

Abstract
Mouse models for the study of cancer immunology provide excellent systems in which to test biological mechanisms of the immune response against cancer. Historically, these models have been designed to have different strengths based on the current major research questions at the time. As such, many mouse models of immunology used today were not originally developed to study questions currently plaguing the relatively new field of cancer immunology, but instead have been adapted for such purposes. In this review, we discuss various mouse model of cancer immunology in a historical context as a means to provide a fuller perspective of each model’s strengths. From this outlook, we discuss the current state of the art and strategies for tackling future modeling challenges.
Keywords: Cancer, Immunology, Laboratory mice, genetically engineered animal
Introduction
The ground-breaking anti-cancer responses to checkpoint inhibitor (CPI) immunotherapy in patients across many cancer types have forced us to reconsider long held assumptions about the anti-tumor immune response. Likewise, this impressive clinical success has revealed many important unanswered questions in cancer immunology regarding the potential of patients’ natural anti-cancer immune responses. The study of human tumor samples, while extremely valuable, is largely correlative by nature. Thus, exploring the mechanisms of these immunotherapies and investigating ways to improve their efficacies requires mouse models that appropriately reflect the natural development of an anti-cancer immune response. The majority of available models have fallen short of being ideal for such studies and this, in large part, is due to the fact that many cancers were previously thought to be non-immunogenic and thus, models of these cancers were not designed for the study of cancer immunology. This is par for the course when it comes to mouse model development, as the combination of technological advancements and new research findings are constantly driving a need for more advanced models, and vice versa. The cyclical nature of this process is apparent when viewing the development of mouse models of cancer through a historical lens, providing context for the current state of available models. The ideal mouse model of cancer immunology will likely be ever-evolving and, as such, remains just out of reach. Therefore, choosing the best model or models for a particular research question should be a decision made based on the current understanding of cancer immunology as well as the requirements of the research project in question. While animal models besides the mouse have been useful for the study of cancer (White et al., 2015, Davis and Ostrander, 2014, Dewi and Cline, 2021, Schachtschneider et al., 2017), this review will exclusively focus on mouse models of cancer immunology. We will first discuss what we have learned so far and reflect on the model evolution that helped us get here. Finally, we will address current modeling challenges and efforts to overcome the limitations of available models. An immense amount of research has contributed to what is now the current state of cancer immunology mouse modeling. Here, rather than provide an exhaustive account of all contributions, we focus on highlights as a means to discuss larger concepts. For those interested in diving deeper into topics mentioned throughout this review, we will reference various in-depth reviews found elsewhere.
Pre-existing anti-tumor immunity drives therapeutic responsiveness
The concept of immune-surveillance, in which a natural anti-tumor adaptive immune response shapes tumor development and cancer progression, was first proposed by Paul Erlich in 1901. Yet, the role of the immune system in cancer development remained under-appreciated and highly debated until the late 1980s. That is not to say that the use of immunotherapies in general was discounted, as the investigation into such therapies date back to a century ago (reviewed elsewhere: (Zhang and Zhang, 2020)), but rather that the presence of a natural anti-tumor immune response in absence of therapy was uncertain. The discovery that tumor-specific antigens in melanomas that could be recognized by T cells helped to establish an active role for the adaptive immune system in anti-tumor responses (Lee et al., 1999). Additionally, the discovery that cytotoxic T-lymphocyte-associated protein 4 (CTLA-4) functioned as an inhibitory molecule on T cells helped to revive the theory of immunosurveillance (Leah et al., 1996, Tivol et al., 1995). At this time only a handful of cancer types were considered to have immunogenic potential such as melanoma and renal cell carcinoma whereas other types of solid tumors were considered “non-immunogenic”. The impressive responses in some patients to PD-1/PD-L1 blocking immunotherapies, across cancer types previously considered both immunogenic and non-immunogenic (Raez et al., 2005, Schreiber et al., 2010), have forced us to reconsider how we think about the relationship between cancer and the immune system (Topalian et al., 2012, Brahmer et al., 2012). CPI therapy targets inhibitory ligands or their cognate receptors, and therefore acts on T cells expressing the specific checkpoint molecules targeted. We now appreciate that tumor-infiltrating T cells (TIL) are heterogeneous, ranging from bystander naive cells (tumor non-responsive) that do not express inhibitory markers, to tumor-specific T cells at various stages of differentiation that have a range of inhibitory marker expression (van der Leun et al., 2020). Importantly, the unexpected success of these therapies across many cancer types confirmed the long debated hypothesis that an ongoing immune response to cancer already exists in patients and, if bolstered therapeutically, has the potential to cure disease.
Early indications of natural anti-tumor immunity came from studies in transplantable and carcinogen-induced mouse models of cancer, as well as from biopsies of patient tumors, which led us to begin classifying tumors either as “hot”, having lymphocytic infiltrate, or as “cold”, having little to no infiltrate (Binnewies et al., 2018). However, recent technological advances in genome sequencing and variant prediction methodologies have shown that tumors traditionally classified as “cold”, and thus thought to be non-immunogenic, possess neoantigens capable of inciting an adaptive antitumor immune response (Vareki, 2018). Likewise, in cancer types traditionally considered to be non-immunogenic, such as certain subtypes of non-small cell lung cancer (NSCLC), CPIs have now become first line therapy (Gardiner et al., 2015, Hui et al., 2017). Yet, long term response rates remain low. Therefore, a new challenge for researchers is to elucidate the factors distinguishing tumors that respond to checkpoint therapy from those that do not. To fill this critical gap in our knowledge, new mouse models of cancer immunology are needed that will allow the careful study of the complicated and evolving antitumor immune response over the progression of disease from initiation to metastasis.
A historical perspective of established mouse models of cancer
Many components of the developing tumor-immune landscape over the course of disease can impact responsiveness at the time of therapy. The goal of early cancer studies was to identify effective therapeutics. Thus, researchers relied heavily on the in vitro study of cancer cells and mouse models that lacked a sufficient immune component, such as human cancer cell xenografts. These models were not designed with the immune system in mind. As a consequence, cancer research has focused primarily on tumor cell-intrinsic characteristics of disease. These intrinsic factors, such as the role of various genetic mutations on therapy responsiveness and disease progression, have been reviewed elsewhere (Ashworth et al., 2011). Comparatively less is understood regarding the contributions of tumor cell-extrinsic factors impacting disease outcomes. As mentioned, we will discuss the complicated dynamics of tumor cell-extrinsic components of cancer immunology through the lens of the historical evolution of mouse models, as depicted in Figure 1. In so doing, we hope to highlight why mouse models of cancer are critical to understanding these factors and their impacts on developing tumors.
Figure 1:

Juxtaposed timelines of notable advances generated in syngeneic cell line transplant models of cancer (dark green), carcinogen-induced models of cancer (purple), virally-induced models of cancer (blue), GEMM of cancer (orange), and humanized PDX models of cancer (light green). These timelines are set in the context of major findings and theoretical advances in the study of cancer immunology, marked above with yellow markers. Created with BioRender.com. Abbreviations: MHC, major histocompatibility complex; CPI, checkpoint inhibitor; KO, knock-out; PAH, polycyclic aromatic hydrocarbons; RAGKO, recombination activating gene kock-out; SCID, severe combined immunodeficiency; GEMM, genetically-engineered mouse model; SV40, simian virus 40; APC, adenomatous polyposis coli; Cre-LoxP, Cre-recombinase-LoxP; PDX, patient-derived xenografts; RAG1, recombination activating gene 1; RAG2, recombination activating gene 2
Syngeneic cell-line transplant models
Syngeneic transplant models of cancer, in which a cancer cell line is injected into a mouse with the same genetic background as the mouse from which it was generated, dominated the field of early cancer immunology. Importantly, these models straddle the line between having adequate genetic similarity to not cause rejection, but harboring enough mutations to generate an anticancer immune response that can be studied. Nevertheless, this largely happened by happenstance and it took many years of trial and error for these concepts to be fully appreciated.
In the mid 1950s, the U.S. National Cancer Institute would begin promoting efforts to produce cancer cell lines in which to perform drug screenings for new cancer treatments. Since then, hundreds of cancer cell lines have been established and propagated. Tumor cell lines isolated from mice in the 1970s and 80s as a part of these efforts (e.g. Colon38, B16 melanoma) continue to be widely used today. Cell lines generated from mouse and human cancer biopsies were used in vitro and in vivo for cancer drug screens (Suggitt and Bibby, 2005). The potential of manipulatable and transplantable systems generated excitement in the field, and a model in which to grow these cell lines with consistency and reproducibility was essential. Thankfully, decades prior, efforts to transplant tumors within and between species had begun. One of the first successful transplantations of a tumor was demonstrated in 1901 between rats (Loeb, 1901), but only a portion of recipients accepted the transplants. Around this time, Leo Loeb and Abbie Lathrop observed that mice with different genetic backgrounds spontaneously developed cancers with varying rates of incidence (Steensma et al., 2010, Lathrop, 1915a, Lathrop, 1915b, Lathrop, 1918). Clarence Cook Little, after carefully mating the progeny of mice obtained from Lathrop, developed some of the first inbred strains of mice: first, the DBA (Dilute, Brown, and non-Agouti) inbred mouse strain and eventually, the C57BL mouse strain widely used today (Little, 1911, Little, 1927, Murray and Little, 1935, Little, 1913). In the years to follow, Little, Leonell Strong, and John Bittner tested various tumor and strain combinations for tumor incidence (Bittner, 1936). Together with Little, Ernest Tyzzer used these inbred strains to provide an explanation for the previous failures of tumor transplantation, and showed a genetic and mendelian basis for rejection of cancer grafts, later described as the major histocompatilbility complex by George Snell and Peter Gorer (Tyzzer, 1909, Little and Tyzzer, 1916, Little, 1914, Snell, 1948, Gorer, 1936).
The generation of inbred mouse strains in the 1900s set the stage for all later mouse models, creating reliable hosts for transplantation of tumor cell lines and later, for chemical and genetic tumorigenesis. Due to ease of availability, presence of an intact host tumor environment, and reproducible growth kinetics, transplanted syngeneic tumors became the most favorable way to screen drugs for therapeutic efficacy. Furthermore, observed changes in the growth rate and invasive characteristics of cancer cells established the idea of tumor progression (Foulds, 1954, Furth, 1959, Klein and Klein, 1957). These properties had not been observed in models of spontaneous disease and were only appreciated after we gained the ability to serially transplant tumor tissue into multiple hosts and study cancers over prolonged periods of time. A genetic basis for tumor progression would be discovered a few decades later (Pradella et al., 2017). While many transplanted tumors were immunogenic, the relevance to human disease was uncertain. Notably, this created a rift in the scientific community and many grew skeptical of the role of immunity in cancer. Further, early experiments by Medawar and colleagues supported Thomas Burnett’s theory of acquired immunological tolerance put forth in 1949, stating that self-reactive cells were deleted prior to adulthood (Parish, 2003, Billingham et al., 1953). These data supported a model in which the immune system could not recognize malignant cells as non-self as they would be indistinguishable from healthy tissues. This was in contrast to findings of Edward Foley, who demonstrated in 1953 with C3H mice that tumor grafts were rejected in genetically identical mice after they had been immunized against the cancer (Foley, 1953). The role of the immune response in cancer would eventually be clarified after many years of studies using carcinogen-induced cancer models.
