvador-Palmer R., López-Blanch R., Jihad-Jebbar A., Vallés S.L., Estrela J.M. The link between oxidative stress, redox status, bioenergetics and mitochondria in the pathophysiology of ALS. Int J Mol Sci. 2021;22:6352. doi: 10.3390/ijms22126352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Bono S., Feligioni M., Corbo M. Impaired antioxidant KEAP1–NRF2 system in amyotrophic lateral sclerosis: NRF2 activation as a potential therapeutic strategy. Mol Neurodegener. 2021;16:71. doi: 10.1186/s13024-021-00479-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Abed D.A., Goldstein M., Albanyan H., Jin H., Hu L. Discovery of direct inhibitors of Keap1–Nrf2 protein–protein interaction as potential therapeutic and preventive agents. Acta Pharm Sin B. 2015;5:285–299. doi: 10.1016/j.apsb.2015.05.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Zhang Y., Shi Z., Zhou Y., Xiao Q., Wang H., Peng Y. Emerging substrate proteins of kelch-like ECH associated protein 1 (Keap1) and potential challenges for the development of small-molecule inhibitors of the Keap1-nuclear factor erythroid 2-related factor 2 (Nrf2) protein–protein interaction. J Med Chem. 2020;63:7986–8002. doi: 10.1021/acs.jmedchem.9b01865. [DOI] [PubMed] [Google Scholar]
- 14.Zhang Y., Zhou Y.J., Tang J.S., Lan J.Q., Kang Y.Y., Wu L., et al. A comparison study between dimethyl itaconate and dimethyl fumarate in electrophilicity, Nrf2 activation, and anti-inflammation in vitro. J Asian Nat Prod Res. 2022;24:577–588. doi: 10.1080/10286020.2021.1949303. [DOI] [PubMed] [Google Scholar]
- 15.Kim K. Glutathione in the nervous system as a potential therapeutic target to control the development and progression of amyotrophic lateral sclerosis. Antioxidants (Basel) 2021;10:1011. doi: 10.3390/antiox10071011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Hemerkova P., Valis M. Role of oxidative stress in the pathogenesis of amyotrophic lateral sclerosis: antioxidant metalloenzymes and therapeutic strategies. Biomolecules. 2021;11:437. doi: 10.3390/biom11030437. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Kowaltowski A.J., Vercesi A.E. Mitochondrial damage induced by conditions of oxidative stress. Free Radic Biol Med. 1999;26:463–471. doi: 10.1016/s0891-5849(98)00216-0. [DOI] [PubMed] [Google Scholar]
- 18.Wang J., Zhou H. Mitochondrial quality control mechanisms as molecular targets in cardiac ischemia–reperfusion injury. Acta Pharm Sin B. 2020;10:1866–1879. doi: 10.1016/j.apsb.2020.03.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Obrador E., Salvador R., Lopez-Blanch R., Jihad-Jebbar A., Valles S.L., Estrela J.M. Oxidative stress, neuroinflammation and mitochondria in the pathophysiology of amyotrophic lateral sclerosis. Antioxidants (Basel) 2020;9:901. doi: 10.3390/antiox9090901. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Calio M.L., Henriques E., Siena A., Bertoncini C.R.A., Gil-Mohapel J., Rosenstock T.R. Mitochondrial dysfunction, neurogenesis, and epigenetics: putative implications for amyotrophic lateral sclerosis neurodegeneration and treatment. Front Neurosci. 2020;14:679. doi: 10.3389/fnins.2020.00679. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Singh T., Jiao Y., Ferrando L.M., Yablonska S., Li F., Horoszko E.C., et al. Neuronal mitochondrial dysfunction in sporadic amyotrophic lateral sclerosis is developmentally regulated. Sci Rep. 2021;11 doi: 10.1038/s41598-021-97928-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Smith E.F., Shaw P.J., De Vos K.J. The role of mitochondria in amyotrophic lateral sclerosis. Neurosci Lett. 2019;710 doi: 10.1016/j.neulet.2017.06.052. [DOI] [PubMed] [Google Scholar]
- 23.Sasaki S., Horie Y., Iwata M. Mitochondrial alterations in dorsal root ganglion cells in sporadic amyotrophic lateral sclerosis. Acta Neuropathol. 2007;114:633–639. doi: 10.1007/s00401-007-0299-1. [DOI] [PubMed] [Google Scholar]
