

Abstract
Objective:
To review the epidemiology, presentation, diagnosis, and management of head and neck paragangliomas.
Methods:
A literature review of english language papers with focus on most current literature.
Results:
Paragangliomas (PGLs) are a group of neuroendocrine tumors that arise in the parasympathetic or sympathetic ganglia. Head and neck PGLs (HNPGLs) comprise 65% to 70% of all PGLs and account for 0.6% of all head and neck cancers. The majority of HNPGLs are benign, and 6% to 19% of all HNPGLs develop metastasis outside the tumor site and significantly compromise survival. PGLs can have a familial etiology with germline sequence variations in different susceptibility genes, with the gene encoding succinate dehydrogenase being the most common sequence variation, or they can arise from somatic sequence variations or fusion genes. Workup includes biochemical testing to rule out secretory components, although it is rare in HNPGLs. In addition, imaging modalities, such as computed tomography and magnetic resonance imaging, help in monitoring in surgical planning. Functional imaging with DOTATATE-positron emission tomography, 18F-fluorodeoxyglucose, or 18F-fluorohydroxyphenylalanine may be necessary to rule out sites of metastases. The management of HNPGLs is complex depending on pathology, location, and aggressiveness of the tumor. Treatment ranges from observation to resection to systemic treatment. Similarly, the prognosis ranges from a normal life expectancy to a 5-year survival of 11.8% in patients with distant metastasis.
Conclusion:
Our review is a comprehensive summary of the incidence, mortality, pathogenesis, presentation, workup and management of HNPGLs.
Keywords: paraganglioma, head and neck neoplasms, neuroendocrine tumors
Incidence and Mortality
Paragangliomas (PGLs) are a group of neuroendocrine tumors that arise in the sympathetic or parasympathetic ganglia. Head and neck PGLs (HNPGLs) comprise of 65% to 70% of all PGLs and account for 0.6% of all head and neck cancers.1–3 The estimated overall incidence of HNPGLs is 0.3 to 1 per 100 000. PGLs of the cervical sympathetic chain have been reported, but most HNPGLs arise from the jugular bulb, glossopharyngeal or vagus nerves, or carotid body.2,4 Carotid body PGLs constitute up to 60% of all PGLs of the head and neck.1 The age of onset is broad; however, hereditary HNPGL presents a decade earlier than sporadic cases.5
The only known acquired risk factor for PGL is chronic hypoxia, with an association of an elevated risk of the disease in patients living at a high altitude.6 This observation may be explained by the linkage between sequence variations in the pseudohypoxia-related pathways and PGLs, has been described in the following.
Although the majority of HNPGLs are benign, 6% to 19% of all HNPGLs develop metastases outside the primary tumor site.7 Malignant HNPGLs also have central necrosis, hypervascularity, or increased mitotic activity but are not completely reliable for discriminating between benign and malignant cases. Although most metastases are limited to the regional lymph nodes, distant metastases can occur, most commonly to the lungs or skeleton occurring in 6% to 13% of cases.8–11 Furthermore, metastasis is less likely to occur in patients with jugulotympanic HNPGLs, and there is no association with SHDx sequence variations.12 Benign HNPGLs have excellent prognosis with local treatment, as evident by Gilbo et al13 who showed a 5-year survival rate of 91% and 5-year distant metastasis–free survival of 91%. On the other hand, a review of 59 cases from the National Cancer Database showed 5-year survival rates of 76.8% in patients with metastasis to regional lymph nodes and 11.8% in patients with distant metastasis.8
Pathogenesis
PGLs can be the result of germline sequence variations in 1 of >15 different PGL susceptibility genes, somatic sequence variations (HRAS, NF1, EPAS1, RET, and CSDE1), or fusion genes (MAML3).2,14,15 In 2000, Baysal et al16 found the first succinate dehydrogenase subunit D (SDHD) gene sequence variation in PGL1 syndrome. These hereditary HNPGLs are mostly associated with germline sequence variations in one of the SDH subunit genes (A through D, collectively referred to as SDHx) or the SDH assembly factor, SDHAF2.15 Sequence variations in other genes (eg, Von Hippel-Lindau/VHL, MYC-associated factor X/MAX, and transmembrane protein 127/TMEM127) are more rarely involved in the pathogenesis of these tumors.2,14,15,17,18 PGL sequence variations and their resultant transcription profiles segregate into 2 main clusters, where cluster 1 comprises genes that are associated with the hypoxic response (sequence variations of VHL, components of the SDH complex, hypoxia-inducible factor [HIF]-2α [HIF2A, also known as EPSA1], prolyl hydroxylase domain 2 gene [PHD2], and fumarate hydroxylase [FH]) and cluster 2 contains tumors that activate kinase signaling and protein translation (RET, NF1, TMEM127, HRAS, BRAF, FGRG, NGRF, and MAX).14,18–22 Other subtypes have also been identified and include the following: (1) wnt-altered subtype (MAML3 and CSDE1) and (2) cortical admixture subtype (CYP11B1, CYP21A2, and STAR).14
Among all known genetic sequence variations in HNPGLs, SDHD-linked patients have a 75% lifetime risk of developing HNPGLs.23,24 Germline SDHAF2 sequence variations are often noted in young patients with multiple HNPGLs.25 SDHD and SDHAF2 genes show maternal imprinting, which means that the allele of the gene inherited from the mother is transcriptionally silent and the paternally inherited allele is active.26 The major predictors of hereditary HNPGLs include a family history of PGL, especially those related to SDHD, a previous history of adrenal or extra-adrenal PGL, and multifocality or a characteristic syndromic presentation that may include renal cell carcinoma, gastrointestinal stromal tumor (GIST), and/or pituitary adenomas, which are also related to SDHx sequence variations.2 In a retrospective study by Baysal et al23, SDHD and SDHB sequence variations account for 70% of familial cases and 8% of nonfamilial cases, with the prevalence of SDHD sequence variations being as high at 50%. The lifetime risks of SDH-deficient malignancies overall among persons with SDHB, SDHD, and SDHC sequence variations are 41%, 4%, and 3%, respectively.27 The proportions of HNPGLs among those malignancies were 12% and 4% with SDHB and SDHD sequence variations, respectively.
