

Abstract
Sphingosine kinase 1 (SK1) converts the pro-death lipid sphingosine to the pro-survival sphingosine-1-phosphate (S1P) and is upregulated in several cancers. DNA damaging agents such as the chemotherapeutic doxorubicin (Dox) have been shown to degrade SK1 protein in cancer cells, a process dependent on wild type p53. As mutations in p53 are very common across several types of cancer, we evaluated the effects of Dox on SK1 in p53 mutant cancer cells. In the p53 mutant breast cancer cell line MDA-MB-231, we show that Dox treatment significantly increases SK1 protein and S1P. Using MDA-MB-231 cells with CRISPR-mediated knockout of SK1 or the selective SK1 inhibitor PF-543, we implicated SK1 in both Dox-induced migration and in a newly uncovered proangiogenic program induced by Dox. Mechanistically, inhibition of SK1 suppressed the induction of the cytokine BMP4 and of the EMT transcription factor Snail in response to Dox. Interestingly, induction of BMP4 by SK1 increased Snail levels following Dox treatment by stabilizing Snail protein. Furthermore, we found that SK1 was required for Dox-induced p38 MAP kinase phosphorylation and that active p38 MAPK in turn upregulated BMP4 and Snail, positioning p38 downstream of SK1 and upstream of BMP4/Snail. Modulating production of S1P by inhibition of de novo sphingolipid synthesis or knockdown of the S1P degrading enzyme lyase identified S1P as the sphingolipid activator of p38 in this model. This work establishes a novel angiogenic pathway in response to a commonly utilized chemotherapeutic and highlights the potential of SK1 as a secondary drug target for patients with p53 mutant cancer.
Keywords: Sphingolipids, angiogenesis, cytokines, MAP kinase, lipidomics, sphingosine phosphate, p53, cell signaling, EMT
Introduction:
Sphingolipids are a class of lipids with important functions in regulation of cellular structure and/or signaling1. Ceramide, sphingosine, and sphingosine-1-phosphate (S1P) are three well-established bioactive sphingolipids, with demonstrated roles as signaling molecules. Ceramide and sphingosine are typically associated with cell death, senescence, differentiation, and growth arrest2. S1P, on the other hand, is associated with increased proliferation, survival, immune response, angiogenesis, and motility in cancer3. The phosphorylation of sphingosine to S1P is catalyzed by sphingosine kinase (SK), which has two isoforms, SK1 and SK2. Due to their relationship to pro-survival and pro-proliferative pathways, S1P and SKs have been extensively studied and shown to be important in cancer development and survival3–5. In fact, SK1 is known to be highly expressed in breast cancer, colon cancer, prostate cancer, head and neck cancer, liver cancer, and glioblastoma6–9. SK1 has been associated with angiogenesis and invasion, both of which are key elements of metastasis10–12. SK1 expression has also been shown to be related to chemoresistance in several cancers, and inhibition of SK1 activity via either RNA interference or small molecule treatment restored chemosensitivity13, 14. High expression of SK1 has also been shown to interfere with immune checkpoint inhibition in melanoma15. Furthermore, elevated SK1, also correlates negatively with overall survival and progression-free survival of breast cancer, colon cancer, glioblastoma, and head and neck cancer6–9, highlighting its importance as a diagnostic marker and a target for therapy.
The pro-survival downstream effects of SK1 have been linked to the changes in its product S1P. S1P is known to signal through two different means. Extracellularly, S1P interacts with one of 5 G-protein coupled receptors, S1PR1–55. S1P interaction with these receptors has been shown to affect migration, invasion, proliferation, and angiogenesis16. However, there is also evidence that S1P can signal intracellularly, including inhibition of histone deacetylase activity to affect gene expression; however, this is typically in response to SK2 activity17. S1P is also shown to bind to and maintain the active state of telomerase, though again this is primarily driven by SK2-generated S1P18.. However, the intracellular response is much less completely understood than receptor driven S1P response.
Previous work in our lab showed that, in vitro, treatment of several different cancer cells with cytotoxic doses of DNA damaging agents like etoposide, actinomycin D, and doxorubicin (Dox) led to proteolysis of SK1, a process that requires wild type p5319. The specific mechanism of SK1 proteolysis remains unknown, however work in our lab has shown that cathepsin B and caspase 2 activities are required to proteolyze SK1 at a currently unknown site20–22.
Breast cancer is the most diagnosed cancer among women, and the fourth deadliest cancer overall23. In some cases, breast cancer can be treated via exploitation of an overexpressed target, such as the estrogen receptor or HER2 receptor. However, in cancers where there is no overexpressed receptor, broad acting chemotherapeutics are the first line option. Dox, one of the agents commonly used to treat Triple Negative Breast Cancer (TNBC), has severe cardiotoxic effects at high doses24. Therefore, Dox is often given at a lower dose in conjunction with one or more drugs to maximize anti-tumor effects while minimizing harmful side effects25. However, treatment with low doses also increases the risk of development of chemoresistance. This, along with TNBC’s higher metastatic potential and fatality rate, highlights the importance of identifying new drug targets.
The aforementioned work showing proteolysis of SK1 in response to cytotoxic doses of DNA damaging agents was conducted in cells containing wild type p53. However, p53 is mutated in 50% of cancers, and 80% of TNBC26 therefore, we investigated the effects of DNA damaging agents in mutant p53 breast cancer cells. In this study, we used MDA-MB-231 cells, a model of breast cancer with mutant p53 to explore the relationship between DNA damage and SK1 regulation. Dox was used as the primary DNA damaging agent due to its status as a frontline drug for TNBC.
This study shows that in a model of p53 mutant breast cancer, treatment with cytostatic doses of Dox lead to substantially elevated SK1 expression and activity. Upregulation of SK1 leads to a Dox-induced migratory and angiogenic response that can be reduced by inactivating SK1. The upregulation of SK1 initiates a novel signaling cascade where SK1 activity leads to the activation of p38 MAP kinase, which upregulates the cytokine BMP4. Active BMP4 stabilizes the EMT protein Snail, and Snail in turn is necessary for the angiogenic response. Therefore, our results implicate the Dox-induced SK1/p38/BMP4/Snail pathway in favoring cancer growth.
Materials and Methods:
Cell Culture and Drug Treatments:
MDA-MB-231 and EAhy926 cells were purchased from the American Type Culture Collection (ATCC Manassas, VA, USA) and cultured in DMEM containing 10% FBS (Invitrogen). Cell lines were authenticated by genotyping and regularly tested for mycoplasma contamination. Doxorubicin (Dox) was purchased from Sigma-Aldrich and diluted to a stock of 10 mM in DMSO. For the drug screen assay, methotrexate and 5-fluorouracil (5FU) were purchased from Sigma-Aldrich, while camptothecin was purchased from Abcam. The p38 inhibitor SB203580 was purchased from Cell Signaling Technologies (product #5633). Noggin was purchased from MilliporeSigma (product #GF173). Bortezomib was purchased from Selleckchem. Myriocin was purchased from MilliporeSigma.
Western Blot:
Cells were plated at a density of 500,000 cells per 6cm plate and treated with the respective concentration of drug or drugs for the respective time point or timepoints. Drug treated cells were washed with ice-cold PBS, lysed in cold RIPA buffer (1M Tris pH 7.4, 1M NaCl, 2% SDS, 10% Triton X100, 5% sodium deoxycholate, protease and phosphatase inhibitor), sonicated, and centrifuged at 14,500xg for 10 minutes to clear out membranes and organelles. Supernatant was transferred to a new tube, and protein concentration was measured via BCA assay. The protein was suspended in Laemelli loading buffer, boiled for 5 minutes, and approximately 20 μg of protein was loaded per well in a Life Technologies 4–20% Tris Glycine Midi-Gel. Protein was transferred to a nitrocellulose membrane (BioRad) for 1 hour, and membranes were blocked for 1 hour in 5% powdered milk dissolved in 0.1% PBS-T. Primary antibodies were added overnight at dilutions of 1:1000 unless otherwise noted. Secondary antibodies were added the next day at dilutions of 1:5000 unless otherwise noted, and blots were developed on radiology film using chemiluminescence. Anti-SK1, anti-phospho-p38, anti-total p38, anti-Snail, anti-Vimentin, anti-phospho-hsp27, anti-total hsp27, and anti-p53 were purchased from Cell Signaling Technology (Danvers, MA, USA). Anti-SK2 antibody was purchased from Abcam (Cambridge, MA, USA). Anti-actin antibody was purchased from Sigma Aldrich (St. Louis, MO, USA). Anti-Slug and anti-Fibronectin were a generous gift from the lab of Dr. Lori Chen.
RNA Interference:
MDA-MB-231 cells were plated at a density of 300,000 cells per 6cm plate. Cells were then transfected with 20 nM of siRNA (Snail and p53) or 40 nM siRNA (SGPL1), with Lipofectamine RNAiMax from Life Technologies (Grand Island, NY, USA) used as the transfection reagent. After 48 hours, media was changed and cells were treated with appropriate drugs. The siRNA and RNAiMax were mixed via the manufacturer’s protocols. siRNA was purchased from Life Technologies, AllStar control siRNA was purchased from Qiagen.
RNA Extraction and cDNA Synthesis:
RNA was extracted from cells using the RNA PureLink Extraction Kit according to the manufacturer’s protocol. RNA concentration and purity were measured via NanoDrop 2000. After isolation of total RNA, cDNA was synthesized using the SuperScript III kit from Invitrogen via manufacturer’s protocol. The cDNA was stored at −20C and used for quantitative real time PCR.
qRT- PCR:
Prior to the reaction, cDNA was diluted 1:10 in RNase free water. A master mix consisting of iTaq (Thermo-Fisher), RNase-free water, and TaqMan primer (Life Technologies) was prepared prior to the addition of cDNA. cDNA was added such that the ratio of all reagents was 10 iTaq:4 RNase free water:1 primer:5 cDNA. 20 μl of this mixture was loaded onto a plate and run on an Applied Biosystems 7500 Real Time PCR System (Applied Biosystems, Foster City, CA, USA). Actin was used as a housekeeping gene unless otherwise indicated. The following TaqMan primers were used: Actin (Hs01060665_g1), SK1 (Hs01116530_g1), SK2 (Hs01016543_g1), BMP4(Hs01041266_m1), VEGFA(Hs0090005_m1), IL6(Hs00174131_m1), SNAI1 (Hs00195591_m1).