- 24.Araujo B.G., Souza E.S.L.F., de Barros Torresi J.L., Siena A., Valerio B.C.O., Brito M.D., et al. Decreased mitochondrial function, biogenesis, and degradation in peripheral blood mononuclear cells from amyotrophic lateral sclerosis patients as a potential tool for biomarker research. Mol Neurobiol. 2020;57:5084–5102. doi: 10.1007/s12035-020-02059-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Muyderman H., Hutson P.G., Matusica D., Rogers M.L., Rush R.A. The human G93A-superoxide dismutase-1 mutation, mitochondrial glutathione and apoptotic cell death. Neurochem Res. 2009;34:1847–1856. doi: 10.1007/s11064-009-9974-z. [DOI] [PubMed] [Google Scholar]
- 26.Turner B.J., Talbot K. Transgenics, toxicity and therapeutics in rodent models of mutant SOD1-mediated familial ALS. Prog Neurobiol. 2008;85:94–134. doi: 10.1016/j.pneurobio.2008.01.001. [DOI] [PubMed] [Google Scholar]
- 27.Walczak J., Debska-Vielhaber G., Vielhaber S., Szymanski J., Charzynska A., Duszynski J., et al. Distinction of sporadic and familial forms of ALS based on mitochondrial characteristics. FASEB J. 2019;33:4388–4403. doi: 10.1096/fj.201801843R. [DOI] [PubMed] [Google Scholar]
- 28.Talarek S., Listos J., Barreca D., Tellone E., Sureda A., Nabavi S.F., et al. Neuroprotective effects of honokiol: from chemistry to medicine. Biofactors. 2017;43:760–769. doi: 10.1002/biof.1385. [DOI] [PubMed] [Google Scholar]
- 29.Woodbury A., Yu S.P., Wei L., Garcia P. Neuro-modulating effects of honokiol: a review. Front Neurol. 2013;4:130. doi: 10.3389/fneur.2013.00130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Yang L., Wang Z.H., Lei H., Chen R.D., Wang X.L., Peng Y., et al. Neuroprotective glucosides of magnolol and honokiol from microbial-specific glycosylation. Tetrahedron. 2014;70:8244–8251. [Google Scholar]
- 31.Tan Y., Yu H., Sun S., Gan S., Gong R., Mou K.J., et al. Honokiol exerts protective effects on neural myelin sheaths after compressed spinal cord injury by inhibiting oligodendrocyte apoptosis through regulation of ER-mitochondrial interactions. J Spinal Cord Med. 2022;45:595–604. doi: 10.1080/10790268.2021.1890878. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Chen H.H., Chang P.C., Chen C., Chan M.H. Protective and therapeutic activity of honokiol in reversing motor deficits and neuronal degeneration in the mouse model of Parkinson's disease. Pharmacol Rep. 2018;70:668–676. doi: 10.1016/j.pharep.2018.01.003. [DOI] [PubMed] [Google Scholar]
- 33.Hou M., Bao W., Gao Y., Chen J., Song G. Honokiol improves cognitive impairment in APP/PS1 mice through activating mitophagy and mitochondrial unfolded protein response. Chem Biol Interact. 2022;351 doi: 10.1016/j.cbi.2021.109741. [DOI] [PubMed] [Google Scholar]
- 34.Wu X., Luo J., Liu H., Cui W., Feng D., Qu Y. SIRT3 protects against early brain injury following subarachnoid hemorrhage via promoting mitochondrial fusion in an AMPK dependent manner. Chin Neurosurg J. 2020;6:1. doi: 10.1186/s41016-019-0182-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Lan J., Gou N., Gao C., He M., Gu A.Z. Comparative and mechanistic genotoxicity assessment of nanomaterials via a quantitative toxicogenomics approach across multiple species. Environ Sci Technol. 2014;48:12937–12945. doi: 10.1021/es503065q. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Tang J., Kang Y., Huang L., Wu L., Peng Y. TIMP1 preserves the blood–brain barrier through interacting with CD63/integrin beta 1 complex and regulating downstream FAK/RhoA signaling. Acta Pharm Sin B. 2020;10:987–1003. doi: 10.1016/j.apsb.2020.02.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Kemmerer Z.A., Ader N.R., Mulroy S.S., Eggler A.L. Comparison of human Nrf2 antibodies: a tale of two proteins. Toxicol Lett. 2015;238:83–89. doi: 10.1016/j.toxlet.2015.07.