SDH is a tumor suppressor in the paraganglionic system. Tumorigenesis requires complete loss of SDH activity, usually because of inheritance of a germline loss of function and subsequent loss of heterozygosity. Less commonly, an independent somatic loss of function sequence variation for the same subunit can result in a compound heterozygous state.2 The 4 SDHX genes encode the 4 subunits of SDH enzyme, which catalyzes the oxidation of succinate to fumarate in the tricarboxylic acid cycle, which results in accumulation of succinate and generation of reactive oxygen species due to disrupted electron transport downstream.28,29 These accumulated oncometabolites inhibit 2-oxoglutarate—dependent dioxygenases, such as the EglN1-3 (also called prolyl hydroxylases 1-3) family, which stabilize HIF-α. This promotes the HIF-α signaling pathway, which leads to tumorigenesis, abnormal angiogenesis, decreased apoptosis, cell migration, and metastatic spread. Accumulation of succinate may cause wide-spread chromatin and DNA hypermethylation, leading to epigenetic programming of cells and tumor development.30 The complete loss of SDH activity is associated with massive, nonrandom, genome-wide epigenetic reprogramming. For example, the number of genome-wide, differentially methylated CpG targets is approximately 85 000 in SDH-deficient GIST versus 8500 in kinase-mutant GIST. Similar results were seen comparing SDH-deficient PGL/pheochromocytoma to SDH-normal versions of these tumors.30 These epigenetic changes are predicted to activate receptor tyrosine kinase signaling and proliferation leading to initiation of SDH-deficient GIST.30 In addition to markedly enhanced DNA hypermethylation, SDH loss is associated with increased histone methylation and activation of the pseudohypoxia pathway by stabilizing HIF1α and HIF2α.31,32 The myriad effects of the loss of SDH activity on cell physiology likely explains why the SDH-deficient tumor typically has very stable genomes and few other acquired somatic sequence variations.30,33
Symptoms/Patient Presentation
The presentation of patients with HNPGLs depends on anatomical location and physiology. Carotid body and vagal PGLs typically present with a neck mass, cough, hoarseness, or dysphagia. Jugular bulb PGLs or PGLs of the nerves around the ear cause pulsatile tinnitus, hearing loss, or lower cranial nerve deficits.34 Up to 95% of HNPGLs are nonsecretory; however, rare secretory cases can cause symptoms of catecholamine excess, such as hypertension, headache, diaphoresis, palpitations, or anxiety.2
Diagnosis and Imaging
The workup of a mass suspicious for PGL should include biochemical tests to rule out a secretory component, although this is rare. These tests should include plasma-free metanephrines or 24-hour urine fractionated metanephrines and catecholamines.3,35 In nonsecretory HNPGLs, imaging modalities, such as computed tomography (CT) or magnetic resonance imaging (MRI), are necessary for monitoring and surgical planning.
HNPGLs usually demonstrate marked enhancement of intratumoral vessels after contrast administration on CT, a low signal on T1-weighted MR images, and an intermediate to high signal on T2-weighted MR images; they also often enhance intensely after gadolinium injection on MRI. Flow signal voids in the tumor are typical of PGLs, with a salt-and-pepper appearance on spin-echo sequences.2 As far as functional imaging is concerned, based on the most current Endocrine Society Guidelines, 18F-fluorohydroxyphenylalanine positron emission tomography (PET)/CT is the imaging modality of choice.36 Iodine-123/iodine-131-metaiodobenzylguanidine (MIBG) is another imaging option but is inferior to Gallium-68 (68Ga)-DOTA-SSTa in the detection of HNPGLs.37 [68Ga]DOTA-SSTa has shown to have the highest sensitivity for the detection of PGL in patients with or without familial sequence variations.38–40 Other forms of functional imaging, such as 18F-fluorodeoxyglucose PET/CT, can be used to assess for metastases given the avidity of metastasis for fluorodeoxyglucose.41 Figure 1 shows CT and DOTATATE-PET in a patient with PGL.
Fig. 1.
