

Abstract
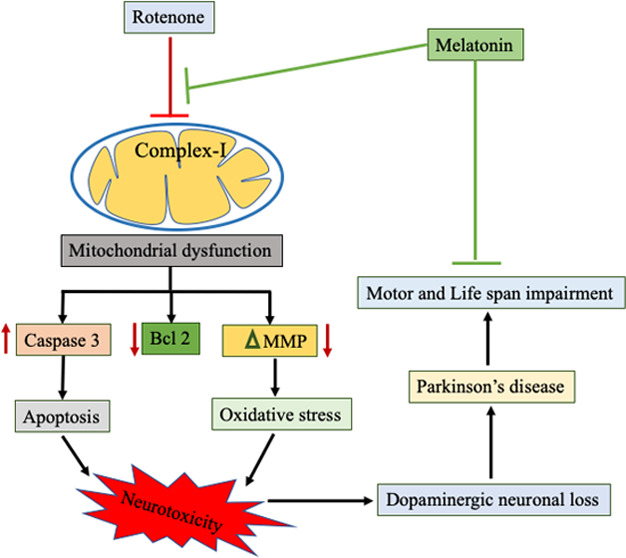
Parkinson’s disease (PD) is the second most common neurodegenerative disorder; however, its etiology remains elusive. Antioxidants are considered to be a promising approach for decelerating neurodegenerative disease progression owing to extensive examination of the relationship between oxidative stress and neurodegenerative diseases. In this study, we investigated the therapeutic effect of melatonin against rotenone-induced toxicity in the Drosophila model of PD. The 3–5 day old flies were divided into four groups: control, melatonin alone, melatonin and rotenone, and rotenone alone groups. According to their respective groups, flies were exposed to a diet containing rotenone and melatonin for 7 days. We found that melatonin significantly reduced the mortality and climbing ability of Drosophila because of its antioxidative potency. It alleviated the expression of Bcl 2, tyrosine hydroxylase (TH), NADH dehydrogenase, mitochondrial membrane potential, and mitochondrial bioenergetics and decreased caspase 3 expression in the Drosophila model of rotenone-induced PD-like symptoms. These results indicate the neuromodulatory effect of melatonin, and that it is likely modulated against rotenone-induced neurotoxicity by suppressing oxidative stress and mitochondrial dysfunctions.
Introduction
Parkinson’s disease (PD) is the second most devastating, late-onset neurological condition next to AD in humans;1,2 it is characterized by the progressive loss of dopaminergic neurons in the substantia nigra pars compacta (SNpc) region and ensuing dopamine (DA) content reduction in the striatum clinically.3,4 DA is an important mammalian neurotransmitter that plays a crucial role in movement control, cognition, and motivation.5 Postural instability, slowness of movement, rigidity, and tremor are the most common pathological conditions in both familial and sporadic cases of PD. The wistful is that these PD symptoms appear at a late stage, i.e., when >70% of the nigrostriatal region’s dopaminergic neurons are already deceased.6 The exact cause of PD remains elusive, but genetic mutations and several environmental risk factors have been implicated in the etiology of the idiopathic form.7 Concerning genetic factors in the onset of PD, parkin, DJ1, and SNCA (α-synuclein) have been identified as causative genes in familial forms. In addition to genetic factors, insecticides have been repeatedly implicated in PD pathogenesis. Rotenone, a commonly used insecticide, is found to be safe at lower concentrations owing to its very short half-life. Because of its lipophilic nature, rotenone readily passes across the blood–brain barrier.8 It is suspected to be a mitochondrial complex I inhibitor that inhibits the transfer of electrons from iron–sulfur centers to ubiquinone and elicits PD-like symptoms.9,10
Rotenone potentially inhibits complex I (NADH: ubiquinone oxidoreductase) of the mitochondrial electron transport chain.11 Rotenone inhibits adenosine triphosphate (ATP) synthesis in mitochondria in vitro by completely preventing the oxidation of pyruvate and many other physiological substrates like malate and glutamate. Pyruvate, malate, and glutamate are complex I substrates and are widely used to initiate mitochondrial oxidative phosphorylation.12,13 Moreover, rotenone and other complex I inhibitors stimulate reactive oxygen species (ROS) production from mitochondria induce oxidative stress14,15 and lead to cell death of dopaminergic neurons.16 There is strong epidemiological evidence that chronic rotenone exposure is associated with PD in humans.17,18
Melatonin (N-acetyl-5-methoxytrptamine), a tryptophan metabolite released by the pineal gland, is a neurohormone that exhibits a circadian pattern of secretion.19 In addition to circadian rhythm regulation, it is also implicated in the regulation of various other important physiological functions including seasonal reproduction, immunity, and modulation of human mood and behavior in mammals.20 Antioxidant responses are strongly linked to melatonin and its metabolites.21 Melatonin ameliorates oxidative stress, and it is initiated by scavenging different oxidative stress-causing agents like hydroxyl radicals, restoring the mitochondrial membrane potential (MMP), and increasing the activity of antioxidant enzymes.22,23 In situations of elevated oxidative stress, it has a regulatory effect on the activities of enzymes involved in the generation of free radicals and on the microsomal membrane fluidity.24 Moreover, its solubility in lipid and aqueous media permits melatonin to function as a highly effective inhibitor of oxidative damage because it allows it to easily cross the morphophysiological barriers and enter subcellular compartments.25,26 It is associated with apoptosis, metastasis, angiogenesis, and inflammatory pathways at the molecular level.27 It inhibits apoptotic cell death through its antiapoptotic properties in various neurological diseases.28−30 Our previous laboratory study indicated that melatonin shows a neuroprotective impact against oxaliplatin-induced neurotoxicity in rodent models by maintaining mitochondrial integrity.31 Hence, based on these actions, it will be very interesting to look at the role of melatonin in rotenone-induced toxicity in the Drosophila model of PD. Further, in the sense of structural and functional similarity with humans, Drosophila melanogaster is the most common genetic model. It showed a human equivalence of the disease gene of 75%.32 Currently, both environmental neurotoxin-intoxicated and transgenic PD models have been developed to understand the pathogenesis.33 The rotenone-exposed Drosophila model showed a wide range of parkinsonian symptoms related to the loss of dopaminergic neurons.34−36 Here, we have studied the behavioral and neurodegenerative effects of rotenone exposure in the Drosophila model. We found that the rotenone-exposed Drosophila displayed severe impairment in locomotion, mortality, and selective loss of dopaminergic neurons in the brain. We also employed the importance of Drosophila as an in vivo model to study the role of melatonin in PD. In our study, we found that melatonin rescued rotenone-induced behavioral deficits and neuronal death, thus displaying the neuroprotective nature of melatonin.