Carcinogen-induced models
In the late 1900s, carcinogen induced tumor models, extensively reviewed elsewhere (McCreery and Balmain, 2017), became critical for the conversations surrounding the role of the immune system in cancer. The ongoing search for environmental carcinogens, pioneered by Yamagiwa and Ichiwaka (Yamagiwa and Ichikawa, 1918), began gaining more traction as genetically similar mice were utilized in experiments. The cancer-causing properties of polycyclic aromatic hydrocarbons (PAHs) were demonstrated in 1930, adding validity to this pursuit (Kennaway and Hieger, 1930). Around this time, Murray Shear set up a drug screening program, the first of its kind, to test thousands of compounds for cancer-causing properties in the murine S37 model of sarcoma (DeVita and Chu, 2008). Shear’s program later served as a model for future cancer drug screening programs. Inbred mice were also used to demonstrate the cancer causing properties of tobacco (Wynder et al., 1953). One of the most reproducible models to come of these efforts was the application of the chemical methylcholanthrene (MCA) to the skin of inbred mice for the induction of tumors, and this model is still used for cancer immunology studies today. Ludwig Gross, followed by E. J. Foley, R. W. Baldwin, R. T. Prehn, J. M. Main and others, all soon showed that chemically induced tumors were recognized by the immune system and, soon after, the rejection of MCA-induced tumors was shown to be immune cell mediated (Schreiber and Podack, 2009, Scott, 1991, Rosenberg et al., 1986). We now appreciate that carcinogen-induction results in many genetic mutations, and thus, resulting tumors contain various neoantigens, or new proteins expressed by transformed cells, that can be recognized by the immune system. Work in these models led, serendipitously, to the discovery of Tumor Specific Antigens (TSAs) not present in healthy cells (Gross, 1943, Old, 1981, Prehn and Main, 1957). This forever shifted the dogma and further legitimized the field of cancer immunology. As a result, Burnet, together with Lewis Thomas, proposed a model of immune surveillance of cancer (Ribatti, 2016). These working models came with defined hypotheses including the existence of TSAs and immune cell-dependent tumor growth restriction. To test these new hypotheses, the field once again looked to new mouse models.
This period of time serves as a cautionary tale about the applicability and drawbacks of chosen models. Experiments aimed at testing the idea of immune surveillance were first conducted in athymic (nude) mice as a way to measure the impact on tumor growth in the absence of adaptive immunity. These experiments erroneously disproved the immune surveillance hypothesis, showing no differences in tumor growth regardless of the presence or absence of adaptive immune cells (Stutman, 1974, Stutman, 1979, Outzen, 1975). Once again it appeared that immunology and cancer biology had little overlap. Later, it was discovered that athymic mice possess macrophages and natural killer cells (NK) with enhanced cytotoxic activity as compensation for the lack of thymically-derived T cells and are therefore not completely immunodeficient (Hasui et al., 1989). Additionally, while these mice lack most mature T cells, an appreciable T cell population remains as a result of extra-thymic maturation, a natural process that was not universally accepted for many years (Torfadottir et al., 2006). The same experiments, repeated in improved immunodeficient mouse models, including RAGKO mice, lacking a gene (RAG) which encodes key enzymes responsible for the generation of all mature T and B cells, as well as severe-combined immunodeficiency (SCID) mice, demonstrated that tumors grow more robustly in immune deficient mice, thus supporting the immune surveillance hypothesis (Shinkai et al., 1992, Shankaran et al., 2001, Engel et al., 1997). Auspiciously, the concept that adaptive immune cells could recognize tumor antigens had been demonstrated by this time using in vitro studies of patient tumor cells and autologous lymphocyte cultures (Brichard et al., 1993, Wölfel et al., 1995, Bruggen et al., 1991). Meanwhile, Robert Schreiber and others developed increasingly sophisticated MCA-induced cancer models (Dunn et al., 2005, Koebel et al., 2007) and, in the late 1990s, were able to demonstrate that CD8 T cells and NK cells produced effector molecules that serve as the primary mediators of the anticancer immune response (Smyth et al., 2001). For more details on immune surveillance in cancer, please refer to the following review: (Swann and Smyth, 2007).
The development of athymic, RAGKO, and SCID mice aided the development of a new model of cancer. Severely weakened immune systems allowed for the transplantation of human cancer tissues without rejection by the immune system, providing a means to quickly screen new anticancer agents in human cancer tissue (Bogden et al., 1984). These were the first xenograft models, named for the transplantation of tissue from one species into another without rejection (see section below on humanized PDX mouse models of cancer).
Virally-induced models
The relationship between infection and cancer has been investigated for well over a century in cancer patients, but could not be proven until the early 1900s, and this line of investigation unintentionally led to the eventual design of another class of mouse models of cancer. Peyton Rous is considered the first to demonstrate the cancer-causing potential of viruses after spreading cancer from one domesticated foul to another via an infectious agent in 1910. Rous would eventually earn the Nobel prize for these findings. Later, the infectious agent discovered by Rous was identified as respiratory syncytial virus (RSV). Notably, the search for an RSV-induced tumor antigen led to the discovery, in the late 1970s, of the phosphoprotein Src. The viral Src gene elicited much excitement in the research community as it was determined to be a protein kinase necessary for malignant transformation but dispensable for viral replication (Bister, 2015). Bridging the divide between the fields of virology and cancer genetics, the discovery of Src created a flurry of excitement for the study of proto-oncogenes; genes that, when mutated, cause normal healthy cells to divide uncontrollably. Soon after the discovery of RSV, other cancer-causing viruses were uncovered in various species including rabbits, mice, cats, non-human primates and humans (Weiss and Vogt, 2011). For example, in the 1920s, after crossing a male DBA mouse with a female mouse from Halsey Bagg’s albino colony, scientists observed that the resulting inbred strain, the C3H strain, developed spontaneous mammary tumors by 6 months of age (Strong, 1935). The cause was soon found to be transmission of mouse mammary tumor virus (MMTV) in breast milk (Bittner, 1937, Bittner, 1939, Visscher et al., 1942). These early findings led to the discovery of human endogenous retroviruses (hERVs) that are homologous to MMTV (Ono et al., 1986). Please find more comprehensive reviews of oncogenic viruses and their contributions to our current understanding of cancer immunology here: (Javier and Butel, 2008).
The fields of virology and cancer genetics overlapped further upon the discovery and genome sequencing of polyomaviruses including simian vacuolating virus 40 (SV40) and murine polyomavirus (MPyV) (Cheng et al., 2009). The discovery of these small DNA viruses led to many years of research around cellular transformation and viral oncogenes (Levine and Oren, 2009, Weiss, 2020). Consequently, viral tools for manipulating genomic DNA were introduced and employed across research disciplines, and in vivo modeling advances proceeded rapidly. For example, the discovery of restriction enzymes in the 1960s quickly led Paul Berg and colleagues to demonstrate that pieces of “foreign” DNA could be inserted into cells in culture (Goff and Berg, 1976, Mulligan et al., 1979, Mulligan and Berg, 1980). In the 1970s, Jaenisch and Mintz microinjected viral oncogenes from SV40 into the blastocoel of mouse embryos, purposefully integrating viral DNA into the mouse genome and consequently generating what are considered by many to be the first transgenic mice (Jaenisch and Mintz, 1974). The resulting growth in recombinant DNA technology led to the wide accessibility and use of lentiviral and adenoviral vectors for the induction cancer. An early example of this, was the transfer of the SV40 Large T antigen (Tag) oncogene linked to the rat insulin promotor (RIP) into fertilized eggs resulting in RIP-Tag mice (Hanahan, 1985). This tissue specific expression of Tag led to proliferation and spontaneous development of tumors in pancreatic beta cells. Similar techniques continue to be used today to build new mouse models of cancer (see section on Genetically engineered mouse models (GEMMs) of Cancer, below) and have contributed to our knowledge of cancer immunology, as the induced expression of viral proteins by transformed cells can facilitate the study of the anti-tumor immune response. Please refer to the detailed review elsewhere of viral oncogene models and how they have been used in the study of cancer immunology (Guerin et al., 2020).
Somewhat counterintuitively, virally-induced genetically engineered mouse models (GEMMs) of cancer have been found, in general, to not elicit robust immune responses in the host and, as such, early GEMMs were less than ideal models for tumor immunology (see GEMMS of Cancer Immunology, below). This can be explained, in part, by the fact that, while viruses are technically foreign or “non-self” to their host organism, once viral DNA integrates into the host genome it, in a sense, becomes “self” and does not elicit a marked immune response. That said, exceptions exist, including the use of SV40 T antigens such as LT which have been shown to induce interferon-stimulated genes which could directly impact immune cell function (Forero et al., 2014).
GEMMs of Cancer
Efforts to understand the mechanisms by which oncogenes caused cancer initiation and progression converged with a growing frustration in the late 1990s and early 2000s over the poor clinical translatability of drugs shown to cure cancer in mice. This led to an increased appreciation for the role of the native tumor microenvironment in cancer development and progression. For these purposes, models were now required that recapitulated the microenvironment seen in patient disease. Carcinogen-induced models, while generated in situ and therefore present in the tissue environment in which they originated, result in genetically heterogeneous tumors within groups of mice, which increase variability and make it difficult to determine biological mechanisms or therapeutic efficacy of candidate drugs. Transplant models, while genetically similar and less variable from tumor to tumor, do not adequately recapitulate the natural tumor microenvironment, due in large part to their rapid growth (Figure 2). Importantly, they also lack the early phases of progression seen in patient disease (Olson et al., 2018). To overcome this, cancer biologists turned to genetically engineered mouse models, or GEMMs.
Figure 2:
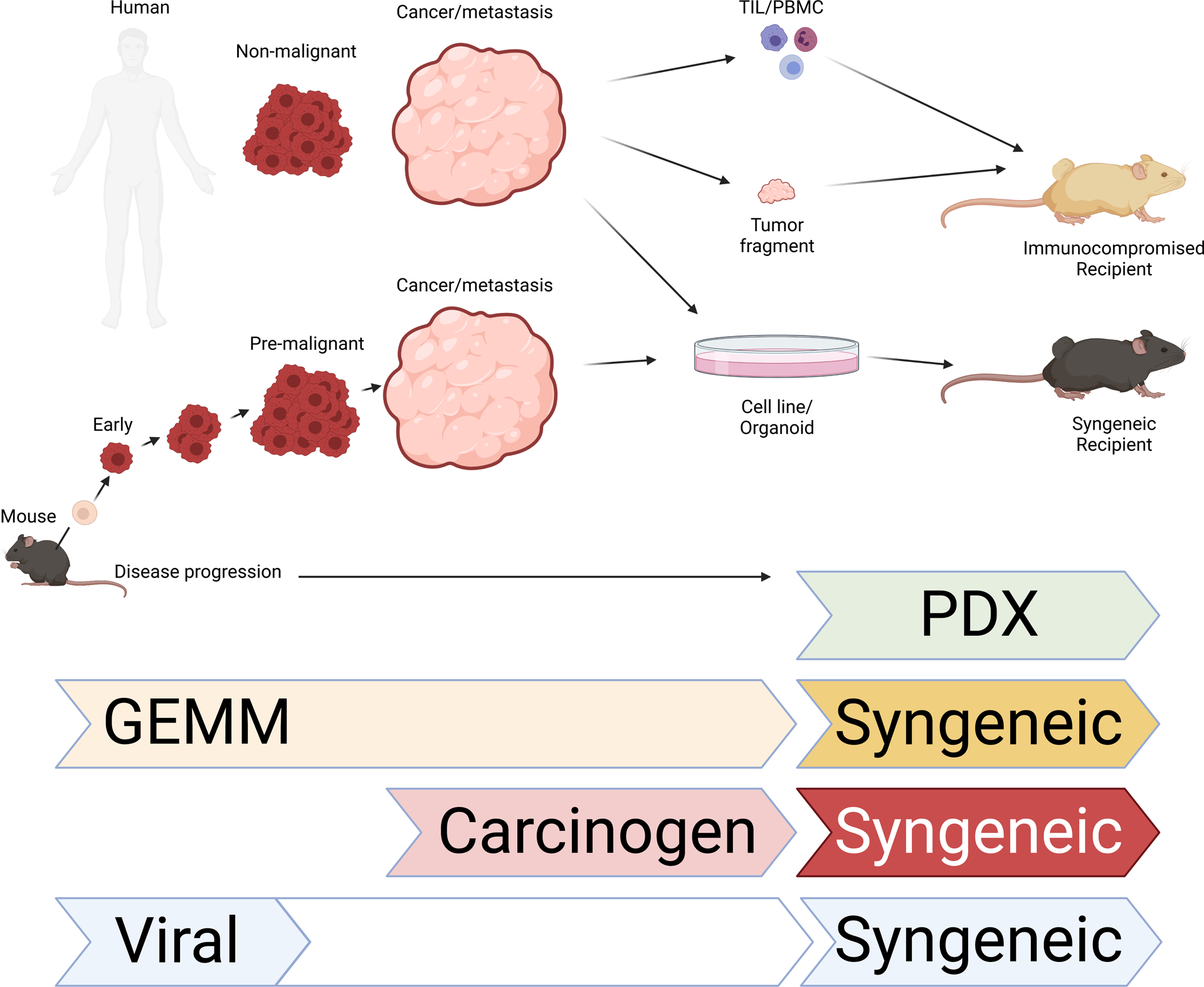
Stages of human (top) and mouse (bottom) cancer that have been observed. On the right, techniques used to transplant tumors into mice are depicted. Mouse models of cancer best able to recapitulate various stages of disease shown below. Created with BioRender.com. Abbreviations: TIL, tumor infiltrating lymphocytes; PBMC, peripheral blood mononuclear cells; PDX, patient-derived xenografts; GEMM, genetically engineered mouse models
There have been many iterations of cancer GEMMs, and advanced GEMMs will continue to be generated in order to meet the needs of novel research questions. They can be broadly categorized as germline or non-germline/conditional (van Dyke and Jacks, 2002). Taking advantage of new viral integration systems and gene recombination, early germline GEMMs were genetically engineered to express dominant oncogenes (e.g. Kras) and provided evidence that oncogene expression in normal cells could lead to cell transformation and tumors (Hanahan et al., 2007). Some systems also use specific recombinases to delete tumor suppressors in desired tissues (e.g. Trp53). These models, comprehensively reviewed elsewhere (Politi and Pao, 2011, Walrath et al., 2010), illustrated the important role that oncogenes play, not just in the initiation of cancer, but also in the maintenance of disease. The ability to turn “on” or “off” mutant oncogenes and tumor suppressors, respectively, thus mimicking what is seen in patient tumor progression, allowed the histopathological recapitulation of early and advanced disease in cancer mouse models (Frese and Tuveson, 2007) and laid the groundwork for current advanced GEMMs. The observation of cancer progression through the stages of disease from transformation to metastasis is a notable strength of these model systems.
