

Abstract
Ni 2,2’–bipyridine (bpy) complexes are commonly employed photoredox catalysts of bond-forming reactions in organic chemistry. However, the mechanisms by which they operate are still under investigation. One potential mode of catalysis is via entry into Ni(I)/Ni(III) cycles, which can be made possible by light-induced, excited state Ni(II)─C bond homolysis. Here we report experimental and computational analyses of a library of Ni(II)-bpy aryl halide complexes, Ni(Rbpy)(R′Ph)Cl (R = MeO, t-Bu, H, MeOOC; R′ = CH3, H, OMe, F, CF3), to illuminate the mechanism of excited state bond homolysis. At given excitation wavelengths, photochemical homolysis rate constants span two orders of magnitude across these structures and correlate linearly with Hammett parameters of both bpy and aryl ligands, reflecting structural control over key metal-to-ligand charge transfer (MLCT) and ligand-to-metal charge transfer (LMCT) excited state potential energy surfaces (PESs). Temperature- and wavelength-dependent investigations reveal moderate excited state barriers (ΔH‡ ~4 kcal mol−1) and a minimum energy excitation threshold (~55 kcal mol−1, 525 nm), respectively. Correlations to electronic structure calculations further support a mechanism in which repulsive triplet excited state PESs featuring a critical aryl-to-Ni LMCT lead to bond rupture. Structural control over excited state PESs provides a rational approach to utilize photonic energy and leverage excited state bond homolysis processes in synthetic chemistry.
Graphical Abstract
1. INTRODUCTION
1.1. Ni(II)─bpy Photoredox Catalysis.
Merging thermal catalysis with photochemistry (i.e., photoredox catalysis) has had a profound influence within organic chemistry, including coupling reactions forging sp2─sp3 and sp3─sp3 C─C or C─X bonds and their applications to medicinal chemistry.1-9 By leveraging photonic energy to drive key catalytic processes and utilizing earth-abundant transition metals, photoredox catalysis provides an attractive and sustainable means to replace precious metal catalysts.10-14 The disparate electron transfer properties of first-row transition metal catalysts can also provide pathways to new reactive intermediates and/or excited state avenues that can unlock synthetic possibilities for drug development and discovery. However, while methodological studies have demonstrated the power of photoredox approaches in achieving bond-forming reactivity, the mechanisms that underlie these processes are largely unknown. In response, recent research has taken key steps towards a deeper mechanistic understanding, utilizing a combination of experiment and theory.4,5,15-19
Mechanistic survey of photoredox catalysts requires thorough exploration due to the numerous possible photophysical pathways present. For example, reactive molecular excited states can be generated photochemically through photosensitized energy transfer17,20-22 or direct excitation.16,23-24 In either case, the ensuing transition metal photophysics will strongly influence the overall catalytic efficacy by directing the photonic energy to specific pathways, only some of which may be productive to the target reaction. This complexity motivates highly detailed studies of the excited state potential energy surfaces (PESs) that govern the important photophysics underlying photoredox catalysis.
Being catalytically active via photosensitization or direct excitation, Ni(II) complexes featuring the bidentate 2,2′–bipyridine (bpy) ligand have received a great deal of attention due to their many applications in photoredox catalysis. For example, MacMillan et al. demonstrated a photosensitized, energy transfer mediated approach to enable Ni(II)─bpy catalyzed coupling of aryl halides with carboxylic acids.20 In particular, an Ir(III) photosensitizer enabled triplet energy transfer to a ground state Ni(II)─bpy aryl acetate complex (formed in situ from a Ni(II)─bpy aryl halide). Energy transfer from the Ir(III) complex generates a long-lived triplet excited state of the Ni(II)-bpy complex, which can subsequently undergo reductive elimination of the aryl and acetate ligands, forming a new C─O bond (Figure 1A, top).25 The mechanism of this photosensitized, energy transfer-mediated reaction is still being investigated. However, ab initio calculations have suggested a triplet metal-to-ligand charge transfer (3MLCT) state may be active for excited state C─O bond formation. 26 Furthermore, chemical oxidation of the ground state Ni(II)─bpy complex also facilitates reductive elimination.25 These ground and excited state pathways are consistent with earlier research from the Hillhouse group demonstrating ground state chemical oxidation of Ni(II) complexes to Ni(III) can trigger reductive elimination and the formation of new C─X bonds.27-28 In addition to energy transfer pathways, photocatalytic cross-couplings can also be driven by direct excitation and can circumvent the need for external photosensitizers, which often contain precious metals. For example, irradiation of the Ni(II)-bpy aryl halide complex in the presence of ancillary ligands enables the downstream formation of new C─O bonds (Figure 1A, bottom).23-24
Figure 1.

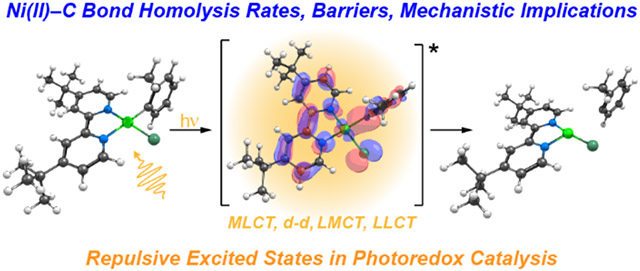
(A) Two possible photocatalytic approaches to Ni(II)─bpy mediated C─O bond formation. (B) Left: Energy diagram showing the direct excitation mechanistic pathway proposed in Reference 16. The structure of the tetrahedral triplet ligand field excited state 3(d-d) is shown. Right: PESs as described in Reference 29 with ab initio calculations showing the 3LMCT-based repulsive surface (in red) responsible for Ni─C bond homolysis. Note that T1 is the 3(d-d) state. The antibonding d(x2-y2)/C(sp2)* orbital is depicted. (C) Summary of this research.
Previous research has noted that direct excitation of the Ni(II)─bpy complex homolytically cleaves the Ni(II)─C(aryl) bond, generating aryl radicals and a formal Ni(I) species. This reduced Ni species may allow access to catalytically active Ni(I)/Ni(III) cycles.16,23-24 While the use of light-induced homolysis to generate reactive Ni species has broad implications for photoredox catalysis, the precise mechanism of this critical bond rupture step is not yet well understood and is the main subject of this study.
1.2. Mechanistic Hypotheses for Excited State Ni(II)─C Bond Homolysis.
There are two proposed excited state Ni(II)─C bond homolysis mechanisms in Ni(II)─bpy aryl halide complexes. Using a combination of transient optical and IR spectroscopies, Doyle et al. demonstrated that excitation of Ni(II)─bpy singlet metal-to-ligand charge transfer (1MLCT) (λpump = 530 nm) resulted in the formation of triplet Ni(II) ligand field excited states (3(d-d)).16 Intersystem crossing occurs in ~5-10 ps, and the 3(d-d) state has a lifetime of ~4 ns. Subsequent correlation to density functional theory (DFT) calculations led to the proposal that Ni(II)─C homolysis occurs thermally from this photochemically formed Ni(II) 3(d-d) state (Figure 1B), which features a tetrahedral coordination geometry and a weakened Ni(II)─C bond. With DFT, the calculated homolytic bond dissociation energy (BDE) is ~25 kcal mol−1. However, no direct experimental evidence was provided to demonstrate homolysis from the 3(d-d) state.
Ab initio multiconfigurational/multireference calculations suggested an alternative mechanism of excited state Ni(II)─C bond homolysis that is also consistent with the experimental data provided by Doyle et al. (Figure 1B).29 This approach yielded larger homolytic BDEs (~90 kcal mol−1 from the S = 0 geometry, ~70 kcal mol−1 from S = 1 geometry) than DFT and highlighted a putative one-photon, two-electron process leading to Ni(II)─C bond homolysis. In this mechanism, initial excitation of the S = 0 complex forms a 1MLCT state (Ni(II)-to-bpy). From this PES (blue curve in Figure 1B), a ligand-to-metal (aryl-to-Ni(III)) charge transfer (LMCT) PES can be accessed. Critically, this LMCT results in the population of the antibonding d(x2-y2)/C(sp2)* orbital (Figure 1B, right), which reduces the bond order and results in a repulsive triplet PES, leading to homolytic bond rupture (red curve in Figure 1B, right).29 Notably, the energy difference between the MLCT/repulsive triplet crossing point (purple circle Figure 1B, right) and the Frank-Condon point of the MLCT state constitutes the energy barrier (Ea) for bond rupture. Thus, it was reasoned that structural and electronic control over the key MLCT/LMCT PESs, and, consequently, the barrier for photolysis, will result in variable rates of excited state Ni(II)─C bond homolysis (Figure 1C), but new experimental data are required to further elucidate the overall mechanism.
Indeed, we demonstrate a direct correlation between experimental rates at given excitation wavelengths and the energies of both of these excited state PESs. Furthermore, we provide an experimental measure of the excited state energetic barrier for homolysis in Ni(II)─bpy aryl halide complexes utilized as photoredox catalysts. The homolysis rate constants are wavelength-dependent, and we have demonstrated a minimum energy threshold for photochemical activation. Coupled to extensive computational analyses, these data provide experimental evidence implicating high energy, repulsive aryl-to-Ni LMCT PESs as being vital to homolytically cleaving the Ni(II)─C bond, a critical process in photocatalytic C─X cross coupling catalysis. The dynamics of the excited states of these Ni(II)-complexes resemble those previously associated with third-row transition metal catalysts (e.g., Re-complexes),30-39 unveiling underexplored reactivity pathways in these earth-abundant transition metal catalysts. Beyond fundamental interest, demonstrating structural and electronic control over the key PESs in photoredox catalysis will, for example, allow chemists to tune the rates of formation of novel reactive intermediates and guide the discovery of new photon-driven organic methodological approaches to coupling reactions.
2. RESULTS AND ANALYSIS
2.1. Experimental Studies.
In Sections 2.1.1-2.1.5, we detail the syntheses and spectroscopic/photochemical characterizations of a matrix of Ni(II)─bpy aryl halide complexes (Figure 2). We demonstrate direct correlations between ligand-based electronic perturbations, observable MLCT transition energies, and rate constants of excited state bond homolysis. Temperature- and wavelength-dependent studies provide experimental barriers and energetic thresholds for excited state Ni(II)─C bond homolysis, respectively.
Figure 2.

Matrix of Ni(Rbpy)(R′Ph)Cl (R = MeO, t-Bu, H, and MeOOC; R′ = ortho- CH3, H, OMe, F, and CF3) complexes examined in this study. The bpy ligand varies down a column, while the aryl ligand varies across a row. Complexes shaded in blue were synthesized and examined both experimentally and computationally, while those in orange were examined only computationally.