Results
Melatonin Supplementation Reduces Rotenone-Induced Mortality, Modulates Climbing Deficits (Negative Geotaxis Assay), and Mitigates Decline in Complex I Enzyme Activity in Drosophila
For this study, concentration of rotenone (250 μM) was employed. We examined the effect of melatonin (1 mM) on the rotenone-induced survival ability of the flies. Exposure of adult male flies to rotenone and melatonin during a 7 day experimental period compared to the control group was observed. The result showed a significant decrease (P < 0.001) in the mortality of the flies over the experimental period compared to the rotenone group, and the number of mortalities each day was counted for 7 days. In general, mortality occurred between 4 and 7 days among rotenone-exposed flies. There was a significant increase (P < 0.001) in the number of deaths in the rotenone-exposed group compared to that in control (Figure 2A).
Figure 2.

Effect of melatonin on (A) mortality assay, (B) climbing assay, and (C) mitochondrial complex I activity. ***P < 0.001 vs unexposed control group, and significant (###P < 0.001) differences were seen between melatonin- and rotenone-exposed groups. One-way analysis of variance (ANOVA) was used to examine differences between the groups, with Tukey’s multiple-comparison test used to examine posthoc pairwise differences. The data is represented as the mean ± standard error of the mean (SEM) of three independent experiments (Mel, melatonin; Rot, rotenone).
The results on climbing ability are shown in Figure 2B. The rotenone-exposed flies show a significant decrease (P < 0.001) in the climbing ability compared to control flies. As evident by the large number of flies staying at the bottom of the glass column, cosupplementation with melatonin significantly improved (P < 0.001) the performances of flies. With melatonin supplementation, a greater number of flies showed negative geotaxis behavior, indicating a neuroprotective effect.
Rotenone-exposed flies displayed significant reduction (P < 0.001) in complex I (NADH dehydrogenase) enzyme activity compared to the control (Figure 2C), while melatonin cosupplementation clearly showed significant improvement (P < 0.001) in complex I activity in isolated head mitochondria of flies compared to the rotenone-treated group.
Effect of Melatonin Supplementation on Rotenone-Induced Alteration in Mitochondrial Oxidative Phosphorylation
We investigated the effect of melatonin on the level of mitochondrial oxygen consumption (state 3 respiration) and the respiratory control ratio (RCR) in rotenone-exposed flies (Figure 3). We observed a significant decline in oxygen consumption and RCR rate (P < 0.001) in the rotenone-exposed flies compared to that in unexposed flies. Melatonin at 1 mM significantly improved (P < 0.05) the oxygen consumption and RCR in the coexposed group compared to that in the rotenone-exposed alone group.
Figure 3.

Effect of melatonin on mitochondrial oxygen consumption (state 3, state 4, and RCR). (A) Effect of melatonin on state 3 concerning the rotenone-exposed group. (B) There was no effect of rotenone and melatonin on state 4. (C) Effect of melatonin on RCR concerning rotenone-exposed group. ***P < 0.001 vs unexposed control group and #P < 0.05 vs rotenone-exposed group. One-way ANOVA was used to examine differences between the groups, with Tukey’s multiple-comparison test used to examine posthoc pairwise differences. The data is represented as the mean ± SEM of three independent experiments (Mel, melatonin; Rot, rotenone).
Effect of Melatonin Exposure on Rotenone-Induced Mitochondrial Membrane Potential
Depolarized or inactive mitochondria have decreased membrane potential, which is vital for mitochondrial integrity or survival. We have done a flow cytometric analysis of tetramethylrhodamine ethyl ester (TMRE) for mitochondrial membrane depolarization. The result has depicted that the rotenone-exposed group of Drosophila had a significant decrease (P < 0.001) in MMP compared to the control group of Drosophila, while the melatonin cosupplemented group of flies showed a significant improvement (P < 0.001) in MMP compared to the rotenone-supplemented group of Drosophila (Figure 4).
Figure 4.

Effect of melatonin on MMP. (A) Pictorial representation of the number of mitochondrial events in different groups. (B) Pictorial representation of the TMRE fluorescence intensity in different groups. (C) Graph of relative changes in the TMRE fluorescence intensity. ***P < 0.001 vs unexposed group, ###P < 0.001 vs rotenone-exposed group. A one-way ANOVA was used to examine differences between the groups, with Tukey’s multiple-comparison test used to examine posthoc pairwise differences. The data is represented as mean ± SEM of three independent experiments (Mel, melatonin; Rot, rotenone).
Melatonin Treatment Reduced the Cell Death by Modulating the Expression of Caspase 3 Protein and Rescued Bcl 2 and TH Expression in the Coexposed Groups
To confirm the antiapoptotic effect of melatonin in the presence of rotenone, we measured the expression of caspase 3 and Bcl 2 proteins. Rotenone-supplemented flies have shown significant activation (P < 0.001) of caspase 3 protein expression compared to the control group, while melatonin cotreated flies significantly reduced (P < 0.05) the expression of caspase 3 protein compared to the flies supplemented with rotenone (Figure 5B). There is another protein called Bcl 2 that has antiapoptotic properties. The rotenone-exposed group had significantly reduced (P < 0.001) expression of Bcl 2 protein, whereas melatonin treatment significantly increased (P < 0.01) the expression of Bcl 2 protein in the coexposed group (Figure 5C).
Figure 5.

Effect of melatonin on expression of caspase 3, Bcl 2, and TH. (A) Representative immunoblots showing expression of caspase 3, Bcl 2, and TH in different groups. (B–D) Relative densitometry data of caspase 3, Bcl 2, and TH analyzed and normalized with β-actin. ***P < 0.001 vs unexposed group, #P < 0.05, ##P < 0.01, and ###P < 0.001 vs rotenone-exposed group. One-way ANOVA was used to examine differences between the groups, with Tukey’s multiple-comparison test used to examine posthoc pairwise differences. The data is represented as the mean ± SEM of three independent experiments (Mel, melatonin; Rot, rotenone).
TH is an enzyme that catalyzes the rate-limiting step in the synthesis of dopamine.46 The result shows that TH expression significantly decreases (P < 0.001) in the rotenone-supplemented group of flies compared to the control group of flies, while the melatonin cosupplemented group of flies shows significantly increased (P < 0.001) expression of TH compared to the rotenone-supplemented flies (Figure 5D).