Gene Transfection and Overexpression:
Tube Formation Assay:
Eahy926 immortalized endothelial cells were plated at 500,000 cells per plate for 24 hours and then serum starved overnight. To prepare the wells, 110 ul of approximately 10 mg/ml of Matrigel (Corning Biosciences, Corning, NY) were added to the wells of a 48-well Falcon plate and set to solidify at 37 C for one hour. The serum starved Eahy926 cells were trypsinized, centrifuged, counted, and seeded in the Matrigel-containing wells at 55,000 cells per well. Conditioned media (150 ul) from each treatment group was added to the wells, and the cells were incubated for 16 hours at 37C to form vessel-like structures. After 16 hours, the tubes were photographed on an EVOS microscope. Tube characteristics were measured and analyzed using the Angiogenesis Analyzer macro for ImageJ27.
MTT and LDH Assay:
Cells were seeded in a 6-well dish at 75,000 cells per dish for 24 hours, and then treated with drugs as indicated for 24 or 48 hours. For LDH assay, 50 ul of media was combined with 50 ul of LDH substrate from ThermoFisher and incubated at 37 degrees C for 30 minutes. Absorbance was then measured at wavelengths of 490 and 680 nm and final value determined by subtracting the former from the latter. For the MTT assay, media was extracted, and cells were treated with 1ml media and 1ml 12 mM MTT and left to incubate for 30 minutes at 37 degrees C. After 30 minutes, the MTT was aspirated and 2 ml of DMSO was added to each well, after which the plate was shaken at RT for 10 minutes. An aliquot of 200 ul was loaded in duplicate wells in a 96 well plate and measured at 570 nm.
Lipid Analysis:
To measure changes in lipid concentration, 500,000 cells were plated on a 10 cm dish and treated with 0.8 μM Dox at 70–80% confluence for 24 hours. After 24 hours, media were extracted in 4 ml of 70% isopropanol: ethyl acetate (15:85, vol/vol), while cells were extracted in 4 ml of 70% isopropanol:ethyl acetate (2:3, vol/vol) with the addition of appropriate internal standards. Lipidomics analysis was performed by the Lipidomics Shared Facility at Stony Brook Cancer Center as previously described28. Measurement of total lipid phosphates was used to normalize lipid concentrations in cells. Media lipid measurements were normalized per milliliters of media.
C17Sph Assay:
After plating and drug treatment, 1 μM of C17Sph (Avanti Polar Lipids) was added to each plate for 1 hour. Cells and media were extracted as described earlier, and samples were processed by the Lipidomics Shared Facility as previously described28. Calculation of total phosphates was conducted to normalize lipid concentrations. Media lipid measurements were standardized per milliliters of media.
Statistical Analysis:
For comparisons between two different conditions, a Student’s t-test was used to determine significant differences in the means of the sample groups. For comparisons between three or more conditions, a one-way ANOVA with Bonferroni post-hoc test was used to determine significant differences between all sample groups. For comparisons involving two or more variables, significance was determined via a two-way ANOVA with Bonferroni post-hoc tests. Graphpad Prism 5 was used to conduct all statistical tests. For all experiments, p < 0.05 was considered statistically significant.
Results:
Dox treatment of MDA-MB-231 leads to upregulation of SK1.
Dox treatment of MDA-MB-231 for 24 hours led to significant induction of both SK1 mRNA and protein levels starting at 0.2 μM and up to 1 μM, with a peak at 0.6–0.8 μM (Figure 1A). At these doses, there was minimal reduction in cell growth at 24 hours, while treatment of dox at all doses led to significantly less growth at 48 hours (Supplemental Figure 1). Time course analysis of 0.8 μM Dox showed that SK1 mRNA and protein were induced at 24 hours post-treatment (Figure 1B). Additionally, these effects were unique to the SK1 isoform, as Dox did not consistently increase either message or protein levels of SK2 at any dose or time point investigated (Figure 1C). Thus, in MDA-MB-231, Dox induced (rather than degraded) SK1.
Figure 1: Dox treatment of MDA-MB-231 cells leads to upregulation of SK1.
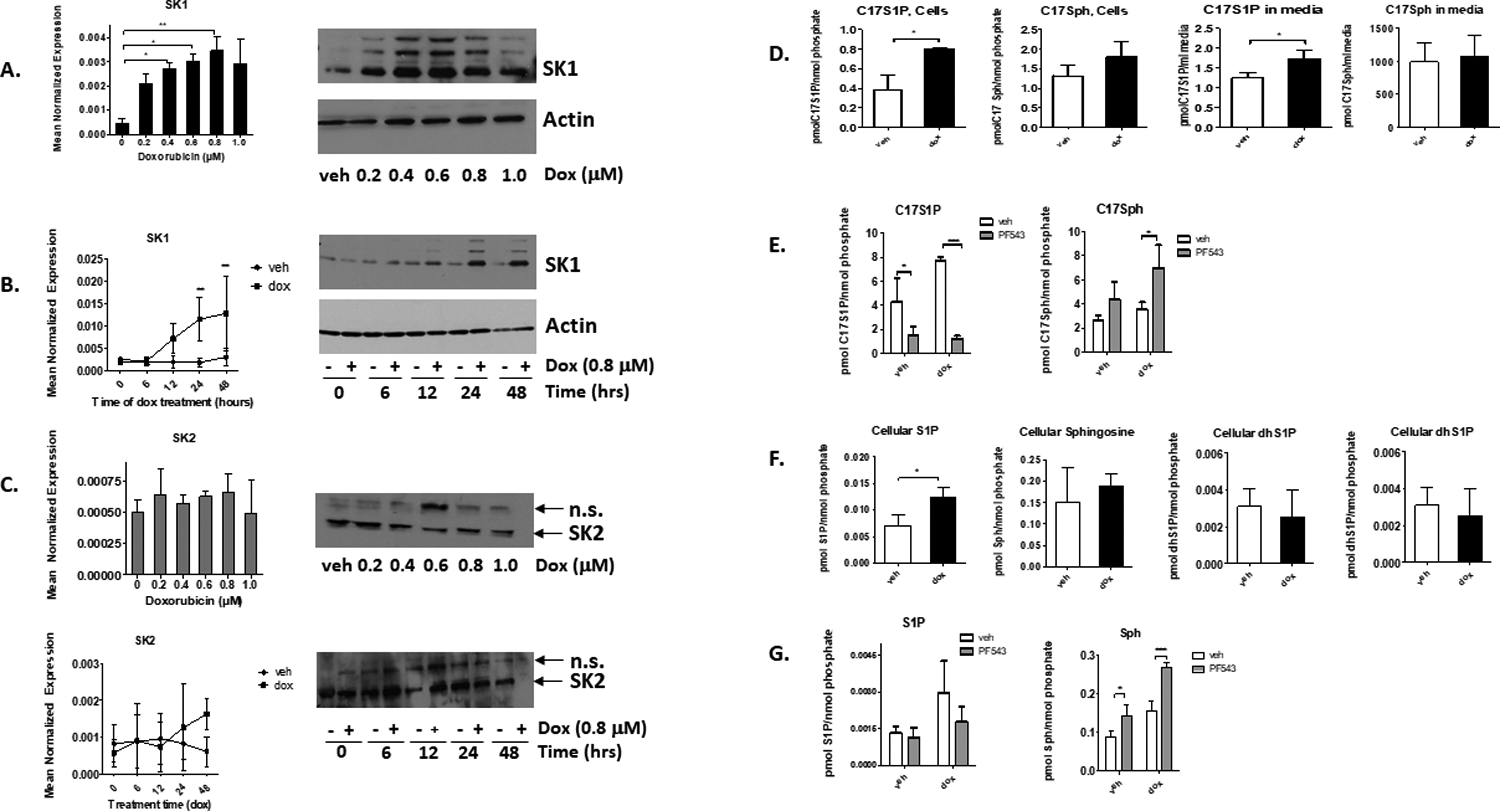
A. qRT-PCR and Western Blot of SK1 and actin in MDA-MB-231 cells treated with increasing doses of Dox for 24 hrs. (*= p<0.05, **<p<0.01, n=3). B. qRT-PCR and Western blot of SK1 and actin in cells treated with 0.8 μM of Dox or DMSO for the times shown (*=p<0.05, **= p<0.01, n=3). C. qRT-PCR and Western blot of SK2 in cells treated with increasing doses of Dox (24 hours, n=3) or increasing time of treatment (0.8 μM, n=4). Actin loading controls are same as in A and B. n.s.= non-specific band. D. Cells were treated with DMSO or 0.8 μM dox for 24 hours and then treated with 1 uM C17 sphingosine (C17Sph) for 1 hr. C17 sphingolipids were measured via mass spectrometry and normalized to nmoles of lipid phosphate in cells or per ml of media (n=3) (*=p<0.05). E. MDA-MB-231 cells were pre-treated with either DMSO or 100 nM PF543 for 2 hours prior to 24-hour treatment with 0.8 μM Dox. After 24 hours, cells were treated with 1 μM C17Sph for 1 hour. C17Sph and C17S1P were measured via tandem mass spectrometry and normalized to cellular lipid phosphates (n=3). F. Cells were treated with either DMSO or 0.8 μM Dox for 24 hours, and total lipids were extracted and measured via mass spectrometry. Lipid levels were normalized to nmol of lipid phosphate per treatment group (n=3, *=p<0.05). G. MDA-MB-231 cells were pre-treated with either DMSO or 100 nM PF543 for 2 hours prior to 24-hour treatment with DMSO or 0.8 μM Dox. Endogenous sphingolipids were measured via tandem mass spectrometry (n=3).
To assess the effects of Dox on SK1 activity, the conversion of C17Sph to C17S1P was measured following Dox treatment of MDA-MB-231 (0.8 μM for 24 hours). Consistent with the increase of SK1 levels, Dox led to increased conversion of C17Sph to C17S1P in cells, along with minor changes in C17Sph (Figure 1D). Analysis of lipids in the media showed similar increase in C17S1P in Dox treated cells (Figure 1D). No changes in total C17Cer were observed; however, there were slight but statistically significant decreases in C24Cer (Supplemental Figure 2). To confirm that this increased activity is due to SK1, cells were pre-treated with the selective SK1 inhibitor PF-543 (100nM, 2 hours) prior to treatment with Dox (0.8 μM, 24 hours) and C17S1P analysis. Results showed that PF-543 treatment led to significantly less C17S1P and significantly more C17Sph (Figure 1E). Thus, in agreement with expression data, SK1 and not SK2 is the isoform affected in this system.
To further assess the metabolic consequences of SK1 upregulation, endogenous lipids were analyzed after Dox treatment in MDA-MB-231 cells (0.8 μM, 24 hours). Endogenous S1P was elevated in cells treated with Dox compared to DMSO vehicle treated cells, with S1P approximately doubled by Dox treatment (Figure 1F), while levels of sphingoid bases in media were mostly unchanged (Supplemental Figure 2). Importantly, pre-treatment of cells with PF-543 (100 nM, 2 hrs) inhibited the Dox-induced S1P levels in cells and increased sphingosine levels in comparison to their vehicle controls (Figure 1G). Taken together, these results show that Dox treatment of MDA-MB-231 cells leads to upregulation of SK1 and increased S1P production.