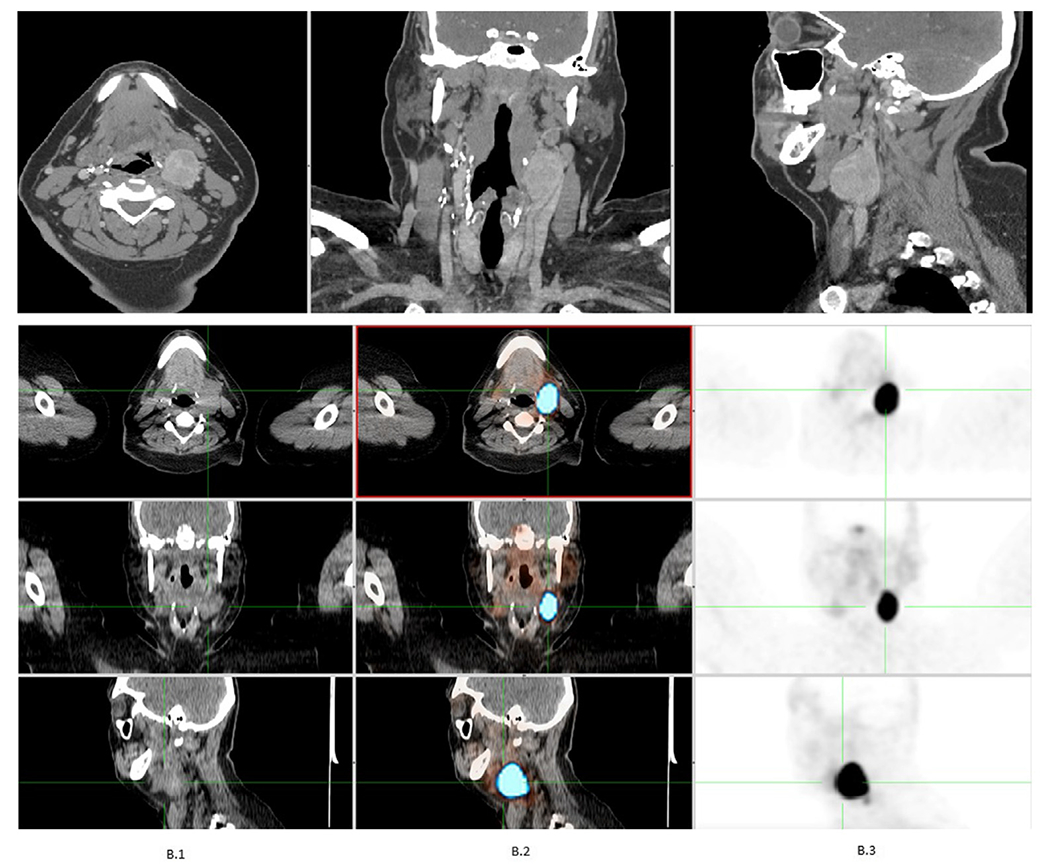
Computed tomography and DOTATATE-positron emission tomography in a patient with paraganglioma.
In vivo metabolomic analysis may serve as an important bridge between molecular genetics and imaging. Proton MR spectroscopy could be added to the classical MR sequences for characterization of various PGLs, especially those related to SDH alterations.
Diagnosis is usually based on functional imaging, with biopsies rarely performed to confirm diagnosis. Histopathological examination based on resected specimens shows a nested alveolar or “zellballen” growth pattern around few to numerous chief cells. These cells are amphophilic to pink; they are usually epithelioid but can also be spindle-shaped. PGL cells have round hyperchromatic nucleus with rare mitosis5 and a lack of mucin or gland formation. A hematoxylin and eosin stained section of PGL is shown in Figure 2. Immunohistochemistry shows expression of neuroendocrine markers (chromogranin, synaptophysin, and neuron-specific enolase) in chief cells and disappearance of S100-positive sustentacular cells; however, these are not required for diagnosis.42 More recent studies have also suggested immunohistochemical staining with antibodies against choline acetyltransferase in which 100% of HNPGLs expressed GATA3, which, on antigen arrival, led to immunoreactivity in all HNPGLs.43,44
Fig. 2.
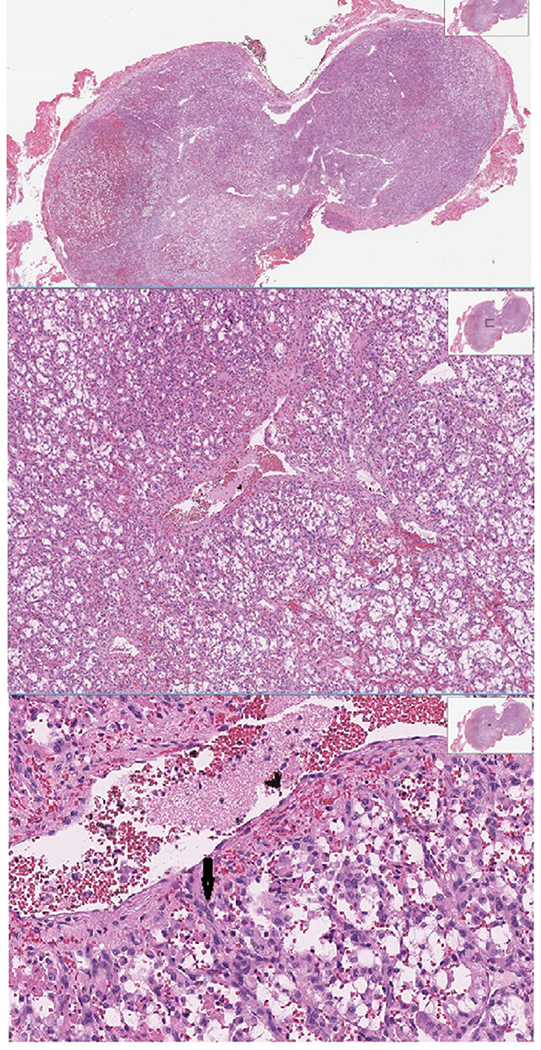
Hematoxylin and eosin staining of paraganglioma showing pink epitheloid cells with round hyperchromatic nucleus (arrow).
In addition to routine testing and imaging, genetic counseling and testing are recommended in all patients with HNPGLs.3,45
Current Treatment
Observation can be considered for asymptomatic patients with a low risk of metastasis based on the absence of signs of local invasion or lack of metastatic disease on functional imaging. These patients are monitored on MRI, with treatment deferred until a finding of progressive disease or the development of symptoms. The surveillance interval should be 6 months initially, followed by annual surveillance. Treatment may include surgery, which is curative in most patients with HNPGLs if complete resection is possible. Ablative stereotactic radiation may be used as a frontline modality in patients; however, there are no head-to-head trials justifying its use over surgery.2
Patients with carotid body PGLs are good candidates for surgical resection when the tumor only partially surrounds the carotid artery. When the tumor encases the entire carotid artery, the surgery is more morbid, and patients may require treatment with radiation therapy. Ellis et al46 demonstrated that patients with carotid body PGLs and SDHB sequence variation should undergo preoperative embolization before resection owing to a higher malignant potential.