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Singh A., Venkannagari S., Oh K.H., Zhang Y.Q., Rohde J.M., Liu L., et al. Small molecule inhibitor of NRF2 selectively intervenes therapeutic resistance in KEAP1-deficient NSCLC tumors. ACS Chem Biol. 2016;11:3214–3225. doi: 10.1021/acschembio.6b00651. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Lin Y., Luo T., Weng A., Huang X., Yao Y., Fu Z., et al. Gallic acid alleviates gouty arthritis by inhibiting NLRP3 inflammasome activation and pyroptosis through enhancing Nrf2 signaling. Front Immunol. 2020;11 doi: 10.3389/fimmu.2020.580593. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Moullan N., Mouchiroud L., Wang X., Ryu D., Williams E.G., Mottis A., et al. Tetracyclines disturb mitochondrial function across eukaryotic models: a call for caution in biomedical research. Cell Rep. 2015;10:1681–1691. doi: 10.1016/j.celrep.2015.02.034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Ozsvari B., Sotgia F., Lisanti M.P. A new mutation-independent approach to cancer therapy: inhibiting oncogenic RAS and MYC, by targeting mitochondrial biogenesis. Aging (Albany NY) 2017;9:2098–2116. doi: 10.18632/aging.101304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Scatena C., Roncella M., Di Paolo A., Aretini P., Menicagli M., Fanelli G., et al. Doxycycline, an inhibitor of mitochondrial biogenesis, effectively reduces cancer stem cells (CSCs) in early breast cancer patients: a clinical pilot study. Front Oncol. 2018;8:452. doi: 10.3389/fonc.2018.00452. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Herkenne S., Ek O., Zamberlan M., Pellattiero A., Chergova M., Chivite I., et al. Developmental and tumor angiogenesis requires the mitochondria-shaping protein Opa1. Cell Metab. 2020;31:987–1003.e8. doi: 10.1016/j.cmet.2020.04.007. [DOI] [PubMed] [Google Scholar]
- 44.Zhuang S., Xia S., Huang P., Wu J., Qu J., Chen R., et al. Targeting P2RX1 alleviates renal ischemia/reperfusion injury by preserving mitochondrial dynamics. Pharmacol Res. 2021;170 doi: 10.1016/j.phrs.2021.105712. [DOI] [PubMed] [Google Scholar]
- 45.Chen C.M., Liu S.H., Lin-Shiau S.Y. Honokiol, a neuroprotectant against mouse cerebral ischaemia, mediated by preserving Na+, K+-ATPase activity and mitochondrial functions. Basic Clin Pharmacol Toxicol. 2007;101:108–116. doi: 10.1111/j.1742-7843.2007.00082.x. [DOI] [PubMed] [Google Scholar]
- 46.Chen H.H., Chang P.C., Wey S.P., Chen P.M., Chen C., Chan M.H. Therapeutic effects of honokiol on motor impairment in hemiparkinsonian mice are associated with reversing neurodegeneration and targeting PPARgamma regulation. Biomed Pharmacother. 2018;108:254–262. doi: 10.1016/j.biopha.2018.07.095. [DOI] [PubMed] [Google Scholar]
- 47.Govindarajulu M., Ramesh S., Neel L., Fabbrini M., Buabeid M., Fujihashi A., et al. Nutraceutical based SIRT3 activators as therapeutic targets in Alzheimer's disease. Neurochem Int. 2021;144 doi: 10.1016/j.neuint.2021.104958. [DOI] [PubMed] [Google Scholar]
- 48.Liou K.T., Shen Y.C., Chen C.F., Tsao C.M., Tsai S.K. Honokiol protects rat brain from focal cerebral ischemia–reperfusion injury by inhibiting neutrophil infiltration and reactive oxygen species production. Brain Res. 2003;992:159–166. doi: 10.1016/j.brainres.2003.08.026. [DOI] [PubMed] [Google Scholar]
- 49.Pacifici F., Rovella V., Pastore D., Bellia A., Abete P., Donadel G., et al. Polyphenols and ischemic stroke: insight into one of the best strategies for prevention and treatment. Nutrients. 2021;13:1967. doi: 10.3390/nu13061967. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Wang X., Duan X., Yang G., Zhang X., Deng L., Zheng H., et al. Honokiol crosses BBB and BCSFB, and inhibits brain tumor growth in rat 9L intracerebral gliosarcoma model and human U251 xenograft glioma model. PLoS One. 2011;6 doi: 10.1371/journal.pone.0018490. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Cashman N.R., Durham H.D., Blusztajn J.K., Oda K., Tabira T., Shaw I.T., et al. Neuroblastoma x spinal cord (NSC) hybrid cell lines resemble developing motor neurons. Dev Dyn. 1992;194:209–221. doi: 10.1002/aja.1001940306. [DOI] [PubMed] [Google Scholar]