2.1.1. Synthetic Approach.
To probe the mechanism of excited state Ni(II)─C bond homolysis, we targeted the matrix of Ni(II)(Rbpy)(R′Ph)Cl complexes (R = CH3O, t-Bu, H, CH3OOC; R′ = ortho- CH3, H, CH3O, F, CF3), 1A–5D, shown in Figure 2. Two primary synthetic approaches were utilized: 1) oxidative addition and 2) ligand substitution (Scheme S1). In the former, bis–(1,5–cyclooctadiene) nickel(0) was pre-stirred with a given bpy ligand; subsequent reaction with the specific aryl halide resulted in the target complex. The latter method called for either bis(triphenylphosphino)(2–methylphenyl)chloronickel(II) or the independent preparation of a pre-catalyst complex, Ni(TMEDA)(R′Ph)Cl, R′ = CH3 or CF3, TMEDA = N,N,N′,N′–tetramethyl ethylenediamine.40 These TMEDA compounds afforded a more labile ligand that could be substituted by bpy.41 The pre-catalyst complexes themselves were prepared by oxidative addition. Ligand substitution was used in cases where oxidative addition proved slow, yielded inconsistent results, or would not produce the desired product. Full synthetic details for both previously prepared16 and novel compounds are available in Supporting Information Section S1.3.
2.1.2. Steady-state UV-vis Spectroscopy.
The steady-state UV-vis spectra of the Ni(II)─bpy aryl halide series in tetrahydrofuran (THF) are given in Figure 3A. Molar absorptivity plots in both THF and toluene are given in Figures S5-S6 and are consistent with previous spectral assignments of dominantly MLCT intensity across the UV-vis range (Table S1). The spectral assignments are discussed further in Section 2.2.1 and given explicitly in Table S10. It is also noted that the MLCT transition energies are generally solvatochromic, with transition energies being lower in toluene relative to THF (Figures S5-S6).
Figure 3.

(A) Normalized UV-vis spectra in THF of complexes studied here (overall Δνmax = 4350 cm−1). (B) Correlation between λmax (dashed lines in A given for the extrema) in THF and the Hammett parameter (σp) for each bpy substituent or (σm) for each aryl substituent. Analogous plots are given in Figure S6 for toluene; molar extinction coefficient data are summarized in Table S1. Legend code used hereafter references the matrix of complexes in Figure 2: red, squares = column 1; blue, triangles = column 5, orange, circles = row B.
Increasing the electron withdrawing effect of the bpy substituents (proceeding down the columns in Figure 2) generally decreases the energies of the 1MLCT transitions. The MLCT λmax for spectra in Figure 3A (extrema denoted by dashed lines) correlate linearly with the Hammett parameter42 (σp) for each bpy substituent (blue and red curves in Figure 3B; Δνmax = 3000 cm−1). Similar Hammett relationships have been shown for Cu and Re bipyridine complexes.43-44
Variation in the aryl ligand (rows in Figure 2) also modulates λmax (Figure 3A) (Δνmax = 1500 cm−1), with increases in electron withdrawing group strength leading to increases in the energy of the 1MLCT transitions. While the aryl ligands are all modified at the ortho- position with respect to the Ni(II)─C bond, the MLCT λmax correlates with the corresponding meta-Hammett parameter (σm) (orange line in Figure 3B). This demonstrates a larger contribution of electrostatic and inductive effects over resonance effects upon variation of the aryl substituent relative to bpy.45-50 Accordingly, this series (1B–5B) also trends with Taft’s field parameter, σF (Figure S7);42 for consistency, we use σm in the main text of this manuscript. No linear trend was observed when using Taft’s steric (Es) parameter,42 as the aryl ligand and its substituent are rotated orthogonal to the plane of the molecule (Figure S7 and S55).45,50
2.1.3. Photochemical Investigations.
We first sought to confirm the formation of aryl radicals upon irradiation (see Supporting Information S1.2 for experimental setup). Irradiating well-characterized 1B and analogous 5B at 390 nm results in distinctive 1H NMR peaks assigned to aryl radical products, 2–(o–tolyl)tetrahydrofuran, and 2,2′–dimethyl-1,1′-biphenyl (Figure S11-12).23 Using 19F NMR, 5B revealed new peaks associated with the free aryl ligand, concomitant with a loss of aryl peaks in the 1H NMR after 390 nm irradiation (Figure S14). As demonstrated previously, homolysis does not occur in the absence of light; only minor degradation is observed when the complex is heated to 55 °C for 60 minutes (Figure S13), but no radical products are observed.23-24 We also noted the formation of radical products upon extended irradiation of the analogous precatalyst complexes, Ni(TMEDA)(R′Ph)Cl (R′ = CH3, CF3), implicating ligand-field excited states as operative for photolysis in the diamine complexes.51 More detailed discussion regarding this result relative to the photochemistry of Ni(II)─bpy complexes is available in Supporting Information Section S1.6.
Time-dependent absorption spectra were obtained during photolyses (390 nm) in THF of all complexes 1A–5D. Photolysis kinetics were monitored at two wavelengths (arrows in Figure 4 and S19-S20). From these kinetics, the observed rate constant (kobs,1) of excited state Ni(II)─C bond homolysis can be obtained. For a representative compound, 1B, concentration dependence studies found negligible change in kobs,1 across the absorbance window tested (Figure S44). The data for series 1A–1D, which varies only the bpy substituent, are provided in Figure 4. Note this series of complexes (1A–1D) has previously been investigated using transient absorption spectroscopy (the only difference being EtOOC versus MeOOC in 1D here).16 Clear rate constant changes are observed upon variation of the bpy ligand (Figure 4 and Table 1). The largest rate constant was found for 1D, which underwent photolysis with an observed rate constant of kobs,1 = (17.0 ± 0.7) x 10−2 min−1. Compounds 1A–1C presented smaller, but noticeable, differences in their decay rate constants (Table 1). For 1A, background scattering from precipitation precluded clear observation of the decay of starting material.
Figure 4.

Photolysis profiles of 1A–1D in THF for 390 nm excitation. Photolysis kinetics were monitored at two wavelengths indicated by the blue and orange arrows in each panel. Insets correspond to the fitted kinetic data (blue curve for the decay of the starting material, orange curve for the formation of the new species). Data were fit using a single exponential; error bars are one standard deviation. For 1A, background scattering from precipitation precluded clear observation of the decay of starting material. This scattering also contributes to the kinetics measured at longer wavelengths for 1A. Photolysis profiles of 5A–5D and 2B–4B are given in Figures S19-S20.
Table 1.
Summary of UV-vis λmax and first-order rate constants. Solvent = THF. Errors are listed as one standard deviation over three trials.
| Compound | λMLCT (nm) |
νMLCT (cm−1) |
kobs,1 (x 10−2 min−1) |
kp (x 10−2 min−1) |
kobs,2 (x 10−2 min−1) |
|---|---|---|---|---|---|
| 1A | 462 | 21645 | n.d. a | 0.9 ± 0.1 | 1.4 ± 0.4 |
| 1B | 475 | 21053 | 2.5 ± 0.2 | 4.1 ± 0.4 | 7.6 ± 0.7 |
| 1C | 483 | 20704 | 3.6 ± 0.6 | 3.3 ± 0.5 | 2.9 ± 0.3 |
| 1D | 532 | 18797 | 17.0 ± 0.7 | 15.0 ± 0.8 | 10.1 ± 3.9 |
| 2B | 471 | 21231 | 2.6 ± 0.1 | 4.4 ± 0.1 | 6.5 ± 0.4 |
| 3B | 464 | 21552 | 0.55 ± 0.02 | 1.6 ± 0.1 | 2.3 ± 0.2 |
| 4B | 447 | 22371 | 0.219 ± 0.002 | 0.14 ± 0.18 | 10.4 ± 5.2 |
| 5A | 432 | 23148 | 0.13 ± 0.01 | 0.08 ± 0.03 | 0.40 ± 0.15 |
| 5B | 443 | 22573 | 0.16 ± 0.03 | n.d. b | 2.3 ± 1.1 |
| 5C | 453 | 22075 | 0.19 ± 0.05 | 0.16 ± 0.01 | 1.4 ± 0.4 |
| 5D | 497 | 20121 | 1.39 ± 0.02 | 3.0 ± 0.5 | 5.1 ± 1.0 |
| 1B–Br | 479 | 20877 | 6.9 ± 0.4 | 8.1 ± 0.8 | 9.2 ± 0.7 |
Because the new species formed upon Ni(II)─C bond homolysis has absorption underneath that of the starting material, we were unable to obtain k1,obs for this compound. The rate is approximated by k2,obs.
Complex 5B was omitted owing to poor convergence of the global kinetics model.
Excited state homolysis of the Ni(II)─C bond yielded a new product with absorbance in the visible region for each compound. Isosbestic points are observed in the photolysis data for all compounds studied here except 1A, where light scattering contributes to the time-dependent spectra. While the general absorption profiles of these new species are similar, the primary low energy features shift from ~650 nm in 1A to 805 nm for 1D (Figure 4). The spectral shift over this series suggests the bpy ligand is present in the new species. The analogous time-dependent UV-vis spectra for compounds 5A–5D are given in Figure S19. There are significant changes in the rate constants of excited state Ni(II)─C bond homolysis across these compounds, with kobs,1 varying over an order of magnitude (Figure S19, Table 1). However, these compounds generally exhibit much smaller rate constants than the complementary 1A─1D series. Thus, the electron withdrawing effect of the aryl ligand also impacts the rate. The growth of a new species was also observed for these complexes (~650 nm in 5A to 805 nm in 5D), albeit at significantly lower quantities. To further investigate the dependence of kobs,1 on variation of the aryl ligand, analogous time-dependent UV-vis data were obtained for complexes 2B–4B (Figure S20). From these data, the full trend is revealed: increasing the electron withdrawing nature of the aryl ligand (left to right in the row of Figure 2) again resulted in smaller rate constants across the series.
This behavior is opposite to that observed for variations in the electron withdrawing effect of the bpy ligand, hence the opposite slopes in Figure 3B. Note also the primary low energy absorption feature of the new species is not dependent on the aryl ligand (λ = 660 nm for 1B–5B, Figures S41-S43).
We note that in certain regions, the absorption spectrum of the photolysis product overlaps with that of the starting material, including where decay kinetics are measured (blue arrows in Figures 4 and S19-S20). Furthermore, kobs,1 is in most cases less than kobs,2. To deconvolute the spectral overlap and rationalize these differences, global kinetics modeling was carried out (full discussion and details of the kinetic modeling are available in Supporting Information Section S1.9). Good agreement is seen between the observed rate constants and those obtained from the global fits, and the kinetic trends across the matrix of compounds are preserved (Figure S40). Comparison between kobs,1 and rate constants from global fitting (kp) are also given in Table 1. Additionally, using the method developed by Gescheidt et al.,52 we calculated the quantum yields for each complex to account for differential absorbance at the 390 nm excitation wavelength (see Supporting Information Section S1.11 for complete details). We found good linear agreement between the observed photolysis rates and the calculated quantum yields (R2 = 0.9730, Figure S53), further supporting our kinetic analysis.