Vagal PGLs can be treated with radiation therapy when indicated, the only exception being rapidly enlarging tumors and/or large tumors, for which surgical resection is indicated for immediate evacuation of tumor to avoid risking vagal nerve injury. The resection is performed using a transcervical approach; although risk of vascular injury is low, the incidence of vagal nerve dysfunction is high.47
Tympanic PGLs should be resected via a retroauricular approach and with ossicular reconstruction; this approach leads to excellent hearing outcomes.48 A retrospective study by Forest et al48 showed excellent disease control after resection with minimal morbidity. Finally, because of their location in the jugular fossa, resection of jugular PGLs is especially difficult, resulting in a high incidence of cranial nerve palsies, especially cranial nerves VII, IX, X, XI, and XII.47 The upfront treatment strategy is usually radiation unless these patients present with catecholamine excess, large tumors causing compression of cranial nerves, or rapidly progressive disease threatening vital organs in which surgery should be considered.47 To minimize the risk when surgery is needed, preoperative embolization to minimize blood loss and decrease surgical morbidity is an option, although Persky et al49 found that 6 of 53 patients experienced adverse effects from embolization.
In patients with bilateral multifocal HNPGLs, resection should be considered carefully and requires an individualized therapeutic approach and shared decision making. A 2-step surgical approach is recommended in patients with bilateral HNPGLs to avoid bilateral nerve palsies. It is recommended to start with the larger tumor, and depending on the outcomes of the first procedure, the second tumor can be observed or treated by surgery or radiation therapy based on symptoms and disease behavior. If the tumors are unresectable, studies have shown that radiotherapy is a successful second-line option, with recent studies arguing for first-line treatment given decreased morbidity compared with surgery.2,50–53 In addition, for HNPGLs with high-risk sequence variations, such as SDHB, which have also been linked with aggressive disease, an upfront aggressive surgical approach may help survival.2,46
Based on the newest World Health Organization guidelines, all PGLs are considered malignant and, thus, at risk for metastasis, although metastasis can be difficult to identify given the wide distribution of paraganglia.54 Metastatic HNPGLs are rare, and patients have a poor prognosis; however, surgery is still recommended to remove the primary tumor, especially in the setting of local metastasis.8,55 In cases where this is not possible, studies have shown that adjuvant radiation can slow tumor growth and prolong survival.2,8 In 2018, the Food and Drug Administration approved iodine-131-MIBG therapy for patients with iobenguane scan—positive, unresectable, locally advanced or metastatic PGL based on a 22% overall tumor response, with 53% achieving a response of at least 6 months, and reduced need for antihypertensive medications in 25% of patients.56
Chemotherapy is reserved for patients with advance disease or following MIBG therapies, given that the regimens are limited and there is the risk of high-grade toxicities. Options for chemotherapy include cardiovascular disease (cyclophosphamide, vincristine, and dacarbazine) or temozolomide and thalidomide, temozolomidecapecitabine, or sunitinib monotherapy.2,57,58 Although studies have shown that it can improve symptoms, chemotherapy does not change overall survival.59 Early-phase studies evaluating the role of somatostatin receptor–targeted radioligand therapies in HNPGLs have showed a high safety profile with promising effectiveness and may be an option when expected surgical morbidity outweighs benefits.60 Targeted therapy with tyrosine kinase inhibitors can also be considered if disease progression on cardiovascular disease.61,62 Currently, there are 3 other phase II clinical trials evaluating the use of tyrosine kinase inhibitors in PGLs although it is important to note that these are not specific to HNPGLs (NCT03008369, NCT01967576, and NCT02302833). Finally, given studies suggesting upregulation of programmed death-ligand 1 in the setting of pseudohypoxia, treatment with immunotherapy is being explored (NCT02834013 and NCT02721732).63,64 Figure 3 summarizes recommendations on the management of PGLs.
Fig. 3.

Algorithm summarizing the management of head and neck paraganglioma (HNPGL). CN = cranial nerve; CT = computed tomography; MRI = magnetic resonance imaging; PGL = paraganglioma; SRS = stereotactic radiosurgery.
Prognosis/Survivorship
Most patients perform well after resection of HNPGLs, unless they have a pathogenic variant of the SDHx susceptibility gene that predisposes them to development of more PGLs or pheochromocytoma. In that case, the patients need annual biochemical screening along with whole-body MRI every 2 to 3 years. This also screens patients for renal cell carcinoma or GIST, in which these patients are predisposed to develop.
As previously mentioned, the new World Health Organization guidelines consider all PGLs to have metastatic potential removing the differentiation between “malignant” and “benign.” There are currently no markers for predicting metastatic spread; however, patients with a tumor size of >5 cm, presence of SDHB sequence variation, and extra-adrenal location are at greater risk of metastasis.61,65–67 With regard to other chemical and histopathologic data, patients with a high Ki-67 index and elevated methoxytyramine levels are at greater risk of metastasis.61,65–67
In addition, scoring systems to predict metastasis have been developed: (1) Pheochromocytoma of the Adrenal Gland Scaled Score and (2) Grading of Adrenal Pheochromocytoma and Paraganglioma. However, the Pheochromocytoma of the Adrenal Gland Scaled Score does not include HNPGLs, and the Grading of Adrenal Pheochromocytoma and Paraganglioma, while including extraadrenal tumors, does not report on whether HNPGLs are included, making their use limited.68,69
The long-term outcome of HNPGLs has not been studied extensively; however, patients with benign HNPGLs not invading adjacent structures have a normal life expectancy.70 Although some patients in the study by de Flines et al.70 show normal expectancy, it does not rule out quality of life (QOL) issues in these patients, especially the ones who underwent surgery or radiation therapy, but they may not alter survival. By contrast, the QOL of these patients is markedly reduced by the local extension and progression of HNPGL. Notably, patients with locally aggressive or metastatic disease showed a significant decrease in their health-related QOL, including anxiety, depression, and fatigue. The major disease-specific factor increasing morbidity is excessive catecholamine in secretory tumors, which can lead to cardiac complications, aspiration secondary to lower cranial motor nerve function loss, compression of the brainstem by local invasion, or metastatic to vital organs.2 In addition, surgery and radiation therapy encompassing critical areas of the head and neck can lead to cranial nerve dysfunctions, which may lead to cure but also significant issues in patients in the future.