- 52.Kim K.Y., Kim Y.R., Choi K.W., Lee M., Lee S., Im W., et al. Downregulated miR-18b-5p triggers apoptosis by inhibition of calcium signaling and neuronal cell differentiation in transgenic SOD1 (G93A) mice and SOD1 (G17S and G86S) ALS patients. Transl Neurodegener. 2020;9:23. doi: 10.1186/s40035-020-00203-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Pinto C., Cardenas P., Osses N., Henriquez J.P. Characterization of Wnt/beta-catenin and BMP/Smad signaling pathways in an in vitro model of amyotrophic lateral sclerosis. Front Cell Neurosci. 2013;7:239. doi: 10.3389/fncel.2013.00239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Scaricamazza S., Salvatori I., Amadio S., Nesci V., Torcinaro A., Giacovazzo G., et al. Repurposing of trimetazidine for amyotrophic lateral sclerosis: a study in SOD1(G93A) mice. Br J Pharmacol. 2022;179:1732–1752. doi: 10.1111/bph.15738. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Lee J.D., McDonald T.S., Fung J.N.T., Woodruff T.M. Absence of receptor for advanced glycation end product (RAGE) reduces inflammation and extends survival in the hSOD1(G93A) mouse model of amyotrophic lateral sclerosis. Mol Neurobiol. 2020;57:4143–4155. doi: 10.1007/s12035-020-02019-9. [DOI] [PubMed] [Google Scholar]
- 56.Winter A.N., Ross E.K., Wilkins H.M., Stankiewicz T.R., Wallace T., Miller K., et al. An anthocyanin-enriched extract from strawberries delays disease onset and extends survival in the hSOD1(G93A) mouse model of amyotrophic lateral sclerosis. Nutr Neurosci. 2018;21:414–426. doi: 10.1080/1028415X.2017.1297023. [DOI] [PubMed] [Google Scholar]
- 57.Hsiao Y.P., Chen H.T., Liang Y.C., Wang T.E., Huang K.H., Hsu C.C., et al. Development of nanosome-encapsulated honokiol for intravenous therapy against experimental autoimmune encephalomyelitis. Int J Nanomedicine. 2020;15:17–29. doi: 10.2147/IJN.S214349. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Qu C., Li Q.P., Su Z.R., Ip S.P., Yuan Q.J., Xie Y.L., et al. Nano-Honokiol ameliorates the cognitive deficits in TgCRND8 mice of Alzheimer's disease via inhibiting neuropathology and modulating gut microbiota. J Adv Res. 2022;35:231–243. doi: 10.1016/j.jare.2021.03.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Loeffler J.P., Picchiarelli G., Dupuis L., Gonzalez De Aguilar J.L. The role of skeletal muscle in amyotrophic lateral sclerosis. Brain Pathol. 2016;26:227–236. doi: 10.1111/bpa.12350. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Marcuzzo S., Zucca I., Mastropietro A., de Rosbo N.K., Cavalcante P., Tartari S., et al. Hind limb muscle atrophy precedes cerebral neuronal degeneration in G93A-SOD1 mouse model of amyotrophic lateral sclerosis: a longitudinal MRI study. Exp Neurol. 2011;231:30–37. doi: 10.1016/j.expneurol.2011.05.007. [DOI] [PubMed] [Google Scholar]
- 61.Xiong L., McCoy M., Komuro H., West X.Z., Yakubenko V., Gao D., et al. Inflammation-dependent oxidative stress metabolites as a hallmark of amyotrophic lateral sclerosis. Free Radic Biol Med. 2021;178:125–133. doi: 10.1016/j.freeradbiomed.2021.11.031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Tam O.H., Rozhkov N.V., Shaw R., Kim D., Hubbard I., Fennessey S., et al. Postmortem cortex samples identify distinct molecular subtypes of ALS: retrotransposon activation, oxidative stress, and activated glia. Cell Rep. 2019;29:1164–11677.e5. doi: 10.1016/j.celrep.2019.09.066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Yang X., Ji Y., Wang W., Zhang L., Chen Z., Yu M., et al. Amyotrophic lateral sclerosis: molecular mechanisms, biomarkers, and therapeutic strategies. Antioxidants (Basel) 2021;10:1012. doi: 10.3390/antiox10071012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Siciliano G., Piazza S., Carlesi C., Del Corona A., Franzini M., Pompella A., et al. Antioxidant capacity and protein oxidation in cerebrospinal fluid of amyotrophic lateral sclerosis. J Neurol. 