Furthermore, the rate constants of excited state Ni(II)─C bond homolysis correlate linearly with specific Hammett parameters of the bpy and aryl ligands. As shown in Figure 5, linear relationships are observed upon plotting log(kobs,1/kobs,1(H)) versus σp or σm (for Rbpy or R′Ph, respectively) (R2 ≥ 0.95) (ρ = ~1.4 for Rbpy and ρ = ~−2.6 for R′Ph). Thus, the rate of excited state Ni(II)─C homolysis is sensitive to electronic structure perturbations from both the bpy and aryl ligands.
Figure 5.

Correlation between normalized rate constants (390 nm excitation) of excited state bond homolysis and specific Hammett σ parameters of the bpy and aryl ligands. Note that in 1A, the rate is approximated by k2,obs. Analogous plot of the relative quantum yields versus specific Hammett σ parameters is given in Figure S54.
Electronically stabilizing the Ni(II)-to-bpy MLCT transition energies by increasing the bpy-based electron withdrawing effect accelerated the rate of photolysis. Conversely, increasing the aryl-based electron withdrawing effect resulted in increased MLCT transition energies and slower rates of photolysis (see the oppositely signed slopes in Figure 3B). These data reflect competing effects on the excited state PESs involved in homolysis and are further described in the computational sections below.
2.1.4. Preliminary Investigations of the Photochemically Generated Species.
The immediate product of Ni(II)─C bond homolysis has been proposed to be a three–coordinate Ni(I)Rbpy(Cl) complex. A recent study by Bird et al. on the related Ni(I)(t-Bubpy)Br reported its UV-vis spectrum as generated by pulse radiolysis or electrolysis.53 For direct comparison, we synthesized Ni(t-Bubpy)(CH3Ph)Br, 1B–Br, and subjected it to the same photolysis conditions as above.
We found a roughly three-fold enhancement in the rate constant of photolysis for 1B–Br (kobs,1 = (6.9 ± 0.4) x 10−2 min−1) relative to 1B (kobs,1 = (2.5 ± 0.2) x 10−2 min−1) and a change in the absorption spectrum of the product species (Figure 6A). The primary low energy absorption feature of the product appears at higher energy when produced from the bromo-complex (653 nm) versus the chloro-complex (660 nm). Thus, there is a halide-dependence on the absorption spectrum of the product compound. A comparison between the long-time spectra of the photoproducts from compounds 1B–Br, 1B, 3B, 1C, and 1D is given in Figure 6B, illustrating a change in peak maxima when changing the bpy or halide ligands, but not the aryl ligand.
Figure 6.

(A) Photolysis profile of 1B–Br in THF (390 nm excitation) monitored at two wavelengths indicated by the blue and orange arrows in each panel. Inset corresponds to the first-order kinetics data (blue curve for the decay of the starting material, orange curve for the formation of the new species). Error bars are one standard deviation. (B) Comparison between long-time UV-vis spectra for 1B–Br (λmax = 653 nm), 1B (λmax = 660 nm), 3B (λmax = 660 nm), 1C (λmax = 673 nm), and 1D (λmax = 805 nm) (C) Comparison between UV-vis spectra for 1B–Br in THF and DMF, before and after irradiation with 390 nm light.
We also followed the photolysis of 1B–Br in dimethylformamide (DMF), the same solvent used by Bird et al. (Figure S51).53 We first note the steady state UV-vis data are solvatochromic, with the main MLCT bands being lower in energy in THF relative to DMF (Figure 6C, blue versus orange lines, respectively). The homolysis product UV-vis spectra are also solvatochromic (Figure 6C, dashed lines). In particular, DMF solutions exhibit the same characteristic UV–vis features for the three-coordinate monomeric species (430, 620, and 860 nm) as observed by Bird et al. (Figure 6C, orange dashed line). We further note that the rate constant of excited state Ni(II)─C bond homolysis is smaller in DMF relative to THF. We tentatively ascribe these differences in rate constants to changes in the MLCT energies and overall excited state PESs (vide infra). These interesting solvent effects on the excited state PESs and, thus, rates of homolysis are currently under more detailed investigation.
Monomeric Ni(I)(t-Bubpy)X, X = Cl, Br has been shown to be active towards the oxidative addition of aryl iodides, while the dimeric form, [Ni(tBubpy)X]2, is unreactive with the same.53-54 We irradiated a sample of 1B–Br in THF, generating the photoproduct. Addition of 2-iodotoluene to this solution revealed rapid reactivity and complete removal of the characteristic absorption feature at 653 nm (Figure S52).
Furthermore, the absorption spectrum of the dimeric species shows peaks only in the UV-region, further implicating the monomeric form as the photoproduct.54
Therefore, we postulate that the new species formed here upon excited state Ni(II)─C homolysis are three-coordinate Ni(I)(Rbpy)X complexes (R = MeO, t-Bu, H, and MeOOC, X = Cl or Br), as they have been shown by steady-state UV-vis spectroscopy to 1) contain the bpy ligand, 2) not contain the aryl ligand, 3) contain the halide, and in the case of 1B–Br, 4) exhibit the same absorption profile as Ni(I)(t-Bubpy)Br, and 5) exhibit oxidative addition reactivity with iodotoluene. A detailed comparative study of the reactivities and further spectroscopic characterizations of these species is currently underway.
2.1.5. Further Examination of the Mechanism of Excited State Bond Homolysis.
To further investigate the mechanism of excited state Ni(II)─C bond homolysis, we carried out temperature-dependent photolyses of 1B and 1B–Br. Among the matrix of complexes studied here, these two are most often utilized for synthetic applications, giving their analyses direct implications for photoredox catalysis.24
Eyring plots of temperature dependent rate constants for these complexes are given in Figure 7. From these data, the enthalpy and entropy of activation for the excited state Ni(II)─C bond homolysis in 1B are ΔH‡ = 4.4 ± 0.6 kcal mol−1 and ΔS‡ = −45.3 ± 1.8 cal mol−1 K−1, with ΔG‡(298 K) = 17.9 ± 0.8 kcal mol−1. Similar analysis of 1B–Br gives ΔH‡ = 2.1 ± 0.1 kcal mol−1 and ΔS‡ = −49.3 ± 0.4 cal mol−1 K−1, with ΔG‡(298 K) = 16.8 ± 0.2 kcal mol−1. At high temperatures (328 K), thermal decay of the starting material occurs for 1B–Br, resulting in a downturn in the temperature-dependent rate constants (dashed yellow line, Figure 7). Because of this, the linear fit utilized a room temperature point. As expected, the barrier for excited state Ni(II)─C bond homolysis is lower in 1B–Br than 1B, consistent with its larger rate constant.
Figure 7.

Eyring analysis of the temperature dependent photolysis rates of 1B (blue, squares) and 1B–Br (orange, circles); error bars are one standard deviation over three trials.
In addition to being dependent on temperature, the rate constant of excited state bond homolysis in 1B is also highly dependent on the excitation wavelength (Figure 8A). Varying incident wavelengths (390, 427, 456, and 525 nm, Figure S21) revealed a minimum energy threshold for excited state Ni(II)─C bond homolysis of ~55 kcal mol−1 (525 nm, 19,050 cm−1) in 1B; below this incident energy, no homolysis is observed (Figure S22). Previous optical transient absorption measurements on 1B were carried out using λpump = 530 nm.16 Laser excitation at this wavelength results in the formation of a Ni(II)-based triplet ligand field excited state, from which homolysis was proposed on the basis of DFT calculations. Notably, however, very limited photolysis occurs here using a 525 nm excitation light source. These results demonstrate that the lower-energy ligand field state is not responsible for excited state Ni(II)─C bond homolysis, but rather indicate the involvement of higher-energy excited states.
Figure 8.

Wavelength-dependent photolysis for 1B, 1D, and 5D. The absorption spectra are shown in blue; the observed photolysis rate constants (squares) and incident wavelengths are given in orange. Analogous plot of wavelength-dependency of quantum yields is given in Figure S25.
To search for general trends across compounds considered here, we also conducted wavelength dependent studies on 1D and 5D (Figure 8B-C). Altogether, these complexes span a wide range of photolysis rate constants, have varying MLCT transition energies, and feature electronic structure differences provided by the bpy and aryl ligands. In each case, a clear wavelength dependence was observed, and high energy incident light was required for homolysis. No appreciable decay was observed using low energy light (Figures S23-24), again implicating high energy excited states in the mechanism of light-induced homolysis. We also evaluated the wavelength-dependency of quantum yields for each complex, accounting for variable LED power and complex absorbance at each wavelength, and found their behavior mirrors the photolysis kinetics (Figure S25). In summary, through experimental analyses of a matrix of Ni(II)─bpy aryl halide complexes, we have demonstrated the following:
a dependence between the MLCT λmax and the Hammett parameters of the bpy and aryl substituents over the 1A–5D series (Figure 3B),
linear correlations between the Hammett parameters of the bpy and aryl substituents and the rate constants of excited state Ni(II)─C bond homolysis over the 1A–5D series, interestingly with oppositely signed slopes (Figure 5),
the barrier for excited state bond homolysis is moderate (e.g., ΔH‡ = 4.4 ± 0.6 kcal mol−1 in 1B using 390 nm excitation; Figure 7), and
excited state bond homolysis is distinctly wavelength dependent (Figure 8); e.g., in 1B, requiring a minimum of ~55 kcal mol−1 (525 nm, 19,050 cm−1).
These experimental observations are discussed below in the context of computational studies, which further aid in the elucidation of the mechanism of excited state Ni(II)─C bond homolysis.
2.2. Computational Studies.
In the following computational Sections 2.2.1-2.2.3, we first compare the ground and excited state properties of 1A–5D computed at different levels of theory. We discuss the possible photoactivation pathways that are accessible in the energy range of the external light sources used in the photolysis experiments, as well as those pathways that are consistent with the experimental barrier. Notably, the incident light energy required for photolysis (as determined from wavelength dependent kinetic experiments) is substantially greater than the energy of the 3(d-d) bands in 1A–5D, and the calculated barriers for homolysis from these states are significantly larger than experiment. These points indicate thermally-driven excited state Ni(II)─C bond homolysis from a spin-forbidden ligand field state is not the operative mechanism. Instead, we focus on the possible photolysis pathways that exploit triplet excited state LMCT-based repulsive PESs. We propose a working mechanism that can ultimately be described as 1MLCT [Ni d → bpy π*(2)] excitation followed by surface hopping to a repulsive 3LMCT (aryl-to-Ni) PES (3MLCT+LMCT). This mechanism is in agreement with the experimentally derived reaction rates and thermodynamic barriers determined herein.
2.2.1. DFT versus CASSCF/QD-NEVPT2 Ground and Excited States.
To evaluate the geometric and electronic structures of 1A–5D, we compared their ground and excited state properties calculated with either DFT/TD-DFT or ab initio complete active space self-consistent field theory with the quasidegenerate N-electron valence state perturbation theory correction (CASSCF/QD-NEVPT2)55-58; full computational details are available in Supporting Information Section S2.1.