Recent or Ongoing Developments/Research
HIF2A inhibitors are being evaluated in several clinical trials for renal cell carcinoma; however, 1 trial is also exploring its role in the treatment of advanced PGL.71 In patients with metastatic pheochromocytomas or PGLs, cabozantinib and lenvatinib are being evaluated for treatment.72,73 In combination with other endocrine tumors, there is an ongoing trial of lutetium 117-DOTATATE (a radioactive drug) enrolling unresectable locally advanced or distantly metastatic HNPGLs, which are somatostatin receptor positive as determined by 68Ga-DOTATATE PET/CT imaging.74 In addition, succinate accumulation has been shown to inhibit homologous recombination DNA repair, increasing the sensitivity of these cells to poly(ADP-ribose)polymerase inhibitors.75,76 Currently, the Alliance is conducting a randomized phase II study comparing temozolomide versus temozolomide + olaparib for advanced PGL.77
PGLs are a group of neuroendocrine tumors that arise outside of the adrenal paraganglia. HNPGLs are the most common PGLs and are extremely rare, with an incidence of 0.3 to 1 per 100 000. Their management is complicated and varies by size, location, growth rate, and genetic background. Multiple agents are under investigation for use in PGL, including lutetium 117-DOTATATE.
Highlights
Paragangliomas are neuroendocrine tumors that are outside of the adrenal paraganglia
Head and neck PGLs are the most common PGLs and rare: incidence 0.3 to 1 per 100000
Management is complicated and varies by grade and location
Multiple agents are under investigation for use in PGL, including lutetium 117-DOTATATE
Clinical Relevance
This review outlines all issues that a practitioner managing head and neck paragangliomas (PGLs) needs to know. We summarize the incidence and mortality of PGLs, including head and neck PGLs. Furthermore, we describe the pathogenesis of PGLs that has implications for patients’ treatment and their family screening. We briefly describe the patient presentation followed by workup, including imaging. Subsequently, we describe the current treatment for PGLs in the head and neck, including the more recently approved metaiodobenzylguanidine therapy, in addition to somatostatin receptor–targeted radioligand therapies.
Acknowledgment
MCH received partial salary support from the following sources: a research grant from the Jonathan David Foundation, a VA Merit Review Grant (I01BX005358), and from NCI R21 grant (R21CA263400).
Abbreviations:
- CT
computed tomography
- GIST
gastrointestinal stromal tumor
- HNPGL
head and neck paraganglioma
- MIBG
metaiodobenzylguanidine
- MRI
magnetic resonance imaging
- PET
positron emission tomography
- PGL
paraganglioma
- QOL
quality of life
- SDHD
succinate dehydrogenase subunit D
References
- 1.Erickson D, Kudva YC, Ebersold MJ, et al. Benign paragangliomas: clinical presentation and treatment outcomes in 236 patients. J Clin Endocrinol Metab. 2001;86(11):5210–5216. [DOI] [PubMed] [Google Scholar]
- 2.Taïeb D, Kaliski A, Boedeker CC, et al. Current approaches and recent developments in the management of head and neck paragangliomas. Endocr Rev. 2014;35:795–819. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Cass ND, Schopper MA, Lubin JA, Fishbein L, Gubbels SP. The changing paradigm of head and neck paragangliomas: what every otolaryngologist needs to know. Ann Otol Rhinol Laryngol. 2020;129(11):1135–1143. [DOI] [PubMed] [Google Scholar]
- 4.Pellitteri PK, Rinaldo A, Myssiorek D, et al. Paragangliomas of the head and neck. Oral Oncol. 2004;40(6):563–575. [DOI] [PubMed] [Google Scholar]
- 5.Williams MD. Paragangliomas of the head and neck: an overview from diagnosis to genetics. Head Neck Pathol. 2017;11(3):278–287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Rodríguez-Cuevas S, López-Garza J, Labastida-Almendaro S. Carotid body tumors in inhabitants of altitudes higher than 2000 meters above sea level. Head Neck. 1998;20(5):374–378. [DOI] [PubMed] [Google Scholar]
- 7.Moskovic DJ, Smolarz JR, Stanley D, et al. Malignant head and neck paragangliomas: is there an optimal treatment strategy? Head Neck Oncol. 2010;2:23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Lee JH, Barich F, Karnell LH, et al. National Cancer Data Base report on malignant paragangliomas of the head and neck. Cancer. 2002;94(3):730–737. [DOI] [PubMed] [Google Scholar]
- 9.Jansen JC, van den Berg R, Kuiper A, van der Mey AG, Zwinderman AH, Cornelisse CJ. Estimation of growth rate in patients with head and neck paragangliomas influences the treatment proposal. Cancer. 2000;88:2811–2816. [PubMed] [Google Scholar]
- 10.Papaspyrou K, Mewes T, Rossmann H, et al. Head and neck paragangliomas: report of 175 patients (1989-2010). Head Neck. 2012;34(5):632–637. [DOI] [PubMed] [Google Scholar]
- 11.Mediouni A, Ammari S, Wassef M, et al. Malignant head/neck paragangliomas. Comparative study. Eur Ann Otorhinolaryngol Head Neck Dis. 2014;131(3):159–166. [DOI] [PubMed] [Google Scholar]