2007;254:575–580. doi: 10.1007/s00415-006-0301-1. [DOI] [PubMed] [Google Scholar]
- 65.DeLeve L.D., Kaplowitz N. Glutathione metabolism and its role in hepatotoxicity. Pharmacol Ther. 1991;52:287–305. doi: 10.1016/0163-7258(91)90029-l. [DOI] [PubMed] [Google Scholar]
- 66.Weiduschat N., Mao X., Hupf J., Armstrong N., Kang G., Lange D.J., et al. Motor cortex glutathione deficit in ALS measured in vivo with the J-editing technique. Neurosci Lett. 2014;570:102–107. doi: 10.1016/j.neulet.2014.04.020. [DOI] [PubMed] [Google Scholar]
- 67.Tohgi H., Abe T., Yamazaki K., Murata T., Ishizaki E., Isobe C. Increase in oxidized NO products and reduction in oxidized glutathione in cerebrospinal fluid from patients with sporadic form of amyotrophic lateral sclerosis. Neurosci Lett. 1999;260:204–206. doi: 10.1016/s0304-3940(98)00986-0. [DOI] [PubMed] [Google Scholar]
- 68.Vargas M.R., Johnson D.A., Johnson J.A. Decreased glutathione accelerates neurological deficit and mitochondrial pathology in familial ALS-linked hSOD1(G93A) mice model. Neurobiol Dis. 2011;43:543–551. doi: 10.1016/j.nbd.2011.04.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Killoy K.M., Harlan B.A., Pehar M., Helke K.L., Johnson J.A., Vargas M.R. Decreased glutathione levels cause overt motor neuron degeneration in hSOD1(WT) over-expressing mice. Exp Neurol. 2018;302:129–135. doi: 10.1016/j.expneurol.2018.01.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Furstenau C.R., de Souza I.C.C., de Oliveira M.R. Tanshinone I induces mitochondrial protection by a mechanism involving the Nrf2/GSH axis in the human neuroblastoma SH-SY5Y cells exposed to methylglyoxal. Neurotox Res. 2019;36:491–502. doi: 10.1007/s12640-019-00091-1. [DOI] [PubMed] [Google Scholar]
- 71.Deng Z., Lim J., Wang Q., Purtell K., Wu S., Palomo G.M., et al. ALS-FTLD-linked mutations of SQSTM1/p62 disrupt selective autophagy and NFE2L2/NRF2 anti-oxidative stress pathway. Autophagy. 2020;16:917–931. doi: 10.1080/15548627.2019.1644076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Mimoto T., Miyazaki K., Morimoto N., Kurata T., Satoh K., Ikeda Y., et al. Impaired antioxydative Keap1/Nrf2 system and the downstream stress protein responses in the motor neuron of ALS model mice. Brain Res. 2012;1446:109–118. doi: 10.1016/j.brainres.2011.12.064. [DOI] [PubMed] [Google Scholar]
- 73.Facchinetti M.M. Heme-oxygenase-1. Antioxid Redox Signal. 2020;32:1239–1242. doi: 10.1089/ars.2020.8065. [DOI] [PubMed] [Google Scholar]
- 74.Ross D., Siegel D. The diverse functionality of NQO1 and its roles in redox control. Redox Biol. 2021;41 doi: 10.1016/j.redox.2021.101950. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Jin Y., Huang Z.L., Li L., Yang Y., Wang C.H., Wang Z.T., et al. Quercetin attenuates toosendanin-induced hepatotoxicity through inducing the Nrf2/GCL/GSH antioxidant signaling pathway. Acta Pharmacol Sin. 2019;40:75–85. doi: 10.1038/s41401-018-0024-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Lee J.M., Calkins M.J., Chan K., Kan Y.W., Johnson J.A. Identification of the NF-E2-related factor-2-dependent genes conferring protection against oxidative stress in primary cortical astrocytes using oligonucleotide microarray analysis. J Biol Chem. 2003;278:12029–12038. doi: 10.1074/jbc.M211558200. [DOI] [PubMed] [Google Scholar]