Discussion
PD is a multifactorial disease and may result due to a combination of reasons, such as environmental exposures, genetic susceptibility, and age-related changes. There has been no perfect parkinsonian model to date that can completely replicate all of the features of human PD, i.e., pathological and pathogenetic. However, most motor and nonmotor symptoms of PD are reproduced by the rotenone-based parkinsonian models. Rotenone is a pesticide that creates oxidative stress by inhibiting complex I of the electron transport chain. Moreover, it downregulates the expression of cytoprotective proteins, TH signaling, and striatal dopamine.47,48 Recently, the reproducibility of the rotenone model of PD has been highly improved.47 Besides, other advantages of rotenone-induced Drosophila models are their inexpensiveness, biocompatibility, and convenient administration, which make them widely applicable in most PD research.49 Mitochondrial complex I dysfunction has long been implicated in dopaminergic neuronal death and the pathogenesis of PD.50 Several rotenone-based toxin cell, rodent, and Drosophila models have been used to study the molecular mechanisms of cell death and potential therapeutics for PD.51 However, little is known about the precise mechanistic action of these agents in Drosophila; the involvement of mitochondrial complex I is well supported by several experimental studies.52,53
In our study, exposure to rotenone in Drosophila reproduces key aspects of PD, for instance, deficits in locomotion, death, and loss of dopaminergic neuronal cells. However, the flies coexposed to melatonin exhibited improvement in geotaxis (climbing), decreased mortality, and restoration of neuronal loss. We observed very high mortality in flies after 7 days exposure to rotenone that was mitigated by the supplementation of melatonin, clearly suggesting the protective effect of melatonin. Furthermore, we performed locomotive assays to confirm the alteration in motor coordination. A significant rate of locomotory deficit was observed in rotenone-exposed flies related to unexposed flies.54 Flies with locomotor deficits seem not to be able to coordinate their legs and wings properly in a normal fashion and have a tendency to stay at the bottom of the glass column. This phenotypic form was clarified earlier because of the high energy demands of ambulatory and flight muscles, which are rich in mitochondria,55 and rotenone is known to inhibit complex I of the same. On the contrary, the melatonin coexposed group of flies showed significant improvement in locomotory ability.19
It has been implicated that mitochondrial dysfunction, ATP depletion, alteration in mitochondrial membrane potential, and oxidative stress are key events involved in the onset of PD.56 Studies report that mitochondria are the powerhouse of cells and make approximately 95% of the cell’s ATP, utilizing oxidative phosphorylation.57 The process of ATP formation is carried out by the oxidative phosphorylation of electron transport chain enzymes.
NADH dehydrogenase introduces electrons to the electron transport chain of complex I.58 In our study, we have observed a significant decrease in the activity of NADH dehydrogenase due to rotenone administration. Interestingly, mitochondria isolated from the head of flies in the melatonin coexposed group showed significantly increased activity of NADH dehydrogenase. Reduced phospholipids content may be the cause of this change, as it may influence the movement of electrons from NADH to ubiquinone, resulting in the enhancement of activity.59 Further investigations are needed for a better understanding of the lipid–protein interactions within complex I and their influence on the kinetics of electron flow. The protective effect of melatonin against neurotoxicants has also been reported earlier.60 Here, we tried to explore the neuroprotective mechanism of melatonin in rotenone-induced toxicity in the Drosophila model of PD. Rotenone-induced mitochondrial ROS production impaired electron flux through complex I, which eventually reduces the MMP. Reduced MMP in PD due to the altered electron flow through complex I led to disturbed pumping of protons across the inner mitochondrial membrane.61 Flow cytometric monitoring of TMRE fluorescence demonstrated that rotenone-exposed flies resulted in a quick decrease in MMP, which is visible as a decline in the fluorescence intensity. This decline in the MMP intensity was significantly enhanced in the melatonin cosupplemented group of flies compared to that in only rotenone-exposed flies. Melatonin-mediated improvement in the preservation of the integrity of mitochondrial membrane has also been studied in vitro and in vivo in the Aβ42-induced AD Drosophila model by other authors.19
To confirm apoptosis, we analyzed the expression of caspase 3 protein that is promptly activated by the mitochondria-mediated stress and cleaved from inactive procaspase-3 protease protein localized in the cytosol.62,63 We found that the rotenone-exposed flies had enhanced expression or activation of caspase 3 compared to the control group. Subsequently, the coexposed group prevented caspase 3 activation compared to the coexposed control group. Moreover, we observed that antiapoptotic protein Bcl 2 expression was downregulated in the rotenone-exposed group and treatment with melatonin enhanced the expression. These observations are also in accordance with those of the previously published reports and, thus, prove that proapoptotic and antiapoptotic proteins are downregulated and upregulated, respectively, by melatonin in rat brains challenged with neurotoxicants.64,65
Further previous studies suggest that TH plays the role of a rate-limiting enzyme in the biosynthesis of catecholamines, such as dopamine, noradrenaline, and adrenaline.66 It is considered an essential biomarker of dopaminergic neurons in the pathogenesis of PD, and its decreased expression results in decreased dopamine synthesis, leading to the onset of PD-like symptoms. The reduction of TH in the striatum is also related to the rotenone-induced reduction of the dopamine content that leads to similar motor impairments.67
Similarly, we found decreased expression of TH in the rotenone-exposed group compared to that in the control group and melatonin supplementation helped to recover the loss in TH. Melatonin has an antioxidative property, which is responsible for the inhibition of neurodegeneration in substantia nigra pars compacta of dopaminergic neuronal cells.68 This might be the reason for the enhanced expression of TH in the melatonin cosupplemented group.
This study can be further extended to the transgenic and knockout Drosophila model of PD. This will help authenticate the present study. However, since Drosophila is an invertebrate some differences might arise between the metabolic processes of humans and Drosophila. Thus, the neuroprotective property of melatonin in the rotenone-induced PD model could be further studied in other model organisms.
Conclusions
Taken together, melatonin exhibited neuroprotective effects against rotenone-induced Drosophila model of PD mainly through its antioxidative property. Melatonin can restore rotenone-induced mitochondrial dysfunction and apoptosis. Additionally, melatonin reduced the behavioral deficit induced by rotenone in the PD model of Drosophila. The current study has explored the mechanism of neuroprotection attributed to melatonin. It could be a promising therapeutic approach in the treatment of PD as melatonin can restore the rotenone-induced decrease in NADH dehydrogenase activity, MMP, and impaired oxidative phosphorylation. Melatonin also retains the normal expression of Bcl 2 and TH. However, future research is required on the transgenic model Drosophila model of PD.