Advances in genetic techniques paved the way for the creation of conditional GEMMs, in which the expression or deletion of oncogenes could be controlled both spatially and temporally. For example, the APCMin mouse model of intestinal and mammary cancer was generated by a germline mutation in the APC gene, originally induced by a mutagen, ethylnitrosourea. Due to the expression of APC in many cell types, this mutation results in multiple spontaneous tumors throughout the intestinal tract as well as in mammary tissue (Moser et al., 1995). Building on this, an inducible model of colorectal cancer was created in which the tumor suppressor APC, flanked by loxP sites, was inactivated following adenovirus-mediated delivery of Cre-recombinase to the colorectal region, resulting in the rapid, controlled onset of colorectal adenomas (Shibata et al., 1997). Further spatiotemporal regulation was achieved following the induction of somatic mutations with Cre-ERT fusion proteins under the control of tissue specific promoters. In this setting, a mutated hormone binding domain of the estrogen receptor is fused to Cre-recombinase. Administration of the estrogen analogue tamoxifen leads to post-translational activation of Cre-recombinase activity and excision of the targeted gene in a tissue specific manner (Vooijis et al., 2001). Conditional GEMMs primarily rely on induced transformation by utilizing tetracycline-enabled promoter accessibility or virally delivered Cre- or flippase (FLP) recombinases to target genomically encoded loxP or FRT sites, respectively. These genetic tools allow for control over when and in which tissues/cell types tumors are induced (Jonkers and Berns, 2002). GEMMs can also take advantage of more diverse methods of tumor induction like RNA interference against tumor suppressor genes and CRISPR/Cas9 mediated somatic mutations (Yang et al., 2013, Livshits and Lowe, 2013). Studies in such models demonstrated that the impact of any single oncogene is heterogeneous, creating genomic diversity similar to what is observed in human disease (Chung et al., 2017). Inducing genetic co-mutations by mimicking multistep carcinogenesis can further increase the relevance of a GEMM to a particular type of human cancer. For this, the combination of the Flp-FRT and Cre-loxP systems allow for the sequential induction of multiple gene changes with unique recombinase actors, allowing for temporal control over each gene change separately (i.e. the inducible dual-recombinase system (Schonhuber et al., 2014)). Overall, cancer GEMMs have supported a rapid expansion of our understanding of development, and in some cases therapeutic vulnerabilities, of diverse cancers (Please refer to (Kersten et al., 2017) for a comprehensive review of GEMM).
GEMMs of Cancer Immunology
In the flood of excitement surrounding CPI therapies in the early 2000s, it was inevitable that the fields of cancer biology and tumor immunology would become permanently intertwined. Together, findings that demonstrated the importance of CD8 T cells for tumor control (first noted in the MCA model and tumor transplants), and the interpretation of CPI therapy as a reversal of T cell exhaustion, created a frenzy of investigation of T cell responses in the traditional cancer GEMMs described above. Some of these experiments were fruitful and deepened our understanding of the interplay between tumor cells and CD8 T cells (Goel et al., 2017, Li et al., 2018). Nevertheless, GEMMs were found to incite very low levels of immune responses to tumors and consequently poor responses to CPI, in contrast to human patients treated in the clinic. Furthermore, it has been shown that many tumor infiltrating immune cells are bystanders, not actually involved in anti-tumor responses, making it necessary to distinguish tumor-specific T cells from the rest (DuPage et al., 2011). The weak immunogenicity of GEMMs is generally thought to be due to the low tumor mutational burden resulting from the precision of oncogene/tumor suppressor mutation (McFadden et al., 2016, Lee et al., 2016). As an example, studies using a GEMM of autochthonous liver cancer, in which SV40 Large T (LT) is expressed after tamoxifen-inducible, albumin-specific production of Cre-recombinase (AST mice), has resulted in a better understanding of T cell dysfunction in cancer (Runge et al., 2014, Schietinger et al., 2016). Nevertheless, for most studies this model requires that transgenic T cells specific for LT be transferred into mice prior to tumor induction, as endogenous T cell responses are minimal.
Several different strategies were used to increase the immunogenicity of cancer GEMMs. Increasing bulk tumor mutational burden by destabilizing the genomes of transformed cells and targeting DNA mismatch repair machinery is effective at mirroring the mutational burdens of human cancer and eliciting immune responses (Sen et al., 2019, Germano et al., 2017). A draw back to this approach is that the variability of antigens in each tumor makes it difficult to determine which immune cells are relevant and specifically responding to tumor antigens. Additional difficulty is involved when one considers the various T cell clones with different receptor specificities that could be at play between mice and experiments. Antigen has also been added into advanced tumors via electroporation. This approach is mainly relevant for studying responses to established tumors and generates a great deal of delivery-related inflammation, but provides a means for studying tumor-specific T cell responses (Radkevich-Brown et al., 2010). Tissue-specific promoters were also bred into GEMM for constitutive expression of antigens in the tissue/cellular location of choice. Unfortunately, the expression of antigens prior to tumor induction leads to immunological tolerance, precluding the study of the endogenous anti-tumor immune response (Huijbers et al., 2012). To avoid tolerance against germline antigens, conditional antigen expression systems have been introduced and most operate similarly to the recombinase-inducible oncogene models described in the previous section (Cheung et al., 2008). However, slight infidelity of the Cre-recombinase on/off switch is a drawback to this approach, as it results in low levels of “leaky” antigen expressed by thymocytes or in peripheral tissues. This low-level, leaky expression can be recognized by T cells and consequently result in the induction of tolerance prior to tumor establishment.
One successful method of generating tumors that trigger a robust immune response against a known antigen are the Lenti-Neoantigen-Cre systems. These utilize established Cre-loxP cancer GEMMs and add neoantigen into the lentiviral vectors for co-delivery with Cre recombinase at the time of tumor induction (DuPage et al., 2011, DuPage and Jacks, 2013). These approaches allow studies of tumor-specific T cells at many stages of tumor development. However, as lentiviruses and adenoviruses are able to infect many types of cells, cell-type specific targeting is an important consideration when programming tumors to express neoantigens in this manner. Neoantigens introduced by non-targeted viruses can be expressed in infected epithelial cells, immune cells, endothelial cells, and more. This creates aberrant sources of neoantigen expression outside of the transformed cells, thus impacting the subsequent neoantigen-specific T cell response. However, the addition of a tissue-specific promoter or micro-RNA to the Cre-recombinase and neoantigen-expressing viruses can substantially limit off-target expression (Alonso et al., 2018). Furthermore, lentiviral silencing can in some cases cause tumors to become neoantigen negative by non-immunological means and thus extinguish any neoantigen-specific anti-tumor immunity (Cherry et al., 2000). Of note, the likelihood of lentiviral silencing seems to vary based on the promoters used and is therefore relatively simple to avoid (Xia et al., 2007). Nevertheless, the controlled introduction of a model neoantigens still results in tumors containing relatively low mutational burdens. As such, models mentioned previously which target DNA mismatch repair machinery may be preferred as they better mirror the mutational burden in human patients. While the Lenti-Neoantigen-Cre vectors have caveats, they successfully generate immunogenic tumors and have proven useful in the investigation of anti-tumor T cell responses, helping expand the field of cancer immunology (DuPage et al., 2012, Joshi et al., 2015).
Like the models that preceded them, neoantigen-expressing GEMM serve the field well for questions within their scope, but can always be improved upon to meet the next scientific challenge. Advanced generations of GEMM with inducible neoantigen tumor models are being developed with multiple lock-and-key induction mechanisms acting in combination to avoid the immunological tolerance and leaky recombinase systems described previously (Damo et al., 2020, Hegde et al., 2020). One such model was designed in our lab to overcome the challenges mentioned above, and is called the iNversion INducible Joined neoAntigen (NINJA) mouse (Damo et al., 2020). The model neoantigen in this case, NINJA, contains GP33 and GP66 derived from the lymphocytic choriomeningitis virus (LCMV). Crossing NINJA mice with the widely used KP (K-rasLSL-G12D/+;p53fl/fl) GEMM of lung adenocarcinoma to produce KP-NINJA mice allows for the study of endogenous tumor-specific T cells in autochthonous lung cancer throughout cancer progression (Fitzgerald et al., 2021, Connolly et al., 2021). A similar model, designed by the lab of David DeNardo, allows for the inducible expression of the neoantigen chicken ovalbumin (OVA), driven by Cre recombinase expression and tetracycline repression. The novel “OG” mouse was crossed to the KPC (K-rasLSL-G12D/+;p53LSL-R172H/+;Pdx-Cre) mouse model of autochthonous pancreatic ductal adenocarcinoma to generate KPC-OG mice (Hegde et al., 2020). The antigens expressed in these models qualify as neoantigens because they are tightly controlled and expressed only by tumor cells following transformation, but they are not strong enough to cause rejection, thus mirroring what is seen in human disease. These genetically programmed, neoantigen-inducible models improve upon existing GEMM while still histopathologically recapitulating human disease, ultimately resulting in anti-tumor immune responses that more closely mirror human cancer physiology.
Humanized PDX models
In sharp contrast to the other mouse models of cancer immunology discussed above, efforts to design a mouse with little to no immune system have been underway since the 1950s. The original objective for these immunodeficient mouse models was to design a suitable host for the implantation and growth of human tumors and these would become known as the first patient-derived xenografts (PDXs). These models allow human tumor tissue to be serially transplanted or made into cell lines or organoids that have been shown to retain the genetic complexity of human disease. Of note, Todd Golub and colleagues have revealed that human tumors grown in mice long term may undergo genetic changes that they would not normally undergo in the human body (Ben-David et al., 2017). Now, after many advances in technology and in our knowledge of immunology, researchers are reconstituting these mouse models with human immune components. This is an emerging area of research with the potential for better translatability than has been seen historically for studies of murine cancer.