- 12.Richter S, Qiu B, Ghering M, et al. Head/neck paragangliomas: focus on tumor location, mutational status and plasma methoxytyramine. Endocr Relat Cancer. 2022;29(4):213–224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Gilbo P, Morris CG, Amdur RJ, et al. Radiotherapy for benign head and neck paragangliomas: a 45-year experience. Cancer. 2014;120(23):3738–3743. [DOI] [PubMed] [Google Scholar]
- 14.Fishbein L, Leshchiner I, Walter V, et al. Comprehensive molecular characterization of pheochromocytoma and paraganglioma. Cancer Cell. 2017;31(2):181–193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Favier J, Amar L, Gimenez-Roqueplo AP. Paraganglioma and phaeochromocytoma: from genetics to personalized medicine. Nat Rev Endocrinol. 2015;11(2):101–111. [DOI] [PubMed] [Google Scholar]
- 16.Baysal BE, Ferrell RE, Willett-Brozick JE, et al. Mutations in SDHD, a mitochondrial complex II gene, in hereditary paraganglioma. Science. 2000;287(5454):848–851. [DOI] [PubMed] [Google Scholar]
- 17.Burnichon N, Buffet A, Parfait B, et al. Somatic NF1 inactivation is a frequent event in sporadic pheochromocytoma. Hum Mol Genet. 2012;21(26):5397–5405. [DOI] [PubMed] [Google Scholar]
- 18.Dahia PL. Pheochromocytoma and paraganglioma pathogenesis: learning from genetic heterogeneity. Nat Rev Cancer. 2014;14(2):108–119. [DOI] [PubMed] [Google Scholar]
- 19.Burnichon N, Vescovo L, Amar L, et al. Integrative genomic analysis reveals somatic mutations in pheochromocytoma and paraganglioma. Hum Mol Genet. 2011;20(20):3974–3985. [DOI] [PubMed] [Google Scholar]
- 20.Castro-Vega LJ, Letouzé E, Burnichon N, et al. Multi-omics analysis defines core genomic alterations in pheochromocytomas and paragangliomas. Nat Commun. 2015;6:6044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Zang YS, Dai C, Xu X, et al. Comprehensive analysis of potential immunotherapy genomic biomarkers in 1000 Chinese patients with cancer. Cancer Med. 2019;8(10):4699–4708. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Comino-Méndez I, de Cubas AA, Bernal C, et al. Tumoral EPAS1 (HIF2A) mutations explain sporadic pheochromocytoma and paraganglioma in the absence of erythrocytosis. Hum Mol Genet. 2013;22(11):2169–2176. [DOI] [PubMed] [Google Scholar]
- 23.Baysal BE, Willett-Brozick JE, Lawrence EC, et al. Prevalence of SDHB, SDHC, and SDHD germline mutations in clinic patients with head and neck paragangliomas. J Med Genet. 2002;39(3):178–183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Snezhkina AV, Fedorova MS, Pavlov VS, et al. Mutation frequency in main susceptibility genes among patients with head and neck paragangliomas. Front Genet. 2020;11, 614908. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Kunst HP, Rutten MH, de Mönnink JP, et al. SDHAF2 (PGL2-SDH5) and hereditary head and neck paraganglioma. Clin Cancer Res. 2011;17(2):247–254. [DOI] [PubMed] [Google Scholar]
- 26.Baysal BE. Mitochondrial complex II and genomic imprinting in inheritance of paraganglioma tumors. Biochim Biophys Acta. 2013;1827(5):573–577. [DOI] [PubMed] [Google Scholar]
- 27.Barbara M, Tsen A, Tenner L, Rosenkranz L. Talking genes in breast and pancreatic malignancies. Mater Sociomed. 2019;31(2):146–149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Rao JU, Engelke UF, Rodenburg RJ, et al. Genotype-specific abnormalities in mitochondrial function associate with distinct profiles of energy metabolism and catecholamine content in pheochromocytoma and paraganglioma. Clin Cancer Res. 2013;19(14):3787–3795. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Hollinshead KE, Tennant DA. Mitochondrial metabolic remodeling in response to genetic and environmental perturbations. Wiley Interdiscip Rev Syst Biol Med. 2016;8(4):272–285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Killian JK, Kim SY, Miettinen M, et al. Succinate dehydrogenase mutation underlies global epigenomic divergence in gastrointestinal stromal tumor. Cancer Discov. 2013;3(6):648–657. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Flavahan WA, Drier Y, Johnstone SE, et al. Altered chromosomal topology drives oncogenic programs in SDH-deficient GISTs. Nature. 2019;575(7781):229–233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Eijkelenkamp K, Osinga TE, Links TP, van der Horst-Schrivers ANA. Clinical implications of the oncometabolite succinate in SDHx-mutation carriers. Clin Genet. 2020;97(1):39–53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Janeway KA, Liegl B, Harlow A, et al. Pediatric KIT wild-type and platelet-derived growth factor receptor alpha-wild-type gastrointestinal stromal tumors share KIT activation but not mechanisms of genetic progression with adult gastrointestinal stromal tumors. Cancer Res. 2007;67(19):9084–9088. [DOI] [PubMed] [Google Scholar]
- 34.Moore MG, Netterville JL, Mendenhall WM, Isaacson B, Nussenbaum B. Head and neck paragangliomas: an update on evaluation and management. Otolaryngol Head Neck Surg. 2016;154(4):597–605. [DOI] [PubMed] [Google Scholar]