Materials and Methods
Drosophila Stock and Culture
The wild-type strain of Drosophila fly (Canton S) was generously gifted by Dr. Yasir Hasan Siddique, Department of Zoology, Faculty of Life Sciences, Aligarh Muslim University, Aligarh, Uttar Pradesh, India. The flies were maintained and reared on a standard cornmeal medium containing yeast granules as the protein source, agar–agar, and nipagin (preservative) at constant temperature (23 ± 2 °C) and 70–80% relative humidity under a 12 h dark/light cycle. Diet was prepared according to a standard protocol. One liter of semisolid diet contained 50 g of corn flour, 35 g of sucrose (carbohydrate source), 10 g of agar–agar (solidifying agent), 5 mL of propionic acid (antifungal agent), and 15 g of yeast granules added to ensure availability of the protein source. After 24 h, Drosophila flies were transferred to stock bottles to avoid sticking flies to the media.
Treatment Regimen and Experimental Design
Adult male Drosophila flies (n = 50 replicate–1; 3 replicates per group) were maintained on rotenone- and melatonin-enriched medium for 7 days. Melatonin was dissolved in 1.0% ethanol in normal saline (0.9%).23 The medium was changed every alternate day. Following are the groups and the treatment schedules we planned:
Group I:Drosophila flies were exposed to an equal volume of vehicle (dimethyl sulfoxide (DMSO)).
Group II: Drosophila flies were exposed to only melatonin (1 mM).
Group III:Drosophila flies were coexposed to rotenone (250 μM) and melatonin (1 mM)
Group IV:Drosophila flies were exposed to only rotenone (250 μM).
Mortality and climbing assays were performed after the completion of 7 day exposure to drugs. Flies’ heads were isolated in ice-cold phosphate-buffered saline (PBS) for molecular and biochemical studies (Figure 1).
Figure 1.

Pictorial representation of the overall experimental design (Mel, melatonin; Rot, rotenone).
Climbing Assay (Negative Geotaxis Assay)
The climbing assay was performed as described by Siddique et al.37 Test flies were anesthetized and placed in a vertical glass column (standard length, 25 cm; diameter, 1.5 cm). After a brief recovery period, flies were gently tapped to the bottom of the column. Following 1 min, flies that reached the top of the column and flies that remained at the bottom were counted separately. Data were expressed as the percent flies escaped beyond a minimum distance of 6 cm in 60 s of interval. Twenty adults per replication were used for the assay. The assays were repeated five times, and the score for each replication was an average of such trials for each group of flies including control. All behavioral studies were performed at 25 °C under standard lighting conditions.
Survival Assay
In this assay, about 150 male flies were separated for each group and transferred to the test food containing rotenone in various concentrations. The test food was changed every alternate day for 7 days, and mortality was noted every day.38 The survival rate was evaluated by counting days of the number of living flies until the end of the experimental period (7 days). The total number of flies represents the sum of three independent experiments (50 flies/each treatment repetition).
Isolation of Mitochondria
Mitochondria were isolated by the differential centrifugation method.39 Briefly, sample were homogenized in 10% volumes (1:10 w/v) by a motor-driven tissue grinder in an ice-cold isolation buffer containing 250 mM sucrose, 10 mM 4-(2-hydroxyethyl)-1-piperazineethanesulfonic acid (HEPES), 1 mM ethylene glycol-bis(β-aminoethyl ether)-N,N,N′,N′-tetraacetic acid (EGTA), and 0.1% fat-free bovine serum albumin (BSA) adjusted by Tris to pH 7.4 and centrifuged at 1000g for 8 min at 4 °C. The supernatant was collected and centrifuged at 10,000g for 10 min at 4 °C. Thereafter, the obtained pellet was resuspended; washed twice with a washing medium containing 250 mM sucrose, 10 mM HEPES, and 0.1 mM EGTA adjusted by Tris to pH 7.4; and centrifuged at 12,300g for 10 min. Finally, the pellet was resuspended in an isolation medium containing 250 mM sucrose, 10 mM HEPES, and 0.1% fat-free BSA adjusted by Tris to pH 7.4 and centrifuged at 12,300g for 10 min.
NADH Dehydrogenase Activity (Complex I)
NADH dehydrogenase activity was measured spectrophotometrically according to Waseem and Parvez.40 The reaction mixture contained 0.6 mM 2,6-dichloroindophenol (DCIP), 2 mM glycyl glycine buffer, 0.6 mM nicotinamide adenine dinucleotide reduced (NADH), and mitochondrial preparations. The reaction was followed by recording the absorbance change at 600 nm against a blank. The enzyme activity was expressed as micromoles of NADH oxidized per minute per milligram protein using a molar extinction coefficient of 21,000 M–1 cm–1.
Assessment of Mitochondrial Bioenergetics
Mitochondrial respiration rates (state 3, ADP-stimulated; state 4, ADP-deleted; and respiratory control ratio) were observed by a Clark-type oxygen electrode system connected to a computer-operated oxygraph control unit (Hansatech Instruments, Norfolk, U.K.).41 About 1.0 mL of total reaction volume was used at 28 °C for all of the experiment group conditions. About 30–60 μg of freshly isolated mitochondria was added to the respiration buffer (120 mM KCl, 5 mM potassium phosphate, 3 mM HEPES, 1 mM ethylene glycol-O,-O′-bis-(2-aminoethyl) tetraacetic acid (EGTA), 1 mM MgCl2, and 0.2% BSA, pH 7.2) and allowed to equilibrate for a minute. FAD-linked substrate (10 mM succinate) was then added to the chamber and allowed to equilibrate for 1 min, followed by the addition of ADP (100 μM final concentration). The measurements involve addition of ADP for state 3 and removal of ADP for state 4. The respiratory control ratio (RCR) was calculated by the ratio of state 3 to state 4 respirations.42
Flow Cytometric Analysis of Mitochondrial Membrane Potential (ΔΨm)
Flow cytometric analysis was carried out using BD-LSR II, and histograms were generated using FACS-DIVA analysis software, as described previously.43 To exclude debris in the side scatter (SSC) and forward scatter (FSC) modes, 50,000 events per sample were collected using the “low” setting for sample flow rate. Mitochondria were selectively stained with tetramethylrhodamine ethyl ester (TMRE) (100 nM), (excitation at 488 nm and emission at 590 nm) accumulated in mitochondria in a membrane potential-dependent manner. Mitochondria (0.1 mg protein mL–1) were stained under dark conditions with TMRE in 1 mL of 3 mM HEPES buffer, pH 7.4, containing 70 mM sucrose, 230 mM mannitol, 1 μM ethylenediaminetetraacetic acid (EDTA) in the presence of 5 mM Pi and 5 mM succinate under dark conditions at 25 °C for 10 min. The mean fluorescent signal intensity was determined by flow cytometry to estimate the mitochondrial membrane potential.