Helene Toolan, an important pioneer of these efforts, established a successful protocol in 1951 for the irradiation of mice and rats prior to transplantation of human tumors and demonstrated superior results (Toolan, 1951, Toolan, 1958). Around the same time, a growing emphasis on screening possible anticancer agents shifted the intended application for these models and concerns grew that preconditioning mice with irradiation might lead to erroneous results (Gallily and Woolley, 1958, Palm et al., 1958). Thus, this field benefitted immensely from inbred mouse strains designed to be immunodeficient including nude, SCID, and RAGKO mice, mentioned previously, that were generated over the next several decades. Aspects of the immune system that had not been appreciated previously were a recurring obstacle for successful transplantation of human tumors. Advanced models of immunodeciency were generated that contained fewer and fewer immunological components and resulted in increasingly successful engraftment (please find a comprehensive review of these models here: (Shultz et al., 2012)). With the use of these advanced models of immunodeficiency, researchers have begun working backwards to rebuild a human immune system within mice in an effort to study human cancer immunology. Various techniques have been used for this purpose including the engraftment of human hematopoietic stem cells (HSCs) or peripheral bone marrow cells (PBMCs) into humanized mice prior to implantation of human patient-derived and cell line-derived xenograft tumors (Verma and Wesa, 2020, Yao et al., 2019, Meraz et al., 2019). These efforts are ongoing (reviewed here: (Mian et al., 2020)). Such humanized PDX models are useful for the purpose of testing cancer therapies (reviewed here: (Guil-Luna et al., 2021)), and provide benefits over other available models. They afford researchers the ability to better model the genetic heterogeneity of human patients in the presence of a functional, humanized, immune response (Cassidy et al., 2015). Furthermore, in comparison to GEMM, tumor development is quick and therefore facilitates the testing of many drug combinations and strategies. Nevertheless, as noted throughout this review, no single model is superior for all purposes. For example, without a known tumor antigen, the study of endogenous T cell responses is challenging and, similar to other transplant models, injected tumor cells are extensively differentiated resulting in tumors that resemble late stage disease.
The current state of the art
Natural biology of the TME
Investigations of the tumor microenvironments (TME) at the earliest stages of transformation and disease could have profound impacts on future diagnoses and therapies by elucidating the early events determining the functionality of the anti-tumor immune response. The TME is highly complex, with cells from both innate and adaptive immune arms coexisting and communicating as tumors progress through various stages of disease. The “Cancer Immunity Cycle” was proposed by Chen and Mellman in 2013 and describes a series of steps necessary for generating a productive anti-tumor immune response (Chen and Mellman, 2013). Still, many important questions regarding the mechanistic details of this cycle remain unanswered. For example, tumor-specific CD8 T cells are critical to anti-tumor responses, but much remains unknown surrounding the process of CD8 T cell cross-priming and activation in situ. The signals received by naive CD8 T cells during cross-priming can vary greatly depending on the type of antigen-presenting cell and local inflammatory milieu (Fu and Jiang, 2018). Consequently, the strength and effectiveness of the resulting anti-tumor T cell response is likely shaped by that initial cross-priming event. Understanding the factors controlling dendritic cell migration and cross-priming dynamics could provide important insights for novel immunotherapies and improve the success of current treatments such as tumor vaccination and CAR-T cell therapy (Ma et al., 2019). Modeling challenges to consider for these studies include (1) the ability to observe the various stages of tumor development, including the earliest stages when priming likely occurs, and (2) physiological immunogenicity of tumor cells. To address the former, a GEMM makes an attractive option as it would recapitulate early stages of disease immediately following transformation of normal cells. In contrast, available transplantable mouse models of cancer, whether syngeneic or PDX, are better suited for studies of advanced cancer (Figure 2).
Beyond this, other aspects of human TMEs have not yet been adequately recapitulated in mouse models. The role of certain immune cells in the development and maintenance of antitumor immunity, such as B cells and follicular helper T cells to name a few, remain poorly understood despite data from human patients suggesting their therapeutic importance (Baumjohann and Brossart, 2021). Relatedly, a mechanistic understanding of tertiary lymphoid structures, which have been observed across many types of human cancer and correlated with improved therapeutic responsiveness, will require mouse cancer models that faithfully recapitulate these phenomena (Munoz-Erazo et al., 2020). As has been demonstrated throughout history (Figure 1), knowledge gained from clinical samples and current mouse models has laid the groundwork for the design of just such emerging models (Engelhard et al., 2018, Joshi et al., 2015, Cui et al., 2020).
Large-scale genetic screens
Emerging cancer mouse models reflect the intersection between our technological capabilities and our current understanding of relevant biology. The rapidly increasing number of mutations identified in human cancer sequencing studies calls for novel mouse modeling strategies that enable accelerated in vivo evaluation of candidate cancer genes. Unfortunately, it is expensive and time consuming to engineer a mouse and thus, until recently, transplant models have been preferred for these purposes as they can be easily mutated (Figure 2). Nevertheless, genetic alteration of embryonic stem cells (ESCs) can be used to produce cohorts of non-germline GEMMS (Heyer et al., 2010). This strategy employs ESCs that are derived from existing multi-allelic GEMMs, allowing for rapid introduction of additional genetic modifications and the generation of chimeric mice with characteristics of both the established GEMM as well as additional genetic modifications (Huijbers et al., 2011, Huijbers et al., 2015). Likewise, CRISPR-Cas9 mice, in combination with lentiviral guide RNA gene libraries and oncogene inducible strains, are being increasingly employed to screen for novel gene contributors to cancer as well as contributors to both immunologic and therapeutic responses (Dong et al., 2019, Chen et al., 2015). Critically, the immunogenicity of a tumor model determines the extent to which an anti-tumor immune response can be generated against it and this can be greatly influenced by the way in which a model was designed. For example, even CRISPR/Cas9, which has become a tool widely used in tumor immunology studies, can potentially affect the immunogenicity of tumors and adversely impact research findings. This was demonstrated recently when increased immunogenicity caused by components of the CRISPR/Cas9 system induced tolerance/anergy to tumor antigens (Dubrot et al., 2021). This study details a method by which to remove the offending CRISPR/Cas9 components and therefore avoid the formation of tolerance. Still, techniques used in the design of new models must be carefully considered with the potential for erroneous immunogenicity in mind. Large scale genetic screens are understandably easier in the setting of syngeneic transplant models and have been designed to manipulate genes in both tumors and immune cells to assess their impact on tumor development (Manguso et al., 2017, Roth et al., 2020, Cortez et al., 2020, Shu et al., 2020, Pan et al., 2018).
Immune-related adverse events
Another challenge currently includes modeling immune-related adverse events (irAEs) in a manner that is physiological and clinically relevant. Notably, the emergence of CPI-induced irAEs was not predicted by available models (Curran et al., 2010). Studying irAEs in human patient tissues is difficult as certain adverse events can be rare, tissues can be difficult to obtain, and manifestations can be heterogeneous. Currently, preclinical models of irAEs for the systematic testing of therapies do not exist and treatment strategies have been largely developed through trial and error in patients, rather than large-scale, controlled clinical trials (Brahmer et al., 2017). irAEs manifest as tissue-specific autoimmunity rather than systemic autoimmunity (Postow et al., 2018), and therefore the pathogenic mechanisms and best treatments may vary depending on the type of manifestation. Early studies of irAEs have relied on autoimmune-prone mouse models and recent work has highlighted the idea that CPI treatments can accelerate the onset of autoimmunity in autoimmune-prone models (mouse models of irAEs reviewed elsewhere: (Liu et al., 2014)). So far, these models have been used to test therapeutics for irAEs. In line with this, mice treated with DSS and anti-CTLA4 have more severe colitis compared with mice treated with just DSS (Perez-Ruiz et al., 2019), and prophylactic administration of TNFa-blocking antibodies ameliorates this phenotype without impacting CPI-induced anti-tumor responses. While promising, the broad utility of autoimmune models in this context is yet to be determined, as a relationship between autoimmune susceptibility and irAEs in human patients remains uncertain. The ideal models for T-cell driven irAEs require three components: (1) Naïve T cell repertoire with TCRs that recognize self-antigens, (2) Inducible expression of target self-antigens in a relevant tissue, and (3) Disease that emerges only in the context of treatment with CPIs. To these points, our lab has employed the NINJA mouse described above, which allows for the induction of a defined set of self-antigens in an organ of choice (Damo et al., 2020, Damo et al., 2021) and thus may be useful for the development of several translationally-relevant irAEs. Of note, modeling the relatively rare antibody-mediated irAEs present additional obstacles, as modeling these would require engineering target epitopes into proteins expressed in specific locations such as desmosomes, extracellular matrix, or in neuronal synapses.
Concluding remarks
Presently, as a result of more than a century of cumulative research, there are many mouse models with which to study cancer immunology. Critically, each model has unique strengths and weaknesses that must be considered to choose the most appropriate model for testing any one hypothesis. The best mouse model of cancer immunology will continue to be a moving target as mouse models are constantly designed that improve upon old models to address new findings.
Future Issues:
Modeling the heterogeneity of human disease. A mouse model of cancer that recapitulates the genetic diversity of human disease could more accurately reflect and inform upon variable responses to therapies seen in the clinic.
Recapitulating the complexities of the tumor microenvironment. Adequately modeling many aspects of the tumor microenvironment will likely require novel findings from, and improved characterization of, the tumor microenvironments of patient cancers. New technologies, such as in situ hybridization techniques allowing for spatially resolved transcriptomes and multiplexed imaging have potential to accelerate these modeling efforts.
Incorporating disease susceptibility factors such as the microbiome. Interplay between the microbiome of various tissues with cancer cells and immune cells in the tumor microenvironment undoubtedly play a role in patient’s response to cancer therapies. Technological advances allowing for improved modeling of these diverse microbiota will be necessary for future studies.
Modeling the relevant aspects of immunotherapy and acquired resistance. Resistance develops across many types of cancer after treatment with various therapies, and there is currently a dearth of models available for studying these phenomena.
Sidebar:
The use of the term “immunogenic”.
In the purest sense, “immunogenic” refers to anything that elicits an immune response in the host. In the context of therapy, “immunogenic” has often been used to refer to a tumor with a clear therapeutic response to immunotherapy. This latter use of the term reflects the historical uncertainty of the existence of natural immune responses in some types of cancers, which were often only revealed following successful immunotherapeutic strategies. Thus, immunotherapeutic responses have been the guide for attempting to discover the presence of immunogenic potential.
Acknowledgements
The authors would like to thank all members of the Joshi lab, past and present. As well as many colleagues that have shared their critical thoughts. Research from the corresponding author’s lab was supported by the Yale Cancer Center (P30 CA016359 40), NCI K22CA200912 (N.S.J.), and NCI 1RO1CA237037-01A1 (N.S.J.), as well as the Interdisciplinary Immunology Training Program: grant number: NIH AI07019 (K.A.C.).
Definitions
- Athymic (nude) mice
A strain of mice homozygous for a mutated Foxn1 gene that do not develop a thymus and lack most mature T cells
- Checkpoint inhibitor (CPI)
a class of blocking antibodies specific for checkpoint receptors such as PD-1/PDL-1 or CTLA-4
- Cold tumor
Tumor with a microenvironment that is largely absent of lymphocytes
- Flippase (FLP) recombinase
Enzyme derived from yeast that recognizes FLP recombinase target (FRT) sequences and recombines DNA at these sites to initiate deletions, inversions, or translocations
- Genetically engineered mouse model (GEMM)
a strain of mouse that has been genomically manipulated using gene editing techniques
- Hot tumor
Tumor with a microenvironment that is highly infiltrated with lymphocytes
- Immune-related adverse event (irAE)
Autoimmune toxicities that can arise in various organ systems as a side effect of treatment with immune checkpoint inhibitors
- Lymphocytic choriomeningitis virus (LCMV)
A virus that results in a well characterized infection in its primary host, the mouse, and is commonly used to study the T cell response to infection
- Methylcholanthrene (MCA)
Chemical carcinogen used in cancer research to induce tumors in mouse models
- P1 bacteriophage cyclization recombinase (Cre)
Enzyme that recognizes loxP sites and recombines DNA between these sites to initiate deletions, inversions, or translocations
- RAGKO mice
A strain of mice lacking one or both of recombination-activating genes (RAG1 and RAG2) resulting in the absence of mature T and B lymphocytes
- Severe combined immunodeficiency (SCID) mice
A strain of mice homozygous for a mutated Prkdc gene causing deficiency of both B and T lymphocytes
- Tumor infiltrating lymphocytes (TIL)
Lymphocytes, including B and T cells, present within the TME
- Tumor microenvironment (TME)
The cellular and molecular composition and structural architecture of the environment surrounding and supporting a tumor
- Tumor-specific antigen (TSA)
Antigens arising from novel peptide sequences representing neoantigens which are expressed only on cancer cells, unlike tumor-associated antigens (TAA) which are also expressed by normal cells
Footnotes
Disclosure statement
The authors are not aware of any affiliations, memberships, funding, or financial holdings that might affect the objectivity of this review.