Increased expression of SK1 may be due to either transcriptional upregulation or stabilization of mRNA. To determine if SK1 upregulation is due to a change in message stability, cells were treated with Dox (0.8 μM, 24 hours) in the presence or absence of actinomycin D (1uM) to block RNA transcription, and SK1 expression was measured at different time points by quantitative real-time PCR. As shown in Supplemental Figure 3, no difference in the rate of mRNA degradation was observed between vehicle and Dox treated cells (with a half-life of 5.92 hours and 5.23 hours, respectively), implying that Dox likely increases SK1 via enhanced transcription rather than altering message stability.
Furthermore, knock down of mutant p53 did not affect the induction of SK1 protein by Dox (Supplemental Figure 4A). These results suggest a mutant p53-independent mechanism for the upregulation of SK1. Overexpression of wild type p53 similarly did not induce degradation of SK1 in response to Dox in MDA-MB-231 cells, implying that regulation of SK1 in these cells is in fact p53 independent (Supplemental Figure 4B).
Since Dox functions primarily (but not exclusively) as a DNA damaging agent, it became important to determine whether the observed changes in SK1 were unique to treatment with Dox or if other drugs would also be able to alter SK1 levels in MDA-MB-231 cells. Cells were treated with the DNA damaging agents etoposide (5 μM), camptothecin (0.8 μM), or Dox (0.8 μM), or with one of the antimetabolites methotrexate (5 μM) or 5-fluorouracil (5 μM) for 24 hours (Supplemental Figure 5). Dox and camptothecin increased SK1 at the message and protein level, while etoposide had a slight effect at the message level. There was no change in SK1 levels among cells treated with antimetabolites (Supplemental Figure 5). We speculated that SK1 upregulation was related to DNA damage effects; however, treatment of cells with increasing doses of ultraviolet radiation did not increase SK1 levels. It is therefore possible that another property of these drugs leads to elevated SK1 expression.
SK1 Is Required for Dox-Induced Directional Migration
After observing substantial upregulation of SK1 in response to Dox, it became important to determine possible roles for SK1 in the biological responses to the drug. SK1 has been connected to survival and chemoresistance in several cancers29. Additionally, in in vivo and in vitro models of breast cancer, SK1 has been positively correlated with metastatic potential, migration, and invasion, with some demonstrating SK1 as being required for metastasis30–32. To evaluate the role of SK1 in regulating this and other biologies, we employed CRISPR-Cas9-mediated deletion of SK1 in MDA-MB-231, as previously reported by our lab33. The cells showed robust knockout of SK1, both with and without treatment of Dox (Supplemental Figure 6A). MTT assays showed significant reduction of viable cells after 48 hours of Dox treatment (Figure 2A), but that was independent from the presence or absence of SK1. Treatment with Dox up to 48h did not cause significant cell cytotoxicity as measured by LDH release or trypan blue exclusion assay (Figure 2B). This shows that 0.8 μM of Dox exerts a cytostatic effect in these cells and that SK1 does not provide a pro-proliferative cell advantage upon exposure to Dox in this model. Wound healing experiments were then conducted on CRISPR cells to determine any possible effect of Dox and SK1 on cell migration. Treatment with Dox enhanced wound healing (expressed as the percentage of the scratched area that closed after 24 or 48 hours compared to the width of the initial area for each treatment group) (Figure 2C). On the other hand, no significant difference in wound healing was found in the SK1KO group between Dox and vehicle control, suggesting a role for SK1 in enabling migration in response to Dox.
Figure 2: SK1 is required for Dox-induced directional migration.
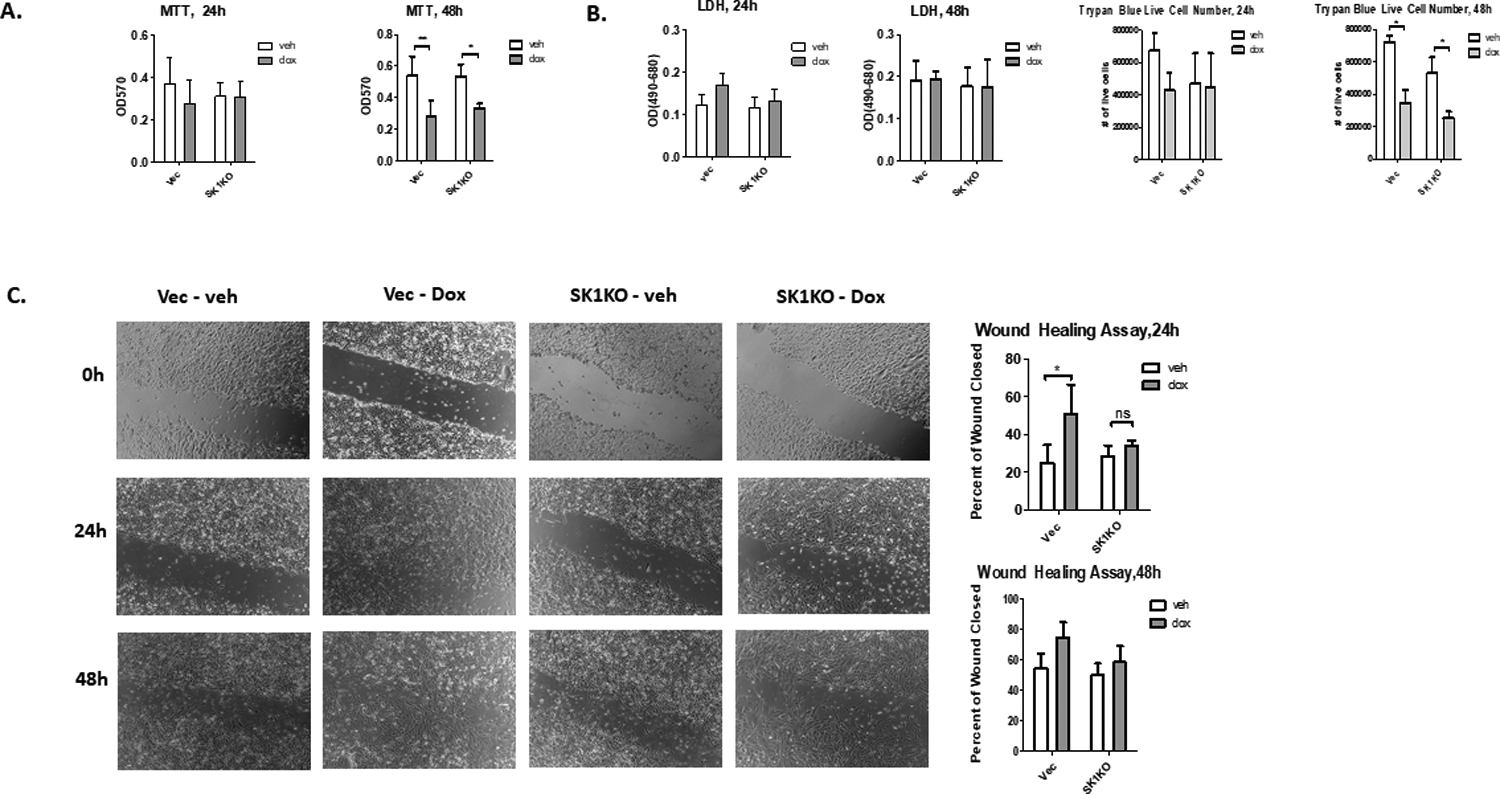
A. MDA-MB-231 CRISPR cells (SK1KO) were treated with 0.8 μM Dox for 24 or 48 hours and the number of viable cells was measured using 12 mM MTT (**=p<0.01, *=p<0.05, n=4). B. Cells were treated with DMSO or 0.8 μM Dox for 24 or 48 hours and cell death was determined by LDH release and trypan blue exclusion assay. For trypan blue assay, both live and dead cells were counted on a hemocytometer and the percentage of live cells over the total number of cells is reported (n=3). C. MDA-MB-231 cells were plated for 24 h, after which a scratch was introduced using a 10 ul pipette tip. Cells were then treated with either DMSO or 0.8 μM Dox, and pictures were taken of each well at 24 and 48 hours. Five pictures per well were taken, and each image is a representative image from an experiment (n=4, **=p<0.01).
SK1 is Required for BMP4 and Snail Upregulation after Dox Treatment
The increase in SK1-dependent migration in response to drug treatment led us to probe for possible mediators for this effect. Prior work in our lab had shown that MDA-MB-231 cells express and release higher levels of certain cytokines and chemokines in response to Dox treatment34. We chose to focus on three cytokines, all of which have been shown to have roles in breast cancer metastasis: VEGF, IL6, and BMP4. VEGF upregulation is known to contribute to angiogenesis and metastasis in breast and other cancers35, 36. IL-6 upregulation has also been associated with invasion and EMT37, 38. BMP4 has been associated with elevated breast cancer migration and metastasis to the bone39, 40. VEGF did not change after Dox treatment, and while IL-6 was modestly upregulated by Dox, its regulation was independent of knockout of SK1 (Figure 3A). BMP4 was upregulated by Dox in a dose dependent manner (Figure 3B), and it was reduced significantly by loss of SK1 (Figure 3C). To corroborate these results, MDA cells were treated with PF543, which leads to degradation of the SK1 protein (Supplemental Figure 6B). Pharmacological inhibition of SK1 with PF543 also reduced BMP4 message in response to Dox (Figure 3C). These results show that BMP4 upregulation in response to Dox is partly dependent on SK1.
Figure 3: Active SK1 is required for BMP4 and Snail upregulation.

A. Control or SK1KO CRISPR cells were treated with DMSO or 0.8 μM Dox for 24 hours, and VEGFA and IL6 mRNA was measured via qRT-PCR (n=6). B. MDA-MB-231 cells were treated with increasing doses of Dox for 24 hours, and BMP4 message was measured via qRT-PCR (n=3, *= p<0.05). C. Either vector and SK1KO CRISPR cells were treated with 0.8 μM dox for 24 hrs (left panel) or wild type cells were pre-treated with either vehicle or 100 nM of PF543 for 2 hours prior to treatment with 0.8 μM dox for 24 hrs (right panel); qRT-PCR was used to determine expression of BMP4 (n=4, *=p<0.05). D. Vector and SK1KO CRISPR cells were treated with DMSO or 0.8 μM Dox for 24 hours (left panel); alternatively, cells were pre-treated with either DMSO or 100 nM PF543 and treated with either vehicle or 0.8 μM dox for 24 hours (right panel); samples were probed for EMT factors by Western blot (n=3). E. Vector and SK1KO CRISPR cells were treated with DMSO or 0.8 μM Dox for 24 hours (left panel, n=6); alternatively, cells were pre-treated with either DMSO or 100 nM PF543 and treated with either vehicle or 0.8 μM dox for 24 hours (right panel, n=4); samples were probed for actin, SK1, and Snail by Western blot.