Western Blotting
Western blot analyses of apoptotic (Bcl2, caspase 3 and TH) protein expression were performed in the brain tissue of control and treated flies according to the standard protocol.44 In brief, an equal number of heads of flies are homogenized in a buffer containing 10 mM Tris–HCl (pH 7.4), 150 mM NaCl, 2 mM EDTA, 1% NP-40, 0.01% sodium dodecyl sulfate (SDS) protease inhibitor cocktail, and phosphatase inhibitor. Equal amounts of proteins (20–40 μg well–1) were resolved in 12–15% SDS gel electrophoresis and transferred onto polyvinylidene fluoride (PVDF) membranes. Briefly, proteins were transferred at 50 mA for 2 h to PVDF membranes using a Mini Trans-Blot Cell apparatus (Bio-Rad, Hercules, California). The procedure for immunodetection includes the transfer and blocking of the membrane for 1 h at room temperature with phosphate-buffered saline-Tween (PBST) (150 mM NaCl, 10 mM KCl, 0.1 M NaH2PO4, 0.1 M KH2PO4, and 0.05% Tween-20, pH 7.4) containing 5% nonfat dried milk. After that, the membranes were incubated with the primary antibodies β-actin (antimouse, 1:1000, Santacruz), Bcl 2 (antimouse 1:1000), TH (tyrosine hydroxylase) (antimouse 1:1000, Santacruz), caspase 3 (antirabbit, 1:1000) overnight at 4 °C. After washing for 5–10 min periods with PBST, the membranes were incubated for 45 min at room temperature with horseradish peroxidase-conjugated secondary antibodies (1:10,000 in PBST). After washing for 5–10 min with PBST, the detection of bound antibodies was visualized by chemiluminescence using the ECL-plus reagent. An anti-β-actin antibody was used to normalize protein loading and transfer. Densitometric analysis was performed by ImageJ software (1.50 version, NIH).
Protein Determination
Protein concentrations were determined by the Bradford method using BSA as a standard.45
Statistical Analysis
All data were analyzed by taking the mean ± standard error of the mean (SEM). All data were analyzed by GraphPad Prism 5 software (GraphPad Software Inc., San Diego, California). All data were analyzed by one-way analysis of variance (ANOVA) followed by Tukey’s test to compare significant differences between different groups. Values of P < 0.05 were considered significant.
Acknowledgments
Prof. Suhel Parvez is appreciative of the support provided by the Government of India for the Promotion of University Research and Scientific Excellence (PURSE) program under the Department of Science & Technology (DST) (No. SR/PURSE Phase 2/39[C]), Fund for Improvement of S&T Infrastructure (FIST) (No. SR/FST/LS-I/2017/05[C]) grant, University Grant Commission (UGC)-XII Plan Innovative Grant (AS/INVRPROJ/XII/JH-11/16), and the Indian Council of Medical Research (ICMR no. 34/6/2020- TOXI/BMS) program. Dr. Zeeshan Rasheed is thankful to the Department of Biotechnology (DBT) for providing a Senior Research Fellowship (No. 09/591(DBT/JRF/14/AL/199).
Author Contributions
∥ M.Z.R. and R.K. contributed equally to the study.
Author Contributions
S.P., M.Z.R., and H.T. designed the study. M.Z.R., R.K., and F.T. conducted the experiments. M.Z.R., R.K., and M.M.A analyzed the data. M.Z.R. wrote the manuscript. All authors have read and approved the final manuscript.
The authors declare no competing financial interest.
References
- Zaltieri M.; Longhena F.; Pizzi M.; Missale C.; Spano P.; Bellucci A. Mitochondrial dysfunction and-synuclein synaptic pathology in Parkinson’s disease: who’s on first?. Parkinson’s Dis. 2015, 2015, 108029 10.1155/2015/108029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kong Y.; Liang X.; Liu L.; Zhang D.; Wan C.; Gan Z.; Yuan L. High throughput sequencing identifies Micro RNAs mediating α-synuclein toxicity by targeting neuroactive-ligand receptor interaction pathway in early stage of Drosophila parkinson’s disease model. PLoS One 2015, 10, e0137432 10.1371/journal.pone.0137432. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Julienne H.; Buhl E.; Leslie D. S.; Hodge J. J. Drosophila PINK1 and parkin loss-of-function mutants display a range of non-motor Parkinson’s disease phenotypes. Neurobiol. Dis. 2017, 104, 15–23. 10.1016/j.nbd.2017.04.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burbulla L. F.; Song P.; Mazzulli J. R.; Zampese E.; Wong Y. C.; Jeon S.; Santos D. P.; Blanz J.; Obermaier C. D.; Strojny C.; Savas J. N.; et al. Dopamine oxidation mediates mitochondrial and lysosomal dysfunction in Parkinson’s disease. Science 2017, 357, 1255–1261. 10.1126/science.aam9080. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fatima A.; Siddique Y. H. Role of tangeritin against cognitive impairments in transgenic Drosophila model of Parkinson’s disease. Neurosci. Lett. 2019, 705, 112–117. 10.1016/j.neulet.2019.04.047. [DOI] [PubMed] [Google Scholar]
- Pagonabarraga J.; Kulisevsky J.; Strafella A. P.; Krack P. Apathy in Parkinson’s disease: clinical features, neural substrates, diagnosis, and treatment. Lancet Neurol. 2015, 14, 518–531. 10.1016/S1474-4422(15)00019-8. [DOI] [PubMed] [Google Scholar]
- Sanders L. H.; Greenamyre J. T. Oxidative damage to macromolecules in human Parkinson disease and the rotenone model. Free Radical Biol. Med. 2013, 62, 111–120. 10.1016/j.freeradbiomed.2013.01.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Y.; Guo H.; Guo X.; Ge D.; Shi Y.; Lu X.; Lu J.; Chen J.; Ding F.; Zhang Q. Involvement of Akt/mTOR in the Neurotoxicity of Rotenone-Induced Parkinson’s Disease Models. Int. J. Environ. Res. Public Health 2019, 16, 3811 10.3390/ijerph16203811. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ryan B. J.; Hoek S.; Fon E. A.; Wade-Martins R. Mitochondrial dysfunction and mitophagy in Parkinson’s: from familial to sporadic disease. Trends Biochem. Sci. 2015, 40, 200–210. 10.1016/j.tibs.2015.02.003. [DOI] [PubMed] [Google Scholar]