With DFT (B3LYP)59-61, all Ni(II) complexes are predicted to have low-spin, singlet (S = 0) ground states with square-planar geometries (note the x-axis is directed along the Ni─halide bond and the y-axis is along the Ni─aryl bond). The fully optimized triplet (S = 1) ligand field excited states are in all cases ~10 kcal mol−1 higher in energy with pseudo-tetrahedral geometries. The valence electronic configuration of the d8 ground state is [d(xy)]2[d(yz)]2[d(xz)]2[d(z2)]2, with three unoccupied bpy-based π* orbitals and a highly covalent antibonding [d(x2-y2)/C(sp2)*]0 orbital (see Figure S59 for an example molecular orbital diagram for 1D). The orbital energies are modulated by the bpy substituents; increasing the electron withdrawing effect of the bpy ligand (columns in Figure 2) decreases the energies of the bpy π* orbitals, reducing the Ni(II)-to-bpy MLCT energy (Figure S60), consistent with the red-shifted experimental λmax features in Figure 3. On the other hand, the HOMO and the bpy π* orbital energies remain essentially unchanged when modulating the aryl substituent (rows in Figure 2), contrasting with the blue shift in Figure 3. The correct behavior can be recovered at the TD-DFT level, which accounts for orbital mixing in the excited states (Figure S62). Interestingly, the changes in orbital energies are not translated into changes in the covalencies of the ground states, which remain ~51–54 % Ni d and ~11-13 % bpy character for 1A–5D (Table S9).
The calculated TD-DFT absorption spectra agree well with the experimental UV-vis data (see overlaid spectra in Figure S62) and also demonstrate a similar linear relationship with the substituent-specific Hammett σ parameters (Figure 9, top). The broad feature at longer wavelengths (~400–600 nm, ~25,000–16,500 cm−1) encompasses all the ‘low-energy’ 1MLCT transitions [Ni d → bpy π*(1)], with [d(yz) → bpy π*(1)] having the highest calculated oscillator strength. The shoulder at ~350–370 nm (~28,500–27,000 cm−1) apparent in most of the experimental UV-vis spectra of 1A–5D can be similarly assigned to a [d(yz) → bpy π*(2)] transition; other ‘high-energy’ 1MLCT [Ni d → bpy π*(2)] transitions are predicted to fall in the ~300–450 nm (~33,000–22,000 cm−1) range. The 1(d-d) transitions are calculated to be comparable in energy to the ‘low-energy’ 1MLCT bands (~400–500 nm, 25,000–20,000 cm−1) and are not visible in the experimental UV-vis spectra of 1A–5D. This assignment is also consistent with the energy of the observable 1(d-d) band [d(yz) → d(x2-y2)/C(sp2)*] in the Ni(II)(TMEDA)(CH3Ph)Cl complex that is detected in the visible range (found at ~470 nm, 21,280 cm−1; calculated at 533 nm, 18,760 cm−1); the less intense bands observed near ~635 nm (15,750 cm−1) can be assigned to the spin-forbidden triplet transitions. For example, the [d(xy) → d(x2-y2)/C(sp2)*] triplet transition is calculated at 642 nm (15,580 cm−1). Others, including [d(xz) → d(x2-y2)/C(sp2)*] and [d(yz) 𡦒 d(x2-y2)/C(sp2)*], are calculated at 847 nm (11,810 cm−1) and 945 nm (10,580 cm−1), respectively. All relevant TD-DFT electronic transition energies are given in Table S10.
Figure 9.

Correlation between the calculated 1MLCT [d(yz) → π*(1)] transition energies (CPCM solvation model, THF) and the specific Hammett σ parameters using both TD-DFT (top) and CASSCF/QD-NEVPT2 (bottom) methods.
Therefore, the light sources exhibiting most appreciable excited state Ni(II)─C bond homolysis (i.e., 390 nm, 25,640 cm−1 and 427 nm, 23,420 cm−1) in 1A–5D favor initial excitation into high-energy 1MLCT bands, [Ni d → bpy π*(2)], while longer wavelength light sources from the wavelength dependence study in Figure 8 (456 nm, 21,930 cm−1 and 525 nm, 19,050 cm−1) are in the range of the low-energy 1MLCT bands, [Ni d → bpy π*(1)]. The minimum energy threshold of ~525 nm, below which no photolysis is observed, is not consistent with the previous DFT-based assignment of bond rupture from a low-lying triplet ligand field state (Figure 1B, left).16 The lack of bond homolysis from the low-lying MLCTs is likely the result of an increased barrier for homolysis (discussed further below in Section 2.2.3).
Multiconfigurational/multireference CASSCF/QD-NEVPT2 calculations were conducted using the DFT-optimized singlet geometries. State-averaged CASSCF calculations (15 singlets, 25 triplets) were performed with an active space of 10 electrons in 9 orbitals (10e, 9o): d(yz), d(z2), d(xy), d(xz), a pair of bonding and antibonding Ni d(x2-y2)/aryl C(sp2) orbitals, and three bpy π* orbitals (Figure S56). The method and active space follow our previous study on 1B.29 For 1A–5D, the ground state wave functions exhibit substantial multiconfigurational character. While the configuration interaction (CI) vectors (Figure S57, Table S9) are primarily comprised of the closed-shell singlet (CSS) configuration (i.e., the configuration also acquired with DFT), both the MLCT and LMCT configurations contribute significantly to the character of the ground state wave function.
The calculated CASSCF/QD-NEVPT2 absorption spectra generally agree with the observed bands in the experimental UV-vis data (Figure S63, tabulated in Table S11). However, the λmax are notably red-shifted (~5000 cm−1) as compared with TD-DFT (Figure S62 and Table S10). The composition of the ground state CI vector, particularly the total 1MLCT contribution, also closely correlates with the transition energy in the calculated UV-vis spectra. As the 1MLCT [Ni d(xz) → bpy π*(1)] weight increases, the calculated energy of λmax in the absorption spectrum decreases (Figure S65). The multiconfigurational/multireference calculations also demonstrate a similar linear relationship with the substituent-specific Hammett σ parameters (Figure 9, bottom).
2.2.3. Investigating the Mechanism of Excited State Ni(II)─C Bond Homolysis.
DFT relaxed PES scans along the Ni(II)─C bond coordinates of 1A–5D revealed a singlet/triplet degenerate homolytic dissociation product ~40 kcal mol−1 above the singlet equilibrium geometry. Thermal dissociation barriers of ~30 kcal mol−1 were calculated from the triplet equilibrium geometries, consistent with a previous study.16 Given the large differences in BDEs between DFT and CASSCF/QD-NEVPT2 observed previously for 1B,27 we have also evaluated the BDEs using different levels of theory. Referencing to the experimental energy of the 3(d-d) state found by Doyle et al. (12 kcal mol−1),16 we find BDEs of ~40 kcal mol−1 (SCS-MP2/QZ), ~41 kcal mol−1 (DLPNO-CCSD(T)/QZ), and ~76 kcal mol−1 (CASSCF/QD-NEVPT2; 10e, 8o) (Tables S6-S7), suggesting that the DFT BDE of ~30 kcal mol−1 represents the lower limit for thermally-driven, excited state Ni(II)─C bond homolysis from the triplet ligand field equilibrium geometry. These calculated barriers are all significantly higher than the experimental value of ~5 kcal mol−1 demonstrated above. Therefore, we again find that thermal dissociation from the lowest energy triplet ligand field state is not consistent with the experimental data provided here and, thus, is not a viable mechanism for excited state Ni(II)─C bond homolysis.
Instead of thermally-driven excited state Ni(II)─C bond homolysis from ligand field states, we propose that the mechanism of bond homolysis exploits excited state triplet repulsive aryl-to-Ni LMCT PESs (outlined in Figure 10).
Figure 10.

Two plausible excited state mechanisms of Ni(II)─C bond homolysis. Initial 1(d-d) or 1MLCT excitation (blue or orange boxes, respectively) is followed by ISC and 3LMCT formation. On the right, this surface hopping mechanism is exemplified with the shaded purple circles connecting the 1(d-d) excited-state surface (purple line) to the one-electron triplet repulsive 3LMCT surface (green line), or 1MLCT excited-state surface (blue line) to the two-electron triplet repulsive 3(MLCT+LMCT) surface (red line). Corresponding plots computed at the TD-DFT and CASSCF/QD-NEVPT2 levels are provided in Figures S68-69. For clarity, exchange splitting between α and β orbitals is neglected. Illustrative orbital energies and ordering on the left are provided from the ground state and are held constant through steps 1-4; right panel gives a more accurate depiction of relative state energies, reflecting a small thermal barrier relative to the excitation energy.
Critically, in these scenarios, the transfer of an electron between the bonding and antibonding d(x2-y2)/C(sp2) orbitals significantly lowers the overall bond order, facilitating bond rupture.
We have found two pathways utilizing either 1(d-d) (blue panel in Figure 10) or 1MLCT (orange panel in Figure 10) transitions as initial entrance to the excited state mechanism. In both cases, these initial excitations are followed by intersystem crossing (ISC) and formation of an aryl-to-Ni 3LMCT state. Accordingly, two principally different triplet repulsive surfaces are viable for homolysis: a ‘one-photon, one-electron’ excited state 3[d(x2-y2)/C(sp2) → d(x2-y2)/C(sp2)*] and a ‘one-photon, two-electron’ excited state 3[d(x2-y2)/C(sp2) + d(yz) → d(x2-y2)/C(sp2)* + π*(1)] (green and red surfaces in Figure 10 (right), respectively). This former pathway expands our initially delineated mechanism in reference 29, wherein the probability for population of the 1(d-d) excited states versus the 1MLCT upon irradiation would contribute to determining the excited-state surface mediating homolysis. Indeed, it is likely this ‘one-photon, one-electron’ repulsive surface that facilitates Ni(II)─C bond homolysis in the Ni(TMEDA) aryl halide complexes, as described in Section 2.1.3 above; a recent report by Park et al. implicated the corresponding surface in the homolysis of related Ni(II) complexes.51
From the singlet ground state, the standard TD-DFT approach (i.e., single-electron excitations) cannot access the two-electron nature of the red surface in Figure 10 (right). Also, the spin-flipped α-to-β excitations (needed for description of the green surface in Figure 10, right) are only available from the singlet ground state using the restricted Kohn-Sham orbitals, which are inadequate for producing the accurate excited state charge transfer states, especially for out-of-equilibrium geometries with increased charge separation. Nevertheless, we were able to identify both of these triplet repulsive PESs by using TD-DFT from the high-spin (S = 1) triplet reference state (i.e., the electronic configuration of the middle structure in the blue panel of Figure 10). From this configuration, the one-electron or two-electron triplet repulsive surfaces can be obtained via β-to-β [d(x2-y2)/C(sp2) → d(z2)] or [d(x2-y2)/C(sp2) → bpy π*(1)] excitations.
However, we note that, due to the triplet reference state containing the singly-occupied Ni d(z2) orbital configuration, our description of the 3MLCT surfaces and two-electron triplet repulsive surface is restricted to those with [Ni d(z2) → bpy π*] MLCT transitions. Detailed discussion on the limitations of this approach is given in Supporting Information Section S2.2.