Several proteins are associated with enhanced migration in cancer cells. Perhaps chief among them are the EMT factors, which are strongly associated with migration, invasion, and metastasis30,31. SK1 has been shown to regulate the expression of several different EMT factors in cancer30,31. To determine whether any of these are upregulated in response to Dox, and whether they are modulated by SK1, we probed for several different EMT factors in both CRISPR cells and cells treated with PF543. Vimentin and Fibronectin did not show any change upon exposure to Dox, while Dox seemed to reduce Slug protein in a SK1-independent manner (Figure 3D). However, there was substantial induction of Snail in response to Dox, which was substantially reduced upon knockout or inhibition of SK1 (Figure 3E). These results indicate that SK1 is part of the process leading to upregulation of Snail in response to Dox.
SK1 and Snail Are Required for Angiogenesis in Response to Dox
In addition to migration, SK1, BMP4 and SNAIL have all been independently associated with angiogenesis in various cancers, such as melanoma, hepatocellular carcinoma, ovarian cancer, and lung cancer10–12, 41–45. Of note, SK1 has been associated with angiogenesis via release of S1P into the media. Hence, we investigated if upregulation of SK1 and Snail in response to Dox led to stimulation of angiogenesis. To evaluate this, we performed a tube formation assay on Eahy926 cell, a cell line derived from HUVEC cells. Conditioned media from MDA-MB-231 cells treated with vehicle or Dox and with either intact SK1, SK1KO, or siRNA-mediated SNAIL knockdown were used to treat the Eahy926 endothelial cells. Results were analyzed using the Angiogenesis Analyzer ImageJ plugin. This plugin measures twenty-one different parameters related to vessel size, branching, and complexity (Figure 4A). Treatment with conditioned media from Dox-treated cells increased the complexity of the vessels as measured by the number of meshes, total mesh area, and the number of junctions and master junctions (Figure 4B and 4C). This treatment also led to a substantial increase in the size of vessels as measured by total segment length and number of segments (Figure 4B and 4C). Conditioned media from cells without SK1 did not show increased vessel length or complexity in response to Dox (Figure 4B and 4C). Additionally, there was a statistically significant difference between the total mesh area of the Dox treated control and SK1KO groups, with other numbers of meshes and junctions trending downward in the SK1KO groups but narrowly missing statistical significance (Figure 4B and 4C). Treatment with conditioned media from cells with or without functional Snail (Figure 5) also showed that loss of Snail countered the effect of Dox on vessel complexity and length as measured by meshes, total mesh area, junctions, segments, and segment lengthIn EAhy926 cells treated with conditioned media from SnailKD cells treated with Dox, there was a statistically significant decline in the number of meshes, total mesh area, number of junctions, and number of segments compared to cells treated with conditioned media from All Star cells treated with Dox (Figure 5B). These data show that both SK1 and Snail are required for enhanced formation and altered architecture of the vessels following Dox treatment of MDA-MB-231 cells.
Figure 4: SK1 is required for angiogenesis in response to Dox.
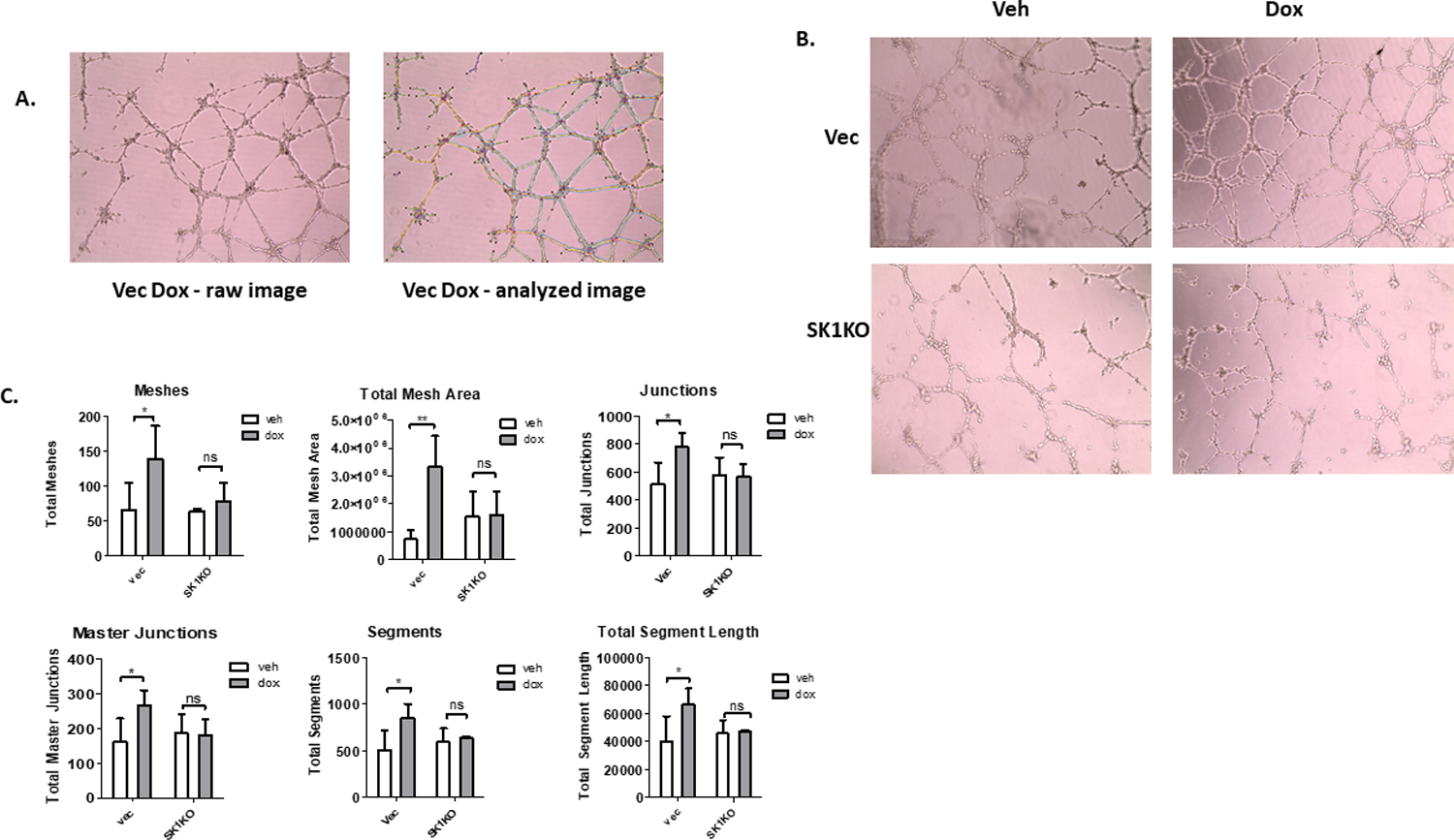
A. Left: image from tube formation assay prior to measurement with the Angiogenesis Analyzer macro from ImageJ. Right: same image after analysis using Angiogenesis Analyzer macro. Sky blue = meshes, orange = master segments, magenta= segments, dark blue= isolated elements, red dot=node, blue surrounding red dot= junction, red surrounding junctions = master junction, green= branches. B. Representative images of tube formation of EAhy926 cells treated with conditioned media from either control vector or SK1KO MDA-MB-231 cells treated with DMSO or 0.8 μM Dox for 16 hours. C. Quantification of vessel length and complexity in EAhy926 cells after treatment with conditioned media from either control vector or SK1KO cells (n=4, *=p<0.05, **= p<0.01).
Figure 5: Requirement of Snail for angiogenesis in response to Dox.

A. Representative images of tube formation of EAhy926 cells after treatment with conditioned media from either MDA-MB-231 cells treated with siRNA Scrambled control (AS) or SNAI1 si-RNA (Snail KD); after 48 hours, cells were treated with vehicle or 0.8 μM Dox for 16 hours. B. Quantification of vessel length and complexity in EAhy926 cells after treatment with conditioned media from either control or Snail KD cells (n=4, *=p<0.05, **= p<0.01, ***=p<0.001).
SK1 and BMP4 are Required for Stabilization of Snail Protein.
To further investigate the molecular mechanism that links SK1 and Snail to regulate angiogenesis in response to Dox, changes in SNAIL expression were determined. While SNAIL message was robustly upregulated after Dox treatment (Figure 6A and B), this response was not affected by SK1 knockout or inhibition (Figures 6A and 6B). The divergence between dependence of Snail protein on SK1 (Figure 3E) versus SK1-independence of SNAIL mRNA (Figure 6) suggested that SK1 may specifically regulate the stability of Snail protein. To probe this, cells were pre-treated with the proteasome inhibitor bortezomib (BTZ, 10nM, 2 hours) prior to 24-hour treatment with Dox. BTZ treatment overcame the inhibitory effect of SK1 knockout on Snail protein levels and led to further increase of Snail protein in response to Dox (Figure 6C). These results suggest that SK1 increases Snail protein upon Dox treatment by protecting it from proteolysis and without affecting its message.
Figure 6: SK1 and BMP4 are required for stabilization of Snail protein.

A. Vector and SK1KO CRISPR cells were treated with 0.8 μM Dox or DMSO for 24 hours, and message level of SNAIL was measured by qRT-PCR (n=4,***=p<0.001). B. SNAIL qRT-PCR of MDA-MB-231 cells pre-treated with DMSO or 100 nM PF543 for 2hrs, and then treated with DMSO of 0.8 μM Dox for 24 hours (n=4,***= p<0.001). C. CRISPR SK1KO cells were pre-treated with 10 nM bortezomib (BTZ) to inhibit proteosome activity for 2 hours prior to 24-hour treatment with either DMSO or 0.8 μM Dox. Western blot was used to determine protein expression (n=3). D. Snail Western blot and qRT-PCR from MDA-MB-231 cells pretreated with 200 ng/ml of BMP4 inhibitor Noggin (nog) for 2 hours prior to 24-hour treatment with 0.8 μM Dox or DMSO (n=3). E. qRT-PCR of SK1 in MDA-MB-231 cells treated with 100 ng/ml Noggin for 2 hours prior to 24-hour treatment with 0.8 μM Dox (n=4).