- 35.Därr R, Kuhn M, Bode C, et al. Accuracy of recommended sampling and assay methods for the determination of plasma-free and urinary fractionated metanephrines in the diagnosis of pheochromocytoma and paraganglioma: a systematic review. Endocrine. 2017;56(3):495–503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Lenders JW, Duh QY, Eisenhofer G, et al. Pheochromocytoma and paraganglioma: an endocrine society clinical practice guideline. J Clin Endocrinol Metab. 2014;99(6):1915–1942. [DOI] [PubMed] [Google Scholar]
- 37.Arora S, Kumar R, Passah A, et al. Prospective evaluation of 68Ga-DOTANOC positron emission tomography/computed tomography and 131I-meta-iodobenzylguanidine single-photon emission computed tomography/computed tomography in extra-adrenal paragangliomas, including uncommon primary sites and to define their diagnostic roles in current scenario. Nucl Med Commun. 2019;40(12):1230–1242. [DOI] [PubMed] [Google Scholar]
- 38.Janssen I, Chen CC, Taieb D, et al. 68Ga-DOTATATE PET/CT in the localization of head and neck paragangliomas compared with other functional imaging modalities and CT/MRI. J Nucl Med. 2016;57(2):186–191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Janssen I, Blanchet EM, Adams K, et al. Superiority of [68Ga]-DOTATATE PET/CT to other functional imaging modalities in the localization of SDHB-associated metastatic pheochromocytoma and paraganglioma. Clin Cancer Res. 2015;21(17):3888–3895. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Janssen I, Chen CC, Millo CM, et al. PET/CT comparing (68)Ga-DOTATATE and other radiopharmaceuticals and in comparison with CT/MRI for the localization of sporadic metastatic pheochromocytoma and paraganglioma. Eur J Nucl Med Mol Imaging. 2016;43(10):1784–1791. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Bozkurt MF, Virgolini I, Balogova S, et al. Guideline for PET/CT imaging of neuroendocrine neoplasms with 68Ga-DOTA-conjugated somatostatin receptor targeting peptides and 18F-DOPA. Eur J Nucl Med Mol Imaging. 2017;44(9):1588–1601. [DOI] [PubMed] [Google Scholar]
- 42.Gimm O, DeMicco C, Perren A, Giammarile F, Walz MK, Brunaud L. Malignant pheochromocytomas and paragangliomas: a diagnostic challenge. Langenbecks Arch Surg. 2012;397(2):155–177. [DOI] [PubMed] [Google Scholar]
- 43.Kimura N, Shiga K, Kaneko K, Sugisawa C, Katabami T, Naruse M. The diagnostic dilemma of GATA3 immunohistochemistry in pheochromocytoma and paraganglioma. Endocr Pathol. 2020;31(2):95–100. [DOI] [PubMed] [Google Scholar]
- 44.Kimura N, Shiga K, Kaneko KI, et al. Immunohistochemical expression of choline acetyltransferase and catecholamine-synthesizing enzymes in head-and-neck and thoracoabdominal paragangliomas and pheochromocytomas. Endocr Pathol. 2021;32(4):442–451. [DOI] [PubMed] [Google Scholar]
- 45.Fishbein L, Merrill S, Fraker DL, Cohen DL, Nathanson KL. Inherited mutations in pheochromocytoma and paraganglioma: why all patients should be offered genetic testing. Ann Surg Oncol. 2013;20(5):1444–1450. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Ellis RJ, Patel D, Prodanov T, Nilubol N, Pacak K, Kebebew E. The presence of SDHB mutations should modify surgical indications for carotid body paragangliomas. Ann Surg. 2014;260(1):158–162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Suárez C, Rodrigo JP, Bödeker CC, et al. Jugular and vagal paragangliomas: Systematic study of management with surgery and radiotherapy. Head Neck. 2013;35(8):1195–1204. [DOI] [PubMed] [Google Scholar]
- 48.Forest JA 3rd, Jackson CG, McGrew BM. Long-term control of surgically treated glomus tympanicum tumors. Otol Neurotol. 2001;22(2):232–236. [DOI] [PubMed] [Google Scholar]
- 49.Persky MS, Setton A, Niimi Y, Hartman J, Frank D, Berenstein A. Combined endovascular and surgical treatment of head and neck paragangliomas–a team approach. Head Neck. 2002;24(5):423–431. [DOI] [PubMed] [Google Scholar]
- 50.Lieberson RE, Adler JR, Soltys SG, Choi C, Gibbs IC, Chang SD. Stereotactic radiosurgery as the primary treatment for new and recurrent paragangliomas: is open surgical resection still the treatment of choice? World Neurosurg. 2012;77(5-6):745–761. [DOI] [PubMed] [Google Scholar]
- 51.Muracciole X, Régis J. Radiosurgery and carcinogenesis risk. Prog Neurol Surg. 2008;21:207–213. [DOI] [PubMed] [Google Scholar]
- 52.Krych AJ, Foote RL, Brown PD, Garces YI, Link MJ. Long-term results of irradiation for paraganglioma. Int J Radiat Oncol Biol Phys. 2006;65(4):1063–1066. [DOI] [PubMed] [Google Scholar]
- 53.Velegrakis G, Kalikakis G, Karampekios S, Stefanaki K, Helidonis E. [Bilateral paraganglioma of the vagus nerve]. HNO. 2001;49(6):471–475. [DOI] [PubMed] [Google Scholar]
- 54.Mete O, Asa SL, Gill AJ, Kimura N, de Krijger RR, Tischler A. Overview of the 2022 WHO classification of paragangliomas and pheochromocytomas. Endocr Pathol. 2022;33(1):90–114. [DOI] [PubMed] [Google Scholar]
- 55.Hescot S, Leboulleux S, Amar L, et al. One-year progression-free survival of therapy-naive patients with malignant pheochromocytoma and paraganglioma. J Clin Endocrinol Metab. 2013;98(10):4006–4012. [DOI] [PubMed] [Google Scholar]
- 56.FDA approves iobenguane I 131 for rare adrenal gland tumors. U.S. Food and Drug Administraction. Accessed September 11, 2022. https://www.fda.gov/drugs/resources-information-approved-drugs/fda-approves-iobenguane-i-131-rare-adrenal-gland-tumors.