Despite these limitations, we were able to obtain an excellent energetic correlation between the ‘one-photon, two-electron’ triplet repulsive surface at the equilibrium geometry and both the experimental ln(kobs,1) and Hammett σ parameters (Figure S66-67), suggesting that this surface (or an analogous surface with another singly-occupied Ni d orbital) is involved in the mechanism of excited state bond homolysis. In contrast, a significantly poorer correlation was found for the energy of the ‘one-photon, one-electron’ 3[d(x2-y2)/C(sp2) → d(x2-y2)/C(sp2)*] repulsive surface (Figure S66). Furthermore, when considering that the calculated 1/3(d-d) transitions are near or below the minimum excitation-energy threshold obtained from the wavelength-dependent experiments in Figure 8, it appears that the one-electron triplet repulsive surface is not likely operative for the excited state Ni(II)─C bond homolysis in 1A–5D.
The activation energies derived from the high-energy 1MLCT excited states (as defined by the crossing of the MLCT/triplet repulsive surface and the Frank Condon point of the MLCT state) agree qualitatively well with the activation enthalpies of ΔH‡ = 4.4 ± 0.6 kcal mol−1 and 2.1 ± 0.3 kcal mol−1 measured for 1B and 1B–Br (Figure 11). The best fit between the calculated activation energies and the experimental ln(kobs,1) was observed for the 1MLCT [d(yz) → π*(2)] excited surface hopping to the two-electron repulsive surface, 3[d(x2-y2)/C(sp2) + d(z2) → d(x2-y2)/C(sp2)* + π*(1)] (Figure 11). It should also be noted that the MLCT transitions from the donor d(yz) orbitals correspond to the absorption bands with greatest oscillator strengths, making the [d(yz) → π*(2)] surface most likely to be populated from initial photon absorption. However, due to the aforementioned limitations of the TD-DFT approach (Section S2.2), we cannot exclude an equal or better correlation for the same pathway utilizing another 3(MLCT+LMCT) excited state instead.
Figure 11.

Computed excited state activation energies (TD-DFT, top; CASSCF/QD-NEVPT2, bottom) for 1A–5D plotted against experimental ln(kobs,1) obtained using 390 nm excitation. Note that for 1A, k1,obs is approximated by k2,obs. The activation energies are estimated from the surface crossing between the high-energy 1MLCT [d(yz) → π*(2)] PES and the ‘one-photon, two-electron’ triplet repulsive surface, 3(MLCT+LMCT) (more details can be found in Supporting Information Section S2.2; see Figure S66 for an analogous plot with the ‘one-photon, one-electron’ triplet repulsive surface). Additionally, we have found an activation energy of ~2.2 kcal mol−1 for 1B–Br complex, which is in an excellent agreement with the experimental ΔH‡.
The CASSCF/QD-NEVPT2 computations provide the same photolysis mechanism for 1A–5D as that obtained from TD-DFT (Figure 10, left). However, the predicted activation energy is increased by ~20-25 kcal mol−1 (Figure 11). This increase is likely the result of the higher Ni(II)─C BDEs found by CASSCF/QD-NEVPT2, as raising the overall BDE also increases the energies of the crossing point between the initial 1MLCT and the repulsive two-electron excited state surfaces. As discussed in detail in Supporting Information Section S2.2, we believe the higher BDEs for these calculations originate from a potentially incomplete description of the CASSCF reference wave function.
From the initial 1MLCT [d(yz) → π*(2)] excitation, we have evaluated the activation energies for this surface crossing from all of the accessible 3(MLCT+LMCT) repulsive states. Although the correlation is satisfactory with the same 3[d(x2-y2)/C(sp2) + d(z2) → d(x2-y2)/C(sp2)* + π*(1)] surface as explored by TD-DFT (with R2 = 0.79), an even better correlation was observed for surface hopping to the 3[d(x2-y2)/C(sp2) + d(yz) → d(x2-y2)/C(sp2)* + π*(1)], characterized by R2 = 0.91 (Figure 11, bottom).
Overall, both TD-DFT and CASSCF/QD-NEVPT2 methods predict analogous photolysis mechanisms that exploit triplet repulsive ‘one-photon, two-electron’ excited state PESs, and the calculations further capture the critical aspects that lead to geometric and electronic structural control over the excited state PES manifold that ultimately governs the photochemical behavior. However, each method is associated with its own limitations, precluding a definitive assignment of which 1/3MLCT is initially operative or at which stage ISC occurs (i.e., before or after the surface hopping between MLCT and MLCT+LMCT surfaces). We also speculate that this mechanism may not be identical for all Ni complexes used in photoredox catalysis. Additional possibilities, including the ‘one-photon, one-electron’ 1/3(d-d) → 3LMCT pathway, might be operative depending on the energetics of the individual excited states, the probability of the initial light-induced transitions, and/or the probability of the surface hopping to the triplet repulsive PESs.
3. DISCUSSION
Light-induced homolysis provides a powerful means to activate ligand–metal bonds for the generation of reactive radical species involved in targeted catalytic bond transformations.5,62-63 Defining the photophysical processes underlying the mechanisms of light-induced homolysis will therefore aid chemists in elucidating the role(s) of photo-generated intermediates in currently established photoredox catalytic cycles, as well as further guide the development of novel bond-formation reactions in organic chemistry. Here we have provided new combined experimental and computational insights that have aided in the elucidation of the mechanism of excited state Ni(II)─C bond homolysis in commonly employed Ni(II)─bpy aryl halide photoredox catalysts.
In particular, rate constants of excited state homolysis depend on temperature (Figure 7) and the wavelength of light excitation (Figure 8). From these observations, we conclude that there exists an energy barrier between the light absorbing and homolytically dissociative excited states. For 1B, ΔH‡ = 4.4 ± 0.6 kcal mol−1 using 390 nm excitation. Note ΔH‡ is the energy parameter that is most relevant for comparisons to calculated excited state PESs. From the wavelength dependence on the rate constants, it is also clear that the lower energy 1MLCTs near ~525 nm (19,050 cm−1, 55 kcal/mol) are not productive for excited state homolysis. Rather, higher energy excitation is required. Overall, these observations are not consistent with thermally-driven Ni(II)─C bond homolysis from excited ligand field states. We further note that similar wavelength dependence trends have been demonstrated for Ni(II)─bpy mediated C─O cross-coupling product yields, directly implicating these high energy 1MLCT states in photoredox catalysis.24 Substituent-driven modulation of the critical PESs and bond homolysis rates may provide a synthetic handle to promote (or discourage) radical formation during catalysis.
In addition to the temperature and wavelength dependence, we have found that the rate constants of homolysis in Ni(II)─bpy aryl halide complexes are also sensitive to variations in both the solvent and the nature of all three ligands. For example, the rate constants of excited state Ni(II)─C bond homolysis for 1B and 1B–Br are slower in toluene and DMF (Figure S48-S51) relative to THF, suggesting a solvent influence on the excited state PESs. Solvent dependence has been observed for other light-induced homolysis reactions that feature activation energies, and similar arguments regarding perturbations to the barrier have been made.30-31 Furthermore, exchanging chloride with bromide (1B versus 1B–Br) increases the observed rate of homolysis from 2.5 ± 0.2 to 6.9 ± 0.4 x 10−2 min−1. The calculated barriers and excited state PESs for these two compounds are similar (Figure S70). However, changing Cl to Br increases the ligand-based spin-orbit coupling constant.64 This increased spin-orbit coupling may increase the rate of surface crossing and suggests spin-vibronic effects may be important to consider (vide infra).65-67
Furthermore, light-induced homolysis results in the production of an additional visible-light absorbing species (Section 2.1.4). Based on the correlation between the UV-vis spectrum of the intermediate formed from 1B–Br via photolysis to that identified recently by Bird et al.,53 we tentatively assign this species as a three-coordinate, formal Ni(I)(bpy)(Br) complex. While further study of this intermediate is outside the scope of this study, it is worth noting that, under controlled air- and moisture-free conditions, excited state homolysis provides a means to generate and isolate putative reactive intermediates for detailed studies of their spectroscopic properties and reactivity patterns. Thus, the rate of formation of these intermediates is tunable via structural and electronic control over key PESs, affording chemists new photon-driven synthetic possibilities in photoredox catalysis.68
Variation of the bpy ligand (columns in Figure 2) modifies the MLCT λmax (Figure 3B) and the rate constant of homolysis at a given excitation wavelength; there is in fact a linear correlation between the normalized logarithm of the rate constant and the σp of the bpy substituent (Figure 5). Increased electron withdrawing strength of the bpy lowers the energies of the MLCT excited state PES manifold (e.g., the MLCT of 1D is 8.1 kcal mol−1 lower relative to 1A, see Figure 12A). If the MLCT surface was the only excited state PES involved in the bond rupture mechanism, one would expect an increased barrier and an accordingly slower rate for homolysis in 1D than for 1A. However, when considering the barrier as governed by the crossing point between the MLCT and 3(MLCT+LMCT) states, the correct rate trend is recovered. Increasing the electron withdrawing strength of the bpy enables more facile aryl-to-Ni(III) charge transfer, lowering the LMCT energies. Furthermore, from the Hammett trends/slopes (Figure 5), we see a greater impact on the rate constant when modulating the aryl substituent (i.e., the LMCT component) versus the bpy substituent.
Figure 12.

Comparison between the change in energy of the MLCT λmax and (A) the experimental ln(kobs,1) obtained with 390 nm excitation and (B) the energetic barrier for photolysis for 1A–1D and 1B–5B. Barriers are estimated using the Arrhenius equation (assuming a uniform preexponential factor) and are normalized to the experimental value obtained for 1B. (C) PES diagram illustrating how the barrier for photolysis is dependent on both the 1MLCT and repulsive 3(MLCT+LMCT) surfaces. Full detailed PESs for 1A-5D are given in Figures S68-S69.
Thus, while the energetic shifts of the 1MLCT and 3(MLCT+LMCT) PESs are of the same sign, they shift at different rates as a function of ligand perturbation. The ligand-based effects on the 3(MLCT+LMCT) repulsive excited state outweigh the drop in the initial MLCT energy. Ultimately, these effects result in a lower barrier for homolysis (e.g., 1D has a barrier 1.4 kcal mol−1 lower than that for 1A, Figure 12B) and, thus, a larger rate constant.
The ligand-based perturbations to the excited state PESs described in Figure 12C demonstrate that the coupled changes to the overall excited state PES manifold, including surface intersections and barriers, govern the barriers and rates of Ni(II)─C bond homolysis.
Electronic structure calculations have provided further insights into these experimental observations, successfully correlating the calculated PES features to the experimental rate constants by capturing the critical aspects that lead to geometric and electronic structure control over the excited state PES manifolds (Figure 9). Also, both TD-DFT and CASSCF/QD-NEVPT2 methods predict analogous mechanisms that exploit ‘one-photon, two-electron’ repulsive 3(MLCT+LMCT) excited state PESs. In particular, Figure 11 demonstrates a strong correlation between the computed excited state activation energies and the experimental ln(kobs,1) obtained using 390 nm excitation. The activation energies are predicted to arise from the surface crossing between the high-energy 1MLCT [d(yz) → π*(2)] PES and the two-electron triplet repulsive surface, 3(MLCT+LMCT). Thus, the calculations are consistent with the overall experimental data and mechanism presented in Figure 12C.