References
- ALONSO R, FLAMENT H, LEMOINE S, SEDLIK C, BOTTASSO E, PEGUILLET I, PREMEL V, DENIZEAU J, SALOU M, DARBOIS A, NUNEZ NG, SALOMON B, GROSS D, PIAGGIO E & LANTZ O 2018. Induction of anergic or regulatory tumor-specific CD4(+) T cells in the tumor-draining lymph node. Nat Commun, 9, 2113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- ASHWORTH A, LORD CJ & REIS-FILHO JS 2011. Genetic interactions in cancer progression and treatment. Cell, 145, 30–8. [DOI] [PubMed] [Google Scholar]
- BAUMJOHANN D & BROSSART P 2021. T follicular helper cells: linking cancer immunotherapy and immune-related adverse events. J Immunother Cancer, 9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- BEN-DAVID U, HA G, TSENG YY, GREENWALD NF, OH C, SHIH J, MCFARLAND JM, WONG B, BOEHM JS, BEROUKHIM R & GOLUB TR 2017. Patient-derived xenografts undergo mouse-specific tumor evolution. Nat Genet, 49, 1567–1575. [DOI] [PMC free article] [PubMed] [Google Scholar]
- BILLINGHAM RE, BRENT L & MEDAWAR PB 1953. ‘ACTIVELY ACQUIRED TOLERANCE’ OF FOREIGN CELLS. Nature, 172. [DOI] [PubMed] [Google Scholar]
- BINNEWIES M, ROBERTS EW, KERSTEN K, CHAN V, FEARON DF, MERAD M, COUSSENS LM, GABRILOVICH DI, OSTRAND-ROSENBERG S, HEDRICK CC, VONDERHEIDE RH, PITTET MJ, JAIN RK, ZOU W, HOWCROFT TK, WOODHOUSE EC, WEINBERG RA & KRUMMEL MF 2018. Understanding the tumor immune microenvironment (TIME) for effective therapy. Nat Med, 24, 541–550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- BISTER K 2015. Discovery of oncogenes: The advent of molecular cancer research. Proc Natl Acad Sci U S A, 112, 15259–60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- BITTNER JJ 1936. The Spontaneous Incidence of Lung Tumors in Relation to the Incidence of Mammary Tumors in an Inbred Strain of Albino Mice (Strain A). Cancer Research, 27, 519–524. [Google Scholar]
- BITTNER JJ 1937. Mammary Tumors in Mice in Relation to Nursing. Cancer Research, 30, 530–538. [Google Scholar]
- BITTNER JJ 1939. Relation of Nursing to the Extra-Chromosomal Theory of Breast Cancer in Mice. Am J Cancer, 35, 90–97. [PubMed] [Google Scholar]
- BOGDEN AE, GRIFFEN W, REICH SD, COSTANZA ME & COBB WR 1984. Predictive testing with the subrenal capsule assay. Cancer Treatment Reviews, 11. [DOI] [PubMed] [Google Scholar]
- BRAHMER J, TYKODI SS, CHOW LQM, HWU W, TOPALIAN SL, HWU P, DRAKE CG, CAMACHO LH, KAUH J, ODUNSI K, PITOT HC, HAMID O, BHATIA S, MARTINS R, EATON K, CHEN S, SALAY TM, ALAPARTHY S, GROSSO JF, KORMAN AJ, PARKER SM, AGRAWAL S, GOLDBERG SM, PARDOLL DM, GUPTA A & WIGGINTON JM 2012. Safety and Activity of Anti–PD-L1 Antibody in Patients with Advanced Cancer. N Engl J Med, 366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- BRAHMER JR, LACCHETTI C, SCHNEIDER BJ, ATKINS MB, BRASSIL KJ, CATERINO JM, CHAU I, ERNSTOFF MS, GARDNER JM, GINEX P, HALLMEYER S, CHAKRABARTY JH, LEIGHL NB, MAMMEN JS, MCDERMOTT DF, NAING A, NASTOUPIL LJ, PHILLIPS T, PORTER LD, PUZANOV I, REICHNER CA, SANTOMASSO BD, SEIGEL C, SPIRA A, SUAREZ-ALMAZOR ME, WANG Y, WEBER JS, WOLCHOK JD & THOMPSON JA 2017. Management of Immune-Related Adverse Events in Patients Treated With Immune Checkpoint Inhibitor Therapy: American Society of Clinical Oncology Clinical Practice Guideline. J Clin Oncol, 36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- BRICHARD V, PEL AV, WÖLFEL T, WÖLFEL C, PLAEN ED, LETHÉ B, COULIE P & BOON T 1993. The Tyrosinase Gene Codes for an Antigen Recognized by Autologous Cytolytic T Lymphocytes on HLA-A2 Melanomas. J Exp Med, 178, 489–495. [DOI] [PMC free article] [PubMed] [Google Scholar]
- BRUGGEN PVD, TRAVERSARI C, CHOMEZ P, LURQUIN C, PLAEN ED, EYNDE BVD, KNUTH A & BOON T 1991. A Gene Encoding an Antigen Recognized by Cytolytic T Lymphocytes on a Human Melanoma. Science, 254. [DOI] [PubMed] [Google Scholar]
- CASSIDY JW, CALDAS C & BRUNA A 2015. Maintaining Tumor Heterogeneity in Patient-Derived Tumor Xenografts. Cancer Res, 75, 2963–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- CHEN DS & MELLMAN I 2013. Oncology meets immunology: the cancer-immunity cycle. Immunity, 39, 1–10. [DOI] [PubMed] [Google Scholar]
- CHEN S, SANJANA NE, ZHENG K, SHALEM O, LEE K, SHI X, SCOTT DA, SONG J, PAN JQ, WEISSLEDER R, LEE H, ZHANG F & SHARP PA 2015. Genome-wide CRISPR screen in a mouse model of tumor growth and metastasis. Cell, 160, 1246–60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- CHENG J, DECAPRIO JA, FLUCK MM & SCHAFFHAUSEN BS 2009. Cellular transformation by Simian Virus 40 and Murine Polyoma Virus T antigens. Semin Cancer Biol, 19, 218–28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- CHERRY SR, BINISZKIEWICZ D, VANPARUS L, BALTIMORE D & JAENISCH R 2000. Retroviral Expression in Embryonic Stem Cells and Hematopoietic Stem Cells. Mol Cell Biol. [DOI] [PMC free article] [PubMed] [Google Scholar]
- CHEUNG AF, DUPAGE MJ, DONG HK, CHEN J & JACKS T 2008. Regulated expression of a tumor-associated antigen reveals multiple levels of T-cell tolerance in a mouse model of lung cancer. Cancer Res, 68, 9459–68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- CHUNG WJ, DAEMEN A, CHENG JH, LONG JE, COOPER JE, WANG BE, TRAN C, SINGH M, GNAD F, MODRUSAN Z, FOREMAN O & JUNTTILA MR 2017. Kras mutant genetically engineered mouse models of human cancers are genomically heterogeneous. Proc Natl Acad Sci U S A, 114, E10947–E10955. [DOI] [PMC free article] [PubMed] [Google Scholar]
- CONNOLLY KA, KUCHROO M, VENKAT A, KHATUN A, WANG J, WILLIAM I, HORNICK N, FITZGERALD B, DAMO M, KASMANI MY, CUI C, FAGERBERG E, MONROY I, HUTCHINS A, CHEUNG JF, FOSTER GG, MARIUZZA DL, ZHAO H, CUI W, KRISHNASWAMY S & JOSHI NS 2021. A reservoir of stem-like CD8 T cells in the tumor-draining lymph node maintains the ongoing anti-tumor immune response. BioRxiv [preprint]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- CORTEZ JT, MONTAUTI E, SHIFRUT E, GATCHALIAN J, ZHANG Y, SHAKED O, XU Y, ROTH TL, SIMEONOV DR, ZHANG Y, CHEN S, LI Z, WOO JM, HO J, VOGEL IA, PRATOR GY, ZHANG B, LEE Y, SUN Z, IFERGAN I, VAN GOOL F, HARGREAVES DC, BLUESTONE JA, MARSON A & FANG D 2020. CRISPR screen in regulatory T cells reveals modulators of Foxp3. Nature, 582, 416–420. [DOI] [PMC free article] [PubMed] [Google Scholar]
- CUI C, WANG J, CHEN P, CONNOLLY KA, DAMO M, FAGERBERG E, CHEN S, EISENBARTH SC, ZHAO H, CRAFT J & JOSHI NS 2020. Neoantigen driven B cell and CD4+ T follicular helper cell collaboration promotes robust anti-tumor CD8+ T cell responses. Available at SSRN: https://ssrn.com/abstract=3751671. [DOI] [PMC free article] [PubMed] [Google Scholar]
- CURRAN MA, MONTALVO W, YAGITA H & ALLISON JP 2010. PD-1 and CTLA-4 combination blockade expands infiltrating T cells and reduces regulatory T and myeloid cells within B16 melanoma tumors. Proc Natl Acad Sci U S A, 107, 4275–80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- DAMO M, CUI C, WILLIAM I, HORNICK NI, KWOK D, CLULO K, DAMSKY WE, LEVENTHAL JS & JOSHI NS 2021. The PD-1 checkpoint receptor maintains tolerance of self-reactive CD8 T cell in skin. Available at BioRxiv: 10.1101/2021.07.09.451765. [DOI] [Google Scholar]
- DAMO M, FITZGERALD B, LU Y, NADER M, WILLIAM I, CHEUNG JF, CONNOLLY KA, FOSTER GG, AKAMA-GARREN E, LEE DY, CHANG GP, GOCHEVA V, SCHMIDT LM, BOILEVE A, WILSON JH, CUI C, MONROY I, GOKARE P, CABECEIRAS P, JACKS T & JOSHI NS 2020. Inducible de novo expression of neoantigens in tumor cells and mice. Nat Biotechnol. [DOI] [PMC free article] [PubMed] [Google Scholar]
- DAVIS BW & OSTRANDER EA 2014. Domestic dogs and cancer research: a breed-based genomics approach. ILAR J, 55, 59–68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- DEVITA VT JR. & CHU E 2008. A history of cancer chemotherapy. Cancer Res, 68, 8643–53. [DOI] [PubMed] [Google Scholar]
- DEWI FN & CLINE JM 2021. Nonhuman primate model in mammary gland biology and neoplasia research. Lab Anim Res, 37, 3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- DONG MB, WANG G, CHOW RD, YE L, ZHU L, DAI X, PARK JJ, KIM HR, ERRAMI Y, GUZMAN CD, ZHOU X, CHEN KY, RENAUER PA, DU Y, SHEN J, LAM SZ, ZHOU JJ, LANNIN DR, HERBST RS & CHEN S 2019. Systematic Immunotherapy Target Discovery Using Genome-Scale In Vivo CRISPR Screens in CD8 T Cells. Cell, 178, 1189–1204 e23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- DUBROT J, LANE-RETICKER SK, KESSLER EA, AYER A, MISHRA G, WOLFE CH, ZIMMER MD, DU PP, MAHAPATRA A, OCKERMAN KM, DAVIS TGR, KOHNLE IC, POPE HW, ALLEN PM, OLANDER KE, IRACHETA-VELLVE A, DOENCH JG, HAINING WN, YATES KB & MANGUSO RT 2021. In vivo screens using a selective CRISPR antigen removal lentiviral vector system reveal immune dependencies in renal cell carcinoma. Immunity, 54, 571–585.e6. [DOI] [PubMed] [Google Scholar]
- DUNN GP, BRUCE AT, SHEEHAN KC, SHANKARAN V, UPPALURI R, BUI JD, DIAMOND MS, KOEBEL CM, ARTHUR C, WHITE JM & SCHREIBER RD 2005. A critical function for type I interferons in cancer immunoediting. Nat Immunol, 6, 722–9. [DOI] [PubMed] [Google Scholar]