Next, we further delineated the causality among SK1, BMP4 and Snail since very limited literature reported functional links and only between Snail and BMP446. To this aim, we probed the relationship between Snail and BMP4 by pre-treating cells with the BMP4 inhibiting peptide Noggin (200 ng/ml, 2 hours) prior to treatment with Dox. After 24 hours Dox, Noggin significantly reduced Snail protein compared to vehicle leaving SNAIL message unaffected (Figure 6D). Furthermore, treatment with Noggin did not alter SK1 induction by Dox (Figure 6E), further establishing upregulation of BMP4 downstream of SK1 and upstream of Snail. All together these results indicate that both SK1 activity and BMP4 expression are required to increase Snail protein level, and they position SK1 upstream of BMP4.
SK1 is Required for Dox-Induced Activation of p38 MAPK
Having ordered BMP4 upregulation downstream of SK1 upregulation, we next investigated how BMP4 signaling may be leading to increase of Snail protein. After interaction with the BMP receptor BMPR1, BMP4 signals through two types of pathways. The “canonical” pathway proceeds via Smad1/5/8 phosphorylation, leading to increased transcription of targets47. In the non-canonical pathway, BMP4 signals through MAP kinases, especially p38 and JNK48. First, the role of SK1 in the canonical pathway was explored. The results showed that neither CRISPR knockout nor inhibition of SK1 with PF543 had any effect on Smad 1/5 phosphorylation (Figure 7A and 7B); in fact, it appeared that Smad 1/5 phosphorylation was reduced in response to Dox regardless of SK1 condition. However, Dox treatment did induce phosphorylation and activation of p38 MAPK, and this was substantially reduced by both SK1 knockout (Figure 7A) and by inhibition with PF543 (Figure 7B). These results argue against a prominent role for the canonical pathway, while they support a role for the p38 MAPK pathway, in the Dox/SK1 response. Next, the role of p38 in induction of Snail was evaluated. MDA-MB-231 cells were treated with 5 μM of the p38 inhibitor SB203580 in addition to Dox. Phosphorylation of Hsp-27, a direct target of p38, was decreased upon treatment with SB203580, showing that p38 activity was in fact inhibited (Figure 7C). Cells treated with SB203580 expressed less Snail protein than those treated with just Dox, with no change in SNAIL gene expression (Figure 7D). P38 therefore takes part in the induction of Snail protein levels in response to Dox.
Figure 7: SK1 is required for Dox-induced activation of p38 MAPK.
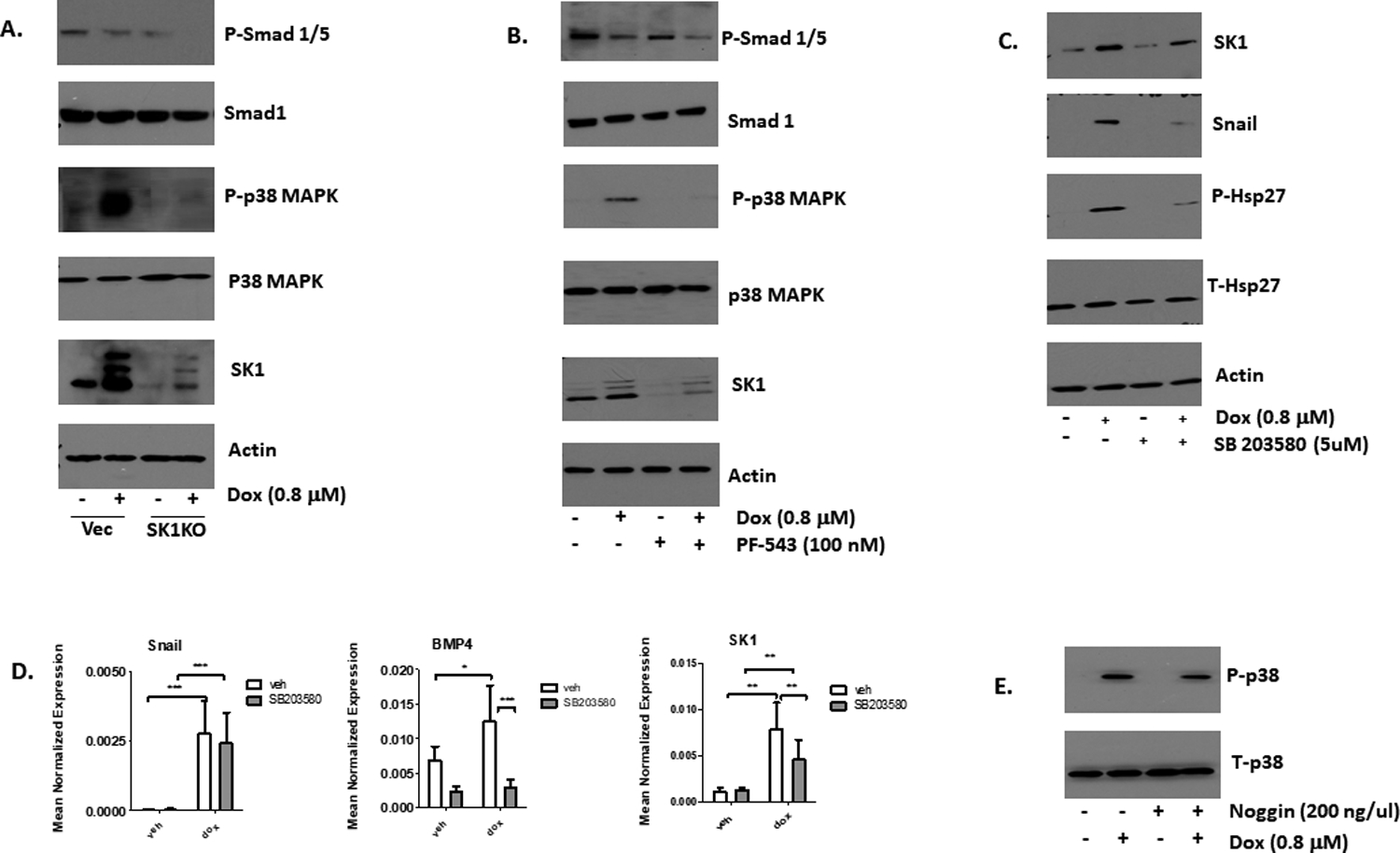
A. Western blot of p-Smad 1/5, Smad 1, p-p38, t-p38, SK1, and actin in vector and CRISPR SK1KO MDA-MB-231 cells after treatment of 24 hours with either DMSO or 0.8 μM Dox (n=4). B. Western blot of p-Smad 1/5, Smad 1, p-p38, t-p38, SK1, and actin in MDA-MB-231 cells pre-treated with either DMSO of 100 nM PF543 for two hours prior to 24-hour treatment with 0.8 μM Dox (n=3). C. MDA-MB-231 cells were treated with either DMSO or 5 μM of SB203580 for 2 hours prior to 24-hour treatment with 0.8 μM Dox. SK1, Snail, Actin, p-Hsp27, and t-Hsp27 were then probed via Western blot (n=4). D. qRT-PCR was performed to measure SNAIL (n=5), BMP4 (n=5) and SK1 (n=7) (*= p<0 .05, **= p<0.01, ***= p<0.001). E. Western blot of phospho- and total p38 from cells treated with noggin as described in Figure 3.
The causal relationship between p38 and BMP4 has not been conclusively established as reports in the literature have p38 either upstream or downstream of BMP449, 50. In our model, treatment with SB203580 reduced basal levels of BMP4 and prevented upregulation of the cytokine in response to Dox (Figure 7D), positioning p38 upstream of BMP4. Interestingly, inhibition of p38 also partly reduced SK1 expression (mRNA and protein) (Figure 7C and 7D). On the other hand, treatment of cells with Noggin had modest effects on p38 phosphorylation (Figure 7E), placing BMP4 downstream of p38. Together the results indicate that p38 activation is required for BMP4 upregulation and stabilization of Snail protein.
Finally, to conclusively order Snail within this signaling module, we knocked down SNAIL via siRNA and probed for changes in SK1, P-p38, and BMP4. Levels of SK1, P-p38, and BMP4 in response to Dox were unaffected by the loss of Snail (Supplemental Figure 7), validating Snail as the most downstream mediator of the Dox-induced SK1/p38/BPM4/Snail signaling module.
P38 is Activated by an Increase in S1P in Response to Dox.
Next, the involvement of a putative lipid mediator regulated by induction of SK1 (i.e. accumulation of S1P) in response to Dox was investigated. To this aim, sphingolipid biosynthesis was inhibited in MDA-MB-231 with 100 nM of the serine palmitoyltransferase inhibitor myriocin, which leads to a general reduction in sphingolipid levels (data not shown). A two-hour pre-treatment with 100 nM myriocin reduced Dox-induced P-p38 and Snail protein levels without altering its message (Figures 8A and 8B). Dox-induced BMP4 expression also declined significantly with myriocin pre-treatment (Figure 8B). These results suggest that activation of p38 and downstream responses following Dox treatment require a sphingolipid mediator.
Figure 8: P38 is activated by an increase in S1P in response to dox.