Further comment on the specific mode of entry into the repulsive 3(MLCT+LMCT) state is warranted. Given that the UV-vis electronic absorption spectra of Ni(II)─bpy compounds exhibit dominantly MLCT intensity, there is a minimum energy threshold for homolysis, and the calculated ligand field transitions are lower than photochemically active higher-energy MLCT excited states, we posit that MLCT surfaces provide an entry into the 3(MLCT+LMCT) state. However, we do not know if intersystem crossing occurs between 1,3MLCT states or the 3(MLCT+LMCT) states. It may also be the case that vibronic coupling between charge transfer and ligand field states may be an important component of the mechanism (vide infra).67 Therefore, experimental determination of the energies of the ligand field transitions across these Ni(II)─bpy aryl halide complexes would be informative. If the 1(d-d) transitions are indeed experimentally obscured by the dominant, photochemically active MLCT intensity, electronic absorption will not be appropriate. Due to the C-term intensity mechanism, low temperature optical magnetic circular dichroism (MCD) would be ideal for this. However, the diamagnetic ground states of these complexes will also likely complicate ligand field assignments. As an alternative, 2p3d resonant inelastic X-ray scattering (RIXS) is a less commonly employed, but powerful, methodology to obtain spectroscopic insights into the spin-allowed and -forbidden ligand field excited state manifolds.69-74 This technique represents a potential approach to observe the ligand field excited state manifold in the presence of charge transfer bands, which will provide important insight into potential 1(d-d) states as a mode of entry to a dissociative 3LMCT state.51
Ni(II)─bpy aryl halide complexes feature a high density of excited states (Figure S68, Tables S10-11), many vibrational degrees of freedom, and large spin-orbit coupling. The combination of these factors can complicate mechanistic analyses that feature discrete processes of vibrational relaxation, internal conversion, and intersystem crossing, especially when they occur on the timescale of molecular vibrations (i.e., sub-picosecond timescale).39,67 Thus, the ultrafast spectroscopic characterization of the homolytically active state in Ni(II)─bpy aryl halide complexes may be hampered by the nature of the excited state homolysis mechanism. That is, the spectral dynamics will be dominated by MLCT and ligand field states, and there may be little to no dynamics detectable for the repulsive 3(MLCT+LMCT) surfaces proposed here, unless they can be accessed in sufficient amounts, perhaps with high energy excitation. Additionally, it may be that the TD-DFT and multiconfigurational/multireference mechanistic pictures provided by the calculated PESs need to be further supplemented to account for more complex aspects of spin-vibronic coupling.67 Further experimental characterization of aspects such as the specific state that provides entry to the repulsive 3(MLCT+LMCT) states, as well as more detailed experimental characterization of potential spin-vibronic coupling effects will be useful for better understanding how they manifest in the mechanism of excited state bond homolysis.
Finally, it is instructive to note that findings reported here resemble results from detailed photochemical investigations of d6 Re-complexes, Re(R)(CO)3(α–diimine), where R = Me, Et, Bz.30-38 For these complexes, it was found that the excited state Re─R bond homolysis is a result of the formation of a repulsive σπ* excited state. In the case of the Re systems, the σπ* excited state refers to an electronic configuration in which one electron is added to a π* orbital of the α-diimine ligand (1MLCT) and one electron is depleted from the Re─R bond (LMCT). The final dissociative state is not accessible via direct excitation, but is instead accessed from excitation of the 1MLCT state followed by the LMCT. Moreover, excited state homolysis in the Re(Me)(CO)3(α–diimine) complex occurs on the sub-picosecond time scale with a small quantum yield, is dependent on temperature and excitation wavelength, and has activation energies between ~1.8–5.5 kcal mol−1 using 458–502 nm excitation (~21,830–19,920 cm−1).31 These photophysical properties and excited state barriers are in the range of those determined for the Ni(II)─bpy aryl halide complexes studied here and are indicative of a dissociative excited state that is higher in energy than the 1MLCT excited state, creating a barrier and surface crossing (Figure 12A-B). It was further noted in the Re-complex that enhanced quantum yields correlated with increasing energy of excitation; this behavior mirrors the rate accelerations we observed herein upon increased excitation energy (Figure S25). This observation was interpreted as arising from excitation into higher energy vibronic levels of the 1MLCT state, which promotes the 1MLCT → σπ* surface hopping.30,36 Based on these similarities, in particular the increased homolysis rate constants with increasing energy of excitation into the higher energy MLCT transitions (Figure 8), we hypothesize that the mechanism of excited state homolysis in Ni(II)─bpy aryl halide complexes may indeed feature a similar vibronically-mediated component, wherein the optically excited 1MLCT is vibronically coupled to dissociative 3(MLCT+LMCT) states, or even potentially to weakly absorbing ligand field excited states as observed by Park et al.51 Overall, we highlight Re-like reactivity in Ni-catalysts, adapting the excited state dynamics of third-row transition metal complexes to first-row, earth abundant metal-based systems.
4. CONCLUSIONS
In summary, we have demonstrated that rate constants of excited state Ni(II)─C bond homolysis are temperature- and wavelength-dependent. Both of these experimental observations point to a thermal barrier involved in the photophysical mechanism. The barrier is moderate and inconsistent with thermally-driven homolysis from a low energy ligand field excited state. Additionally, we have demonstrated a linear correlation between bpy and aryl perturbations and the observed rate constants of homolysis using a consistent energy of irradiation. In this way, pinning the excitation wavelength reveals the ligand-induced electronic perturbations to the energetic barrier for Ni(II)─C bond homolysis by controlling the key excited state MLCT/LMCT surfaces. In accordance with this, electronic structure calculations predict a mechanism that exploits ‘one-photon, two-electron’ repulsive 3(MLCT+LMCT) excited state PESs and reveal a strong correlation between the computed excited state activation energies and the experimental ln(kobs,1) obtained using 390 nm excitation. The activation energies are predicted to arise from the surface crossing between the high-energy 1MLCT [d(yz) → π*(2)] and the two-electron triplet repulsive surfaces, highlighting the specific excited state PESs that contribute to Ni(II)─bpy-mediated photoredox catalysis. This study provides insights into the electronic structural control over light-induced homolysis and, thus, guides the use of photonic energy as a sustainable alternative to coupling catalysis as carried out by precious metals.
Supplementary Material
ACKNOWLEDGMENTS
D.A.C. is a National Science Foundation Graduate Research Fellow (DGE-1745301) and is supported by a National Academies of Science, Engineering, and Medicine Ford Foundation Predoctoral Fellowship. B.S. acknowledges funding through a Southern California Edison WAVE fellowship at Caltech. N.P.K. acknowledges support from the Hertz Fellowship and from the National Science Foundation Graduate Research Fellowship under Grant No. DGE-1745301. This project has received funding from the European Union’s Horizon 2020 research and innovation programme under the Marie Sklodowska-Curie grant agreement No. 883987 (D.B.). Support has been provided by the National Institutes of Health (National Institute of General Medical Sciences, R35─GM142595). We also acknowledge M. K. Takase in the Beckman Institute X-ray crystallography facility. The computations presented here were conducted in the Resnick High Performance Computing Center, a facility supported by Resnick Sustainability Institute at the California Institute of Technology.
Footnotes
SUPPORTING INFORMATION
The Supporting Information is available free of charge on the ACS Publications website at DOI: . Experimental and computational methods, UV-vis/photochemical data, X-ray crystallography, NMR spectra, calculated spectra/properties, global analysis modeling, and additional comments.
REFERENCES
- (1).Nicewicz DA; MacMillan DWC Merging Photoredox Catalysis with Organocatalysis: The Direct Asymmetric Alkylation of Aldehydes. Science 2008, 322 (5898), 77–80. 10.1126/science.1161976. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (2).Shaw MH; Twilton J; MacMillan DWC Photoredox Catalysis in Organic Chemistry. J. Org. Chem 2016, 81 (16), 6898–6926. 10.1021/acs.joc.6b01449. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (3).Cavalcanti LN; Molander GA Photoredox Catalysis in Nickel-Catalyzed Cross-Coupling. Top. Curr. Chem 2016, 374 (4), 39. 10.1007/s41061-016-0037-z. [DOI] [PubMed] [Google Scholar]