- DUPAGE M, CHEUNG AF, MAZUMDAR C, WINSLOW MM, BRONSON R, SCHMIDT LM, CROWLEY D, CHEN J & JACKS T 2011. Endogenous T cell responses to antigens expressed in lung adenocarcinomas delay malignant tumor progression. Cancer Cell, 19, 72–85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- DUPAGE M & JACKS T 2013. Genetically engineered mouse models of cancer reveal new insights about the antitumor immune response. Curr Opin Immunol, 25, 192–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- DUPAGE M, MAZUMDAR C, SCHMIDT LM, CHEUNG AF & JACKS T 2012. Expression of tumour-specific antigens underlies cancer immunoediting. Nature, 482, 405–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- ENGEL AM, SVANE IM, RYGAARD J & WERDELIN O 1997. MCA sarcomas induced in scid mice are more immunogenic than MCA sarcomas induced in congenic, immunocompetent mice. Scand J Immunol, 45. [DOI] [PubMed] [Google Scholar]
- ENGELHARD VH, RODRIGUEZ AB, MAULDIN IS, WOODS AN, PESKE JD & SLINGLUFF CL 2018. Immune Cell Infiltration and Tertiary Lymphoid Structures as Determinants of Antitumor Immunity. The Journal of Immunology, 200, 432–442. [DOI] [PMC free article] [PubMed] [Google Scholar]
- FITZGERALD B, CONNOLLY KA, CUI C, MARIUZZA DL, FAGERBERG E, HORNICK NI, FOSTER GG, WILLIAM I, CHEUNG JF & JOSHI NS 2021. A novel mouse model for the study of anti-tumor T cell responses in Kras driven lung adenocarcinoma. Available at SSRN: 10.2139/ssrn.3835424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- FOLEY EJ 1953. Antigenic Properties of Methylcholanthrene-induced Tumors in Mice of the Strain of Origin. Cancer Res, 13. [PubMed] [Google Scholar]
- FORERO A, GIACOBBI NS, MCCORMICK KD, GJOERUP OV, BAKKENIST CJ, PIPAS JM & SARKAR SN 2014. Simian virus 40 large T antigen induces IFN-stimulated genes through ATR kinase. J Immunol, 192, 5933–42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- FOULDS L 1954. The Experimental Study of Tumor Progression: A Review. Cancer Res, 14. [PubMed] [Google Scholar]
- FRESE KK & TUVESON DA 2007. Maximizing mouse cancer models. Nat Rev Cancer, 7, 645–58. [DOI] [PubMed] [Google Scholar]
- FU C & JIANG A 2018. Dendritic Cells and CD8 T Cell Immunity in Tumor Microenvironment. Front Immunol, 9, 3059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- FURTH J 1959. A Meeting of Ways in Cancer Research: Thoughts on the Evolution and Nature of Neoplasms. Cancer Res, 19. [PubMed] [Google Scholar]
- GALLILY R & WOOLLEY GW 1958. THE HUMAN TUMOR IN THE MOUSE*. Ann. N. Y. Acad. Sci, 76, 791–796. [DOI] [PubMed] [Google Scholar]
- GARDINER RE, JAHANGEER S, FORDE P, ARIFFIN AB, BIRD B, SODEN D & HINCHION J 2015. Low immunogenicity in non-small cell lung cancer; do new developments and novel treatments have a role? Cancer Metastasis Rev, 34, 129–44. [DOI] [PubMed] [Google Scholar]
- GERMANO G, LAMBA S, ROSPO G, BARAULT L, MAGRI A, MAIONE F, RUSSO M, CRISAFULLI G, BARTOLINI A, LERDA G, SIRAVEGNA G, MUSSOLIN B, FRAPOLLI R, MONTONE M, MORANO F, DE BRAUD F, AMIROUCHENE-ANGELOZZI N, MARSONI S, D’INCALCI M, ORLANDI A, GIRAUDO E, SARTORE-BIANCHI A, SIENA S, PIETRANTONIO F, DI NICOLANTONIO F & BARDELLI A 2017. Inactivation of DNA repair triggers neoantigen generation and impairs tumour growth. Nature, 552, 116120. [DOI] [PubMed] [Google Scholar]
- GOEL S, DECRISTO MJ, WATT AC, BRINJONES H, SCENEAY J, LI BB, KHAN N, UBELLACKER JM, XIE S, METZGER-FILHO O, HOOG J, ELLIS MJ, MA CX, RAMM S, KROP IE, WINER EP, ROBERTS TM, KIM HJ, MCALLISTER SS & ZHAO JJ 2017. CDK4/6 inhibition triggers anti-tumour immunity. Nature, 548, 471–475. [DOI] [PMC free article] [PubMed] [Google Scholar]
- GOFF SP & BERG P 1976. Construction of Hybrid Viruses Containing SV40 and A Phage DNA Segments and Their Propagation in Cultured Monkey Cells. Cell, 9. [DOI] [PubMed] [Google Scholar]
- GORER PA 1936. The Detection of Antigenic Differences in Mouse Erythrocytes by the Employment of Immune Sera. Br J Exp Pathol, 17. [Google Scholar]
- GROSS L 1943. Intradermal Immunization of C3H Mice against a Sarcoma That Originated in an Animal of the Same Line. Cancer Res, 3. [Google Scholar]
- GUERIN MV, FINISGUERRA V, VAN DEN EYNDE BJ, BERCOVICI N & TRAUTMANN A 2020. Preclinical murine tumor models: a structural and functional perspective. Elife, 9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- GUIL-LUNA S, SEDLIK C & PIAGGIO E 2021. Humanized Mouse Models to Evaluate Cancer Immunotherapeutics. Annu. Rev. Cancer Biol, 5. [Google Scholar]
- HANAHAN D 1985. Heritable formation of pancreatic Beta cell tumours in transgenic mice expressing recombinant insulin/simian virus 40 oncogenes. Nature, 315, 115–122. [DOI] [PubMed] [Google Scholar]
- HANAHAN D, WAGNER EF & PALMITER RD 2007. The origins of oncomice: a history of the first transgenic mice genetically engineered to develop cancer. Genes Dev, 21, 2258–70. [DOI] [PubMed] [Google Scholar]
- HASUI M, SAIKAWA Y, MIURA M, TAKANO N, UENO Y, YACHIE A, MIYAWAKI T & TANIGUCHI N 1989. Effector and Precursor Phenotypes of Lymphokine-Activated Killer Cells in Mice with Severe Combined lmmunodeficiency (Scid) and Athymic (Nude) Mice. Cellular Immunology, 120. [DOI] [PubMed] [Google Scholar]
- HEGDE S, KRISNAWAN VE, HERZOG BH, ZUO C, BREDEN MA, KNOLHOFF BL, HOGG GD, TANG JP, BAER JM, MPOY C, LEE KB, ALEXANDER KA, ROGERS BE, MURPHY KM, HAWKINS WG, FIELDS RC, DESELM CJ, SCHWARZ JK & DENARDO DG 2020. Dendritic Cell Paucity Leads to Dysfunctional Immune Surveillance in Pancreatic Cancer. Cancer Cell, 37, 289–307 e9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- HEYER J, KWONG LN, LOWE SW & CHIN L 2010. Non-germline genetically engineered mouse models for translational cancer research. Nat Rev Cancer, 10, 470–80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- HUI R, GARON EB, GOLDMAN JW, LEIGHL NB, HELLMANN MD, PATNAIK A, GANDHI L, EDER JP, AHN MJ, HORN L, FELIP E, CARCERENY E, RANGWALA R, LUBINIECKI GM, ZHANG J, EMANCIPATOR K, ROACH C & RIZVI NA 2017. Pembrolizumab as first-line therapy for patients with PD-L1-positive advanced non-small cell lung cancer: a phase 1 trial. Ann Oncol, 28, 874–881. [DOI] [PMC free article] [PubMed] [Google Scholar]
- HUIJBERS IJ, DEL BRAVO J, BIN ALI R, PRITCHARD C, BRAUMULLER TM, VAN MILTENBURG MH, HENNEMAN L, MICHALAK EM, BERNS A & JONKERS J 2015. Using the GEMM-ESC strategy to study gene function in mouse models. Nat Protoc, 10, 1755–85. [DOI] [PubMed] [Google Scholar]
- HUIJBERS IJ, KRIMPENFORT P, BERNS A & JONKERS J 2011. Rapid validation of cancer genes in chimeras derived from established genetically engineered mouse models. Bioessays, 33, 701–10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- HUIJBERS IJ, SOUDJA SM, UYTTENHOVE C, BUFERNE M, INDERBERG-SUSO EM, COLAU D, PILOTTE L, POWIS DE TENBOSSCHE CG, CHOMEZ P, BRASSEUR F, SCHMITT-VERHULST AM & VAN DEN EYNDE BJ 2012. Minimal tolerance to a tumor antigen encoded by a cancer-germline gene. J Immunol, 188, 111–21. [DOI] [PubMed] [Google Scholar]
- JAENISCH R & MINTZ B 1974. Simian Virus 40 DNA Sequences in DNA of Healthy Adult Mice Derived from Preimplantation Blastocysts Injected with Viral DNA. Proc Natl Acad Sci U S A, 71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- JAVIER RT & BUTEL JS 2008. The history of tumor virology. Cancer Res, 68, 7693–706. [DOI] [PMC free article] [PubMed] [Google Scholar]
- JONKERS J & BERNS A 2002. Conditional mouse models of sporadic cancer. Nat Rev Cancer, 2, 251–65. [DOI] [PubMed] [Google Scholar]
- JOSHI NS, AKAMA-GARREN EH, LU Y, LEE DY, CHANG GP, LI A, DUPAGE M, TAMMELA T, KERPER NR, FARAGO AF, ROBBINS R, CROWLEY DM, BRONSON RT & JACKS T 2015. Regulatory T Cells in Tumor-Associated Tertiary Lymphoid Structures Suppress Anti-tumor T Cell Responses. Immunity, 43, 579–90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- KENNAWAY EL & HIEGER I 1930. CARCINOGENIC SUBSTANCES AND THEIR FLUORESCENCE SPECTRA. Br Med J, 1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- KERSTEN K, DE VISSER KE, VAN MILTENBURG MH & JONKERS J 2017. Genetically engineered mouse models in oncology research and cancer medicine. EMBO Mol Med, 9, 137–153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- KLEIN G & KLEIN E 1957. The evolution of independence from specific growth stimulation and inhibition in mammalian tumour-cell populations. Symp Soc Exp Biol, 11. [PubMed] [Google Scholar]
- KOEBEL CM, VERMI W, SWANN JB, ZERAFA N, RODIG SJ, OLD LJ, SMYTH MJ & SCHREIBER RD 2007. Adaptive immunity maintains occult cancer in an equilibrium state. Nature, 450, 903–7. [DOI] [PubMed] [Google Scholar]
- LATHROP, A. E. C. L., LEO 1915a. Further Investifation on the Origin of Tumors in Mice: I. Tumor Incidence and Tumor Age in Various Strains of Mice. J Exp Med, 22, 646–673. [DOI] [PMC free article] [PubMed] [Google Scholar]
- LATHROP, A. E. C. L., LEO 1915b. Further Investigation on the Origin of Tumors in Mice: II. Tumor Incidence and Tumor Age in Hybrids. J Exp Med, 22, 713–731. [DOI] [PMC free article] [PubMed] [Google Scholar]
- LATHROP, A. E. C. L., LEO 1918. Further Investigation on the Origin of Tumors in Mice: V. The Tumor Rate in Hybrid Strains. J Exp Med, 28, 475–500. [DOI] [PMC free article] [PubMed] [Google Scholar]
- LEAH DR, KRUMMEL MF & ALLISON JP 1996. Enhancement of Antitumor Immunity by CTLA-4 Blockade. Science, 271. [DOI] [PubMed] [Google Scholar]