- 57.Jimenez C, Rohren E, Habra MA, et al. Current and future treatments for malignant pheochromocytoma and sympathetic paraganglioma. Curr Oncol Rep. 2013;15(4):356–371. [DOI] [PubMed] [Google Scholar]
- 58.Averbuch SD, Steakley CS, Young RC, et al. Malignant pheochromocytoma: effective treatment with a combination of cyclophosphamide, vincristine, and dacarbazine. Ann Intern Med. 1988;109(4):267–273. [DOI] [PubMed] [Google Scholar]
- 59.Huang H, Abraham J, Hung E, et al. Treatment of malignant pheochromocytoma/paraganglioma with cyclophosphamide, vincristine, and dacarbazine: recommendation from a 22-year follow-up of 18 patients. Cancer. 2008;113(8):2020–2028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Ramsay HA, Kairemo KJ, Jekunen AP. Somatostatin receptor imaging of olfactory neuroblastoma. J Laryngol Otol. 1996;110(12):1161–1163. [DOI] [PubMed] [Google Scholar]
- 61.Nölting S, Ullrich M, Pietzsch J, et al. Current management of pheochromocytoma/paraganglioma: a guide for the practicing clinician in the era of precision medicine. Cancers (Basel). 2019;11(10):1505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.O’Kane GM, Ezzat S, Joshua AM, et al. A phase 2 trial of sunitinib in patients with progressive paraganglioma or pheochromocytoma: the SNIPP trial. Br J Cancer. 2019;120(12):1113–1119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Chouaib S, Noman MZ, Kosmatopoulos K, Curran MA. Hypoxic stress: obstacles and opportunities for innovative immunotherapy of cancer. Oncogene. 2017;36(4):439–445. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Hatfield SM, Sitkovsky M. A2A adenosine receptor antagonists to weaken the hypoxia-HIF-1± driven immunosuppression and improve immunotherapies of cancer. Curr Opin Pharmacol. 2016;29:90–96. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Schovanek J, Martucci V, Wesley R, et al. The size of the primary tumor and age at initial diagnosis are independent predictors of the metastatic behavior and survival of patients with SDHB-related pheochromocytoma and paraganglioma: a retrospective cohort study. BMC Cancer. 2014;14:523. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Eisenhofer G, Lenders JW, Siegert G, et al. Plasma methoxytyramine: a novel biomarker of metastatic pheochromocytoma and paraganglioma in relation to established risk factors of tumour size, location and SDHB mutation status. Eur J Cancer. 2012;48(11):1739–1749. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Fassnacht M, Assie G, Baudin E, et al. Adrenocortical carcinomas and malignant phaeochromocytomas: ESMO-EURACAN Clinical Practice Guidelines for diagnosis, treatment and follow-up. Ann Oncol. 2020;31(11):1476–1490. [DOI] [PubMed] [Google Scholar]
- 68.Kimura N, Takayanagi R, Takizawa N, et al. Pathological grading for predicting metastasis in phaeochromocytoma and paraganglioma. Endocr Relat Cancer. 2014;21(3):405–414. [DOI] [PubMed] [Google Scholar]
- 69.Thompson LD. Pheochromocytoma of the Adrenal gland Scaled Score (PASS) to separate benign from malignant neoplasms: a clinicopathologic and immunophenotypic study of 100 cases. Am J Surg Pathol. 2002;26(5):551–566. [DOI] [PubMed] [Google Scholar]
- 70.de Flines J, Jansen J, Elders R, et al. Normal life expectancy for paraganglioma patients: a 50-year-old cohort revisited. Skull Base. 2011;21(6):385–388. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.A phase 2 study to evaluate the efficacy and safety of belzutifan (MK-6482, formerly PT2977) monotherapy in participants with advanced pheochromocytoma/paraganglioma (PPGL), pancreatic eeuroendocrine tumor (pNET), or Von Hippel-Lindau (VHL) disease-associated tumors. ClinicalTrials.gov. Accessed September 11, 2022. https://www.clinicaltrials.gov/ct2/show/NCT04924075.
- 72.A phase II study to evaluate the effects of cabozantinib in patients with unresectable metastatic pheochromocytomas and paragangliomas. ClinicalTrials.gov. Accessed September 11, 2022. https://www.clinicaltrials.gov/ct2/show/NCT02302833.
- 73.Lenvatinib in treating patients with metastatic or advanced pheochromocytoma or paraganglioma that cannot be removed by surgery. ClinicalTrials.gov. Accessed September 11, 2022. https://clinicaltrials.gov/ct2/show/results/NCT03008369.
- 74.M.D. Anderson Cancer Center. A Phase II study to evaluate the effects of 177Lu-DOTATATE in patients with unresectable and progressive rare metastatic endocrine carcinomas: medullary thyroid cancer. ClinicalTrials.gov. Accessed September 11, 2022. https://clinicaltrials.gov/ct2/show/NCT04106843.
- 75.Sulkowski PL, Sundaram RK, Oeck S, et al. Krebs-cycle-deficient hereditary cancer syndromes are defined by defects in homologous-recombination DNA repair. Nat Genet. 2018;50(8):1086–1092. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Pang Y, Lu Y, Caisova V, et al. Targeting NAD+/PARP DNA repair pathway as a novel therapeutic approach to SDHB-mutated cluster I pheochromocytoma and paraganglioma. Clin Cancer Res. 2018;24(14):3423–3432. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Testing the addition of an anticancer drug, olaparib, to the usual chemotherapy (temozolomide) for advanced neuroendocrine cancer. ClinicalTrials.gov. Accessed September 11, 2022. https://clinicaltrials.gov/ct2/show/NCT04394858.