- (4).Till NA; Tian L; Dong Z; Scholes GD; MacMillan DWC Mechanistic Analysis of Metallaphotoredox C─N Coupling: Photocatalysis Initiates and Perpetuates Ni(I)/Ni(III) Coupling Activity. J. Am. Chem. Soc 2020, 142 (37), 15830–15841. 10.1021/jacs.0c05901. [DOI] [PubMed] [Google Scholar]
- (5).Qin Y; Sun R; Gianoulis NP; Nocera DG Photoredox Nickel-Catalyzed C─S Cross-Coupling: Mechanism, Kinetics, and Generalization. J. Am. Chem. Soc 2021, 143 (4), 2005–2015. 10.1021/jacs.0c11937. [DOI] [PubMed] [Google Scholar]
- (6).Huang L; Ji T; Rueping M Remote Nickel-Catalyzed Cross-Coupling Arylation via Proton-Coupled Electron Transfer-Enabled C─C Bond Cleavage. J. Am. Chem. Soc 2020, 142 (7), 3532–3539. 10.1021/jacs.9b12490. [DOI] [PubMed] [Google Scholar]
- (7).Ackerman LKG; Martinez Alvarado JI; Doyle AG Direct C─C Bond Formation from Alkanes Using Ni-Photoredox Catalysis. J. Am. Chem. Soc 2018, 140 (43), 14059–14063. 10.1021/jacs.8b09191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (8).Kim T; McCarver SJ; Lee C; MacMillan DWC Sulfonamidation of Aryl and Heteroaryl Halides through Photosensitized Nickel Catalysis. Angew. Chem. Int. Ed 2018, 57 (13), 3488–3492. 10.1002/anie.201800699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (9).Zhang R; Li G; Wismer M; Vachal P; Colletti SL; Shi Z-C Profiling and Application of Photoredox C(sp3)─C(sp2) Cross-Coupling in Medicinal Chemistry. ACS Med. Chem. Lett 2018, 9 (7), 773–777. 10.1021/acsmedchemlett.8b00183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (10).Schultz DM; Yoon TP Solar Synthesis: Prospects in Visible Light Photocatalysis. Science 2014, 343 (6174), 1239176. 10.1126/science.1239176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (11).Arias-Rotondo DM; McCusker JK. The Photophysics of Photoredox Catalysis: A Roadmap for Catalyst Design. Chem. Soc. Rev 2016, 45 (21), 5803–5820. 10.1039/C6CS00526H. [DOI] [PubMed] [Google Scholar]
- (12).Larsen CB; Wenger OS Photoredox Catalysis with Metal Complexes Made from Earth-Abundant Elements. Chem. – Eur. J 2018, 24 (9), 2039–2058. 10.1002/chem.201703602. [DOI] [PubMed] [Google Scholar]
- (13).Yoon TP; Ischay MA; Du J Visible Light Photocatalysis as a Greener Approach to Photochemical Synthesis. Nat. Chem 2010, 2 (7), 527–532. 10.1038/nchem.687. [DOI] [PubMed] [Google Scholar]
- (14).Chan AY; Perry IB; Bissonnette NB; Buksh BF; Edwards GA; Frye LI; Garry OL; Lavagnino MN; Li BX; Liang Y; Mao E; Millet A; Oakley JV; Reed NL; Sakai HA; Seath CP; MacMillan DWC Metallaphotoredox: The Merger of Photoredox and Transition Metal Catalysis. Chem. Rev 2021. 10.1021/acs.chemrev.1c00383. [DOI] [PubMed] [Google Scholar]
- (15).Diccianni JB; Diao T Mechanisms of Nickel-Catalyzed Cross-Coupling Reactions. Trends Chem. 2019, 1 (9), 830–844. 10.1016/j.trechm.2019.08.004. [DOI] [Google Scholar]
- (16).Ting SI; Garakyaraghi S; Taliaferro CM; Shields BJ; Scholes GD; Castellano FN; Doyle AG 3d-d Excited States of Ni(II) Complexes Relevant to Photoredox Catalysis: Spectroscopic Identification and Mechanistic Implications. J. Am. Chem. Soc 2020, 142 (12), 5800–5810. 10.1021/jacs.0c00781. [DOI] [PubMed] [Google Scholar]
- (17).Yuan M; Song Z; Badir SO; Molander GA; Gutierrez O On the Nature of C(sp3)─C(sp2) Bond Formation in Nickel-Catalyzed Tertiary Radical Cross-Couplings: A Case Study of Ni/Photoredox Catalytic Cross-Coupling of Alkyl Radicals and Aryl Halides. J. Am. Chem. Soc 2020, 142 (15), 7225–7234. 10.1021/jacs.0c02355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (18).Gonzalez MI; Gygi D; Qin Y; Zhu Q; Johnson EJ; Chen Y-S; Nocera DG Taming the Chlorine Radical: Enforcing Steric Control over Chlorine-Radical-Mediated C─H Activation. J. Am. Chem. Soc 2022, 144 (3), 1464–1472. 10.1021/jacs.1c13333. [DOI] [PubMed] [Google Scholar]
- (19).Treacy SM; Rovis T Copper Catalyzed C(sp3)─H Bond Alkylation via Photoinduced Ligand-to-Metal Charge Transfer. J. Am. Chem. Soc 2021, 143 (7), 2729–2735. 10.1021/jacs.1c00687. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (20).Welin ER; Le C; Arias-Rotondo DM; McCusker JK; MacMillan DWC Photosensitized, Energy Transfer-Mediated Organometallic Catalysis through Electronically Excited Nickel(II). Science 2017, 355 (6323), 380–385. 10.1126/science.aal2490. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (21).Strieth-Kalthoff F; Glorius F Triplet Energy Transfer Photocatalysis: Unlocking the Next Level. Chem 2020, 6 (8), 1888–1903. 10.1016/j.chempr.2020.07.010. [DOI] [Google Scholar]
- (22).Sun R; Qin Y; Ruccolo S; Schnedermann C; Costentin C; Nocera DG Elucidation of a Redox-Mediated Reaction Cycle for Nickel-Catalyzed Cross Coupling. J. Am. Chem. Soc 2019, 141 (1), 89–93. 10.1021/jacs.8b11262. [DOI] [PubMed] [Google Scholar]
- (23).Shields BJ; Kudisch B; Scholes GD; Doyle AG Long-Lived Charge-Transfer States of Nickel(II) Aryl Halide Complexes Facilitate Bimolecular Photoinduced Electron Transfer. J. Am. Chem. Soc 2018, 140 (8), 3035–3039. 10.1021/jacs.7b13281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (24).Yang L; Lu H-H; Lai C-H; Li G; Zhang W; Cao R; Liu F; Wang C; Xiao J; Xue D Light-Promoted Nickel Catalysis: Etherification of Aryl Electrophiles with Alcohols Catalyzed by a NiII-Aryl Complex. Angew. Chem. Int. Ed 2020, 59 (31), 12714–12719. 10.1002/anie.202003359. [DOI] [PubMed] [Google Scholar]
- (25).Tian L; Till NA; Kudisch B; MacMillan DWC; Scholes GD Transient Absorption Spectroscopy Offers Mechanistic Insights for an Iridium/Nickel-Catalyzed C─O Coupling. J. Am. Chem. Soc 2020, 142 (10), 4555–4559. 10.1021/jacs.9b12835. [DOI] [PubMed] [Google Scholar]
- (26).Ma P; Wang S; Chen H Reactivity of Transition-Metal Complexes in Excited States: C─O Bond Coupling Reductive Elimination of a Ni(II) Complex Is Elicited by the Metal-to-Ligand Charge Transfer State. ACS Catal. 2020, 10 (1), 1–6. 10.1021/acscatal.9b03827. [DOI] [Google Scholar]
- (27).Han R; Hillhouse GL Carbon─Oxygen Reductive-Elimination from Nickel(II) Oxametallacycles and Factors That Control Formation of Ether, Aldehyde, Alcohol, or Ester Products. J. Am. Chem. Soc 1997, 119 (34), 8135–8136. 10.1021/ja9714999. [DOI] [Google Scholar]
- (28).Koo K; Hillhouse GL Carbon-Nitrogen Bond Formation by Reductive Elimination from Nickel(II) Amido Alkyl Complexes. Organometallics 1995, 14 (9), 4421–4423. 10.1021/om00009a054. [DOI] [Google Scholar]
- (29).Cagan DA; Stroscio GD; Cusumano AQ; Hadt RG Multireference Description of Nickel─Aryl Homolytic Bond Dissociation Processes in Photoredox Catalysis. J. Phys. Chem. A 2020, 124 (48), 9915–9922. 10.1021/acs.jpca.0c08646. [DOI] [PubMed] [Google Scholar]
- (30).Kleverlaan CJ; Stufkens DJ; Clark IP; George MW; Turner JJ; Martino DM; van Willigen H; Vlček A Photoinduced Radical Formation from the Complexes [Re(R)(CO)3(4,4′-Me2-bpy)] (R = CH3, CD3, Et, iPr, Bz): A Nanosecond Time-Resolved Emission, UV–Vis and IR Absorption, and FT-EPR Study. J. Am. Chem. Soc 1998, 120 (42), 10871–10879. 10.1021/ja980540k. [DOI] [Google Scholar]
- (31).Rossenaar BD; Kleverlaan CJ; Van De Ven MCE; Stufkens DJ; Vlček A Jr Mechanism of an Alkyl-Dependent Photochemical Homolysis of the Re─Alkyl Bond in [Re(R)(CO)3(α-diimine)] Complexes via a Reactive σπ* Excited State. Chem. – Eur. J 1996, 2 (2), 228–237. 10.1002/chem.19960020216. [DOI] [Google Scholar]
- (32).Rossenaar BD; Kleverlaan CJ; Stufkens DJ; Oskam A Photochemistry of ReR(CO)3(Pri-Dab)(R = Me, Et, Bn; Dab = 1,4-Diazabuta-1,3-Diene): Homolysis of the Re─R Bond, Its Dependence on R and Evidence for the Reactive σbπ* State from Transient Absorption Spectra. J. Chem. Soc. Chem. Commun 1994, No. 1, 63–64. 10.1039/C39940000063. [DOI] [Google Scholar]
- (33).Stufkens DJ; Vlček A Ligand-Dependent Excited State Behaviour of Re(I) and Ru(II) Carbonyl─Diimine Complexes. Coord. Chem. Rev 1998, 177 (1), 127–179. 10.1016/S0010-8545(98)00132-5. [DOI] [Google Scholar]
- (34).Weinstein JA; van Slageren J; Stufkens DJ; Záliš S; George MW, A Time-Resolved Infrared Spectroscopic Study of [M(SnR3)2(CO)2(α-Diimine)] (M = Ru, Os; R = Ph, Me): Evidence of Charge Redistribution in the Lowest-Excited State. J. Chem. Soc. Dalton Trans 2001, 18, 2587–2592. 10.1039/B102450G. [DOI] [Google Scholar]
- (35).Stufkens DJ; Aarnts MP; Rossenaar BD; Vlcek A A new series of Re- and Ru-complexes having a lowest σπ* excited state that varies from reactive to stable and long lived. Pure Appl. Chem 1997, 69 (4), 831–836. 10.1351/pac199769040831. [DOI] [Google Scholar]
- (36).Aarnts MP; Wilms MP; Stufkens DJ; Baerends EJ; Vlček A σ─π* Electronic Transition of the Di- and Trinuclear Complexes Ru(E)(E′)(CO)2(iPr-DAB): Resonance Raman, Electronic Absorption, Emission, and Density Functional Study (E = Me, SnPh3, M(CO)5; E′ = M(CO)5; M = Mn, Re; iPr-DAB = N,N’-Diisopropyl-1,4-Diaza-1,3-Butadiene). Organometallics 1997, 16 (10), 2055–2062. 10.1021/om9608667. [DOI] [Google Scholar]
- (37).Nieuwenhuis HA; van de Ven MCE; Stufkens DJ; Oskam A; Goubitz K Photochemistry of [Ru(I)(iPr)(CO)2(iPr-DAB)] (iPr-DAB = N,N’-Diisopropyl-1,4-Diaza-1,3-Butadiene): Homolysis of the Metal-Alkyl Bond from the σb(Ru-iPr)π* State. Crystal Structure of the Photoproduct [Ru(I)2(CO)2(iPr-DAB)]. Organometallics 1995, 14 (2), 780–788. 10.1021/om00002a027. [DOI] [Google Scholar]