- LEE JW, KOMAR CA, BENGSCH F, GRAHAM K & BEATTY GL 2016. Genetically Engineered Mouse Models of Pancreatic Cancer: The KPC Model (LSL-Kras(G12D/+) ;LSL-Trp53(R172H/+) ;Pdx-1-Cre), Its Variants, and Their Application in Immuno-oncology Drug Discovery. Curr Protoc Pharmacol, 73, 14 39 1–14 39 20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- LEE PP, YEE C, SAVAGE PA, FONG L, BROCKSTEDT DG, WEBER JS, JOHNSON D, SWETTER S, THOMPSON J, GREENBERG PD, ROEDERER M & DAVIS MM 1999. Characterization of circulating T cells specific for tumor- associated antigens in melanoma patients. Nat Med, 5. [DOI] [PubMed] [Google Scholar]
- LEVINE AJ & OREN M 2009. The first 30 years of p53: growing ever more complex. Nat Rev Cancer, 9, 749–58. [DOI] [PMC free article] [PubMed] [Google Scholar]
- LI J, LEE Y, LI Y, JIANG Y, LU H, ZANG W, ZHAO X, LIU L, CHEN Y, TAN H, YANG Z, ZHANG MQ, MAK TW, NI L & DONG C 2018. Co-inhibitory Molecule B7 Superfamily Member 1 Expressed by Tumor-Infiltrating Myeloid Cells Induces Dysfunction of Anti-tumor CD8(+) T Cells. Immunity, 48, 773–786 e5. [DOI] [PubMed] [Google Scholar]
- LITTLE CC 1911. The “Dilute” Forms of Yellow Mice. Science, 33, 896–897. [DOI] [PubMed] [Google Scholar]
- LITTLE CC 1913. “Yellow” and “Agouti” Factors in Mice. Science, 38, 205. [DOI] [PubMed] [Google Scholar]
- LITTLE CC 1914. A POSSIBLE MENDELIAN EXPLANATION FOR A TYPE OF INHERITANCE APPARENTLY NON-MENDELIAN IN NATURE. Science, 40. [DOI] [PubMed] [Google Scholar]
- LITTLE CC 1927. Notes on a Species Cross in Mice and on an Hypothesis Concerning the Quantitative Potentiality of Genes. Science, 66, 542–543. [DOI] [PubMed] [Google Scholar]
- LITTLE CC & TYZZER EE 1916. Further Experimental Studies on the Inheritance of Susceptibility to a Transplantable Tumor, Carcinoma (J. w. A.) of the Japanese Waltzing Mouse. J Med Res, 33, 393–453. [PMC free article] [PubMed] [Google Scholar]
- LIU J, BLAKE SJ, SMYTH MJ & TENG MW 2014. Improved mouse models to assess tumour immunity and irAEs after combination cancer immunotherapies. Clin Transl Immunology, 3, e22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- LIVSHITS G & LOWE SW 2013. Accelerating cancer modeling with RNAi and nongermline genetically engineered mouse models. Cold Spring Harb Protoc, 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- LOEB L 1901. On Transplantation of Tumors. J Med Res, 6, 28–38. [PMC free article] [PubMed] [Google Scholar]
- MA L, DICHWALKAR T, CHANG JYH, COSSETTE B, GARAFOLA D, ZHANG AQ, FICHTER M, WANG C, LIANG S, SILVA M, KUMARI S, MEHTA NK, ABRAHAM W, THAI N, LI N, WITTRUP KD & IRVINE DJ 2019. Enhanced CAR–T cell activity against solid tumors by vaccine boosting through the chimeric receptor. Science, 365, 162–168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- MANGUSO RT, POPE HW, ZIMMER MD, BROWN FD, YATES KB, MILLER BC, COLLINS NB, BI K, LAFLEUR MW, JUNEJA VR, WEISS SA, LO J, FISHER DE, MIAO D, VAN ALLEN E, ROOT DE, SHARPE AH, DOENCH JG & HAINING WN 2017. In vivo CRISPR screening identifies Ptpn2 as a cancer immunotherapy target. Nature, 547, 413–418. [DOI] [PMC free article] [PubMed] [Google Scholar]
- MCCREERY MQ & BALMAIN A 2017. Chemical Carcinogenesis Models of Cancer: Back to the Future. Annu. Rev. Cancer Biol, 1, 295–310. [Google Scholar]
- MCFADDEN DG, POLITI K, BHUTKAR A, CHEN FK, SONG X, PIRUN M, SANTIAGO PM, KIM-KISELAK C, PLATT JT, LEE E, HODGES E, ROSEBROCK AP, BRONSON RT, SOCCI ND, HANNON GJ, JACKS T & VARMUS H 2016. Mutational landscape of EGFR-, MYC-, and Kras-driven genetically engineered mouse models of lung adenocarcinoma. Proc Natl Acad Sci U S A, 113, E6409–E6417. [DOI] [PMC free article] [PubMed] [Google Scholar]
- MERAZ IM, MAJIDI M, MENG F, SHAO R, HA MJ, NERI S, FANG B, LIN SH, TINKEY PT, SHPALL EJ, MORRIS J & ROTH JA 2019. An Improved Patient-Derived Xenograft Humanized Mouse Model for Evaluation of Lung Cancer Immune Responses. Cancer Immunol Res, 7, 1267–1279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- MIAN SA, ANJOS-AFONSO F & BONNET D 2020. Advances in Human Immune System Mouse Models for Studying Human Hematopoiesis and Cancer Immunotherapy. Front Immunol, 11, 619236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- MOSER AR, LUONGO C, GOULD KA, MCNELEY MK, SHOEMAKER AR & DOVE WF 1995. ApcMin: A Mouse Model for Intestinal and Mammary Tumorigenesis. Eur J Cancer, 31A, 1061–1064. [DOI] [PubMed] [Google Scholar]
- MULLIGAN RC & BERG P 1980. Expression of a bacterial gene in mammalian cells. Science, 209. [DOI] [PubMed] [Google Scholar]
- MULLIGAN RC, HOWARD BH & BERG P 1979. Synthesis of rabbit 13-globin in cultured monkey kidney cells following infection with a SV40 13-globin recombinant genome. Nature, 277. [DOI] [PubMed] [Google Scholar]
- MUNOZ-ERAZO L, RHODES JL, MARION VC & KEMP RA 2020. Tertiary lymphoid structures in cancer - considerations for patient prognosis. Cell Mol Immunol, 17, 570–575. [DOI] [PMC free article] [PubMed] [Google Scholar]
- MURRAY WS & LITTLE CC 1935. Further data on the existence of extra-chromosomal influence on the incidence of mammary tumors in mice. Science, 82. [DOI] [PubMed] [Google Scholar]
- OLD LJ 1981. Cancer Immunology: The Search for Specificity — G. H. A. Clowes Memorial Lecture. Cancer Res, 41. [PubMed] [Google Scholar]
- OLSON B, LI Y, LIN Y, LIU ET & PATNAIK A 2018. Mouse Models for Cancer Immunotherapy Research. Cancer Discov, 8, 1358–1365. [DOI] [PMC free article] [PubMed] [Google Scholar]
- ONO M, YASUNAGA T, MIYATA T & USHIKUBO H 1986. Nucleotide Sequence of Human Endogenous Retrovirus Genome Related to the Mouse Mammary Tumor Virus Genome. J. Virol, 60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- OUTZEN HC 1975. Spontaneous and induced tumor incidence in germfree “nude” mice. J Reticuloendothel Soc, 17. [PubMed] [Google Scholar]
- PALM JE, TELLER MN, MERKER PC & WOOLLEY GW 1958. HOST CONDITIONING IN EXPERIMENTAL CHEMOTHERAPY*. Ann N Y Acad Sci, 76, 812–820. [DOI] [PubMed] [Google Scholar]
- PAN D, KOBAYASHI A, JIANG P, DE ANDRADE LF, TAY RE, LUOMA AM, TSOUCAS D, QIU X, LIM K, RAO P, LONG HM, YUAN G, DOENCH JG, BROWN M, LIU XS & WUCHERPFENNIG KW 2018. A major chromatin regulator determines resistance of tumor cells to T cell–mediated killing. Science, 359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- PARISH CR 2003. Cancer immunotherapy: the past, the present and the future. Immunol Cell Biol, 81, 106–13. [DOI] [PubMed] [Google Scholar]
- PEREZ-RUIZ E, MINUTE L, OTANO I, ALVAREZ M, OCHOA MC, BELSUE V, DE ANDREA C, RODRIGUEZ-RUIZ ME, PEREZ-GRACIA JL, MARQUEZ-RODAS I, LLACER C, ALVAREZ M, DE LUQUE V, MOLINA C, TEIJEIRA A, BERRAONDO P & MELERO I 2019. Prophylactic TNF blockade uncouples efficacy and toxicity in dual CTLA-4 and PD-1 immunotherapy. Nature, 569, 428–432. [DOI] [PubMed] [Google Scholar]
- POLITI K & PAO W 2011. How genetically engineered mouse tumor models provide insights into human cancers. J Clin Oncol, 29, 2273–81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- POSTOW MA, SIDLOW R & HELLMANN MD 2018. Immune-Related Adverse Events Associated with Immune Checkpoint Blockade. N Engl J Med, 378, 158–168. [DOI] [PubMed] [Google Scholar]
- PRADELLA D, NARO C, SETTE C & GHIGNA C 2017. EMT and stemness: flexible processes tuned by alternative splicing in development and cancer progression. Mol Cancer, 16, 8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- PREHN RT & MAIN JM 1957. Immunity to Methylcholanthrene-In- duced Sarcomas. Journal of the National Cancer Institute, 18. [PubMed] [Google Scholar]
- RADKEVICH-BROWN O, PIECHOCKI MP, BACK JB, WEISE AM, PILON-THOMAS S & WEI WZ 2010. Intratumoral DNA electroporation induces anti-tumor immunity and tumor regression. Cancer Immunol Immunother, 59, 409–17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- RAEZ LE, FEIN S & PODACK ER 2005. Lung Cancer Immunotherapy. Clin. Med. Res, 3, 221–228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- RIBATTI D 2016. The concept of immune surveillance against tumors: The first theories. Oncotarget, 8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- ROSENBERG SA, SPIESS P & LAFRENIERE R 1986. A New Approach to the Adoptive Immunotherapy of Cancer with Tumor-Infiltrating Lymphocytes. Science, 233. [DOI] [PubMed] [Google Scholar]
- ROTH TL, LI PJ, BLAESCHKE F, NIES JF, APATHY R, MOWERY C, YU R, NGUYEN MLT, LEE Y, TRUONG A, HIATT J, WU D, NGUYEN DN, GOODMAN D, BLUESTONE JA, YE CJ, ROYBAL K, SHIFRUT E & MARSON A 2020. Pooled Knockin Targeting for Genome Engineering of Cellular Immunotherapies. Cell, 181, 728–744 e21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- RUNGE A, HU J, WIELAND M, BERGEEST JP, MOGLER C, NEUMANN A, GERAUD C, ARNOLD B, ROHR K, KOMLJENOVIC D, SCHIRMACHER P, GOERDT S & AUGUSTIN HG 2014. An inducible hepatocellular carcinoma model for preclinical evaluation of antiangiogenic therapy in adult mice. Cancer Res, 74, 4157–69. [DOI] [PubMed] [Google Scholar]
- SCHACHTSCHNEIDER KM, SCHWIND RM, NEWSON J, KINACHTCHOUK N, RIZKO M, MENDOZA-ELIAS N, GRIPPO P, PRINCIPE DR, PARK A, OVERGAARD NH, JUNGERSEN G, GARCIA KD, MAKER AV, RUND LA, OZER H, GABA RC & SCHOOK LB 2017. The Oncopig Cancer Model: An Innovative Large Animal Translational Oncology Platform. Front Oncol, 7, 190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- SCHIETINGER A, PHILIP M, KRISNAWAN VE, CHIU EY, DELROW JJ, BASOM RS, LAUER P, BROCKSTEDT DG, KNOBLAUGH SE, HAMMERLING GJ, SCHELL TD, GARBI N & GREENBERG PD 2016. Tumor-Specific T Cell Dysfunction Is a Dynamic Antigen-Driven Differentiation Program Initiated Early during Tumorigenesis. Immunity, 45, 389–401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- SCHONHUBER N, SEIDLER B, SCHUCK K, VELTKAMP C, SCHACHTLER C, ZUKOWSKA M, ESER S, FEYERABEND TB, PAUL MC, ESER P, KLEIN S, LOWY AM, BANERJEE R, YANG F, LEE CL, MODING EJ, KIRSCH DG, SCHEIDELER A, ALESSI DR, VARELA I, BRADLEY A, KIND A, SCHNIEKE AE, RODEWALD HR, RAD R, SCHMID RM, SCHNEIDER G & SAUR D 2014. A next-generation dual-recombinase system for time- and host-specific targeting of pancreatic cancer. Nat Med, 20, 1340–1347. [DOI] [PMC free article] [PubMed] [Google Scholar]