- 77.Scibior A., Wojda I., Wnuk E., Pietrzyk L., Plewa Z. Response of cytoprotective and detoxifying proteins to vanadate and/or magnesium in the rat liver: the Nrf2–Keap1 system. Oxid Med Cell Longev. 2021;2021 doi: 10.1155/2021/8447456. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Thimmulappa R.K., Mai K.H., Srisuma S., Kensler T.W., Yamamoto M., Biswal S. Identification of Nrf2-regulated genes induced by the chemopreventive agent sulforaphane by oligonucleotide microarray. Cancer Res. 2002;62:5196–5203. [PubMed] [Google Scholar]
- 79.Rysava A., Cizkova K., Frankova J., Roubalova L., Ulrichova J., Vostalova J., et al. Effect of UVA radiation on the Nrf2 signalling pathway in human skin cells. J Photochem Photobiol B. 2020;209 doi: 10.1016/j.jphotobiol.2020.111948. [DOI] [PubMed] [Google Scholar]
- 80.Xia S., Lin H., Liu H., Lu Z., Wang H., Fan S., et al. Honokiol attenuates sepsis-associated acute kidney injury via the inhibition of oxidative stress and inflammation. Inflammation. 2019;42:826–834. doi: 10.1007/s10753-018-0937-x. [DOI] [PubMed] [Google Scholar]
- 81.Park E.J., Dusabimana T., Je J., Jeong K., Yun S.P., Kim H.J., et al. Honokiol protects the kidney from renal ischemia and reperfusion injury by upregulating the glutathione biosynthetic enzymes. Biomedicines. 2020;8:352. doi: 10.3390/biomedicines8090352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Yu F.L., Wu J.W., Zhu H. Honokiol alleviates acetaminophen-induced hepatotoxicity via decreasing generation of acetaminophen–protein adducts in liver. Life Sci. 2019;230:97–103. doi: 10.1016/j.lfs.2019.05.062. [DOI] [PubMed] [Google Scholar]
- 83.Cui H.S., Huang L.S., Sok D.E., Shin J., Kwon B.M., Youn U.J., et al. Protective action of honokiol, administered orally, against oxidative stress in brain of mice challenged with NMDA. Phytomedicine. 2007;14:696–700. doi: 10.1016/j.phymed.2007.03.005. [DOI] [PubMed] [Google Scholar]
- 84.Greco V., Longone P., Spalloni A., Pieroni L., Urbani A. Crosstalk between oxidative stress and mitochondrial damage: focus on amyotrophic lateral sclerosis. Adv Exp Med Biol. 2019;1158:71–82. doi: 10.1007/978-981-13-8367-0_5. [DOI] [PubMed] [Google Scholar]
- 85.Draper A.C.E., Wilson Z., Maile C., Faccenda D., Campanella M., Piercy R.J. Species-specific consequences of an E40K missense mutation in superoxide dismutase 1 (SOD1) FASEB J. 2020;34:458–473. doi: 10.1096/fj.201901455R. [DOI] [PubMed] [Google Scholar]
- 86.Gao J., Wang L., Liu J., Xie F., Su B., Wang X. Abnormalities of mitochondrial dynamics in neurodegenerative diseases. Antioxidants (Basel) 2017;6:25. doi: 10.3390/antiox6020025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Luo G., Yi J., Ma C., Xiao Y., Yi F., Yu T., et al. Defective mitochondrial dynamics is an early event in skeletal muscle of an amyotrophic lateral sclerosis mouse model. PLoS One. 2013;8 doi: 10.1371/journal.pone.0082112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Song W., Song Y., Kincaid B., Bossy B., Bossy-Wetzel E. Mutant SOD1G93A triggers mitochondrial fragmentation in spinal cord motor neurons: neuroprotection by SIRT3 and PGC-1alpha. Neurobiol Dis. 2013;51:72–81. doi: 10.1016/j.nbd.2012.07.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Garcia I., Calderon F., la Torre P., Vallier S.S., Rodriguez C., Agarwala D., et al. Mitochondrial OPA1 cleavage is reversibly activated by differentiation of H9c2 cardiomyoblasts. Mitochondrion. 2021;57:88–96. doi: 10.1016/j.mito.2020.12.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Gilkerson R., De La Torre P., St Vallier S. Mitochondrial OMA1 and OPA1 as gatekeepers of organellar structure/function and cellular stress response. Front Cell Dev Biol. 2021;9 doi: 10.3389/fcell.2021.626117. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.