- (38).Rossenaar BD; George MW; Johnson FPA; Stufkens DJ; Turner JJ; Vlcek A First Direct Structural Information on a Reactive σπ* Excited State: Time-Resolved UV-Vis and IR Spectroscopic Study of Re(Benzyl)(CO)3(iPr-DAB). J. Am. Chem. Soc 1995, 117 (46), 11582–11583. 10.1021/ja00151a025. [DOI] [Google Scholar]
- (39).Vlček A; Víchová J; Hartl F Bond Activation by MLCT Excitation of Organometallic Compounds: Prompt CO-Photodissociation from [Cr(CO)4bpy]. Coord. Chem. Rev 1994, 132, 167–174. 10.1016/0010-8545(94)80037-5. [DOI] [Google Scholar]
- (40).Shields JD; Gray EE; Doyle AG A Modular, Air-Stable Nickel Precatalyst. Org. Lett 2015, 17 (9), 2166–2169. 10.1021/acs.orglett.5b00766. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (41).Marshall WJ; Grushin VV Activation of Chlorobenzene with Ni(0) N,N-Chelates – A Remarkably Profound Effect of a Minuscule Change in Ligand Structure. Can. J. Chem 2005, 83 (6–7), 640–645. 10.1139/v05-022. [DOI] [Google Scholar]
- (42).Hansch Corwin.; Leo, A.; Taft, R. W. A Survey of Hammett Substituent Constants and Resonance and Field Parameters. Chem. Rev 1991, 91 (2), 165–195. 10.1021/cr00002a004. [DOI] [Google Scholar]
- (43).Magenau AJD; Kwak Y; Schröder K; Matyjaszewski K Highly Active Bipyridine-Based Ligands for Atom Transfer Radical Polymerization. ACS Macro Lett. 2012, 1 (4), 508–512. 10.1021/mz3000489. [DOI] [PubMed] [Google Scholar]
- (44).Clark ML; Cheung PL; Lessio M; Carter EA; Kubiak CP Kinetic and Mechanistic Effects of Bipyridine (Bpy) Substituent, Labile Ligand, and Brønsted Acid on Electrocatalytic CO2 Reduction by Re(bpy) Complexes. ACS Catal. 2018, 8 (3), 2021–2029. 10.1021/acscatal.7b03971. [DOI] [Google Scholar]
- (45).Sinnokrot MO; Sherrill CD Substituent Effects in Π–π Interactions: Sandwich and T-Shaped Configurations. J. Am. Chem. Soc 2004, 126 (24), 7690–7697. 10.1021/ja049434a. [DOI] [PubMed] [Google Scholar]
- (46).Ringer AL; Sinnokrot MO; Lively RP; Sherrill CD The Effect of Multiple Substituents on Sandwich and T-Shaped π–π Interactions. Chem. – Eur. J 2006, 12 (14), 3821–3828. 10.1002/chem.200501316. [DOI] [PubMed] [Google Scholar]
- (47).Gung BW; Patel M; Xue X A Threshold for Charge Transfer in Aromatic Interactions? A Quantitative Study of π-Stacking Interactions. J. Org. Chem 2005, 70 (25), 10532–10537. 10.1021/jo051808a. [DOI] [PubMed] [Google Scholar]
- (48).Wheeler SE; Houk KN Substituent Effects in the Benzene Dimer Are Due to Direct Interactions of the Substituents with the Unsubstituted Benzene. J. Am. Chem. Soc 2008, 130 (33), 10854–10855. 10.1021/ja802849j. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (49).Santiago CB; Milo A; Sigman MS Developing a Modern Approach To Account for Steric Effects in Hammett-Type Correlations. J. Am. Chem. Soc 2016, 138 (40), 13424–13430. 10.1021/jacs.6b08799. [DOI] [PubMed] [Google Scholar]
- (50).Lewis M; Bagwill C; Hardebeck LKE; Wireduaah S The Use of Hammett Constants to Understand the Non-Covalent Binding of Aromatics. Comput. Struct. Biotechnol. J 2012, 1, e201204004. 10.5936/csbj.201204004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (51).Shin J; Lee J; Suh J-M; Park K Ligand-Field Transition-Induced C─S Bond Formation from Nickelacycles. Chem. Sci 2021, 12 (48), 15908–15915. 10.1039/D1SC05113J. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (52).Stadler E; Eibel A; Fast D; Freißmuth H; Holly C; Wiech M; Moszner N; Gescheidt G A Versatile Method for the Determination of Photochemical Quantum Yields via Online UV-Vis Spectroscopy. Photochem. Photobiol. Sci 2018, 17 (5), 660–669. 10.1039/C7PP00401J. [DOI] [PubMed] [Google Scholar]
- (53).Till NA; Oh S; MacMillan DWC; Bird MJ The Application of Pulse Radiolysis to the Study of Ni(I) Intermediates in Ni-Catalyzed Cross-Coupling Reactions. J. Am. Chem. Soc 2021, 143 (25), 9332–9337. 10.1021/jacs.1c04652. [DOI] [PubMed] [Google Scholar]
- (54).Mohadjer Beromi M; Brudvig GW; Hazari N; Lant HMC; Mercado BQ Synthesis and Reactivity of Paramagnetic Nickel Polypyridyl Complexes Relevant to C(sp2)─C(sp3)Coupling Reactions. Angew. Chem. Int. Ed 2019, 58 (18), 6094–6098. 10.1002/anie.201901866. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (55).Roos BO; Taylor PR; Sigbahn PEM A Complete Active Space SCF Method (CASSCF) Using a Density Matrix Formulated Super-CI Approach. Chem. Phys 1980, 48 (2), 157–173. 10.1016/0301-0104(80)80045-0. [DOI] [Google Scholar]
- (56).Siegbahn PEM; Almlöf J; Heiberg A; Roos BO The Complete Active Space SCF (CASSCF) Method in a Newton–Raphson Formulation with Application to the HNO Molecule. J. Chem. Phys 1981, 74 (4), 2384–2396. 10.1063/1.441359. [DOI] [Google Scholar]
- (57).Angeli C; Borini S; Cestari M; Cimiraglia R A Quasidegenerate Formulation of the Second Order N-Electron Valence State Perturbation Theory Approach. J. Chem. Phys 2004, 121 (9), 4043–4049. 10.1063/1.1778711. [DOI] [PubMed] [Google Scholar]
- (58).Lang L; Sivalingam K; Neese F The Combination of Multipartitioning of the Hamiltonian with Canonical Van Vleck Perturbation Theory Leads to a Hermitian Variant of Quasidegenerate N-Electron Valence Perturbation Theory. J. Chem. Phys 2020, 152 (1), 014109. 10.1063/1.5133746. [DOI] [PubMed] [Google Scholar]
- (59).Becke AD Density-functional Thermochemistry. III. The Role of Exact Exchange. J. Chem. Phys 1993, 98 (7), 5648–5652. 10.1063/1.464913. [DOI] [Google Scholar]
- (60).Lee C; Yang W; Parr RG Development of the Colle-Salvetti Correlation-Energy Formula into a Functional of the Electron Density. Phys. Rev. B 1988, 37 (2), 785–789. 10.1103/PhysRevB.37.785. [DOI] [PubMed] [Google Scholar]
- (61).Becke AD Density-Functional Exchange-Energy Approximation with Correct Asymptotic Behavior. Phys. Rev. A 1988, 38 (6), 3098–3100. 10.1103/PhysRevA.38.3098. [DOI] [PubMed] [Google Scholar]
- (62).Abderrazak Y; Bhattacharyya A; Reiser O Visible-Light-Induced Homolysis of Earth-Abundant Metal-Substrate Complexes: A Complementary Activation Strategy in Photoredox Catalysis. Angew. Chem. Int. Ed 2021, 60 (39), 21100–21115. 10.1002/anie.202100270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (63).Hwang SJ; Powers DC; Maher AG; Anderson BL; Hadt RG; Zheng S-L; Chen Y-S; Nocera DG Trap-Free Halogen Photoelimination from Mononuclear Ni(III) Complexes. J. Am. Chem. Soc 2015, 137 (20), 6472–6475. 10.1021/jacs.5b03192. [DOI] [PubMed] [Google Scholar]
- (64).Richards WG; Trivedi HP; Cooper DL Spin-Orbit Coupling in Molecules. In The International series of monographs on chemistry, Clarendon Press, 1981, pp 17. [Google Scholar]
- (65).Harvey JN Understanding the Kinetics of Spin-Forbidden Chemical Reactions. Phys. Chem. Chem. Phys 2007, 9 (3), 331–343. 10.1039/B614390C. [DOI] [PubMed] [Google Scholar]
- (66).Harvey JN Spin-Forbidden Reactions: Computational Insight into Mechanisms and Kinetics. WIREs Comput. Mol. Sci 2014, 4 (1), 1–14. 10.1002/wcms.1154. [DOI] [Google Scholar]
- (67).Penfold TJ; Gindensperger E; Daniel C; Marian CM Spin-Vibronic Mechanism for Intersystem Crossing. Chem. Rev 2018, 118 (15), 6975–7025. 10.1021/acs.chemrev.7b00617. [DOI] [PubMed] [Google Scholar]
- (68).Gisbertz S; Reischauer S; Pieber B Overcoming Limitations in Dual Photoredox/Nickel-Catalysed C─N Cross-Couplings Due to Catalyst Deactivation. Nat. Catal 2020, 3 (8), 611–620. 10.1038/s41929-020-0473-6. [DOI] [Google Scholar]
- (69).Wang R-P; Liu B; Green RJ; Delgado-Jaime MU; Ghiasi M; Schmitt T; van Schooneveld MM; de Groot FMF Charge-Transfer Analysis of 2p3d Resonant Inelastic X-Ray Scattering of Cobalt Sulfide and Halides. J. Phys. Chem. C 2017, 121 (45), 24919–24928. 10.1021/acs.jpcc.7b06882. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (70).Al Samarai M; Hahn AW; Beheshti Askari A; Cui Y-T; Yamazoe K; Miyawaki J; Harada Y; Rüdiger O; DeBeer S Elucidation of Structure-Activity Correlations in a Nickel Manganese Oxide Oxygen Evolution Reaction Catalyst by Operando Ni L-Edge X-Ray Absorption Spectroscopy and 2p3d Resonant Inelastic X-Ray Scattering. ACS Appl. Mater. Interfaces 2019, 11 (42), 38595–38605. 10.1021/acsami.9b06752. [DOI] [PubMed] [Google Scholar]
- (71).de Groot FMF; Fuggle JC; Thole BT; Sawatzky GA 2p X-Ray Absorption of 3d Transition-Metal Compounds: An Atomic Multiplet Description Including the Crystal Field. Phys. Rev. B 1990, 42 (9), 5459–5468. 10.1103/PhysRevB.42.5459. [DOI] [PubMed] [Google Scholar]
- (72).Van Kuiken BE; Hahn AW; Maganas D; DeBeer S Measuring Spin-Allowed and Spin-Forbidden d–d Excitations in Vanadium Complexes with 2p3d Resonant Inelastic X-Ray Scattering. Inorg. Chem 2016, 55 (21), 11497–11501. 10.1021/acs.inorgchem.6b02053. [DOI] [PubMed] [Google Scholar]
- (73).Hahn AW; Van Kuiken BE; Chilkuri VG; Levin N; Bill E; Weyhermüller T; Nicolaou A; Miyawaki J; Harada Y; DeBeer S Probing the Valence Electronic Structure of Low-Spin Ferrous and Ferric Complexes Using 2p3d Resonant Inelastic X-Ray Scattering (RIXS). Inorg. Chem 2018, 57 (15), 9515–9530. 10.1021/acs.inorgchem.8b01550. [DOI] [PubMed] [Google Scholar]
- (74).Hahn AW; Van Kuiken BE; al Samarai M; Atanasov M; Weyhermüller T; Cui Y-T; Miyawaki J; Harada Y; Nicolaou A; DeBeer S Measurement of the Ligand Field Spectra of Ferrous and Ferric Iron Chlorides Using 2p3d RIXS. Inorg. Chem 2017, 56 (14), 8203–8211. 10.1021/acs.inorgchem.7b00940. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.