- SCHREIBER TH & PODACK ER 2009. A critical analysis of the tumour immunosurveillance controversy for 3-MCA-induced sarcomas. Br J Cancer, 101, 381–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- SCHREIBER TH, RAEZ L, ROSENBLATT JD & PODACK ER 2010. Tumor immunogenicity and responsiveness to cancer vaccine therapy: the state of the art. Semin Immunol, 22, 105–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- SCOTT OCA 1991. Tumor Transplantation and Tumor Immunity: A Personal View. Cancer Res, 51. [PubMed] [Google Scholar]
- SEN T, RODRIGUEZ BL, CHEN L, CORTE CMD, MORIKAWA N, FUJIMOTO J, CRISTEA S, NGUYEN T, DIAO L, LI L, FAN Y, YANG Y, WANG J, GLISSON BS, WISTUBA II, SAGE J, HEYMACH JV, GIBBONS DL & BYERS LA 2019. Targeting DNA Damage Response Promotes Antitumor Immunity through STING-Mediated T-cell Activation in Small Cell Lung Cancer. Cancer Discov, 9, 646–661. [DOI] [PMC free article] [PubMed] [Google Scholar]
- SHANKARAN V, IKEDA H, BRUCE AT, WHITE JM, SWANSON PE, OLD LJ & SCHREIBER RD 2001. IFNg and lymphocytes prevent primary tumour development and shape tumour immunogenicity. Nature, 410. [DOI] [PubMed] [Google Scholar]
- SHIBATA H, TOYAMA K, SHIOYA H, ITO M, HIROTA M, HASEGAWA S, MATSUMOTO H, TAKANO H, AKIYAMA T, TOYOSHIMA K, KANAMARU R, KANEGAE Y, SAITO I, NAKAMURA Y, SHIBA K & NODA T 1997. Rapid Colorectal Adenoma Formation Initiated by Conditional Targeting of the Apc Gene. Science, 278. [DOI] [PubMed] [Google Scholar]
- SHINKAI Y, RATHBUN G, LAM K, OLTZ EM, STEWART V, MENDELSOHN M, CHARRON J, DATTA M, YOUNG F, STALL AM & ALT FW 1992. RAG-2-Deficient Mice Lack Mature Lymphocytes Owing to Inability to Initiate V(D)J Rearrangement. Cell, 68. [DOI] [PubMed] [Google Scholar]
- SHU S, WU H-J, GE JY, ZEID R, HARRIS IS, JOVANOVIĆ B, MURPHY K, WANG B, QIU X, ENDRESS JE, REYES J, LIM K, FONT-TELLO A, SYAMALA S, XIAO T, REDDY CHILAMAKURI CS, PAPACHRISTOU EK, D’SANTOS C, ANAND J, HINOHARA K, LI W, MCDONALD TO, LUOMA A, MODISTE RJ, NGUYEN Q-D, MICHEL B, CEJAS P, KADOCH C, JAFFE JD, WUCHERPFENNIG KW, QI J, LIU XS, LONG H, BROWN M, CARROLL JS, BRUGGE JS, BRADNER J, MICHOR F & POLYAK K 2020. Synthetic Lethal and Resistance Interactions with BET Bromodomain Inhibitors in Triple-Negative Breast Cancer. Molecular Cell, 78, 1096–1113.e8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- SHULTZ LD, BREHM MA, GARCIA-MARTINEZ JV & GREINER DL 2012. Humanized mice for immune system investigation: progress, promise and challenges. Nat Rev Immunol, 12, 786–98. [DOI] [PMC free article] [PubMed] [Google Scholar]
- SMYTH MJ, GODFREY DI & TRAPANI JA 2001. A fresh look at tumor immunosurveillance and immunotherapy. Nat Immunol 2. [DOI] [PubMed] [Google Scholar]
- SNELL GD 1948. Methods for the study of histocompatibility genes*. J Genet, 49. [DOI] [PubMed] [Google Scholar]
- STEENSMA DP, KYLE RA & SHAMPO MA 2010. Abbie Lathrop, the “mouse woman of Granby”: rodent fancier and accidental genetics pioneer. Mayo Clin Proc, 85, e83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- STRONG LC 1935. The Establishment of the C3H Inbred Strain of Mice for the Study of Spontaneous Carcinoma of the Mammary Gland. Genetics, 20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- STUTMAN O 1974. Tumor Development after 3-Methylcholanthrene in Immunologically Deficient Athymic-Nude Mice. Science, 183. [DOI] [PubMed] [Google Scholar]
- STUTMAN O 1979. Spontaneous tumors in nude mice: effect of the viable yellow gene. Exp Cell Biol, 47. [DOI] [PubMed] [Google Scholar]
- SUGGITT M & BIBBY MC 2005. 50 Years of Preclinical Anticancer Drug Screening: Empirical to Target-Driven Approaches. Clin Cancer Res, 11. [PubMed] [Google Scholar]
- SWANN JB & SMYTH MJ 2007. Immune surveillance of tumors. J Clin Invest, 117, 1137–46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- TIVOL EA, BORRIELLO F, SCHWEITZER AN, LYNCH WP, BLUESTONE JA & SHARPE AH 1995. Loss of CTLA-4 Leads to Massive Lymphproliferation and Fatal Multiorgan Tissue Destruction, Revealing a Critical Negative Regulatory Role of CTLA-4. Immunity, 3, 541–547. [DOI] [PubMed] [Google Scholar]
- TOOLAN HW 1951. Successful subcutaneous growth and transplantation of human tumors in X-irradiated laboratory animals. Proc Soc Exp Biol Med, 77, 572–578. [DOI] [PubMed] [Google Scholar]
- TOOLAN HW 1958. THE TRANSPLANTABLE HUMAN TUMOR*. Ann N Y Acad Sci, 76, 733–741. [DOI] [PubMed] [Google Scholar]
- TOPALIAN SL, HODI FS, BRAHMER JR, GETTINGER SN, SMITH DC, MCDERMOTT DF, POWDERLY JD, CARVAJAL RD, SOSMAN JA, ATKINS MB, LEMING PD, SPIGEL DR, ANTONIA SJ, HORN L, DRAKE CG, PARDOLL DM, CHEN L, SHARFMAN WH, ANDERS RA, TAUBE JM, MCMILLER TL, XU H, KORMAN AJ, JURE-KUNKEL M, AGRAWAL S, MCDONALD D, KOLLIA GD, GUPTA A, WIGGINTON JM & SZNOL M 2012. Safety, Activity, and Immune Correlates of Anti–PD-1 Antibody in Cancer. N Engl J Med, 366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- TORFADOTTIR H, FREYSDOTTIR J, SKAFTADOTTIR I, HARALDSSON A, SIGFUSSON G & OGMUNDSDOTTIR HM 2006. Evidence for extrathymic T cell maturation after thymectomy in infancy. Clin Exp Immunol, 145, 407–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- TYZZER EE 1909. A Study of Inheritance in Mice with Reference to their Susceptibility to Transplantable Tumors. J Med Res, 21, 519–573. [PMC free article] [PubMed] [Google Scholar]
- VAN DER LEUN AM, THOMMEN DS & SCHUMACHER TN 2020. CD8(+) T cell states in human cancer: insights from single-cell analysis. Nat Rev Cancer, 20, 218–232. [DOI] [PMC free article] [PubMed] [Google Scholar]
- VAN DYKE T & JACKS T 2002. Cancer Modeling in the Modern Era: Progress and Challenges. Cell, 108. [DOI] [PubMed] [Google Scholar]
- VAREKI SM 2018. High and low mutational burden tumors versus immunologically hot and cold tumors and response to immune checkpoint inhibitors. J Immunother Cancer, 6, 157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- VERMA B & WESA A 2020. Establishment of Humanized Mice from Peripheral Blood Mononuclear Cells or Cord Blood CD34+ Hematopoietic Stem Cells for Immune-Oncology Studies Evaluating New Therapeutic Agents. Curr Protoc Pharmacol, 89. [DOI] [PubMed] [Google Scholar]
- VISSCHER MB, GREEN RG & BITTNER JJ 1942. Characterization of Milk Influence in Spontaneous Mammary Carcinoma. Proc. Soc. Exp. Biol. Med, 49. [Google Scholar]
- VOOIJIS M, JONKERS J & BERNS A 2001. A highly efficient ligand-regulated Cre recombinase mouse line shows that LoxP recombination is position dependent. EMBO reports, 2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- WALRATH JC, HAWES JJ, VAN DYKE T & REILLY KM 2010. Genetically Engineered Mouse Models in Cancer Research. [DOI] [PMC free article] [PubMed]
- WEISS RA 2020. A perspective on the early days of RAS research. Cancer Metastasis Rev, 39, 1023–1028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- WEISS RA & VOGT PK 2011. 100 years of Rous sarcoma virus. J Exp Med, 208, 2351–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- WHITE SB, CHEN J, GORDON AC, HARRIS KR, NICOLAI JR, WEST DL & LARSON AC 2015. Percutaneous ultrasound guided implantation of VX2 for creation of a rabbit hepatic tumor model. PLoS One, 10, e0123888. [DOI] [PMC free article] [PubMed] [Google Scholar]
- WÖLFEL T, HAUER M, SCHNEIDER J, SERRANO M, WÖLFEL C, KLEHMANN-HIEB E, PLAEN ED, HANKELN T, BÜSCHENFELDE K-HMZ & BEACH D 1995. A p16-Insensitive CDK4 Mutant Targeted by Cytolytic T Lymphocytes in a Human Melanoma. Science, 269. [DOI] [PubMed] [Google Scholar]
- WYNDER EL, GRAHAM EA & CRONINGER AB 1953. Experimental Production of Carcinoma with Cigarette Tar. Cancer Res, 13. [PubMed] [Google Scholar]
- XIA X, ZHANG Y, ZIETH CR & ZHANG S 2007. Transgenes Delivered by Lentiviral Vector Are Suppressed in Human Embryonic Stem Cells in a Promoter-Dependent Manner. Stem Cells Dev, 16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- YAMAGIWA K & ICHIKAWA K 1918. Experimental study of the pathogenesis of carcinoma. The Journal of Cancer Research, III. [DOI] [PubMed] [Google Scholar]
- YANG H, WANG H, SHIVALILA CS, CHENG AW, SHI L & JAENISCH R 2013. One-step generation of mice carrying reporter and conditional alleles by CRISPR/Cas-mediated genome engineering. Cell, 154, 1370–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- YAO LC, ARYEE KE, CHENG M, KAUR P, KECK JG & BREHM MA 2019. Creation of PDX-Bearing Humanized Mice to Study Immuno-oncology. Methods Mol Biol, 1953, 241–252. [DOI] [PubMed] [Google Scholar]
- ZHANG Y & ZHANG Z 2020. The history and advances in cancer immunotherapy: understanding the characteristics of tumor-infiltrating immune cells and their therapeutic implications. Cell Mol Immunol, 17, 807–821. [DOI] [PMC free article] [PubMed] [Google Scholar]