- Klein C.; Westenberger A. Genetics of Parkinson’s disease. Cold Spring Harbor Perspect. Med. 2012, 2, a008888 10.1101/cshperspect.a008888. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang T.; Li C.; Han B.; Wang Z.; Meng X.; Zhang L.; He J.; Fu F. Neuroprotective effects of Danshensu on rotenone-induced Parkinson’s disease models in vitro and in vivo. BMC Complementary Med. Ther. 2020, 20, 20 10.1186/s12906-019-2738-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao R. Z.; Jiang S.; Zhang L.; Yu Z. B. Mitochondrial electron transport chain, ROS generation and uncoupling (Review). Int. J. Mol. Med. 2019, 44, 3–15. 10.3892/ijmm.2019.4188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maruyama D.; Hirata N.; Miyashita R.; Kawaguchi R.; Yamakage M. Substrate-dependent modulation of oxidative phosphorylation in isolated mitochondria following in vitro hypoxia and reoxygenation injury. Exp. Clin. Cardiol. 2013, 18, 158–160. [PMC free article] [PubMed] [Google Scholar]
- Tyurina Y. Y.; Polimova A. M.; Maciel E.; Tyurin V. A.; Kapralova V. I.; Winnica D. E.; Vikulina A. S.; Domingues M. R. M.; McCoy J.; Sanders L. H.; Bayır H.; et al. LC/MS analysis of cardiolipins in substantia nigra and plasma of rotenone-treated rats: implication for mitochondrial dysfunction in Parkinson’s disease. Free Radical Res. 2015, 49, 681–691. 10.3109/10715762.2015.1005085. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sarrafchi A.; Bahmani M.; Shirzad H.; Rafieian-Kopaei M. Oxidative stress and Parkinson’s disease: new hopes in treatment with herbal antioxidants. Curr. Pharm. Des. 2015, 22, 238–246. 10.2174/1381612822666151112151653. [DOI] [PubMed] [Google Scholar]
- Chia S. J.; Tan E. K.; Chao Y. X. Historical perspective: models of Parkinson’s Disease. Int. J. Mol. Sci. 2020, 21, 2464 10.3390/ijms21072464. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kamel F.; Goldman S. M.; Umbach D. M.; Chen H.; Richardson G.; Barber M. R.; Meng C.; Marras C.; Korell M.; Kasten M.; Hoppin J. A.; et al. Dietary fat intake, pesticide use, and Parkinson’s disease. Parkinsonism Relat. Disord. 2014, 20, 82–87. 10.1016/j.parkreldis.2013.09.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goldman S. M.; Kamel F.; Meng C.; Korell M.; Umbach D. M.; Hoppin J.; Ross G. W.; Marras C.; Kasten M.; Chade A.; Comyns K.. Rotenone and Parkinson’s disease (pd): Effect modification by membrane transporter variants. In Movement Disorders, WILEY-BLACKWELL, 2016; Vol. 31, pp S148–S149. [Google Scholar]
- Khatoon R.; Rasheed M. Z.; Rawat M.; Alam M. M.; Tabassum H.; Parvez S. Effect of melatonin on Aβ42 induced changes in the mitochondrial function related to Alzheimer’s disease in Drosophila melanogaster. Neurosci. Lett. 2019, 711, 134376 10.1016/j.neulet.2019.134376. [DOI] [PubMed] [Google Scholar]
- Pandi-Perumal S. R.; BaHammam A. S.; Brown G. M.; Spence D. W.; Bharti V. K.; Kaur C.; Hardeland R.; Cardinali D. P. Melatonin anti oxidative defense: therapeutical implications for aging and neurodegenerative processes. Neurotox. Res. 2013, 23, 267–300. 10.1007/s12640-012-9337-4. [DOI] [PubMed] [Google Scholar]
- Mendivil-Perez M.; Soto-Mercado V.; Guerra-Librero A.; et al. Melatonin enhances neural stem cell differentiation and engraftment by increasing mitochondrial function. J. Pineal Res. 2017, 63, e12415 10.1111/jpi.12415. [DOI] [PubMed] [Google Scholar]
- Jung Y. J.; Choi H.; Oh E. Melatonin attenuates MPP+-induced apoptosis via heat shock protein in a Parkinson’s disease model. Biochem. Biophys. Res. Commun. 2022, 621, 59–66. 10.1016/j.bbrc.2022.06.099. [DOI] [PubMed] [Google Scholar]
- Salman M.; Kaushik P.; Tabassum H.; Parvez S. Melatonin provides neuroprotection following traumatic brain injury-promoted mitochondrial perturbation in wistar rat. Cell Mol. Neurobiol. 2021, 41, 765–781. 10.1007/s10571-020-00884-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- García J. J.; López-Pingarrón L.; Almeida-Souza P.; Tres A.; Escudero P.; García-Gil F. A.; Tan D. X.; Reiter R. J.; Ramírez J. M.; Bernal-Pérez M. Protective effects of melatonin in reducing oxidative stress and in preserving the fluidity of biological membranes: a review. J. Pineal Res. 2014, 56, 225–237. 10.1111/jpi.12128. [DOI] [PubMed] [Google Scholar]
- Dies H.; Cheung B.; Tang J.; Rheinstädter M. C. The organization of melatonin in lipid membranes. Biochim. Biophys. Acta, Biomembr. 2015, 1848, 1032–1040. 10.1016/j.bbamem.2015.01.006. [DOI] [PubMed] [Google Scholar]
- Álvarez-Diduk R.; Galano A.; Tan D. X.; Reiter R. J. N-Acetylserotonin and 6-hydroxymelatonin against oxidative stress: Implications for the overall protection exerted by melatonin. J. Phys. Chem. B 2015, 119, 8535–8543. 10.1021/acs.jpcb.5b04920. [DOI] [PubMed] [Google Scholar]
- Singhal N. K.; Srivastava G.; Agrawal S.; Jain S. K.; Singh M. P. Melatonin as a neuroprotective agent in the rodent models of Parkinson’s disease: is it all set to irrefutable clinical translation?. Mol. Neurobiol. 2012, 45, 186–199. 10.1007/s12035-011-8225-x. [DOI] [PubMed] [Google Scholar]