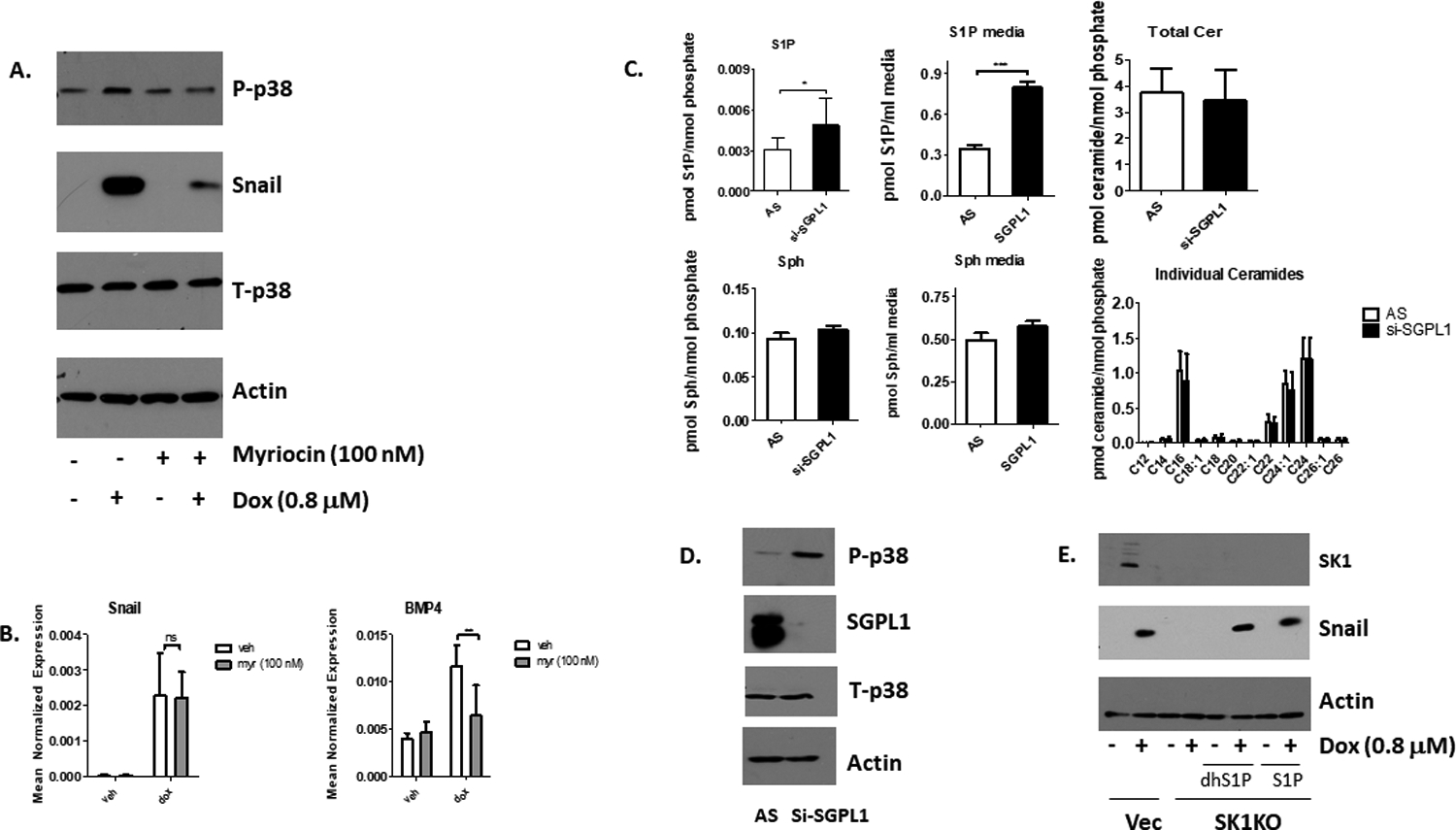
A. MDA-MB-231 cells were pre-treated with 100 nM myriocin for 2 hours, and then treated with 0.8 μM Dox for 24 hours (n=3). Western blots were conducted on phosphor- and total p38, Snail, and Actin. B. MDA-MB-231 cells were pretreated with 100 nM myriocin (Myr) for 2 hours, followed by a 24-hour treatment of 0.8 μM Dox (n=3). qRT-PCR was conducted on SNAIL and BMP4 and normalized to actin (n=4, **=p<0.01). C. Cells were transfected with 40 nM sphingosine-1-phosphate lyase (SGPL1) or scrambled control (AS) siRNA for 48 hours, and lipids in cells and media were measured (n=6, *=p<0.05, ***= p<0.001). D. MDA-MB-231 cells were transfected with 40 nM SGPL1 or scrambled control (AS) siRNA for 48 hours, and p38 phosphorylation, and levels of Sgpl1 were measured via Western Blot (n=3). E. SK1KO MDA-MB-231 cells were treated with 0.8 uM Dox for 12 hours and then treated with either 100 nM dhS1P or 500 nM S1P for 12 hours for a total of 24 hour Dox treatment (n=2). Levels of SK1, Actin, Snail, p-p38, and total p-38 were measured by Western blots.
To determine whether an endogenous increase in S1P is associated with p38 activation, we knocked down the S1P lyase (SGPL1) which cleaves S1P into hexadecenal and phosphoethanolamine51, clearing lipids from the sphingolipid pathway. Lipid measurements showed that knockdown of SGPL1 led to a buildup of S1P both in cells and in the media while other major sphingolipids remained unaltered (Figure 8C). Functionally, knockdown of SGPL1 via siRNA led to substantial phosphorylation of p38, indicating a role for the buildup of S1P in p38 activation (Figure 8D). These results support a role for endogenous S1P in regulating the p38 pathway.
Finally, we investigated whether adding exogenous S1P or dhS1P could rescue the effect of SK1 knockout on Snail. We added either 100 nM of dhS1P or 500 nM S1P to CRISPR SK1 knockout cells 12 hours after treatment with 0.8 μM of Dox, and then collected cells 12 hours later for a total treatment time of 24 hours with Dox and 12 hours with S1P or dhS1P. The results showed that dhS1P and S1P did not upregulate Snail in DMSO treated groups, but restored Snail protein in Dox treated SK1 knockout cells (Figure 8E). This shows that S1P is necessary but not sufficient for Snail protein stabilization.
Discussion
In this study, we show that treatment of a mutant p53 cell line with Dox induces the pro-metastatic sphingolipid enzyme SK1, which stimulates a tumor intrinsic promigratory response and an extrinsic pro-angiogenic response mediated by p38 MAPK activation, BMP4 production, and stabilization of the transcription factor Snail (Figure 9). Neither knockdown nor inhibition of SK1 affected cell survival in response to Dox. Notably, targeting SK1 was able to block these Dox-induced effects, while induction of SK1 was shown in response to other DNA damaging agents, suggesting it might be part of a broader response to DNA damaging chemotherapies. Collectively, these results define a novel pro-angiogenic pathway stimulated by chemotherapy treatment and suggest SK1 inhibitors might be useful for blocking this potential adverse effect of Dox without interfering with its anti-tumor efficacy.
Figure 9: SK1 is required for Snail stability and an angiogenic response to Dox.

Dox separately induces SNAIL message and SK1 message and protein. Snail protein induction requires SK1 and S1P activates p38 MAPK. Activated p38 MAPK is necessary for the upregulation of BMP4, which is required to stabilize Snail protein. Stabilized Snail then initiates an angiogenic response.
Importantly, this represents a novel means of Snail stabilization, as sphingolipid metabolism has not previously been connected to maintaining Snail protein levels. This also provides intriguing insight into the mechanisms of SK1 driven angiogenesis. While the relationship between SK1 and angiogenesis in tumors is known10,12, some of the exact mechanisms of this process are unknown. Angiogenesis driven by SK1 via stabilization of Snail in tumors has not been previously described. The lack of targeted therapies available for TNBC means that systemic DNA damaging agents such as Dox are still the first line of defense. However, clinical outcomes for this aggressive breast cancer subtype remain poor. Our results implicating SK1 as a driver of angiogenesis and migration in response to sublethal doses of Dox may provide insight as to why outcomes remain poor despite Dox treatment. This study looked at cytostatic rather than cytotoxic doses of Dox, as clinically Dox is often given at sublethal doses to minimize its severe side effects25. However, this study illustrates a potential danger of this treatment regimen, as it is upregulating a key enzyme governing cancer survival and progression. Treatment with lethal doses of Dox degrades SK119−22, but at the cost of severe clinical side effects. It is therefore important to find a balance between Dox treatment dose and risk of SK1 driven cancer progression, and our study provides a further rationale for pharmacological targeting of SK1.
Prior work from our lab has shown the degradation of SK1 in cells treated with DNA damaging agents including Dox, etoposide, actinomycin D, and ionizing radiation19–22. This was mediated by induction of wild-type p53. While this suggested that loss of SK1 is a key part of the chemotherapy response, a caveat to these findings is that a number of cancers harbor p53 mutations which lose normal p53 functions and some of which display gain of function. The results from the current study extend this prior work and clearly show that Dox induces SK1 mRNA and protein in a breast cancer cell line with mutant p53, a sharp contrast with the previous work with wild type p5319,22. This was functionally important as it led to increased SK1 activity, and higher levels of its downstream metabolite S1P. Importantly, the observed induction was isoform-specific, as SK2 expression and protein were unchanged. Furthermore, SK1 was observed to increase in response to several DNA damaging agents suggesting this is a consequence of the broader DNA damage response. Mechanistically, while our prior studies showed the degradation of SK1 protein required wild type p53, this study found that Dox-induced SK1 induction was not dependent on mutant p53 (Supplemental Figure 4A), even though its induction is transcriptional in nature. Thus, in the context of p53/SK1, the difference is likely due primarily to loss of wild type p53 function in MDA-MB-231 cells. Interestingly, treatment of BT-549 cells, another breast cancer cell line with mutant p53, with Dox also led to increased expression of SK1 (data not shown). The specific mechanisms by which DNA damage induces SK1 in the context of p53 are currently under investigation. Nonetheless, given that p53 mutations are common in aggressive breast cancer subtypes where chemotherapy is often the first line of defense, the current findings are of relevance to these cancers.
Biologically, Dox-induction of SK1 led to the initiation of a pro-angiogenic response. Intriguingly, Dox’s connection to angiogenesis is not well defined with some studies showing Dox to be pro-angiogenic, at least at low doses52, 53; others conclude that Dox is anti-angiogenic52–55. Here, our results support a pro-angiogenic effect of sublethal doses of Dox, and they implicate upregulation of SK1 as a novel mechanism through which Dox may exert this effect. Notably, while the connection of S1P and S1PRs to angiogenesis is well established [10–12], the specific roles of SK1 and SK2 to angiogenesis are less well defined. Here, we establish a role for SK1 in drug-induced angiogenesis by tumor cells and we connect it to the effects of SK1 on Snail.
Although traditionally associated with EMT, a connection between angiogenesis and Snail was recently suggested where knockout of Snail reduced expression of several factors key to tumor neoangiogenesis, such as ID1 and VEGFA45. Snail can do so via transcriptional regulation of mi-RNAs, as it was demonstrated in gastric cancer, or through direct transcriptional regulation of angiogenic chemokines44, 56. Snail knockout has also been associated with decreased angiogenic properties in mouse skin carcinoma cell lines57, confirming Snail’s link to angiogenesis across different cancers. This study marks the first time, however, that Snail has been connected to a pro-angiogenic response to drug treatment. Thus, this study defines important roles for sublethal Dox, SK1, and Snail in regulating tumor-cell induced angiogenesis.
Mechanistically, we identify robust regulation of Snail by SK1 at the level of protein stabilization as SNAIL mRNA was unaffected by the loss or inhibition of SK1. Prior studies showed overexpression of SK1 in lung cancer increasing expression of Snail and stimulating migration and invasion58. However, this work is the first to establish the importance of SK1 in Snail protein stabilization providing a more mechanistic insight. The post translational regulation of Snail is consistent with research showing that Snail has a short half-life in cells59, and regulation of Snail often involves the expression of certain deubiquitinases such as USP13, USP28, USP37, OTUD6A, and DUB3 to stabilize Snail protein60. Therefore, it is possible that SK1 may influence deubiquitinase activity and our results suggest that activation of p38 is involved. This is in line with the ability of p38 to phosphorylate and stabilize Snail in a way to prevent ubiquitination61.