- Zhao L.; An R.; Yang Y.; Yang X.; Liu H.; Yue L.; Li X.; Lin Y.; Reiter R. J.; Qu Y. Melatonin alleviates brain injury in mice subjected to cecal ligation and puncture via attenuating inflammation, apoptosis, and oxidative stress: the role of SIRT 1 signaling. J. Pineal Res. 2015, 59, 230–239. 10.1111/jpi.12254. [DOI] [PubMed] [Google Scholar]
- Andrabi S. S.; Parvez S.; Tabassum H. Melatonin and ischemic stroke: mechanistic roles and action. Adv. Pharmacol. Sci. 2015, 2015, 384750 10.1155/2015/384750. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang X. The antiapoptotic activity of melatonin in neurodegenerative diseases. CNS Neurosci. Ther. 2009, 15, 345–357. 10.1111/j.1755-5949.2009.00105.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Waseem M.; Sahu U.; Salman M.; Choudhury A.; Kar S.; Tabassum H.; Parvez S. Melatonin pre-treatment mitigates SHSY-5Y cells against oxaliplatin induced mitochondrial stress and apoptotic cell death. PLoS One 2017, 12, e0180953 10.1371/journal.pone.0180953. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Naz F.; Siddique Y. H. Drosophila melanogaster a versatile model of Parkinson’s disease. CNS Neurol. Disord.: Drug Targets 2021, 20, 487–530. 10.2174/1871527320666210208125912. [DOI] [PubMed] [Google Scholar]
- Bezard E.; Yue Z. Y.; Kirik D.; Spillantini M. G. Animal models of Parkinson’s disease: limits and relevance to neuro protection studies. Mov. Disord. 2013, 28, 61–70. 10.1002/mds.25108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mizuno H.; Fujikake N.; Wada K.; Nagai Y. α-synuclein transgenic Drosophila as a model of Parkinson’s disease and related synucleinopathies. Parkinson’s Dis. 2011, 2011, 212706 10.4061/2011/212706. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dehay B.; Bourdenx M.; Gorry P.; Przedborski S.; Vila M.; Hunot S.; Singleton A.; Olanow C. W.; Merchant K. M.; Bezard E.; Petsko G. A.; Meissner W. G. Targeting α-synuclein for treatment of Parkinson’s disease: mechanistic and therapeutic considerations. Lancet Neurol. 2015, 14, 855–866. 10.1016/S1474-4422(15)00006-X. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Siddique Y. H.; Naz F.; Jyoti S. Effect of capsaicin on the oxidative stress and dopamine content in the transgenic Drosophila model of Parkinson’s disease. Acta Biol. Hung. 2018, 69, 115–124. 10.1556/018.69.2018.2.1. [DOI] [PubMed] [Google Scholar]
- Siddique Y. H.; Naz F.; Jyoti S.; Ali F.; Fatima A.; Rahul; Khanam S. Protective effect of Geraniol on the transgenic Drosophila model of Parkinson’s disease. Environ. Toxicol. Pharmacol. 2016, 43, 225–231. 10.1016/j.etap.2016.03.018. [DOI] [PubMed] [Google Scholar]
- Shaposhnikov M.; Proshkina E.; Shilova L.; Zhavoronkov A.; Moskalev A. Lifespan and stress resistance in Drosophila with overexpressed DNA repair genes. Sci. Rep. 2015, 5, 15299 10.1038/srep15299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Salman M.; Tabassum H.; Parvez S. Nrf2/HO-1 mediates the neuroprotective effects of pramipexole by attenuating oxidative damage and mitochondrial perturbation after traumatic brain injury in rats. Dis. Model. Mech. 2020, 13, 045021 10.1242/dmm.045021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Waseem M.; Parvez S. Neuroprotective activities of curcumin and quercetin with potential relevance to mitochondrial dysfunction induced by oxaliplatin. Protoplasma 2016, 253, 417–430. 10.1007/s00709-015-0821-6. [DOI] [PubMed] [Google Scholar]
- Ali M.; Tabassum H.; Alam M. M.; Parvez S. N-acetyl-L-cysteine ameliorates mitochondrial dysfunction in ischemia/reperfusion injury via attenuating Drp-1 mediated mitochondrial autophagy. Life Sci. 2022, 293, 120338 10.1016/j.lfs.2022.120338. [DOI] [PubMed] [Google Scholar]
- Andrabi S. S.; Ali M.; Tabassum H.; Parveen S.; Parvez S. Pramipexole prevents ischemic cell death via mitochondrial pathways in ischemic stroke. Dis. Models Mech. 2019, 12, dmm033860 10.1242/dmm.033860. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Salman M.; Tabassum H.; Parvez S. Tannic acid provides neuroprotective effects against traumatic brain injury through the PGC-1α/Nrf2/HO-1 pathway. Mol. Neurobiol. 2020, 57, 2870–2885. 10.1007/s12035-020-01924-3. [DOI] [PubMed] [Google Scholar]
- Chaudhary S.; Sahu U.; Kar S.; Parvez S. Phytanic acid-induced neurotoxicological manifestations and apoptosis ameliorated by mitochondria-mediated actions of melatonin. Mol. Neurobiol. 2017, 54, 6960–6969. 10.1007/s12035-016-0209-4. [DOI] [PubMed] [Google Scholar]
- Bradford M. M. A rapid and sensitive method for the quantitation of microgram quantities of protein utilizing the principle of protein-dye binding. Anal. Biochem. 1976, 72, 248–254. 10.1016/0003-2697(76)90527-3. [DOI] [PubMed] [Google Scholar]
- Kawahata I.; Fukunaga K. Degradation of tyrosine hydroxylase by the ubiquitin-proteasome system in the pathogenesis of Parkinson’s disease and dopa-responsive dystonia. Int. J. Mol. Sci. 2020, 21, 3779 10.3390/ijms21113779. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cannon J. R.; Tapias V.; Na H. M.; Honick A. S.; Drolet R. E.; Greenamyre J. T. A highly reproducible rotenone model of Parkinson’s disease. Neurobiol. Dis. 2009, 34, 279–290. 10.1016/j.nbd.2009.01.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sonia Angeline M.; Chaterjee P.; Anand K.; Ambasta R. K.; Kumar P. Rotenone-induced parkinsonism elicits behavioral impairments and differential expression of parkin, heat shock proteins and caspases in the rat. Neuroscience 2012, 220, 291–301. 10.1016/j.neuroscience.2012.06.021. [DOI] [PubMed] [Google Scholar]