The current results implicate S1P as the lipid mediator of SK1’s actions on Snail. The results clearly showed that treatment with S1P and dhS1P in SK1KO cells was able to restore Snail stabilization in response to Dox. However, S1P was not sufficient to induce Snail in the absence of Dox. The ability of low concentrations of S1P to restore Snail effects argue for receptor-mediated effects of S1P; however, the identity of the putative receptor(s) is currently not known. On the other hand, one of the few known S1P intracellular signaling pathways involves direct binding of S1P to the ubiquitinase TRAF2 and subsequent stimulation of its ubiquitinase activity62. Thus, it cannot be excluded that regulation of Snail stabilization may be a unique and newly discovered effect of intracellular S1P signaling.
The current results also revealed that Dox activates p38 MAPK and that this occurs in an SK1 dependent manner. These results are consistent with work observing several sphingolipids regulating p38 in response to stress in diverse systems63–65. In fact, a prior study in our own lab showed that SK1 itself is required for the activation of p38 in response to TNFα treatment63. While p38 is known to be phosphorylated in response to cellular stress, including DNA damage63, 66, 67, this is the first time that sphingolipids and sphingolipid enzymes have been implicated in the activation of p38 in response to DNA damage. Additionally, sphingolipid activation of p38 has largely been linked to cell death pathways68,69. Thus, the current work proposes a novel role for SK1 as a regulator of p38 activation in response to DNA damage, leading to a more aggressive cancer phenotype rather than apoptosis. Defining a role for SK1 in activating p38 in the two distinct conditions of DNA damage and TNF treatment raises the possibility that active SK1 and S1P signaling are central components of the p38 stress response. Our data showed that simple buildup of S1P in cells, absent of other known cellular stresses, was enough to activate p38, consistent with evidence that exogenous S1P can activate p38 in certain systems. Interestingly, inhibition of p38 also affected SK1 protein levels and message, implying there may be a feedback loop between SK1 activity and p38 activation68–70. This would put sphingolipid metabolism at a key nexus in cell stress response, and future work should explore more in depth the connection between SK1 and p38.
Given the results demonstrating the connection between SK1, Snail, and angiogenesis, it is possible that this pathway may be also involved in stimulating metastasis. Snail is best known for being upregulated during initiation of EMT, an important first step in metastasis. Since MDA-MB-231 is a mesenchymal like cell line with high metastatic potential, Snail has been previously shown to be basally expressed71 although, in our model its expression was further induced by Dox. Interestingly, some work has shown that SK1 expression correlates positively with EMT score in breast cancer cell lines72. Other work has also shown that SK1 is required for the metastasis of TNBC30–32. Further research is required to confirm whether elevated SK1 in response to Dox leads to a more general pro-metastatic response. Due to the expansive side effect profile of Dox, the drug is often given at sublethal doses in the clinic to reduce tumor growth while limiting the drug’s side effects. To make up for this reduced efficacy, Dox is typically given in combination with other chemotherapeutic agents. The effects of additional chemotherapeutics on the Dox responses were not evaluated in this study, but should be considered in future studies
In conclusion, the current results establish SK1 as a key component in a pro-angiogenic response to drug treatment in a model of mutant p53 TNBC and defines a novel signaling pathway downstream of S1P. Given the prevalence of p53 mutations in TNBC (>50%) and the association of angiogenesis with adverse effects, this suggests that SK1 may have utility as a potential drug target in conjunction with traditional chemotherapy in p53 mutant TNBC. Furthermore, as p53 mutations are common in several other cancers, exploring the relevance of the SK1-p38 MAPK-Snail pathway in such cancers could expand the scope of these findings, particularly as our studies suggest that it occurs in response to other chemotherapies and DNA damaging agents.
Supplementary Material
Acknowledgements.
The authors wish to acknowledge Izolda Mileva and the Biological Mass Spectrometry Shared Resource at Stony Brook Cancer Center for expert assistance with lipidomics analysis. We thank Dr. Daniel Canals for reviewing this manuscript. We thank Dr. Magali Trayssac for help with the C17Sph activity assay (Stony Brook University). We thank Jeffery Stith for his support with S1P and dhS1P signaling experiments. This research was supported by NIH grants R01 GM130878 and PO1 CA97132.
Abbreviations
- S1P
Sphingosine-1-Phosphate
- SK1
Sphingosine Kinase 1
- SK2
Sphingosine Kinase 2
- Dox
Doxorubicin
- S1PR
S1P receptor
- VEGF
vascular endothelial growth factor
- IL-6
interleukin-6
- BMP4
bone morphogenic protein 4
- EMT
epithelial-to-mesenchymal transition
- P38 MAPK
p38 mitogen associated protein kinase
- dhS1P
dihydrosphingosine-1-phosphate
- dhSph
dihydrosphingosine
Footnotes
Conflict of Interest Statement
The authors declare no conflicts of interest.
• This work is dedicated to the memory of Lina M. Obeid.
Data Availability Statement
All data for this manuscript are available in the figures, materials and methods section, or supplementary figures.
References:
- 1.Hannun YA and Obeid LM, Principles of bioactive lipid signalling: lessons from sphingolipids. Nature Reviews Molecular Cell Biology, 2008. 9(2): p. 139–150. [DOI] [PubMed] [Google Scholar]
- 2.Mullen TD and Obeid LM, Ceramide and apoptosis: exploring the enigmatic connections between sphingolipid metabolism and programmed cell death. Anticancer Agents Med Chem, 2012. 12(4): p. 340–63. [DOI] [PubMed] [Google Scholar]
- 3.Pyne NJ and Pyne S, Sphingosine 1-phosphate and cancer. Nat Rev Cancer, 2010. 10(7): p. 489–503. [DOI] [PubMed] [Google Scholar]
- 4.Heffernan-Stroud LA and Obeid LM, Sphingosine kinase 1 in cancer. Adv Cancer Res, 2013. 117: p. 201–35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Maceyka M, et al. , Sphingosine-1-phosphate signaling and its role in disease. Trends in cell biology, 2012. 22(1): p. 50–60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Kawamori T, et al. , Role for sphingosine kinase 1 in colon carcinogenesis. FASEB journal : official publication of the Federation of American Societies for Experimental Biology, 2009. 23(2): p. 405–414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Ruckhäberle E, et al. , Microarray analysis of altered sphingolipid metabolism reveals prognostic significance of sphingosine kinase 1 in breast cancer. Breast Cancer Research and Treatment, 2008. 112(1): p. 41–52. [DOI] [PubMed] [Google Scholar]
- 8.Shirai K, et al. , A Role of Sphingosine Kinase 1 in Head and Neck Carcinogenesis. 2011. 4(3): p. 454–462. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Van Brocklyn JR, et al. , Sphingosine Kinase-1 Expression Correlates With Poor Survival of Patients With Glioblastoma Multiforme: Roles of Sphingosine Kinase Isoforms in Growth of Glioblastoma Cell Lines. Journal of Neuropathology & Experimental Neurology, 2005. 64(8): p. 695–705. [DOI] [PubMed] [Google Scholar]
- 10.Anelli V, et al. , Role of sphingosine kinase-1 in paracrine/transcellular angiogenesis and lymphangiogenesis in vitro. 2010. 24(8): p. 2727–2738. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Nagahashi M, et al. , Sphingosine-1-Phosphate Produced by Sphingosine Kinase 1 Promotes Breast Cancer Progression by Stimulating Angiogenesis and Lymphangiogenesis. 2012. 72(3): p. 726–735. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Salama MF, et al. , A novel role of sphingosine kinase-1 in the invasion and angiogenesis of VHL mutant clear cell renal cell carcinoma. The FASEB Journal, 2015. 29(7): p. 2803–2813. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Baran Y, et al. , Alterations of ceramide/sphingosine 1-phosphate rheostat involved in the regulation of resistance to imatinib-induced apoptosis in K562 human chronic myeloid leukemia cells. J Biol Chem, 2007. 282(15): p. 10922–34. [DOI] [PubMed] [Google Scholar]
- 14.Li J, et al. , Downregulated miR-506 expression facilitates pancreatic cancer progression and chemoresistance via SPHK1/Akt/NF-κB signaling. Oncogene, 2016. 35(42): p. 5501–5514. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Imbert C, et al. , Resistance of melanoma to immune checkpoint inhibitors is overcome by targeting the sphingosine kinase-1. Nature Communications, 2020. 11(1): p. 437. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Ogretmen B, Sphingolipid metabolism in cancer signalling and therapy. Nature reviews. Cancer, 2018. 18(1): p. 33–50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Hait NC, et al. , Regulation of Histone Acetylation in the Nucleus by Sphingosine-1-Phosphate. 2009. 325(5945): p. 1254–1257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Panneer Selvam S, et al. ,. Binding of the sphingolipid S1P to hTERT stabilizes telomerase at the nuclear periphery by allosterically mimicking protein phosphorylation. Sci Signal. 2015. Jun 16;8(381):ra58. doi: 10.1126/scisignal.aaa4998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Taha TA, et al. , Down-regulation of sphingosine kinase-1 by DNA damage: dependence on proteases and p53. J Biol Chem, 2004. 279(19): p. 20546–54. [DOI] [PubMed] [Google Scholar]
- 20.Carroll BL, et al. , A role for caspase-2 in sphingosine kinase 1 proteolysis in response to doxorubicin in breast cancer cells – implications for the CHK1-suppressed pathway. 2018. 8(1): p. 27–40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Taha TA, et al. , Sphingosine kinase-1 is cleaved by cathepsin B in vitro: Identification of the initial cleavage sites for the protease. FEBS Letters, 2006. 580(26): p. 6047–6054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Taha TA, et al. , Tumor Necrosis Factor Induces the Loss of Sphingosine Kinase-1 by a Cathepsin B-dependent Mechanism. Journal of Biological Chemistry, 2005. 280(17): p. 17196–17202. [DOI] [PubMed] [Google Scholar]