- Xiong N.; Long X.; Xiong J.; Jia M.; Chen C.; Huang J.; Ghoorah D.; Kong X.; Lin Z.; Wang T. Mitochondrial complex I inhibitor rotenone induced toxicity and it’s potential mechanism in Parkinson’s disease models. Crit. Rev. Toxicol. 2012, 42, 613–632. 10.3109/10408444.2012.680431. [DOI] [PubMed] [Google Scholar]
- Choi W. S.; Palmiter D. R.; Xia Z. Loss of mitochondrial complex I activity potentiates dopamine neuron death induced by microtubule dysfunction in a Parkinson’s disease model. J. Cell Biol. 2011, 192, 873–882. 10.1083/jcb.201009132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu Z.; Li T.; Yang D.; Smith W. W. Curcumin protects against rotenone-induced neurotoxicity in cell and drosophila models of Parkinson’s disease. Adv. Parkinson’s Dis. 2013, 02, 27827 10.4236/apd.2013.21004. [DOI] [Google Scholar]
- Cicchetti F.; Drouin-Ouellet J.; Gross R. E. Environmental toxins and Parkinson’s disease: what have we learned from pesticide induced animal models?. Trends Pharmacol. Sci. 2009, 30, 475–483. 10.1016/j.tips.2009.06.005. [DOI] [PubMed] [Google Scholar]
- Sudati J. H.; Vieira F. A.; Pavin S. S.; Dias G. R. M.; Seeger R. L.; Golombieski R.; Athaydeb M. L.; Soares F. A.; Rochaa J. B. T. R.; Barbosa N. V. Valerianaofficinalis attenuates the rotenone-induced toxicity in Drosophila melanogaster. NeuroToxicology 2013, 37, 118–126. 10.1016/j.neuro.2013.04.006. [DOI] [PubMed] [Google Scholar]
- Rao S. V.; Yenisettic S. C.; Rajini P. S. Evidence of neuro protective effects of saffron and crocin in a Drosophila model of parkinsonism. Neurotoxicology 2016, 52, 230–242. 10.1016/j.neuro.2015.12.010. [DOI] [PubMed] [Google Scholar]
- Doktór B.; Damulewicz M.; Pyza E. Overexpression of Mitochondrial Ligases Reverses Rotenone-Induced Effects in a Drosophila Model of Parkinson’s Disease. Front. Neurosci. 2019, 13, 94 10.3389/fnins.2019.00094. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aryal B.; Lee Y. Disease model organism for Parkinson disease: Drosophila melanogaster. BMB Rep. 2019, 52, 250–258. 10.5483/BMBRep.2019.52.4.204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- LeBleu V. S.; O’Connell J. T.; Gonzalez Herrera K. N.; Herrera K. N.; Wikman H.; Pantel K.; Haigis M. C.; de Carvalho F. M.; De Carvalho F. M.; Damascena A.; Domingos Chinen L. T.; Rocha R. M.; Asara J. M.; et al. PGC-1α mediates mitochondrial biogenesis and oxidative phosphorylation in cancer cells to promote metastasis. Nat. Cell Biol. 2014, 16, 992–1003. 10.1038/ncb3039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Waseem M.; Bhardwaj M.; Tabassum H.; Raisuddin S.; Parvez S. Cisplatin hepatotoxicity mediated by mitochondrial stress. Drug Chem. Toxicol. 2015, 38, 452–459. 10.3109/01480545.2014.992437. [DOI] [PubMed] [Google Scholar]
- Kumari K. K.; Setty O. H. Protective effect of Phyllanthus fraternus against mitochondrial dysfunction induced by co-administration of cisplatin and cyclophosphamide. J. Bioenerg. Biomembr. 2012, 44, 179–188. 10.1007/s10863-012-9423-6. [DOI] [PubMed] [Google Scholar]
- Waseem M.; Tabassum H.; Parvez S. Neuroprotective effects of melatonin as evidenced by abrogation of oxaliplatin induced behavioral alterations, mitochondrial dysfunction and neurotoxicity in rat brain. Mitochondrion 2016, 30, 168–176. 10.1016/j.mito.2016.08.001. [DOI] [PubMed] [Google Scholar]
- Kalogeris T.; Bao Y.; Korthuis R. J. Mitochondrial reactive oxygen species: a double-edged sword in ischemia/reperfusion vs pre conditioning. Redox Biol. 2014, 2, 702–714. 10.1016/j.redox.2014.05.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Babbitt S. E.; Sutherland M. C.; Francisco B. S.; Mendez D. L.; Kranz R. G. Mitochondrial cytochrome c biogenesis: no longer an enigma. Trends Biochem. Sci. 2015, 40, 446–455. 10.1016/j.tibs.2015.05.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun H.; Huang F. F.; Qu S. Melatonin: a potential intervention for hepatic. Lipids Health Dis. 2015, 14, 75 10.1186/s12944-015-0081-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chetsawang B.; Putthaprasart C.; Pujito P. P.; Govitrapong P. Melatonin protects against hydrogen peroxide induced cell death signaling in SH-SY5Y cultured cells: involvement of nuclear factor kappa B, Bax and Bcl-2. J. Pineal Res. 2006, 41, 116–123. 10.1111/j.1600-079X.2006.00335.x. [DOI] [PubMed] [Google Scholar]
- Maddirala Y.; Tobwala S.; Ercal N. N-acetylcysteine amide protects against manganese-induced toxicity in SHSY5Y cell line. Brain Res. 2015, 1608, 157–166. 10.1016/j.brainres.2015.02.006. [DOI] [PubMed] [Google Scholar]
- Nagatsu T.; Nakashima A.; Ichinose H.; Kobayashi K. Human tyrosine hydroxylase in Parkinson’s disease and in related disorders. J. Neural Transm. 2019, 126, 397–409. 10.1007/s00702-018-1903-3. [DOI] [PubMed] [Google Scholar]
- Rasheed M. Z.; Andrabi S. S.; Salman M.; Tabassum H.; Shaquiquzzaman M.; Parveen S.; Parvez S. Melatonin improves behavioral and biochemical outcomes in a rotenone-induced rat model of Parkinson’s disease. J. Environ. Pathol. Toxicol. Oncol. 2018, 37, 139–150. 10.1615/JEnvironPatholToxicolOncol.2018025666. [DOI] [PubMed] [Google Scholar]
- Mayo J. C.; Sainz R. M.; Tan D. X.; Antolín I.; Rodríguez C.; Reiter R. J. Melatonin and Parkinson’s disease. Endocrine 2005, 27, 169–178. 10.1385/ENDO:27:2:169. [DOI] [PubMed] [Google Scholar]