- 23.Collaboration, G.B.o.D.C., The Global Burden of Cancer 2013. JAMA Oncology, 2015. 1(4): p. 505–527. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Chatterjee K, et al. , Doxorubicin cardiomyopathy. Cardiology, 2010. 115(2): p. 155–62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Wen S. h., et al. , Sulbactam-enhanced cytotoxicity of doxorubicin in breast cancer cells. Cancer Cell International, 2018. 18(1): p. 128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Duffy MJ, Synnott NC, and Crown J, Mutant p53 in breast cancer: potential as a therapeutic target and biomarker. Breast Cancer Research and Treatment, 2018. 170(2): p. 213–219. [DOI] [PubMed] [Google Scholar]
- 27.Carpentier G, et al. , Angiogenesis Analyzer for ImageJ — A comparative morphometric analysis of “Endothelial Tube Formation Assay” and “Fibrin Bead Assay”. Scientific Reports, 2020. 10(1): p. 11568. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Snider JM, Trayssac M, Clarke CJ, Schwartz N, Snider AJ, Obeid LM, Luberto C, Hannun YA. Multiple actions of doxorubicin on the sphingolipid network revealed by flux analysis. J Lipid Res. 2019. Apr;60(4):819–831. doi: 10.1194/jlr.M089714. Epub 2018 Dec 20. Erratum in: J Lipid Res. 2021;62:100009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Velazquez FN, et al. , Bioactive sphingolipids: Advancements and contributions from the laboratory of Dr. Lina M. Obeid. Cell Signal, 2021. 79: p. 109875. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Acharya S, et al. , Sphingosine Kinase 1 Signaling Promotes Metastasis of Triple-Negative Breast Cancer. Cancer Res, 2019. 79(16): p. 4211–4226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Sarkar S, et al. , Sphingosine kinase 1 is required for migration, proliferation and survival of MCF-7 human breast cancer cells. FEBS Lett, 2005. 579(24): p. 5313–7. [DOI] [PubMed] [Google Scholar]
- 32.Wang S, et al. , Triple Negative Breast Cancer Depends on Sphingosine Kinase 1 (SphK1)/Sphingosine-1-Phosphate (S1P)/Sphingosine 1-Phosphate Receptor 3 (S1PR3)/Notch Signaling for Metastasis. Medical science monitor : international medical journal of experimental and clinical research, 2018. 24: p. 1912–1923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Pulkoski-Gross MJ, et al. , An intrinsic lipid-binding interface controls sphingosine kinase 1 function. J Lipid Res, 2018. 59(3): p. 462–474. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Carroll BL, et al. , CHK1 regulates NF-κB signaling upon DNA damage in p53- deficient cells and associated tumor-derived microvesicles. Oncotarget, 2016. 7(14): p. 18159–70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Bachelder RE, Wendt MA, and Mercurio AM, Vascular endothelial growth factor promotes breast carcinoma invasion in an autocrine manner by regulating the chemokine receptor CXCR4. Cancer Res, 2002. 62(24): p. 7203–6. [PubMed] [Google Scholar]
- 36.Dvorak HF, et al. , Vascular permeability factor/vascular endothelial growth factor and the significance of microvascular hyperpermeability in angiogenesis. Curr Top Microbiol Immunol, 1999. 237: p. 97–132. [DOI] [PubMed] [Google Scholar]
- 37.Sullivan NJ, et al. , Interleukin-6 induces an epithelial–mesenchymal transition phenotype in human breast cancer cells. Oncogene, 2009. 28(33): p. 2940–2947. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Zhang GJ and Adachi I, Serum interleukin-6 levels correlate to tumor progression and prognosis in metastatic breast carcinoma. Anticancer research, 1999. 19(2B): p. 1427–1432. [PubMed] [Google Scholar]
- 39.Ampuja M, et al. , The impact of bone morphogenetic protein 4 (BMP4) on breast cancer metastasis in a mouse xenograft model. Cancer Letters, 2016. 375(2): p. 238–244. [DOI] [PubMed] [Google Scholar]
- 40.Ampuja M, et al. , BMP4 inhibits the proliferation of breast cancer cells and induces an MMP-dependent migratory phenotype in MDA-MB-231 cells in 3D environment. BMC Cancer, 2013. 13(1): p. 429. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Rothhammer T, et al. , Functional implication of BMP4 expression on angiogenesis in malignant melanoma. Oncogene, 2007. 26(28): p. 4158–4170. [DOI] [PubMed] [Google Scholar]
- 42.Tang Y, et al. , BMP4 mediates the interplay between adipogenesis and angiogenesis during expansion of subcutaneous white adipose tissue. Journal of Molecular Cell Biology, 2016. 8(4): p. 302–312. [DOI] [PubMed] [Google Scholar]
- 43.Li X, et al. , Function of BMP4 in the Formation of Vasculogenic Mimicry in Hepatocellular Carcinoma. J Cancer, 2020. 11(9): p. 2560–2571. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Taki M, et al. , Snail promotes ovarian cancer progression by recruiting myeloid-derived suppressor cells via CXCR2 ligand upregulation. Nature Communications, 2018. 9(1): p. 1685. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Zhang Y-K, et al. , Novel role of Snail 1 in promoting tumor neoangiogenesis. Bioscience Reports, 2019. 39(5). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Caja L, et al. , Snail regulates BMP and TGFβ pathways to control the differentiation status of glioma-initiating cells. Oncogene, 2018. 37(19): p. 2515–2531. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Su D, et al. , BMP4-Smad signaling pathway mediates adriamycin-induced premature senescence in lung cancer cells. J Biol Chem, 2009. 284(18): p. 12153–64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Sieber C, et al. , Recent advances in BMP receptor signaling. Cytokine Growth Factor Rev, 2009. 20(5–6): p. 343–55. [DOI] [PubMed] [Google Scholar]
- 49.Tan T-W, et al. , CCN3 increases BMP-4 expression and bone mineralization in osteoblasts. 2012. 227(6): p. 2531–2541. [DOI] [PubMed] [Google Scholar]
- 50.Wang L, et al. , Involvement of p38MAPK/NF-κB Signaling Pathways in Osteoblasts Differentiation in Response to Mechanical Stretch. Annals of Biomedical Engineering, 2012. 40(9): p. 1884–1894. [DOI] [PubMed] [Google Scholar]
- 51.Shin KO, et al. , A Bioassay Using a Pentadecanal Derivative to Measure S1P Lyase Activity. Int J Mol Sci, 2021. 22(3). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Cao Y, et al. , Tumor cells upregulate normoxic HIF-1α in response to doxorubicin. Cancer research, 2013. 73(20): p. 6230–6242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Michaelis M, et al. , Chemotherapy-associated angiogenesis in neuroblastoma tumors. Am J Pathol, 2012. 180(4): p. 1370–7. [DOI] [PubMed] [Google Scholar]
- 54.Park M, et al. , REDD1 is a determinant of low-dose metronomic doxorubicin-elicited endothelial cell dysfunction through downregulation of VEGFR-2/3 expression. Experimental & Molecular Medicine, 2021. 53(10): p. 1612–1622. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Quesada AJ, et al. , In vivo upregulation of CD95 and CD95L causes synergistic inhibition of angiogenesis by TSP1 peptide and metronomic doxorubicin treatment. Cell Death & Differentiation, 2005. 12(6): p. 649–658. [DOI] [PubMed] [Google Scholar]
- 56.Faget J, et al. , Neutrophils and Snail Orchestrate the Establishment of a Pro-tumor Microenvironment in Lung Cancer. Cell Reports, 2017. 21(11): p. 3190–3204. [DOI] [PubMed] [Google Scholar]
- 57.Olmeda D, et al. , Snai1 and Snai2 collaborate on tumor growth and metastasis properties of mouse skin carcinoma cell lines. Oncogene, 2008. 27(34): p. 4690–4701. [DOI] [PubMed] [Google Scholar]
- 58.Zhu L, et al. , Sphingosine kinase 1 enhances the invasion and migration of non-small cell lung cancer cells via the AKT pathway. Oncol Rep, 2015. 33(3): p. 1257–63. [DOI] [PubMed] [Google Scholar]
- 59.Zhou BP, et al. , Dual regulation of Snail by GSK-3β-mediated phosphorylation in control of epithelial–mesenchymal transition. Nature Cell Biology, 2004. 6(10): p. 931–940. [DOI] [PubMed] [Google Scholar]
- 60.Qian W, et al. , Deubiquitinase USP29 promotes gastric cancer cell migration by cooperating with phosphatase SCP1 to stabilize Snail protein. Oncogene, 2020. 39(44): p. 6802–6815. [DOI] [PubMed] [Google Scholar]
- 61.Ryu K-J, et al. , p38 Stabilizes Snail by Suppressing DYRK2-Mediated Phosphorylation That Is Required for GSK3β-βTrCP–Induced Snail Degradation. 2019. 79(16): p. 4135–4148. [DOI] [PubMed] [Google Scholar]
- 62.Alvarez SE, et al. , Sphingosine-1-phosphate is a missing cofactor for the E3 ubiquitin ligase TRAF2. Nature, 2010. 465(7301): p. 1084–1088. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Adada MM, et al. , Sphingosine kinase 1 regulates tumor necrosis factor-mediated RANTES induction through p38 mitogen-activated protein kinase but independently of nuclear factor κB activation. J Biol Chem, 2013. 288(38): p. 27667–27679. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Ahn EH and Schroeder JJ, Sphinganine Causes Early Activation of JNK and p38 MAPK and Inhibition of AKT Activation in HT-29 Human Colon Cancer Cells. Anticancer Research, 2006. 26(1A): p. 121. [PubMed] [Google Scholar]
- 65.Chen C-L, et al. , Ceramide induces p38 MAPK and JNK activation through a mechanism involving a thioredoxin-interacting protein-mediated pathway. Blood, 2008. 111(8): p. 4365–4374. [DOI] [PubMed] [Google Scholar]
- 66.Borodkina A, et al. , Interaction between ROS dependent DNA damage, mitochondria and p38 MAPK underlies senescence of human adult stem cells. Aging, 2014. 6(6): p. 481–495. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Wood CD, et al. , Nuclear localization of p38 MAPK in response to DNA damage. Int J Biol Sci, 2009. 5(5): p. 428–37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Fegley AJ, et al. , Sphingosine-1-phosphate stimulates smooth muscle cell migration through galpha(i)- and pi3-kinase-dependent p38(MAPK) activation. J Surg Res, 2003. 113(1): p. 32–41. [DOI] [PubMed] [Google Scholar]
- 69.Kozawa O, et al. , Sphingosine 1-Phosphate Induces Heat Shock Protein 27 via p38 Mitogen-Activated Protein Kinase Activation in Osteoblasts. 1999. 14(10): p. 1761–1767. [DOI] [PubMed] [Google Scholar]
- 70.Lin CC, et al. , Sphingosine-1-phosphate mediates ICAM-1-dependent monocyte adhesion through p38 MAPK and p42/p44 MAPK-dependent Akt activation. PLoS One, 2015. 10(3): p. e0118473. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Smith BN, et al. , Snail Promotes Epithelial Mesenchymal Transition in Breast Cancer Cells in Part via Activation of Nuclear ERK2. PLOS ONE, 2014. 9(8): p. e104987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Wang W, et al. , Sphingosine 1–phosphate signaling induces SNAI2 expression to promote cell invasion in breast cancer cells. 2019. 33(6): p. 7180–7191. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
All data for this manuscript are available in the figures, materials and methods section, or supplementary figures.